
Abstract
Narcolepsy–cataplexy is a disabling sleep disorder marked by excessive daytime sleepiness and sudden, uncontrollable decreases in muscle tone (cataplexy). It is believed to be caused by the autoimmune destruction of hypocretin (orexin) neurons in the hypothalamus. Neuroimaging studies, both structural and functional, have investigated the neural correlates of narcolepsy using a variety of imaging modalities, namely, magnetic resonance imaging, magnetic resonance spectroscopy, positron emission tomography, single-photon emission computed tomography and functional magnetic resonance imaging. Hypothalamic damage is a common, though not ubiquitous, finding among anatomical and spectroscopic studies, in support of the hypocretinergic depletion theory of narcoleptic pathophysiology. Alterations to fronto-temporal cortices and the thalamus were also found in a number of anatomical studies, possibly relating to cognitive, mood and sleep disturbances. As for functional imaging, several blood flow and metabolic studies have revealed hypoactivation in the hypothalamus, thalamus and fronto-temporal cortices during resting wakefulness, in agreement with structural data, but functional data during sleep are lacking. Emotional reactivity was probed in several functional imaging paradigms, the results of which converge with anatomical findings of hypothalamic and amygdalar changes in supporting an altered emotional network response in narcolepsy, possibly underlying cataplectic episodes. Lastly, extensive research has examined neurotransmission in narcolepsy, mostly dopaminergic, but no consistent difference was found between narcoleptic patients and controls. Overall, neuroimaging findings in narcolepsy constitute a substantial contribution to our knowledge of the pathophysiology of the disease.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Narcolepsy
- Cataplexy
- Neuroimaging
- Magnetic resonance imaging
- Magnetic resonance spectroscopy
- Positron emission tomography
- Single photon emission computed tomography
- Functional magnetic resonance imaging
- Hypocretin (orexin)
Introduction
Narcolepsy is a life-long, debilitating sleep disorder characterized by excessive daytime sleepiness. Most often, it is also accompanied by cataplectic attacks , which are sudden, transient decreases in muscle tone that can be triggered by strong, usually positive emotions (such as laughter or pleasant surprise). Another core feature of narcolepsy is early entry into rapid eye movement (REM) sleep after sleep onset, known as sleep-onset REM periods (SOREMPs). REM sleep-like phenomena may also intrude in the narcoleptic waking state, such as hypnagogic hallucination and sleep paralysis [1]. Nocturnal sleep patterns are typically fragmented in narcolepsy, such that individuals with narcolepsy are chronically sleep-deprived and take frequent naps throughout the day, which are refreshing only for a few hours. When these naps occur uncontrollably, they are known as sleep attacks. Their daily occurrence in narcolepsy impedes quality of life and increases the risk of accidents [2].
The diagnosis of narcolepsy is usually based on clinical interview and polysomnography (PSG ) coupled with the multiple sleep latency test (MLST), during which the patient is invited to take 5 daytime naps in succession [3]. Rapid sleep onset and two or more SOREMPs during naps, as measured by PSG/MSLT , are suggestive of narcolepsy. The prevalence of narcolepsy follows a bimodal age distribution at 15 and 35 years old and affects males and females equally [4]. When first detected, symptoms can be mistaken for epilepsy or depression [1]. Treatment options mostly consist of central nervous system stimulants (such as methylphenidate and modafinil), but antidepressants and sodium oxybate are sometimes administered, especially in cases with frequent cataplexy [5]. These medications are purely symptomatic and do not provide a cure for the disease.
Early research into the pathophysiology of narcolepsy identified a genetic autoimmune factor ; blood typing revealed that 90 % of narcoleptic patients with cataplexy possessed the DQB1*0602 subtype of the human leukocyte antigen (HLA) [6]. More recently, narcolepsy has been linked to a central deficiency in hypocretin (otherwise known as orexin) [7–10], as evidenced by low levels of the neuropeptide (specifically, hypocretin-1) in the cerebrospinal fluid (CSF) [11]. Since hypocretin-1 neurons are implicated in the arousal system [12, 13] and control motor function (e.g. muscle tone) [14], their deficiency might explain the abnormal sleep–wake patterns and sleep attacks that are characteristic of narcolepsy. One major hypothesis today is thus that narcolepsy with cataplexy is an autoimmune disease, associated with the HLA-DQB1*0602 genotype, specifically targeting hypocretin-secreting cells situated mainly in the lateral hypothalamus [1].
A growing body of literature is now using neuroimaging techniques to reveal the neural correlates of narcolepsy and further inform its neurological causes and consequences. Structural neuroimaging using magnetic resonance imaging (MRI), in concert with analytical techniques such as voxel-based morphometry (VBM) and diffusion tensor imaging (DTI ), has identified alterations in brain anatomy that are associated with the disorder (see Table 13.1). Large-scale metabolic changes have been probed by magnetic resonance spectroscopy (MRS) (Table 13.1). Functional neuroimaging studies have examined changes in brain activity during wakefulness and cataplexy in narcoleptic patients, using positron emission tomography (PET), single-photon emission computed tomography (SPECT) and functional MRI (fMRI ) (see Table 13.2). The present review will synthesize the findings of these neuroimaging studies of narcolepsy. Neuroimaging has also been used to assess the effect of therapeutic drugs on the narcoleptic brain; these studies exceed the scope of this chapter and are reviewed elsewhere [15].
Structural Imaging in Narcolepsy
Grey Matter Alterations
Early etiological theories of narcolepsy proposed that an impairment of the pontine tegmentum, which is involved in the modulation of vigilance states, is at the root of narcoleptic pathophysiology. Anatomical alterations of the pontine tegmentum were reported in a first MRI study of three narcoleptic patients by Plazzi and colleagues [16], but subsequent MRI studies did not reproduce these results [17, 18]. In one of these latter studies [18], pontine lesions were detected in 2 of 12 narcoleptic patients, but these patients suffered from long-standing hypertension, and the lesions were indistinguishable from ischemic damage. Similar age-related pontine lesions have been reported by Pullicino and colleagues [19], and so it may be that the results of Plazzi’s study were also due to age-related cardiovascular conditions.
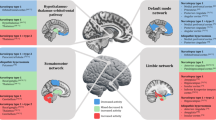
Whereas early studies visually assessed anatomical changes in MRIs of narcoleptic patients, more recent studies have used VBM to systematically quantify small-scale structural alterations in grey matter density (Fig. 13.1). Among these, several reported grey matter volume decreases in the hypothalamus [20–23], in line with the hypocretinergic dysfunction theory of narcoleptic pathophysiology. In contrast, an equal number of VBM studies found no hypothalamic differences between narcoleptic patients and healthy controls [24–27]. Hypothalamic grey matter reductions in narcoleptic patients were also correlated with scores on a subjective scale of disease severity, the Ullanlinna Narcolepsy Scale [23]. Other notable brain areas showing decreased volume in narcolepsy were the nucleus accumbens [21, 22], a major projection site of hypocretinergic neurons, and the thalamus [22, 23], which is an important modulator of sleep oscillations. Fronto-temporal cortices have also shown consistent decreases in several VBM studies [21–23, 25–27]. This result has been interpreted as mirroring attentional deficits reported by narcoleptic patients, but this remains speculative. Widespread volumetric decreases in the narcoleptic brain may together reflect secondary neuronal loss from the destruction of afferent hypocretinergic neurons in the hypothalamus. However, global grey matter reduction was not correlated with disease duration [25]. A single VBM study reported no significant structural difference between narcoleptic patients and controls [24]. Divergent findings among VBM studies may be due to differences in data preprocessing among studies, as well as patient sample heterogeneities (e.g. age, disease severity).

Grey matter volumetric alterations in narcolepsy. VBM and cortical thickness studies have most consistently shown grey matter decreases in the hypothalamus [20–23] and fronto-mesial regions [22, 23, 26, 28]. Other cortical areas showing consistent volumetric decreases are the cingulate [23, 27, 28], inferior temporal [25, 27, 28] and orbitofrontal [25, 28, 29] cortices. One study found a volumetric increase in narcoleptic patients with respect to healthy controls, in the dorsolateral prefrontal cortex [29]. Prepared with illustrations by Patrick J. Lynch and C. Carl Jaffe. http://creativecommons.org/licenses/by/2.5/
Cortical thickness measures have also been applied to MRI scans to detect subtle changes in the narcoleptic cerebral cortex compared to controls (Fig. 13.1). A first study found cortical thinning in the cingulate, inferior parietal and fronto-temporal cortices [28], while a second study showed thinning in the paracentral lobule and the orbitofrontal cortex [29]. Additionally, thinning of the inferior parietal and orbitofrontal cortices was inversely correlated with subjective disease severity in the respective studies. These results are in accordance with the aforementioned VBM data showing grey matter decrements in fronto-temporal cortices, possibly related to narcoleptic cognitive dysfunction. Interestingly, the latter study also detected a cortical thickening of the dorsolateral prefrontal cortex in narcoleptic patients with respect to controls; these data were instead proposed to reflect compensatory activity in reaction to cognitive challenges. Also of interest, global cortical thinning was greater for early-onset (<16 y.o.) than for late-onset (>16 y.o.) narcoleptic patients, which may represent a chronic effect of the disease or, alternatively, a differentiation between two etiological subtypes of narcolepsy [29]. The global cortical decrements observed in structural studies led Joo and colleagues [30] to hypothesize that memory function may be affected in narcolepsy, and they turned their attention to the hippocampus. Using manual volumetry, they determined that hippocampal volumes were indeed smaller in a sample of 36 narcoleptic patients, even though their visual or verbal memory performances were not significantly different from 36 age- and sex-matched controls (Fig. 13.1). Furthermore, shorter sleep and REM sleep latencies on the MSLT (which are objective markers of disease severity) were associated with smaller hippocampal volume in narcoleptic patients. Narcolepsy may thus have a detrimental effect on cognition in the hippocampus and fronto-temporal cortices, which is masked by compensation from other brain areas, such as the dorsolateral prefrontal cortex. Manual volumetry was also applied to describe structural changes in the amygdala [31] (Fig. 13.1). This MRI study found that the amygdala was reduced in volume in narcoleptic patients, which is possibly related to emotional dysregulation in narcolepsy (see functional studies below).
White Matter Alterations
Microstructural changes in axonal integrity can be quantified from MRI scans using DTI . This analytical technique is able to track the direction of diffusion of water molecules through the brain volume [32]. The directional organization of white matter tracts constrains water molecule diffusivity in an anisotropic (uneven) manner, the extent of which can be measured by a scalar value, fractional anisotropy (FA). FA is sensitive to the organization, integrity and myelination of white matter axons . In addition, mean diffusivity (MD) of water molecules provides an indirect measure of neuronal loss by quantifying the extracellular fluid space in grey and white matter. Applied to narcoleptic patients, DTI revealed decreases in FA [33] and increases in MD [27] in the hypothalamus compared to controls, in corroboration with VBM studies implicating this area in narcoleptic pathophysiology. FA decreases likely signified a pathological reduction in the number and coherence of axons in the area, while increased MD signified neuronal loss. FA decreases concomitant with MD increases provide strong evidence for neurodegenerative effects, since the erosion of white matter should be followed by an increase in extracellular fluid space. Combined MD increases and FA decreases were found in the fronto-orbital and anterior cingulate cortices , possibly explaining attentional deficits in narcolepsy [27]. Other brain areas showing reduced FA only were the midbrain and medulla oblongata [33], as well as fronto-temporal cortices [27, 33], areas which receive hypocretinergic projections from the hypothalamus and which may thus have undergone secondary neuronal loss. MD increases without FA decreases were found in the dorsal raphe nuclei and the ventral tegmental area, which are also afferented by hypothalamic hypocretin neurons [27]. Curiously, FA increases were found in the pons, pre- and postcentral gyri and corona radiata, which may be explained by compensatory processes [33]. A number of objective and subjective measures of disease severity were associated with FA decreases in the hypothalamus and brainstem, but none of these correlations survived Bonferroni correction for multiple comparisons [33]. Still, trends towards significance were found in the positive correlation between FA values in the hypothalamus and the Epworth Sleepiness Scale (ESS), a subjective marker of disease severity, and in the negative correlation between the ESS and FA values in the medulla oblongata [33]. A recent DTI study examined structural differences between two narcoleptic subtypes: narcolepsy with cataplexy and narcolepsy without cataplexy [34]. While narcolepsy with cataplexy showed differences with respect to healthy controls, notably in the inferior frontal gyrus and the amygdala, narcolepsy without cataplexy displayed no significant differences from controls. The authors concluded that narcolepsy with and without cataplexy may constitute two distinct etiologies.
Spectroscopy
Proton MRS (1H-MRS) allows a noninvasive assay of specific molecular concentrations in the brain. By measuring localized ratios of cell metabolites, 1H-MRS can provide evidence of neuronal damage in specific areas of the brain. An early 1H-MRS study examined concentrations of N-acetylaspartate (NAA) and creatine-phosphocreatine (Cr-PCr) in the ventral pons [35]. NAA is thought to provide a sensitive index of cell mass, while Cr-PCr, metabolites of oxidative phosphorylation, are believed to be relatively constant in the brain [36]. A decrease in the NAA/Cr-PCr ratio can thus be understood as net neuronal loss in the observed brain region. Examining this ratio in the pons revealed no change between narcoleptic patients and healthy controls, anticipating results later obtained in MRI studies of the pons [17, 18]. With the eventual discovery of hypocretin’s role in narcolepsy, two more 1H-MRS studies from Plazzi and colleagues targeted the hypothalamus and indeed found that NAA/Cr-PCr ratios were lower in this region for narcoleptic patients than for healthy controls [37, 38]. Reduced NAA/Cr-PCr in the hypothalamus is evidence of neuronal loss, in agreement with the autoimmune destruction of hypocretinergic neurons in this area. In the latter of these two studies [38], researchers hypothesized that brain regions innervated by hypothalamic hypocretin neurons may show signs of secondary neuronal loss (a common finding in the aforementioned VBM studies). They examined two of these regions, the thalamus and parietal-occipital cortex, but found no difference between narcoleptic patients and controls. A recent 1H-MRS study from another research group sought to corroborate findings in the narcoleptic hypothalamus, while also examining the amygdala and ponto-mesencephalic junction, but contrary to previous results, no change in NAA/Cr-PCr ratios was detected in either of these regions when compared to controls [39]. Instead, they found that myoinositol/Cr-PCr ratios were lower in the narcoleptic amygdala, which the authors propose may be due to changes in amygdalar cell signalling, possibly in relationship with emotional dysregulation (see functional studies). Results from these MRS studies corroborate some MRI findings in the pons and hypothalamus, but also add to the inconsistencies in the literature concerning these regions, as well as the thalamus and cerebral cortex. Furthermore , the apparent reversibility of NAA /Cr-PCr ratio decreases in the recovery from acute brain pathology [40] has called into question its link with neuronal loss, instead suggesting it may be indicative of neuronal dysfunction [41]. Neurotransmitter concentrations have also been studied with MRS. Using a 3.0-T MRS scanner, absolute gamma-aminobutyric acid (GABA ) concentrations were assessed in a sample of 17 narcolepsy–cataplexy patients versus 17 healthy controls [42]. It was found that patients had significantly higher GABA concentrations in the medial prefrontal cortex (mPFC) than did normal controls. Moreover, narcoleptic patients without nocturnal sleep disturbances had higher mPFC GABA levels than those with nocturnal sleep disturbances, and both groups had higher mPFC GABA levels than controls. According to the authors’ interpretation, elevated GABA may act as a compensatory mechanism in narcolepsy, whereby nocturnal sleep disturbances are alleviated. In line with this interpretation, GABA levels were reduced in MRS studies of insomniacs, for whom nocturnal sleep disturbances are chronic [43, 44].
In sum, while definite trends pervade the structural neuroimaging literature for narcolepsy, more evidence is needed to stem the still widespread inconsistencies, even within the same imaging modalities (Table 13.1). Nonetheless, a number of anatomical studies convergently support hypothalamic damage in narcolepsy–cataplexy, which is compatible with a loss of hypocretinergic neurons, whose cell bodies are located exclusively in the hypothalamus [13]. Alterations in fronto-temporal regions were another common finding, which may relate to cognitive and mood disturbances in narcolepsy. Also of note, correlations between subjective disease severity and neuroanatomical alterations were discovered, including a correlation between hypothalamic damage and the Ullanlinna Narcolepsy Scale score [23] and between ESS and cortical thickness measurements [28, 29]. These results, though still uncertain, can be considered in tandem with functional neuroimaging data to provide a clearer picture of narcoleptic neuropathology.
Functional Imaging in Narcolepsy
Baseline Conditions
Few neuroimaging studies have described baseline activity in the narcoleptic brain (Table 13.2). An early SPECT study scanned narcoleptic patients and healthy controls after radioactive 133Xe inhalation, during both resting wakefulness and sleep [45]. Waking activity in narcolepsy displayed decreased regional cerebral blood flow (rCBF) in the brainstem area, in comparison with controls. After sleep onset (SOREMP in 3 out of 13 cases), rCBF increased in all regions, which the authors interpreted as increased dreaming activity specific to narcolepsy. More recent SPECT studies have also characterized rCBF during baseline states in narcolepsy (Fig. 13.2). A study using 99mTc-hexamethylpropyleneamineoxime (99mTc-HMPAO) compared wakefulness and SOREMPs in six narcoleptic patients [46]. No significant difference was detected in 99mTc-HMPAO uptake between states, but interestingly, the researchers reported high perfusion in parietal regions, which is uncharacteristic of normal REM sleep [47]. Further functional studies during narcoleptic REM sleep are needed to explore this phenomenon. Another SPECT study used 99m-technetium ethyl cysteinate dimer (99mTc-ECD), to evaluate rCBF differences in waking activity between 25 patients with narcolepsy–cataplexy and 25 healthy controls [48]. Decreased perfusion was observed throughout the brain, notably in bilateral hypothalami, caudate nuclei, pulvinar nuclei of the thalamus, cingulate gyrus and fronto-parietal cortices. These areas of decreased activity correspond to regions of hypocretinergic projection and are thus in line with structural studies, indicating a loss of hypothalamic neurons in narcolepsy.

Blood flow and metabolic differences during resting wakefulness in narcolepsy. Few studies have examined narcoleptic brain activity at baseline. Among them, recurrent findings are activity decreases in the dorsal thalamus and the hypothalamus during resting wakefulness [48, 49]. Contrary to these studies, Dauvilliers et al. [50] detected activity increases during resting wakefulness, in the cingulate cortex and lingual gyrus. Prepared with illustrations by Patrick J. Lynch and C. Carl Jaffe. http://creativecommons.org/ licenses/by/2.5/
Two PET studies have also examined waking activity in narcolepsy compared to controls, using 18F-fluorodeoxy glucose (18F-FDG ) as a radiotracer for the cerebral metabolic rate of glucose utilization (CMRglu) (Fig. 13.2). The first study of 24 narcoleptic patients and 24 healthy controls found reduced CMRglu in the hypothalami, thalami and fronto-parietal cortices [49]. In stark contrast, the second study found no areas of hypometabolism in 21 narcoleptics compared to 21 controls, but instead found hypermetabolism of the cingulate and visual association cortices during wakefulness [50]. The authors of this latter study attribute the discrepancy between both PET studies to differences in scanning conditions, sample inclusion criteria and patient treatment history.
As an interim summary, imaging studies examining baseline narcoleptic brain activity have been few, but some concordance exists between observations of reduced activity in the hypothalamus, thalamus and fronto-parietal cortices. Strikingly, there is a lack of well-characterized and recent imaging data for sleep in narcolepsy. New studies of narcoleptic sleep using modern imaging techniques will be crucial to understanding the disease, particularly its disruptive effects on nocturnal sleep.
Cataplexy
Cataplectic attacks, by their nature, are unpredictable and difficult to capture with neuroimaging. Researchers have resorted to eliciting cataplexy with emotional stimulation, for instance, by telling a funny story, but they have had limited success [50]. Consequently, neuroimaging studies of cataplexy are few and have diminutive sample sizes (Table 13.2). A first such study captured cataplectic episodes in two narcoleptic patients using 99mTc-ECD SPECT [51]. Compared with baseline wakefulness and REM sleep, the cataplectic state was characterized by hyperperfusion in limbic areas (including the right amygdala), thalami, basal ganglia, brainstem and parietal cortices, and hypoperfusion in prefrontal and occipital cortices. A case study using the same imaging modality instead found hyperperfusion throughout the cerebral cortex, specifically in the orbitofrontal, temporal, cingulate and right putamen [52]. The authors likened this activation to a REM sleep state, albeit without usual pontine, occipital or amygdalar activation. An 18F-FDG PET study managed to capture cataplectic episodes in two narcoleptic patients and found metabolic increases in the pre-postcentral gyri and the somatosensory cortex as well as a decrease in hypothalamic metabolism [50]. Lastly, a cataplectic attack was captured during an fMRI scan and showed marked hypoactivation of the hypothalamus [53]. Little can be concluded from these limited data, but the repeat finding of hypothalamic hypoactivity [50, 53] is promising, in light of structural data. It will be important for researchers to develop improved procedures for eliciting cataplectic attacks in the laboratory.
Emotional Stimulation
Because cataplexy is often triggered by strong emotions, it can also be informative to examine how narcoleptic patients respond differently to emotional stimuli. A few studies have employed emotional stimulation paradigms during fMRI scans to capture these differences (Table 13.2). The fMRI scanner measures changes in the blood-oxygen-level-dependent (BOLD ) signal between task and baseline conditions as an index of event-related brain activity. In two independent studies, funny pictures or cartoons were displayed to narcoleptic–cataplectic patients and healthy controls undergoing fMRI scans. One of these studies showed enhanced activity in the emotional network, including the hypothalamus and nucleus accumbens, when comparing patients to controls [53]. The second study, in contrast, showed reduced activity in the hypothalamus, concomitant with heightened amygdalar activity [54]. Together, these studies suggest a dysregulation of the hypothalamic-amygdalar emotional network in narcolepsy with cataplexy. Interestingly, the first study reported that narcoleptic–cataplectic patients were less likely than controls to rate a cartoon as funny, although this effect was not replicated in the second study.
The neuropeptide hypocretin modulates more than just sleep–wake patterns. It is also believed to be involved in reward-related behaviours, motivation, feeding and addiction [55–57]. Hence, hypocretin deficiency in narcolepsy may also lead to anomalous responses to reward and aversive stimulation. Following this rationale, Ponz and colleagues probed both the narcoleptic reward and fear conditioning systems in two fMRI studies. In the reward study [58], researchers presented patients and controls with the monetary incentive-delay task, which is known to recruit the mesolimbic and midbrain reward system in normal subjects. While performing this task in the scanner, patients recruited vastly different regions from healthy controls. Instead of activating the ventral pathway (ventral tegmentum, ventromedial prefrontal cortex and nucleus accumbens), narcoleptic patients showed enhanced activity in the amygdala and dorsal striatum in response to positive outcomes, in agreement with previous studies showing increased amygdalar activation in intense emotional states [51, 54]. Interestingly, ventral-medial prefrontal cortex activation positively correlated with disease duration in the narcoleptic group, suggesting that alternate pathways may be learned by patients over the years to compensate for the lack of input from the ventral midbrain pathway. In the fear conditioning study [59], patients and controls were presented with pictures in synchronization with a painful electric shock, in a classical conditioning paradigm. It was found that normal amygdalar response to aversive conditioned stimuli was reduced in narcoleptic patients. Furthermore, the normal functional coupling between the amygdala and medial prefrontal cortex during conditioning was observed in controls but not in patients.
Data from fMRI studies, though still sparse, are well aligned in demonstrating abnormal emotional processing in narcolepsy. Reward conditioning, aversive conditioning and humour judgement are three paradigms which represent various facets of emotional reactivity, and each has been shown to be altered in narcolepsy ([58], [59] and [54], respectively). These studies, buttressed by anatomical studies reporting alterations in the amygdala [31, 34, 39], support dysfunctional emotional network activity in narcolepsy-cataplexy, which may be linked to global hypocretin depletion.
Neurotransmission
Acetylcholine (ACh ) is known to be an important neurotransmitter in the control of REM sleep [60, 61] and was hypothesized to be dysregulated in narcolepsy. However, a PET study using the radioligand 11C-N-methyl-4-piperidyl-benzilate (11C-MPB) found no difference in cholinergic binding between narcoleptic patients and healthy controls [62].
Serotonin (5-HT ) has also been proposed as showing disturbance in narcolepsy, based on animal studies linking it with the suppression of REM sleep [63]. A PET study using 4-(2′-methoxyphenyl)-1-[2′-(N-2″-pyridinyl)-p-18F-fluorobenzamido]ethylpiperazine (18F-MPPF) to study 5-HT1A receptor binding reported increased 18F-MPPF binding affinity in the anterior cingulate and temporal cortices during sleep compared to wakefulness in patients with narcolepsy [64]. However, the omission of a control group limits the study’s implications for narcoleptic pathophysiology.
Dopamine became a neurotransmitter of interest in narcolepsy research when increased dopamine receptor D2 binding was shown in postmortem studies of deceased narcoleptic patients [65, 66]. A series of PET and SPECT studies ensued, using various radioligands to characterize presynaptic and postsynaptic dopamine binding (e.g. [123I](N)-(3-iodopropene-2-yl)-2β-carbomethoxy-3β-(4-chlorophenyl)tropane [123IPT] with SPECT, and 11C-raclopride in PET, respectively). Only one SPECT study of seven patients found a correlation between D2 binding in the striatum (using [123I](S)-2-hydroxy-3-iodo-6-methoxy-([1-ethyl-2-pyrrolidinyl]methyl)benzamide [123IBZM]) and the frequency of cataplectic and sleep attacks [67]. Six other PET and SPECT studies were unable to replicate this finding [68–73]. It may be that increased dopamine binding in the postmortem studies was the result of life-long medication use on behalf of the deceased patients rather than constituting an intrinsic feature of pathophysiology of narcolepsy [41]. Altogether, neurotransmission studies of narcolepsy have not uncovered any neurotransmitter-specific abnormalities. A more detailed description of these neurotransmission studies can be found in [74]
Summary
The expanding brain imaging literature in narcolepsy is showing remarkable convergences. Of prime interest, functional, anatomical and spectroscopic data concordantly show atrophy and dysfunction of the hypothalamus in narcolepsy–cataplexy, in agreement with the pathophysiological theory of narcolepsy–cataplexy as a disease affecting hypocretinergic neurons in the hypothalamus. In line with this theory, several brain regions receiving major or moderate hypocretinergic projections from the hypothalamus [13] also show signs of neurodegeneration and altered function, notably the thalamus, amygdala and fronto-temporal cortices. The limbic system, particularly the amygdala, shows altered responding in functional studies, paralleled by signs of damage from some structural studies. These changes may relate to the emotional disturbances observed in narcolepsy–cataplexy as well as to the triggering of cataplectic attacks by intense emotional events. Cortical and subcortical anatomical changes, notably in the hippocampus and prefrontal cortices, may explain specific cognitive dysfunctions in narcolepsy. Lastly, reduced thalamic volume and activity may be responsible for narcoleptic sleep fragmentation.
A paucity of functional research has characterized brain activity in narcolepsy during sleep. Further research in this area may provide valuable insights into normal and pathological sleep–wake neurophysiology and may help to explain the little-researched but debilitating symptom of sleep fragmentation in narcolepsy. Studies of neurotransmission in narcolepsy have yielded few positive results, having largely focused on the dopaminergic system. New techniques for imaging hypocretin binding in PET (see Wang et al. [75] for hypocretin-2) or SPECT will be invaluable in understanding the extent of narcoleptic pathophysiology. Finally, some methodological considerations may curb the variability of results in brain imaging studies. Given that narcolepsy is a relatively rare condition and participants are difficult to find, strict controls on sampling and procedures are crucial for obtaining reliable results. In structural studies, this may be achieved by standardized data preprocessing, to facilitate valid cross-study comparisons. Furthermore, in light of abundant associations between the extent of neural abnormalities and indices of disease severity [23, 28–30, 33], age of disease onset [29] and symptomatology [34], it will be important to consider possible phenotypic and etiological heterogeneities within the narcolepsy population, with particular attention to the differentiation between narcolepsy with and without cataplexy.
References
Dauvilliers Y, Arnulf I, Mignot E. Narcolepsy with cataplexy. Lancet. 2007;369(9560):499–511. PubMed Epub 2007/02/13. eng.
Findley L, Unverzagt M, Guchu R, Fabrizio M, Buckner J, Suratt P. Vigilance and automobile accidents in patients with sleep apnea or narcolepsy. Chest. 1995;108(3):619–24. PubMed.
Carskadon MA, Dement WC, Mitler MM, Roth T, Westbrook PR, Keenan S. Guidelines for the multiple sleep latency test (MSLT): a standard measure of sleepiness. Sleep. 1986;9(4):519–24. PubMed Epub 1986/12/01. eng.
Dauvilliers Y, Montplaisir J, Molinari N, Carlander B, Ondze B, Besset A, et al. Age at onset of narcolepsy in two large populations of patients in France and Quebec. Neurology. 2001;57(11):2029–33. PubMed.
Black J, Houghton WC. Sodium oxybate improves excessive daytime sleepiness in narcolepsy. Sleep. 2006;29(7):939–46. PubMed.
Mignot E, Hayduk R, Black J, Grumet FC, Guilleminault C. HLA DQB1*0602 is associated with cataplexy in 509 narcoleptic patients. Sleep. 1997;20(11):1012–20. PubMed.
Lin L, Faraco J, Li R, Kadotani H, Rogers W, Lin X, et al. The sleep disorder canine narcolepsy is caused by a mutation in the hypocretin (orexin) receptor 2 gene. Cell. 1999;98(3):365–76. PubMed.
Nishino S, Ripley B, Overeem S, Lammers GJ, Mignot E. Hypocretin (orexin) deficiency in human narcolepsy. Lancet. 2000;355(9197):39–40. PubMed.
Thannickal TC, Moore RY, Nienhuis R, Ramanathan L, Gulyani S, Aldrich M, et al. Reduced number of hypocretin neurons in human narcolepsy. Neuron. 2000;27(3):469–74. PubMed.
Mignot E, Lammers GJ, Ripley B, Okun M, Nevsimalova S, Overeem S, et al. The role of cerebrospinal fluid hypocretin measurement in the diagnosis of narcolepsy and other hypersomnias. Arch Neurol. 2002;59(10):1553–62. PubMed Epub 2002/10/11. Eng.
Baumann CR, Khatami R, Werth E, Bassetti CL. Hypocretin (orexin) deficiency predicts severe objective excessive daytime sleepiness in narcolepsy with cataplexy. J Neurol Neurosurg Psychiatry. 2006;77(3):402–4. PubMed Pubmed Central PMCID: 2077721. Epub 2006/02/18. eng.
Govindaiah G, Cox CL. Modulation of thalamic neuron excitability by orexins. Neuropharmacology. 2006;51(3):414–25. PubMed.
Peyron C, Tighe DK, van den Pol AN, de Lecea L, Heller HC, Sutcliffe JG, et al. Neurons containing hypocretin (orexin) project to multiple neuronal systems. J Neurosci. 1998;18(23):9996–10015. PubMed.
Baumann CR, Bassetti CL. Hypocretins (orexins): clinical impact of the discovery of a neurotransmitter. Sleep Med Rev. 2005;9(4):253–68. PubMed.
Dang-Vu TT. Neuroimaging of treatment response in narcolepsy. In: Nofzinger EA, Maquet P, Thorpy M, editors. Neuroimaging of sleep and sleep disorders. Cambridge: Cambridge University Press; 2013. p. 228–30.
Plazzi G, Montagna P, Provini F, Bizzi A, Cohen M, Lugaresi E. Pontine lesions in idiopathic narcolepsy. Neurology. 1996;46(5):1250–4. PubMed.
Bassetti C, Aldrich MS, Quint DJ. MRI findings in narcolepsy. Sleep. 1997;20(8):630–1. PubMed.
Frey JL, Heiserman JE. Absence of pontine lesions in narcolepsy. Neurology. 1997;48(4):1097–9. PubMed.
Pullicino P, Ostrow P, Miller L, Snyder W, Munschauer F. Pontine ischemic rarefaction. Ann Neurol. 1995;37(4):460–6. PubMed.
Buskova J, Vaneckova M, Sonka K, Seidl Z, Nevsimalova S. Reduced hypothalamic gray matter in narcolepsy with cataplexy. Neuro Endocrinol Lett. 2006;27(6):769–72. PubMed eng.
Draganski B, Geisler P, Hajak G, Schuierer G, Bogdahn U, Winkler J, et al. Hypothalamic gray matter changes in narcoleptic patients. Nat Med. 2002;8(11):1186–8. PubMed.
Joo EY, Tae WS, Kim ST, Hong SB. Gray matter concentration abnormality in brains of narcolepsy patients. Korean J Radiol. 2009;10(6):552–8. PubMed Pubmed Central PMCID: 2770823. Epub 2009/11/04. eng.
Kim SJ, Lyoo IK, Lee YS, Lee JY, Yoon SJ, Kim JE, et al. Gray matter deficits in young adults with narcolepsy. Acta Neurol Scand. 2009;119(1):61–7. PubMed Epub 2008/07/16. eng.
Overeem S, Steens SC, Good CD, Ferrari MD, Mignot E, Frackowiak RS, et al. Voxel-based morphometry in hypocretin-deficient narcolepsy. Sleep. 2003;26(1):44–6. PubMed.
Kaufmann C, Schuld A, Pollmacher T, Auer DP. Reduced cortical gray matter in narcolepsy: preliminary findings with voxel-based morphometry. Neurology. 2002;58(12):1852–5. PubMed.
Brenneis C, Brandauer E, Frauscher B, Schocke M, Trieb T, Poewe W, et al. Voxel-based morphometry in narcolepsy. Sleep Med. 2005;30. PubMed.
Scherfler C, Frauscher B, Schocke M, Nocker M, Gschliesser V, Ehrmann L, et al. White and gray matter abnormalities in narcolepsy with cataplexy. Sleep. 2012;35(3):345–51. PubMed Epub 2012/03/02. eng.
Joo EY, Jeon S, Lee M, Kim ST, Yoon U, Koo DL, et al. Analysis of cortical thickness in narcolepsy patients with cataplexy. Sleep. 2011;34(10):1357–64. PubMed Pubmed Central PMCID: 3174837. Epub 2011/10/04. eng.
Schaer M, Poryazova R, Schwartz S, Bassetti CL, Baumann CR. Cortical morphometry in narcolepsy with cataplexy. J Sleep Res. 2012;21(5):487–94. PubMed Epub 2012/02/09. Eng.
Joo EY, Kim SH, Kim ST, Hong SB. Hippocampal volume and memory in narcoleptics with cataplexy. Sleep Med. 2012;13(4):396–401. PubMed Epub 2012/03/01. eng.
Brabec J, Rulseh A, Horinek D, Pala A, Guerreiro H, Buskova J, et al. Volume of the amygdala is reduced in patients with narcolepsy—a structural MRI study. Neuro Endocrinol Lett. 2011;32(5):652–6. PubMed Epub 2011/12/15. eng.
Schaefer PW, Grant PE, Gonzalez RG. Diffusion-weighted MR imaging of the brain. Radiology. 2000;217(2):331–45. PubMed.
Menzler K, Belke M, Unger MM, Ohletz T, Keil B, Heverhagen JT, et al. DTI reveals hypothalamic and brainstem white matter lesions in patients with idiopathic narcolepsy. Sleep Med. 2012;13(6):736–42. PubMed Epub 2012/05/01. eng.
Nakamura M, Nishida S, Hayashida K, Ueki Y, Dauvilliers Y, Inoue Y. Differences in brain morphological findings between narcolepsy with and without cataplexy. PLoS One. 2013;8(11), e81059. PubMed Pubmed Central PMCID: 3842956.
Ellis CM, Simmons A, Lemmens G, Williams SC, Parkes JD. Proton spectroscopy in the narcoleptic syndrome. Is there evidence of a brainstem lesion? Neurology. 1998;50(2 Suppl 1):S23–6. PubMed eng.
Howe FA, Maxwell RJ, Saunders DE, Brown MM, Griffiths JR. Proton spectroscopy in vivo. Magn Reson Q. 1993;9(1):31–59. PubMed.
Lodi R, Tonon C, Vignatelli L, Iotti S, Montagna P, Barbiroli B, et al. In vivo evidence of neuronal loss in the hypothalamus of narcoleptic patients. Neurology. 2004;63(8):1513–5. PubMed.
Tonon C, Franceschini C, Testa C, Manners DN, Poli F, Mostacci B, et al. Distribution of neurochemical abnormalities in patients with narcolepsy with cataplexy: an in vivo brain proton MR spectroscopy study. Brain Res Bull. 2009;80(3):147–50. PubMed.
Poryazova R, Schnepf B, Werth E, Khatami R, Dydak U, Meier D, et al. Evidence for metabolic hypothalamo-amygdala dysfunction in narcolepsy. Sleep. 2009;32(5):607–13. PubMed Pubmed Central PMCID: 2675895. Epub 2009/06/02. eng.
De Stefano N, Matthews PM, Arnold DL. Reversible decreases in N-acetylaspartate after acute brain injury. Magn Reson Med. 1995;34(5):721–7. PubMed.
Desseilles M, Dang-Vu T, Schabus M, Sterpenich V, Maquet P, Schwartz S. Neuroimaging insights into the pathophysiology of sleep disorders. Sleep. 2008;31(6):777–94. PubMed Pubmed Central PMCID: 2442420.
Kim SJ, Lyoo IK, Lee YS, Sung YH, Kim HJ, Kim JH, et al. Increased GABA levels in medial prefrontal cortex of young adults with narcolepsy. Sleep. 2008;31(3):342–7. PubMed eng.
Plante DT, Jensen JE, Schoerning L, Winkelman JW. Reduced gamma-aminobutyric acid in occipital and anterior cingulate cortices in primary insomnia: a link to major depressive disorder? Neuropsychopharmacology. 2012;37(6):1548–57. PubMed Pubmed Central PMCID: 3327859.
Winkelman JW, Buxton OM, Jensen JE, Benson KL, O’Connor SP, Wang W, et al. Reduced brain GABA in primary insomnia: preliminary data from 4T proton magnetic resonance spectroscopy (1H-MRS). Sleep. 2008;31(11):1499–506. PubMed Pubmed Central PMCID: 2579978.
Meyer JS, Sakai F, Karacan I, Derman S, Yamamoto M. Sleep apnea, narcolepsy, and dreaming: regional cerebral hemodynamics. Ann Neurol. 1980;7(5):479–85. PubMed eng.
Asenbaum S, Zeithofer J, Saletu B, Frey R, Brucke T, Podreka I, et al. Technetium-99m-HMPAO SPECT imaging of cerebral blood flow during REM sleep in narcoleptics. J Nucl Med. 1995;36(7):1150–5. PubMed eng.
Maquet P. Functional neuroimaging of normal human sleep by positron emission tomography. J Sleep Res. 2000;9(3):207–31. PubMed.
Joo YE, Hong SB, Tae WS, Kim JH, Han SJ, Cho YW, et al. Cerebral perfusion abnormality in narcolepsy with cataplexy. Neuroimage. 2005;28(2):410–6. PubMed.
Joo EY, Tae WS, Kim JH, Kim BT, Hong SB. Glucose hypometabolism of hypothalamus and thalamus in narcolepsy. Ann Neurol. 2004;56(3):437–40. PubMed.
Dauvilliers Y, Comte F, Bayard S, Carlander B, Zanca M, Touchon J. A brain PET study in patients with narcolepsy-cataplexy. J Neurol Neurosurg Psychiatry. 2010;81(3):344–8. PubMed Epub 2009/10/24. eng.
Hong SB, Tae WS, Joo EY. Cerebral perfusion changes during cataplexy in narcolepsy patients. Neurology. 2006;66(11):1747–9. PubMed.
Chabas D, Habert MO, Maksud P, Tourbah A, Minz M, Willer JC, et al. Functional imaging of cataplexy during status cataplecticus. Sleep. 2007;30(2):153–6. PubMed eng.
Reiss AL, Hoeft F, Tenforde AS, Chen W, Mobbs D, Mignot EJ. Anomalous hypothalamic responses to humor in cataplexy. PLoS One. 2008;3(5):e2225. PubMed eng.
Schwartz S, Ponz A, Poryazova R, Werth E, Boesiger P, Khatami R, et al. Abnormal activity in hypothalamus and amygdala during humour processing in human narcolepsy with cataplexy. Brain. 2008;131(Pt 2):514–22. PubMed.
Schultz W. Multiple reward signals in the brain. Nat Rev Neurosci. 2000;1(3):199–207. PubMed.
Harris GC, Wimmer M, Aston-Jones G. A role for lateral hypothalamic orexin neurons in reward seeking. Nature. 2005;437(7058):556–9. PubMed.
Boutrel B, Kenny PJ, Specio SE, Martin-Fardon R, Markou A, Koob GF, et al. Role for hypocretin in mediating stress-induced reinstatement of cocaine-seeking behavior. Proc Natl Acad Sci U S A. 2005;102(52):19168–73. PubMed Pubmed Central PMCID: 1323172.
Ponz A, Khatami R, Poryazova R, Werth E, Boesiger P, Bassetti CL, et al. Abnormal activity in reward brain circuits in human narcolepsy with cataplexy. Ann Neurol. 2010;67(2):190–200. PubMed Epub 2010/03/13. eng.
Ponz A, Khatami R, Poryazova R, Werth E, Boesiger P, Schwartz S, et al. Reduced amygdala activity during aversive conditioning in human narcolepsy. Ann Neurol. 2010;67(3):394–8. PubMed Epub 2010/04/08. eng.
Steriade M, McCarley RW. Brainstem Control of Wakefulness and Sleep. New York: Plenum Press; 1990.
Hobson JA, McCarley RW, Wyzinski PW. Sleep cycle oscillation: reciprocal discharge by two brainstem neuronal groups. Science. 1975;189(4196):55–8. PubMed.
Sudo Y, Suhara T, Honda Y, Nakajima T, Okubo Y, Suzuki K, et al. Muscarinic cholinergic receptors in human narcolepsy: a PET study. Neurology. 1998;51(5):1297–302. PubMed.
Portas CM, McCarley RW. Behavioral state-related changes of extracellular serotonin concentration in the dorsal raphe nucleus: a microdialysis study in the freely moving cat. Brain Res. 1994;648(2):306–12. PubMed.
Derry C, Benjamin C, Bladin P, le Bars D, Tochon-Danguy H, Berkovic SF, et al. Increased serotonin receptor availability in human sleep: evidence from an [18F]MPPF PET study in narcolepsy. NeuroImage. 2006;30(2):341–8. PubMed Epub 2005/11/09. eng.
Aldrich MS, Hollingsworth Z, Penney JB. Dopamine-receptor autoradiography of human narcoleptic brain. Neurology. 1992;42(2):410–5. PubMed.
Kish SJ, Mamelak M, Slimovitch C, Dixon LM, Lewis A, Shannak K, et al. Brain neurotransmitter changes in human narcolepsy. Neurology. 1992;42(1):229–34. PubMed.
Eisensehr I, Linke R, Tatsch K, von Lindeiner H, Kharraz B, Gildehaus FJ, et al. Alteration of the striatal dopaminergic system in human narcolepsy. Neurology. 2003;60(11):1817–9. PubMed.
Rinne JO, Hublin C, Partinen M, Ruottinen H, Nagren K, Lehikoinen P, et al. Striatal dopamine D1 receptors in narcolepsy: a PET study with [11C]NNC 756. J Sleep Res. 1996;5(4):262–4. PubMed.
Rinne JO, Hublin C, Nagren K, Helenius H, Partinen M. Unchanged striatal dopamine transporter availability in narcolepsy: a PET study with [11C]-CFT. Acta Neurol Scand. 2004;109(1):52–5. PubMed.
Staedt J, Stoppe G, Kogler A, Riemann H, Hajak G, Rodenbeck A, et al. [123I]IBZM SPET analysis of dopamine D2 receptor occupancy in narcoleptic patients in the course of treatment. Biol Psychiatry. 1996;39(2):107–11. PubMed.
Hublin C, Launes J, Nikkinen P, Partinen M. Dopamine D2-receptors in human narcolepsy: a SPECT study with 123I-IBZM. Acta Neurol Scand. 1994;90(3):186–9. PubMed.
Khan N, Antonini A, Parkes D, Dahlitz MJ, Meier-Ewert K, Weindl A, et al. Striatal dopamine D2 receptors in patients with narcolepsy measured with PET and 11C-raclopride. Neurology. 1994;44(11):2102–4. PubMed.
MacFarlane JG, List SJ, Moldofsky H, Firnau G, Chen JJ, Szechtman H, et al. Dopamine D2 receptors quantified in vivo in human narcolepsy. Biol Psychiatry. 1997;41(3):305–10. PubMed eng.
Dang-Vu TT, Desseilles M, Schwartz S, Maquet P. Neuroimaging of narcolepsy. CNS Neurol Disord Drug Targets. 2009;8(4):254–63.
Wang C, Wilson CM, Moseley CK, Carlin SM, Hsu S, Arabasz G, et al. Evaluation of potential PET imaging probes for the orexin 2 receptors. Nucl Med Biol. 2013;40(8):1000–5. PubMed Pubmed Central PMCID: 3812298.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing Switzerland
About this chapter
Cite this chapter
O’Byrne, J.N., Salimi, A., Dang-Vu, T.T. (2016). Neuroimaging of Narcolepsy. In: Goswami, M., Thorpy, M., Pandi-Perumal, S. (eds) Narcolepsy. Springer, Cham. https://doi.org/10.1007/978-3-319-23739-8_13
Download citation
DOI: https://doi.org/10.1007/978-3-319-23739-8_13
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-23738-1
Online ISBN: 978-3-319-23739-8
eBook Packages: MedicineMedicine (R0)