
Abstract
This chapter summarizes the nonneoplastic (inflammatory processes as well as congenital and acquired cysts) and neoplastic lesions (thymoma, thymic carcinomas, lymphomas, germ cell tumors, neurogenic tumors, and other soft tissue tumors) involving the mediastinum and thymus. Lymphoproliferative lesions and germ cell tumors resemble their counterparts outside the mediastinum. Thymomas are uncommon tumors which arise from the thymic epithelium. Although their behavior is generally indolent, they are capable of invasive growth and metastasis. The most commonly used system for thymoma classification (WHO) was refined in 2014 by the International Thymic Malignancy Interest Group (ITMIG), which introduced major and minor criteria to accurately classify subtypes.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others



Keywords
- Fibrosing mediastinitis
- Mediastinal congenital and acquired cysts
- Thymoma
- Thymic carcinoma
- Mediastinal lymphoproliferative disorders
- Thymic and extrathymic mesenchymal tumors
- Mediastinal germ cell tumors
- Mediastinal neuroendocrine tumors
Nonneoplastic Disorders
Inflammatory/Infectious
Acute Mediastinitis
Clinical
-
♦
Acute mediastinitis is a life-threatening acute infection. Primary acute mediastinitis is rare; nearly all cases are the result of direct inoculation or spread from another compartment. The presenting clinical symptoms vary depending on the site of origin of infection. As mediastinitis evolves, patients experience chest pain, fever, and dysphagia
-
♦
The most common causes of acute mediastinitis are secondary to cardiac surgery, following esophageal perforation, spread from infections of the head and neck, and spread to the mediastinum from other sites such as lungs or meninges
-
Acute mediastinitis has an incidence rate of 1–4% following cardiac surgery. The infection localizes in the anterior mediastinum
-
Esophageal perforation typically involves the posterior mediastinum. Boerhaave syndrome represents spontaneous rupture of the esophagus following bouts of vomiting. The most common cause of esophageal rupture is iatrogenic
-
Descending necrotizing mediastinitis is secondary to head and neck infections which track through fascial planes from deep cervical infections originating in the oropharynx. Typically infections are due to mixed aerobic and anaerobic organisms and locate in the superior or posterior mediastinum
-
Direct extension from lung or pleural infections is an infrequent cause of mediastinitis
-
Macroscopic
-
♦
Acute mediastinitis is characterized by purulent exudate layered on mediastinal structures with or without abscess formation. Hemorrhagic necrosis is characteristic of mediastinitis due to inhalational anthrax. Air crepitance is the result of pneumomediastinum following esophageal rupture or pneumothorax
Microscopic
-
♦
Acute fibrinopurulent inflammation is the major histological pattern. Infectious agents are identified by appropriate stains for fungi and bacteria
Differential Diagnosis
-
♦
There are variable potential organisms, depending on the source of the infection and immune status of the patient. Microbiologic cultures and special stains for microorganisms are helpful in determining the causative agent
Granulomatous Mediastinitis (Localized Fibrosing Mediastinitis)
Clinical
-
♦
Granulomatous mediastinitis is a localized process due to chronic mediastinal adenitis. Lymph nodes enlarge and become confluent, and the capsules thicken with fibrotic reaction in the immediate vicinity of the lymph nodes
-
♦
The most common cause of granulomatous mediastinitis is histoplasmosis, followed in frequency by tuberculosis
-
♦
Complications of this process include perforation and fistula formation, esophageal diverticula, or compression of a bronchus. Lymph nodes frequently calcify and may erode into the airways as broncholiths causing bronchial obstruction
Macroscopic
-
♦
Localized fibrosing mediastinitisThe appearance is that of a large, calcified, and fibrocaseous mass of coalesced lymph nodes (mediastinal granuloma) (Fig. 26.1A)
Fig. 26.1. 
(A) Mediastinal granuloma . Enlarged, matted lymph nodes replaced by fibrocaseous granulomas (scale = 1 cm). (B) Fibrocaseous granuloma with central cavitation within lymph node. (C) Budding yeast-like organisms consistent with Histoplasma capsulatum (GMS stain).

Microscopic
-
♦
Lymph node architecture is partially or completely effaced by necrotizing granulomatous inflammation, fibrosis, and calcified caseonecrotic debris. The histological pattern is similar in both fungal and mycobacterial infections (Fig. 26.1B)
-
♦
Organisms may be visualized by acid fast stains for mycobacteria or Gomori methenamine silver for yeast forms of Histoplasma capsulatum (Fig. 26.1C)
Differential Diagnosis
-
♦
Mycobacterial vs. fungal infection
-
The histopathological features are similar as discussed above. Organisms may be demonstrated by acid fast or fungal stains
-
-
♦
Sarcoidosis
-
In sarcoidosis, mediastinal lymph nodes are enlarged by nonnecrotizing granulomas. Stains for microorganisms are negative, and granulomas are confined within the capsule of the lymph node
-
-
♦
Infected teratoma or bronchogenic cyst
-
Necrosis may obscure typical identifying features of teratoma or bronchogenic cyst simulating the appearance of a fibrotic cavitated mass of lymph nodes
-
-
♦
Fibrosing mediastinitis
-
Fibrosing mediastinitis (see below) is a more generalized process within the mediastinum, extending well beyond infected lymph nodes. Caseous necrosis is usually absent. The major histological feature is coarse, ropy, sparsely cellular collagen. Obstruction of major mediastinal structures such as the superior vena cava is commonly present
-
Fibrosing Mediastinitis
Clinical
-
♦
Fibrosing mediastinitis is a form of chronic mediastinitis characterized by compressive and obstructive involvement of mediastinal structures. The presenting manifestation is often that of an asymptomatic mediastinal mass. There is a predominance of young adult females
-
♦
A variety of compression syndromes include:
-
Superior vena cava syndrome
-
Tracheobronchial compression (hilar fibrosis)
-
Pulmonary artery or venous obstruction
-
-
♦
Mediastinal fibrosis may be associated with collagen vascular disease, pseudotumor of orbit, Riedel struma, retroperitoneal fibrosis, and pulmonary hyalinizing granuloma. An association with elevated IgG4 levels has also been described
-
The etiology of fibrosing mediastinitis is unknown; however, an exaggerated response to histoplasma infection is thought to be involved in many, if not most, cases in the United States
-
Macroscopic
-
♦
Macroscopically a firm, gray, ill-defined mass of fibrous tissue invades and compresses mediastinal structures
Microscopic
-
♦
Histologically fibrosing mediastinitis is characterized by dense paucicellular hyalinized collagenous tissue (ropy collagen) (Fig. 26.2), with scant lymphocytic and plasmacytic inflammation. Fibromyxoid granulation tissue may represent an early stage of the process. Rare granulomas, if present, may suggest an infectious etiology
Fig. 26.2. 
Fibrosing mediastinitis . Dense, ropy, collagenous fibrosis infiltrates the adjacent lung tissue (right and upper).
-
♦
Secondary effects seen in open lung biopsy include hyalinizing granulomas or are the result of central pulmonary vein occlusion including pulmonary hemosiderosis, venous infarcts, and venous and arterial sclerosis which can simulate pulmonary venoocclusive disease or pulmonary venous hypertension

Immunohistochemistry
-
♦
CD45, CD3, vimentin, or muscle actins are nonspecific markers of lymphocytes or mesenchymal cells in early cellular lesions. There are few reactive cells in sclerotic lesions
Differential Diagnosis
-
♦
Nodular sclerosis Hodgkin disease; other lymphomas with sclerosis
-
Reed-Sternberg cells in Hodgkin disease are positive for CD30 and CD15. Sclerosing lymphomas of the mediastinum are often of B-cell type (see section on “Lymphoproliferative Disorders”)
-
-
♦
Fungal/mycobacterial infection (granulomatous mediastinitis)
-
Histoplasmosis is the most common cause of granulomatous mediastinitis, although mycobacteria are sometimes responsible. Histologically fibrocaseous granulomas form a localized mass (see “Granulomatous Mediastinitis”)
-
-
♦
Amyloidosis
-
Amyloidosis is more amorphous than the ropey collagen of fibrosing mediastinitis. Stains for amyloid include Congo red with apple-green birefringence on polarization or metachromasia with crystal violet stain
-
-
♦
Progressive massive fibrosis (PMF )
-
PMF refers to large conglomerate masses due to the inhalation of coal dust and/or silica. PMF usually resides within the lung parenchyma although lesions may extend to mediastinal structures. Coarse collagen may have a whorled appearance especially when silica exposure has occurred. Abundant black pigment and birefringent crystals consistent with silica and silicates appear throughout the lesion
-
-
♦
Desmoplastic mesothelioma (rare)
-
Desmoplastic mesothelioma is a variant of sarcomatous mesothelioma in which coarse paucicellular collagen is a major histological feature. Mesothelioma may extend from the pleura or pericardium into the mediastinum. Pleomorphic sarcomatoid tumor cells which stain positively for keratin suggest the diagnosis of desmoplastic mesothelioma
-
Cystic Lesions
Bronchogenic Cyst
Clinical
-
♦
Bronchogenic cyst , the most common congenital cyst of the mediastinum, is derived from the embryonic foregut and occurs mainly in children and young adults. Symptoms occur when the cyst becomes infected or compresses bronchi. The usual location is the middle mediastinum
Macroscopic
-
♦
The gross appearance is that of a smooth, thin-walled spherical, unilocular or multilocular, and clear or gelatinous fluid-filled cyst (Fig. 26.3A)
Fig. 26.3. 
(A) Bisected bronchogenic cyst showing inner trabeculations of cyst wall and mucoid content (scale = 1 cm). (B) Epithelial lining of bronchogenic cyst consists of ciliated respiratory epithelium.

Microscopic
-
♦
Respiratory or squamous epithelium forms the inner lining which overlays a wall in which the bronchial glands, smooth muscle, and cartilage may be present (Fig. 26.3B)
Immunohistochemistry
-
♦
The epithelium of bronchogenic cyst stains positively for CK7 and TTF1
Differential Diagnosis
-
♦
Esophageal cyst
-
The esophageal cyst usually occurs on the right side. Histologically it is lined by squamous epithelium and has a wall structure devoid of the cartilage with a prominent double muscle layer
-
-
♦
Metastatic teratoma
-
Metastatic testicular teratoma may occur in the mediastinum. The distinction from bronchogenic cyst includes better differentiated architecture in bronchogenic cyst with predominantly respiratory-type epithelium that is positive for CK7 and TTF1. Metastatic teratoma has mixed enteric and respiratory-type epithelium and variably stains positively for CK20, CDX-2, CK7, and TTF1, respectively
-
Esophageal Cyst
Clinical
-
♦
Esophageal duplication cyst is most frequently seen in infancy and early childhood. The cyst closely approximates the wall of the esophagus in the middle or posterior mediastinum. Symptoms of respiratory distress occur due to compression of bronchopulmonary structures
Macroscopic
-
♦
The gross appearance is that of a smooth-walled spherical, unilocular fluid-filled cyst
Microscopic
-
♦
Histologically the inner lining consists of squamous and/or ciliated epithelium or columnar epithelium (Fig. 26.4A)
Fig. 26.4. 
(A) Esophageal duplication cyst . Thick double-layered smooth muscle. Cyst wall devoid of cartilage. (B) Squamous epithelium lines surface of cyst.
-
♦
Esophageal glands may be present, but the cartilage is absent. The most definitive feature is a double layer of the smooth muscle in the wall (Fig. 26.4B)

Differential Diagnosis
-
♦
Bronchogenic cyst
-
The cartilage is often present in the wall and there is no double muscular layer. Bronchogenic cysts may occur in the wall of the esophagus
-
Gastroenteric Cyst
Clinical
-
♦
Gastroenteric cyst is most frequently seen in infancy and early childhood. The cyst is usually located in the posterior mediastinum and is frequently connected to the vertebral column. Symptoms occur due to compression of bronchopulmonary structures or to nerve compression, peptic ulceration, or perforation
-
♦
Gastroenteric cysts are often associated with malformations of thoracic vertebrae (neurenteric cyst)
Macroscopic
-
♦
The gross appearance is that of a unilocular cyst attached to the vertebral column or within the esophageal wall
Microscopic
-
♦
The inner lining is variable: gastric, duodenal, small intestinal, large intestinal, squamous, or respiratory epithelium (or mixed epithelium). Occasionally pancreatic tissue is present. There is usually a well-developed muscularis propria
Differential Diagnosis
-
♦
Gastroenteric cysts are differentiated from bronchogenic cysts by the absence of the cartilage and the presence of the columnar epithelium. The distinction from esophageal cyst is by location and the presence of gastric or intestinal epithelium
Pericardial Cyst (Coelomic Cyst)
Clinical
-
♦
Pericardial cysts usually occur in adulthood and have a typical location in the right cardiophrenic angle, often an asymptomatic finding on chest X-ray. However, large cysts may cause hemodynamic compromise
Macroscopic
-
♦
The gross appearance is that of a roughly spherical, unilocular, and thin-walled cyst filled with serous fluid (Fig. 26.5A). There is usually no communication with the pericardial sac
Fig. 26.5. 
(A) Pericardial (coelomic) cyst . (B) Cyst lined by flattened mesothelial cells. (C) Cyst-lining cells show nuclear and cytoplasmic staining for calretinin.

Microscopic
-
♦
The inner lining is composed of a single layer of mesothelial cells overlying loose connective tissue (Fig. 26.5B)
Immunohistochemistry
-
♦
Lining mesothelial cells are strongly keratin and calretinin positive (Fig. 26.5C)
Differential Diagnosis
-
♦
See “Bronchogenic Cyst,” “Esophageal Cyst,” and “Gastroenteric Cyst”
-
♦
Thymic cyst
-
Thymic cyst is located in the superior mediastinum, and there is thymic tissue in its wall
-
Unilocular Thymic Cyst (Developmental Origin)
Clinical
-
♦
Unilocular thymic cyst is of developmental origin. It is a small cyst located more often in the lateral neck than in the (anterosuperior) mediastinum
Macroscopic
-
♦
The gross appearance is that of a unilocular cyst with a thin, translucent wall
Microscopic
-
♦
The histological appearance shows flat, cuboidal, columnar, or (rarely) squamous epithelial lining of the inner cyst wall. Usually there is a lack of inflammation and thymic tissue is present in the cyst wall (Fig. 26.6)
Fig. 26.6. 
Unilocular thymic cyst . Thin-walled cyst lined by flat epithelium. Thymic remnant adjacent to cyst. Incidental finding at autopsy.

Differential Diagnosis
-
♦
Pericardial cyst
-
Pericardial cyst is located at the cardiophrenic angles and lacks thymic tissue in its wall
-
Multilocular Thymic Cyst (Acquired [Reactive] Process)
Clinical
-
♦
Multilocular thymic cyst is regarded as an acquired, reactive lesion that clinically presents most frequently as an asymptomatic large tumorlike mass in the anterosuperior mediastinum
-
♦
It is usually an incidental finding on chest X-ray
-
♦
It is frequently associated with thymic neoplasms, such as germ cell tumors, lymphoma, thymoma, or thymic carcinoma
-
♦
Multilocular thymic cyst may arise in setting of HIV infection
Macroscopic
-
♦
The macroscopic appearance is that of a multiloculated cyst with thick fibrous septa forming compartments that contain cloudy to blood-tinged fluid
-
♦
Large cysts may be densely adherent to adjacent mediastinal structures, simulating a neoplasm
Microscopic
-
♦
The lining epithelium of the cysts is usually squamous; however, flat cuboidal or ciliated columnar cells may occur either as a single layer or as a stratified epithelial lining
-
♦
Acute and chronic inflammation with fibrovascular proliferation, necrosis, hemorrhage, cholesterol granulomas, and reactive lymphoid hyperplasia is also common (Fig. 26.7)
Fig. 26.7. 
Multilocular thymic cyst . Multiloculated cyst with dense chronic inflammation associated with thymoma.
-
♦
Thymic tissue is present in the cyst wall

Differential Diagnosis
-
♦
Cystic thymoma
-
As opposed to multilocular thymic cyst, cystic thymoma maintains features of thymoma with nodular proliferation of thymic epithelial cells in addition to prominent cystic changes
-
Cystic teratoma (see “Germ Cell Tumors”)
-
-
♦
Cystic lymphangioma
-
Prominent ectatic lymphatic channels are the hallmark of cystic lymphangioma
-
-
♦
Nodular sclerosis Hodgkin disease and large cell lymphoma with cystic changes (see “Lymphoproliferative Disorders”)
-
♦
Seminoma (germinoma) with cystic changes (see “Germ Cell Tumors”)
-
♦
Thymic carcinoma
-
Thymic carcinoma can be distinguished from multilocular thymic cyst by its malignant cytology showing invasive growth
-
Nonneoplastic Lesions of Thymus
Thymic Dysplasia
Clinical
-
♦
Thymic dysplasia is a condition present at birth, thought to represent failure or arrest in thymic development
-
♦
Thymic dysplasia is associated with various immunodeficiency syndromes, including usual X-linked or autosomal recessive form of severe combined immunodeficiency, ataxia telangiectasia, and related chromosomal instability syndromes, Nezelof syndrome, and incomplete form of DiGeorge syndrome (the thymus is usually located ectopically in DiGeorge syndrome)
Macroscopic
-
♦
The thymus is of very small size (<5 g)
Microscopic
-
♦
The histological features include primitive-appearing epithelium without segregation into cortical and medullary regions
-
♦
Absence of Hassall corpuscles
-
♦
Almost total absence of lymphocytes
-
♦
Small-sized vessels
Differential Diagnosis
-
♦
The major differential diagnosis of thymic dysplasia is acute thymic involution, often seen at autopsy. The discriminating features of acute thymic involution are:
-
Acute thymic involution results from stress and superimposed infections
-
There is marked lymphocytic depletion with preservation of lobular architecture and of Hassall corpuscles
-
Vessels are disproportionately large compared to the size of lobules
-
Inflammatory cells (particularly plasma cells) are scattered within interlobular and perilobular tissue
-
Thymic Aplasia
Clinical
-
♦
Thymic aplasia refers to the complete absence of the thymus
-
♦
Thymic aplasia is present from birth and is most notably associated with the complete form of DiGeorge syndrome
Macroscopic and Microscopic
-
♦
There is a complete absence of the thymus gland, accompanied by parathyroid developmental failures in DiGeorge syndrome
True Thymic Hyperplasia
Clinical
-
♦
True thymic hyperplasia refers to the enlargement of the thymus gland (by weight and volume) beyond upper limit of normal for age
-
♦
Seen mainly in children, the clinical significance of true thymic hyperplasia is unknown. It may represent an immunologic rebound phenomenon (e.g., post-cessation of chemotherapy) or a sequela of other therapeutic manipulation
Macroscopic
-
♦
Enlarged thymus gland by weight and volume
Microscopic
-
♦
Microscopically the thymic architecture is normal
Differential Diagnosis
-
♦
Thymoma
-
True thymic hyperplasia is discerned from thymoma by the preservation of thymic architecture in hyperplasia
-
Lymphoid Hyperplasia (Thymic Follicular Hyperplasia)
Clinical
-
♦
Lymphoid hyperplasia refers to the increased presence of lymphoid follicles within the thymus gland. Lymphoid hyperplasia is associated with myasthenia gravis (in majority of cases), systemic lupus erythematosus, rheumatoid arthritis, scleroderma, allergic vasculitis, thyrotoxicosis, and other autoimmune diseases
Macroscopic
-
♦
The thymus gland is usually of normal size and weight for age
Microscopic
-
♦
There are an increased number of lymphoid follicles with prominent germinal centers (Fig. 26.8)
Fig. 26.8. 
Thymic lymphoid hyperplasia . Prominent lymphoid follicles are present in the thymus gland. Note adjacent Hassall corpuscle (arrow).

Differential Diagnosis
-
♦
Thymoma
-
There is preservation of thymic architecture in lymphoid hyperplasia
-
-
♦
Reactive lymph node hyperplasia or lymphoproliferative lesions
-
Reactive germinal centers with preserved thymic architecture distinguish thymic lymphoid hyperplasia from reactive lymph nodes and from lymphoproliferative disorders involving the thymus
-
-
♦
Mediastinal seminoma with florid follicular lymphoid hyperplasia
-
Variant of mediastinal seminoma in which lymphoid follicular hyperplasia may obscure the germ cell tumor
-
May be underdiagnosed as reactive lymphoid hyperplasia
-
Lesions Mimicking Primary Mediastinal Neoplasms
Substernal Thyroid (Mediastinal Goiter)
Clinical
-
♦
Substernal goiter refers to enlarged thyroid due to nodular hyperplasia usually located in the anterior-superior mediastinum. Mediastinal goiter is usually connected to a cervical goiter and receives its blood supply from cervical arteries
-
♦
The main presentation is an asymptomatic neck mass. Tracheal compression with associated dyspnea is the major complication in about 25% of cases
-
♦
Radioactive iodine scanning is positive in over 50% of cases
-
♦
Associated thyroid malignancy ranges from 2 to 20%
-
♦
Anterior substernal goiter can usually be excised via a neck incision
Macroscopic
-
♦
Features of adenomatous goiter frequently extend from the inferior pole of the thyroid in the neck (Fig. 26.9) (see Chapter 20)
Fig. 26.9. 
Large substernal goiter arising from the lower pole of the left lobe of the thyroid gland (scale = 1 cm).

Microscopic
-
♦
The histopathology is that of typical nodular thyroid hyperplasia as seen in cervical adenomatous goiter (see Chapter 20)
Differential Diagnosis
-
♦
Other mediastinal thyroid lesions such as adenoma or carcinoma are much less frequent
Mediastinal Parathyroid Lesions
Clinical
-
♦
Parathyroid lesions arise from ectopic parathyroid tissue in the superior mediastinum
-
♦
Clinically parathyroid adenomas and hyperplasia are associated with hyperparathyroidism and hypercalcemia
-
♦
Mediastinal parathyroid lesions may assume a larger size than cervical lesions
Macroscopic
-
♦
The macroscopic appearance is that of an enlarged parathyroid gland. Parathyroid cysts are rare
Microscopic
-
♦
The features of parathyroid adenoma or hyperplasia are similar to cervical lesions (Chapter 20)
-
♦
Parathyroid cyst is a simple cyst with parathyroid tissue in its wall
Differential Diagnosis
-
♦
The major differential considerations in the mediastinum are paraganglioma or thymic carcinoid tumor
-
Neither of these lesions is associated with hypercalcemia
-
Histologically chief cells and/or water clear cells indicate parathyroid lesions. Sustentacular cells are seen in paragangliomas
-
Other Conditions
Pneumomediastinum
Clinical
-
♦
Pneumomediastinum refers to air within the fascial planes of the mediastinum, often in association with pulmonary interstitial emphysema, pneumopericardium, pneumothorax, and subcutaneous emphysema
-
♦
The major causes of pneumomediastinum are mechanical ventilation (barotrauma), esophageal perforation, acute mediastinitis, and increased intrathoracic pressure (e.g., straining, Valsalva maneuver)
-
♦
Chest X-ray and CT scan show mediastinal air outlining the aorta, esophagus, and left heart border
Macroscopic
-
♦
Grossly, air bubbles and crepitance in mediastinal tissue are seen at autopsy
-
♦
Associated tension pneumothorax is often also seen in patients receiving mechanical ventilation
Microscopic
-
♦
The microscopic appearance is that of widened, empty interstitial spaces in the mediastinal fat (Fig. 26.10)
Fig. 26.10. 
Pneumomediastinum . Empty-appearing cystic spaces displace the mediastinal fat and fibrovascular tissue (Elastic van Gieson).
-
♦
Associated interstitial pulmonary emphysema shows similar widened spaces which track along bronchovascular bundles within the lung

Differential Diagnosis
-
♦
The major histological differential diagnosis is lymphangiectasia
-
Lymphangiectasia has the same distribution as interstitial air dissection
-
Look for endothelial lining in lymphangiectasia vs. acute hemorrhage and empty spaces in air dissection
-
Neoplasms
Thymus
Histologic Features of Thymus
-
♦
The thymus is a lobulated and encapsulated organ and is subdivided into cortex and medulla
-
♦
The different cell types of the thymus include epithelial cells, lymphocytes (traditionally known as thymocytes), and other (minor) cell types
-
♦
Epithelial cells are endodermally derived cells. They modulate the differentiation of T lymphocytes and are keratin and HLA-DR positive. The different subtypes of epithelial cells include cortical cells that are medium to large, round or polygonal with conspicuous nucleoli, and medullary cells with spindled nuclei and epithelium forming Hassall corpuscles
-
♦
Lymphocytes (thymocytes) are bone marrow-derived cells. The different subtypes include (a) cortical thymocytes which are immature T cells (cytoplasmic CD3+/surface CD3−, CD1a+, terminal deoxynucleotidyl transferase (TdT)+, and double negative for CD4/8 or coexpressing CD4/8) and (b) medullary thymocytes which are mature T lymphocytes (surface CD3+, either CD4+ or CD8+)
-
♦
Other (minor) cell types of the thymus are interdigitating reticulum cells, Langerhans cells, mast cells, eosinophils (especially in neonates), and mesenchymal stromal cells
Thymoma
-
♦
Thymomas are tumors of the thymic epithelium that are cytologically bland and demonstrate the presence of associated cortical-type T cells (cortical thymocytes)
-
♦
Classification is difficult and has been controversial, and systems have been criticized for poor interobserver reproducibility
-
♦
Classification systems for thymoma include:
-
Lattes-Bernatz classification (1961)
-
Marino and Muller-Hermelink classification (1985)
-
Suster and Moran classification (1999)
-
WHO classification (2004 and 2015): most widely used system (Table 26.1)
Table 26.1. The Definitions of the WHO Classification Categories of Thymic Epithelial Tumors -
The International Thymic Malignancy Interest Group (ITMIG) refined guidelines and definitions to WHO classification, including major and minor criteria, and proposed new type A variant (2014)
-
Clinical
-
♦
Thymomas are mainly seen in adults in their fifth, sixth, and seventh decades. They are usually located in the anterosuperior mediastinum. The most common symptoms include dyspnea, chest pain, and cough
-
♦
On chest X-ray, CT, and MRI, a thymoma appears as a lobulated mediastinal mass
-
♦
Thymomas can be seen in association with certain diseases:
-
Myasthenia gravis (mainly associated with type AB, B2, and B3 thymoma)
-
Hypogammaglobulinemia (mainly associated with type A thymoma)
-
Red cell aplasia (mainly associated with type A thymoma)
-
Treatment
-
♦
Surgery alone can be the treatment for an entirely encapsulated thymoma; however, there is a 2–10% recurrence rate. Surgery with radiation is used for invasive thymomas and chemotherapy is used for metastatic disease
Prognosis
-
♦
Thymomas are generally indolent. However, all are considered malignant (regardless of histologic subtype)
-
♦
The degree of tumor invasion (stage of disease) is the most important prognostic factor. WHO histologic subtype may be an independent prognostic factor, particularly in Stage I and II thymomas, among which WHO type A, AB, and B1 thymomas form a low-risk group. All histologic types are capable of invasive growth and metastasis. The proportion of invasive tumors increases by type – from A to AB, B1, B2, and B3. Myasthenia gravis has no prognostic significance
Macroscopic
-
♦
Circumscribed thymomas can range from 2 to 20 cm in size. They are predominantly solid, white to light brown or yellowish gray, lobulated by connective tissue septa, and encapsulated (Fig. 26.11A). Cystic degeneration and necrosis may be seen in larger tumors. Invasive thymomas are grossly similar to circumscribed thymomas but, in addition, infiltrate surrounding structures
Fig. 26.11. 
(A) Circumscribed thymoma (mixed type). Note lobules of tumor separated by bands of connective tissue and central scar. (B) Thymoma , general features. Cellular lobules separated by fibrous septa. (C) Thymoma, general features. Note intact capsule. (D) General features. Invasive thymoma. The thymoma breaks through the capsule and invades the lung parenchyma. (E) Thymoma, general features. Perivascular spaces (serum lakes) containing lymphocytes, proteinaceous fluid, red blood cells, and foamy macrophages.

Microscopic
-
♦
In general (applies to all histologic subtypes), thymomas show cellular lobules separated by fibrous septa (Fig. 26.11B, C). The fibrous capsule is complete in circumscribed thymomas and incomplete with capsular invasion in invasive thymomas (Fig. 26.11D). Varying proportion of lymphocytes and neoplastic epithelial cells is seen. Perivascular spaces containing lymphocytes, proteinaceous fluid, red blood cells, foamy macrophages, or fibrous tissue are also seen (Fig. 26.11E). Occasional gland or pseudogland structures or Hassall corpuscle-like structures can be present
-
♦
Type A and AB thymomas have spindled or oval-shaped epithelial cells, while type B thymomas have epithelioid epithelial cells
-
♦
Type B thymomas are subclassified based on lymphoid cell density, with type B1 having a dense lymphoid component, B2 being intermediate, and B3 having low-absent lymphoid component
-
♦
Most thymomas demonstrate a mixture of different histologic types
-
♦
Histologic subtypes (WHO/ITMIG):
-
Major criteria: required for diagnosis
-
Minor criteria: supportive/helpful, but not required for diagnosis
-
-
♦
Type A (Fig. 26.12A–D):
Fig. 26.12. 
(A–C) Type A thymoma . (D) Epithelial cells in type A thymoma demonstrating immunoreactivity for CD20.
-
Predominantly spindle or oval-shaped epithelial cells are present with few or no mature lymphocytes. This type of thymoma can show variable patterns including storiform, hemangiopericytoma-like, rosette-like, and glandular formation. Perivascular spaces are uncommon. Capsular invasion is rare
-
Major criteria: high epithelial cell content, spindled or oval-shaped tumor cells lacking nuclear atypia, paucity or absence of TdT+ T cells, and absence of medullary islands
-
Minor criteria: large lobular growth pattern, rosettes, subcapsular cysts, focal glandular formations, pericytomatous vascular pattern, paucity or absence of perivascular spaces (PVS), lack of Hassall corpuscles, complete or major encapsulation, expression of CD20 in epithelial cells (Fig. 26.12D), and absence of cortex-specific markers (beta5t, PRSS16, and cathepsin V)
-
-
♦
Atypical type A (new subtype proposed by ITMIG): In addition to the above criteria for type A, also demonstrate increased mitotic activity (four or more mitoses per ten HPF) and coagulative necrosis (tumor necrosis). Necrosis may predict aggressive behavior
-
♦
Type AB :
-
Spindled epithelial cells with focal or diffuse abundance of immature lymphocytes. Perivascular spaces are occasionally seen. Capsular invasion is rare
-
Major criteria: high epithelial cell content, spindled or oval-shaped tumor cells, and moderate to high TdT+ T cells
-
Minor criteria: biphasic pattern at low magnification (due to variable lymphocyte content), medullary islands rare, small lobular growth pattern rare, usually large lobular growth pattern, paucity or absence of perivascular spaces (PVS), expression of CD20 in epithelial cells, and presence of cortex-specific markers (beta5t, PRSS16, and cathepsin V)
-
-
♦
Type B1 (Fig. 26.13A):
Fig. 26.13. 
(A) Type B1 thymoma . (B) Type B2 thymoma. (C) Type B3 thymoma.
-
Closely resembles the normal thymus with cortical areas and medullary islands. Well-formed lobules are seen with intervening sclerotic septa. Scattered large polygonal epithelial cells (keratin+), obscured by a diffuse, small, and round lymphocytic background (CD1a+, TdT+, CD99+, coexpress CD4/8), are present. Medullary islands appear as pale-staining areas with epithelial cells, mature T cells (TdT−), and B cells (CD20+), with or without Hassall corpuscles, surrounded by dark cortical areas. Perivascular spaces are often present. Capsular invasion is rare
-
Major criteria: thymus-like pattern throughout, medullary islands (+/− Hassall corpuscles), no confluence of epithelial cells in cortical areas (clusters of three or more epithelial cells should not be present), and absence of any type A areas
-
Minor criteria: large lobular growth pattern is common (small lobular growth pattern is rare), perivascular spaces commonly present, and keratin+ network (as in the normal thymus)
-
-
♦
Type B2 (Fig. 26.13B):
-
Sheets of polygonal epithelial cells (with large nuclei and prominent nucleoli) are intermixed with lymphocytes. Lobulation is often seen. There is minimal medullary differentiation. Capsular invasion may be present
-
Major criteria: confluence of epithelial cells (at least three adjacent cells) in cortical areas and absence of any type A areas
-
Minor criteria: thymus-like pattern is rare, medullary islands (+/− Hassall corpuscles) occasionally present, small lobular growth pattern is common (large lobular growth pattern is rare), perivascular spaces commonly present, and keratin+ network (denser than in the normal thymus)
-
-
♦
Type B3 (Fig. 26.13C):
-
Sheets of polygonal epithelial cells with sparse or absent immature lymphocytes. Epithelial cells typically have irregular “raisinoid” nuclei, inconspicuous nucleoli, and eosinophilic to clear cytoplasm. In other cases, cells may show large vesicular, hyperchromatic granular nuclei and conspicuous nucleoli. Slight to moderate cellular atypia may be present, and mitotic figures (up to 10/10 HPF) can be seen. They usually show an invasive growth pattern
-
-
♦
Invasive (malignant) thymomas are defined as thymomas with any of the above histologic subtypes with evidence of invasion (Fig. 26.11D)
-
♦
Very rare thymoma subtypes include micronodular thymoma, metaplastic thymoma, microscopic thymoma, sclerosing thymoma, and lipofibroadenoma (for more information on these very rare subtypes, refer to the WHO)


Electron Microscopy
-
♦
Neoplastic epithelial cells show branching tonofilaments, complete desmosomes, elongated cell processes, and basal lamina
Immunohistochemistry (see Table 26.2)
-
♦
Some commonly used antibodies, including pancytokeratins, p63, CD1a, TdT, CD99, CD20, CD5, and CD117, can be very helpful for thymoma classification (see Table 26.2)
-
♦
Specific antibodies for thymoma such as AIRE, FoxN1, and CD205 are available but typically only used in the research setting or at referral centers
Differential Diagnosis
-
♦
Thymic carcinoma (in differential with type B2 and B3 thymoma)
-
Cytologically malignant with high mitotic rate, atypical mitoses, invasive growth, sclerosis in center of tumor, and coagulative necrosis. GLUT-1, c-kit (CD117), and CD5 are positive in thymic carcinoma, but negative in thymoma. Infiltrating lymphocytes are mature lymphocytes and negative for CD1a and TdT (very useful criteria in needle biopsy). Usually thymic carcinomas are not associated with myasthenia gravis
-
-
♦
Neuroendocrine tumor and paraganglioma (in differential with type A thymoma)
-
Unencapsulated; monomorphous cell population with true rosettes, ribbons, or festoons are seen. These tumors show positive staining for neuroendocrine markers
-
-
♦
Thymic Hodgkin lymphoma
-
There is extensive fibrosis with rounded (as opposed to angulated) lobules. Prominent cysts can be seen. Reed-Sternberg cells or lacunar cells (CD15+, CD30+) and mixed inflammatory cells are also seen
-
-
♦
Precursor B or T lymphoblastic lymphoma (LBL)
-
Diffuse growth or thin, separated lobules with numerous mitoses in lymphoid cells. They may be positive for T-cell receptor or immunoglobulin chain gene rearrangement
-
-
♦
Primary mediastinal (thymic) large B-cell lymphoma (diffuse large B-cell lymphoma [DLBCL] with sclerosis)
-
Diffuse growth pattern with variable fibrosis with occasional compartmentalization. Residual cystic thymus can be seen. Lymphocytes have vesicular nuclei, prominent nucleoli, and variable cytoplasm. B-cell markers (CD20) are positive
-
-
♦
Thymic seminoma
-
Subdivided by fine fibrous trabeculae into variable-sized compartments. Placental-like alkaline phosphatase (PLAP) and c-kit positive and keratin negative
-
-
♦
Localized lymphoid hyperplasia (vs. type B1 thymoma)
-
Retention of normal cortical and medullary structure and the presence of lymphoid follicles with germinal centers are seen
-
-
♦
Fibrous histiocytoma and hemangiopericytoma (vs. type A thymoma with storiform and hemangiopericytoma-like growth pattern)
-
Keratin negative
-
-
♦
Spindle epithelial tumor with thymus-like elements (SETTLE )
-
Seen at young age with occurrence in the thyroid gland. Mucous glands are frequently present. Lymphocytes are absent
-
Thymic Carcinoma
Clinical
-
♦
Thymic carcinomas are rarely associated with myasthenia gravis or other thymoma-related paraneoplastic syndromes. Patients are usually asymptomatic or have nonspecific symptoms found by routine chest X-ray. Occasionally they may present with superior vena cava syndrome. The usual metastatic sites are the lymph nodes (mediastinal, cervical, and axillary), bone, lung, liver, and brain. Treatment is surgery and radiation with or without chemotherapy. Prognosis depends on the histologic subtype. Nonkeratinizing carcinomas (including lymphoepithelioma-like tumors), sarcomatoid carcinoma, clear cell carcinoma, and undifferentiated (anaplastic) carcinoma (Fig. 26.14A, B) are very aggressive. Squamous cell carcinomas (SCC) are of intermediate category, and mucoepidermoid and basaloid carcinomas are relatively indolent
Fig. 26.14. 
Thymic carcinoma , undifferentiated (nonkeratinizing type) (A, B). Note infiltrative malignant cells with prominent nucleoli and mitotic figures.

Macroscopic
-
♦
Thymic carcinomas show a homogeneous, yellow to gray cut surface with hemorrhage, necrosis, and infiltrating borders
Microscopic
-
♦
Morphologically these tumors are similar to carcinoma in other organ systems and demonstrate cytologic features of malignancy. They usually lack cortical-type immature T lymphocytes
-
♦
The International Thymic Malignancy Interest Group (ITMIG) proposed criteria for the diagnosis of thymic carcinoma including major criteria (required for diagnosis) and minor criteria (supportive/helpful, but not required for diagnosis)
-
Major criteria: clear-cut atypia of tumor epithelial cells with the severity typical of carcinoma, exclusion of “thymoma with atypia and/or anaplasia” and of typical or atypical carcinoids, and exclusion of metastasis to the thymus and germ cell and mesenchymal tumors with epithelial features
-
Minor criteria: infiltrative growth pattern, small tumor cell nests within desmoplastic stroma, absence of TdT+ T cells (with rare exceptions), epithelial expression of CD5 and CD117, and extensive expression of GLUT1 and MUC1a
-
Features compatible with thymic carcinoma (which are also characteristic of thymoma): invasion with pushing borders, occurrence of perivascular spaces, occurrence of “Hassall-like” epidermoid whorls and/or of myoid cells, and occurrence of (usually rare) TdT+ T cells
-
Subtypes
-
•
Common
-
◦
Keratinizing or nonkeratinizing SCC
-
◦
Lymphoepithelioma-like carcinoma
-
◦
Epstein-Barr virus associated (some cases)
-
◦
-
•
Rare
-
◦
Mucoepidermoid carcinoma
-
◦
Adenosquamous carcinoma
-
◦
Basaloid carcinoma
-
◦
Papillary adenocarcinoma
-
◦
Adenocarcinoma
-
◦
Small cell undifferentiated carcinoma
-
◦
Large cell undifferentiated (anaplastic) carcinoma
-
◦
Sarcomatoid carcinoma, spindle cell carcinoma, and carcinosarcoma
-
◦
Clear cell carcinoma
-
◦
Undifferentiated carcinoma (Fig. 26.14A, B)
-
◦
Hepatoid carcinoma
-
◦
Rhabdoid carcinoma
-
◦
-
•
-
Immunohistochemistry
-
♦
Neoplastic epithelial cells are positive for keratin, EMA, CEA, B72.3, and infrequently Leu-7 (CD57)
-
♦
The epithelial cells in thymic carcinoma cells are usually positive for CD5 (CD5 usually negative in carcinomas other than thymic primary), GLUT1, and CD117
-
♦
Infiltrating lymphocytes are CD1a, CD99, and TdT negative
-
♦
Foxn1 and CD205 are markers for thymic carcinoma but typically only used in the research setting or at referral centers
Differential Diagnosis
-
♦
Thymoma (see above)
-
♦
Carcinoma metastatic to or invading anterior mediastinum (particularly carcinoma of the lung) and malignant mesothelioma
-
♦
Thymic squamous carcinoma often exhibits lobulated growth pattern, central sclerosis, and abrupt keratinization simulating Hassall corpuscles. Other subtypes of thymic carcinoma cannot be differentiated based solely on histology (thymic carcinoma is a diagnosis of exclusion – in the presence of malignant epithelial tumor located in the thymic region in the absence of known primary). CD5 positivity for tumor cells supports the diagnosis of primary thymic carcinoma
-
♦
Germ cell tumors
-
PLAP+, human chorionic gonadotropin (HCG) (see section on “Germ Cell Tumors”)
-
-
♦
Primary mediastinal (thymic) large B-cell lymphoma (diffuse large cell lymphoma with sclerosis)
-
Leukocyte common antigen (LCA)+, CD20 (B-cell marker)+, keratin−, and EMA−
-
-
♦
Carcinoma showing thymus-like elements (CASTLE)
-
Extremely rare tumor of the thyroid gland or soft tissues of the neck morphologically and immunohistochemically similar to thymic carcinoma
-
Generally has better prognosis with more indolent course
-
Thymic Carcinoid (Including Atypical Carcinoid)
Clinical
-
♦
Thymic carcinoids are commonly seen in adults, with a male predominance. Radiologically nonfunctional carcinoids are large, radiopaque, noncystic anterior mediastinal masses with/without fine calcification. Functional carcinoids (associated with Cushing syndrome) are usually of small size and detected by CT scan
-
♦
Carcinoid syndrome is extremely rare. Functional tumors have a more aggressive course, invade locally, but metastasize infrequently. Occasionally they are associated with multiple endocrine neoplasia syndrome (MEN) types 1 or 2a or carcinoid tumors of other sites, such as bronchus and ileum. Overall thymic carcinoids have an approximately 50% 5-year survival. Atypical carcinoids have increased metastatic potential and a survival of approximately 25%. Treatment often consists of multimodal therapy
Macroscopic
-
♦
Thymic carcinoids are solid, well circumscribed, but not encapsulated (Fig. 26.15A)
Fig. 26.15. 
(A) Thymic carcinoid tumor . The hemorrhagic appearance denotes prominent tumor vascularity (scale = 1 cm). (B) Typical carcinoid tumor. Note uniform appearance of cells in a neuroendocrine nesting pattern.
-
♦
Appear as a lobulated, yellow-tan, or hemorrhagic mass

Microscopic
-
♦
Typical carcinoid shows rosette-like glands with central lumina, ribbon, and festoon formation (Fig. 26.15B)
-
♦
Uniform nuclei with low mitotic rate and absence of necrosis
-
♦
Marked vascularization is usually seen
-
♦
Atypical carcinoids (majority of tumors) show nuclear pleomorphism with increased mitotic rate (>2 mitoses per high-power field) and necrosis
Electron Microscopic
-
♦
Dense-core neuroendocrine granules
Immunohistochemistry
-
♦
Keratin+, neuron-specific enolase+, synaptophysin, chromogranin+, and CD56+
-
♦
PAX-8 is positive in about one-third of thymic carcinoids; TTF-1 is infrequently expressed; GATA3 is usually negative
Differential Diagnosis
-
♦
Small cell undifferentiated carcinoma (vs. atypical carcinoid) shows high mitotic rate and small nuclear size, with homogenous chromatin pattern
-
♦
Metastatic neuroendocrine carcinoma
-
♦
Paraganglioma (keratin−, GATA3+)
-
♦
Epithelial cell predominant thymoma may show rosette-like structures frequently associated with lymphocytes
-
Type A thymoma resembles spindle cell carcinoid
-
Thymoma is negative for neuroendocrine markers
-
Thymic Stromal Tumors
Thymolipoma
-
Clinical
-
♦
Seen in young adults (mean age = 20–30 years) with a male to female ratio of 2.3:1
-
♦
Present as a benign, large, and asymptomatic mass (>500 g) of anterior mediastinum
-
♦
Associated with myasthenia gravis, aplastic anemia, and Graves disease
-
♦
Radiographically resemble cardiomegaly or pulmonary sequestration
-
♦
-
Macroscopic
-
♦
These are encapsulated tumors and they can be very large in size. They have the appearance of a lipoma with focal presence of whitish solid areas
-
♦
-
Microscopic
-
♦
There is an admixture in various proportions of mature adipose tissue and unremarkable thymic tissue being in excess of that normally expected for age (Fig. 26.16)
Fig. 26.16. 
Thymolipoma . Thymic tissue is separated by lobules of mature adipose tissue.
-
♦
Myoid cells may rarely be present
-
♦
-
Differential Diagnosis
-
♦
Lipoma
-
Lacks thymic component
-
-
♦
Lymphoid hyperplasia
-
They are smaller size with less adipose tissue and prominent germinal centers
-
-
♦
Thymic stromal sarcomas
-
These are rare, low-grade malignant mesenchymal tumors arising from thymic stroma. Well-differentiated liposarcoma is the predominant component in these tumors (“thymoliposarcoma”)
-
-
♦

Neoplasms Not Limited to Thymus
Neurogenic Tumors
-
♦
Account for approximately 19% of primary mediastinal masses; the majority occur in the posterior mediastinum
-
♦
May be of either sympathetic nervous system origin (neuroblastoma, ganglioneuroblastoma) or peripheral nerve sheath origin (neurilemmoma, neurofibroma)
Peripheral Nerve Sheath Tumors
-
Schwannoma (Neurilemmoma)
-
Clinical
-
♦
Schwannoma is the most common neurogenic tumor of the mediastinum, typically occurring in young adults
-
♦
It is usually asymptomatic, occasionally presenting as a “dumbbell” tumor with extension through intervertebral foramen causing nerve root and spinal cord compression
-
♦
-
Radiology
-
♦
Schwannoma is seen radiographically as a well-circumscribed posterior mediastinal mass, usually single
-
♦
Multiple tumors are associated with von Recklinghausen disease
-
♦
Schwannomas usually have a good prognosis; recurrence is rare
-
♦
-
Macroscopic, Microscopic, and Differential Diagnosis
-
♦
See Chapter 22
-
♦
Mediastinal schwannomas frequently attain large size and are associated with degenerative changes including sclerosis, fatty degeneration, hemorrhage, and cyst formation
-
♦
-
-
Neurofibroma
-
Clinical
-
♦
The clinical presentation of neurofibroma is similar to that of schwannoma. Neurofibroma is second to schwannoma in frequency
-
♦
-
Macroscopic, Microscopic, and Differential Diagnosis
-
♦
See Chapter 22
-
♦
Mediastinal neurofibroma is often enclosed by a complete fibrous capsule
-
♦
-
-
Malignant Peripheral Nerve Sheath Tumor
-
Clinical
-
♦
Malignant peripheral nerve sheath tumor (MPNST ) occurs in the age range of 20–50 years. It is rare in the mediastinum (<10% of thoracic neurogenic tumors). The prognosis of MPNST is poor and characterized by local invasion, recurrence, and metastasis
-
♦
MPNST is associated with von Recklinghausen disease and prior radiation
-
♦
-
Macroscopic, Microscopic, and Differential Diagnosis
-
♦
See Chapter 22
-
♦
-
Tumors of Sympathetic Nervous System
-
Neuroblastoma
-
Clinical
-
♦
Neuroblastoma is the most common mediastinal neurogenic neoplasm in children, particularly <1 year of age. It is usually symptomatic. Prognosis is better than for retroperitoneal neuroblastoma
-
♦
Direct extension may lead to esophageal or spinal nerve root compression or erosion of contiguous vertebral bones. Some patients may develop a paraneoplastic neurologic syndrome known as opsoclonus myoclonus (“dancing feet and dancing eyes”). Failure to thrive is common. The elevated level of catecholamine metabolites in urine or blood may be helpful in establishing the clinical diagnosis
-
♦
More than half of patients present with metastasis at the time of diagnosis. Frequent sites of metastasis include the brain, liver, bone, or regional lymph nodes
-
♦
-
Macroscopic, Microscopic, and Differential Diagnosis
-
♦
See Chapter 20
-
♦
-
-
Ganglioneuroblastoma
-
Clinical
-
♦
Ganglioneuroblastoma usually occurs in older infants and children. It tends to be less aggressive than neuroblastoma
-
♦
-
Macroscopic, Microscopic, and Differential Diagnosis
-
♦
See Chapter 20
-
♦
-
-
Ganglioneuroma
Paraganglioma
Clinical
-
♦
There are two major clinical presentations of mediastinal paragangliomas based on the location of the tumor:
-
Paragangliomas of the anterosuperior mediastinum:
-
•
Associated with aorticopulmonary paraganglia
-
•
Average age = 49 years
-
•
Slight predilection for women
-
•
3% of cases synthesize catecholamines
-
•
-
Paragangliomas of the posterior mediastinum:
-
•
Associated with the aortico-sympathetic ganglia in a paravertebral location
-
•
Average age = 29 years
-
•
Men dominant
-
•
50% of cases synthesize catecholamines
-
•
Secretory paragangliomas produce clinical symptoms similar to those of pheochromocytoma
-
•
-
-
♦
Approximately 50% of cases of mediastinal paraganglioma are associated with significant morbidity or mortality due to infiltrative growth and unresectability due to close association with great vessels. This rate of aggressive behavior is higher than that seen with paragangliomas of other locations
-
There are no well-defined histological criteria which separate benign from malignant paragangliomas
-
A high mitotic rate correlates with clinical malignant behavior
-
-
♦
Carney triad
-
The Carney triad includes functional paragangliomas, pulmonary chondromas, and gastrointestinal malignant stromal tumors
-
The syndrome is most common in young women
-
Macroscopic
-
♦
Paragangliomas are typically solitary and tan to red-brown on cross section. Hemorrhage is common. Larger tumors undergo fibrosis or cyst formation
Microscopic
-
♦
The characteristic microscopic organoid pattern consists of cell nests (zellballen) of cuboidal cells (Fig. 26.17)
Fig. 26.17 
Paraganglioma . Uniform neuroendocrine cells in a zellballen pattern.
-
♦
Nuclear pleomorphism may be considerable with nuclear pseudoinclusions
-
♦
Mitotic figures are scarce
-
♦
Sustentacular cells are seen at the periphery of tumor nests
-
♦
There is a highly vascularized fibrous stroma
-
♦
Stromal hemorrhage or fibrosis may be prominent

Electron Microscopic
-
♦
The dominant chief cells contain abundant cytoplasmic dense-core neurosecretory granules (120–250 nm in diameter). Each granule is round and regular in shape with a clear halo
-
♦
Junctions are a constant feature but true desmosomes are not typically seen
Immunohistochemistry
-
♦
Positivity for peptide hormones including neuron-specific enolase, synaptophysin, chromogranin, somatostatin, neuro-filament, and other peptide hormones
-
♦
GATA3 positive in approximately 50% of paragangliomas
-
♦
Keratin and TTF-1 are negative
-
♦
Sustentacular cells are positive for S-100 protein
Differential Diagnosis
-
♦
Carcinoid tumors are usually keratin positive, are GATA3 negative, and lack sustentacular cells (some carcinoid tumors, however, do have S-100-positive sustentacular cells). The zellballen pattern is usually less pronounced in carcinoid tumor
Germ Cell Tumors
General Features
-
♦
Account for approximately 20% of mediastinal tumors and cysts
-
♦
Germ cell tumors tend to be located in the anterior and superior mediastinum
-
♦
They are usually associated with the thymus
-
♦
In postpubertal adolescents and adults, mature cystic teratoma has an equal sex distribution, while malignant mediastinal germ cell tumors almost exclusively occur in males
-
♦
In prepubertal children, there is an equal sex distribution for teratomas and a female predilection for yolk sac tumors
-
♦
There is an association between germ cell tumors and Klinefelter syndrome
-
♦
There is also an association with hematologic neoplasia (e.g., leukemia, anaplastic large cell [Ki-1] lymphoma)
-
♦
Symptoms of germ cell tumors are due to compression. Some tumors are detected incidentally as an asymptomatic mass
-
♦
Isolated mediastinal metastasis from a testicular or retroperitoneal germ cell tumor must always be excluded but is a rare occurrence
Teratomas
-
Mature Cystic Teratoma
-
Clinical
-
♦
Most common mediastinal germ cell tumor
-
♦
Affects adolescents and young adults
-
♦
There is an equal sex distribution
-
♦
Mature cystic teratoma is often asymptomatic
-
♦
Rarely mature cystic teratoma may erode a bronchus, followed by expectoration of tumor content
-
♦
Surgical excision is curative
-
♦
-
Macroscopic
-
♦
Grossly, mature cystic teratoma appears as a cystic, well-circumscribed mass with fibrous encapsulation; it may contain fat, oily liquid, and hair
-
♦
-
Microscopic
-
♦
The histological picture is similar to gonadal teratoma (see Chapter 32) (Fig. 26.18)
Fig. 26.18. 
Mature cystic teratoma of the thymus . Note teratomatous elements alternating with the thymic tissue (right). A keratinous cyst with adjacent sebaceous glands is a major component of the neoplasm.
-
♦
Pancreatic and gastric tissue is more common than in gonadal teratomas. Thyroid elements are rare
-
♦
Frequently there is a prominent inflammatory reaction with xanthogranulomatous features on the periphery of the neoplasm
-
♦
-
Differential Diagnosis
-
♦
Metastatic teratoma of gonadal origin
-
♦
Immature teratoma
-
Embryonic tissue components
-
-
♦
Teratoma with additional malignant components
-
Mixed germ cell or non-germ cell malignant components
-
-
♦
Multilocular thymic cyst
-
Lacks heterogeneity of tissue components
-
-
♦
Enteric or bronchogenic cyst
-
Enteric cyst situated in the posterior mediastinum
-
Lack heterogeneous tissue components
-
-
♦
-
-
Immature Teratoma
-
Clinical
-
♦
Immature teratoma of the mediastinum is a rare variant
-
♦
Like mature cystic teratoma, immature teratoma has a benign clinical course in children and a variable prognosis in adults
-
♦
-
Macroscopic
-
♦
Macroscopically immature teratoma is typically a large, solid mass which is adherent to mediastinal structures, similar to mature cystic teratoma
-
♦
The cut surface is variegated
-
♦
-
Microscopic
-
♦
Contains immature epithelial, mesenchymal, or neural elements (most common), with or without elements of mature teratoma
-
♦
Embryonal carcinoma is not present
-
♦
-
Differential Diagnosis
-
♦
See mature teratoma (above)
-
♦
-
-
Teratoma with Additional Malignant Components
-
Clinical
-
♦
This variant has an aggressive course with invasion of adjacent structures
-
♦
-
Macroscopic
-
♦
Grossly the lesion appears solid with areas of hemorrhage and necrosis
-
♦
-
Microscopic
-
♦
Microscopically one sees mature or immature teratoma with components of other germ cell tumors (mixed germ cell tumor with teratomatous component) (Fig. 26.19)
-
Teratocarcinoma refers to the combination of embryonal carcinoma and teratoma
Fig. 26.19. 
Mixed germ cell tumor . (A) Mature teratoma component. (B) Embryonal cell carcinoma showing sheets of pleomorphic cells. (C) OCT4 positivity. (D) CD30 positivity.
-
-
♦
Teratoma with somatic malignant transformation (carcinoma, sarcoma). This microscopic pattern is associated with a very poor prognosis and lack of response to germ cell-type chemotherapy protocols
-
Sarcoma is the most frequent somatic component, followed by peripheral neuroectodermal tumor and adenocarcinoma
-
-
♦
-


Seminoma (Germinoma )
-
Clinical
-
♦
Males predominant, exceedingly rare in females
-
♦
Occurs in the second to fourth decades
-
♦
Responsive to radiation therapy (up to 100% 5-year survival)
-
♦
Serum HCG may be positive
-
♦
-
Macroscopic
-
♦
Solid, lobulated, and homogeneous mass
-
♦
-
Microscopic
-
Immunohistochemistry
-
♦
Immunopositive for OCT4, SALL4, CD117, SOX17, and PLAP
-
♦
Immunoreactive for low molecular weight keratin
-
♦
PAX-8 inconsistently positive
-
♦
Glypican-3 and CD30 negative
-
♦
-
Cytogenetics
-
♦
80% of mediastinal germ cell tumors of all histological types show gain of 12p [i(12p)], detectable by FISH
-
♦
-
Differential Diagnosis
-
♦
Lymphoepithelioma-like carcinoma of the thymus
-
Immunopositive for EMA and cytokeratin; negative for PLAP
-
-
♦
Diffuse large cell lymphoma
-
Immunopositive for leukocytic common antigen
-
-
♦
Hodgkin disease
-
Reed-Sternberg cells are present, positive for CD15 and CD30
-
Negative for PLAP
-
-
♦
Non-seminomatous germ cell tumors (see below)
-
♦
Metastatic melanoma
-
Immunopositive for HMB-45, S-100, and Melan A
-
-
♦

Embryonal Carcinoma
-
Clinical
-
♦
Embryonal carcinoma is a rare, highly malignant mediastinal tumor
-
♦
Embryonal carcinoma almost exclusively affects males
-
♦
-
Macroscopic
-
♦
Grossly embryonal carcinoma appears as a large, solid, hemorrhagic, and necrotic mass
-
♦
-
Microscopic
-
♦
Poorly differentiated, with necrosis (identical to gonadal embryonal carcinoma)
-
♦
Usually mixed with other germ cell tumor components (mixed germ cell tumor) (Fig. 26.19)
-
♦
-
Immunohistochemistry
-
♦
Embryonal carcinoma is immunoreactive for keratin, SALL4, OCT4, and CD30. Variable staining for PLAP and AFP. CD117 is usually negative
-
♦
-
Differential Diagnosis
-
♦
Undifferentiated carcinoma (e.g., thymic carcinoma) of the mediastinum
-
Occurrence in a young male should raise concern of possible embryonal carcinoma
-
Undifferentiated carcinoma is immunopositive for keratin, but negative for CD30 and PLAP
-
-
♦
Metastatic adenocarcinoma
-
Adenocarcinoma is negative for OCT4, PLAP, and CD30
-
-
♦
Yolk sac tumor
-
Histologically yolk sac tumor includes the presence of Schiller-Duval bodies and a reticular pattern (see below)
-
-
♦
Yolk Sac Tumor
-
Clinical
-
♦
Yolk sac tumor affects males in the third and fourth decades
-
♦
It is associated with high levels of serum alpha-fetoprotein
-
♦
Yolk sac tumor may be associated with hematologic neoplasia
-
♦
There is a poor prognosis for pure mediastinal yolk sac tumor
-
♦
-
Macroscopic
-
♦
Large, necrotic, and invasive mass
-
♦
-
Microscopic
-
♦
Mediastinal yolk sac tumor is histologically similar to gonadal yolk sac tumor but may have prominent spindle cell or hepatoid features
-
Solid pattern may mimic seminoma
-
-
♦
Yolk sac tumor is usually mixed with teratoma or other germ cell tumor elements (i.e., mixed germ cell tumor)
-
♦
-
Immunohistochemistry
-
♦
Yolk sac tumor is positive for AE1/3, SALL4, Glypican-3, and AFP
-
♦
Negative for OCT4 and CD30
-
♦
-
Differential Diagnosis
-
♦
Embryonal carcinoma
-
Absence of Schiller-Duval bodies or a reticular growth pattern
-
Embryonal carcinoma is immunopositive for OCT4 and CD30
-
-
♦
Seminoma (vs. solid pattern of yolk sac tumor)
-
AE1/3 usually negative and Glypican-3 negative in seminoma
-
AFP may be negative in solid pattern yolk sac tumor
-
-
♦
Choriocarcinoma
-
Clinical
-
♦
Choriocarcinoma affects males in their third decade
-
♦
Pure choriocarcinoma is a very rare subtype in the mediastinum
-
♦
In evaluating a possible mediastinal choriocarcinoma, it is important to exclude metastasis from an occult testicular primary choriocarcinoma. Testicular choriocarcinoma may regress in the testis as metastases develop
-
♦
Serum HCG is usually elevated
-
♦
Gynecomastia is a common symptom
-
♦
Mediastinal choriocarcinoma has a poor prognosis
-
♦
-
Macroscopic
-
♦
Choriocarcinoma appears grossly as a necrotic, hemorrhagic mass
-
♦
-
Microscopic
-
♦
Identical to gonadal choriocarcinoma (see Chapter 39)
-
♦
-
Immunohistochemistry
-
♦
Choriocarcinoma is positive for HCG, AE1/AE3, Glypican-3, PLAP, and SALL4 (variable)
-
♦
Negative for OCT4
-
♦
-
Differential Diagnosis
-
♦
Metastatic choriocarcinoma from a gonadal primary must always be excluded for mediastinal choriocarcinomas. Testicular choriocarcinomas may regress as metastases develop
-
♦
Mixed germ cell tumor includes other components in addition to choriocarcinoma
-
♦
Anaplastic carcinoma
-
Immunoreactive for keratin and negative for HCG (however, some large cell carcinomas of lung are immunoreactive for HCG)
-
-
♦
Lymphoproliferative Disorders
General
-
♦
Lymphoproliferative disorders can occur in all mediastinal compartments. Lymphomas are the most common primary neoplasms of the middle portion of the mediastinum. Lymphoma may be a primary mediastinal process or a manifestation of disseminated disease (see Chapter 15)
Hodgkin Lymphoma
-
Clinical
-
♦
Hodgkin lymphoma involves the thymus and/or lymph nodes. Hodgkin lymphoma occurring in the mediastinum is usually seen in young adults with a female predominance. Patients may present with local pressure symptoms (dyspnea, cough, or chest pain) or an incidental finding on chest X-ray. Mediastinal Hodgkin lymphoma is nearly always nodular sclerosis type and frequently also involves the cervical lymph nodes
-
♦
-
Macroscopic
-
♦
These are well-circumscribed tumors with a thick capsule. They can mimic a thymoma. They are composed of single or multiple hard nodules with a lobulated pattern. Occasionally they can be cystic (within the thymus)
-
♦
-
Microscopic
-
♦
Cellular nodules surrounded by fibrous bands are seen (Fig. 26.21A). They are composed of a polymorphic cell population with diagnostic lacunar cells (Fig. 26.21B) and Reed-Sternberg (R-S) cells with a mixed inflammatory background
Fig. 26.21. 
Hodgkin lymphoma . (A) Cellular nodules surrounded by fibrous bands. (B) Polymorphic cell population with diagnostic lacunar cells. (C) CD15 stain. (D) CD30 stain. Note targetoid staining pattern in large neoplastic cells in (C) and (D).
-
♦
-
Immunohistochemistry
-
Differential Diagnosis
-
♦
Thymoma (see “Thymoma”)
-
♦
Thymic cysts (see “Thymic Cyst”)
-
♦

Primary Mediastinal (Thymic) Large B-Cell Lymphoma (Pmbl)
-
Clinical
-
♦
Large B-cell lymphoma presents as a mass in the thymus with or without lymph node involvement. It is usually seen in young adult females (<35 years old). Patients frequently present with superior vena cava syndrome. On radiology a large mass in the anterior mediastinum is seen. The mass frequently invades adjacent structures such as the large vessels, pericardium, pleura, lung, and chest wall. These tumors may recur (usually involving the kidney) after initial good response to chemotherapy and radiotherapy
-
♦
-
Macroscopic
-
♦
These are large tumors (>10 cm) with grossly invasive features and show extension into the pericardium, pleura, lung, sternum, and chest wall. They are firm with lobulation and foci of necrosis
-
♦
-
Microscopic
-
♦
These tumors have lobules separated by wide fibrous bands (Fig. 26.22A). Neoplastic lymphocytes with large, vesicular, and irregularly shaped nuclei (indented, kidney-shaped, and polylobated) and abundant pale to basophilic cytoplasm (Fig. 26.22B) are also present. Frequent mitotic figures are seen. There is entrapment of intrathymic and perithymic fat. Invasion of blood vessel wall, pleura, or lung can be seen. Sometimes abundant reactive histiocytic cells are also present
Fig. 26.22. 
Primary mediastinal (thymic) large B-cell lymphoma . (A) Lobules separated by wide fibrous bands. (B) Neoplastic lymphocytes with large, vesicular, and irregularly shaped nuclei (indented, kidney-shaped, polylobated) and abundant pale to basophilic cytoplasm. (C) CD20 stain.
-
♦
-
Immunophenotyping and Genotyping
-
♦
Majority of cases have B-cell phenotype (CD19+, CD20+ (Fig. 26.22C), CD5−, CD23+/−, CD30+ (weak and heterogeneous)/−, MPL+/−, CD15−, EBER (EBV-encoded RNA)−, CD10+/−, Bcl-6+/−, MUM1+/−)
-
♦
Surface immunoglobulins are often negative
-
♦
Positive for heavy chain and light chain gene rearrangements
-
♦
Molecular signature of PMBL is more closely related to classical Hodgkin lymphoma than to the other diffuse large B-cell lymphoma subtypes
-
♦
-
Differential Diagnosis
-
♦
Malignant thymoma: keratin+
-
♦
Germinoma: PLAP+
-
♦
Hodgkin lymphoma: CD15+, CD30+
-
♦
Metastatic undifferentiated carcinoma: keratin+
-
♦
Metastatic amelanotic melanoma: S-100 protein+, HMB45+
-
♦
Other systemic lymphomas
-
♦

B-Cell Lymphoma , Unclassifiable, with Features Intermediate Between Diffuse Large B-Cell Lymphoma And Classic Hodgkin Lymphoma
-
-
♦
These are B-cell lymphomas that show clinical, morphological, and/or immunophenotypic features of both DLBCL and classic Hodgkin lymphoma (HL)
-
♦
They are usually seen in young males (a median age of 29.5 years, range 16–51 years) but may be seen in younger or older individuals. In about 20% or fewer cases, Epstein-Barr viral sequences have been identified. These tumors most commonly present as a large anterior mediastinal mass with or without supraclavicular lymph node involvement. Patients may present with superior vena cava syndrome. These lymphomas have an aggressive clinical course and relatively poorer prognosis compared to either primary mediastinal B-cell lymphoma or classic HL. Some cases have also been reported in peripheral lymph node groups without mediastinal involvement. These cases tend to occur in older adults (a median age of 55 years, range, 24–91 years)
-
♦
-
Microscopic
-
♦
Sheets of pleomorphic cells within a diffusely fibrotic stroma are seen. There is variation in the cytomorphological characteristics in different areas of the tumor with some areas showing features of classic HL and other areas showing features of DLBCL. Mild inflammatory infiltrate can be seen. Necrosis is frequent
-
♦
-
Immunophenotyping and genotyping
-
♦
In cases with overall morphologic features more closely resembled classical Hodgkin lymphoma, the tumor cells usually show CD30 expression and positive staining for CD20 (strong and homogenous), CD79a, BOB1, Oct-2, and/or CD45. CD15 can be positive in about 50% of cases
-
♦
In cases with morphologic features more characteristic of large B-cell lymphoma, the tumor cells show consistent positivity for CD30 and frequent positivity for CD15 (80% of cases). CD20 and/or CD79a are strongly expressed in the vast majority of cases. BOB.1 and Oct-2 are positive in the majority of cases
-
♦
Amplifications in 2p16.1 (REL/BCL11A locus) and gains of 8q24 (MYC) are observed in 33 and 27% of cases, and alterations affecting the JAK2/PDL2 locus in 9p24.1 are present in 55%. The rearrangement of the CIITA locus at 16p13.13 is noted in 27% of cases
-
♦
-
Differential Diagnosis
-
♦
Classic Hodgkin lymphoma
-
♦
Primary mediastinal (thymic) large B-cell lymphoma
-
♦
ALK-negative anaplastic large cell lymphoma
-
♦
Precursor B Or T Lymphoblastic Lymphoma/Leukemia (Lbl)
-
Clinical
-
♦
LBLs have a predilection for the thymic region. Children and adolescents present with acute respiratory distress due to a large mediastinal mass. There is a male predominance. The bone marrow, lymph node, central nervous system, and gonads are also usually involved
-
♦
-
Macroscopic
-
♦
These are solid, soft, and nonencapsulated masses
-
♦
-
Microscopic
-
♦
There is a diffuse and infiltrative pattern of atypical lymphocytic growth involving thymic parenchyma. The neoplastic lymphocytes are medium sized with very fine chromatin pattern with frequent nuclear convolutions (Fig. 26.23A). Numerous mitotic figures and necrotic/apoptotic cells are seen. A starry sky pattern similar to that seen in Burkitt lymphoma is seen. The neoplastic lymphocytes extend into perithymic fat and blood vessels
Fig. 26.23. 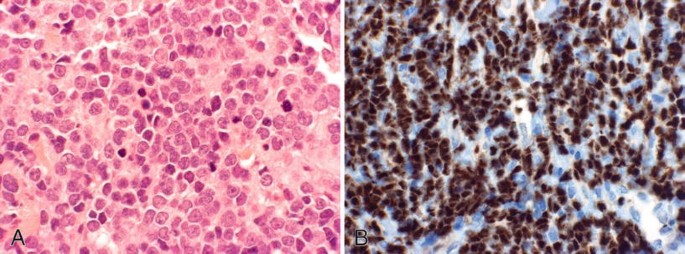
Lymphoblastic lymphoma . (A) Neoplastic lymphocytes with a medium-sized, very fine chromatin pattern with frequent nuclear convolutions. Frequent mitotic figures are present. (B) TdT stain. Note the nuclear stain pattern.
-
♦
-
Immunophenotyping and Genotyping
-
♦
T cells (80% of cases)
-
Immunophenotyping pattern is similar to cortical thymocytes (TdT+ (Fig. 26.23B), CD1a+, coexpression of CD4/CD8). There usually is positive T-cell receptor gene rearrangement
-
-
♦
B cells (20% of cases)
-
TdT and B-cell marker (CD19, PAX5) are positive. Usually, positive immunoglobulin heavy chain and light chain gene rearrangement is also detected
-
-
♦
-
Differential Diagnosis
-
♦
Type B1 thymoma
-
Residual thymic lobules and Hassall corpuscles in LBL can mimic thymomas. Lymphocytes can be frequent in some thymoma subtypes. Other features of thymoma such as perivascular spaces are not present. True thymomas are rare in children
-
-
♦
Other lymphomas
-
♦

Low-Grade B-Cell Lymphoma of Malt Type
-
Clinical
-
♦
The tumor is localized at diagnosis. T his lymphoma is more common in Asians and has a female predilection (male/female = 1:4), with a mean age of 55 years at diagnosis. The tumor has an indolent clinical course and is usually cured by excision. Some cases are associated with autoimmune diseases such as Sjögren’s syndrome, rheumatoid arthritis, and systemic lupus erythematosus
-
♦
-
Macroscopic
-
♦
They are encapsulated masses ranging from 3 to 17 cm (average, 9.2 cm) in largest diameter with a pale, tan, homogenous cut surface, many with fluid-filled cysts
-
♦
-
Microscopic
-
♦
Normal architecture of the thymus is obscured by lymphoid infiltrate, in which Hassall corpuscles are seen. Lymphoid infiltrate consists of reactive type of follicles and diffuse growth of predominantly “centrocyte-like lymphocytes” (monocytoid B cells), with abundant, clear, faintly granular cytoplasm and sharp cell borders, admixed with scattered large transformed lymphocytes and scattered plasma cells. Scattered cysts lined by attenuated epithelium and/or Hassall corpuscles are infiltrated by neoplastic lymphocytes forming prominent lymphoepithelial lesions . Plasmacytic differentiation is usually apparent
-
♦
-
Immunophenotyping
-
♦
The neoplastic lymphocytes are CD20+, CD22+, CD5−, CD10−, and T-cell markers
-
♦
The plasma cells express immunoglobulin (Ig) A phenotype, in striking contrast with the IgM phenotype observed in most of MALT lymphomas at other sites
-
♦
Granulocytic Sarcoma (Myeloblastoma, Chloroma)
-
Clinical
-
♦
Granulocytic sarcoma may occur de novo, as a manifestation of acute myeloid leukemias at presentation or relapse or as a manifestation of blastic transformation of a myeloproliferative and/or myelodysplastic neoplasm such as chronic myeloid leukemia. They rarely present in the mediastinum
-
♦
-
Macroscopic
-
♦
These tumors show a greenish hue on cut surface
-
♦
-
Microscopic
-
♦
Blastic lesions are composed of predominantly myeloblasts. Immature lesions are composed of myeloblasts and promyelocytes. Differentiated lesions are composed of promyelocytes and cells at later stages of maturation
-
♦
-
Immunophenotyping and Enzyme Histochemical Stains
-
♦
CD43+, myeloperoxidase+, lysozyme+, chloroacetate esterase+
-
♦
-
Differential Diagnosis
-
♦
Large cell lymphoma and LBL: see “Lymphoproliferative Disorders” and Chapter 15
-
♦
Undifferentiated carcinomas are keratin+
-
♦
Extramedullary Plasmacytoma
-
Clinical
-
♦
Extramedullary plasmacytomas rarely involve the mediastinum
-
♦
-
Macroscopic, Microscopic, and Differential Diagnosis
-
♦
See Chapter 15
-
♦
Castleman Disease (Cd )
-
Hyaline-Vascular Type
-
Clinical
-
♦
This type of CD usually presents unicentrically, involving a single node or localized group of nodes (75–90% of localized CD) and occurring in equal numbers of young adult men and women with the median age of the fourth decade. This form of CD most frequently occurs in a mediastinal lymph node although various extranodal presentations also occur. Most cases of localized hyaline-vascular CD present with a mass lesion and lack the systemic signs and/or symptoms
-
♦
-
Macroscopic
-
♦
Castleman disease presents as a single encapsulated mass with a solid, homogenous, gray nodular and sometimes hemorrhagic appearance
-
♦
-
Microscopic
-
♦
The tumors show abnormal follicles and striking interfollicular vasculature (Fig. 26.24A). Follicles with expanded mantle zones of homogenous small lymphocytes forming concentric rings (“onion skinning”) and atrophic germinal centers are seen. One or more small blood vessels are seen entering from the perifollicular tissue into follicles creating the appearance of a lollipop (Fig. 26.24B), and the vessels have hyalinized and thickened walls. More than one small germinal center within a single follicle may be seen
Fig. 26.24. 
Castleman disease (angiofollicular lymphoid hyperplasia), hyaline-vascular type. (A) Abnormal follicles with atrophic germinal centers and expanded mantle zones and striking interfollicular vasculature. (B) One or more small blood vessels entering from the perifollicular tissue into follicles.
-
♦
-
Clinical Association
-
♦
The association between hyaline-vascular CD and follicular dendritic cell tumors has been observed, with some cases behaving in a malignant fashion
-
♦
-
-
Plasma Cell Type
-
Clinical
-
♦
This type of CD is more commonly multicentric than unicentric. The unicentric form usually affects an aggregate mass of lymph nodes, as opposed to a dominant lymph node in hyaline-vascular unicentric CD. The unicentric form occurs in a similar patient population as the hyaline-vascular type but is often associated with systemic symptoms and signs, such as fevers, night sweats, malaise, splenomegaly, hypergammaglobulinemias, anemia, and thrombocytopenia. The localized form likely represents an early stage of the multicentric form since cases of unicentric form seem to be associated with an increase in serum interleukin (IL)-6, similar to multicentric form. The patient population of multicentric form without HHV-8 (human herpes virus-8) infection is generally older than that of localized form, with the median age being in the sixth decade. Of note, surgical excision may be curative in unicentric form, as opposed to the multicentric form, which requires systemic therapy
-
♦
-
Macroscopic
-
♦
This type presents as several discrete matted nodes or a mass with adjacent smaller nodes
-
♦
-
Microscopic
-
♦
There is preserved lymph node architecture, with variable germinal center hyperplasia showing expanded mantle zones and a marked paracortical plasmacytosis (Fig. 26.25). Hyaline-vascular type follicles may occur in some cases providing a link to the hyaline-vascular type, and some have considered these cases as mixed or transitional types of CD. Plasma cells may be monotypic and usually express λ-restricted light chains, especially in cases associated with POEMS syndrome. As these histologic features are nonspecific, exclusion of other entities that may simulate plasma cell CD is important
Fig. 26.25. 
Castleman disease, plasma cell type. Interfollicular areas are densely infiltrated by plasma cells.
-
♦
Immunohistochemical stain for Human herpes virus-8 (HHV-8) latency-associated nuclear antigen-1 (LANA-1) is recommend to exclude the HHV-8-associated (plasmablastic) multicentric Castleman disease (see below)
-
♦
-
Clinical Association
-
♦
Both the unicentric and multicentric forms of plasma cell CD may be associated with POEMS syndrome (polyneuropathy, organomegaly, endocrinopathy, M protein, and skin changes)
-
♦
-


Human Herpes Virus-8-Associated (Plasmablastic) Multicentric Type
-
Clinical
-
♦
Multicentric CD most frequently affects immunosuppressed individuals, especially the HIV-positive patient population, and has been referred as HHV-8-associated or plasmablastic variant of CD. HHV-8-associated multicentric CD has a risk of progression to large B-cell lymphoma (see below). The prognosis of HHV-8-associated multicentric CD is usually poor, with average survival on the order of months
-
♦
-
Microscopic
-
♦
The morphology is similar to that of plasma cell variant described above. Additionally, there is usually a population of larger plasmablasts (plasmacytoid immunoblasts) that localize to the mantle zone or paracortical regions. These plasmablasts express HHV-8-related antigen (specifically, latency-associated nuclear antigen-1), cytoplasmic IgM, and λ-restricted light chains by immunohistochemistry. In most cases of HHV-8-positive CD, these “plasmablasts” are scattered throughout the interfollicular regions among the plasma cell infiltrate and may form microscopic collections (microlymphomas). Initially the IgM λ-restricted plasmablasts are monotypic, but polyclonal, and may progress to monoclonal proliferations later in the disease course
-
♦
-
Clinical Association
-
♦
In some patients, this type of CD may progress to large B-cell lymphoma arising in HHV-8-associated multicentric CD. Association with Kaposi sarcoma has also been reported
-
♦
-
Differential Diagnosis
-
♦
For hyaline-vascular type
-
Other lymphomas including mantle cell lymphoma, follicle center lymphoma, and angioimmunoblastic T-cell lymphoma
-
-
♦
For plasma cell type
-
Lymphadenopathy in patients with rheumatoid arthritis
-
•
Clinical correlation
-
•
-
Lymphadenopathy in patients with syphilis
-
•
Nonnecrotizing (sarcoidal) or necrotizing granulomas are seen
-
•
Spirochetes may be found
-
•
-
Lymphoplasmacytic lymphoma
-
Marginal zone lymphoma
-
-
♦
For both types
-
Lymphadenopathy in patients with HIV infections
-
-
♦
Mesenchymal Tumors (Other than Nerve Sheath Tumors)
-
♦
Mesenchymal tumors account for <2% of primary tumors of the mediastinum
-
♦
Many types of mesenchymal tumor have been described
-
♦
Most mesenchymal tumors have similar clinical findings, including mediastinal mass and/or compressive symptoms
Benign (Low-Grade) Mesenchymal Tumors
-
Lipoma
-
Clinical
-
♦
Usually located in anterior compartment
-
♦
Tends to be bulky with extension to bilateral pleural cavities
-
♦
Lipoma can cause respiratory embarrassment
-
♦
-
Differential Diagnosis
-
♦
Thymolipoma
-
In thymolipoma , there is the presence of thymic tissue in addition to mature adipose tissue
-
-
♦
Lipomatosis
-
Refers to a diffuse accumulation of adipose tissue seen in association with obesity, Cushing syndrome, and steroid therapy
-
Associated with “saber-sheath” tracheal deformity
-
-
♦
Other benign adipose tissue tumors and low-grade liposarcoma (see Chapter 22)
-
♦
-
-
Lymphangioma
-
Clinical
-
♦
Lymphangiomas occur most frequently in the anterosuperior mediastinum
-
♦
Lymphangiomas usually occur in children; often, there is a cervical component (i.e., cystic hygroma)
-
♦
-
Macroscopic
-
♦
The gross appearance of lymphangioma is that of a circumscribed mass with large, cystic, and smooth-walled spaces
-
♦
-
Microscopic
-
♦
The histologic appearance of lymphangioma includes endothelial-lined lymphatic channels accompanied by a lymphocytic infiltrate
-
♦
-
Immunohistochemistry
-
♦
D2-40 highlights lymphatic endothelial cells
-
♦
-
Differential Diagnosis
-
♦
Hemangioma
-
In hemangioma, the cystic spaces are blood filled
-
CD31 positive in vascular endothelial cells. D2-40 negative
-
-
♦
Lymphangioleiomyomatosis (LAM )
-
Prominent smooth muscle component (immunoreactive for HMB 45)
-
LAM is infiltrative and occurs almost exclusively in females
-
-
♦
-
-
Hemangioma
-
Clinical
-
♦
Rare. Comprise less than 0.5% of mediastinal tumors
-
♦
Usually occur in anterior mediastinum
-
♦
May be complicated by Kasabach-Merritt syndrome
-
♦
-
Macroscopic
-
♦
Poor circumscription, large cystic spaces filled with blood
-
♦
-
Microscopic
-
♦
Cavernous hemangioma is more common in adults. Histologically dilated blood-filled spaces are separated by thin fibrous trabeculae
-
Thrombosis, calcification, and cholesterol granulomas are commonly present
-
-
♦
Cellular hemangiomas are more commonly seen in children
-
♦
-
Differential Diagnosis
-
♦
Lymphangioma (see above)
-
♦
Other vascular tumors include hemangiopericytoma, epithelioid hemangioendothelioma, and angiosarcoma (see Chapter 22)
-
♦
-
-
Solitary Fibrous Tumor
-
Clinical
-
♦
Solitary fibrous tumor of the mediastinum is analogous to solitary fibrous tumor of the pleura. Solitary fibrous tumor of the mediastinum accounts for approximately 15% of all solitary fibrous tumors. Some tumors may extend from the mediastinal pleura; however, most solitary fibrous tumors probably originate from mediastinal fibrous stroma. Some cases appear to arise from the thymus
-
♦
Solitary fibrous tumor of the mediastinum tends to be locally infiltrative and may run an aggressive course. There is a higher rate of sarcomatous transformation than in solitary fibrous tumors of the pleura
-
♦
Some mediastinal solitary fibrous tumors are associated with hyperglycemia, like their pleural counterparts
-
♦
-
Macroscopic
-
♦
Solitary fibrous tumor tends to be large, tan, firm, and well circumscribed
-
♦
-
Microscopic
-
♦
Solitary fibrous tumor is histologically represented by a variable combination of bland-appearing short spindle cells and fibrous stroma with thick collagen bundles
-
♦
-
Immunohistochemistry
-
♦
Solitary fibrous tumors are immunoreactive for CD34 and bcl-2 and are negative for cytokeratin
-
♦
-
Differential Diagnosis
-
♦
Thymoma (hemangiopericytoma-like) is immunoreactive for cytokeratin and negative for CD34
-
♦
Synovial sarcoma features malignant spindle cells which are immunoreactive (focally) for cytokeratin and bcl-2 and negative for CD34
-
♦
Sarcomatous mesothelioma features malignant-appearing spindle cells which are immunoreactive for keratin and negative for CD34
-
♦
Fibromatosis shows poor circumscription and is immunohistochemically negative for CD34 and bcl-2
-
♦
Spindle cell liposarcoma lacks the dense collagen of solitary fibrous tumor. Spindle cell liposarcoma is immunoreactive for CD34 and may weakly express bcl-2 and S-100
-
♦
Leiomyomas are immunoreactive for smooth muscle markers (smooth muscle actin and desmin) and are negative for CD34 and bcl-2
-
♦
Nerve sheath tumors are immunoreactive for bcl-2 and S-100 but are negative for CD34
-
♦
-
Malignant Mesenchymal Tumors
-
Liposarcoma
-
Clinical
-
♦
Most common malignant mesenchymal tumor of the mediastinum and may be associated with liposarcoma at other body sites
-
♦
Liposarcoma usually occurs in adults and is rare in children
-
♦
All mediastinal compartments may be involved
-
♦
Some liposarcomas may be cured by surgical excision; however, recurrence rate is high
-
♦
The overall prognosis is poor
-
♦
-
Microscopic
-
♦
The histological appearance of mediastinal liposarcoma is similar to that in other soft tissue locations, including well-differentiated liposarcoma or dedifferentiated liposarcoma (58% of cases), low-grade myxoid liposarcoma (8%), and pleomorphic liposarcoma (29%)
-
♦
Some liposarcomas may include a heavy lymphoid infiltrate mimicking lymphoma
-
♦
Uncommon patterns such as myxoid change in well-differentiated liposarcoma and pleomorphic liposarcoma may be more frequent in the mediastinum
-
♦
-
Differential Diagnosis
-
♦
Metastatic liposarcoma or direct extension from the retroperitoneum
-
♦
Lipoma
-
In lipoma, lipoblasts are absent
-
-
♦
Thymolipoma
-
Remnants of the thymus gland are present in association with lipomatous neoplasm
-
-
♦
-
-
Synovial Sarcoma
-
Clinical
-
♦
Synovial sarcoma is rare in the mediastinum where it tends to occur in the superior and middle compartments
-
♦
Synovial sarcoma is usually a tumor of adults. Symptoms often include chest and shoulder pain, with or without dyspnea. Pericardial effusion is relatively common (approximately 22% of cases). Superior vena cava syndrome is rare (3%)
-
♦
Most cases follow a highly aggressive course
-
♦
-
Macroscopic and Microscopic
-
♦
Similar to soft tissue counterpart (see Chapter 22)
-
♦
Monophasic synovial sarcoma is slightly more frequent than biphasic synovial sarcoma in the mediastinum
-
♦
-
Immunohistochemistry
-
♦
Synovial sarcoma is focally reactive for cytokeratin and bcl-2 and variably reactive for CD99
-
♦
-
Cytogenetics
-
♦
Synovial sarcoma is characterized by the t(X;18) translocation
-
♦
-
Differential Diagnosis
-
♦
Other spindle cell sarcomas (see “Differential Diagnosis” for “Solitary Fibrous Tumor”)
-
♦
-
-
Rhabdomyosarcoma
-
Clinical
-
♦
Rhabdomyosarcoma occurs in adults or children, usually as a component of germ cell tumors or sarcomatoid thymic carcinoma
-
♦
Pure rhabdomyosarcoma is very rare in the mediastinum
-
♦
Rhabdomyosarcoma follows a highly aggressive course
-
♦
-
Macroscopic and Microscopic
-
♦
Similar to soft tissue counterparts (see Chapter 22)
-
♦
-
Metastatic Tumors
Small Cell Undifferentiated Carcinoma of the Lung
-
Clinical
-
♦
Frequently presents with mediastinal widening due to lymph node metastases that overshadows lung involvement
-
♦
-
Differential Diagnosis
-
♦
Thymic small cell undifferentiated carcinoma
-
Absence of recognizable lung primary, more solid mass (as opposed to lymph node involvement), and lobulated appearance
-
Primary small cell carcinoma of lung is usually TTF1 positive. Primary thymic small cell carcinoma is often TTF1 negative
-
-
♦
Lymphoma
-
Tends to show less cell cohesion and a coarse chromatin pattern
-
Lymphomas are immunoreactive for LCA and negative for cytokeratin and neuroendocrine markers
-
-
♦
Metastasis From Distal (Nonthoracic) Sites
-
Clinical
-
♦
Distinguishing metastatic from primary mediastinal tumors is especially problematic for metastatic germ cell tumors, melanoma, and prostatic carcinoma
-
♦
Clinical data and radiographic imaging may suggest an extrathoracic primary site
-
♦
-
Macroscopic
-
♦
Lymph node involvement often predominates in metastatic lesions with lesser degree of extranodal soft tissue spread
-
♦
-
Differential Diagnosis
-
♦
In the consideration of thymic carcinoma and primary germ cell tumors, metastases from an extramediastinal source must always be excluded
-
♦
In distinguishing thymic carcinoma from histologically similar tumors of the lung or head and neck, thymic carcinoma tends to have a lobulated appearance and may express CD5 (approximately 50% of cases), CD70 (50%), or CD117 (40–100%). These markers are usually negative in metastatic tumors of lung or head and neck origin
-
♦
Direct extension of tumors from the esophagus, chest wall, vertebrae, pleura, lung, and trachea (Fig. 26.26)
Fig. 26.26. 
Mediastinal structures are encased by direct extension from a non-small cell lung cancer. Tumor replaces the lymph node (LN), invades and compresses SVC (arrows), and surrounds the aortic arch (AO) and pulmonary arteries (PA).
-
♦

TNM Classification of Thymoma
-
♦
Primary Tumor (T)
-
T1: Macroscopically completely encapsulated and microscopically no capsular invasion
-
T2: Macroscopic adhesion or invasion into the surrounding fatty tissue or mediastinal pleura or microscopic invasion into capsule
-
T3: Invasion into neighboring organs, such as the pericardium, great vessels, and lung
-
T4: Pleural or pericardial dissemination
-
-
♦
Regional Lymph Node (N)
-
N0: No lymph node metastasis
-
N1: Metastasis to anterior mediastinal lymph nodes
-
N2: Metastasis to intrathoracic lymph nodes other than anterior mediastinal lymph nodes
-
N3: Metastasis to extrathoracic lymph nodes
-
-
♦
Distant Metastasis (M)
-
M0: No distal metastasis
-
M1: Distal metastasis
-
-
♦
Clinical Staging
-
Stage I: Completely encapsulated without microscopic capsular invasion (benign thymoma)
-
Stage II: Macroscopic invasion into the surrounding fatty tissue or mediastinal pleura or microscopic invasion into capsule
-
Stage III: Macroscopic invasion into neighboring organs (i.e., the pericardium, great vessels, or lung)
-
Stage IVa: Diffuse pleural or pericardial involvement
-
Stage IVb: Distal metastasis
-
SUGGESTED READING
General
Carter BW, Tomiyama N, Bhora FY, et al. A modern definition of mediastinal compartments. J Thorac Oncol. 2014;9:S97–101.
Marchevsky AM, Wick MR, editors. Pathology of the mediastinum. New York: Cambridge University Press; 2014
Rosai J. Mediastinum. In: Rosai J, editor. Rosai and Ackerman’s surgical pathology. 10th ed. St Louis: Mosby-Elsevier; 2011. p. 437–86.
Strollo DC, Rosado-de-Christenson ML. State of the art—tumors of the thymus. J Thorac Imaging. 1999;14:152–71.
Suster S, Rosai J. Histology of normal thymus. Am J Surg Pathol. 1990;14:284–303.
Travis WD, Brambilla E, Burke AP, Marx A, Nicholson AG. WHO Classification of Tumours of the Lung, Pleura, Thymus and Heart. WHO Classification of Tumours. Lyon: IARC Press; 2015. pp. 183–298.
Inflammation/Fibrosing Mediastinitis
Dines DE, Payne WS, Bernatz PW, et al. Mediastinal granuloma and fibrosing mediastinitis. Chest. 1979;75:320–4.
Flieder DB, Suster S, Moran CA. Idiopathic fibroinflammatory (fibrosing/sclerosing) lesions of the mediastinum: a study of 30 cases with emphasis on morphologic heterogeneity. Mod Pathol. 1999;12:257–64.
Lloyd JE, Tillman BF, Atkinson JB, DesPrez RM. Mediastinal fibrosis complicating histoplasmosis. Medicine. 1988;67:295–310.
McNeeley MF, Chung JH, Bhalla S, Godwin JD. Imaging of granulomatous fibrosing mediastinitis. AJR Am J Roentgenol. 2012;199:319–27.
Peikert T, Shrestha B, Aubry MC, et al. Histopathologic overlap between fibrosing mediastinitis and IgG4-related disease. Int J Rheumatol. 2012;2012:207056.
Rossi SE, McAdams P, Rosado-de-Christenson ML, Franks TJ, Galvin JR. Fibrosing mediastinitis. Radiographics. 2001;21:737–57.
Schroeyers P, Wellens F, Degrieck I, et al. Aggressive primary treatment for poststernotomy acute mediastinitis: our experience with omental- and muscle flaps surgery. Eur J Cardiothorac Surg. 2001;20:743–6.
Cysts General
Wick MR. Mediastinal cysts and intrathoracic thyroid tumors. Semin Diagn Pathol. 1990;7:285–94.
Developmental
Nobuhara KK, Gorski YC, La Quaglia MP, Shamberger RC. Bronchogenic cysts and esophageal duplications: common origins and treatment. J Pediatr Surg. 1997;32:1408–13.
Roma A, Varsegi M, Magi-Galluzzi C, et al. The distinction of bronchogenic cyst from metastatic testicular teratoma. Am J Clin Pathol. 2008;130:265–73.
Sarper A, Ayten A, Golbasi I, et al. Bronchogenic cyst. Texas Heart Inst J. 2003;30:105–8.
Pericardial
Lillie WI, McDonald JR, Clagett OT. Pericardial coelomic cysts and pericardial diverticula. A concept of etiology and report of cases. J Thorac Surg. 1950;20:494–504.
Thymic
Suster S, Barbato D, Carlson S, et al. Multilocular thymic cysts with pseudoepitheliomatous hyperplasia. Hum Pathol. 1991;22:455–60.
Suster S, Rosai J. Multilocular thymic cyst. An acquired reactive process: study of 18 cases. Am J Surg Pathol. 1991a;15:388–98.
Weissferdt A, Moran CA. Thymic carcinoma associated with multilocular thymic cyst: a clinicopathologic study of 7 cases. Am J Surg Pathol. 2011;35:1074–9.
Nonneoplastic Lesions of Thymus
Berry CL, Thompson EN. Clinico-pathological study of thymic dysplasia. Arch Dis Child. 1968;43:579–84.
Parrens M, Dubus P, Danjoux M, et al. Mucosa-associated lymphoid tissue of the thymus – hyperplasia vs lymphoma. Am J Clin Pathol. 2002;117:51–6.
Singla S, Litzky LA, Kaiser LR, Shrager JB. Should asymptomatic enlarged thymus glands be resected? J Thorac Cardiovasc Surg. 2010;140:977–83.
Van Baarlen J, Schuurman H-J, Huber J. Acute thymus involution in infancy and childhood: reliable marker for duration of acute illness. Hum Pathol. 1988;19:1155–60.
Lesions Mimicking Mediastinal Tumors
Hedayati N, McHenry CR. The clinical presentation and operative management of nodular and diffuse substernal thyroid disease. Am Surg. 2002;68:245–51.
Takashima S, Nakano H, Minamoto K, et al. A thoracoscopically resected case of mediastinal parathyroid cyst. Acta Med Okayama. 2005;59:165–70.
Pneumomediastinum
Bejvan SM, Godwin JW. Pneumomediastinum: old signs and new signs. AJR Am J Roentgenol. 1996;166:1041–8.
Thymoma
Addis BJ, den Bakker MA. Classifying thymomas. Histopathology. 2008;52:759–66.
Chalabreysse L, Roy P, Cordier JF, Loire R, Gamondes JP, Thivolet-Bejui F. Correlation of the WHO schema for the classification of thymic epithelial neoplasms with prognosis: a retrospective study of 90 tumors. Am J Surg Pathol. 2002;26:1605–11.
Chen G, Marx A, Wen-Hu C, Yong J, Puppe B, Stroebel P, et al. New WHO histologic classification predicts prognosis of thymic epithelial tumors: a clinicopathologic study of 200 thymoma cases from China. Cancer. 2002;95:420–9.
Den Bakker MA, Roden AC, Marx A, Marino M. Histologic classification of thymoma: a practical guide for routine cases. J Thorac Oncol. 2014;9:S125–30.
Detterbeck FC, Stratton K, Giroux D, et al. The IASLC/ITMIG Thymic Epithelial Tumors Staging Project: proposal for an evidence-based stage classification system for the forthcoming (8th) edition of the TNM classification of malignant tumors. J Thorac Oncol. 2014;9:S65–72.
Evoli A, Lancaster E. Paraneoplastic disorders in thymoma patients. J Thorac Oncol. 2014;9:S143–7.
Fukai I, Masaoka A, Hashimoto T, et al. Immunohistologic study of epithelial components of 81 cases of thymoma. Cancer. 1992;69:2463–8.
Kirchner T, Schalke D, Buchwald J, et al. Well-differentiated thymic carcinoma. An organotypical low grade carcinoma with relationship to cortical thymoma. Am J Surg Pathol. 1992;16:1153–69.
Kondo K, Van Schil P, Detterbeck FC, et al. The IASLC/ITMIG thymic epithelial tumors staging project: proposals for the N and M components for the forthcoming (8th) edition of the TNM classification of malignant tumors. J Thorac Oncol. 2014;9:S81–7.
Kuo T, Lo S. Thymoma: study of pathologic classification of 71 cases with evaluation of Muller-Hermelink system. Hum Pathol. 1993;24:766–71.
Marx A, Muller-Hermelink HK. From basic immunobiology to the upcoming WHO-classification of tumors of the thymus. The second conference on biological and clinical aspects of thymic epithelial tumors and related recent developments. Pathol Res Pract. 1999;195:515–33.
Marx A, Strobel P, Badve S, et al. ITMIG consensus statement on the use of the WHO histological classification of thymoma and thymic carcinoma: refined definitions, histological criteria and reporting. J Thorac Oncol. 2014a;9:596–611.
Masaoka A. Staging system of thymoma. J Thorac Oncol. 2010;5:S304–12.
Moran CA, Walsh G, Suster S, Kaiser L. Thymomas II: a clinicopathologic correlation of 250 cases with emphasis on pathologic assessment. Am J Clin Pathol. 2012a;137:451–61.
Moran CA, Weissferdt A, Kalhor N, et al. Thymomas I: the clinicopathologic correlation of 250 cases with emphasis on the World Health Organization Schema. Am J Clin Pathol. 2012b;137:444–50.
Morgenthaler TI, Brown LR, Colby TV, et al. Thymoma. Mayo Clin Proc. 1993;68:1110–23.
Muller-Hermelink HK, Marx A. Pathological aspects of malignant and benign thymic disorders. Ann Med. 1999;31:5–14.
Nicholson AG, Detterbeck FC, Marino M, et al. The IASLC/ITMIG Thymic Epithelial Tumors Staging Project: proposals for the T component for the forthcoming (8th) edition of the TNM classification of malignant tumors. J Thorac Oncol. 2014;9:S73–80.
Nonaka D, Henley JD, Chiriboga L, Yee H. Diagnostic utility of thymic epithelial markers CD205 (DEC205) and Foxn1 in thymic epithelial neoplasms. Am J Surg Pathol. 2007;31:1038–44.
Okumura M, Miyoshi S, Fujii Y, Takeuchi Y, Shiono H, Inoue M, et al. Clinical and functional significance of WHO classification on human thymic epithelial neoplasms: a study of 146 consecutive tumors. Am J Surg Pathol. 2001;25:103–10.
Pescarmona E, Rendina EA, Venuta F, et al. The prognostic implication of thymoma histologic subtyping: study of 80 consecutive cases. Am J Clin Pathol. 1990;93:190–5.
Quintanilla-Martinez L, Wilkins Jr EW, Ferry JA, et al. Thymomamorphologic subclassification correlates with invasiveness and immuno-histologic features: study of 122 cases. Hum Pathol. 1993;24:958–69.
Rajan A, Girard N, Marx A. State of the art of genetic alterations in thymic epithelial tumors. J Thorac Oncol. 2014;9:S131–6.
Shimosato Y. Controversies surrounding subclassification of thymoma. Cancer. 1994;74:542–4.
Suster S, Rosai J. Cystic thymomas. A clinicopathologic study of ten cases. Cancer. 1992;69:92–7.
Suster S, Moran CA. Histologic classification of thymoma: the World Health Organization and beyond. Hematol Oncol Clin North Am. 2008;22:381–92.
Tomaszek S, Wigle DA, Keshavjee S, Fischer S. Thymomas: review of current clinical practice. Ann Thorac Surg. 2009;87:1973–80.
Wu M, Sun K, Gil J, Gan L, Burstein DE. Immunohistochemical detection of p63 and XIAP in thymic hyperplasia and thymomas. Am J Clin Pathol. 2009;131:689–93.
Thymic Carcinoma
Berezowski K, Grimes MM, Gal A, et al. CD5 immunoreactivity of epithelial cells in thymic carcinoma and CASTLE using paraffin-embedded tissue. Am J Clin Pathol. 1996;106:483–6.
Brown JG, Familiari U, Papotti M, Rosai J. Thymic basaloid carcinoma: a clinicopathologic study of 12 cases. With a general discussion of basa-loid carcinoma and its relationship with adenoid cystic carcinoma. Am J Surg Pathol. 2009;33:1113–24.
Dorfman DM, Shahsafaei A, Chan JK. Thymic carcinomas, but not thymomas and carcinomas of other sites, show CD5 immunoreactivity. Am J Surg Pathol. 1997;21:936–40.
Hasserjian RP, Klimstra DS, Rosai J. Carcinoma of thymus with clear cell features: report of eight cases and review of literature. Am J Surg Pathol. 1995;19:835–41.
Kornstein MJ, Rosai J. CD5 labeling of thymic carcinomas and other non-lymphoid neoplasms. Am J Clin Pathol. 1998;109:722–6.
Marx A, Strobel P, Badve S, et al. ITMIG consensus statement on the use of the WHO histological classification of thymoma and thymic carcinoma: refined definitions, histological criteria and reporting. J Thorac Oncol. 2014b;9:596–611.
Marx A, Strobel P, Zettl A, et al. Thymomas. In: Travis WD, Brambilla E, Muller-Hermelink HK, et al, editors. Pathology and genetics. Tumours of the lung, pleura, thymus and heart. WHO classification of tumours. Lyon: IARC Press; 2004. p. 152–153.
Moran CA, Suster S. Mucoepidermoid carcinomas of thymus: a clinico-pathologic study of six cases. Am J Surg Pathol. 1995a;19:826–34.
Moran CA, Suster S. Thymic carcinoma: current concepts and histologic features. Hematol Oncol Clin North Am. 2008;22:393–407.
Muller-Hermelink HK, Engel P, Kuo TT, et al. Tumours of the thymus: introduction. In: Travis WD, Brambilla E, Muller-Hermelink HK, et al, editors. Pathology and genetics. Tumours of the lung, pleura, thymus and heart. WHO classification of tumours. Lyon: IARC Press; 2004. p. 148–51.
Ritter JH, Wick MR. Primary carcinomas of the thymus gland. Semin Diagn Pathol. 1999;16:18–31.
Suster S. Thymic carcinoma: update of current diagnostic criteria and histologic types. Semin Diagn Pathol. 2005;22:198–212.
Suster S, Rosai J. Thymic carcinoma clinicopathologic study of 60 cases. Cancer. 1991b;67:1025–32.
Wick MR, Scheithauer BW. Oat-cell carcinoma of thymus. Cancer. 1982;49:1652–7.
Thymic Carcinoid Tumor
Moran CA. Primary neuroendocrine carcinomas of the mediastinum: review of current criteria for histopathologic diagnosis and classification. Semin Diagn Pathol. 2005;22:223–9.
Weissferdt A, Kalhor N, Liu H, et al. Thymic neuroendocrine tumors (paragangliomas and carcinoid tumors): a comparative immunohistochemical study of 46 cases. Hum Pathol. 2014;45:2463–70.
Thymic Stromal Tumors
Moran CA, Rosado-de-christenson M, Suster S. Thymolipoma: clinico-pathologic review of 33 cases. Mod Pathol. 1995;8:741–4.
Neurogenic Tumors
Lonergan GF, Schwab CM, Suarez ES, Carlson CL. From the archives of the AFIP. Neuroblastoma, ganglioneuroblastoma, and ganglioneuroma: radiologic-pathologic correlation. Radiographics. 2012;22:911–34.
Paragangliomas
Brown ML, Zayas GE, Abel MD, et al. Mediastinal paragangliomas: the Mayo Clinic experience. Ann Thorac Surg. 2008;86:946–51.
Odze R, Begin LR. Malignant paraganglioma of posterior mediastinum case report and review of literature. Cancer. 1990;65:564–9.
Mediastinum Germ Cell Tumors
den Bakker MA, Oosterhuis JW. Tumours and tumour-like conditions of the thymus other than thymoma; a practical approach. Histopathology. 2009;54:69–89.
de Ment SH. Association between mediastinal germ cell tumors and hematologic malignancies: update. Hum Pathol. 1990;21:699–703.
Kao C-S, Idrees MT, Young RH, Ulbright TM. Solid pattern yolk sac tumor: a morphologic and immunohistochemistry study of 52 cases. Am J Surg Pathol. 2012;36:360–7.
Moran CA, Suster S. Mediastinal seminomas with prominent cystic changes. A clinicopathologic study of 10 cases. Am J Surg Pathol. 1995b;19:1047–53.
Moran CA, Suster S. Primary germ cell tumors of the mediastinum: I. Analysis of 322 cases with special emphasis on teratomatous lesions and a proposal for histopathologic classification and clinical staging. Cancer. 1997;80:681–90.
Moran CA, Suster S, Koss MN. Primary germ cell tumors of the mediastinum. III. Yolk sac tumor, embryonal carcinoma, choriocarcinoma, and combined nonteratomatous germ cell tumors of the mediastinum—a clinicopathologic and immunohistochemical study of 64 cases. Cancer. 1997;80:699–707.
Patel IJ, Hsiao E, Ahmad AH, et al. AIRP best cases in radiologic-pathologic correlation. Mediastinal mature cystic teratoma. Radiographics. 2013;33:797–801.
Sung M-T, MacLennan GT, Lopez-Beltran A, Zhang S, Montironi R, Cheng L. Primary mediastinal seminoma: a comprehensive assessment integrated with histology, immunohistochemistry, and fluorescence in situ hybridization for chromosome 12p abnormalities in 23 cases. Am J Surg Pathol. 2008;21:146–55.
Weissferdt A, Moran CA. Mediastinal seminoma with florid follicular lymphoid hyperplasia: a clinicopathological and immunohistochemical study of six cases. Virchows Arch. 2015;466:209–15.
Weissferdt A, Suster S, Moran CA. Primary mediastinal “thymic” seminomas. Adv Anat Pathol. 2012;19:75–80.
Mediastinum Lymphoproliferative and Myeloproliferative Disorders
Cronin DMP, Warnke RA. Castleman disease: an update on classification and the spectrum of associated lesions. Adv Anat Pathol. 2009;16:236–46.
Inagaki H, Chan JKC, Ng JWM, et al. Primary thymic extranodal marginal-zone B-cell lymphoma of mucosa-associated lymphoid tissue type exhibits distinctive clinicopathological and molecular features. Am J Pathol. 2002;160:1435–43.
Isaacson PG, Chan JK, Tang C, et al. Low grade B-cell lymphoma of mucosa-associated lymphoid tissue arising in the thymus. A thymic lymphoma mimicking myoepithelial sialadenitis. Am J Surg Pathol. 1990;14:342–51.
Lamarre L, Jacobson JO, Aisenberg AC, et al. Primary large cell lymphoma of mediastinum. A histologic and immunophenotypic study of 29 cases. Am J Surg Pathol. 1989;13:730–9.
Pileri SA, Gaidano G, Zinzani PL, Falini B, Gaulard P, Zucca E, et al. Primary mediastinal B-cell lymphoma: high frequency of BCL-6 mutations and consistent expression of the transcription factors OCT-2, BOB.1, and PU.1 in the absence of immunoglobulins. Am J Pathol. 2003;162:243–53.
Quintanilla-Martinez L, Fend F. Mediastinal gray zone lymphoma. Haematologica. 2011;96:496–9.
Swerdlow SH, Campo E, Harris NL, et al, editors. WHO classification of tumours of haematopoietic and lymphoid tissues. WHO classification of tumours. Lyon: IARC Press; 2008.
Mediastinum Mesenchymal Tumors
Boland JM, Colby TV, Folpe AL. Liposarcomas of the mediastinum and thorax: a clinicopathologic and molecular cytogenetic study of 24 cases, emphasizing unusual and diverse histologic features. Am J Surg Pathol. 2012;36:1395–403.
Brown LR, Reiman HM, Rosenow III EC, et al. Intrathoracic lymphangioma. Mayo Clin Proc. 1986;61:882–92.
Cheng LSL, Tse GMK, Li WWL, Lee TW, Yim APC. Mediastinal synovial sarcoma: a case report and literature review. Can Respir J. 2003;10:393–5.
Hanau CA, Miettinen M. Solitary fibrous tumor: histological and immunohistochemical spectrum of benign and malignant variants presenting at different sites. Hum Pathol. 1995;26:440–9.
Kubokura H, Okamoto J, Hoshina H, et al. Mediastinal cystic hemangioma presenting as bilateral bloody pleural effusion: a case study. J Nippon Med Sch. 2012;79:381–4.
Ortega P, Suster D, Falconieri G, et al. Liposarcomas of the posterior mediastinum: clinicopathologic study of 18 cases. Mod Pathol. 2014;28:721–31.
Ryu Y-J, Yoo S, Jung M, et al. Embryonal rhabdomyosarcoma arising from a mediastinal teratoma: an unusual case report. J Korean Med Sci. 2013;28:476–9.
Salah S, Salem A. Primary synovial sarcomas of the mediastinum: a systematic review and pooled analysis of the published literature. ISRN Oncol. 2014;2014:412527.
Witkin GB, Rosai J. Solitary fibrous tumor of the mediastinum. A report of 14 cases. Am J Surg Pathol. 1989;13:547–57.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Chang, CC.(., Fanaian, N., Tomashefski, J.F. (2016). Mediastinum and Thymus. In: Cheng, L., Bostwick, D. (eds) Essentials of Anatomic Pathology. Springer, Cham. https://doi.org/10.1007/978-3-319-23380-2_26
Download citation
DOI: https://doi.org/10.1007/978-3-319-23380-2_26
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-23379-6
Online ISBN: 978-3-319-23380-2
eBook Packages: MedicineMedicine (R0)