
Abstract
The field of genetics continues to evolve at a rapid pace. The completion of the Human Genome Project coupled with advancing scientific techniques has led to the explosion of genetic information and testing capabilities ranging from rare genetic conditions to common and sometimes preventable conditions. This chapter reviews a variety of genetic conditions categorized by primary mode of inheritance and is further subcategorized by disease process. For each disorder, the chromosome, gene location, inheritance pattern, incidence, clinical phenotype, laboratory findings, and treatment options are reviewed, providing a general overview of a wide range of genetic conditions and various inheritance patterns extending across multiple subspecialties.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others



Keywords
- Chromosome
- Aneuploidy
- Microdeletion
- Deletion
- Point mutation
- Chromosome breakage
- Gene
- Molecular
- Trinucleotide repeat
- Biochemical
- Metabolic
- Familial
- Inheritance
- Mitochondrial
Chromosomal Disorders
Chromosomal Aneuploidy
Trisomy 21 (Down Syndrome)
Chromosome and Gene Location
-
♦
Additional chromosome 21
-
♦
Critical region at 21q22
Inheritance
-
♦
95% meiotic nondisjun ction (47 chromosomes present)
-
80–90% maternal, increases with maternal age
-
10–20% paternal
-
-
♦
3–4% unbalanced transl ocation (46 chromosomes with an extra chromosome 21 fused to another acrocentric chromosome)
-
66% sporadic (not inherited)
-
33% balanced translocation in one parent
-
-
♦
1–2% mosaicism (at least two different cell lines)
-
Results from po stzygotic nondisjunction or loss of the extra 21 in one cell line
-
Incidence
-
♦
1/800 live births
Clinical Manifestations
-
♦
Flat facial p rofile, thick nuchal fold, upward slanting palpebral fissures, depressed nasal bridge, epicanthal folds, Brushfield spots of iris (Fig. 13.1A), single transverse palmar crease (Fig. 13.1B), gap between the first and second toes (Fig. 13.1C), and low-set c-shaped ears
Fig. 13.1. 
Down syndrome – brushfield spots on irides (A). Single transverse palmar creases and fifth finger clinodactyly (B). Gap between first and second toe (C).
-
♦
Developmental delay, learning disabled (IQ = 40–80), poor Moro reflex, infantile hypotonia, joint hyperflexibility, and increased risk for Alzheimer disease
-
♦
Congenital heart defects (endocardial cushion defects including atrioventricular canal defect and ventricular septal defects), increased risk for childhood acute leukemia, hypothyroidism, obesity, short stature, increased incidence of pulmonary hypoplasia, and duodenal atresia
-
♦
Ultrasound findings may include increased nuchal translucency, thickened nuchal fold, congenital heart defects, duodenal atresia, short humerus and femur, short middle phalanx of fifth finger, and echogenic bowel
-
♦
Seventy-five percent spontaneously abort in first trimester. Of those live born, 80% survival at age 30

Laboratory
-
♦
Chromosome analysis identifies extra chromosome 21
-
♦
Prenatal diagnosis through chromosome analysis of chorionic villi or amniocentesis
-
♦
An increased risk could be indicated by altered prenatal maternal serum markers or non-invasive prenatal screening (NIPS) using cell-free fetal nucleic acids
Treatment
-
♦
Not curable, supportive/symptomatic
Trisomy 13 (Patau Syndrome)
Chromosome and Gene Location
-
♦
Additional chromosome 13
Inheritance
-
♦
80% meiotic nondisjuncti on (47 chromosomes present)
-
80–90% maternal, increases with maternal age
-
10–20% paternal
-
-
♦
20% unbalanced translocation (46 chromosomes with an extra chromosome 13 fused to another acrocentric chromosome)
Incidence
-
♦
1/10,000 births
Clinical Manifestations
-
♦
Midline abnormalities ranging from simple ocular hypotelorism to cyclopia to complete absence of eyes, prominent occiput, microcephaly, malformed low-set ears, cl eft lip and palate, polydactyly, and transverse palmar crease (Fig. 13.2)
Fig. 13.2. 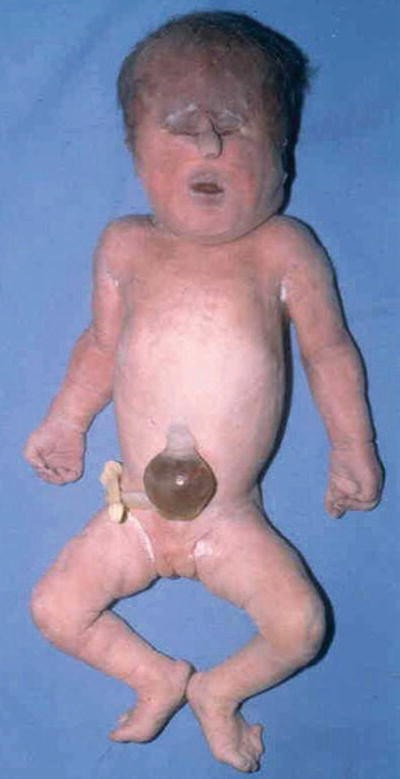
Trisomy 13 – showing hypertelorism and tubelike nasal structure. Polydactyly on foot.
-
♦
Complete or incomplete holoprosencephaly, severe intellectual disability, seizures, deafness, hypotonia, and apneic spells
-
♦
Congenital heart defects (hypoplastic left heart and ventricular septal defects), urogenital defects, cryptorchidism, bicornuate uterus and hypoplastic ovaries, polycystic kidneys, umbilical hernia, and omphalocele
-
♦
Ultrasound findings may include holoprosencephaly, cleft lip and palate, cystic hygroma, polydactyly, congenital heart defects, cystic kidneys, and omphalocele
-
♦
95% spontaneously abort. Of those live born, 90% die within first year of life

Laboratory
-
♦
Chromosome a nalysis identifies extra chromosome 13
-
♦
Prenatal diagnosis through chromosome analysis of chorionic villi or amniocentesis
-
♦
An increased risk could be indicated by altered prenatal maternal serum markers or non-invasive p renatal screening (NIPS) using cell-free fetal nucleic acids
Treatment
-
♦
Not curable, supportive/symptomatic
Trisomy 18 (Edward Syndrome)
Chromosome and Gene Location
-
♦
Additional chromosome 18
Inheritance
-
♦
Meiotic nondisjunction
-
95% maternal, increases with maternal age
-
5% paternal
-
Incidence
-
♦
1/5,000–1/10,000 births
Clinical Manifestations
-
♦
Microcephaly with prominent occiput, micrognathia, malformed ears, clenched hands, second and fifth digits overlapping third and fourth (Fig. 13.3), rocker bottom feet, single transverse palmar crease, and hypoplastic nails
Fig. 13.3. 
Trisomy 18 – clenched hand and overlapping fingers.
-
♦
Severe intellect ual disability, seizures, and hypertonia
-
♦
Severe intrauterine growth retardation, congenital heart defects (ventricular septal defects), urogenital defects, cryptorchidism, horseshoe kidney, diaphragmatic hernia, and omphalocele
-
♦
Ultrasound findings may include clenched hands, club and rocker bottom feet, micrognathia, congenital heart defects, omphalocele, diaphragmatic hernia, neural tube defects, ch oroid plexus cysts, and cystic hygroma

Life Expectancy
-
♦
95% spontaneously abort. Of those live born, 90% die within first year of life
Laboratory
-
♦
Chromosome analysis identifies extra chromosome 18
-
♦
Prenatal diagnosis through chromosome analysis of chorionic villi or amniocentesis
-
♦
An increased risk could be indicated by altered prenatal maternal serum markers or non-invasive pr enatal screening (NIPS) using cell-free fetal nucleic acids
Treatment
-
♦
Not curable, supportive/symptomatic
Klinefelter Syndrome (XXY)
Chromosome and Gene Location
-
♦
Extra X chromosome in a male
Inheritance
-
♦
Meiotic nondisjunction
-
55% maternal nondisjunction
-
45% paternal nondisju nction
-
-
♦
Also may be mosaic XY/XXY or rarely XX/XXY
Incidence
-
♦
1/800 males
Clinical Manifestations
-
♦
Tall habitus, undervirilized, small testes, gynecomastia, and poor musculature
-
♦
Mild delay, behavioral immaturity, shyness, learning disabilities (reading) speech delay
-
♦
Infertility
-
♦
Normal life expectancy
Laboratory
-
♦
Chromosome analysis identifies XXY
-
♦
Prenatal diagnosis through chromosome analysis of chorionic villi or amniocentesis
-
♦
Maternal serum screening and ultrasound findings are not useful
-
♦
An increased risk could be indicated by non-invasive prenatal screening (NIPS) using cell-free fetal nucleic acids
Treatment
-
♦
Testosterone suppleme ntation for the development of secondary sexual characteristics
Turner Syndrome (45, X)
Chromosome and Gene Location
-
♦
Missing or structurally abnormal X chromosome
Inheritance
-
♦
55% 45,X
-
80% loss of paternal X chromosome
-
20% loss of maternal X (no maternal age effect)
-
-
♦
25% 46,XX
-
Structural alt eration in one X chromosome
-
-
♦
15% mosaic
-
45,X with 46,XX, 46,XY, or others
-
Incidence
-
♦
1/2,000–1/5,000 female births
-
♦
Most common chr omosome finding in spontaneous abortions
Clinical Manifestations
-
♦
Short stature, webbed neck (Fig. 13.4), lymphedema of hands and feet, high arched palate, cystic hygroma, low posterior hairline, and hypoplastic widely spaced nipples
Fig. 13.4. 
Turner syndrome – webbing of the neck and low posterior hairline.
-
♦
Normal or near normal intelligence; may have delay in speech, neuromotor skills, and learning abilities
-
♦
Gonadal dysgenesis (infertility, primary amenorrhea), renal malformations (horseshoe kidney), cardiovascular malformations (coarctation of aorta, hypoplastic left heart), and increased risk f or gonadoblastoma if mosaic for some Y chromatin
-
♦
Ultrasound findings include cystic hygroma (detectable after 10 weeks), lymph collections (ascites, pleural effusion), congenital heart disease, and renal anomalies
-
♦
99% spontaneously abort; those who survive infancy usually reach adulthood

Laboratory
-
♦
Chromosome analysis indicates monosomy X or other variants
-
♦
Prenatal diagnosis thro ugh chromosome analysis of chorionic villi or amniocentesis
-
♦
An increased risk could be indicated by altered prenatal maternal serum markers or non-invasive prenatal screening (NIPS) using cell-free fetal nucleic acids
Treatment
-
♦
Estrogen, thyroid, and growth hormone replacement therapy for development of secondary sexual characteristics and growth
Microdeletion Syndromes
-
♦
See Table 13.1
Table 13.1. Microdeletion Syndromes
Angelman Syndrome
Chromosome and Gene Location
-
♦
15q11.2
Inheritance
-
♦
Angelman syndrome results from the loss of the maternally imprinted region at chromosome 15q11.2. Loss can occur via numerous mechanisms. Recurrence is dependent on mechanism of loss
-
60–70% deletion of maternal 15q11.2
-
5% paternal uniparental disomy (UPD) (two copies of the paternal chromosome)
-
3% imprinting defect
-
11% ubiquitin-protein ligase E3A (UBE3A) mutation
-
11% unknown
-
Incidence
-
♦
1/20,000
Clinical Manifestations
-
♦
Prominent mandibl e, open-mouthed expression, hyperreflexia, microcephaly, brachycephaly, and optic atrophy
-
♦
Ataxia, severe intellectual disability, seizures, and gross motor developmental delay
-
♦
Absent speech, inappropriate laughter, arm flapping, and feeding difficulties
-
♦
Most survive into adulthood
Laboratory
-
♦
First-tier testing
-
Molecular methylation anal ysis identifies missing maternal allele
-
Methylation-sensitive multiple ligation-dependent probe amplification (MLPA) detects missing maternal allele or deletion
-
-
♦
Second-tier testing
-
Fluorescent in situ hybridization (FISH) or chromosomal microarray (CMA) identifies deletion
-
Molecular UPD studies identify two copies of paternal allele
-
Sequence analysis identifies m utations in UBE3A gene
-
Mutation analysis of imprinting center
-
Treatment
-
♦
Not curable, supportive/symptomatic
Prader–Willi Syndrome
Chromosome and Gene Location
-
♦
15q11.2
Inheritance
-
♦
Prader–Willi syndrome results from the loss of the paternally imprinted region at chromosome 15q11.2. Loss can occur via numerous mechanisms. Recurrence is dependent on mechanism of loss
-
75% deletion of paternal 15q11.2
-
20–25% maternal UPD (two copies of maternal chromosome 15)
-
Remainder is thought to be due to imprinting defects
-
Incidence
-
♦
1/10,000–1/25,000
Clinical Manifestations
-
♦
Hypotonia, hypogon adism, obesity, small hands and feet, almond-shaped eyes, and short stature
-
♦
Mild to moderate intellectual disability
-
♦
Failure to thrive and hyperphagia
-
♦
Most survive into adulthood
Laboratory
-
♦
First-tier testing
-
Molecular methylation analysis id entifies missing paternal allele
-
Methylation-sensitive MLPA detects missing paternal allele or deletion
-
-
♦
Second-tier testing
-
FISH and/or CMA identifies deletion
-
Molecular UPD studies ident ify two copies of maternal allele if either first-tier test is abnormal
-
Mutation analysis of imprinting center
-
Treatment
-
♦
Not cu rable, supportive/symptomatic
1p36 Microdeletion Syndrome
Chromosome and Gene Location
-
♦
1p36
Inheritance
-
♦
90–95% sporadic
-
♦
5–10% familial translocations
Incidence
-
♦
1/5,000–10,000
Clinical Manifestations
-
♦
Microcephaly, deep-set eyes, flat nasal bridge, cardiac malformations, and hypotonia
-
♦
Intellectual disa bility and seizures
-
♦
Growth retardation
-
♦
Most survive into adulthood
Laboratory
-
♦
Deletion may or may not be detected on chromosome analysis depending upon resolution of the study and size of the deletion
-
♦
FISH and/or CMA identifies deletion
Treatment
-
♦
Not curable, supportive/sympto matic
Cri Du Chat Syndrome
Chromosome and Gene Location
-
♦
5p15
Inheritance
-
♦
85–90% sporadic (85% deletion, 4% mosaics, 3% ring chromosomes, 4% translocations)
-
♦
10–15% familial trans locations, inversions, and parental mosaicism
Incidence
-
♦
1/50,000
Clinical Manifestations
-
♦
“Cat-like” cry, microcephaly, hypertelorism, micrognathia, transverse palmar crease, and hypotonia
-
♦
Intellectual di sability
-
♦
50% no speech, growth failure
-
♦
Most survive into adulthood
Laboratory
-
♦
Deletion may or may not be detected on chromosome analysis depending upon resolution of the study an d size of the deletion
-
♦
FISH and/or CMA identifies deletion
Treatment
-
♦
Not curable, supportive/sympto matic
22q Deletion Syndrome (DiGeorge/Velocardiofacial Syndrome)
Chromosome and Gene Location
-
♦
22q11.2
Inheritance
-
♦
90% are deletions involving multiple genes
-
♦
Approximately 10–15% are familial (autosomal dominant)
Incidence
-
♦
1/4,000
Clinical Manifestations
-
♦
The disturbance of neural crest migration of pharyngeal pouches is thought to cause clinical features
-
♦
Interpatien t variability may be dependent on extent of deletion
-
♦
Hypertelorism, down-slanting eyes, high arched palate, micrognathia, low-set ears, bulbous nose, square nasal tip, cleft palate (velocardiofacial (VCF)), small open mouth, retrognathia, microcephaly, and slender hands and digits
-
♦
Cardiac manifestations include tetralogy of Fallot, outflow tract defect, right-sided aortic arch, interrupted aortic arch, and ventricular septal defect
-
♦
Mild-moderate learning dif ficulties, seizures, tetany, and emotional and behavioral problems
-
♦
Hypoparathyroidism, neonatal hypocalcemia (DiGeorge (DGS)), immune/T-cell deficit (DGS), hypernasal speech, hypospadius, and short stature
-
♦
Most reach ad ulthood if cardiac lesion is not life threatening
Laboratory
-
♦
Hypocalcemia, decreased T cells
-
♦
Deletion of 22q11 is usually not visible on chromosome analysis
-
♦
FISH and/or CMA identifies deletion
Treatment
-
♦
Cardiac surg ery, calcium supplements, and supportive care
Smith–Magenis Syndrome
Chromosome and Gene Location
-
♦
17p11.2
Inheritance
-
♦
Most are sporadic interstitial deletions
-
♦
A few cases of pericentric inversions with breakpoints in 17p11
Incidence
-
♦
1/50,000
Clinical Manifestations
-
♦
Brachycephaly, flat, broad midface, and prominent forehead
-
♦
Seizures and intellectual disability
-
♦
Hyperactivity, sleep disturbances, behavioral problems including screaming outbursts and self- mutilation behaviors, and speech delay
-
♦
Most survive into adulthood
Laboratory
-
♦
Chromosome analysis detects deletion in most cases
-
♦
FISH and/or CMA identifies deletion
Treatment
-
♦
Not curable, supportive/sympto matic
Miller–Dieker Syndrome
Chromosome and Gene Location
-
♦
17p13.3
Inheritance
-
♦
90% are sporadic deletions
-
♦
Approximately 10–15% are familial
Incidence
-
♦
1/100,000
Clinical Manifestations
-
♦
Lissencephaly, microcephaly, anteverted nostrils, carp mouth, and agenesis of corpus callosum
-
♦
Seizures and intellectual disability
-
♦
Failure to thrive and absent speech
-
♦
Life expectancy is variable, but most die in childhood
Laboratory
-
♦
Deletion may or may not be detected on chromosome analysis depending upon resolution of the st udy and size of the deletion
-
♦
FISH and/or CMA identifies deletion
Treatment
-
♦
Not curable, supportive/sympt omatic
Williams Syndrome
Chromosome and Gene Location
-
♦
7q11.23
Inheritance
-
♦
Mostly sporadic
-
♦
Few cases of autoso mal dominant inheritance have been reported
Incidence
-
♦
1/20,000 live births
Clinical Manifestations
-
♦
Broad forehead, bitemporal narrowness, periorbital fullness, wide mouth, broad nasal tip, long philtrum, micrognathia, growth retardation, and small widely spaced teeth
-
♦
Cardiac manifestations include supravalvular aortic stenosis and peripheral pulmonary stenosis
-
♦
Intellectual disability
-
♦
Gregarious personality, joint limitations, and hypercalcemia
-
♦
Most survive in to adulthood
Laboratory
-
♦
Elevated serum calcium
-
♦
Deletion is usua lly not visible on chromosome analysis
-
♦
FISH and/or CMA identifies deletion
Treatment
-
♦
Not curable, supportive/symptomatic
-
♦
Eliminatio n of vitamin D and calcium from the diet
Wolf–Hirschhorn Syndrome
Chromosome and Gene Location
-
♦
4p16.3
Inheritance
-
♦
85–90% sporadic
-
♦
10–15% familial translocations
Incidence
-
♦
1/50,000
Clinical Manifestations
-
♦
Microcephal y, hypertelorism, micrognathia, and hypotonia
-
♦
Seizures and intellectual disability
-
♦
Congenital heart defects, renal malformations, and genital malformations
-
♦
Most survive into adulthood
Laboratory
-
♦
Deletion may or may not be detected on chromosome analysis depending upon resolution of the study and size of the deletion
-
♦
FISH and/or CMA identifies deletion
Treatment
-
♦
Not curable, supportive/symptoma tic
Chromosome Breakage Syndromes
Fanconi Anemia
Chromosome and Gene Location
-
♦
Genetically heterogeneous (multiple gene loci involved)
Inheritance
-
♦
Autosomal recessive
Incidence
-
♦
1/22,000
Clinical Manifestations
-
♦
Short stature, ab sent or hypoplastic radii and thumbs, brown skin pigmentation, cryptorchidism, and renal anomalies
-
♦
Pancytopenia, anemia, increased incidence of leukemia, and solid tumors
Molecular Basis of Disease
-
♦
Multiple complementation groups have been identified and appear to be involved in the formation of a protein complex that participates in a DNA damage response pathway. The exact molecular mechanism that leads to impaired genomic stability has not been elucidated
Laboratory
-
♦
Increased chromosomal breakage, gaps, and rearrangements after exposure to diepoxybutan e or mitomycin C (DNA alkylating agents)
-
♦
Mutation analysis is available for specific mutations
Treatment
-
♦
Transfusions and bone marrow transplantation
-
♦
Not cura ble, supportive/symptomatic
Bloom Syndrome
Chromosome and Gene Location
-
♦
15q26.1
Inheritance
-
♦
Autosomal recessive
Incidence
-
♦
Rare
Clinical Manifestations
-
♦
Short s tature
-
♦
Facial erythema
-
♦
Increased susceptibility to infections
-
♦
Increase incidence of leukemia
-
♦
High pitched voice
Molecular Basis of Disease
-
♦
Decreased activity of DNA ligase I leads to genomic instability and multisystem anomalies
Laboratory
-
♦
Increased sister chromatid exchange (12× normal)
-
♦
Quadrilateral formation is increased as are random breaks and translocations between nonhomologous chromosomes
Treatment
-
♦
Not curable , supportive/symptomatic
-
♦
Minimize exposure to radiation/mutagenic agents
Ataxia Telangiectasia
Chromosome and Gene Location
-
♦
11q22–q23, ATM gene
Inheritance
-
♦
Autosomal recessive
Incidence
-
♦
1/40,000–1/100,000
Clinical Manifestations
-
♦
Cerebellar a taxia, conjunctival telangiectasia, and IgA deficiency
-
♦
Predisposition to malignancy
-
♦
Increased infections
-
♦
Growth failure, onset first 2 years of life
Molecular Basis of Disease
-
♦
Defect in DNA repair mechanisms leading to chromosomal breakage, increased intrachromosomal recombination, sensitivity to ionizing radiation (IR), and abnormal resistance to inhibition of DNA synthesis by IR
Laboratory
-
♦
Increased chromosomal breakage and X-radiation sensitivity; especially with breakpoints at sites of immunoglobulin genes or receptors
-
♦
7;14 chromosomal translocation is identified in 5–15% of cells
-
♦
Molecular analysis of ATM gene identifies mutation in approximately 95% of patients
Treatment
-
♦
Not curable, supportive/symptomatic
-
♦
Treatment of i nfections and neoplasms and avoidance of radiation
Xeroderma Pigmentosum
Chromosome and Gene Location
-
♦
Multiple loci
Inheritance
-
♦
Autosomal recessive
Incidence
-
♦
1/250,000
Clinical Manifestations
-
♦
Sensitivity to sunlight (blistering and freckling with little exposure, beginning in childhood)
-
♦
Predisposition to malignancy (especially skin cancer)
-
♦
Mental deterioration in some
Molecular Basis of Disease
-
♦
Defect in ult raviolet-induced DNA repair mechanisms
-
♦
Skin cells unable to repair sunlight-induced DNA damage
Laboratory
-
♦
Diagnosis is based on clinical criteria; no routine clinical laboratory abnormality is observed in patients with XP
-
♦
Cytogenetic analysis can identify clones of cells with chromosome abnormalities, increased ultraviolet-induced chromosome breaks, and sister chromatin exchanges
-
♦
Fibroblasts show UV hyp ersensitivity and abnormal unscheduled DNA synthesis
Treatment
-
♦
Not curable, supportive/symptomatic
-
♦
Avoidance of u ltraviolet light
Trinucleotide Repeat Disorders
-
♦
A growing number of inherited disease syndromes (primarily neurologic) are known to be caused by the abnormal presence of an expanded tract of trinucleotide repeats within disease-specific genes (Table 13.2)
Table 13.2. Trinucleotide Repeat Disorders -
♦
Typically, the clinical hallmark of these trinucleotide repeat diseases is anticipation. Anticipation is defined as the clinical observance of an earlier age of onset and increased rate of disease progression due to amplification in the number of expanded repeats in successive generations
-
♦
The diagnosis is made by determining the number of trinucleotide repeats within a specific disease-causing expandable allele (within the proper clinical context). The normal allele will have a “normal range” of trinucleotide repeats, whereas the disease-associated (expanded) allele will contain an increased number of repeats (in the hundreds and thousands, for some diseases)
Fragile X Syndrome
Chromosome and Gene Location
-
♦
Xq27.3
Inheritance
-
♦
X-linked d ominant
Incidence
-
♦
1/5,000 males (accounts for up to 5% of male intellectual disability)
-
♦
1/2,500 females
Clinical Manifestations
-
♦
Intellectual disability
-
♦
Large narrow face, prominent forehead and jaw, with moderately increased head circumference, and large ears
-
♦
Macroorc hidism (80%)
Molecular Basis of Disease
-
♦
Expanded CGG repetitive element within FMR1 gene
-
5–44 normal
-
45–54 intermediate (minimal expansion due to meiotic instability in a small percentage of cases), no associated phenotype
-
55–200 premutation (meiotic instability with potential to expand to full mutation), fragile X-associated tremor/ataxia syndrome (FXTAS) characterized by cerebellar ataxia with the observation of white matter lesions on MRI and intentional tremor is observed in both males and females with almost half of all male premutation carriers affected by age 79; other clinical manifes tations of FXTAS include memory loss, cognitive deficit, parkinsonism, and neuropathy; premature ovarian failure (POF) is also observed in an average 21% of female premutation carriers; however, penetrance is inversely correlated with the number of repeats in the intermediate and premutation carrier ranges
-
>200 full mutation, leads to abnormal methylation and transcriptional suppression of FMR1 gene and absence of FMRP (RNA binding protein)
-
CGG repeat expansion is through female germ line (males transmit premutation repeat without much change)
-
-
♦
Sherman paradox
-
Describes apparent deviation from traditional Mendelian inheritance and varies according to whether the causative gene is transmitted through a male or female
-
In fragile X, this is the result of expansion from premutation to full mutation through the female germ line only
-
Laboratory
-
♦
Molecular analysis (PCR and Southern blot with methylation analysis) detects expanded CGG repeat and is the recommended first-tier molecular test, as expanded CGG repe ats account for ~99% of disease-causing mutations
-
♦
Molecular analysis by sequencing and dosage analysis detect the remaining 1% of FMR1 mutations
-
♦
Chromosomal fragile site may be apparent by standard chromosome analysis when culture d in folate-deficient/thymidine inhibiting media
Treatment
-
♦
Not curable
-
♦
Supportive/symptomatic
Myotonic Dystrophy Type 1
Chromosome and Gene Location
-
♦
19q13.2
Inheritance
-
♦
Autosomal dominant
Incidence
-
♦
1/8,000 (DM1 and DM2, together, represent the most common muscular dystrophy in adults)
Clinical Manifestations
-
♦
Myotonia (sustained muscle contraction), muscle wasting, and facial weakness
-
♦
Cataracts
-
♦
Hypogonadism
-
♦
Frontal balding
-
♦
Cardiac conduction disturbances
-
♦
Diabetes mellitus (5%)
-
♦
Swallowing and speech disability
-
♦
Respiratory failure
-
♦
Neonatal hypotonia
-
♦
Delayed motor development
-
♦
Wide phenotypic range and age of onset from severely affected infants (congenital DM1) to classically affected adults (classic DM1) to minimally symptomatic elderly individuals (mild DM1) with mildly affected adults having myotonia and cataracts vs congenital DM1 which is characterized by severe weakness and often fatal respirato ry insufficiency
Molecular Basis of Disease
-
♦
Expansion of a trinucleotide CTG repeat in t he myotonic dystrophy protein kinase (DMPK) gene
-
5–34 normal allele size
-
35–49 mutable normal (premutation) allele; not associated with disease
-
>50 full penetrance allele; severity of phenotype increases as repeat size increases (mild DM1: 50–150 repeats; classic DM1: 100–1,000 repeats)
-
>1,000 full penetrance allele, severely affected (congenital form)
-
Expansion occurs preferentially through mate rnal transmission but can also occur through paternal transmission
-
Laboratory
-
♦
Molecular analysis identifies expanded repeat in nearly 100% of cases
-
♦
Electromyogram demonstrates characteristic electrical myotonic discharges
-
♦
Muscle biopsy ma y show marked proliferation of fibers within the central nuclei, sacroplasmic masses, and type 1 muscle fiber atrophy
Treatment
-
♦
Not curable, supportive/symptomatic
-
♦
Monitor and treat for cataracts, cardiac conduction disturbances, diabetes, sleep apnea, hyp ogonadism, and other endocrine problems
Myotonic Dystrophy Type 2 (Proximal Myotonic Myopathy)
Chromosome and Gene Location
-
♦
3q21.3
Inheritance
-
♦
Autosomal dominant
Incidence
-
♦
1/8,000 (DM1 and DM2; together, th e most common muscular dystrophy in adults)
Clinical Manifestations
-
♦
Myotonia (sustained muscle contraction), proximal weakness primarily in the thighs, and muscle pain
-
♦
Cataracts
-
♦
Cardiac conduction disturbances
-
♦
Insulin insensitivity predisposing to hyperglycemia and diabetes mellitus
-
♦
Hypogonadism
-
♦
Onset typically occurs in the third decade of life
-
♦
Myotonic dystrophy type 2 includes DM2 and proximal myotonic myopathy (PROMM)
Molecular Basis of Disease
-
♦
Expansion of a trinucleotide CCTG repeat in intron 1 of the CNBP/ZNF9 gene
-
Reported allele sizes usually include the total number of repeats/base pairs in the (TG) n (TCTG) n (CCTG) n repeat tract all present at the DM2 locus
-
Tetranucleotides (TCTG or GCTG) commonly interrupt the CCTG repeats in the normal range; however, these sequence interruptions are not found in fully expanded pathogenic alleles
-
11–26 CCTG repeats (104–176 bp total allele length) normal allele
-
27–74 CCTG repeats (177–372 bp total allele length) intermediate normal (intermediate/borderline alleles of unclear significance) allele; never been reported
-
75–11,000 CCTG repeats (372–44,000 bp total allele length) full penetrance allele
-
CCGT repeat size typically co ntracts through either maternal or paternal transmissions and somatic instability causes CCGT repeat sizes to increase as the affected individual ages
-
Phenotype and age of onset cannot be predicted by CCGT repeat size as there is no correlation between repeat size and se verity of disease
-
Laboratory
-
♦
Molecular analysis identifies expanded repeat in 99% of cases
-
♦
Electromyogram demonstrates characteristic electrical myotonic changes
-
♦
Muscle biopsy may show marked proliferation of fibers within the central nuclei and ty pe 2 muscle fiber atrophy
Treatment
-
♦
Not curable, supportive/symptomatic
-
♦
Monitor a nd treat for cataracts, cardiac conduction disturbances, diabetes, and hypogonadism
Friedreich Ataxia
Chromosome and Gene Location
-
♦
9q21.11
Inheritance
-
♦
Autosomal recessive
Incidence
-
♦
1/25,000–1/50,000
Clinical Manifestations
-
♦
Progressive ataxia, muscle weakness, dysarthria, and dysphagia
-
♦
Absent deep tendon reflexes and decreased vibration and/or joint-position sense
-
♦
Optic nerve atrophy (25%)
-
♦
Deafness (13%)
-
♦
Scoliosis
-
♦
Hypertrophic cardiomyopathy (66%)
-
♦
Bladder dysfunctio n (urinary frequency and urgency)
-
♦
Hammer toes, pes cavus (foot deformity marked by high arch), and restless leg syndrome
-
♦
Diabetes (30%) or glucose intolerance (50%)
-
♦
Obstructive sleep apnea (20%)
-
♦
Age of onset is approximately 10–15 years for classic Friedreich ataxia (FRDA); however, ~15% of cases are associated with delayed onset (late-onset FRDA, LOFA, and very late-onset FRDA, VLOFA), and ~12% are associated with delayed-on set and intact tendon reflexes (FRDA with retained reflexes, FARR)
Molecular Basis of Disease
-
♦
Expansion of a trinucleotide GAA repeat in intron 1 of the frataxin (FXN) gene
-
5–33 normal allele
-
34–65 premutation (meiotic instability with potential to expand to full penetrance allele) allele. (Note: the term borderline alleles, defined as 44–66, is sometimes used because the shortest repeat le ngth associated with disease has not yet been clearly determined)
-
66–1,700 full penetrance allele
-
Carriers of permutation alleles are very rare; therefore, expansion of alleles from one generation to the next is typically not observed, and anticipation is not a hallmark characteristic of this disease
-
Statistically significant correlations have been observed between the size of the smaller of the two expanded alleles and age of onset (i.e., LOFA and VLOFA), presence of leg muscle weakness, duration of wheelchair use, and presence of other clinical manifestations
-
Laboratory
-
♦
Molecular analysis identifies two expanded repeat sizes in 90–94% of cases
-
♦
Molecular analysis by sequencing and dosage analysis detects the remaining FXN mutations
-
♦
Nerve conduction studies generally show slow or absent sensory nerve conduction velociti es but normal motor nerve conduction
Treatment
-
♦
Not curable, supportive/symptomatic
-
♦
Monitor for hypertrophic cardiomyopathy (by echocardiography and electrocardiogram), diabetes, and deafness
-
♦
Treat with assistive devices for weakness, antispasmodic agents for bladder dysfunction, and speech therapy to maximize communication
Huntington Disease
Chromosome and Gene Location
-
♦
4p16.3
Inheritance
-
♦
Autosomal dominant
Incidence
-
♦
3/100,000–7/100,000 (Western European ancestry)
-
♦
0.1/100,000–15/100,000 (range, all ethnicities)
Clinical Manifestations
-
♦
Slowly progress ive fatal disorder with onset of neurologic or psychiatric changes usually in mid-life (35–44 years)
-
♦
Life expectancy is approximately 50–60 years or about 15–18 years after initial onset of disease
-
♦
Early (preclinical)
-
Behavioral/mood changes and personality changes (72%)
-
Depression
-
Anxiety
-
Delusions and hallucinations
-
Abnormal eye movements
-
Minor changes in coor dination and movement
-
-
♦
Middle (clinical)
-
Chorea (90%)
-
Involuntary movements
-
Difficulty with voluntary movements (slow, difficult to initiate or control, impaired reaction time, trouble with balance and walking)
-
Weakness
-
Dysarthria and dysphagia
-
Cognitive decline
-
-
♦
Late
-
Inability to walk and speak
-
Incontinence
-
Rigidity and dystonia
-
Total dependence on others
-
-
♦
Juvenile Huntington disease (onset prior to age 20 years) is characterized by motor, cognitive, and psychiatric changes; however, the clinical presentation differs and is much more severe than in adults; epileptic seizures are also common
Molecular Basis of Disease
-
♦
Disease results from an expansion of a trinucleotide CAG repeat in exon 1 of the HTT gene
-
10–26 normal allele
-
27–35 intermediate allele (meiotic instability, expansion to full penetrance allele may occur with transmission); not associated with disease
-
36–39 reduced penetrance allele (at risk but not certain to develop symptoms of disease)
-
>40 full penetrance allele
-
>60 full penetrance allele is associated with juvenile onset Huntington disease
-
Expansion can occur through both maternal and paternal transmission; however, large expansions are more frequentl y observed through paternal transmission
-
Inverse correlation between number of CAG repeats and age of onset
-
Laboratory
-
♦
Molecula r analysis identifies expanded repeat in 100% of cases
-
♦
Imaging studies such as magnetic resonance imaging (MRI) identifies atrophy of the caudate nucleus and putamen
Treatment
-
♦
Not curable, supportive/symptom atic
Autosomal Dominant Cerebellar Ataxias
Chromosome and Gene Location
-
♦
Numerous loci have been identified, and more than 30 subtypes have been delineated (commonly referred to as spinocerebellar ataxias, SCAs are numbered by subtype with the exception of DRPLA; dentatorubropallidoluysian atrophy)
Inheritance
-
♦
Autosomal d ominant
Incidence
-
♦
1–5/100,000
Clinical Manifestations
-
♦
Clinical manifestations vary by subtype; however, the most common features include adult-onset gait ataxia, dysarthria, ataxia (NOS), visual disturbance and diplopia, and dizziness
-
♦
Peripheral neuropathy
-
♦
Death usually occurs 10–20 years following age of onset
Molecular Basis of Disease
-
♦
Many subtypes of spinocerebellar ataxia (SCA) result from expansion of trinucleotide CAG repeat, a lthough some (e.g., SCA5) result from nonrepeat mutations
-
♦
SCA3 (also known as Machado–Joseph disease) is the most common subtype followed by SCA 1, 2, 6, and 7 (represented in Table 13.2)
Laboratory
-
♦
Molecular an alysis identifies expanded trinucleotide repeat in most subtypes
-
♦
Neuroimaging may be necessary to distinguish hereditary forms of ataxia from acquired forms
Treatment
-
♦
Not curable, supportive/symptoma tic
Spinal and Bulbar Muscular Atrophy (Kennedy Disease)
Chromosome and Gene Location
-
♦
Xq12
Inheritance
-
♦
X-linked recessive
Incidence
-
♦
1/50,000 males
Clinical Manifestations
-
♦
Teen to adult onset, usually in the third to fifth decades of life
-
♦
Slowly progressive proximal muscle weakness, muscle atrophy, and fasciculations with bulbar involvement, dysarthria, and dysphagia
-
♦
Androgen insensitivity (gynecomastia, reduced fertility, testicular atrophy)
-
♦
Normal life expectancy
Molecular Basis of Disease
-
♦
Expansion of trinucleotide CAG repeat in t he human androgen receptor (AR) gene
-
11–34 normal allele size
-
35 unknown clinical significance
-
36–37 reduced penetrance allele (at risk but not certain to develop symptoms of disease)
-
>37 full penetrance allele
-
The larger the expansion, the earlier the age at onset of disease and the more rapid the disease progression
-
Laboratory
-
♦
Molecula r analysis identifies expanded repeat in 100% of cases
Treatment
-
♦
Not curable, supportive/symptomatic
-
♦
Hormone replacement as needed
-
♦
Monitor with annual strength testing and pulmonary function
Neuromuscular Disorders
Duchenne/Becker Muscular Dystrophy
Chromosome and Gene Location
-
♦
Xp21.2
Inheritance
-
♦
X-linked r ecessive
Incidence
-
♦
1/3,500 (male) 1/1,500 (female carriers)
-
30% of cases are new mutations
-
5–15% of sporadic cases are result of gonadal mosaicism (mother carries a subpopulation of oocytes with the mutation)
-
Clinical Manifestations
-
♦
Duchenne muscular dystrophy
-
Proximal muscle weakness le ading to difficulties involving gait, jumping, and climbing stairs
-
Positive for Gower sign (use of arms to push oneself into standing position by moving hands up one’s thighs, indicative of hip weakness)
-
Pseudohypertrophy of the calf muscles and proximal muscle weakness
-
Rapidly progressive with loss of ability to walk before age 13
-
Dilated cardiomyopathy (100% after by age 18)
-
Intellectual disability (25–35%)
-
Death usually occurs by the third decade in most
-
-
♦
Becker muscular dystrophy
-
Milder course of muscle involve ment with later-onset skeletal muscle weakness
-
Slower progression, wheelchair dependent after age 16
-
Survival into the 30–40 s, with dilated cardiomyopathy being the most common cause of death
-
Cognitive problems ar e rare
-
-
♦
Female carriers of Duchenne/Becker muscular dystrophy (DMD or BMD)
-
Muscle weakness (19% DMD carriers, 14% BMD carriers)
-
Myalgia cramps (5%)
-
Left ventricle dilation (19% DMD carriers, 16% BMD carriers)
-
Dilated cardiom yopathy (8% of DMD carriers)
-
Molecular Basis of Disease
-
♦
Dystrophin is a protein found in the sarcolemma of normal muscle. It is thought to be involved in the anchorin g of the cytoskeleton of the muscle cell to extracellular proteins
-
♦
DMD and BMD result from alterations within the dystrophin (DMD) gene
-
♦
Deletions in 50–70% of DMD and BMD
-
Deletions that disrupt the reading frame of th e triplet code (frameshift mutations) lead to DMD
-
Deletions that do not disrupt the reading frame of the triplet code (in-frame mutations) most often lead to BMD
-
-
♦
Duplications in 5–10% of DMD and BMD
-
♦
Point mutati ons in 20–35% of DMD and 10–20% of BMD
Laboratory
-
♦
Pathology
-
Variability in size of muscle fiber s, degeneration, atrophy of individual fibers, and proliferation of endomysial and perimysial connective tissue
-
-
♦
Antidystrophin antibodies detect
-
<5% of normal dystrophin in DMD
-
5–20% in mild DMD or severe BMD
-
>20% normal dystrophin co rrelates with BMD
-
-
♦
Serum creatinine phosphokinase (CK) concentration
-
>5–10× the normal range (100% of affected individuals)
-
50% of DMD carriers and 33% of BMD carriers have elevated CK levels (2–10× normal)
-
Caution must be used as CK levels vary with age, pregnancy, and activity
-
-
♦
Genetics
-
Deletions and duplications detected directly by molecular analysis
-
Linkage analysis is available when deletion/duplication analysis negative
-
Treatment
-
♦
Not curable, su pportive/symptomatic
-
♦
Treatment of dilated cardiomyopathy with medication and transplantation in rare cases
-
♦
Corticoste roid therapy (prednisone) in affected individuals to improve strength and function
Spinal Muscular Atrophy, Types I–IV
Chromosome and Gene Location
-
♦
5q13.1
Inheritance
-
♦
Aut osomal recessive
Incidence
-
♦
1/25,000
Clinical Manifestations
-
♦
See Table 13.3
Table 13.3. Spinal Muscular Atrophy (SMA) Subtypes
Molecular Basis of Disease
-
♦
Survival motor neuron (SMN1) gene is homozygously deleted in nearly all of types I and II and ab out 80% of type III spinal muscular atrophy (SMA)
-
♦
2–5% of type I SMA is caused by compound heterozygosity for an SMN1 deletion and point mutation
-
♦
The presence of three or more copies of SMN2, a gene just adjacent to SMN1, is associated with a milder SMA phenotype in individuals with two SMN1 deletions and/or point mutations
Laboratory
-
♦
The primary test used to diagnose individuals with SMA is molecular analysis for deletions in exon 7 and/or exon 8 of SMN1 gene
-
♦
Muscle biopsy reveals group atrophy of type 1 and type 2 muscle fibers
Treatment
-
♦
Not curable, supportive/symptomat ic
Charcot–Marie–Tooth Disease
Chromosome and Gene Location
-
♦
Numerous loci have been identified
Inheritance
-
♦
Autosomal dominant, recessive and X-linked forms (see Table 13.4)
Table 13.4. Charcot–Marie–Tooth ( CMT) Subtypes
Incidence
-
♦
1/3,300 (the mos t common genetic cause of neuropathy)
Clinical Manifestations
-
♦
Hereditary neuropathy resulting in progressive distal muscular atrophy and weakness of arms and legs (presenting in first to third decade)
-
♦
Pes cavus
-
♦
CMT1 – Charcot–Marie–Tooth type 1 (50%)
-
Demyelinating per ipheral neuropathy
-
Distal muscle weakness and atrophy
-
Decreased nerve conduction velocities
-
Absent deep tendon reflexes
-
Onset 5–25 years
-
Six subtypes (CMT1A–CMT1F) are dis tinguishable only by molecular analysis
-
Autosomal dominant inheritance
-
-
♦
CMT2 – Charcot–Marie–Tooth type 2 (20–40%)
-
Axonal (non-demyelinat ing) neuropathy
-
Distal muscle weakness and atrophy
-
Normal or slightly decreased nerve conduction velocities
-
Deep tendon reflexes are preserved
-
Milder phenotype than CMT1
-
15 subtypes distinguishable by molecular analysis
-
Autosomal dominan t inheritance
-
-
♦
Autosomal dominant intermediate CMT
-
Demyelinating and axonal neuropathy
-
Decreased nerve conduction velocities
-
-
♦
CMT4 – Charcot–Marie–Tooth type 4
-
Demyelinating or a xonal neuropathy
-
Distal muscle weakness and atrophy
-
Pes cavus
-
Autosomal recessive inheritance
-
-
♦
CMTX – X-linked Charcot–Marie–Tooth (10–15%)
-
Axonal neuropathy in males, intellec tual disability, deafness, and spasticity
-
Five subtypes
-
Molecular Basis of Disease
-
♦
Multiple gene s at different loci are involved (see Table 13.4). Molecular analysis is available for many subtypes. Some subtypes are clinically indistinguishable and are designated solely on molecular findings
Laboratory
-
♦
Molecular testing is available for many of the CMT subtypes
-
♦
Electrophysiological studies and nerve biopsy may be helpful in distinguishing it from other acquired and hereditary forms of peripheral neuropathy and in establishing a diag nosis of CMT
Treatment
-
♦
Not curable, supportive/symptomatic
Hereditary Neuropathy with Liability to Pressure Palsies
Chromosome and Gene Location
-
♦
17p11.2
Inheritance
-
♦
Autosoma l dominant
Incidence
-
♦
Unknown
Clinical Manifestations
-
♦
Recurrent transient palsies (i.e., carpel tunnel syndrome and foot drop)
-
♦
Sensory dysfunction (result of compression to peripheral nerve)
-
♦
Pes cavus (20%)
-
♦
Absent ank le reflexes (50–80%) and reduced tendon reflexes (15–30%)
-
♦
Scoliosis
Molecular Basis of Disease
-
♦
Deletion at 17p11.2 involving the peripheral myelin protein 22 (PMP22) gene (80%) or poin t mutations in the PMP22 gene (20%)
-
♦
Unequal crossing over between misaligned repetitive elements leads to the hereditary neuropathy with liability to pressure palsies (HNPP) deletion and the CMT1A duplication syndromes
-
♦
De novo mutations are most often paternally derived
Laboratory
-
♦
Cytogene tics using FISH or molecular genetics analysis identifies deletion at 17p11.2 involving the PMP22 gene or a point mutation
-
♦
Nerve biopsy reveals sausage-shaped swellings of myelin sheath
-
♦
Increase in distal motor latency of the median nerve at the wrist in both symptomatic and asymptomatic individuals
-
♦
Reduced motor and sensory nerve conduction velocity
Treatment
-
♦
Not curable, supportive/sympt omatic
Skeletal Disorders
Craniosynostosis
Apert Syndrome
Chromosome and Gene Location
-
♦
10q24 (FGFR2)
Inheritance
-
♦
Autosomal dominant, vast majority due to new mutation
Incidence
-
♦
1/65,000–1/100,000
Clinical Manifestations
-
♦
Brac hycephaly due to coronal craniosynostosis
-
♦
Hypoplasia of midface and hypertelorism
-
♦
“Mitten hand” deformity; symmetric and severe osseous or cutaneous syndactyly affecting hands and feet, and broad thumbs and great toes
-
♦
Fused ce rvical vertebrae, typically C5–C6
-
♦
Genitourinary anomalies in 10%
-
♦
Cardiac anomalies in 10%
-
♦
Intellectual disability
Molecular Basis of Disease
-
♦
Mutation in fibroblast growth factor receptor 2 (FGFR2), tyrosine kinase receptor involved in regulation of osteogenesis
-
♦
Two common gain-of-function mutations account for >98% of cases
Laboratory
-
♦
Targeted m olecular testing for common mutations, whole-gene sequencing, and deletion/duplication studies of FGFR2 are all available
Treatment
-
♦
Not curable, sup portive/symptomatic and surgical management
-
♦
Early surgical consultation and correction are important to reduce the risk of intracranial pressure
Crouzon Syndrome
Chromosome and Gene Location
-
♦
10q24 (FGFR2)
Inheritance
-
♦
Autosomal dominant with variable expressivity
Incidence
-
♦
1/25,000
Clinical Manifestations
-
♦
Coron al, lambdoid, or sagittal craniosynostosis
-
♦
Hypoplasia of midface
-
♦
Hypertelorism
-
♦
Proptosis
-
♦
Beaked nose
-
♦
Normal intelligence
Molecular Basis of Disease
-
♦
Mutation in FGFR2, tyrosine ki nase receptor involved in regulation of osteogenesis
Laboratory
-
♦
Targeted molecular testing for common mutations, sequencing of select exons or the entire FGFR2 gene, and deletion/duplication studies of FGFR2 are all available
Treatment
-
♦
Not curable, supp ortive/symptomatic and surgical management
Pfeiffer Syndrome (Types I–III)
Chromosome and Gene Location
-
♦
8p11.2 (FGFR1) type I
-
♦
10q24 (FGFR2) types I–III
Inheritance
-
♦
Autosom al dominant with variable expressivity
Incidence
-
♦
1/100,000
Clinical Manifestations
-
♦
See Table 13.5
Table 13.5. Clinical Findings of Pfeiffer Syndrome
Molecular Basis of Disease
-
♦
Type I: mutation in fibroblast growth factor receptor (FGFR) 1 (5% of cases) or 2 (95% of cases), tyro sine kinase receptors involved in regulation of osteogenesis
-
♦
Type II or III: mutation in FGFR2, tyrosine kinase receptor involved in regulation of osteogenesis
Laboratory
-
♦
Targeted molecular testing for common mutations, sequencing of select exons or the entire FGFR1/FGFR2 genes, and deletion/duplication studies of FGFR1/ FG FR2 are all available
Treatment
-
♦
Not curable, supportive/symptomatic and surgical management
-
♦
Types II and III are typically more severe and have an increased risk for early death, but early and aggressive surgical and medical treatment may increase the possibility of a positive outcome
Saethre–Chotzen Syndrome
Chromosome and Gene Location
-
♦
7p21.1 (TWIST1)
Inheritance
-
♦
Autosomal dominant
Incidence
-
♦
1/25,000–1/50,000
Clinical Manifestations
-
♦
Brachyc ephaly due to coronal craniosynostosis
-
♦
Ptosis
-
♦
Maxillary hypoplasia
-
♦
Small ears with prominent crus
-
♦
Cutaneous syndactyly
-
♦
Normal intelligence typically
-
♦
Patients wi th microdeletions may have intellectual disability
Molecular Basis of Disease
-
♦
Typically caused by loss of function of the TWIST gene, which negatively regulates FGFR and osteogenic transcription factors. Loss-of-function mutations, exonic or whole-gene deletions, and translocations/inversions involving 7p21 have all been reported
Laboratory
-
♦
Targeted molecular testing for common mutations, sequencing of select exons or the entire gene, and deletion/duplication studies of TWIST1 are all available
Treatment
-
♦
Not curab le, supportive/symptomatic and surgical management
Muenke Syndrome
Chromosome and Gene Location
-
♦
4p16.3 (FGFR3)
Inheritance
-
♦
Autosomal dominant
Incidence
-
♦
1/30,000
Clinical Manifestations
-
♦
Brachyc ephaly due to bilateral coronal craniosynostosis or anterior plagiocephaly due to unilateral coronal craniosynostosis
-
♦
Intellectual disabilities
-
♦
Strabismus
-
♦
Hearing loss
-
♦
Carpal bone and/or tarsal bone fusions
-
♦
Broad toes and/or thumbs
-
♦
High-arche d palate or cleft lip and/or palate
Molecular Basis of Disease
-
♦
Specific pathogenic mutation p.Pro250Arg in FGFR3 which accelerates bone differentiation
Laboratory
-
♦
Targeted molecular testing for the common mutation, sequencing of select exons or the entire gene, and deletion/duplication studies of FGFR3 are all available
Treatment
-
♦
Not curable, supportive/sym tomatic
Achondroplasia
Chromosome and Gene Location
-
♦
4p16.3 (FGFR3)
Inheritance
-
♦
Autosoma l dominant; 80% result from a new mutation
Incidence
-
♦
1/25,000
Clinical Manifestations
-
♦
Short stature, proximal shortening of long bones, disproportionate shortening of limbs, and gen u varum
-
♦
Large head, frontal bossing, and hypoplasia of midface
-
♦
Infantile hypotonia
-
♦
Gross motor developmental delay
-
♦
Normal int elligence
-
♦
Normal life expectancy
-
♦
Also at risk for cord compression due to odontoid hypoplasia
Molecular Basis of Disease
-
♦
Mutation in transmembrane domain of fibroblast growth factor transmembrane receptor (FGFR3)
-
♦
>99% caused by same mutation
Laboratory
-
♦
X-ray implicates skeletal involvement
-
♦
Targeted mutation testing for the common mutations in FGFR3 that account for >99% of cases is available. Sequencing of select exons or the entire FGFR3 gene is also avai lable
Treatment
-
♦
Not curable, supportive/sympto matic
Osteogenesis Imperfecta (Types I–VII)
Chromosome and Gene Location
-
♦
See Table 13.6
Table 13.6. Clinical Findings of Osteogenesis Imperfecta
Inheritance
-
♦
See Table 13.6
Incidence
-
♦
1/20,000–30,000
Clinical Manifestations
-
♦
See Table 13.6
-
♦
A genetic consulta tion is recommended given clinical and molecular genetic diagnosis of one of the many types may be quite complex
Molecular Basis of Disease
-
♦
Collagen is the major protein of the white fibers of connective tissue, cartilage, and bone
-
♦
There have been nu merous types of collagen identified
-
♦
Mutations in the procollagen genes, whose products make up the triple helix of type 1 collagen, lead to the various types of osteogenesis imperfecta
-
♦
Clinical presentation is dependent on the extent to which the mutation alters the protein product
Laboratory
-
♦
X-ray implicates s keletal involvement
-
♦
Molecular genetic testing of the procollagen and some of the other disease-causing genes is available
-
♦
Collagen testing on cultured fibroblasts available
Treatment
-
♦
Not curable, supportive/symptomatic
-
♦
Surgical inte rvention when indicated
Connective Tissue Disorders
Marfan Syndrome
Chromosome and Gene Location
-
♦
15q21.1 (FBN1)
Inheritance
-
♦
Autosomal dominant
-
♦
15–30% result from a new mutation
Incidence
-
♦
1 in 5,000–10,000
Clinical Manifestations
-
♦
Diagno sis based on clinical criteria
-
♦
Tall, thin habitus, long extremities, and arachnodactyly (Fig. 13.5A)
Fig. 13.5. 
Marfan syndrome – arachnodactyly, wrist sign (A). Pectus deformity and striae on shoulders (B). Intraoperative view of dilated aortic root (C).
-
♦
Pectus deformities (Fig. 13.5B) and scoliosis
-
♦
Ectopia lentis and retinal detachment
-
♦
Mitral valve prolapse, aortic dilation (Fig. 13.5C), and aortic aneurysm
-
♦
Without treatment, life expectancy is reduced to about two-thirds normal life span. With proper management of cardiovascular manifestations, life expectancy approximates that of the general population

Molecular Basis of Disease
-
♦
FBN1 codes for fibrillin, a structural protein, which is the major constituent of microfibrils
Laboratory
-
♦
Sanger sequencing a nd deletion/duplication testing via array comparative genomic hybridization (aCGH) or multiplex ligation-dependent probe amplification (MLPA) is available for the FBN1 gene
-
♦
FBN1 analysis is also available as a part of several clinically available multigene panels which utilize next-generation sequencing
Treatment
-
♦
Surgical inte rvention when indicated
-
♦
Close monitoring of heart defects as they can lead to sudden death
-
♦
Use of beta-adrenergic blockade
-
♦
Clinical trials underway to assess the potential benefits of angiotensin II receptor antagonist drugs (losartan) on slowing aneurysm progression
-
♦
Referral to genetics for personal and family history evaluation, detailed medical exam, and app ropriate health management
Loeys–Dietz Syndrome
Chromosome and Gene Location
-
♦
9q22 (TGFBR1), 3p22 (TGFBR2), 15q22.33 (SMAD3), and 1q41 (TGFB2)
Inheritance
-
♦
Autosomal dominant
-
♦
~75% result from new mutation
Incidence
-
♦
Unknown, no apparent enrichment in any ethnic group
Clinical Manifestations
-
♦
Minimal clinical diagnostic criteria not established
-
♦
Two clinical phenotypes described. LDS type I has vascular, skeletal, and craniofacial findings. LDS type II has vascular, skeletal, and cutaneous findings
-
♦
Dilatation or dissection of the aorta. Aortic dilatation presents in >95% of probands. Arterial aneurysms tend to be more aggressive and dissect at smaller aortic dimensions than in Marf an syndrome
-
♦
Other arterial aneurysms and tortuosity throughout the arterial tree
-
♦
Pectus excavatum or pectus carinatum and scoliosis
-
♦
Joint laxity
-
♦
Arachnodactyly
-
♦
Talipes equinovarus
-
♦
Ocular hyper telorism, bifid uvula/cleft palate, and craniosynostosis (most commonly sagittal, but also coronal and metopic) in LDS type I
-
♦
Translucent skin, easy bruising, and dystrophic scars in LDS type II
Molecular Basis of Disease
-
♦
Mutations in genes coding for a transforming growth factor ligand, cofactor, and two receptors, which regulate a variety of cellular processes, including proliferation, differentiation, cell cycle arrest, apoptosis, and formation of the extrac ellular matrix
Laboratory
-
♦
Sanger sequencing and deletion/duplication testing via array comparative genomic hybridization (aCGH) or multiplex ligation-dependent probe amplification (MLPA) is a vailable for TGFBR1, TGFBR2, SMAD3, and TGFB2
-
♦
TGFBR1, TGFBR2, SMAD3, and TGFB2 analysis is also available as a part of several clinically available multigene panels which utilize next-generation sequencing
Treatment
-
♦
Echocardiography at frequent intervals to monitor ascending aorta, MRA, or CT as indicated
-
♦
Early and aggressive surgical intervention
-
♦
Use of beta-adrenergic blockade
-
♦
Referral to genetics for personal and family history evaluation, detailed medical exam, and ap propriate health management
Thoracic Aortic Aneurysm and Dissection
Chromosome and Gene Location
-
♦
Seven genes and two loci have been associated with thoracic aortic aneurysm and dissection (TAAD):
-
TGFBR2 on 3p22
-
TGFBR1 on 9q33–q34
-
MYH11 on 16p13.13–p13, 12
-
ACTA2 on 10q22–q24
-
MYLK on 3q21
-
SMAD3 on 15q22.33
-
AAT2 (TAAD1) locus on 5q13–q14 (causative gene unknown)
-
AAT1 (FAA1) locus on 11q23.3–q24 (causative gene unknown)
-
Rare cases of FBN1 mutations, 15q21.1, have been associated with isolated TAAD (incidence unkn own)
-
Inheritance
-
♦
Autosomal dominant
Incidence
-
♦
Aortic aneurysms account for approximately 1–2% of deaths in industrialized society
-
♦
Approximately 20% of familial TAAD is accounted for by mutations in known genes. It is likely that other genes with variable penetrance have yet to be discovered
Clinical Manifestations
-
♦
Major diagnostic criteria for TAAD are the following:
-
♦
Presence of dilatation and/or dissection of the ascending thoracic aorta or dissection of the descending aorta just distal to the origin of the subclavian artery
-
♦
Exclusion of Marfan syndrome, Loeys–Dietz syndrome, and other connective tissue abnormalities
-
♦
Other occasional manifestations include inguinal hernia, scoliosis, aneurysms in other portions of the aorta, cerebral aneurysms, livedo reticularis (ACTA2), iris flocculi (ACTA2), bicuspid aor tic valve (ACTA2), and patent ductus arteriosus (MYH11)
Molecular Basis of Disease
-
♦
TGFBR1, TGFBR2, and SMAD3 code for proteins that regulate a variety of cellular processes, including proliferation, differentiation, cell cycle arrest, apoptosis, and formation of the extracellular matrix. Mutations in TAAD families are most often found in the kinase domain of these proteins, though rarely other mutations have been reported
-
♦
MYH11 codes for smooth muscle heavy chain protein. Smooth muscle myosin is the major contractile protein of smooth muscle and is composed of a MYH11 dimer and two pairs of nonidentical light chains
-
♦
ACTA2 codes for smooth muscle cell alpha-actin, a major contractile protein in smooth muscle cells
-
♦
MYLK codes for a phosphokinase which facilitates interactions between myosin and actin filaments, important in contractile activity
Laboratory
-
♦
Sanger sequencing and deletion/duplication testing via array comparative genomic hybridization (aCGH) or multiplex ligation-dependent probe amplification (MLPA) is available fo r TGFBR1, TGFBR2, MYH11, ACTA2, FBN1, and SMAD3
-
♦
Sanger sequencing is available for MYLK
-
♦
TGFBR1, TGFBR2, MYH11, ACTA2, MYLK, FBN1, and SMAD3 analysis is also available as a part of several clinically available multigene panels which utilize next-generation sequencing
Treatment
-
♦
Echocardio graphy to monitor aortic root, consider imaging entire aorta and other vasculature
-
♦
Prophylactic surgical repair of the aorta (timing depends on rate of progression, causative gene, etc)
-
♦
Medications that reduce hemodynamic stress, such as beta-adrenergic blockade
-
♦
Aggressive treatment of hypertension
-
♦
Assessmen t and standard treatment for hernias and scoliosis
-
♦
Referral to genetics for personal and family history evaluation, detailed medical exam, a nd appropriate health management
Ehlers–Danlos Syndrome
Chromosome and Gene Location
-
♦
Classic type: 9q34 (COL5A1), and 2q31 (COL5A2)
-
♦
Vascular type: 2q31 (COL3A1)
-
♦
Others
-
Hypermobility type: c ausative gene is mostly unknown; there are some cases of TNXB mutations (6p21)
-
Kyphoscoliotic type: 1p36 (PLOD1)
-
Arthrochalasia type: 17q21–q22 (COL1A1), and 7q22 (COL1A2)
-
Spondylocheiro dy splastic form: 11p11.2 (SLC39A13)
-
Dermatosparactic type: 5q35.3 (ADAMTS2)
-
Musculocontractural type 1: 15q15.1 (CHST14)
-
Progressive kyphoscoliosis, myopathy, and hearing loss type: 7p15 (FKBP14)
-
Inheritance
-
♦
Classic type, arthrochalasia type, and vascular type are autosomal dominant
-
♦
Most rare forms of Ehlers–Danlos syndrome are autosomal recessive
-
♦
Mutations in TNXB can be inherited in an autosomal dominant or autosomal recessive manner
Incidence
-
♦
Classi c type: estimated 1/20,000
-
♦
Vascular type: estimated 1/250,000
Clinical Manifestations
-
♦
Classic type
-
Skin hyperex tensibility
-
Smooth, velvety skin
-
Widened atrophic scarring
-
Abnormal, delayed wound healing
-
Joint hypermobility
-
Joint dislocations
-
Easy bruising
-
Generalized tissue fragility
-
Less common ly, mitral and tricuspid valve prolapse, aortic root dilatation, and spontaneous rupture of large arteries
-
-
♦
Vascular type
-
Thin, translucent skin
-
Characteristi c facial appearance (thin nose and lips, emaciated face with prominent cheekbones, eyes appear sunken or bulging, often with coloring around them and thin telangiectasia on the eyelids)
-
Easy bruising
-
Arterial, intestinal, and/or uterine fragility
-
Vascular rupture or dissection
-
GI perforation
-
Org an rupture
-
Molecular Basis of Disease
-
♦
Collagen is the major protein of the white fibers of connective tissue, cartilage, and bone
-
♦
Mutations in collagen genes lead to decreased synthesis, altered secretion, and instability of collagen
-
♦
The defect for some types is unknown
Laboratory
-
♦
Protein analysis is available for some subtypes
-
♦
Histological features are nondiagnostic
-
♦
Sanger sequencing and deletion/duplication testing via array comparative genomic hybridization (aCGH) or multiplex ligation-dependent probe amplification (MLPA) is available for COL5A1, COL5A2, COL3A1, COL1A1, and COL1A2
-
♦
Sanger sequencing is available for PLOD1
-
♦
ADAMTS2 , CHST14, COL1A1, COL1A2, COL3A1, COL5A1, COL5A2, FKBP14, PLOD1, and SLC39A13 analysis is also available as a part of several clinically available multigene panels which utilize next-generation sequencing
Treatment
-
♦
Not curable, supportive/symptomatic
-
♦
Referral to genetics for personal and family history evaluation, detailed medical exam, and a ppropriate health management
Stickler Syndrome
Chromosome and Gene Location
-
♦
12q13 ( COL2A 1) and 1p21 (COL11A1) account for the majority of individuals with Stickler syndrome
-
♦
Also associated with 6p21.3 (COL11A2), 6q13 (COL9A1), 1p33–p32 (COL9A2), and 20q13.3 (COL9A3)
Inheritance
-
♦
Autosomal dominant for COL2A1, COL11A1, and COL11A2
-
♦
Autosomal recessive for COL9A1, COL9A2, and COL9A3
Incidence
-
♦
While the exact prevalence is unknown, estimations based on incidence of Pierre Robin sequence in newborns and the percent of these newborns who develop signs or symptoms of Stickler syndrome are approximately 1 in 7,500–9,000
Clinical Manifestations
-
♦
Progressive myopia, retinal detachment, and blindness
-
♦
Pierre Robin sequence (micrognathia and abnormal smallness of the tongue, often with cleft palate)
-
♦
Severe myo pia, congenital glaucoma, retinal detachment, and cataracts
-
♦
Premature degenerative changes in various joints with abnormal epiphyseal development
-
♦
Mitral valve prolapse
Molecular Basis of Disease
-
♦
The COL2A1 gene encodes the chains of type II collagen, a major structural component of cartilag inous tissues
-
♦
The COL11A1 gene encodes the alpha 1 chain of type XI collagen, thought to play an important role in fibrillogenesis by controlling lateral growth of collagen II fibrils
-
♦
The COL11A2 gene encodes the alpha 2 chain of type XI collagen expressed in cartilage but not in adult liver, skin, tendon, or vitreous
-
♦
The COL9A1, COL9A2, and COL9A3 genes code for type IX collagen which, together, form a subunit of three alpha chains found in tissues containing type II collagen
Laboratory
-
♦
Skeletal X-ra ys show changes of a skeletal dysplasia
-
♦
Sanger sequencing and deletion/duplication testing via array comparative genomic hybridization (aCGH) or multiplex ligation-dependent probe amplification (MLPA) is available for COL2A1, COL11A1, COL11A2, COL9A1, COL9A2, and COL9A3
-
♦
COL2A1, COL11A1, COL11A2, COL9A1, COL9A2, and COL9A3 analysis is also available as a part of several clinically available multigene panels which utilize next-generation sequencing
Treatment
-
♦
Not curable, supportive/symptomatic
-
♦
Referral to geneti cs for personal and family history evaluation, detailed medical exam, and appropriate health management
Alport Syndrome
Chromosome and Gene Location
-
♦
Xq22.3 (COL4A5), 2q36–q37 (COL4A3), and 2q35–q37 (COL4A4)
Inheritance
-
♦
COL4A5 mutations are inherited in an X-linked pattern (80% total AS cases)
-
♦
COL4A3 and COL4A4 cases are inherited in both autosomal recessive (15% total AS cases) and autosomal dominant (5% total AS cases) patterns
Incidence
-
♦
Estimated to be 1 in 50,000 live births
Clinical Manifestations
-
♦
Renal failure
-
♦
Sensorineural deafness
-
♦
Lenticonus
-
♦
Macular changes
Molecular Basis of Disease
-
♦
Mutations in the typ e IV collagen genes result in abnormalities in expression of the collagen chains and absent or defective structure and function in the collagen networks of the basement membranes
-
♦
Molecular testing on clinical basis
Laboratory
-
♦
Microscopic hematuria
-
♦
Urinary red cell casts
-
♦
Proteinuria
-
♦
Leukocyturia
-
♦
Abnormal glomer ular basement membrane on electron microscopy
Treatment
-
♦
Referral to genetics for personal and family history evaluation, detailed medical exam, and appropriate health management
-
♦
Not curable, sup portive/symptomatic
-
♦
Kidney transplant as indicated
Hematologic Disorders
Alpha Thalassemia
-
♦
Normal adult hemoglobin (Hb A) is a tetramer of two alpha and two beta globin chains. The alpha thalassemias are a group of inherited conditions characterized by decreased synthesis of alpha globin chains, resulting in an imbalance of globin chains in the formation of the hemoglobin (Hb) tetramer
Chromosome and Gene Location
-
♦
16p13.3-pt er (there are two alpha globin genes present at this locus on both copies of chromosome 16 for total normal complement of four alpha globin genes)
Inheritance
-
♦
Complex: individuals with alpha thalassemia may have alterations in two, three, or four alpha globin genes
Incidence
-
♦
Varies by po pulation; most common in African-American, Southeast Asian, Mediterranean, and Indian populations
-
♦
Severe forms occur predominantly in Asians
Clinical Manifestations
-
♦
An individual with one altered alpha globin gene is a “silent” carrier and typically does not have any clinical symptoms (α+-thalassemia)
-
♦
Individuals with two altered alpha globin genes, either of different chromosomes (i.e., in trans) or on the same chromosome (i.e., in cis), have alpha thalassemia trait (α0-thalassemia), which manifests as minimal anemia with microcytosis
-
♦
Hemoglobin H (Hb H) disease results when three alpha globin genes are altered. Hb H is an abnormal tetramer of four beta chains and is unstable. Thus, Hb H disease is a form of hemolytic anemia. This is characterized by moderate anemia, enlarged liver and spleen, and erythroid hyperplasia in the bone marrow
-
♦
Bart hydro ps fetalis results when all four alpha globin genes are not functional. Hb Bart is a tetramer of four gamma chains. The oxygen affinity of hemoglobin Bart is so high that it cannot release oxygen to the tissues. Onset is in the fetal period and includes severe hypochromic anemia, extramedullary erythropoiesis, generalized edema, pleural and pericardial effusions, hepatosplenomegaly, and hydrocephalus. This condition is not compatible with life; death occurs from anoxia in utero
Molecular Basis of Disease
-
♦
Alpha thalassemia results from mutations in the HBA1 and HBA2 genes that encode α1-globin and α2-globin, respectively. Mutations result in reduced production of the alpha globin chains. The clinical severity is determined by the degree of alpha globin chain deficiency relative to beta globin production. Numerous mutations have been found
-
♦
The most common types of mutations are deletions of one or both alpha globin genes, which results from the misalignment and subsequent recombination of the alpha globin genes during meiosis (i.e., nonhomologous crossover)
Laboratory
-
♦
Red blood cell indices reveal microcytic anemia in Hb H and α-thalassemia trait. Silent carriers generally have normal red blood cell indices. Hb Bart disease shows macrocytic red blood cells
-
♦
Peripheral blood s mear demonstrates hypochromic red cells and anisopoikilocytosis in Hb Bart syndrome. In Hb H disease, microcytosis, hypochromia, anisocytosis, and poikilocytosis are apparent. Carriers have morphologic changes that are less severe than affected individuals and may also have reduced MCV and MCH
-
♦
Inclusion bodies can be shown in Hb H disease using supravital stain; carriers and those with alpha thalassemia trait can also show small amounts of inclusions
-
♦
Hemoglobin analysis (e.g., electrophoresis, HPLC, and isoelectric focusing) detects the presence of Hb H (the abnormal β-globin tetramer) in adults and Hb Bart (an abnormal ϒ-globin tetramer) in infants
-
♦
Molecular genetic analysis is clinically available
Treatment
-
♦
Hemolytic or aplastic crisis caused by Hb H disease can be treated with red blood cell transfusions
-
♦
Splenect omy may be performed in cases of massive splenomegaly
-
♦
Iron chelation therapy is used if chronic blood transfusions are necessary
-
♦
Surveillance includes hematologic evaluation and growth and development assessment. Iro n overload should be monitored in individuals requiring transfusions
Beta Thalassemia
Chromosome and Gene Location
-
♦
11p15.5
Inheritance
-
♦
Typically autosomal recessive
Incidence
-
♦
Most commo nly found in Mediterranean and Southeast Asian populations but also in Middle Easterners, Africans, and African Americans
Clinical Manifestations
-
♦
The clinical presentation and classification of beta thalassemia is dependent on the extent to which beta chain production is decreased and the resulting imbalance of alpha globin chains to non-alpha globin chains
-
♦
Beta thalasse mia becomes clinically evident as fetal hemoglobin production decreases after birth and adult hemoglobin fails to replace it (around 6 months of age)
-
♦
Beta thalassemia major results from the homozygous or compound heterozygous mutations that severely reduce beta chain production
-
Onset between 6 and 24 months
-
Characterized by microcytic anemia, failure to thrive, fever, diarrhea, liver and spleen enlargement, and progressive pallor
-
Complications from iron overload include growth retardation, failure of sexual maturation, dilated cardiomy opathy, pericarditis, liver fibrosis and cirrhosis, diabetes, and parathyroid, thyroid, and pituitary insufficiency
-
Death often occurs in second or third decade due to cardiac complications
-
Untreated individuals exhibit severe failure to thrive, jaundice, increased skin pigmentation, liver and spleen enlargement, poor musculature, “chipmunk face,” skeletal deformities, and stunted growth and have a shortened life expectancy
-
-
♦
Beta thalassemia intermedia has m ilder phenotype and results from homozygosity or compound heterozygosity of alleles with reduced beta chain production; it may result from coinheritance of alpha and beta thalassemia
-
Variable presentation may include milder anemia, liver and spleen enlargement, moderate skeletal features, pallor, jaundice, cholelithiasis, osteopenia and osteoporosis, and extramedullary erythropoietic masses
-
There is an increased risk for iron overload due to increased absorption of iron in the intestine
-
Transfusions are performed as needed
-
-
♦
Beta thalassemia minor or thalassemia trait refers to a heterozygous carrier state. Carriers generally do not exhibit clinical symptoms other than possible mild anemia
Molecular Basis of Disease
-
♦
Beta thalassemia results from mutations in the HBB gene, which encodes for the beta hemoglo bin subunits
-
♦
The clinical presentation of beta thalassemia is dependent on the extent to which the genetic alteration disrupts beta chain production ranging from complete absence of production (i.e., β0 mutations) to “silent” or mild production disturbances (β+ mutations) and subsequently imbalance in the alpha to non-alpha globin chain ratio
-
♦
Normally, both alpha and beta globin chains are produced in roughly equal amounts. When beta globin synthesis is decreased, there are more free alpha chains. Free alpha chain s are very unstable and precipitate in the red cell. This results in ineffective erythropoiesis and anemia. Subsequently, there is a phenomenal increase in erythropoiesis with marrow expansion and persistence of erythropoiesis in the liver and spleen
-
♦
More than 200 genetic alterations hav e been described in the HBB gene, including:
-
Nucleotide substitutions
-
Deletions
-
Transcriptional mutations
-
RNA processing mutations
-
-
♦
Molecular analysis may be helpful in determining clinical phenotype and identifying at-risk relatives
Laboratory
-
♦
Red blood cell indices reveal microcytic anemia
-
♦
Peripheral blood smears show nucleated red blood cells, microcytosis, hypochromia, anisocytosis, and poikilocytosis
-
Carriers have reduced MCV and M CH, and RBC morphology is less severe than in affected individuals. Nucleated red blood cells are generally not seen in carriers
-
-
♦
Elevated iron
-
♦
Hyperplastic marrow with hyperplasia of red cell precursors
-
♦
Hemoglob in analysis shows reduced amounts of Hb A (normal adult hemoglobin) and increases in normal minor hemoglobins, such as Hb A2
-
♦
Molecular analysis is clinically available
Treatment
-
♦
Thalassemia major
-
Regular transfusions to correc t anemia, suppress erythropoiesis, and inhibit gastrointestinal absorption of iron
-
Iron chelation to prevent iron overload
-
Bone marrow transplantation from HLA-identical sib or cord blood transplantation from related donor
-
Ongoing surveillanc e is necessary to monitor effectiveness of transfusion and chelation therapy
-
•
Regular physical exams including growth and development assessment
-
•
Liver function tests; serum ferritin
-
•
Ophthalmologic, audiologic, and cardiac exams
-
•
Endocrine function and liver ultrasound
-
•
In adults, bone densitometry, serum AFP, and gallbladder echography are also assessed
-
•
-
-
♦
Thalassemia intermedia
-
Symptom atic therapy
-
Splenectomy
-
Folic acid supplementation
-
Sporadic red cell transfusions
-
Radiotherapy, transfusions, and hydroxyurea for treatment of extramedullary erythropoietic masses
-
Chelation therapy if iron overload develops
-
Sickle Cell Anemia
Chromosome and Gene Location
-
♦
11p15.5
Inheritance
-
♦
Autosomal recessive
Incidence
-
♦
1/500 African-Ame rican births
Clinical Manifestations
-
♦
Severe anemia
-
♦
Painful swelling and crisis due to vaso-occlusive episodes usually affecting the limbs, back, abdomen, and chest
-
♦
Pneumococcal sepsis
-
♦
Leg ulcerations
-
♦
Enlarged spleen
-
♦
Pallor
-
♦
Acute chest syndrome (e.g., the presence of infiltrate on chest radiograph, sometimes including lower respiratory tract symptoms, fever, and hypoxemia)
-
♦
Autosplenect omy (i.e., the reduction in spleen size due to continual splenic infarctions resulting from the filtration of abnormal red blood cells)
-
♦
Chronic organ damage (e.g., renal or hepatic damage; retinopathy)
-
♦
Delayed growth and sexual development
-
♦
Repeated infections
Molecular Basis of Disease
-
♦
Sickle cell disease most commonly results from inheritance of a glutamic acid to valine substitution in position six of the HBB gene encoding the beta globin
-
♦
This mutation le ads to the formation of hemoglobin S (Hb S), which alters the configuration of the hemoglobin molecule and causes the cells to sickle and obstruct blood flow in small vessels leading to ischemia of tissues and organs. Variable degrees of hemolysis also occur
-
♦
When inherited in a homozygous fashion, the condition is referred to as homozygous sickle cell disease or Hb SS
-
♦
Other forms of sickle cell disease result from inheritance of one Hb S allele along with anoth er abnormal beta chain variant, such as Hb C
Laboratory
-
♦
Significant quantities of Hb S are identified by HPLC, isoelectric focusing, or electrophoresis
-
♦
Sickling and reduced oxygen tension of red cells and nucleated red blood cells are apparent in a peripheral blood smear
-
♦
There is also an increase in neutrophils and platelets
-
♦
All 50 states provide newborn screening for sickle cell disease
-
♦
Direct molecular testing for nucleotide substitution is clinically available and can be used for confirmatory testing, carrier testing for at-risk relatives, and prenatal diagnosis
Treatment
-
♦
Vaso-occlusive pain is treated with oral hydration and analgesics
-
♦
Transfusion is used d uring splenic or hepatic sequestration crises
-
♦
Acute chest syndrome is treated with oxygen, spirometry, analgesics, antibiotics, and transfusions
-
♦
Pulmonary hypertension is treated with transfusions, oxygen, hydroxyurea, and hypertension-specific investigational agents
-
♦
Preventative measures include hydration, prophylactic antibiotics, and immunizations
-
♦
Stem cell transplantation has been used on a limited basis
-
♦
A number of investigative therapies including gene therapy, nitric oxide (and its precursor l-arginine), therapies that increase the percentage of fetal hemoglobin, and therapies involvi ng membrane cation transport systems are being studied
-
♦
Regular surveillance is necessary to assess development and for the early detection a nd treat organ damage, stroke risks, and cardiovascular manifestations
Hemophilia A
Chromosome and Gene Location
-
♦
Xq28
Inheritance
-
♦
X-linked recessive
Incidence
-
♦
1 in 10,000 live male births, among all ethnic populations
-
♦
About 30% of cas es result from new mutations occurring in families in whom there is no apparent family history of hemophilia A (HA)
Clinical Manifestations
-
♦
HA predominantly affects males
-
♦
Bleeding is main manifestation and occurs spontaneously in severe hemophilia, but only after major trauma or surgery in mild to moderate hemophilia
-
♦
Some female carr iers have reduced FVIII levels and may have clinically significant bleeding, for example, lyonization of the normal X chromosome, Turner syndrome (XO karyotype)
Molecular Basis of Disease
-
♦
Reported deleterious mutations and polymorphisms are cataloged in an international database available on the Internet and updated periodically: (http://www.europium.csc.mrc.ac. uk/usr/WWW/WebPages/main.dir/main.htm)
-
♦
The most common deleterious mutation is an inversion within intron 22 found in approximately 40% of severe HA patients
-
♦
An additional inversi on of exon 1 of the FVIII gene affecting up to 5% of patients with severe HA has also been described
-
♦
Deletions are common and account for about 5% of characterized mutations
-
♦
The remaining patients typically have single base pair changes (resulting in missense, frameshift splice junction mutations), insertions, or duplications spread throughout the factor VIII gene
-
♦
X-autosome translocation involving a breakpoint within the factor VIII gene and uniparental isodi somy have been implicated
Laboratory
-
♦
The diagnosis of HA in males is established by assaying plasma FVIII coagulant activity (FVIII:C) for reduced or absent activity
-
♦
Classificat ion (based on FVIII:C)
-
Severe FVIII:C <1%
-
Moderate FVIII:C 1–5%
-
Mild FVIII:C >5–40%
-
Estimated prevalence of severe, moderate, and mild are 43, 26, and 31%, respectively
-
-
♦
For carrier testing of at risk females, initially performing FVIII:C is reasonable; if it is reduced, carrier status is established; however, in the large majority of carriers, FVIII:C is normal; thus, carrier status is established by genetic testing
-
♦
Other laborato ry findings include normal prothrombin time (PT), variably prolonged activated partial thromboplastin time (APTT), and normal von Willebrand factor (VWF) levels
Treatment
-
♦
Plasma-derived or recombinant FVIII concentrate is infused intravenously for prevention or treatment of bleeding episodes
-
♦
Current standard practice for severe hemophilia is to provide prophylactic clotting factor concen trate two to three times a week from childhood, so these patients currently lead relatively normal lives
-
♦
Patients with large deletions, nonsense mutations, and inversion mutations are susceptible to formation of FVIII inhibitor (antibodies) in response to therapy with FVIII c oncentrates
Hemophilia B
Chromosome and Gene Location
-
♦
Long arm of X chromosome (Xq27.1)
Inheritance
-
♦
X-linked recessive
Incidence
-
♦
Approximately 1 i n 30,000 live male births across all ethnic groups
-
♦
Up to 30% of hemophilia B (HB) cases occur in families with no prior family history of HB
Clinical Manifestations
-
♦
HB is a bleeding disorder due to a deficiency in factor IX (FIX) and is clinically indistinguishable from HA
-
♦
HB predominantly affects males
-
♦
Bleeding is main manifestation and occurs spontaneously in severe hemophilia, but only after major trauma or surgery in mild to moderate hemophilia
-
♦
Some female carriers have reduced FIX levels and may have clinically significant bleeding, e.g., lyonization of the normal X chromosome, Turner syndrome (XO karyotype)
Molecular Basis of Disease
-
♦
Reported deleterious mutations and polymorphisms are cataloged in an international database availa ble on the Internet and updated periodically (http://www.kcl.ac.uk/ip/peter-green/haemBdatabase.html)
-
♦
The majority of the mutations are missense, frameshift, or nonsense mutations. Short deletions (<30 nucleotides) account for ~7%, larger deletions ~3%, and insertions ~2% of mutations
-
♦
Mutations have been detected in all regions including the poly(A) signal
-
♦
An unusual FIX variant, due to mutation at Ala-10, is characterized by normal baseline FIX activity. However, warfarin therapy results in a severe and disproportionate reduction in FIX activity (typically <1%) and causes bleeding in patients being treated with warfar in who have an apparently therapeutic International Normalized Ratio (INR). An indication of such a situation is a disproportionate prolongation of the APTT, which should prompt clotting factor assays
Laboratory
-
♦
The diagnosis of HB in males is established by assaying plasma FIX coagulant activity (FIX:C) for reduced or absent activity
-
♦
Classification (based on FIX:C)
-
Severe FIX:C <1%
-
Moderate FIX:C 1–5%
-
Mild FIX:C >5–40%
-
-
♦
For carrier testing of at risk females, initially performing FIX:C is reasonable, if it is reduced, carrier status is established; however, in the large majority of carriers FIX:C is norm al; thus, carrier status is established by genetic testing
-
♦
Other laboratory findings include normal PT, variably prolonged APTT, and normal VWF levels
Treatment
-
♦
Plasmas-derived or recombinant FIX concentrate is infused intravenously for prevention or treatment of bleeding episodes
-
♦
Current standard practice for severe hemophilia is to provide prophylactic clotting factor concentrate two to three times a week from childhood, so these patients currently need relatively normal lives
-
♦
Patients w ith large deletions are susceptible to formation of FIX inhibitors (antibodies) in response to therapy with FIX concentrates and may experience anaphylactic reactions in response to factor IX concentrate infusions
Von Willebrand Disease
Chromosome and Gene Location
-
♦
12p
-
♦
A pseudogene is located on chromosome 22q11.2
Inheritance
-
♦
Autosomal domina nt and autosomal recessive
Incidence
-
♦
Von Willebrand disease (VWD) is the most commonly recognized congenital bleeding disorder, with a prevalence varying from 0.82 to 2%
Clinical Manifestations
-
♦
Diagnosis is established based on a personal and family history of abnormal clinical bleeding and reduced plasma VWF antigen levels and/or functional activity (ristocetin cofactor activity) and a nalysis of distribution of the VWF multimers
-
♦
VWD is classified into quantitative (types 1 and 3) or qualitative (type 2) abnormalities of plasma VWF
-
♦
Quantitative abnormalities include a mild reduction of qualitatively normal VWF (type 1) or absent VWF (type 3)
-
♦
Qualitative abnormalities (type 2 VWD) types 2A, B, M, and N VWD
-
♦
Mild reductions in VWF do not typically result in spontaneous bleeding; however, patient will bleed after trauma or surgery
-
♦
Type 3 VWD patients ha ve a severe reduction in a VWF levels, thus may bleed spontaneously
Molecular Basis of Disease
-
♦
Reported deleterious mutations and polymorphisms are cataloged in an international database available on the Internet and updated periodically: http://www.sheffield.ac.uk/VWF/
-
♦
Type 1 VWD
-
Autosomal dominant with variable penetrance
-
In the large majority of patients, mutations are not found, implying locus heterogeneity as an explanation for the mild quantitative deficiency of VWF associated with type 1 VWD, e.g., defects in glycosylation o f the VWF protein
-
However, selected patients with type 1 VWD had missense mutations spread throughout VWF gene
-
-
♦
Type 3 VWD
-
Autosomal recessive
-
Deleterious mutations currently characterized include frameshifts, deletions, or nonsense mutations
-
Although most patients are typically compound heterozygous for such VWF mutations, homozygosity has been demons trated in a few consanguineous families
-
-
♦
Type 2A VWD
-
Autosomal dominant, accounts for ~75% of all type 2 VWD
-
Missense mutations res ulting in type 2A VWD occur predominantly in the A2 domain
-
-
♦
Type 2B VWD
-
Autosomal dominant, accounts ~20% of all type 2 VWD
-
This variant is distinguished from type 2A VWD by the presence of mild to moderate thrombocytopenia in type 2B
-
Causative missense mutations are found in the A domain and result in a dominant gain-of-function p henotype
-
-
♦
Type 2M VWD
-
Mutations in A1 domain can result in decreased binding affinity for platelet GPIb
-
-
♦
Type 2N (Normandy) VWD
-
Mutati ons in the FVIII binding domain of VWF result in suboptimal binding of FVIII to VWF. This binding defect results in a shorter half-life of plasma FVIII, and thus plasma FVIII activity is reduced
-
This subtype mimics mild HA; however, it is distinguished by its autosomal recessive pattern of inheritance rather than the X-linked recessive pattern of HA
-
Three VWF gene mutations Thr791Met, Arg816Trp, and Arg854Gln accounted for 96% of type 2N patients
-
Type 2N VWD should be considered in patients with a diagnosis of “mild HA” with a non-X-linked inheritance
-
Typically, heterozygotes have normal FVIII levels, and homozygotes have reduced FVIII activity. However, apparent heterozygotes with low FVIII levels typically have inherited a second allele result ing in VWD type 1 (compound heterozygotes)
-
Laboratory
-
♦
See Table 13.7
Table 13.7. Typical laboratory Values in von Willebrand Disease -
Currently, the most significant im pact on clinical management and genetic counseling is the differentiation of VWD type 2N and mild HA. Both have a mild to moderate reduction in FVIII and normal levels of VWF antigen and ristocetin cofactor activity. The autosomal inheritance pattern and the need for the use of VWF concentrates rather than pure FVIII concentrates make this an important distinction
-
Differentiation of VWD types 2 A and B provides useful information that would alter clinical management. Although patients with type 2B are characterized by the presence of variable degrees of thrombocytopenia, such a clear distinction is not always possible. The importance of such a distinction affects management given that the therapeutic use of vasopressin (DDAVP) is contraindicated in patients with type 2B given the potential for worsening the thromb ocytopenia
-
Treatment
-
♦
Intravenous infusion of plasma-derived VWF concentrate is used for prevention and treatment of bleeding
-
♦
Occasionally, patients with type 1 VWD are treated with intravenous infusion of desmopressin (DDAVP) if th ey have had a documented response at a DDAVP trial
Factor V Leiden
Chromosome and Gene Location
-
♦
1q21–25
Inheritance
-
♦
Autosomal dominant with variable penetrance
Incidence
-
♦
Factor V Leide n is considered to be a founder mutation
-
♦
Prevalence of the mutation varies with ethnic population and ranges from 2% in southern Europe to 15% in southern Sweden
-
♦
In the USA, approximately 3–7% of asymptomatic white populations of Northern European or Scandinavian ancestry are heterozygous carriers
-
♦
Prevalence varies from 1.2% in African Americans, 2.2% in Hispanics, 1.2% in Native Americans, and 0.45% in Asian Americans
Clinical Manifestations
-
♦
Large majority of patients are asymptomatic
-
♦
Patients may develo p venous thromboembolism or pregnancy-related complications
-
♦
The large majority of asymptomatic carriers may never develop any clinical manifestations attributable to the presence of factor V Leiden mutation
-
♦
Selective patients m ay develop clinical manifestations of venous thromboembolism, especially when exposed to other acquired risk factors for venous thrombosis
-
♦
The incidence of recurrent venous thromboembolism attributable to heterozygous factor V Leiden mutation is debated; however, homozygotes are higher risk for recurrent venous thromboembolism
Molecular Basis of Disease
-
♦
The factor V Leiden mutat ion encodes for a single nucleotide substitution 1,691G>A within exon 10 of the factor V gene. This results in a missense mutation of a glutamine (Q) for arginine (R) at amino acid position 506 (506R>Q)
Laboratory
-
♦
Laboratory testing is used to evaluate for inherited risk factors for patients with venous thromboembolism (deep vein thrombosis and pulmonary embolism), recurrent fetal loss, and complications of pregnancy
-
♦
Initial screeni ng test consists of a clot-based functional assay to detect activated protein C resistance (APC-R)
-
♦
If patient plasma is found to be resistant to activated protein C, reflexive molecular testing for the factor V Leiden mutation could be performed to determine genotype
-
♦
Alternatively, direct DNA-based testing could be considered
Treatment
-
♦
Carriers of the factor V Leiden mutation who have not had any venous thromboembolism are counseled regarding risks of venous thrombosis, and empiric anticoagulation is not indicated
-
♦
Heterozygous carriers of the factor V Leiden who have had venous thrombosis typically are treated with anticoagulants for 3–6 months
-
♦
Homozygous carriers of the factor V Leiden mutation who have had venous thrombosis are typically treated with long-term anticoagulation
-
♦
Patients who experience pregnancy-related complications attributable to the presence of the factor V Leiden typically receive heparin anticoagulation during subsequ ent pregnancies
Prothrombin Thrombophilia
Chromosome and Gene Location
-
♦
Chromosome 11p
Inheritance
-
♦
Autosomal dominant with variable penetrance
Incidence
-
♦
Overall pre valence in Europe is approximately 2% (1.7% in northern Europe to 3.0% in southern Europe)
-
♦
Prevalence among US Caucasians is 1–2%
-
♦
The prothrombin 20210G>A mutation is uncommon among African Americans, Asian Americans, and Native Americans
Clinical Manifestations
-
♦
Large major ity of patients are asymptomatic
-
♦
Patients may develop venous thromboembolism or pregnancy-related complications
-
♦
Young women on estrogen-based oral contraceptives who carried the prothrombin G20210A mutation are at higher risk for cerebral venous sinus thrombosis
-
♦
The large majority of asymptomatic carriers may never develop any clinical manifestations attributable to the presence of prothrombin 20210G>A
-
♦
Selective patien ts may develop clinical manifestations of venous thromboembolism, especially when exposed to other acquired risk factors for venous thrombosis
-
♦
The incidence of recurrent venous thromboembolism attributable to heterozygous prothrombin 20210G>A mutation is debated; however, homozygotes are at higher risk fo r recurrent venous thromboembolism
Molecular Basis of Disease
-
♦
Prothrombin 20210G>A mutation is a common mutation in the gene encoding prothrombin (FII gene)
-
♦
The prothrombin 20210G>A mutation is located at 3′-untranslated region
-
♦
Presence of this mutation increases recognition of the polyadenylation cleavage signal, 3′ end RNA proces sing, and mRNA accumulation, thus increasing protein synthesis
Laboratory
-
♦
Direct DNA-based testing for the specific mutation is required for diagnosis
-
♦
Testing allows evaluation of inherited risk factors for patients with venous thromboembolism (deep vein thrombosis and pulmonary embolism), recurrent fetal loss, and complications of pregnancy
Treatment
-
♦
Carriers of the PT20210A mutation who have not had any venous thromboembolism are counseled regarding risks of venous thrombosis, and empiric anticoagulation is not in dicated
-
♦
Heterozygous carriers of the PT20210A who have had venous thrombosis typically are treated with anticoagulants for 3–6 months
-
♦
Homozygous carriers of the PT20210A mutation who have had venous thrombosis are typically treated with long-term anticoagulation
-
♦
Patients who experience pregnancy-related complications attributable to the presence of the PT20210A typically receive heparin anticoagulation during subsequent p regnancies
Other Genetic Disorders
Cystic Fibrosis
Chromosome and Gene Location
-
♦
7q31.2 (CFTR)
Inheritance
-
♦
Autosomal recessive
Incidence
-
♦
1/2,500 in the Caucasian population, varies in other ethnic groups
Clinical Manifestations
-
♦
Chronic pulmonary disease (inflammation and infection)
-
♦
Pancreatic insufficiency, failure to thrive
-
♦
Hepatobiliary disease
-
♦
Male infertility due to congenital absence of the vas deferens (CAVD)
-
♦
Meconium ileus in neonate (15–20%)
-
♦
Median survival age for classic CF is about 35 years
-
♦
Other CFTR-related disorders include isolated CAVD, CFTR-related pancreatitis, bronchiectasis, allergic bronchopulmonary aspergillosis, and chronic rhino-sinusitis
Molecular Basis of Disease
-
♦
Point mutati ons and large deletions in the cystic fibrosis transmembrane conductance regulator (CFTR) gene
-
♦
Over 1,000 mutations described are associated with a wide range of clinical severity and presentation of classic CF and CFTR-related disorders
-
♦
DeltaF508 most common mutation in Caucasian (75% of carriers)
-
♦
W1282X most common mutation in Ashkenazi Jews (60% of carriers)
-
♦
Polymorphic poly-T tract in intron 8 of the CFTR gene modifies the phenotype associated with certain CFTR genotypes; the number of repeats within the TG tract, just upstream of the poly-T tract, also modifies the splicing efficiency of intron 8
-
♦
5T allele is a ssociated with decreased efficiency in splicing of intron 8
-
♦
R117H in trans with 5T allele causes CAVD; therefore, evaluation of the poly-T tract shou ld be considered in males being evaluated for CAVD
Laboratory
-
♦
Molecular analysis (sequencing and dosage) identifies mutations in CFTR gene
-
♦
Elevated sweat chloride (>60 mEql/L)
-
♦
Abnormal tra nsepithelial nasal potential difference
-
♦
Semen analysis to evaluate for azoospermia
Treatment
-
♦
Antibiotics for control of respiratory infections
-
♦
Postural drainage and inhalation therapy
-
♦
Chest physioth erapy for airway clearance
-
♦
Pancreatic enzyme replacement therapy and supplemental feedings
-
♦
Oral ursodiol for biliary buildup/obstruction
-
♦
Assisted reproductive technologies for infertility
-
♦
Lung transplant for end-stage lung disease
-
♦
Surveillance
-
Blood glucose testing for diabetes
-
Chest radiograph and pulmonary function testing
-
Cultures of respiratory tract secretion s to evaluate for the presence of Pseudomonas aeruginosa
-
Bone density
-
Albinism (Oculocutaneous Albinism Type 1)
Chromosome and Gene Location
-
♦
11q14.3 (oculocutaneous albinism type 1 (OCA1))
-
♦
There are numerous other types of albinism
Inheritance
-
♦
Autosomal recessive
Incidence
-
♦
1/40,000
Clinical Manifestations
-
♦
Hypopig mented skin and hair
-
♦
Nystagmus
-
♦
Reduced iris and retinal pigment
-
♦
Foveal hypoplasia with significantly reduced visual acuity
-
♦
Two subtypes
-
OCA1A – no melanin synthesis in any tissue
-
OCA1B – variable amounts of melanin synthesis in some tissues
-
Molecular Basis of Disease
-
♦
Point mutations and large deletions in the tyrosinase gene (TYR) alter melanin production
-
♦
Mutations in other genes involved in melanocyte production, transportation, or proliferati on lead to various other types of albinism
Laboratory
-
♦
Molecular analysis (sequencing and dosage) identifies mutations in the TYR gene
-
♦
Deficient tyrosinase activity in hair bulb
Treatment
-
♦
Not curable, s upportive/symptomatic
-
♦
Avoidance of direct sunlight
-
♦
Ophthalm ologic care and correction
Neurofibromatosis Type 1 (von Recklinghausen)
Chromosome and Gene Location
-
♦
17q11.2
Inheritance
-
♦
Autosomal dominant
-
50% are new mut ations (de novo)
-
50% inherited from a parent
-
Incidence
-
♦
1/3,000
Clinical Manifestations
-
♦
Diagnosis based on established clinical criteria
-
♦
Neurofib romas (cutaneous and plexiform)
-
♦
Café au lait spots (Fig. 13.6A)
Fig. 13.6. 
Neurofibromatosis type 1 – Café au lait macule and axillary freckling (A). Lisch nodules (B).
-
♦
Axillary and inguinal freckling (Fig. 13.6A)
-
♦
Lisch nodules (Fig. 13.6B)
-
♦
Optic gliomas
-
♦
Scoliosis
-
♦
Tibial dysplasia
-
♦
Learning disabilities (50%)
-
♦
Peripheral nerve sheath tumors
-
♦
Osseous lesions
-
♦
Hypertension
-
♦
Noonan syndrome phenotype (12%)
-
Ocular hypertelorism , down-slanting palpebral fissures, low-set ears, webbed neck, and pulmonary stenosis
-

Molecular Basis of Disease
-
♦
Point mutations and large deletions in the NFI gene; large chromosomal rearrangements are als o a rare cause of NF1
-
♦
Homozygous or compound heterozygous mutations in the four mismatch repair genes, MLHI, MSH2, MSH6, and PMS2 result in childhood malignancies and features of NF1
Laboratory
-
♦
Molecular analysis (sequencing and dosage), in addition to cytogenetic analysis (standard chromosome analysis, FISH, chromosomal microarray), identifies mutations involving the NFI gene in approximately 95% of individuals
Treatment
-
♦
Not curable, supportive/symptomatic
-
♦
Surgical intervention when indicated
-
♦
Monitor by ophthalmologi c examination and blood pressure measurements
Neurofibromatosis Type 2
Chromosome and Gene Location
-
♦
22q12.2
Inheritance
-
♦
Autosomal dominant
-
50% are new mutations (de novo)
-
50% inherited fr om a parent
-
Incidence
-
♦
1/35,000
Clinical Manifestations
-
♦
Diagnosis based on established clinical criteria
-
♦
Vestibular schwannomas (bilateral acoustic neuromas)
-
♦
Spinal tu mors (schwannoma, astrocytoma, ependymoma)
-
♦
Meningiomas (50%)
-
♦
Loss of hearing
-
♦
Balance disturbance
-
♦
Ocular manifestations include subcapsular lens opacities that may progress to cataract, reti nal hamartoma, and epiretinal membrane
-
♦
Childhood mononeuropathy and adult polyneuropathy
-
♦
Intellectual disability (associated with large deletions)
Molecular Basis of Disease
-
♦
Point mutations and large deletions in the NF2 gene; large chromosomal rearrangements are also a rare cause of NF2
-
♦
Submicrosc opic (10–600 kb) deletions (10–15%)
-
♦
Ring chromosome 22 (NF2 phenotype)
Laboratory
-
♦
Molecular analysis (sequencing and dosage), in addition to cytogenetic analysis involvi ng the NF2 gene in up to 93% of individuals with NF2
Treatment
-
♦
Not curable, supportive/symptomatic
-
♦
Surgical intervention when indicated
-
♦
Radiation therapy can induce or accelerate tumors
-
♦
Monitor b y MRI for presence of tumors
Tuberous Sclerosis Complex
Chromosome and Gene Location
-
♦
9q34.13 (TSC1)
-
♦
16p13.3 (TSC2)
Inheritance
-
♦
Autosomal dominant
-
Two-thirds are new mutations (de novo)
-
One-third inherited fr om a parent
-
Incidence
-
♦
1/6,000
Clinical Manifestations
-
♦
Diagnosis based on established clinical criteria
-
♦
Highly variable
-
♦
Skin abnormalities (~100%)
-
♦
Central nervous system abnormalities
-
Cortic al tubers
-
Subependymal nodules
-
Subependymal giant cell astrocytomas
-
Cerebral white matter radial migration lines
-
Seizures and epilepsy
-
Intellectual disability/learning difficulties (50%)
-
Behavioral and psychiatric disorders
-
-
♦
Renal abnormalities
-
Renal lesions (angiomyolipomas)
-
Renal cysts
-
Polycystic renal disease
-
Renal cell carcinoma
-
-
♦
Cardiac rhabdomyomas, retinal hamartomas, hamartomatous rectal polyps, and neuroendocrine tumors
-
♦
Bone cysts
-
♦
Lymphangiomyomatosis
-
♦
Dental enamel pits

Laboratory
-
♦
Molecular analysis (sequencing and dosage) for mutations in the TSC1 and TSC2 genes in ab out 85% of individuals with a definite diagnosis of TSC
Treatment
-
♦
Not curable, supportive/symptomatic
-
♦
Surgical removal of tumors
-
♦
Monitor for renal abnormalities by renal ultrasound
-
♦
Monitor f or brain abnormalities by CT/MRI
-
♦
Chest CT for adult females to monitor for lymphangio-myomatosis
Beckwith–Wiedemann Syndrome
Chromosome and Gene Location
-
♦
11p15.5 (includes multiple genes: IGF2 and H19 in domain 1 and LIT1/KCNQ1OT1, CDKN1C, and KCNQ1 in domain 2)
Inheritance
-
♦
85% sporadic
-
♦
10–15% autosomal do minant (inherited from a parent)
Incidence
-
♦
1/14,000
Clinical Manifestations
-
♦
Overgrowth syndrome: macroglossia, macrosomia, nephromegaly, splenomegaly, cardiomegaly, and hepatomegaly
-
♦
Hemihyperplasia and midface hypoplasia
-
♦
Ear creases and pos terior helical ear pits
-
♦
Cryptorchidism
-
♦
Omphalocele observed by fetal ultrasound
-
♦
Neonatal hypoglycemia
-
♦
Cytomegaly of the fetal adrenal cortex
-
♦
Increased risk for embryonic tumors (Wilms tumor, hepatoblastoma, rhabdomyosarcoma, neuroblast oma)
Molecular Basis of Disease
-
♦
Beckwith–Wiedemann Syndrome (BWS) is due to dysregulation of imprinted genes at 11p15
-
♦
Imprinting refers to the difference in gene expression based on parent of origin
-
♦
Imprinted genes are usually regulated by methylation, which prevents a gene from being expressed
-
♦
Imprinted genes associated with BWS are re gulated by imprinting center 1 (IC1) and imprinting center 2 (IC2)
-
IGF2 (paternally expressed)
-
KCNQ1OT1 (paternally expressed)
-
H19 (maternally expressed)
-
CDKN1C/p57(KIP2) (mat ernally expressed)
-
-
♦
Molecular etiology of sporadic cases of BWS
-
Hypomethylation of LIT1 (IC2) (50%)
-
Paternal UPD of chromosome 11 (20%)
-
Hypermethylation of H19 (IC1) (5%)
-
Point mutation in CDKN1C (5–10%)
-
Cytogenetic abnormality (1–2%)
-
Unknown (10%)
-
-
♦
Molecular etiology of inh erited cases of BWS
-
Point mutations in CDKN1C (40%)
-
Unknown
-
Laboratory
-
♦
Methyla tion analysis to detect methylation abnormalities of LIT1 (IC2) and H19 (IC1)
-
♦
UPD analysis of chromosome 11
-
♦
Molecular analysis of CDKN1C for point mutations
-
♦
Cytogenetic analysis identifies paternal duplications, translocations, or inversions of 11p15
Treatment
-
♦
Not curable, supportive/symptomatic
-
♦
Abdominal wall repair for abdominal wall defects in neonates
-
♦
Treatment o f hypoglycemia
-
♦
Monitor for embryonic tumors by ultrasound and serum alpha-fetoprotein measurement
Metabolic Disorders: Inborn Errors of Metabolism
Amino Acid Disorders
Phenylketonuria
Chromosome and Gene Location
-
♦
12q24
Inheritance
-
♦
Autosomal recessive
Incidence
-
♦
1/10,000–1/25,00 0 (lower in African Americans and Ashkenazi Jews)
Clinical Manifestations
-
♦
Intellectual disability
-
♦
Seizures
-
♦
Hyperactivity
-
♦
Eczema
-
♦
Hypopigmentation
-
♦
“Mousey” odor
-
♦
Maternal phenylket onuria (PKU) – children born to mothers with PKU have a high risk (90%) of having intellectual disability, microcephaly, impaired growth, and cardiac malformations, if mothers have not received dietary restriction of phenylalanine during pregnancy
Molecular Basis of Disease
-
♦
Mutations in phenylalanine hydro xylase (PAH) gene cause:
-
Inactivation of enzyme (more than 500 described)
-
Disruption of phenylalanine to tyrosine conversion with subsequent hyperphenylalaninemia
-
Laboratory
-
♦
New born screening detects blood phenylalanine (>0.13–1.2 mM) and phenylalanine/tyrosine ratio (>2.5) using tandem mass spectrometry
-
♦
Molecular genetic analysis available for positive cases
Treatment
-
♦
Low phe nylalanine diet supplemented with tyrosine throughout life
-
♦
Sapropter in dihydrochloride (BH4) at 20 mg/kg administration for BH4-responsive forms of PKU
Tyrosinemia (Type 1 Most Prevalent)
Chromosome and Gene Location
-
♦
15q23–q25
Inheritance
-
♦
Autosomal recessive
Incidence
-
♦
Varies by p opulation, highest incidence along the St. Laurence waterway in Canada (1/1,846)
Clinical Manifestations
-
♦
Vomiting, acidosis, diarrhea, and failure to thrive
-
♦
Rickets
-
♦
Hepatic cirrhosis
-
♦
Fanconi renal tubular syndrome
-
♦
Urine odor of boiled cabbage (caused from methionine metabolites)
-
♦
Increased risk for hepatocellular carcinoma
Molecular Basis of Disease
-
♦
Mutations in the gene for tyrosinemia inactivate the enzyme fumarylacetoacetate hydrolase (FAH), which results in the accumulation of tyrosine and its metabolites (succinylacetoacetate, succinylacetone, and fumarylacetone) in the liver and kidney
-
♦
Numerous mutations have been found in the FAH gene and are generally family or population specific
Laboratory
-
♦
Increased urine tyrosine metabolites, serum alpha-fetoprotein concentration, and succinylacetone excretion. Reduced delta-ALA-dehydratase (PBG synthase) activity in erythrocytes due to inhibition by succinylacetone. Plasma or serum tyrosin e is not 100% sensitive or specific
-
♦
Newborn screening detects succinylacetone by tandem mass spectrometry (an analyte not widely included in all newborn screening laboratories)
-
♦
Decreased fumarylacetoacetate hydrolase activity in liver biopsy specimens or cultured fibroblasts or chorionic villi sample
-
♦
Molecular analysis for targeted mutations and entire coding region is commercially available
Treatment
-
♦
Low ph enylalanine, tyrosine, and methionine diet delays but does not stop progression of disease
-
♦
NTBC (2-(2-nitro-4-trifluoro-methylbenzoyl)-1,3-cyclohexa-nedione; nitisinone treatment reduces the accumulation of toxic metabolites by blocking the second step in the tyrosine degradation pathway and ameliorates the severe clinical manifestation s
Maple Syrup Urine Disease
Chromosome and Gene Location
-
♦
19q13.1–q13.2, (type 1-E1α subunit)
-
♦
1p31 (type 2-E2 subunit)
-
♦
6p14 (type 3-E1β subunit)
-
♦
7q31–q32 (E3 subunit)
Inheritance
-
♦
Autosom al recessive
Incidence
-
♦
1/125,000–1/300,000
Clinical Manifestations
-
♦
Variable based on type, level of enzyme deficiency, and management of acute episodes
-
♦
Features include
-
Apnea
-
Hypoglycemia
-
Cerebral edema
-
Poor feeding, vomitin g, and lethargy
-
Maple syrup odor of urine and cerumen
-
Hypertonicity/hypotonicity
-
Muscle rigidity
-
Seizures
-
Molecular Basis of Disease
-
♦
The branched ch ain alpha keto acid dehydrogenase is a mitochondrial enzyme consisting of four subunits: E1α, E1β, E2, and E3 (each subunit is located on a different chromosome; see above)
-
♦
The enzyme system is responsible for the decarboxylation of the branched chain amino acids: leucine, isoleucine, and valine
-
♦
Mutations in the genes encoding the various subunits result in defective expression and buildup of the branched chain amino acids and their metabolites in the blood, urine, and cerebral spi nal fluid
Laboratory
-
♦
Deficiency of branched chain alpha keto acid dehydrogenase activity in leukocytes and cultured skin fibroblasts (prenatal diagnosis also available)
-
♦
Elevation of leucine, isoleucine, valine, and alloisoleucine in plasma or serum. Elevation of branch-chain keto acids in urine, serum, or plasma
Treatment
-
♦
Thiamine for B1-responsive forms
-
♦
Not curable, supportive/symptomatic
-
♦
Manage acute episodes
-
♦
Reduce intake of branched chain amino acids by using special formulas and redu cing protein intake
Homocystinuria
Chromosome and Gene Location
-
♦
21q22.3
Inheritance
-
♦
Autosomal recessive
Incidence
-
♦
1/100,000
-
♦
Clinical Manifestations
-
♦
Dislocati on of ocular lens, myopia, strabismus, cataracts, and glaucoma
-
♦
Thinning and lengthening of the long bones, osteoporosis, and scoliosis
-
♦
High-arched palate, pes cavus, genu valgum, and biconcave vertebrae
-
♦
Thromboembolism
-
♦
Bluish mo ttling of the skin on legs and hands
-
♦
Developmental delay and seizures
Molecular Basis of Disease
-
♦
The enzyme, cystathionine-β-synthase (CBS), converts homocysteine to serine and cystathionine
-
♦
Mutations in the CBS gene result in inactivation of enzyme function, which leads to elevated m ethionine and homocysteine levels
Laboratory
-
♦
Increased levels of homocysteine in urine or plasma
-
♦
Hyperhomocysteinemia with increased methionine in plasma or serum. Decreased enzyme activity i n fibroblasts, liver biopsy specimens, or PHA-stimulated lymphocytes (prenatal diagnosis available)
-
♦
Molecular analysis available for mutation detection
Treatment
-
♦
High doses of p yridoxine (vitamin B6), which is a cofactor of CBS, are given to decrease homocysteine levels (increases conversion of homocysteine to cysteine)
-
♦
Folic acid permits response to pyridoxine and optimizes conversion of homocysteine to methionine
-
♦
Betaine is used in pyridoxine-unresponsive patients to lower plasma homocysteine levels by allowing conversion to methionine
-
♦
Low-methionine diets reduce accumulation of methionine and homocysteine and their metabolites
-
♦
Patients treated from newborn period have fewer complications than those treated late or untr eated
Urea Cycle Disorders
-
♦
The urea cycle functions to prevent the accumulation of toxic nitrogenous compounds and also contain several reactions required for the synthesis of arginine. The urea cycle consists of five biochemical reactions. Defects in the biosynthesis or function of any of the five enzymes in this pathway lead to di sease
Chromosome and Gene Location
-
♦
See Table 13.8
Table 13.8. Urea Cycle Disorders
Inheritance
-
♦
See Table 13.8
Incidence
-
♦
See Table 13.8
Clinical Manifestations
-
♦
All defects with the exception of argin ase deficiency result in a similar clinical phenotype which includes:
-
Lethargy, persistent vomiting, and poor feeding coma
-
Hypotonia
-
Seizures
-
Hepatomegaly
-
-
♦
The onset usua lly occurs after feeding in newborn period or after infections or protein overload
-
♦
In arginase deficiency, children are often asymptomatic for the first few months to years of life
-
♦
Clinical features of arginase deficiency include:
-
Loss of developmental milestones
-
Increased clumsiness
-
Spastic quadriplegia with loss o f ambulation
-
Loss of speech
-
Seizures
-
Progressive inte llectual disability
-
Molecular Basis of Disease
-
♦
See Table 13.8 for enzyme involved. Enzyme defects lead to the accumulation of ammonia and alter levels of other amino acids
-
♦
Ammonia i s a neurotoxin that adversely affects the CNS
Laboratory
-
♦
Hyperammonemia, altered amino acid levels in plasma, which are specific to each reaction
-
♦
Deficient enzyme activity, molecular testing available for carrier and prenatal testing
Treatment
-
♦
Control of and avoidance of acute attacks, including supportive care during symptomatic episodes
-
♦
Restrict protein, high-caloric diet to minimize breakdown of protein
-
♦
Prognosis is better if disorder is treated promptly
Organic Acidemias
Isovaleric Acidemia
Chromosome and Gene Location
-
♦
15q14–q15
Inheritance
-
♦
Autosomal recessive
Incidence
-
♦
Rare
Clinical Manifestations
-
♦
Usually presents within the f irst few days of life, symptoms include:
-
Acute attacks of vomiting and lethargy
-
Acidosis
-
Ataxia
-
Coma
-
Seizures
-
Intellectual disability
-
-
♦
Attacks are u sually triggered by stresses such as infections or surgery
Molecular Basis of Disease
-
♦
Isovaleryl CoA dehydrogenase catalyzes the conversion of isovaleric acid to 3-methylcrotonic acid in the branch-chain amino acid, leucine, and degradation pathway
-
♦
Deficient isovaleryl CoA dehydrogenase results in the accumulation of isovaleric acid and its metabolites, which are toxic to the body
-
♦
A recurrin g mutation, A282V, is frequently found in asymptomatic and relatively mild IVA cases detected by newborn screening with tandem mass spectrometry
Laboratory
-
♦
Elevated isovaleric acid
-
♦
Hyperammonemia
-
♦
Hypocalcemia pancytopenia
-
♦
Neutropenia
-
♦
Thrombocytopenia
-
♦
Anemia
-
♦
Elevated urine isovalerylglycine during an acute attack
-
♦
Deficien cy of isovaleryl CoA dehydrogenase in leukocytes or cultured skin fibroblasts (prenatal diagnosis available)
Treatment
-
♦
Not curable, supportive/symptomatic
-
♦
Avoidance of, and symptomatic control of, acute episodes
-
♦
Correct dehydration, electrolyte disturbances, and metabolic acidosis
-
♦
Reduce protein intake
-
♦
Remove e xcess isovalerylic acid by use of glycine and carnitine, which allows for urina ry excretion
Propionic Acidemia
Chromosome and Gene Location
-
♦
13q32
-
♦
3q21–q22
Inheritance
-
♦
Autosomal recessive
Incidence
-
♦
Rare
Clinical Manifestations
-
♦
Onset may o ccur within the first few weeks of life
-
♦
Symptoms include:
-
Apnea
-
Hypoglycemia
-
Poor feeding, vomiting, and lethargy
-
Coma
-
Hypotonia
-
Seizures
-
Frequent infections
-
Osteoporosis
-
Intellectual di sability
-
-
♦
Symptoms may be triggered by infections, constipation, and protein overload
Molecular Basis of Disease
-
♦
Propionyl-CoA-carboxylase (PCC) is a multimeric, mitochondrial enzyme consisting of alpha subunits, encoded by PCCA, and beta subunits, encoded by PCCB
-
♦
The gene for each subunit is located on different chromosomes: chromosome 13 (alpha) and chromosome 3 (beta)
-
♦
Propionic acidemia can arise from mutations in either the alpha or beta subunit
-
♦
PCC is involved in the catabolic pathway for the odd-chain length fatty acids: threonine, methionine, isoleucine, and valine
-
♦
Deficiencies of PCC lead to the accumulation of propionic acid, which results in the inhibition of citric acid cycle enzymes, acetylglutamate synthetase, granulocytes, and T- and B-cell development
-
♦
Numerous mutatio ns have been described in the beta and alpha subunits
Laboratory
-
♦
Metabolic acidosis
-
♦
Hypoglycemia
-
♦
Hyperammonemia
-
♦
Carnitine deficiency
-
♦
Elevated glycine
-
♦
Elevated propionic and methylcitric acids
-
♦
Neutropenia
-
♦
Thrombocytopenia
-
♦
Deficiency of PCC activity in leukocytes or cultured skin fibroblasts (prenatal diagnosis ava ilable)
Treatment
-
♦
Not curative. Avoidance of, and symptomatic control of, acute episodes
-
♦
Correct dehydration, electrolytes disturbances, and metabolic acidosis (bicarbonate)
-
♦
Decrease protein intake
-
♦
Antibiotics prevent production of propionic acid by intestinal bacteria
-
♦
Some individu als respond to biotin treatment, as it is a cofactor for PCC
Methylmalonic Acidemia (Methylmalonyl-CoA Mutase and Other Forms)
Chromosome and Gene Location
-
♦
6p21
Inheritance
-
♦
Autosomal recessive
Incidence
-
♦
1/50,000
Clinical Manifestations
-
♦
Lethargy, failure to thrive, recurrent vomiting, and dehydration
-
♦
Respiratory distress
-
♦
Muscular hypotonia
-
♦
Growth retardation
-
♦
Psychomotor retardation
-
♦
Impairment of renal function
-
♦
Neurological abnormalities
Molecular Basis of Disease
-
♦
Methylmalonic acid is derived from propionic acid as part of the catabolic pathway of isoleucine, valine, threonine, methionine, cholesterol, and odd-chain fatty acids
-
♦
Methylmalonic acid is converted to succinic acid by methylmalonyl-CoA mutase and cobalamine (vitamin B12), which is a cofactor
-
♦
Defects in the mutase or reduced levels of its cofactor result in the accumulation of methylmalonic acid and its precursors
-
♦
The number of different genes involved in cobalamine metabolism and methylmalonic acidemia is uncertain; however, six complementation groups have been identified indicating multiple alleles
-
♦
Numerous mutati ons within the mutase gene on chromosome 6 have been reported
Laboratory
-
♦
Ketosis, acidosis, anemia, elevated urinary, and serum methylmalonic acid
-
♦
Decreased or absent mutase activity in cultured fibroblasts
-
♦
Prenatal diagnosis available
Treatment
-
♦
Restricted protein
-
♦
Vitamin B12 supplementation to lower MMA levels
Mitochondrial Fatty Acid Oxidation Disorders
-
♦
Mitochondrial fatty acid oxidation fuels ketogenesis in the liver and energy production during an unfed state or during activities requiring a high energetic demand. Thus, many disorders of mitochondrial fatty acid oxidation present with sudden death that is often triggered by an acute illness or other episode. In addition, several of these disorders affect liver function and may also be complicated by cardiac dysfunction
-
♦
Mitochondrial fatty acid oxidation consists of multiple biochemical reactions. Defects in the function of several of the enzymes in this pathway lead to disease
Medium-Chain Acetyl-CoA Dehydrogenase Deficiency
Chromosome and Gene Location
-
♦
1p31
Inheritance
-
♦
Autoso mal recessive
Incidence
-
♦
1/15,000
Clinical Manifestations
-
♦
Onset may occur within the first few days of life if triggered by an illness or fasting
-
♦
Symptoms include:
-
Sudden, unexp lained death
-
Hypoketotic hypoglycemia
-
Vomiting and lethargy
-
Seizures
-
Coma
-
Reye-like syndrome
-
Hepatomegaly and acute liver disease
-
Molecular Basis of Disease
-
♦
Mediu m-chain acetyl-CoA dehydrogenase (MCAD) is an enzyme encoded by ACADM on chromosome 1, and it is required for the beta oxidation of medium-chain fatty acids in the mitochondria
-
♦
A defect in MCAD results in the reduction of acetyl-CoA levels and in the accumulation of medium-chain fatty acids, which are further conjugated to glycine and carnitine–esters or metabolized to dicarboxylic acids
-
♦
The accumulation of medium-chain fats and their conjugates can be detected in the blood, urine, and tissue of patients with MCAD deficiency. The medium-chain, carnitine–ester conjugates are used as biomarkers for newborn screening of this disorder by tandem mass spectrometry
-
♦
A frequent cause of MCAD deficiency is homozygosity for the 985A>G mutation in exon 11 of ACADM, a mutation that has an estimated carrier frequency of 1/40 in pe oples of Northern European descent
-
♦
Numerous mutations within the ACADM gene have been identified, and several may lead to a mild biochemical phenotype
Laboratory
-
♦
Hypoketotic hypoglycemia
-
♦
Hyperammonemia
-
♦
Carnitine deficiency
-
♦
Elevated medium-chain acylcarnitines with prominent octanoylcarnitine
-
♦
Elevated medium-chain acylglycines with prominent hexanoylglycine and suberylglycine
-
♦
Deficiency of MCAD activity in cultured skin fibroblasts
-
♦
ACADM mutations, such as 985A>G
Treatment
-
♦
Avoidance of fasting for more than 12 h
-
♦
Control of acute illnesses by intravenous glucose
-
♦
Uncooked cornstarch for toddlers at bedtime
-
♦
l-carnitine su pplementation (100 mg/kg/day)
-
♦
Low-fat diet
Very Long-Chain Acetyl-CoA Dehydrogenase Deficiency
Chromosome and Gene Location
-
♦
17p13
Inheritance
-
♦
Autosomal recessive
Incidence
-
♦
1/40,000
Clinical Manifestations
-
♦
Onset often occ urs during infancy where it can present with cardiomyopathy and hepatic dysfunction. It also occurs in childhood where it presents with hepatic symptoms and hypoglycemia. A few adult patients have been described with muscle symptoms and without hepatic or cardiac involvement
-
♦
Symptoms include:
-
Sudden, unexp lained death
-
Hypoketotic hypoglycemia
-
Vomiting, lethargy
-
Seizures
-
Rhabdomyolysis
-
Myoglobinuria
-
Dilated or hypertrophic cardiomyopathy
-
Hepatomegaly and acute liver disease
-
Molecular Basis of Disease
-
♦
Very long-chain acetyl-CoA dehydrogenase (VLCAD) is an enzyme encoded by ACADVL on chromosome 17 that is essential for the beta oxidation of long-chain fatty acids in the mitochondria
-
♦
VLCAD leads to the accumulation of long-chain fatty acids, particularly the unsaturated C12–C14 chain-length fats, which are further conjugated to carnitine–esters or metabolized to dicarboxylic acids
-
♦
Accumulation of long-chain fats (unsaturated C12–C14) are found in VLCAD patient tissues and th eir carnitine–ester conjugates are used as biomarkers for newborn screening of this disorder by tandem mass spectrometry
-
♦
Numerous private mutations within the VLCAD gene account for this disorder
Laboratory
-
♦
Hypoketotic hypoglycemia
-
♦
Hyperammonemia
-
♦
Carnitine deficiency
-
♦
Elevated very long-chain pla sma acylcarnitines with prominent unsaturated C14 species
-
♦
Elevated hepatic enzymes and creatine kinase (CK)
-
♦
Myoglobinuria
-
♦
Deficiency of VLCAD activity in cultured skin fibroblasts
-
♦
ACADVL mutation analysis is available for confirmation and prenatal diagnosis
Treatment
-
♦
Avoidance of fasting or prolonged exercise
-
♦
Control of acute illnesses by intravenous fluids
-
♦
High-carbohydrate and low-fat diet
-
♦
Supplementat ion with MCT-oil
-
♦
l-carnitine supplementation is controversial
Long-Chain 3-Hydroxylacetyl-CoA Dehydrogenase/Mitochondrial Trifunctional Protein Deficiency
Chromosome and Gene Location
-
♦
2p23
Inheritance
-
♦
Autosomal recessive
Incidence
-
♦
Rare
Clinical Manifestations
-
♦
This presents as neonatal hypoglycemia, lactic acidosis, and sudden, unexplained death. It also occurs in childhood where it presents with additional symptoms, such as progressive neuropathy, myopathy, and ophthalmologic abnormalities. It is also associated with maternal, acute fatty liver of pregnancy or HELLP (hemolysis, elevated liver enzymes, and low platelets) syndrome in mothers of affected children
-
♦
Symptoms include:
-
Sudden, unexplained death
-
Hypoketotic hypoglycemia
-
Coma
-
Lactic academia
-
Hypotonia
-
Seizures
-
Progressive retin opathy
-
Peripheral neuropathy
-
Cardiomyopathy
-
Hepatic dysfunction
-
Molecular Basis of Disease
-
♦
Long-chain 3-hydroxylacetyl-CoA dehydrogenase/mitochondrial trifunc tional protein deficiency (LCHAD/TFP) is caused by the deficiency of proteins encoded by HADHA and HADHB. Both genes are located in tandem on chromosome 2p23, and their protein products form a heteromer that is essential for the beta oxidation of long-chain fatty acids in the mitochondria. The heteromer functions as a long-chain 3-hydroxylacetyl-CoA dehydrogenase (LCHAD, encoded by HADHA), a long-chain enoyl-CoA hydratase, and a long-chain thiolase, hence the name, mitochondrial trifunctional protein (TFP)
-
♦
LCHAD/TFP leads to the accumulation of 3-hydroxyl long-chain fatty acids, particularly the hydroxylated C14–C18 chain-length fats, which are further conjugated to carnitine–esters or metabolized to dicarboxylic acids
-
♦
Accumulation of long-chain (hydroxylated C14–C18), fatty acid; carnitine–ester conjugates are used as biomarkers for newborn screening of this disorder by tandem mass spectrometry
-
♦
Private mu tations are frequent within HADHA and HADHB genes
Laboratory
-
♦
Hypoketotic hypoglycemia
-
♦
Hyperammonemia and lactic acidemia
-
♦
Carnitine deficiency
-
♦
Elevated, long-chain, hydroxylated plasma acylcarnitines with prominent C14–C18 species
-
♦
Elevated hepatic enzymes and CK
-
♦
Deficiency of LCHAD/TFP activity in cultured skin fibroblasts
-
♦
HADH A and HADHB mutation analysis is available for confirmation and prenatal diagnosis
Treatment
-
♦
Avoidance of fasting or prolonged exercise
-
♦
Control of acute illnesses by intravenous fluids
-
♦
High-car bohydrate and low-fat diet
-
♦
Supplementation of diet with MCT-oil
-
♦
DHA supplementation is also commonplace with the aim to stabilize the retinopathy
-
♦
l-carnitin e supplementation is controversial
Short-Chain Acetyl-CoA Dehydrogenase Deficiency
Chromosome and Gene Location
-
♦
12q22–qter
Inheritance
-
♦
Autosomal recessive
Incidence
-
♦
1/50,000
Clinical Manifestations
-
♦
Clinical heter ogeneity of this disorder has complicated a complete understanding of its natural history. Several case reports have associated short-chain acetyl-CoA dehydrogenase (SCAD) deficiency with neonatal hypotonia and developmental delay, with or without seizures. However, with the advent of newborn screening by tandem mass spectrometry, of which SCAD deficiency is a secondary target, numerous SCAD-deficient patients have been found to be asymptomatic exclusive of persistent ethylmalonic aciduria. The presence of two common SCAD gene variants may account for the biochemical and clinical variability of this disorder
-
♦
Reported symptoms include:
-
Hypotonia
-
Seizures
-
Developmental and speec h delay
-
Lethargy
-
Failure to thrive
-
Ethylmalonic aciduria without other symptoms
-
Molecular Basis of Disease
-
♦
Short-chain acetyl-CoA dehydrogenase (SCAD) is an enzyme encoded by ACADS on chromosome 12 and is required for the last chain-shortening step of mitochondrial fatty acid oxidation
-
♦
Reduction of SCAD function leads to the accumulation of short-chain fatty acids (of the C4 chain-length), which are further conjugated to glycine and carnitine–esters, or metabolized to dicarboxylic acids, ethylmalonic acid, and methylsuccinic acid
-
♦
Accumulation of short-chain fats and their conjugates can be detected in the blood and urine of patients with SCAD deficiency, and the carnitine–ester conjugate (C4 acyl carnitine) is used as a biomarker for newborn screening of this disorder by tandem mass spectrometry
-
♦
Two common SCAD gene variants, 625G>A and 511C>T, are frequently found in the normal population, lead to mild ethylmalonic aciduria, but do not confer disease
-
♦
Numerous SCAD-deficient patients who are compound heterozygous for an SCAD gene variant and a single mutation have been identified by newborn screening. It doe s not appear that compound heterozygosity for a mutation and a variant will lead to a clinical phenotype. However, additional studies are needed to understand the possible complex multifactorial components that lead to a phenotype
Laboratory
-
♦
Hypoglycemia
-
♦
Elevated short-chain plasma acylcarnitines with prominent butyrylcarnitine
-
♦
Elevated ethylmalo nic acid excretion detectable by urine organic acids or acylglycines
-
♦
Deficiency of SCAD activity in cultured skin fibroblasts
-
♦
SCAD gene sequence analysis is commercially available and will detect gene variants and most missense mutations
Treatment
-
♦
Avoidance of fasting
-
♦
Control of acu te illnesses by intravenous fluids
-
♦
l-carnitine supplementation (100 mg/kg/day)
-
♦
Low-fat diet
Carbohydrate Metabolism Disorders
Glycogen Storage Disease (Type I)
Chromosome and Gene Location
-
♦
17q21
Inheritance
-
♦
Autosomal recessive
Incidence
-
♦
1/100,000
Clinical Manifestations
-
♦
Hypogl ycemia
-
♦
Hypertension
-
♦
Excessive perspiration
-
♦
Bruising and nosebleeds
-
♦
Short stature
-
♦
Delayed puberty
-
♦
Protuberant abdomen
-
♦
Liver adenomas, hepatomegaly, hepatoblastoma, and hepatocellular carcinoma
-
♦
Chronic pancreatitis
-
♦
Renal insufficiency
Molecular Basis of Disease
-
♦
The enzyme glucose-6-phosphatase catalyzes the conversion of glucose-6-phosphate to glucose
-
♦
Mutations in the gene encoding glucose-6-phosphatase result in a deficiency of this enzyme and the subsequent inability to free glucose for use in the body
-
♦
Mutations in the glucose-6-phosphatase gene (G6PC) have been found in about 80% of patients studied
-
♦
There are various types of glycogen storage diseases, which result from deficiencies of other enzymes involved in the glycogen metabolism pathway
Laboratory
-
♦
Deficiency of glucose-6-phosphatase in liver biopsy (enzyme not present in skin fibroblasts)
-
♦
Lipidemia
-
♦
Hyperuricemia
-
♦
Lactic acidemia
-
♦
Ketonemia
-
♦
Molecular analysis available
Treatment
-
♦
Maintenance of normal blood glucose concentration, by supplementation of glucose, pancreatic enzymes, and cornstarch
-
♦
Limit intake of fructose and galactose
-
♦
Low-fat diet
-
♦
Liver tra nsplantation if necessary
Galactosemia
Chromosome and Gene Location
-
♦
9p13
Inheritance
-
♦
Autosomal recessive
Incidence
-
♦
1/30,000
Clinical Manifestations
-
♦
Babies with galactosemia may app ear normal at birth
-
Symptoms begin soon after milk feedings have begun
-
-
♦
Symptoms include:
-
Lethargy, irritability, vomitin g, feeding difficulties, poor weight gain, and failure to thrive
-
Seizures
-
Jaundice
-
Hepatomegaly, hepa tic cirrhosis, and splenomegaly
-
Hypoglycemia
-
Ascites
-
Lens opacities
-
Increased suscep tibility to infections
-
-
♦
Long-term complications include:
-
Speech deficits
-
Ataxia
-
Dysmetria
-
Diminished bone density
-
Premature ovarian failure
-
Molecular Basis of Disease
-
♦
Galactose-1-P uridyltransferase (GALT) catalyzes the conversion of galactose-1-phosphate to glucose-1-phosphate
-
♦
Deficiency of GALT results in the accumulation of galactose-1-phosphate, which causes injury to the parenchymal cells of the kidney, liver, brain, ovaries, and eyes
-
♦
A single mutation (Q188R) accounts for about 70% of galactosemia alleles in Northern Europeans
-
♦
12% carry the Duarte variant, which decreases enzyme activity, but does not result in phenotypic consequence
Laboratory
-
♦
Newborn scre ening uses an enzyme fluorescent assay and/or measurement of total galactose in dried blood spots
-
♦
Classic and variant alleles can be detected with isoelectric focusing
-
♦
Molecular analysis is used in conjunction with biochemical testing for prognostication, heterozygote detection, and prenatal diagnosis
-
♦
Serum in affected individual shows elevated transaminase, hyperbilirubinemia, follicular-stimulating hormone, luteinizing hormone, decreased estrogen, and anemia
-
♦
E. coli septicemia frequently occurs
Treatment
-
♦
Eliminate la ctose and galactose from the diet; special formula is necessary
-
♦
Even with good dietary control, there may be poor intellectual function, speech proble ms, and ovarian dysfunction
Galactokinase Deficiency
Chromosome and Gene Location
-
♦
17q24
Inheritance
-
♦
Autosomal recessive
Incidence
-
♦
1/40,000
Clinical Manifestations
-
♦
Catara cts
-
♦
Neonatal jaundice
-
♦
Normal intelligence
Molecular Basis of Disease
-
♦
Galactokinase catalyzes the conversion of galactose to galactose-1-phosphate
-
♦
Galactoki nase deficiency results in the accumulation of galactose
-
♦
Some galactose is converted to galactitol, which may be responsible for the cataract formation
Laboratory
-
♦
Deficiency of galactokinase activity in red blood cells
Treatment
-
♦
Eliminatio n of lactose and galactose from the diet
Transport Disorders
Familial Hypophosphatemic Rickets
Chromosome and Gene Location
-
♦
Xp22.2–p22.1
Inheritance
-
♦
X-linked dominant (also autosomal recessive and sporadic forms)
Incidence
-
♦
1/1,000,000
Clinical Manifestations
-
♦
Bowing of lower extremities
-
♦
Waddling gait
-
♦
Short stature
-
♦
Decreased femur/shaft angle
-
♦
Dolichocephaly
-
♦
Too th deformities
Molecular Basis of Disease
-
♦
Deficiency interferes with phosphate reabsorption in kidney and conversion of 25-hydroxy-D to 1,25-hydroxy-2D
Laboratory
-
♦
Hyperphosphaturia
-
♦
Normal amino acids
-
♦
X-rays show metaphyseal widening and fraying and cupping of metaphyses at tibia, femur, radius, and ulna
-
♦
Molecular analysis of the PHEX gene is available
Treatment
-
♦
Phosphate s upplements and surgery for limb deformities
Cystinuria
Chromosome and Gene Location
-
♦
2p16.3 and 19q13.1
Inheritance
-
♦
Complex inheritance that is mostly autosomal recessive with genetic components from multiple alleles producing three clinical types
Incidence
-
♦
1/2,000 in E ngland to 1/100,000 in Sweden
-
♦
Overall approximately 1/7,000
Clinical Manifestations
-
♦
Urinary tract calculus
-
♦
Cystine stones are formed and crystals appear in the urine
-
♦
Increased risk for impaired cerebral function
Molecular Basis of Disease
-
♦
Mutations in the SLC3A1 gene on chromosome 2p are responsible for type I, which is recessive; mutations in SLC7A9 are responsible for types II and III, which show incomplete penetrance
-
♦
The relationship between SLC3A1 and SLC7A9 gene products and their influence on the complex inheritance of cystinuria is not completely understood
Laboratory
-
♦
Dibasic ami noaciduria (excess excretion of cystine) and increased urinary lysine, arginine, and ornithine
-
♦
Molecular analysis of SLC3A1 and SLC7A is available
Treatment
-
♦
Dietary ther apy to reduce cystine excretion and increase solubility
-
♦
Decreased methionine
-
♦
Low-sodium diets
-
♦
High fluid intake
-
♦
Alkalization of urine by sodium bicarbonate or sodium citrate to increase solubility of cystine
-
♦
Surgery to remove stones
-
♦
Penicillamine treatment for dissolving stones
Hartnup Disease
Chromosome and Gene Location
-
♦
5p15
Inheritance
-
♦
Autosomal recessive
Incidence
-
♦
1/30,000
Clinical Manifestations
-
♦
Cerebellar ataxia
-
♦
Emotional instability, psychosis
-
♦
Delayed development
-
♦
Severe retardation
-
♦
Photosensitive skin
Molecular Basis of Disease
-
♦
Caused by m utations in SLC6A19, which leads to a defect in neutral amino acid transport across the brush-border membrane of the renal and intestinal epithelium
Laboratory
-
♦
Characteristic pattern of increased secretion of neutral amino acids (alanine, serine, threonine, valine, leucine, isoleucine, phenylalanine, tyrosine, tryptophan, histidine, glutamine, and asparagine)
Treatment
-
♦
Nicotin ic acid or nicotinamide for deficiency of this vitamin reduces rash, ataxia, and psychotic behavior
-
♦
High-protein diet or supplementation
Cystinosis
Chromosome and Gene Location
-
♦
17p13
Inheritance
-
♦
Autosomal recessive
Incidence
-
♦
1/100,0 00–1/200,000
Clinical Manifestations
-
♦
There appears to be three types of cystinosis with varying degrees of involvement:
-
Nephrotic-classic renal and systemic disease in which children develop dehydration, acidosis, vomiting, electrolyte imbalances, hypophosphatemic rickets, failure to grow, photophobia, co rneal crystals, hypothyroidism, myopathy, decreased ability to sweat, and renal failure by age 9–10
-
Intermediate late onset of nephrotic cystinosis
-
Non-nephrotic, clinically affecting only the corneas
-
Molecular Basis of Disease
-
♦
Defective carrier-mediated transport of cystine causes accumulation of cystine and formation of crystals in the lysosomes of most tissues
-
♦
Mutations in t he CTNS gene are responsible for all three types of cystinosis
Laboratory
-
♦
Generalized aminoaciduria
-
♦
Glucosuria with normal blood glucose
-
♦
Elevated leukocyte cystine
-
♦
Cystine crysta ls in tissue biopsy of bone marrow, conjunctiva, kidney, liver, intestine, and other tissues (not present in skin biopsy) and cystine storage in pancreatic islet cells, aorta, atrophic ovaries, and brain
-
♦
Molecular mutation analysis and prenatal diagnosis are available
Treatment
-
♦
Management of electrolyte imbalance
-
♦
Renal allografts
-
♦
Growth hormones
-
♦
Therapy with cysteamine averts the otherwise inevitable renal failure, but systemic therapy does not improve the corneal keratopathy; cysteamine eye drops may prevent photophobia
Metal Metabolism Disorders
Wilson Disease
Chromosome and Gene Location
-
♦
13q14
Inheritance
-
♦
Autosomal recessive
Incidence
-
♦
1/30,000
Clinical Manifestations
-
♦
Liver d isease
-
♦
Neurologic disturbance
-
♦
Acute jaundice
-
♦
Hemolysis
-
♦
Dysarthria
-
♦
Involuntary movement
-
♦
Renal tubular acidosis
-
♦
Osteoarthropathy
-
♦
Cardiomyopathy
-
♦
Golde n-brown granular pigmentation is seen in the outer crescent of the iris (“Kayser–Fleischer” rings)
Molecular Basis of Disease
-
♦
Wilson disease is caused by mutations in the ATP7B gene, which encodes a copper-binding P-type ATPase
-
♦
Expression occurs primarily in the liver, kidney, and placenta
-
♦
Defects in the gene lead to defective liver-specific copper transport and defective copper excretion into the bile
-
♦
Copper accumulates in the liver and other tissues and inhibits various enzymatic reactions
-
♦
Numerous mutations in ATP7B have been identified
Laboratory
-
♦
Reduced se rum copper level and ceruloplasmin and increased urinary copper excretion
-
♦
Elevated copper in liver biopsy
-
♦
Diagnosis by quantitating hepatic copper stores which are increased
-
♦
Mole cular analysis clinically available
Treatment
-
♦
Zinc acetate, which inhibits intestinal absorption of copper
-
♦
Penicill amine, tetrathiomolybdate, or trientine (which may have fewer side effects) increases urinary copper excretion
-
♦
Liver tra nsplantation when necessary
Acrodermatitis Enteropathica
Chromosome and Gene Location
-
♦
8q24.3
Inheritance
-
♦
Autosoma l recessive
Incidence
-
♦
Rare
Clinical Manifestations
-
♦
Symptoms most often occur early in infancy after transition from breast milk to cow milk
-
♦
Features include:
-
Dry, scaly, reddish skin lesions on the face, knees, elbows, and perineal areas
-
Reddish hair
-
Hair loss
-
Photophobia
-
Conjunctivitis
-
Corneal dystrophy
-
Chronic diarrhea
-
Growth retardation
-
Delayed wound healing
-
Immune defects
-
Molecular Basis of Disease
-
♦
Impaired zinc absorption due to an absent or defective zinc-specific transporter, which is encoded by SLC39A4 and facilitates zinc absorption
-
♦
Variety of clinical features is likely because zinc plays a role in numerous metabolic pathways
-
♦
Without therapy, plasma zinc concentration and serum alkaline phosphatase, as well as urinary excretion of zinc, are very low
Treatment
-
♦
Oral zinc compo unds abolish the manifestations of the disease
Mnkes Disease
Chromosome and Gene Location
-
♦
Xq13
Inheritance
-
♦
X-linked recessive
Incidence
-
♦
1/40,000–1/350,000
Clinical Manifestations
-
♦
Hypot hermia
-
♦
Hypotonia
-
♦
Myoclonic seizures
-
♦
Chubby, rosy cheeks
-
♦
Kinky, colorless friable hair (Fig. 13.8)
Fig. 13.8. 
Menkes disease – magnified image of pili torti of hair.
-
♦
Failure to thrive
-
♦
Severe intellectual disability
-
♦
Optic atrophy
-
♦
Osteopenia with pathologic fractures (may be mistaken for child abuse)

Molecular Basis of Disease
-
♦
Caused by mutations in the copper-transporting ATPase, ATP7A
-
♦
Mutations in ATP7A result in defective copper absorption and transport
-
♦
A wide spectrum of mutations has been identified
Laboratory
-
♦
Low serum copper and ceruloplasmin levels
-
♦
Intracellular accumulation of copper in cultured cells
-
♦
Molecular analysis of ATP7A is available; prenatal diagnosis is also available
Treatment
-
♦
Copper hi stidine treatment has had limited success. Many patients deteriorate and die of complications from seizures before 2 years of age
Hemochromatosis
Chromosome and Gene Location
-
♦
6p21.3
Inheritance
-
♦
Autosomal recessive
Incidence
-
♦
Varies by population
-
♦
1/200–1/300 in Northern European populations
-
♦
Lowe r in others
Clinical Manifestations
-
♦
Fatigue
-
♦
Palpitations
-
♦
Premature arthritis
-
♦
Impotence in males
-
♦
Amenorrhea in females
-
♦
Cardiac arrhythmias
-
♦
Congestive heart failure due to cardiomyopathy
-
♦
Cirrhos is with hepatosplenomegaly
-
♦
Ascites
-
♦
Hyperpigmentation
-
♦
Onset is generally between ages 40 and 60
-
♦
Females have l ater onset due to menses/child birthing
Molecular Basis of Disease
-
♦
A gene called HLA-H or HFE has been shown to be altered in the majority of hemochromatosis patients
-
♦
The most common mutation causes an amino acid substitution at codon 282 (C282Y)
-
♦
Approximately 85% of hemochromatosis patients of Northern European descent are homozygous, 5–10% are heterozygous, and an additional 5–10% do not carry the C282Y mutation
-
♦
A second les s frequent alteration is an amino acid substitution at codon 63 (H63D); its role in this disease is controversial, but may increase one’s risk for developing hemochromatosis
-
♦
It has been hypothesized that the C282Y mutation results in an abnormal protein trafficking or cell surface expression of the HFE gene
Laboratory
-
♦
Elevate d serum transferrin saturation and concentration
-
♦
Elevated hepatic iron concentration on liver biopsy
-
♦
Molecular mutation analysis is available
Treatment
-
♦
Phleboto my reduces iron stores to normal
-
♦
Chelating agents remove smaller amounts of iron, but are not as effective as phlebotomy
Purine Metabolism Disorders
Lesch–Nyhan Syndrome
Chromosome and Gene Location
-
♦
Xq26–q27
Inheritance
-
♦
X-linked recessive
Incidence
-
♦
1/300,000
Clinical Manifestations
-
♦
Hypoto nia
-
♦
Delayed motor development
-
♦
Choreoathetosis
-
♦
Spastic movements
-
♦
Hyperreflexia
-
♦
Self-injurious behavior
-
♦
Gouty arthritis
-
♦
Kidney stones composed of uric acid
-
♦
Carrier females develop gout in later years
Molecular Basis of Disease
-
♦
The enzyme, hypoxanthine-guanine phosphoribosyltransferase, converts hypoxanthine a nd xanthine to nucleotides, inosinic acid, and guanylic acid
-
♦
Deficiency in enzyme activity results in accelerated purine production de novo and increased uric acid
-
♦
Deficiencies may also result in decreased synthesis of nucleotides
Laboratory
-
♦
Elevated serum uric acid concentration
-
♦
Elevated urate to creatinine ratio in urine
-
♦
Marked increases in the production and excretion of uric acid
-
♦
Absence of hypoxanthine-guanine phosphoribosyltransferase activity in erythrocytes and fibroblasts (prenatal diagnosis available)
-
♦
Molecular mutation analysis is available, including prenatal diagnosis
Treatment
-
♦
Avoidanc e of dehydration
-
♦
Adequate nutrition
-
♦
Behavior control and modification
-
♦
Allopurinol to prevent damage to kidneys
-
♦
Experim ental gene therapy
Adenosine Deaminase
Chromosome and Gene Location
-
♦
20q13
Inheritance
-
♦
Autosomal recessive
Incidence
-
♦
1/200,000–1,000,000
Clinical Manifestations
-
♦
Severe immunodeficiency
-
♦
Complete impairment of T-cell function
-
♦
Rib cage abnormalities
-
♦
Chondro-osseous dysplasia
Molecular Basis of Disease
-
♦
Mutations in the ADA gene encoding the enzyme adenosine deaminase result in the accumulation of adenosine, 2′ deoxyadenosine and 2′-O-methyladenosine, which leads to lymphocyte toxicity and subsequent immunodeficiency
Laboratory
-
♦
Absence of adenosine deaminase in red blood cells or cultured fibroblasts
Treatment
-
♦
Enzyme replace ment therapy has resulted in clinical and immunologic improvement in some patients
-
♦
Bone marrow transplantation
Peroxisomal Disease
Zellweger Syndrome, Neonatal Adrenoleukodystrophy, and Infantile Refsum Disease
Chromosome and Gene Location
-
♦
7q11.23 (also chromosomes 1, 2, 6, 8, 11, 12, 17, and 22)
Inheritance
-
♦
Autosomal recessive
Incidence
-
♦
1/25,000–1/50,000
Clinical Manifestations
-
♦
Clinical spect rum with Zellweger syndrome representing the most severe presentation; infantile Refsum disease is the least severe phenotype
-
♦
High forehead
-
♦
Upslanting palpebral fissures
-
♦
Hypoplastic supraorbital ridges
-
♦
Epicanthal folds
-
♦
Severe weakness
-
♦
Hypotonia
-
♦
Seizures
-
♦
Eye abnormalities
-
♦
Profound int ellectual disability
-
♦
Early lethality
-
♦
Liver cysts
-
♦
Chondrodysplasia punctata
-
♦
Hearing loss and visual impairment
Molecular Basis of Disease
-
♦
Peroxisome bi ogenesis disorders are caused by mutations in several different genes involved in peroxisome biogenesis
-
♦
Mutations have been identified in numerous peroxisome biogenesis factor (PEX) ge nes as well as the peroxisomal membrane protein-3 gene
Laboratory
-
♦
Reduced or absent peroxisomes
-
♦
Catalase in cytosol
-
♦
Reduced plasmalogens
-
♦
Accum ulation of very long-chain fatty acids
-
♦
Increased phytanic acid
-
♦
Bile acid precursors are elevated
-
♦
Increased l-pipecolic acid
-
♦
Increased urinary excretion of dicarboxylic acids
-
♦
Molecular mutation analysis available
Treatment
-
♦
Not curable, supportive/sympto matic
Adrenoleukodystrophy (X linked)
Chromosome and Gene Location
-
♦
Xq28
Inheritance
-
♦
X-linked recessive
Incidence
-
♦
1/42,000
Clinical Manifestations
-
♦
Learning deficits (attention deficit disorder, hyperactivity), followed by regression of academic performance
-
♦
Leg weakness, loss of sphincter control, and sexual dysfunction
-
♦
Progressive spastic paresis
-
♦
Loss of hearing and vision
-
♦
Dementia
-
♦
Demyelination of cerebral hemispheres
Molecular Basis of Disease
-
♦
Mutations in the ABCD1 gene result in defective peroxisomal beta oxidation of very long-chain fatty acids (VLCFAs); the protein is an ATP-binding cassette (ABC) protein transporter
-
♦
Deficiency results in the accumulation of VLCFAs in the adrenal cortex and brain gangliosides, plasma, blood cells, and cultured fibroblasts
-
♦
Numerous mutations have been identified in the ABCD1 gene
Laboratory
-
♦
Accumulation of unbranched saturated fatty acids with a chain length of 24–30 carbons in plasma, cultured skin fibroblasts, and prenatal diagnosis available
-
♦
Molecular mutation analysis is available
Treatment
-
♦
VLCFA-restricted diet
-
♦
Hematopoiet ic stem cell transplantation for affected males
-
♦
Corticosteroi d replacement therapy for adrenal insufficiency
Lysosomal Storage Disorders
Mucopolysaccharidoses
Chromosome and Gene Location
-
♦
See Table 13.9
Table 13.9. Mucopolysaccharidoses
Inheritance
-
♦
See Table 13.9
Incidence
-
♦
See Table 13.9
Clinical Manifestations
-
♦
Onset ty pically by 2 years
-
♦
Coarse facial features
-
♦
Corneal clouding
-
♦
Organomegaly
-
♦
Skeletal dysplasia (dysostosis multiplex)
-
♦
Intellectual disability (not observed in MPS IV or MPS VI)
-
♦
Loss of developmental milestones
-
♦
Behavioral problems (MPS III)
-
♦
Hearing loss
-
♦
Decrease d range of motion in joints
-
♦
Heart valve abnormalities
-
♦
Inguinal or umbilical hernias
Molecular Basis of Disease
-
♦
The mucop olysaccharidoses (MPS) are a group of lysosomal storage disorders caused by the deficiency of any of the enzymes involved in the stepwise degradation of dermatan sulfate, heparan sulfate, keratan sulfate, or chondroitin sulfate (glycosaminoglycans)
-
♦
Accumulation of glycosaminoglycans (previously called mucopolysaccharides) in the lysosomes interferes with the normal functioning of cells, tissues, and organs
Laboratory
-
♦
Increased urinary excretion of glycosaminoglycans
-
♦
Decreased enzyme activity in cultured fibroblasts, leukocytes, serum, or plasma
-
♦
Radiographs show skeletal abnormalities
-
♦
Molecular sequencing is available for most types
-
♦
Prenatal diagnosis available for known familial mutations
Treatment
-
♦
Hematopoi etic stem cell transplantation (bone marrow or cord blood) can improve somatic features for some patients with severe forms of MPS I and MPS VI; neurological outcomes are still being investigated
-
♦
Enzyme replacement therapy is available for patients with MPS I, MPS II, and MPS VI and in clinical trials for other MPS disorders
-
♦
Treatment remains primarily supportive/symptomatic
-
♦
Bacterial endocarditis prophylaxis for patients with cardiac abnormalities
-
♦
Orthopedic sur gery for skeletal complications
Mucolipidosis
-
♦
Mucolipidosis consists of a group of disorders, which result from abnormal lysosomal enzyme transport
I-Cell Disease (Mucolipidosis II Alpha/Beta)
Chromosome and Gene Location
-
♦
12q23.3
Inheritance
-
♦
Autoso mal recessive
Incidence
-
♦
Rare
Clinical Manifestations
-
♦
Mucolipidosis II is a severe type of mucolipidosis. Presentation is similar to the mucopolysaccharidoses
Molecular Basis of Disease
-
♦
GNPTAB (N-acetylglucosamine-1-phosphotransferase, alpha/beta subunits gen e) is the only gene known to be associated with ML II alpha/beta
-
♦
Mutations in the GNPTAB gene disrupt interactions and recognition of the phosphotransferase, resulting in secretion of lysosomal enzymes into the extracellular medium instead of being correctly targeted to the lysosomes
Laboratory
-
♦
Increased a ctivity of lysosomal hydrolases in plasma and other body fluids (in particular: beta-d-hexosaminidase, beta-d-glucuronidase, beta-d-galactosidase, alpha-l-fucosidase, and arylsulfatase A); diagnosis cannot be made in leukocytes
-
♦
Increased urinary excretion of oligosaccharides
-
♦
Cytoplasmic granular inclusions in skin fibroblasts (swollen lysosomes called I-cells)
-
♦
Molecular s equencing of the GNPTAB gene is available
-
♦
Prenatal diagnosis available for known familial mutations
Treatment
-
♦
Not curable, supportive/sympto matic
Pseudo-Hurler Polydystrophy (Mucolipidosis III Alpha/Beta)
Chromosome and Gene Location
-
♦
12q23.3
Inheritance
-
♦
Autos omal recessive
Incidence
-
♦
Rare
Clinical Manifestations
-
♦
Onset is typic ally between 2 and 4 years. Clinical presentation resembles the mucopolysaccharidoses
Molecular Basis of Disease
-
♦
GNPTAB (N-acetylglucosamine-1-phosphotransferase, alpha/beta subunits ge ne) is the only gene known to be associated with ML III alpha/beta
-
♦
Mutations within the gene disrupt interactions and recognition of the phosphotransferase, resulting in secretion of lysosomal enzymes into the extracellular medium instead of being targeted correctly to the lysosomes
-
♦
A milder variant of I-cell disease (ML II alpha/beta) is caused by the same enzyme deficiency
Laboratory
-
♦
Incre ased activ ity of lysosomal hydrolases in plasma and other body fluids (in particular: beta-d-hexosaminidase, beta-d-glucuronidase, beta-d-galactosidase, and beta-d-mannosidase); the diagnosis cannot be made in leukocytes
-
♦
Increased urinary excretion of oligosaccharides
-
♦
Cytoplasmic granular inclusions in skin fibroblasts (swollen lysosomes)
-
♦
Molecular sequencing of the GNPTAB gene is available
-
♦
Prenatal diagnosis available for known familial mutations
Treatment
-
♦
Not curable, supportive/sympto matic
Sphingolipidoses
GM1 Gangliosidosis
Chromosome and Gene Location
-
♦
3p21.33
Inheritance
-
♦
Autosomal recessive
Incidence
-
♦
1 in 100,000–200,000
Clinical Manifestations
-
♦
Onset
-
Type 1 (infantile) – between birth and 6 months
-
Type 2 (late infantile or juv enile) – between 7 months and 3 years
-
Type 3 (adult or late-onset) – between 3 and 30 years
-
-
♦
Psychomotor deterioration
-
♦
Hepatosplenomegaly
-
♦
Skeletal abnormalities (widening of the long bones and ribs, broad short stubby fingers, contractures)
-
♦
Coarse facial features
-
♦
Failure to thrive
-
♦
Gum h ypertrophy
-
♦
Macroglossia
-
♦
Macrocephaly
-
♦
Protuberant abdomen
-
♦
Frontal bossing
-
♦
Depressed nasal bridge
-
♦
Hypertelorism
-
♦
Cherry red spot on macula
-
♦
Cardiomyopathy
-
♦
Exag gerated startle response
Molecular Basis of Disease
-
♦
GLBI is the only gene known to be associated with GM1 gangliosidosis
-
♦
Mutations in the GLBI gene lead to a deficiency of beta galactosidase enzyme activity and subsequently results in an accumulation of GM1gangliosides, oligosaccharides, and keratan sulfate derivatives
Laboratory
-
♦
Deficient beta galactosidase in leukocytes or cultured fibroblasts
-
♦
Empty-appearing vacuoles on electron microscopy
-
♦
Large foam cells in bone marrow
-
♦
Radiographs s how skeletal abnormalities
-
♦
Molecular sequencing of the GLBI gene is available
-
♦
Prenatal diagnosis available for known familial mutations
Treatment
-
♦
Not curable, supportive/sympto matic
GM2 Gangliosidoses (Tay–Sachs Disease and Sandhoff Disease)
-
♦
The GM2 gangliosidoses are a group of disorders caused by excessive accumulation of GM2 ganglioside and other glycolipids in the lysosomes, particularly in neuronal cells
Tay–Sachs Disease
Chromosome and Gene Location
-
♦
15q23–q24
Inheritance
-
♦
Autosomal recessive
Incidence
-
♦
The incid en ce of Tay–Sachs has been substantially reduced in the Ashkenazi Jewish population in North America as a result of population-based carrier screening programs, education, counseling, and subsequent monitoring and testing of at-risk pregnancies
-
♦
Carrier frequency in Ashkenazi Jewish population: 1 in 30
-
♦
Carrier frequency in non-Ashkenazi Jewish population: ~1 in 300
Clinical Manifestations
-
♦
Onset typically between 3 and 6 months
-
♦
Other forms may present with later onset (through adulthood) and have a more mild and vari e d clinical presentation
-
♦
Progressive mental and motor deterioration
-
♦
Exaggerate d startle response
-
♦
Cherry red spot on macula
-
♦
Macrocephaly
-
♦
Decreased attentiveness
-
♦
Convulsions
-
♦
Blindness
-
♦
Usually f atal by 4 years of age
Molecular Basis of Disease
-
♦
HEXA, the gene encoding the alpha subunit of the HEX A enzyme, is the only gene associated with hexosaminidase A deficiency
-
♦
Hexosami ni dase A is made up of one alpha subunit (encoded by HEXA) and one beta subunit (encoded by HEXB)
-
♦
Mutations in the alpha chain lead to a deficiency of hexosaminidase A, and mutations in the beta chain lead to Sandhoff disease (see below). Six common mutations include four deleterious mutations (three null alleles (+TATC1278, +1IVS12, and +1IVS9), one adult-onset allele (G269S)), and two pseudodeficiency alleles: R247W and R249W. Pseudodeficiency alleles result in low enzyme level on biochemical testing, but are not true deficiency alleles
-
♦
Hexo saminid ase A deficiency results in the accumulation of GM2 gangliosides in the brain, nervous system, liver, spleen, and heart, leading to the disruption of cell function and cell death
Laboratory
-
♦
Decreased hexosaminidase A in serum, leukocytes, or cultured fibroblasts
-
♦
Pseudodeficiency alleles account for approximately 35% of non-Ashkenazi Jewish indiv iduals and 2% of Ashkenazi Jewish individuals identified as carriers through enzyme-based testing and may falsely indicate carrier status
-
♦
Carrier screening can be performed by molecular analysis, biochemical analysis, or both
-
♦
Targeted mutation analysis for common mutations and sequence analysis are available
-
♦
Prenatal diagnosis available for known familial mutations
Treatment
-
♦
Not curable, su pportive/symptomatic
-
♦
Hematopoie tic stem cell transplantation (bone marrow or cord blood) is investiga tional
Sandhoff Disease
Chromosome and Gene Location
-
♦
5q13
Inheritance
-
♦
Autosomal recessive
Incidence
-
♦
~1/300,000 (non-Ashkenazi Jewish)
-
♦
~1/1,000,000 (Ashkenazi Jewish)
Clinical Manifestations
-
♦
Neurological presentat ion and clinical course similar to Tay–Sachs (note: Sandhoff does not demonstrate increased incidence in the Ashkenazi Jewish population)
-
♦
Hepatosplenomegaly
-
♦
Skeletal abnormalities
-
♦
Macrocephaly
Molecular Basis of Disease
-
♦
Mutations in HEXB, the gene encoding the beta subunit of the HEX B enzyme, result in deficiencies of both hexosaminidase A and B enzymes
-
♦
Deficiency result s in the accumulation of GM2 gangliosides in the brain, nervous system, liver, spleen, and heart, leading to the disruption of cell function and cell death
Laboratory
-
♦
Deficient hexosaminidase A and hexosaminidase B in serum, leukocytes, or cultured fibroblasts
-
♦
Molecular sequence analysis is available
-
♦
Prenatal diagnos is available for known familial mutations
Treatment
-
♦
Not curable, supportive/sym ptomatic
Niemann–Pick Disease, Types A and B
Chromosome and Gene Location
-
♦
11p15.4–p15.1
Inheritance
-
♦
Autosomal recessive
Incidence
-
♦
1/250,000
-
♦
Carrier frequ ency for type A is increased in the Ashkenazi Jewish population (1/80–1/100), although carrier screening and subsequent prenatal diagnosis have resulted in a decreased incidence in this population
Clinical Manifestations
-
♦
Type A
-
Onset typically by 3 months
-
Hepatosplenomegaly
-
Progressive neurodegeneration
-
Interstitial lung disease
-
Cherry red spot on macula
-
Usually fatal b y 3 years of age
-
-
♦
Type B
-
Typically later onset with milder presentation
-
Hepatosplenomegaly
-
Respiratory insufficiency
-
A minority have neurologic involvement
-
Life expectancy may extend into adolescence or adulthood
-
Molecular Basis of Disease
-
♦
Mutations in the SMPD1 gene coding for acid sphingomyelinase result in an accumulation of sphingomyelin in cells and tissues
-
♦
Three common mutations causing type A have been described in the Ashkenazi Jewish population (L302P, R496L, and fsP330) and account for >90% of the mutations in this population
-
♦
Only one common mutation for type B has been described (R608del) and is more common in i ndividuals of North African background
Laboratory
-
♦
Deficiency of acid sphingomyelinase in leukocytes (or lymphocytes) or cultured fibroblasts
-
♦
Targeted mutation analysis for common mutations and sequence analysis are available
-
♦
Prenatal diagnosis available for known familial mutations
Treatment
-
♦
Bone marrow transplantation may be able to improve blood counts and reduce liver and spleen volum es. Improvement in neurologic features has not been reported
Niemann–Pick Disease Type C
Chromosome and Gene Location
-
♦
18q11–q12 (Niemann–Pick disease type C [NPC1]), 14q24.3 (NPC2)
Inheritance
-
♦
Autosomal recessive
Incidence
-
♦
1/150,000
Clinical Manifestations
-
♦
Typically late in fantile or juvenile onset (ranges from infantile to adult)
-
♦
Neonatal jaundice
-
♦
Sea-blue histiocytes in marrow
-
♦
Hepatosplenomegaly
-
♦
Progressive neurological deterioration: ataxia, dysarthria, dystonia, dementia, and seizures
-
♦
Vertical supranuclear gaze palsy
Molecular Basis of Disease
-
♦
Mutations in two genes (NPC1 and NPC2) result in a complex defect involving cellular lipid trafficking and subsequent cholesterol and glycosphingolipid accumulation in the lysosomes
-
♦
Most pati ents (~90%) have mutations in the NPC1 gene
Laboratory
-
♦
Delayed induction of cholesterol esterification following LDL uptake
-
♦
Increased filipin staining of accumulated cholesterol in fibroblasts
-
♦
Molecular sequencing of the NPC1 and NPC2 genes is available
-
♦
Prenatal diagnos is available for known familial mutations
Treatment
-
♦
Not curable, supportive/symp tomatic
Gaucher Disease
Chromosome and Gene Location
-
♦
1q21
Inheritance
-
♦
Autosomal recessive
Incidence
-
♦
Type 1: 1/855 (Ashkenazi Jewish)
-
♦
The prevalence is difficult to ascertain in non-Ashkenazi Jewish populations and f or types 2 and 3
Clinical Manifestations
-
♦
Type 1
-
Adult onset is most common, though can present at any age
-
Disease severity is variable
-
Bone disease (osteopenia, bone pain, pathologic fractures)
-
Hepatosplenomegaly
-
Anemia
-
Thrombocytopenia
-
Acquired coagulation deficiencies of factor IX and XI and von Willebrand
-
-
♦
Type 2
-
Onset is typ ically within first few months of life
-
Strabismus
-
Hypertonia of neck muscles results in extreme arching of the neck
-
Loss of head control
-
Hepatosplenomegaly
-
Poor sucking and swallowing
-
Failure to thrive
-
Spasticity
-
Seizures
-
Usually fatal by 2 years of age
-
-
♦
Type 3
-
Clinically heterogeneous
-
Onset typically in adolescence or early adulthood
-
Strabismus
-
Myoclonus
-
Ataxia
-
Seizures
-
Hepatosplenomegaly
-
Life expe ctancy is extremely variable (5–50 years) and dependent on severity of clinical manifestations
-
Molecular Basis of Disease
-
♦
Mutations in the GBA gene result in the deficiency of glucocerebrosidase (glucosylceramidase or acid β-glucosidase) and subsequent accumulation of glucocerebr oside (glucosylceramide) in the lysosomes
-
♦
There are four common mutations: N370S, L444P, 84GG, and IVS2(+1G>A), which account for >90% of the mutations in the Ashkenazi Jewish population
-
♦
Individuals with at least one N370S allele are not predicted to have central nervous system involvement (it is not found in types 2 or 3) and are thought to confer a less severe phenotype when compounded with other mutations
Laboratory
-
♦
Defi ciency of glucocerebrosidase activity in leukocytes or cultured fibroblasts (not accurate for carrier determination)
-
♦
Elevated glucopsychosine (glucosylsphingosine) level in blood
-
♦
Presence of Gaucher storage cells in bone marrow
-
♦
Targeted mutation analysis for common mutations and sequence analysis are available
-
♦
Prenatal diagnosis available for known familial mutations
Treatment
-
♦
Enzy me replacement therapy available since l994 (effective for somatic symptoms but not CNS)
-
♦
Skeletal problems may be treated with limitation of physical activity, prostheses, analgesics, and surgical inter vention
Krabbe Disease
Chromosome and Gene Location
-
♦
14q31
Inheritance
-
♦
Autosomal recessive
Incidence
-
♦
1/100,000
Clinical Manifestations
-
♦
Onset typically within first 6 months
-
♦
Extreme irritability and hypersensitivity to external stimuli
-
♦
Feeding difficulties
-
♦
Spasticity
-
♦
Progressive neurologic deterioration
-
♦
Cortical blindness with optic atrophy
-
♦
Deafness
-
♦
Usually fatal by 2 years of age
-
♦
Also late-on set variants
Molecular Basis of Disease
-
♦
The GALC gene codes for galactosylceramidase (galactocerebrosidase), a lysosomal enzyme, which degrades galactocerebroside to ceramide and galactose
-
♦
Mutations in the gene cause deficient enzyme activity resulting in an accumulation of enzyme substrates and early destruction of oligodendroglia (type of glial cell that forms the myelin sheath of nerve fibers in the central nervous system)
Laboratory
-
♦
Deficient galactosylceramide beta galactosidase activity in leukocytes or cultured fibroblasts
-
♦
Elevated protein in cerebral spinal fluid
-
♦
Elevated psychosine level in blood
-
♦
Neuroimaging d emonstrates progressive, diffuse, and symmetrical, cerebral atrophy
-
♦
Targeted mutation analysis for common mutations and sequence analysis are available
-
♦
Prenatal diagnosis available for known familial mutations
-
♦
Newborn screen ing has been instituted in some states despite lack of effective treatment
Treatment
-
♦
Hematopoietic stem cell transplantation may slow the course of disease progression in asymptomatic patients and individuals with only minimal neurologic invol vement
-
♦
Treatment remains primarily supportive/symptomatic
Metachromatic Leukodystrophy
Chromosome and Gene Location
-
♦
22q13.31–qter
Inheritance
-
♦
Autosomal recessive
Incidence
-
♦
1/40,000–1/160,000
Clinical Manifestations
-
♦
Late infantile (50–60% of cases)
-
Onset typically between 1 and 2 years
-
Hypotonia, weakness, cl umsiness, frequent falls, toe walking, and inability to stand
-
Progressive neurologic deterioration
-
Increased muscle tone
-
Slurred speech
-
Blindness
-
Unaware of surroundings
-
Death typically occurs app roximately 5 years after the onset of symptoms
-
-
♦
Juvenile (20–30% of cases)
-
Onset typically between 4 and 14 years
-
Decline in school performance and behavioral problems
-
Clumsiness a nd gait problems
-
Slurred speech
-
Seizures
-
Death typically by 20 years of age
-
-
♦
Adult (15–20% of cases)
-
Onset typically after puberty and into the sixth decade of life
-
Problems in school or job performance and personality changes
-
Weakness, loss of coordination, and peripheral neuropathy
-
Seizures
-
Duration of disease may range from a few years to several decades before the patient reaches a vegetative state and, ultimately, death
-
Molecular Basis of Disease
-
♦
The ARSA (arylsulfatas e A) gene is the only known gene associated with MLD
-
♦
Mutations in this gene lead to a deficiency in the enzymatic hydrolysis of sulfatide and subsequent accumulation of sulfatides, mainly within the lysosomes
Laboratory
-
♦
Deficient arylsulfatase A enzyme activity in leukocytes or cultured fibroblasts
-
♦
The presence of indiv iduals with pseudodeficiency (unaffected individuals whose ARSA activity in leukocytes is 5–20% of controls) makes establishing a diagnosis of true ARSA deficiency difficult by biochemical analysis alone
-
♦
Increased protein in the cerebrospinal fluid
-
♦
Metachromatic lipid deposits in a nerve or brain biopsy
-
♦
Increased urinary sulfatide excretion
-
♦
Targeted mutation analysis for common mutations and sequence analysis are available
-
♦
Prenatal diagnosis available for known familial mutations
Treatment
-
♦
Not curable, suppor tive/symptomatic
-
♦
Bone marrow transplantation may be able to slow disease progression when performed prior to symptoms occurring in juvenile and adult-onset forms. BMT does not appear to alleviate peripheral nervous system manifestations and has not been shown to be effective in symptomatic patients with the late infantile form. BMT remains controversial due to risks and uncertain long-term effects
Fabry Disease
Chromosome and Gene Location
-
♦
Xq22.1
Inheritance
-
♦
X linked
Incidence
-
♦
1/40,000 males; newborn screening studies including late-onset variants are finding a prevalence closer to 1/5,000 males
Clinical Manifestations
-
♦
Onset typicall y in childhood or adolescence
-
♦
Also cardiac and renal variants with variable presentation; late-onset variants are likely underdiagnosed
-
♦
Pain in distal extremities (acroparesthesias)
-
♦
Fever due to hypohid rosis (reduced sweating)
-
♦
Corneal opacities
-
♦
Angiokeratomas (Fig. 13.9)
Fig. 13.9. 
Fabry disease – angiokeratomas.
-
♦
Proteinuria; this can lead to end-stage renal disease typically occurs in the third to fifth decades
-
♦
Cardiovascular disease
-
♦
Carrier females are us ually asymptomatic but may be as severely affected as males or have an attenuated form of the disease due to random X-inactivation

Molecular Basis of Disease
-
♦
The GLA gene codes for alpha galactosidase A, an enzyme involved in the degradation of glycosphingolipids
-
♦
Mutations in the GLA gene result in the accumulation of globotriaosylceramide (Gb3) and other glycosphingolipids in bodily fluids, lysosomes, and cell types of various other tissue s in organs
Laboratory
-
♦
Deficient alpha galactosidase A activity in leukocytes, tears, cultured fibroblasts, plasma, or serum (unreliable for carrier determination)
-
♦
Molecular sequence analysis is available
-
♦
Prenatal diagnosis available for known familial mutations
-
♦
Newborn screening has been instituted in some states
Treatment
-
♦
Enzyme replacem ent therapy
-
♦
Symptoms of peripheral neuropathy may respond to carbamazepine or diphenylhydantoin; however, the neuropathy does not diminish
-
♦
Renal dialysi s and transplantation
Familial Cancer Syndromes
Familial Adenomatous Polyposis
Chromosome and Gene Location
-
♦
5q22.2 (APC)
Inheritance
-
♦
Autosomal dominant
-
♦
75–80% have an affected parent
-
♦
20–25% are the result of new (de novo) mutations
Incidence
-
♦
3 in 100,000
Clinical Manifestations
-
♦
Multiple (>100) polyps early in life, on average at 16 years of age with a range of 7–36 (by age 35 over 95% of gene carriers will develop polyps)
-
♦
Polyps progress to colonic carcinoma on average by age 39 (range 34–43)
-
♦
Other tu mors and/or carcinomas may occur including duodenal, thyroid, brain, liver, and pancreatic desmoid tumors (Fig. 13.10), and childhood hepatoblastomas
Fig. 13.10. 
Familial adenomatous polyposis – desmoid tumor.
-
♦
Additional features include congenital hypertrophy of the retinal pigment epithelium (CHRPE) and dental anomalies, such as supernumerary teeth
-
♦
“Gardner syndrome” refers to individuals with familial adenomatous polyposis (FAP) who have extracolonic manifestations such as tumors of the jaw (osteomas), lipomas, and fibromas
-
♦
“Turcot syndrome” refers to individuals with FAP and central nervous tumors (CNS), specifically medulloblastoma
-
♦
Attenuated FAP refers to individuals who still have a significant increased risk for colonic carcinom a, with fewer than 100 polyps (on average 30)

Molecular Basis of Disease
-
♦
Numerous mutations have been identified in the adenomatous polyposis coli (APC) gene; most mutations lead to truncated protein product
-
♦
The protein is thought to be involved in maintaining normal apoptosis and decreasing cell proliferation and may also be involved in maintaining chromosomal stability
Laboratory
-
♦
Direct mutation analysis is available. Linkage testing is also available for families without an identifiable mutation
-
♦
Less than 30% of individuals clinically diagnosed with attenuated FAP will have an identifiabl e APC mutation
Treatment/Surveillance
-
♦
Genetic consultation is necessary to determine the utility of genetic testing in these families
-
♦
For individuals with a diagnosis of FAP, colectomy is advised when polyps become too numerous to re move or watch, to prevent colorectal carcinoma
-
♦
For individuals who have a positive gene test, flexible sigmoidoscopy or colonoscopy every year beginning at age 10–15 years, colonoscopy when polyps are detected
-
♦
For individuals who have not had genetic testing and are at risk, flexible sigmoidoscopy or colonoscopy every year beginning at 10–15 years to 24 years, every 2 years until age 34 if polyps are not detected, every 3 years to age 44 if polyps are not detected, and e very 3–5 years thereafter
-
♦
Esophagogastroduodenoscopy should be initiated when colonoscopies are started or by age 25
-
♦
Annual thyroid palpation beginning in late teens
-
♦
Ophthalmologic examination for CHRPE is done if confirmation of diagnosis is sought; however, note these lesions are harmless
-
♦
For children at risk, liver palpitation, abdominal ultrasound, and alpha-fetoprotein testing are recommended for hepatoblastoma every 3–6 months until age 5
MYH-Associated Polyposis
Chromosome and Gene Location
-
♦
1p34.1 (MUTYH)
Inheritance
-
♦
Autosomal recessive
Incidence
-
♦
1 in 20,000 to 1 in 40,000
-
♦
Approximately 1–2% of the general population are carriers of one MUTYH m utation
Clinical Manifestations
-
♦
Multiple (>10–100) adenomatous polyps early in life, similar to the presentation of attenuated FAP, evident by the mean age of 50
-
♦
Polyps are likely to progress to carcinoma if left untreated (80% cumulative risk to age 80 years)
-
♦
Additional features include similar extracolonic manifestations exhibited with FAP; app roximately 65% of individuals will have at least one
-
♦
Duodenal adenomas occur in 17–25% of individuals with a lifetime duodenal cancer risk of approximately 4%
-
♦
Additional risk for colon cancer remains unclear related to carriers of one MUTYH mutation; some suggest a 2–3 time increased risk over the general population; however, the evi dence has not been enough to warrant additional screening
Molecular Basis of Disease
-
♦
Two mutations account for ~90% of mutations within the Northern European population (Y179C, G396D). Therefore, only ~2% of people with biallelic mutations will have a negative DNA test, when only these two mutations are studied
-
♦
Other founder m utations exist in certain ethnic groups (i.e., Indian, Dutch, Pakistani)
-
♦
The MUTYH protein repairs DNA by removing adenine residues that are mispaired with 8-oxoguanine during replication of oxidized DNA. Deficiency in this base excision repair leads to somatic mutations in APC, hence the clinical similarity to FAP
-
♦
Biallelic MUTYH mutations are identified in up to 30% of individuals with 10–100 polyps and 7–14% with greater than 100 polyps
Laboratory
-
♦
Direct mutation analysis for the two common mutations is clinical available; full gene se quencing can also be performed
-
♦
Predictive genetic testing can be offered once mutations have been identified within a family
Treatment/Surveillance
-
♦
Genetic consultation is necessary to determine the utility of genetic testing in these families
-
♦
For individuals with a diagnosis of attenuated FAP without clear evidence of dominant inh eritance, testing for MUTYH should be offered
-
♦
MUTYH testing should be offered for those suspected to have FAP or attenuated FAP with negative APC testing
-
♦
Treatment/surveillance of families with biallelic MUTYH mutations should include colonoscopy every 2–3 years beginning at 25–30 years and every 1–2 years if polyps are noted
-
♦
Esophagogastroduodenoscopy is advised at 30–35 years and repeated based on findings, at minimum every 4 years. If adenomas are found, management proceeds akin to that of clas sical FAP
Nevoid Basal Cell Carcinoma Syndrome (Gorlin Syndrome, Basal Cell Nevus Syndrome)
Chromosome and Gene Location
-
♦
9q22.32 (PTCH1)
Inheritance
-
♦
Autosomal dominant
Incidence
-
♦
1 in 30–50,000 (in United Kingdom)
Clinical Manifestations
-
♦
The following criteria have been proposed for making a diagnosis. (The sensitivity and specificity are not known) A diagno si s is made when two major or two minor and one major criteria are fulfilled:
-
Major criteria
-
•
More than 2 basal cell carcinomas (BCC) or 1 in an individual before 20
-
•
Odontogenic keratocyst of the jaw
-
•
Three or more palmar or plantar pits
-
•
Bilamellar calcification of falx cerebri (90% of individuals have by age 20)
-
•
Bifid, fused, or markedly splayed ribs
-
•
Family hist ory of NBCC syndrome
-
•
-
Minor criteria
-
•
Macrocephaly (head circumference >97th percentile)
-
•
Congenital skeletal malformations (cleft lip/palate, frontal bossing, “coarse facies,” ocular hypert elo rism, pectus, polydactyly, syndactyly)
-
•
Radiologic abnormalities (hemivertebrae, fusion or elongation of the vertebral bodies, bridging of the sella turcica)
-
•
Cardiac or ovarian fibroma
-
•
Medulloblastoma
-
•
-
-
♦
Rate of BCC risk increases with age; 51% risk for age >20 and 71% risk for age >40
-
♦
Variable express ivity is noted
Molecular Basis of Disease
-
♦
Majority of mu tations are nonsense mutations leading to a premature stop codon resulting in a truncated protein
-
♦
20–30% are the result of de novo mutations
-
♦
The protein is thought to be involved in controlling cell fates, patterning, and growth in numerous tissues
Laboratory
-
♦
Molecular genet ic testing is available
-
♦
Failure to detect a PTCH1 mutation does not exclude a clinical diagnosis of NBCCS
-
♦
9q22.3 microdeletion syndrome involves PTCH1; however, beyond NBCCS causes developmental and int ellectual delays, metopic craniosynostosis, obstructive hydrocephalus, pre- and postnatal macrosomia and seizures
Treatment/Surveillance
-
♦
Advice regarding avoidance of sun exposure and radiation
-
♦
Developmental and physical assessment every 6 months in the first few years of life due to increa sed medulloblastoma risk
-
♦
Screening by dermatologist in puberty and every 4–12 months thereafter with early excision of basal cell cancers
-
♦
Jaw radiograph every 12–18 months after the age of 8 for keratocysts
-
♦
Gynecolo gic exam done annually in adulthood
Birt–Hogg–Dube Syndrome
Chromosome and Gene Location
-
♦
17p11.2 (FLCN)
Inheritance
-
♦
Autosomal dominant
Incidence
-
♦
Unkn own; under recognized, current estimate is 1 in 200,000
Clinical Manifestations
-
♦
The clinical triad of BHDS is co mprised of:
-
♦
Cutaneous lesions including multiple fibrofolliculomas (specific for BHDS) as well as trichodiscomas and acrochordons (associated with BHDS). These skin lesions are easily overlooked and usually initially develop in the third to fourth decades of life
-
♦
Pulmonary cysts (present in 90%) with spontaneous pneumothorax (~25%) may be the presenting feature
-
♦
Bilateral and multiple renal tumors, developing at a median age of 48 years (25–35% with BDH S have lesions before 50). The histology is variable including oncocytomas, chromophobe, clear cell, or papillary renal cell carcinomas and hybrid oncocytic neoplasms
-
♦
-
♦
A range of 1–28 renal tum ors have been reported with an average number of 5.3
-
♦
Nonrenal carcinomas have been described but are rare
Molecular Basis of Disease
-
♦
FLCN encodes a protein called folliculin, which appears to be involved in AMPK and mTOR signaling
-
♦
Approximately 90% fulfilling diagnostic criteria will have an identifiable FLCN mutation
-
♦
Approximately 50% will have a mutation identified in the gene’s hotspot, exon 11
Laboratory
-
♦
Skin biopsy is required to confirm a diagnosis of fibrofolliculomas and trichodi scomas, both of which are fibroepithelial hamartomas of the hair follicles
Treatment/Surveillance
-
♦
Genetic consultation is necessary to determine if clinical genetic testing will be of utility in a given family
-
♦
Due to concern for an increased risk for melanoma, dermatology visits may be considered and an a voidance of radiation exposure advised
-
♦
Surveillance is not indicated for the cutaneous lesions as they are not premalignant
-
♦
Annual renal imaging by MRI (optimal) or abdominal/pelvic CT scan with contrast is appropriate beginning at age 18, when the diagnosis is made or if a FLCN mutation is confirmed
-
♦
Renal sparing surgery should be offered due to the propensity for multifocal tumor development
-
♦
Affected individuals should be counseled on the signs, symptoms, and management of spontaneous pneu mothorax and to avoid smoking and high ambient pressures
Hereditary Breast and Ovarian Cancer (HBOC) Syndrome
Chromosome and Gene Location
-
♦
17q21.31 (BRCA1)
-
♦
13q13.1 (BRCA2)
Inheritance
-
♦
Autos omal dominant
Incidence
-
♦
Varies by population
-
♦
Approximately 1/400 non-Ashkenazi Caucasian individuals carry a BRCA1 or BRCA2 mutation
-
♦
Approximately 1/40 Ashkenazi Jewish individuals carry one of three founder mutations in BRCA1 or BRCA2
-
♦
Less is known about mutation frequency in other populations
-
♦
Clinical Manifestations
-
♦
Early onset (prem enopausal) adenocarcinoma of the breast, often bilateral disease
-
♦
Additionally, ovarian, fallopian, and primary peritoneal cancer occurs less frequently for BRCA2 families (11–18%) than BRCA1 families (24–40%)
-
♦
BRCA2 is known to have an ovarian cancer cluster region (OCCR) in exon 11 in which the risk for ovarian cancer is higher than that for any other region in which a BRCA2 mutation is found
-
♦
Breast cancer in males is more common in BRCA2 families (5–10%) but also present in BRCA1 families (1–2%)
-
♦
Males carryin g a BRCA1 or BRCA2 mutation are at an increased risk of developing prostate cancer (up to 39%)
-
♦
Males and females carrying a BRCA1 or BRCA2 mutation are at an increased risk for pancreatic cancer (1–3% and 2–7%, respectively)
-
♦
BRCA1 mutations are estimated to account for 50% of all familial early-onset cases (2.5–5% of all breast cancer) and about 80% of inherited breast and ovarian cancer
-
♦
BRCA2 mutations are estimated to account for 17–35% of hereditary breast cancer
-
♦
It is estimated that individuals who carry a BRCA1 or BRCA2 gene mutation may have as high as 40–80% risk of developing breast cancer and 11–40% risk of developing ovarian cancer by age 70, compared with the lifetime general population risk of 12.5% for breast cancer and 1.4% for ovarian cancer
-
♦
The following criteria have been esta blished to determine when further evaluation for a mutation is warranted
-
Breast cancer at or below the age of 45
-
Breast cancer at or below the age of 50 with:
-
An additional primary
-
One or more close blood relatives with breast cancer at any age
-
Unknown or limited family history
-
-
Ovarian, fallopian, or primary peritoneal cancer at any age
-
Diagnosed at or under 60 with a triple negative (ER-, PR-, Her2-) breast cancer
-
Breast cancer at any age and:
-
One or more close bloo d relatives with breast cancer at or under 50
-
One or more close blood relative with ovarian cancer at any age
-
Two or more close blood relatives with breast, pancreatic, and/or prostate cancer at any age
-
Close male blood relative with breast cancer
-
-
Male breast cancer
-
Pancreatic or prostate cancer at any age with two or more close blood relatives with breast and/or ovarian cancer and or pancreatic or prostate cancer on the same side of the family
-
Previous BRCA mutation identified in the family
-
Molecular Basis of Disease
-
♦
Various mutations (deletions, insertion, point mutations) have been identified; most result in truncation or absence of BRCA1 and BRCA2 proteins
-
♦
Both are thought to be tumor suppressor genes
Laboratory
-
♦
Molecular testing ava ilable for BRCA1 and BRCA2
Treatment/Surveillance
-
♦
Breast awar eness starting at age 18 and clinical breast exams every 6–12 months starting at age 25
-
♦
Breast screening recommendations include:
-
Annual breast MRI (mammogram if MRI is unavailable) beginning at age 25–29 or individualized based on earliest age of onset in family
-
Annual breast MRI and mam mogram age 30–75
-
-
♦
Additional screening beyond age 75 should be considered on an individual basis
-
♦
Discuss risk-reducing mastectomy including degree of protection, reconstruction options, and risks
-
♦
Recommend risk-reducing salpingo-oophorectomy between 35 and 40 or upon completion of childbearing or individualized based on earliest age of onset in family; counseling includes discussion of reproductive desires, extent of risk, degree of protection, and management of related medical issues
-
♦
For those electing not to undergo risk-reduc ing BSO, consideration of semiannual screening using transvaginal ultrasound and serum CA-125 starting at age 30 or 5–10 years before earliest ovarian cancer diagnosis in the family
-
♦
Caution: less than half of early stage ovarian tumors produce elevated levels of CA-125; there is no evidence of efficacy of screening for ovarian cancer
-
♦
Consider chemoprevention options for breast and ovarian cancer; tamoxifen or related compounds confer up to a 50% reduction in risk contralaterally
-
♦
For males, self-breast exam training and education, as well clinical breast exams every 6–12 months should begin at age 35 with consideration of baseline mammography at age 40
-
♦
Prostate cancer screening should begin at age 40 for BRCA2 mutation carriers and can be considered for BRCA1 mutation carriers
-
♦
Addition al screening guidelines should be considered and individualized based on cancers observed in the family (i.e., pancreatic, melanoma, etc.)
Lynch Syndrome
-
♦
Previously referred to as hereditary nonpolyposis colon cancer syndrome (HNPCC)
Chromosome and Gene Location
-
♦
Caused by of mutations in one of four DNA mismatch repair genes
-
3p22.1 (MLH1)
-
2p21 (MSH2)
-
2p16.3 (MSH6)
-
7p22.1 (PMS2)
-
2p21 (EPCAM, upstream of MSH2)
-
Inheritance
-
♦
Autosomal dominant
Incidence
-
♦
Estimated at 1 in 440
-
♦
Accounts for 2–3% of all colon cancer
Clinical Manifestations
-
♦
Risks vary dependent upon the underlying genetic cause
-
♦
Colorectal cancer
-
70–85% noted to occur in the proximal region (right sided)
-
Average age at diagnosis is 44 (range 44–61)
-
80% lifetime risk for colon cancer
-
-
♦
Endometrial adenocarcinoma lifetime risk may be up to 60%; average age of diagnosis is 62
-
♦
Increased risk for ovarian, transitional cell renal collecting system, ureter, stomach, small bowel, hepatobiliary tract, and pancreatic cancers. Cumulative lifetime risk for wo men is 47% and for men is 27%
-
♦
Increased risk for sebaceous carcinomas and basal and squamous cell carcinomas of the skin
-
♦
Muir–Torre syndrome is a variant form of Lynch syndrome characterized by a combination of benign or malignant sebaceous skin tumors and internal malignancy most commonly associated with a mutation in MSH2
-
♦
Turcot syndrome is characterized by brain tumor (glioblastoma multiforme) and colorectal t umors and is also associated with Lynch syndrome
-
♦
While reports have indicated a potential associated breast cancer risk, the relationship remains unclear
-
♦
Biallelic pathogenic mutations are possible and likely incur an increased risk
-
♦
The following Amsterdam II criteria were developed to define HNPCC. All four conditions need to be fulfilled to meet the Amsterdam criterion. However, the criteria have been found to be over-restrictive for clini cal use:
-
Three or more family members with a HNPCC-related cancer, one of which is a first degree relative of the other two
-
Two successive generations affected
-
One or more HNPCC-related cancers diagnosed before age 50
-
The family does not have FAP
-
-
♦
Note that only abou t half of families that fulfill these strict criteria actually have defective DNA mismatch repair (i.e., have Lynch syndrome)
-
♦
50% of families that meet criteria and do not have a detected Lynch syndrome mutation have a familial risk of uncertain etiology
Molecular Basis of Disease
-
♦
The products of the genes MLH1, MSH2, MSH6, and PMS2 participate in a DNA mismatch repair (MMR) complex
-
♦
EPCAM is not part of th e MMR complex; however, it can disrupt it due to its inactivation of MSH2
-
♦
MLH1 and MSH2 account for 50% and 40%, respectively, of the mutations found in Lynch syndrome to date
-
♦
Colon cancer is a multistep process, meaning that a number of mutations in different genes need to occur before the onset of colon cancer
-
♦
When a mutation occurs in a mismatch repair complex, the ability to repair other random mutations is compromised, thus leading to an accumulation of mutations (microsatellite instability [MSI])
-
♦
The mutations may inactivate tumor suppresser genes and lead to the subsequent penetrance of colon cancer
Laboratory
-
♦
Nearly all colorec tal tumors in Lynch syndrome show widespread MSI, and Bethesda guidelines have been developed to assist in what tumors should be tested
-
♦
MSI in combination with immunostaining is a useful tool to identify which gene may be involved
-
♦
Immunohistochemistry (IHC) can be utilized on the tumor tissue to identify the presence or absence of protein expression for MLH1, MSH2, MSH6, and PMS2 proteins, helping to determine the gene of interest for a given individual
-
♦
BRAF and MLH1 hypermethylation are useful to distinguish sporadic cancers from those that may be due to a MMR gene; mutational analysis is available
Treatment/Surveillance
-
♦
Surveillan ce depends upon the gene involved and the family history
-
♦
For asymptomatic, suspected, or known gene carriers, a colonoscopy every beginning between ages 20–30 or 2–5 years prior to the earliest diagnosis, repeating every 1–2 years
-
♦
Prophylactic total abdominal hysterectomy and bilateral salpingo-oophorectomy considered after childbearing complete
-
♦
Annual screening for endometrial and ovarian cancer may be considered; however, there is no c lear evidence supporting its use
-
♦
Consider annual urinalysis and cytology beginning at age 25–30
-
♦
Consideration of upper gastrointestinal endoscopy every 3–5 years beginning at age 30–35 for families in which gastric cancer has occurred
-
♦
Annual physical/neurological exam beginning at age 25–30 for CNS tumors
-
♦
Breast cancer surveillance has been suggested; however, no guidelines exist at this time; suggest to base upon family history
Juvenile Polyposis Syndrome (JPS)
Chromosome and Gene Location
-
♦
10q23.2 (BMPR1A)
-
♦
18q21.2 (SMAD4)
-
♦
Each accounts for approximately 20–25% of mutations detected in families who meet criteria; 50–60% of families do not have a detected mutation in either
Inheritance
-
♦
Autosomal dominant
-
♦
Estima ted 25% are due to de novo mutations
Incidence
-
♦
Estimated to range between 1 in 16,000 to 1 in 100,000
Clinical Manifestations
-
♦
Increased risk for gastrointestinal and pancreatic cancer
-
♦
Note: juve nile polyps are a type of polyp, not the age of onset of polyps. They are typically less than 3 cm in diameter and comprised of focal malformations of mucosa and lamina propria
-
♦
Individuals typically have between 50 and 200 polyps, but the number can be variable
-
♦
Solitary juvenile polyps occur in approximately 2% of adults and children
-
♦
JPS is clinically diagnosed if an individual has:
-
5 or more juvenile polyps in the colon or rectum or
-
Multiple juvenile polyps throughout the gastrointestinal tract or
-
Any number of juvenile polyps and a family history of JPS
-
-
♦
Lifetime esti mates of GI cancers range from 9 to 50%
-
♦
Incident of colon cancer is 17–22% by 35 years, approaching 68% by age 60 with a median age of diagnosis of 42
-
♦
15–22% of individuals with SMAD4 mutations are likely to have JPS and hereditary hemorrhagic telangiectasia (HHT); recent discussion includes the suggestion that those with a SMAD4 mutation and JPS also have HHT
-
♦
HHT include s mucocutaneous telangiectases, pulmonary, hepatic, cerebral, and/or GI arteriovenous malformations, epistaxis, and intracranial bleeding
-
♦
Myhre syndrome and thoracic aortic disease have also been noted to be caused by SMAD4 mutations
Molecular Basis of Disease
-
♦
It is unclear how germ line mutations of SMAD4 or BMPR1A cause juvenile polyps to form; SMAD4 is a tumor suppressor gene, BMPR1A’s protein product mediates intracellular signaling through SMAD4
Laboratory
-
♦
Pathologic confirmation of the type of polyp necessary to apply clinical diagnosis
-
♦
Molecular mutation analysis is available
-
♦
If molecular testing is negative, consideration of PTEN testing is appropriate to determine if an individual has PTEN hamartoma tumor syndrome rather than JPS
Treatment/Surveillance
-
♦
Colonoscopy a nd upper endoscopy to begin at approximately age 15 with repeat annually if polyps are noted and every 2–3 years if polyps are not noted
-
♦
If SMAD4 mutation, screen for vascular lesions associated with HHT within the first 6 months of life
-
♦
Due to the rare and undefined nature of small intestine and pancreatic cancer risk, no recommendation s have been made at this time
Cowden Syndrome (One of the PTEN Hamartoma Tumor Syndromes)
Chromosome and Gene Location
-
♦
10q23.31 (PTEN)
Inheritance
-
♦
Autosomal dominant
Incidence
-
♦
Esti mated to be 1 in 200,000, likely underestimated
Clinical Manifestations
-
♦
Clinical criteria exist to determine those at risk to have Cowden syndrome
-
Major criteria include breast cancer, endometrial cancer, follicular thyroid cancer, multiple GI hamartomas or ganglioneuromas, macrocephaly, macular pigmentation of glans penis, and mucocutaneous lesions
-
Minor criteria include colon cancer, autism spectrum disorder, lipomas, intellectual delay, esophageal glycogenic acanthuses, papillary or follicular variants of papillary thyroid cancer, structural thyroid lesions, renal cell carcinoma, testicular lipomatosis, single GI hamartoma or ganglioneuroma, and vascular anomalies
-
-
♦
85% of individuals meeting clinical diagnostic criteria have an identified PTEN mutation
-
♦
80% of individuals will have dermatologic findings, and these pathognomic criteria include tr ichilemmomas, acral keratoses, papillomatous lesions, and mucosal lesions
-
♦
Hamartomatous polyps of the stomach, small bowel, and colon occur in 35–60%; the intestinal polyps are indistinguishable from those noted in JPS
-
♦
Breast cancer occurs in 30–50% of female mutation carriers; 85% lifetime risk
-
♦
Increased ri sk for endometrial cancer (28%) and thyroid adenoma and carcinomas (mainly follicular, 35%)
-
♦
Condition overlaps with Bannayan–Ruvalcaba–Riley syndrome
Molecular Basis of Disease
-
♦
Mutations in PTEN gene thought to disrupt the tyrosine/dual-specificity phosphatase domain of this gene
Laboratory
-
♦
Molecular m utation analysis is available
Treatment/Surveillance
-
♦
Annual thyroid ultrasound beginning at age 18
-
♦
Breast awareness beginning at age 18, clinical breast exams every 6–12 months beginning at age 25, and a nnual mammography and breast MRI beginning at age 30–35
-
♦
Consideration of endome trial biopsy and/or ultrasound beginning at age 30–35
-
♦
Consideration of risk-reducing mastectomy and hysterectomy
-
♦
Colonoscopy every 5 years or as findings appropriate beginning at age 35
-
♦
Consider renal ultrasound every 1–2 years beginning at age 40
-
♦
Additional screening guidelines should be considered and individualized based upon cancers observe d in the family
Li–Fraumeni Syndrome
Chromosome and Gene Location
-
♦
17p13.1 (Tp53)
Inheritance
-
♦
Autosomal dominant
-
♦
Approximately 7–20% due to de novo mutations
Incidence
-
♦
Approx imately 1 in 5,000 to 1 in 20,000
Clinical Manifestations
-
♦
Soft tissue sarcomas
-
♦
Early onset breast cancer; estimated that 4–8% of individuals diagnosed under age 30 who test negative for BRCA1 and BRCA2 mutations test positive for a Tp53 mutation
-
♦
Adrenocortical and brain tumors
-
♦
Osteosarcomas
-
♦
Leuke mia
-
♦
Risk of developing invasive cancer is approximately 50% by age 30 and 90% by age 60
-
♦
Classical definition requires:
-
One patient with sarcoma under age 45
-
A first degree relative under age 45 with any cancer
-
A third affected first- or second-degree relative with either sarcoma at any age or any cancer under age 45
-
-
♦
About 70–83% of families meeting these criteria have identifiable mutation in Tp53
-
♦
Several additional guidelines exist, such as Chompret
Molecular Basis of Disease
-
♦
Mutation types include nonsense mutations and splice site mutations, which generate truncated pro tein products
Laboratory
-
♦
Mutation analysis available for Tp53
Treatment/Surveillance
-
♦
Efficacy not well established
-
♦
Annual compr ehensive physical exam and promptly seek medical care for the evaluation of lingering symptoms or illnesses
-
♦
Breast surveillance includes clinical breast exams every 6 months beginning at 20–25 years, annual breast MRI beginning at 20–29 years, and annual mammography and breast MRI after age 30
-
♦
Consider colonoscopy screening no later than 25 years with repeat every 2–5 years
-
♦
Consider orga n-targeted screening based upon family history
-
♦
Whole-body MRI protocols are currently under investigation but could be considered
-
♦
Additional surveillance strategies have been published, and discussion to participate in novel screening approaches using technologies such as whole-body MRI, abdomi nal ultrasound, and brain MRI should be offered
Multiple Endocrine Neoplasia Type 1 (MEN1)
Chromosome and Gene Location
-
♦
11q13.1 (MEN1)
Inheritance
-
♦
Autosomal dominant
Incidence
-
♦
1 in 20,000 to 1 in 40,000
-
♦
10% are the result of de novo mutations
Clinical Manifestations (Also See Chap. 20 )
-
♦
Parath yroid adenomas
-
♦
Pituitary adenoma (prolactinoma)
-
♦
Pancreatic islet cell tumors (most commonly gastrinomas)
-
♦
Insulinoma
-
♦
Peptic ulcer disease
-
♦
Hyperpa rathyroidism
-
♦
Other findings include lipoma and leiomyoma and dermatologic findings such as facial angiofibroma and collagenoma
-
♦
Features are variable within and between families
-
♦
80% penetrance by age 50
-
♦
Diagnostic criteria include the pres ence of two of the following three endocrine tumors:
-
Parathyroid tumor (hypercalcemia, primary hyper-parathyroidism)
-
Pituitary tumors (prolactinomas)
-
Well-differentiated endocrine tumors of the gastroenteropancreatic (GEP) tract (gastro ma, insulinoma, VIPoma)
-
Molecular Basis of Disease
-
♦
MEN1 encodes a transcript called menin
-
♦
Numerous frameshift, nonsense, missense, and in-frame deletions have been reported
-
♦
Function of protein is not known but thought to have a role in DNA replication and repair, transcription, and chromatin modification
Laboratory
-
♦
Elevated ACTH
-
♦
Abnormal secretion test
-
♦
Hypoglycemia
-
♦
Hypergastrinemia
-
♦
Hyperparathyroidism
-
♦
Glucose intolerance
-
♦
Molecular analysis is available
Treatment/Surveillance
-
♦
Screening recomm endations begin at age 5 and include an annual evaluation for prolactin, cortisol, glucose, calcium, and phosphorus
-
♦
Physical exam with endocrinologic review of systems
-
♦
Annual head MRI begi nning at age 5 and abdominal CT or MRI beginning at age 20
Multiple Endocrine Neoplasia Types 2A and 2B and Familial Medullary Thyroid Carcinoma
Chromosome and Gene Location
-
♦
10q11.21 (RET)
Inheritance
-
♦
Autosomal dominant
Incidence
-
♦
1 in 35,000
-
♦
About 10% of thy roid cancers are of the medullary type; of these, 25–30% are due to germ line RET mutations
-
♦
Approximately 5% of MEN2A diagnoses are the result of de novo RET mutations; approximately 50% of MEN2B diagnoses are the result of de novo RET mutations
Clinical Manifestations (Also See Chap. 20 )
-
♦
MEN2A
-
Medullary thyroid carcinoma (>95%)
-
Pheochromocytoma (50%)
-
-
♦
Hyperparathyroidism (parathyroid hyperplasia or adenoma) (20–30%)
-
♦
MEN2B
-
Same features as MEN2A but earlier onset (10 years; 100%)
-
Parathyroid dise ase is rare
-
Additional findings include
-
Mucosal neuromas (Fig. 13.11)
Fig. 13.11. 
Multiple endocrine neoplasia type 2b – mucosal ganglioneuroma.
-
Thickened corneal nerves
-
Marfanoid habitus (75%)
-
Gastr ointestinal involvement
-
-
-
♦
Familial medullary thyroid carcinoma (FMTC)
-
Medullary thyroid carcinoma only
-
More indolent than M EN2A or 2B
-

Molecular Basis of Disease
-
♦
Mutations in cysteine residues of extracellular binding domain in exons 10, 11, and 13–16 of the RET proto-oncogene account for most of the cases of MEN2A and FMTC; mutation s can occur in exon 5 and 8 but are rare
-
♦
A single point mutation in RET exon 16 (M918T) is the most common cause for MEN2B
-
♦
Mutations of codon 634Cys in exon 11 account for over 85% of MEN2A mutations
-
♦
Mutations within these exons of the RET proto-oncogene account for ~98% of MEN2A families, 95% of FMTC, and 98% of MEN2B families
-
♦
Strong genotype/phenotype correlations exist
Laboratory
-
♦
Mutations in RET
-
♦
Elevated calcitonin after stimulation
-
♦
Metanephr ines may be elevated if pheochromocytoma is present
-
♦
Parathyroid hormone and calcium elevation may also be present
Treatment/Surveillance
-
♦
Screening rec ommendations include genetic testing for at risk individuals
-
♦
For positive individuals, prophylactic thyroidectomy and annual screening for pheochromocytoma and hyperparathyroidism are indicated
-
♦
Those at risk fo r pheochromocytoma should avoid dopamine (D2) receptor antagonists
Peutz–Jeghers Syndrome
Chromosome and Gene Location
-
♦
19p13.3 (STK11, previously known as LKB1)
Inheritance
-
♦
Autosomal dominant
Incidence
-
♦
Estimate ranges from 1 in 25,000 to 1 in 280,000
Clinical Manifestations
-
♦
Numerous p igmented spots found on lips and buccal mucosa (more rarely on face, forearms hands, feet, and perianal area) (Fig. 13.12). Many fade over time, and some individuals do not have pigmentation
Fig. 13.12. 
Peutz–Jeghers syndrome with freckles on lips.
-
♦
Multiple gastrointestinal hamartomatous polyps found in the jejunum (malignant transformation not common) can also occur in the stomach and colon
-
♦
Most individuals present with intussusception, small bowel obstruction, or rectal bleeding
-
♦
Colon cancer risk estimated to be 39% by age 70 and breast cancer risk estimated to be 54% by age 65
-
♦
Cancers of the ovary, cervix, testis, pancreas, uterus, and lung also reported
-
♦
Risk of any malignancy estimated as 67–85% by age 70 years
-
♦
Clinical diagnosis may be made when any one of the following is present:
-
Two or more histologically confirmed PJS-type hamartomatous polyps
-
Any number of PJS-type polyps detected in one individual with a PJS family history
-
Characteristic mucocutan eous pigmentation in an individual with a PJS family history
-
Any number of PJS-type polyps in an individual who also has characteristic mucocutaneous pigmentation
-
Individual found to have STK11 mutation
-
-
♦
80–94% of those with clinical diagnosis will have identifiable STK11 mutation

Molecular Basis of Disease
-
♦
Disorder is due to mutations in the serine threonine kinase, STK11
-
♦
About 45% thought due to a de novo mutation, however, and may also be underdiagnosed within a family
Laboratory
-
♦
Polyps can have a unique cellular morphology imparting branching treelike appearance of mucosa with interdigitating smooth muscle
-
♦
Molecular testing available
Treatment/Surveillance
-
♦
CT or MRI of small bowel beginning at age 8–10, following up based upon findings, and then every 2–3 years beginning at age 18
-
♦
Colonoscopy and upper endoscopy every 2–3 years beginning in the late teens
-
♦
Removal of polyps if feasible to prevent small bowel intussusception
-
♦
Clinical breast exams every 6 months and annual mammography and breast MRI beginning at age 25
-
♦
MR cholangiopancreatography or endoscopic ultrasound every 1–2 years beginning at age 30–35
-
♦
Annual pelvic ultrasound and pap smear with consideration of transvaginal ultrasound beginning at 18–20 years
-
♦
Annual testic ular exam with observation for feminizing changes beginning at age 10
Retinoblastoma
Chromosome and Gene Location
-
♦
13q14.2 (RB1)
Inheritance
-
♦
Autosomal dominant with incomplete penetrance
-
♦
Approximately 65–75% are unilateral and sporadic with a mean diagnosis age of 24 months
-
♦
25–35% are bilateral a nd hereditary with a mean diagnosis age of 15 months
Incidence
-
♦
1 in 15,000–1 in 20,000
Clinical Manifestations
-
♦
Strabismus and/or leukocoria, multifocal, and bilateral tumors of the retina
-
♦
Other secondary cancers include:
-
Osteosar coma
-
Fibrosarcoma
-
Melanoma
-
Pineoblastoma
-
-
♦
May also be at an increased risk for lung, prostate, and breast cancer
Molecular Basis of Disease
-
♦
RB1 pro tein is involved in cell cycle and cell growth regulation
Laboratory
-
♦
Probability of detecting RB1 mutation in WBC DNA of index case will depend upon whether the tumors are bilateral or unilateral and whether there is positive family history or index case and the sensitivity of the testing method
-
♦
Deletions and point mutations are noted
-
♦
Chromosome analysis to detect large rearrangements and deletions
-
♦
Linkage analysis is available
Treatment/Surveillance
-
♦
Surgical options include enucleation and cryosurgery; additional options of chemotherapy and radiation also utilized
-
♦
Screening includ es ophthalmologic examination at birth and every 4–6 weeks for 6 months and then at increasing intervals (3–6 months) until age 7 and then every 6–12 months thereafter
Von Hippel–Lindau
Chromosome and Gene Location
-
♦
3p25.3 (VHL)
Inheritance
-
♦
Autosomal dominant
-
♦
Approximately 100% penetrant by age 65
Incidence
-
♦
1 in 36,000
-
♦
20% due to de novo mutations
Clinical Manifestations (Used Prior to Advent of Molecular Testing)
-
♦
Hemangioblastoma of the brain, spi nal cord, and retina:
-
Retinal hemangioblastomas (25–60%)
-
Cerebellar hemangioma (44–72%)
-
Spinal hemangiomas (13–50%)
-
-
♦
Pancreatic cysts (35–70%)
-
♦
Pancreatic neuroendocrine tumors
-
♦
Renal cysts
-
♦
Clear cell renal cell carcinoma (occurs in approximately 70% by age 60), leading cause of mortality
-
♦
Endolymphatic sac tumors and epididymal tumors are also common
-
♦
Four VHL disease phenotypes:
-
Type 1: low risk for pheochromocytoma, high risk for renal cell carcinoma
-
Type 2A: high risk for pheochromocytoma, low risk for renal cell carcinoma
-
Type 2B: high risk for pheochromocytoma, high risk for renal cell carcinoma
-
Type 2C: high risk for pheochromocytoma, very low risk for renal cell carcinoma
-
Molecular Basis of Disease
-
♦
Numerous deletions, insertions, nonsense, and missense mutations in VHL
-
♦
VHL is a t umor suppressor gene, the gene product functions to negatively regulate transcription
-
♦
Genotype/phenotype correlations
Laboratory
-
♦
Mutation analysis of VHL available
Treatment/Surveillance
-
♦
Annual eye a nd retinal ophthalmologic exam beginning at age 1
-
♦
Annual blood pressure evaluation at age 1
-
♦
Annual labs including catecholamines and metanephrines beginning at age 5
-
♦
Annual abdominal ultrasound beginning at age 16
-
♦
Complete audiologic assessment every 2–3 years, with MRI if recurrent ear infections, beginning at age 5
-
♦
Brain, cervic al spine, and abdominal MRI every 2 years beginning at age 16
Mitochondrial Disorders
-
♦
Mitoc hondria are cellular organelles, which generate energy for cellular processes by producing ATP by means of the electron transport chain and the oxidative phosphorylation system (the “respiratory chain”). The respiratory chain is located in the inner mitochondrial membrane. It consists of five multimeric protein complexes:
-
Reduced nicotinamide adenine dinucleotide (NADH) dehydrogenase–ubiquinone oxidoreductase (complex I, 45 subunits)
-
Succinate dehydrogenase–ubiquinone oxidoreductase (complex II, four subunits)
-
Ubiquinone–cytochrome c oxidoreductase (complex III, 11 subunits)
-
Cytochrome c oxidase (complex IV, 13 subunits)
-
ATP synthase (co mplex V, 16 subunits)
-
Chromosome and Gene Location
-
♦
Apart from the nucleus, mitochondria are the only cellular organelles that contain their own DNA (called mtDNA) for synthesizing RNA and proteins. Cells contain hundreds to thousands mitochondria, and each has approximately five mitochondrial genomes
-
♦
The mtDNA contains 37 genes, but most of the gene products (approximately 1,000) required for mitochondrial functioning are encoded by nuclear DNA (nDNA) and imported from the cytoplasm
-
♦
Mitochondrial DNA is a double-stranded circular molecule, which encodes 13 protein respiratory chain subunits and 24 RNAs (two ribosomal RNAs and 22 transfer RNAs) required within the mitochondria for translation of the protein-coding units
Inheritance
-
♦
Mitochondrial inheritance (mtDNA) (maternal inheritance)
-
Generally, all mitochondria in the z ygote are derived from the ovum. Therefore, a mother carrying an mtDNA mutation may pass it onto each of her children, but only her daughters will transmit it to their offspring. Most mtDNA-related diseases are maternally inherited, but there are exceptions. The occurrence of large deletions is almost always sporadic, probably taking place in oogenesis or early embryogenesis
-
Homoplasmy refers to the presence of all normal or all mutant mitochondrial DNA
-
Heteroplasmy: mtDNA pathogenic mutations are present in some, but not all mtDNA molecules in each cell. As a result, cells and tissues harbor both normal (wild type) and mutant mtDNA
-
A minimal number of mutant mtDNAs must be present before oxidative dysfunction occurs and clinical signs become apparent: threshold effect
-
During mitotic segregation, the ran dom redistribution of organelles during cell division might alter the proportion of mutant mtDNAs received by daughter cells; if the pathogenic threshold in a previously unaffected tissue is surpassed, the phenotype might also change. This underlies the age-related and tissue-related variability of clinical features in mtDNA-related diseases
-
-
♦
Mendelian inheritance
-
Most mitochondrial d ysfunction is due to alterations in nuclear genes and is inherited according to Mendelian laws with autosomal recessive pattern thought to be the most common
-
Incidence
-
♦
Mitochondrial diseases are not rare; incidence 1:10,000 live births
Clinical Manifestations
-
♦
Nearly all tissues depend on oxidative metabolism. Therefore, mitochondrial diseases are complex multisystem disorders, which include a variety of neurologic, ophthalmologic, cardiac, endocrine, gastrointestinal, renal, hematologic, audiologic, and pulmonary manifestations. Specific examples are provided below
Molecular Basis of Disease
-
♦
Mutations in mtDNA can affect specific structural respiratory chain proteins or the synthesis of all/m any mitochondrial proteins (mutations in RNA genes or mtDNA rearrangements)
-
♦
Examples of the clinical manifestation of various mutation types are provided below
Respiratory chain disorders due to defects in mtDNA
-
♦
Point mutations in protein-encoding genes
-
Neurogenic weakness, atax ia, and retinitis pigmentosa (NARP)
-
-
♦
Mutation in transfer RNA
-
Mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes (MELAS)
-
•
Seizures
-
•
Stroke-like episodes
-
•
Psychomotor regression
-
•
Weakness and exercise intolerance
-
•
Intestinal pseudo-obstruction
-
•
Sensorineural hearing loss
-
•
-
Myoclonic epilepsy with ragged-red fibe rs (MERRF)
-
•
Myoclonus
-
•
Seizures
-
•
Cerebellar ataxia
-
•
Mitochondrial myopathy
-
•
-
-
♦
Mutation in messenger RNA
-
Leber hereditary optic neuropathy (LHON)
-
•
Bilateral visual loss
-
•
Central scotomas
-
•
Abnormal c olor vision
-
•
-
-
♦
Mutation in ribosomal RNA
-
Aminoglycoside-induced non- syndromic deafness
-
-
♦
Rearrangements/del etions in mtDNA
-
Kearns–Sayre syndrome (KSS)
-
•
Chronic progressive external ophthalmoplegia
-
•
Ptosis
-
•
Pigmentary retinopathy
-
•
Heart block
-
•
Elevated CSF protein greater than 100 mg/dL
-
•
Ataxia
-
•
-
Nuclear-encoded mito chondrial diseases, which exhibit Mendelian inheritance (examples)
-
♦
Structural OXPHOS gene defects
-
Complex I deficiency
-
•
Leigh (−like) syndrome – NDUFS4 (NADH dehydrog enase (ubiquinone) Fe–S protein), NDUFS7, NDUFS8 genes
-
◦
Progressive central nervous system neurological deterioration
-
◦
Characteristic radiographic features with symmetric involvement of brainstem and basal ganglia
-
◦
-
•
Hypertrophic cardiomyopathy and encephalomyopathy – NDUFS2 gene
-
•
Macrocephaly, leukodystro phy, and myoclonic epilepsy – NDUFV1 gene
-
•
-
Complex II deficiency (all complex II enzymes are nuclear encoded [none are in the maternally inherited genome])
-
•
Leigh (−like) syndrome – flavoprotein (Fp) gene
-
•
-
-
♦
Nonstructural OXPHOS gen e defects
-
Intergenomic (mitochondrial and nuclear DNA) communication defects
-
•
Mitochondrial neurogastrointestinal encephalomyopathy syndrome (MNGIE)
-
◦
Mutations in thymidine phosphorylase gene
-
◦
Ophthalmoparesis
-
◦
Peripheral neuropathy
-
◦
Leukoencephalopathy
-
◦
Gastrointestinal dysmotility
-
◦
Myopathy
-
◦
-
•
-
Assembling d efects
-
•
Complex IV deficiencies (Leigh syndrome, mutations in SURF1 gene)
-
•
-
Homeostasis and import defects
-
•
Deafness–dystonia syndrome – DDP gene (involved in protein transport)
-
◦
X-linked recessive
-
◦
Sensorineural hearing loss
-
◦
Dystonia
-
◦
Dementia
-
◦
Ps ychotic features
-
◦
Optic atrophy
-
◦
-
•
-
Laboratory
-
♦
Biochemic al investigations – elevated serum and cerebrospinal fluid lactate
-
♦
Tissue evaluation – ragged-red fibers and ragged-blue cox negative fibers in skeletal muscle; defective oxidative phosphorylation enzyme activity on biopsied (affected) tissue
-
♦
Electromyography, nerve conduction studies – myopathic potentials; axonal and demyelinating peripheral neuropathy
-
♦
ECG and ECHO – cardiac conduction defects and cardiomyopathy
-
♦
Mitochondrial and nuclear gene mutations
Treatment
-
♦
Supportive/s ymptomatic
Suggested Reading
Online Resources
GeneReviews. http://www.geneclinics.org.
Online Mendelian Inheritance in Man (OMIM). http://www.ncbi.nlm.nih.gov.
General Genetic Resources
Jones KL. Smith’s recognizable patterns of human malformation. 6th ed. Philadelphia: Elsevier; 2006.
Rimoin DL, Connor JM, Pyeritz RE, Korf BR, editors. Emery and Rimoin’s principles and practice of medical genetics. 5th ed. New York: Churchill Livingstone; 2007.
Scriver CR, et al., editors. The metabolic and molecular basis of inherited disease. 8th ed. New York: McGraw-Hill; 2001.
Chromosomal Disorders
ACOG Practice Bulletin – Clinical Management Guidelines for Obstetrician-Gynecologists. ACOG Practice Bulletin No. 77: screening for fetal chromosomal abnormalities. Obstet Gynecol. 2007;109:217–27.
De Decker HP, Lawrenson JB. The 22q11.2 deletion: from diversity to a single gene theory. Genet Med. 2001;3:2–5.
Duker NJ. Chromosome breakage syndromes and cancer. Am J Med Genet. 2002;115:125–9.
Gardner RJ, Sutherland G. Chromosome abnormalities and genetic counseling. New York: Oxford University Press; 2004.
Horsthemke B, Wagstaff J. Mechanisms of imprinting of the Prader-Willi/Angelman region. Am J Med Genet A. 2008;146A:2041–52.
Joenje H, Patel KJ. The emerging genetic and molecular basis of Fanconi anaemia. Nat Rev Genet. 2001;2:446–57.
Mitchell EB, et al. Ready reference to common segmental aneusomies by syndromic names, major features and chromosomal locations. J Assoc Genet Technol. 2008;34:5–7.
Trinucleotide Repeat Disorders
Evidente VG, Gwinn-Hardy KA, Caviness JN, Gilman S. Hereditary ataxias. Mayo Clin Proc. 2000;75:475–90.
Shao J, Diamond MI. Polyglutamine diseases: emerging concepts in pathogenesis and therapy. Hum Mol Genet. 2007;16:R115–23.
Wenstrom KD. Fragile X, and other trinucleotide repeat diseases. Obstet Gynecol Clin North Am. 2002;29:367–88.
Neuromuscular Disorders
Ogino S, Wilson RB. Genetic testing and risk assessment for spinal muscular atrophy (SMA). Hum Genet. 2002;111:477–500.
Saifi GM, Szigeti K, Snipes GJ, et al. Molecular mechanisms, diagnosis, and rational approaches to management of and therapy for Charcot-Marie-Tooth disease and related peripheral neuropathies. J Invest Med. 2003;51:261–8.
Zatz M, de Paula F, Starling A, et al. The 10 autosomal recessive limb-girdle muscular dystrophies. Neuromuscul Disord. 2003;13:532–44.
Skeletal Disorders
Kimonis V, et al. Genetics of craniosynostosis. Semin Pediatr Neurol. 2007;14:150–61.
Valadares ER, Carneiro TB, Santos PM, et al. What is new in genetics and osteogenesis imperfecta classification? J Pediatr (RioJ). 2014;90:536–41.
Warman ML, Cormier-Daire V, Hall C, et al. Nosology and classification of genetic skeletal disorders: 2010 revision. Am J Med Genet A. 2011;115:943–68.
Wilkie AO, Patey SJ, Kan SH, et al. FGFs, their receptors, and human limb malformations: clinical and molecular correlations. Am J Med Genet. 2002;112:266–78.
Connective Tissue Disorders
Colige A, Nuytinck L, et al. Novel types of mutation responsible for the dermatosparactic type of Ehlers-Danlos syndrome (Type VIIC) and common polymorphisms in the ADAMTS2 gene. J Invest Dermatol. 2004;123:656–63.
Kashtan CE. Alport syndrome and thin basement membrane nephropathy. 2001 Aug 28 [Updated 2013 Feb 28]. In: Pagon RA, Adam MP, Ardinger HH, et al., editors. GeneReviews [Internet]. Seattle, WA: University of Washington, Seattle; 1993–2014. http://www.ncbi.nlm.nih.gov/books/NBK1207/.
Levy M, Feingold J. Estimating prevalence in single-gene kidney diseases progressing to renal failure. Kidney Int. 2000;58:925–43.
Loeys BL, Schwarze U, Holm T, et al. Aneurysm syndromes caused by mutations in the TGF-B receptor. N Engl J Med. 2006;355:788–98.
MacCarrick G, Black III J, et al. Loeys–Dietz syndrome: a primer for diagnosis and management. Genet Med. 2014;16:576–87.
Milewicz DM, Guo DC, Tran-Fandulu V, et al. Genetic basis of thoracic aortic aneurysms and dissections: focus on smooth muscle cell contractile dysfunction. Annu Rev Genomics Hum Genet. 2008;9:283–302.
Muller T, Mizumoto S, et al. Loss of dermatan sulfate epimerase (DSE) function results in musculocontractural Ehlers-Danlos syndrome. Hum Mol Genet. 2013;22:3761–72.
Paterick T, Humphries J, et al. Aortopathies: etiologies, genetics, differential diagnosis, prognosis and management. Am J Med. 2013;126:670–8.
Robin NH, Moran RT, Ala-Kokko L. Stickler syndrome. 2000 Jun 9 [Updated 2014 Sep 11]. In: Pagon RA, Adam MP, Ardinger HH, et al., editors. GeneReviews [Internet]. Seattle, WA: University of Washington, Seattle; 1993–2014. http://www.ncbi.nlm.nih.gov/books/NBK1302/.
Hematologic Disorders
Old JM. Screening and genetic diagnosis of haemoglobin disorders. Blood Rev. 2003;17:43–5.
Pruthi RK. Hemophilia: a practical approach to genetic testing. Mayo Clin Proc. 2005;80:1485–99.
Schrier SL. Pathophysiology of thalassemia. Curr Opin Hematol. 2002;9:123–6.
Thein SL. Genetic insights into the clinical diversity of beta thalassaemia. Br J Haematol. 2004;124:264–74.
Other Genetic Disorders
Coffee B, Keith K, Albizua I, Malone T, Mowrey J, Sherman SL, Warren ST. Incidence of fragile X syndrome by newborn screening for methylated FMR1 DNA. Am J Hum Genet. 2009;85:503–14.
Kulczycki LL, Kostuch M, Bellanti JA. A clinical perspective of cystic fibrosis and new genetic findings: relationship of CFTR mutations to genotype-phenotype manifestations. Am J Med Genet A. 2003;116A:262–7.
Roach ES, Sparagana SP. Diagnosis of tuberous sclerosis complex. J Child Neurol. 2004;19:643–9.
Rossi M, Perez-Lloret S, Doldan L, et al. Autosomal dominant cerebellar ataxias: a systematic review of clinical features. Eur J Neurol. 2004;21:607–15.
Suominen T, Bachinski LL, Auvinen S, et al. Population frequency of myotonic dystrophy: higher than expected frequency of myotonic dystrophy type 2 (DM2) mutation in Finland. Eur J Hum Genet. 2011;19:776–82.
Turner C, Hilton-Jones D. Myotonic dystrophy: diagnosis, management and new therapies. Curr Opin Neurol. 2014;27:599–606.
Udd B, Meola G, Krahe R, et al. 140th ENMC International Workshop: myotonic dystrophy DM2/PROMM and other myotonic dystrophies with guidelines on management. Neuromuscul Disord. 2006;16:403–13.
Watson MS, Cutting GR, Desnick RJ, et al. Cystic fibrosis population carrier screening: 2004 revision of American College of Medical Genetics mutation panel. Genet Med. 2004;6:387–91.
Metabolic Disorders: Inborn Errors of Metabolism
Blau N, Duran M, Gibson MK. Laboratory guide to the methods in biochemical genetics. Berlin: Springer; 2008.
Blau N, Duran M, Gibson KM, Dionisi-Vici C. Physician’s guide to the diagnosis, treatment, and follow-up of inherited metabolic diseases. Berlin: Springer; 2014.
Fearing MK, Levy HL. Expanded newborn screening using tandem mass spectrometry. Adv Pediatr. 2003;50:81–111.
Fernandes J, Saudubray JM, van den Berghe G, Walter JH, editors. Inborn metabolic diseases diagnosis and treatment. 4th ed. Heidelberg: Springer; 2006.
Rinaldo P, Lim JS, Tortorelli S, Gavrilov D, Matern D. Newborn screening of metabolic disorders: recent progress and future developments. Nestle Nutr Workshop Ser Paediatr Program. 2008;62:81–93. discussion 93–96.
Lysosomal Storage Diseases
Beck M. New therapeutic options for lysosomal storage disorders: enzyme replacement, small molecules and gene therapy. Hum Genet. 2007;121:1–22.
Staretz-Chacham O, Lang TC, LaMarca ME, Krasnewich D, Sidransky E. Lysosomal storage disorders in the newborn (review). Pediatrics. 2009;123:1191–207.
Familial Cancer Syndromes
Alsanea O, Clark OH. Familial thyroid cancer. Curr Opin Oncol. 2001;13:44–51.
Bausch B, Jilg C, Glasker S, et al. Renal cancer in von Hippel-Lindau disease and related syndromes. Nat Rev Nephrol. 2013;9:529–38.
Boardman LA. Heritable colorectal cancer syndromes: recognition and preventive management. Gastroenterol Clin North Am. 2002;31:1107–31.
Callender GG, Rich TA, Perrier ND. Multiple endocrine neoplasia syndromes. Surg Clin North Am. 2008;88:863–95. viii.
Classon M, Harlow E. The retinoblastoma tumour suppressor in development and cancer. Nat Rev Cancer. 2002;2:910–7.
Cohen SA, Leininger A. The genetic basis of Lynch syndrome and its implications for clinical practice and risk management. Appl Clin Genet. 2014;7:147–58.
Economopoulou P, Dimitriadis G, Psyrri A. Beyond BRCA: new hereditary breast cancer susceptibility genes. Cancer Treat Rev. 2015;41:1–8.
Eng C. PTEN: one gene, many syndromes. Hum Mutat. 2003;22:183–98.
Fleischmann C, Peto J, Cheadle J, Shah B, Sampson J, Houlston RS. Comprehensive analysis of the contribution of germline MYH variation to early-onset colorectal cancer. Int J Cancer. 2004;109:554–8.
Frank TS, Deffenbaugh AM, Reid JE, et al. Clinical characteristics of individuals with germline mutations in BRCA1 and BRCA2: analysis of 10,000 individuals. J Clin Oncol. 2002;20:1480–90.
Giardiello FM, Allen JI, Axilbund JE, et al. Guidelines on genetic evaluation and management of Lynch syndrome: a consensus statement by the US Multi-society Task Force on colorectal cancer. Am J Gastroenterol. 2014;109:1159–79.
Kuroda N, Furuya M, Nagashima Y, et al. Review of renal tumors associated with Birt-Hogg-Dube syndrome with focus on clinical and pathobiological aspects. Pol J Pathol. 2014;65:93–9.
Larsen MJ, Thomassen M, Gerdes AM, Kruse TA. Hereditary breast cancer: clinical, pathological and molecular characteristics. Breast Cancer (Auckl). 2014;8:145–55.
Leggett BA, Young JP, Barker M. Peutz-Jeghers syndrome: genetic screening. Expert Rev Anticancer Ther. 2003;3:518–24.
Lindor NM, McMaster ML, Lindor CJ, Greene MH, National Cancer Institute DoCPCO, Prevention Trials Research Group. Concise handbook of familial cancer susceptibility syndromes – second edition. J Natl Cancer Inst Monogr. 2008;1–93.
Markey K, Axel L, Ahnen D. Basic concepts for genetic testing in common hereditary colorectal cancer syndromes. Curr Gastroenterol Rep. 2002;4:404–13.
Mester J, Eng C. Cowden syndrome: recognizing and managing a not-so-rare hereditary cancer syndrome. J Surg Oncol. 2015;111:125–30.
Metzger R, Milas M. Inherited cancer syndromes and the thyroid: an update. Curr Opin Oncol. 2014;26:51–61.
Robbins DH, Itzkowitz SH. The molecular and genetic basis of colon cancer. Med Clin North Am. 2002;86:1467–95.
Sims KB. Von Hippel-Lindau disease: gene to bedside. Curr Opin Neurol. 2001;14:695–703.
Stoffel EM, Mangu PB, Gruber SB, et al. Hereditary colorectal cancer syndromes: American Society of Clinical Oncology Clinical Practice Guideline endorsement of the familial risk-colorectal cancer: European Society for Medical Oncology Clinical Practice Guidelines. J Clin Oncol. 2015;33:209–17.
Varley JM. Germline TP53 mutations and Li-Fraumeni syndrome. Hum Mutat. 2003;21:313–20.
Villani A, Tabori U, Schiffman J, et al. Biochemical and imaging surveillance in germline TP53 mutation carriers with Li-Fraumeni syndrome: a prospective observational study. Lancet Oncol. 2011;12:559–67.
Wooster R, Weber BL. Breast and ovarian cancer. N Engl J Med. 2003;348:2339–47.
Mitochondrial Disorders
DiMauro S, Schon EA. Mitochondrial respiratory-chain diseases. N Engl J Med. 2003;348:2656–68.
Haas RH, Parikh S, Falk MJ, et al. The in-depth evaluation of suspected mitochondrial disease. Mol Genet Metab. 2008;94:16–37.
Smeitink J, van den Heuvel L, DiMauro S. The genetics and pathology of oxidative phosphorylation. Nat Rev Genet. 2007;2:342–52.
Van den Heuvel L, Smeitink J. The oxidative phosphorylation (OXPHOS) system: nuclear genes and human genetic diseases. Bioessays. 2001;23:518–25.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Volk, A.K. et al. (2016). Human Genetic Disorders. In: Cheng, L., Bostwick, D. (eds) Essentials of Anatomic Pathology. Springer, Cham. https://doi.org/10.1007/978-3-319-23380-2_13
Download citation
DOI: https://doi.org/10.1007/978-3-319-23380-2_13
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-23379-6
Online ISBN: 978-3-319-23380-2
eBook Packages: MedicineMedicine (R0)