
Abstract
Heart failure (HF) is associated with significant morbidity and mortality attributable largely to structural changes in the heart and with associated cardiac dysfunction. Remodeling is defined as alteration of the mass, dimensions, or shape of the heart (termed cardiac or ventricular remodeling) and vessels (vascular remodeling) in response to hemodynamic load and/or cardiovascular injury in association with neurohormonal activation. Remodeling may be described as physiologic or pathologic; alternatively, remodeling may be classified as adaptive or maladaptive. The importance of remodeling as a pathogenic mechanism has been controversial because factors leading to remodeling as well as the remodeling itself may be major determinants of patients’ prognosis. The basic mechanisms of cardiovascular remodeling, and especially the roles of microRNAs in HF progression and vascular diseases, will be reviewed here.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
Introduction
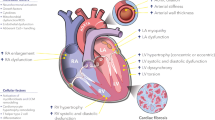
Cardiovascular disease is the leading cause of morbidity and mortality in developed countries. Cardiovascular remodeling is thought to be an important aspect of disease progression in heart failure (HF), regardless of cause. It is manifested clinically by changes in cardiac size, shape, and function in response to aging, cardiac injury, or increased load. The importance of remodeling as a pathogenic mechanism is not completely understood because the factors leading to remodeling have not been fully investigated. Generally, pathological processes of the heart are associated with an altered expression profile of genes that are important for cardiac function (Fig. 10.1) [1].

Pathological processes of the heart under stress
The regulation of cardiac gene expression is complex, with individual genes controlled by multiple transcription factors associated with their regulatory enhancer/promoter sequences to activate gene expression [2]. Moreover, epigenetic regulation of gene expression and alternative splicing mechanisms also further complicate the patterns of gene expression. microRNAs (miRNAs; miRs) have reshaped our view of how gene expression is regulated by adding another layer of regulation at the posttranscriptional level. Cardiovascular remodeling encompassed many pathologies including cardiac hypertrophy, myocardial ischemia/myocardial infarction (MI), cardiac fibrosis, arrhythmia, and vascular diseases that will be discussed in more detail in the following sections (Fig. 10.2).

Dysregulated microRNAs in cardiovascular disease
The implications of miRNA-derived regulation in cardiovascular pathology have only been recognized very recently, and research on miRNAs in relation to such diseases has now become a rapidly evolving field. In this chapter, we will summarize the current understanding of miRNA function in the pathogenesis of cardiovascular remodeling.
Cardiac Hypertrophy
Left ventricular hypertrophy is a common finding in patients with hypertension and it can be diagnosed either using an electrocardiogram or by echocardiography. Because cardiac hypertrophy, an increase in heart size, is associated with nearly all forms of HF, it is of great clinical importance that we understand the mechanisms responsible for cardiac hypertrophy. Therefore, the regulation of hypertrophy-associated genes has attracted great interest from many researchers.
Pathological hypertrophy is mainly caused by hypertension, loss of myocytes following ischemic damage, and genetic alterations that cause cardiomyopathy. Moreover, metabolic abnormality or neurohormonal activation can also lead to hypertrophy [3]. Pathological hypertrophy is the phenotypic endpoint that has been most studied in relation to miRNAs in the heart to date.
Clinical studies in human hearts have indicated that the fetal gene expression program is reactivated in pathologic hypertrophy and failing hearts, which results in a switching of structural proteins from adult to fetal isoforms [4]. It is well known that there is a decrease in the fast-shortening-velocity isoform (α-myosin heavy chain) coupled with an increase in the slow-shortening-velocity isoform (β-myosin heavy chain [β-MHC]) during the transition from cardiac hypertrophy to HF [5]. This contributes to the decrease in contractile function in failing human hearts. Interestingly, transcriptome analysis of failing and fetal hearts revealed a similar pattern of miRNA expression. More than 80 % of the deregulated miRNAs in failing hearts displayed a similar expression pattern as in fetal hearts, suggesting that reactivation of a fetal miRNA program may contribute to the gene expression pattern of failing hearts [6]. The most consistent changes were upregulation of miR-21, miR-29b, miR-129, miR-210, miR-211, miR-212, and miR-423, with downregulation of miR-30, miR-182, and miR-526. Interestingly, gene expression analysis revealed that most of the upregulated genes were characterized by the presence of a significant number of the predicted binding sites for downregulated miRNAs and vice versa.
In animal models of cardiac hypertrophy, whole arrays of miRNAs have indicated that separate miRNAs are upregulated, downregulated, or remain unchanged with respect to their levels in a normal heart [6–12]. In these studies, some miRNAs have been more frequently reported than others, indicating the possibility that these miRNAs might have common roles in hypertrophy pathogenesis.
Tissue-specific expression of miRNAs was first reported in 2002 [13]. It is known that there is a family of so-called myomiRs that are encoded within the introns of the separate myosin heavy chain genes. miR-208a, miR-208b, and miR-499 are located within the Myh6, Myh7, and Myh7b genes, respectively. It was reported that miR-208−/− mice show reduced cardiac hypertrophy in response to pressure overload [14]. Targets of miR-208a include thyroid hormone receptor-associated protein 1 [14, 15], suggesting that miR-208a initiates cardiomyocyte hypertrophy by regulating triiodothyronine-dependent repression of β-MHC expression. miR-27a also regulates β-MHC gene expression by targeting TRβ1 in cardiomyocytes [16]. Overexpression of miR-208a was sufficient to upregulate Myh7 and to elicit cardiac hypertrophy, resulting in systolic dysfunction [15]. Although miR-208a is required for cardiac hypertrophy, the role of miR-208b in these pathologic conditions remains to be elucidated. miR-499 is encoded in an intron of the myh7 gene and is considered likely to play a role in myosin gene regulation [17, 18].
miR-1 is also a cardiac and skeletal muscle-specific miRNA, and it is probably one of the most abundant miRNAs in the heart. It was reported to target a cytoskeletal regulatory protein, twinfilin 1 (Twf1) , which binds to actin monomers and prevents their assembly into filaments [19]. Downregulation of miR-1 induced by hypertrophic stimuli, such as transverse aortic constriction or α-adrenergic stimulation with phenylephrine, results in increased Twf1 expression, and overexpression of Twf1 is sufficient to induce cardiac hypertrophy. Another target of miR-1 is insulin-like growth factor (IGF-1), IGF-1 receptor [20], calmodulin 1 and 2, Mef2a [21], and sodium calcium exchanger [22]. Repression of miR-1 and upregulation of IGF-1 was also demonstrated in models of cardiac hypertrophy [20]. miR-1 is downregulated in patients with aortic stenosis [11] and acromegaly associated cardiac hypertrophy [20].
miR-1 is encoded by two bicistronic clusters—miR-133a-1/miR-1-2 and miR-133a-2/miR-1-1. As well as miR-1, miR-133 also has the potential to attenuate agonist-induced hypertrophy [23, 24], whereas repression of miR-133 sensitized the myocardium to excessive cardiac growth. Therefore, these clusters generate antagonizing effects on the stimulation of cardiac hypertrophy.
In contrast, miR-195 was sufficient to drive pathologic cardiac growth when overexpressed in neonatal cardiac myocytes and in transgenic mice [7]. These results suggested that miR-195 is a pro-hypertrophic factor that actively participates in the hypertrophic process; however, no direct targets of miR-195 have been reported in the context of cardiac hypertrophy.
Myocardial Ischemia and Cell Death
A rapidly increasing number of studies have shown that cardiac and circulating miRNAs are markedly altered in myocardial ischemia or MI. These novel findings shed new light on the mechanisms that lead to MI complications, post-MI ventricular remodeling, and cardiac repair. Furthermore, recent studies showed that circulating miRNAs may represent novel and sensitive biomarkers of MI and, possibly, also function as an intercellular signaling mechanism (see Chap. 7 of the volume “microRNA: Basic Science” for a detailed discussion of miRNA and cardiac regeneration).
Cardiomyocyte death/apoptosis is a key cellular event in ischemic hearts. There are miRNAs that have been shown to exert proapoptotic effects by targeting key cardioprotective proteins. It was found that miR-320 expression was consistently dysregulated in ischemic hearts [25]. Ren et al. identified heat-shock protein 20 (HSP20), a known cardioprotective protein, as a target of miR-320. Knockdown of endogenous miR-320 provided protection against cardiomyocyte apoptosis through the upregulation of HSP20. miR-34 family members promote growth arrest and apoptosis [26]. Therapeutic inhibition of miR-34 attenuated ischemia-induced remodeling and improved cardiac recovery [27]. One of the targets of miR-34 was shown to be a protein phosphatase 1 nuclear targeting subunit (Pnuts) [28].
On the other hand, there are a number of miRNAs that exert an antiapoptotic function by targeting important proapoptotic proteins. The miRNA expression signature in rat hearts at 6 h after MI revealed that miR-21 expression was significantly downregulated in infracted areas but was upregulated in border areas [29]. Adenoviral transfer of miR-21 in vivo decreased cell apoptosis in the border and infracted areas through its target gene, programmed cell death 4 (PDCD4), and the activator protein 1 (AP1) pathway. miR-24 also inhibited cardiomyocyte apoptosis via repression of the proapoptotic protein Bim [30]. Ex vivo miR-24 enrichment, together with miR-21 and miR-221, improved the therapeutic potential of cardiac progenitor cells upon transplantation in ischemic rodents [31]. Similarly, miR-499 and miR-30 family members diminished apoptosis in injured hearts by attenuating activation of dynamin-related protein-1 and thus inhibiting mitochondrial fission [32, 33].
Early reperfusion of the ischemic heart remains the most effective intervention for improving clinical outcomes after a MI. However, abnormal increases in intracellular Ca2+ during myocardial reperfusion can cause cardiomyocyte death, known as ischemia-reperfusion (I/R) injury. Cardiac I/R injury is also accompanied by dynamic changes in the expression of miRNAs; for example, miR-214 is upregulated during ischemic injury. Genetic deletion of miR-214 in mice caused a loss of cardiac contractility, increased apoptosis, and excessive fibrosis in response to I/R injury [34]. The cardioprotective roles of miR-214 during I/R injury were attributed to repression of the mRNA encoding sodium/calcium exchanger 1, a key regulator of Ca2+ influx; and to repression of several downstream effectors of Ca2+ signaling that mediate cell death. These results suggested a pivotal role for miR-214 as a regulator of cardiomyocyte Ca2+ homeostasis and survival during cardiac injury. Moreover, CaMKIIδ is a shared target of both miR-214 and miR-145 [35]. miR-145 concomitantly protects cardiomyocytes from reactive oxygen species by targeting Bnip3 [36]. Boosting miR-214 and miR-145 levels to attenuate Ca2+ overload and cardiac cell death may provide a valuable therapeutic benefit for the treatment or prevention of heart failure after I/R injury.
Recent studies have shown that some miRNAs are present in circulating blood and that they are included in exosomes and microparticles [37, 38]. Recently, results obtained in studies of cancer suggest that the profiles of blood circulating miRNAs might reflect the changes observed in cancerous tissue [39]. This concept has also proved valid in cardiovascular disease [40], and circulating specific miRNAs have been reported in patients with MI [41, 42]. Moreover, plasma levels of endothelial cell-enriched miRNAs, such as miR-126, miR-17, and miR-92a, inflammation-associated miR-155, and smooth muscle-enriched miR-145 were reported to be significantly reduced in coronary artery disease (CAD) patients compared with healthy controls. These results also indicated that they can be used as biomarker candidates for CAD [43]. Therefore, the source and the mechanism of the change determined the set of miRNAs that can be used for myocardial ischemia/MI.
Cardiac Fibrosis
Cardiac fibrosis is a major aspect of myocardial remodeling and an important contributor to the development of cardiac dysfunction in diverse pathologic conditions, such as MI, in ischemic, dilated, and hypertrophic cardiomyopathies, and HF [44–49]. The extracellular deposition of collagen by fibroblasts contributes to this adverse remodeling. Cardiac fibrosis leads to an increased mechanical stiffness, initially causing diastolic dysfunction, and eventually resulting in systolic dysfunction and overt HF. In addition, fibrosis can also disturb the electrical continuity between cardiomyocytes, leading to conduction slowing and hence an increase in the chance of arrhythmias. It is also possible that the enhanced diffusion distance for cardiac substrates and oxygen to cardiac myocytes, caused by fibrosis, negatively influences the myocardial balance between energy demand and supply [46, 47].
miR-21 is expressed in all cell types of the cardiovascular system, most prominently in cardiac fibroblasts but rather weakly in cardiomyocytes. Furthermore, miR-21 is among the most strongly upregulated miRNAs in response to a variety of forms of cardiac stress [7, 50, 51]. Thum et al. showed that miR-21 is upregulated in cardiac fibroblasts in the failing heart, where it represses the expression of Sprouty homolog 1 (SPRY1) , a negative regulator of the extracellular signal-regulated kinase/mitogen-activated protein kinase (ERK-MAPK) signaling pathway [52]. Upregulation of miR-21 in response to cardiac injury was shown to enhance ERK-MAPK signaling, leading to fibroblast proliferation and fibrosis. Moreover, miR-21-dependent targeting of SPRY1 and PDCD4 was shown to promote the fibroblastoid phenotype in epicardial-to-mesenchymal transition [53]. Phosphatase and tensin homologue (PTEN) has also been demonstrated to be a direct target of miR-21 in cardiac fibroblasts [54]. Previous reports characterized PTEN as a suppressor of matrix metalloprotease-2 (MMP-2) expression [55, 56]. I/R injury in the heart induced miR-21 in cardiac fibroblasts in the infracted region. Thus, I/R injury-induced miR-21-limited PTEN function and caused activation of the Akt pathway and increased MMP-2 expression in cardiac fibroblasts.
On the other hand, miR-29 family members, miR-133, and miR-30 directly downregulated key profibrotic proteins. The miR-29 family is composed of three members, miR-29a, b, and c. It was shown that the miR-29 family, which is highly expressed in fibroblasts, targets mRNAs encoding a multitude of extracellular matrix (ECM)-related proteins involved in fibrosis, including col1a1, col3a1, elastin, and fibrillin [50]. miR-29 was dramatically repressed in the border zone flanking the infracted area in a mouse model of MI. Downregulation of miR-29 would be predicted to counter the repression of these mRNAs and enhance fibrotic responses. Therefore, it is tempting to speculate that upregulation of miR-29 may be a therapeutic option for MI.
Connective tissue growth factor (CTGF) , a key molecule involved in fibrosis, was shown to be regulated by miR-133 and miR-30, which are both consistently downregulated in several models of pathologic hypertrophy and HF [57]. The authors indicated that miR-133 and miR-30 are downregulated during cardiac disease, which inversely correlated with the upregulation of CTGF. In vitro experiments designed to overexpress or inhibit these miRNAs can effectively repress CTGF expression by interacting directly with the 3′-untranslated region (UTR) region of the CTGF mRNA.
Together, these data indicate that miRNAs are important regulators of cardiac fibrosis and are involved in structural heart disease.
Arrhythmias
One of the earliest reports of involvement of miRNA regulation of cardiac repolarization came from Zhou et al. in 2007 with the targeted deletion of miR-1-2 in mice. Surface electrocardiography in mutant mice demonstrated reduced average heart rate, accelerated atrioventricular conduction, and slowed ventricular conduction [58]. They found Irx5 as a target for miR-1-2, which belongs to the Iroquois family of homeodomain-containing transcription factors that regulate cardiac repolarization by repressing transcription of KCND2. KCND2 encodes a K+ channel subunit (Kv4.2) responsible for the transient outward K+ current (Ito) that is the major determinant of the transmural repolarization gradient in the ventricular wall. The increase in Irx5 protein levels in miR-1-2 mutants corresponded with a decrease in KCND2 expression.
Additional evidence supporting a role for miR-1 in cardiac repolarization and arrhythmogenesis came from a rat model of MI induced by occlusion of the coronary artery. It was established that gap junction protein a1 (GJA1; encoding connexin43 [Cx43]) and potassium inwardly rectifying channel, subfamily J, member 2 (KCNJ2; encoding the K+ channel subunit Kir2.1) are target genes for miR-1 [59]. Cx43 is critical for inter-cell conductance of excitation [60–62], and Kir2.1 governs cardiac membrane potential [63, 64], both of which are important determinants for cardiac excitability.
To date, the cardiac ion channel genes that have been confirmed experimentally to be targets of miR-1 or miR-133 include GJA1/Cx43/IJ [59], KCNJ2/Kir2.1/IK1 [59], potassium voltage-gated channel, subfamily H (eag-related) member 2 (KCNH2)/human ether-à-go-go-related gene (HERG)/IKr [65], potassium voltage-gated channel, KQT-like subfamily, member 1 (KCNQ1)/KvLQT1/IKs [66], and potassium voltage-gated channel, Isk-related family, member 1 (KCNE1)/mink/IKs [66]. The fact that altered expression of miRNAs can deregulate expression of cardiac ion channels provided novel insight into the molecular understanding of cardiac excitability.
miR-212 has been found to be upregulated in both animal models and human HF [6]. KCNJ2/Kir2.1 3′-UTR contains potential miR-212 binding sites and transfection of HeLa cells with miR-212 reduced inward rectifier K+ current density, as demonstrated by whole-cell patch-clamp recordings [67].
It was also reported that miR-328 is upregulated in the atria of dogs with induced atrial fibrillation (AF) and targets the L-type calcium channel [68]. Strikingly, inhibition of miR-328 levels with an antagomir reversed the conditions. The fact that genetic knockdown of endogenous miR-328 reduced AF vulnerability also suggests the potential of miR-328 as a target for AF treatment.
Circulating miRNAs, which can be potential biomarkers for AF, were also sought. Plasma miR-150 levels from AF patients were substantially lower than that from healthy people in a cohort of 105 participants [69]. miRNAs may serve as molecular diagnostic markers for AF in the future.
Angiogenesis and Vascular Disease
miRNAs are also important in vascular development, physiology, and disease. Initial evidence for the functional roles of miRNAs in vascular development was provided by the observation that mice carrying a Dicer hypomorphic allele died prenatally with severely disrupted blood vessel formation [70].
Profiling of endothelially expressed miRNAs has been performed using human umbilical vein endothelial cells. These results revealed high expression levels of miR-221/222, miR-21, let-7 family, miR-17-92 cluster, miR-23-24 cluster, and miR-126 in vascular endothelial cells. Among them, miR-126 is the only miRNA considered to be expressed specifically in endothelial cells [71].
Studies focusing on individual miRNAs or miRNA clusters suggested the importance of miRNAs in endothelial cell function and angiogenesis. The miR-17-92 cluster is one of the most important miRNAs for the regulation of angiogenesis. It encodes six miRNAs (miR-17, miR-18a, miR-19a, miR-20a, miR-19b-1, and miR-92-1), which are tightly grouped within an 800 base-pair region, and it is transcriptionally regulated by c-Myc [72]. In particular, miR-18 preferentially suppressed CTGF, whereas miR-19 targeted the potent angiogenesis-inhibitor thrombospondin-1 to promote tumor angiogenesis [73]. On the other hand, miR-92a controlled the growth of new blood vessels (angiogenesis) [74]. Forced overexpression of miR-92a in endothelial cells blocked angiogenesis, and systemic administration of an antagomir to inhibit miR-92a led to enhanced blood vessel growth and functional recovery of damaged tissue in mouse models of limb ischemia and MI. Therefore, miR-92a may serve as a valuable therapeutic target in the setting of ischemic disease.
miR-126 is an abundant, endothelial cell-enriched miRNA that is encoded in the second intron of an endothelial cell-specific gene, Egfl7, and mechano-sensitive zinc finger transcription factor Klf2a was shown to induce miR-126 expression to activate vascular endothelial growth factor signaling [75]. This work described a novel genetic mechanism in which a miRNA facilitated integration of a physiological stimulus with growth factor signaling in endothelial cells to guide angiogenesis. On the other hand, transfection of endothelial cells with an oligonucleotide that decreased miR-126 permitted an increase in tumor necrosis factor-α stimulated vascular adhesion molecule 1 expression and increased leukocyte adherence to endothelial cells [76]. The apparent role of miR-126 in angiogenesis has led to increasing interest in miR-126 overexpression as a therapeutic approach. It has been reported that systemic delivery of miR-126 by miRNA-loaded bubble liposomes improved blood flow and may be useful for the treatment of hind-limb ischemia [77].
There is increasing evidence that specific miRNAs are involved in angiogenesis. So far, pro-angiogenic miRNAs include let7f and miR-27b [78], miR-17-92 cluster [73], miR-126 [79, 80], miR-130a [81], miR-210, and miR-378 [82, 83]. miRNAs that exert anti-angiogenic effects include miR-15/16 [84, 85], miR-20a/b [84], miR-92a [74], and miR-221/222 [86, 87].
In the context of vascular remodeling, Ji et al. identified miRNAs that are aberrantly expressed in the vascular walls after balloon injury [88]. Modulating an aberrantly overexpressed miR-21 via antisense-mediated depletion had a significant negative effect on neointimal lesion formation. They also demonstrated that Pten and Bcl2 were involved in miR-21-mediated cellular effects. The same group also revealed that miR-221 and miR-222 expression levels were elevated in rat carotid arteries after angioplasty [89]. Moreover, they found that p27 (Kip1) and p57 (Kip2) were target genes involved in miR-221- and miR-222-mediated effects on vascular smooth muscle cell (VSMC) growth. Knockdown of miR-221 and miR-222 resulted in decreased VSMC proliferation both in vitro and in vivo.
miR-145 is selectively expressed in VSMCs of the vascular wall, and its expression was significantly downregulated in vascular walls with neointimal lesion formation. The target of miR-145 is KLF5 and its downstream signaling molecule, myocardin. Restoration of miR-145 in balloon-injured arteries via Ad-miR-145 inhibited neointimal growth and might be used for treatment of a variety of proliferative vascular disorders.
Aortic aneurysms are a common clinical condition that can cause death due to aortic dissection or rupture. The association between aortic aneurysm pathogenesis and altered TGF-β signaling, inflammation and apoptosis has been the subject of numerous investigations. Recently, a TGF-β-responsive miR-29 [90, 91] and miR-21 [92] whose targets include Pten, Spry1, Pdcd4, and Bcl2 have been identified to play roles in cellular phenotypic modulation during aortic development. It was demonstrated that decreasing the levels of miR-29b or increasing the levels of miR-21 in the aortic wall could attenuate aortic aneurysm progression in a porcine pancreatic elastase infusion and angiotensin II infusion model of abdominal aortic aneurysms in mice [90, 92].
Heart Failure
Because all of the previously described pathologies, i.e. cardiac hypertrophy, fibrosis, arrhythmia, and CAD can cause HF, all of the miRNAs discussed so far are also relevant to this disease entity.
Many profiling studies have been conducted and revealed a large number of miRNAs that are differentially expressed in HF, pointing to a new mode of regulation of cardiovascular diseases [9, 11, 12, 21, 57, 93].
A diverse range of circulating miRNAs have been studied for the detection of HF. Tijsen et al. tried to determine whether miRNAs make it possible to distinguish clinical HF not only from healthy controls but also from non-HF forms of dyspnea [40]. They revealed that miR423-5p was most strongly related to the clinical diagnosis of HF and receiver-operator-characteristics curve analysis showed miR423-5p to be a diagnostic predictor of HF, with an area under the curve of 0.91 (p < 0.001).
From a diagnostic perspective, Goren et al. tried to evaluate a multimarker approach to HF diagnosis [94]. They measured the levels of 186 miRNAs in the sera of 30 stable chronic systolic HF patients and 30 controls. The differences in miRNA levels between the two groups were characterized, and a score, based on the levels of four specific miRNAs with the most significant increase in the HF group (miR-423-5p, miR-320a, miR-22, and miR-92b) was defined. Interestingly, the score was utilized to discriminate HF patients from controls with a sensitivity and specificity of 90 %. Moreover, in the HF group, there was a significant association between the score and important clinical parameters such as elevated serum natriuretic peptide levels, a wide QRS, and dilatation of the left ventricle and left atrium. These results suggested that a multimarker approach is useful for the detection of not only HF but also left ventricular structure and function.
miRNAs are also related to a more specific cause of HF, such as chemotherapy-induced HF or obesity-related HF. It has been proposed that miRNAs can exert their roles in response to treatment with chemotherapeutic agents. For example, it was suggested that upregulation of miR-146a after doxorubicin (Dox) treatment is involved in acute Dox-induced cardiotoxicity by targeting ErbB4 [95]. Inhibition of both ErbB2 and ErbB4 signaling may be one of the reasons why those patients who receive concurrent therapy with Dox and trastuzumab suffer from HF.
miRNA microarray analyses and real-time polymerase chain reaction have revealed that miR-451 levels were significantly increased in type 2 diabetes mellitus mouse hearts [96]. Calcium-binding protein 39 (Cab39) is a scaffold protein of liver kinase B1 (LKB1), an upstream kinase of AMP-activated protein kinase (AMPK). Cab39 is a direct target of miR-451 in neonatal rat cardiac myocytes, and Cab39 overexpression rescued lipotoxicity. Protein levels of Cab39 and phosphorylated AMPK were increased and phosphorylated mammalian target of rapamycin was reduced in cardiomyocyte-specific miR-451 knockout mouse hearts compared with control mouse hearts. Thus, these results demonstrated that miR-451 is involved in diabetic cardiomyopathy through suppression of the LKB1/AMPK pathway.
Conclusion
miRNAs have emerged as powerful and dynamic modifiers of cardiovascular diseases. The miRNA species discussed above are able to directly regulate the expression of transcription factors, signaling molecules, contractile proteins, and play critical roles in cardiovascular remodeling. Work from several investigators have demonstrated the ability of exogenously administered miRNA inhibitors or miRNA mimics to modulate these pathological processes, thereby ameliorating cardiovascular diseases, which is promising and potentially opens the door for novel therapeutic approaches in the future. The potential of circulating miRNAs as biomarkers for cardiovascular diseases is also in its early stages. Their roles as prognostic biomarkers have yet to be elucidated, and larger studies with longer follow-up periods will be needed.
References
Kairouz V, Lipskaia L, Hajjar RJ, Chemaly ER. Molecular targets in heart failure gene therapy: current controversies and translational perspectives. Ann N Y Acad Sci. 2012;1254:42–50.
Olson EN. Gene regulatory networks in the evolution and development of the heart. Science. 2006;313:1922–7.
Rajabi M, Kassiotis C, Razeghi P, Taegtmeyer H. Return to the fetal gene program protects the stressed heart: a strong hypothesis. Heart Fail Rev. 2007;12:331–43.
Nakao K, Minobe W, Roden R, Bristow MR, Leinwand LA. Myosin heavy chain gene expression in human heart failure. J Clin Invest. 1997;100:2362–70.
Miyata S, Minobe W, Bristow MR, Leinwand LA. Myosin heavy chain isoform expression in the failing and nonfailing human heart. Circ Res. 2000;86:386–90.
Thum T, Galuppo P, Wolf C, Fiedler J, Kneitz S, van Laake LW, Doevendans PA, Mummery CL, Borlak J, Haverich A, Gross C, Engelhardt S, Ertl G, Bauersachs J. MicroRNAs in the human heart: a clue to fetal gene reprogramming in heart failure. Circulation. 2007;116:258–67.
van Rooij E, Sutherland LB, Liu N, Williams AH, McAnally J, Gerard RD, Richardson JA, Olson EN. A signature pattern of stress-responsive microRNAs that can evoke cardiac hypertrophy and heart failure. Proc Natl Acad Sci U S A. 2006;103:18255–60.
Chen JF, Murchison EP, Tang R, Callis TE, Tatsuguchi M, Deng Z, Rojas M, Hammond SM, Schneider MD, Selzman CH, Meissner G, Patterson C, Hannon GJ, Wang DZ. Targeted deletion of Dicer in the heart leads to dilated cardiomyopathy and heart failure. Proc Natl Acad Sci U S A. 2008;105:2111–6.
Cheng Y, Ji R, Yue J, Yang J, Liu X, Chen H, Dean DB, Zhang C. MicroRNAs are aberrantly expressed in hypertrophic heart: do they play a role in cardiac hypertrophy? Am J Pathol. 2007;170:1831–40.
Sayed D, Hong C, Chen IY, Lypowy J, Abdellatif M. MicroRNAs play an essential role in the development of cardiac hypertrophy. Circ Res. 2007;100:416–24.
Ikeda S, Kong SW, Lu J, Bisping E, Zhang H, Allen PD, Golub TR, Pieske B, Pu WT. Altered microRNA expression in human heart disease. Physiol Genomics. 2007;31:367–73.
Tatsuguchi M, Seok HY, Callis TE, Thomson JM, Chen JF, Newman M, Rojas M, Hammond SM, Wang DZ. Expression of microRNAs is dynamically regulated during cardiomyocyte hypertrophy. J Mol Cell Cardiol. 2007;42:1137–41.
Lagos-Quintana M, Rauhut R, Yalcin A, Meyer J, Lendeckel W, Tuschl T. Identification of tissue-specific microRNAs from mouse. Curr Biol. 2002;12:735–9.
van Rooij E, Sutherland LB, Qi X, Richardson JA, Hill J, Olson EN. Control of stress-dependent cardiac growth and gene expression by a microRNA. Science. 2007;316:575–9.
Callis TE, Pandya K, Seok HY, Tang RH, Tatsuguchi M, Huang ZP, Chen JF, Deng Z, Gunn B, Shumate J, Willis MS, Selzman CH, Wang DZ. MicroRNA-208a is a regulator of cardiac hypertrophy and conduction in mice. J Clin Invest. 2009;119:2772–86.
Nishi H, Ono K, Horie T, Nagao K, Kinoshita M, Kuwabara Y, Watanabe S, Takaya T, Tamaki Y, Takanabe-Mori R, Wada H, Hasegawa K, Iwanaga Y, Kawamura T, Kita T, Kimura T. MicroRNA-27a regulates beta cardiac myosin heavy chain gene expression by targeting thyroid hormone receptor {beta}1 in neonatal rat ventricular myocytes. Mol Cell Biol. 2011;31:744–55.
van Rooij E, Quiat D, Johnson BA, Sutherland LB, Qi X, Richardson JA, Kelm Jr RJ, Olson EN. A family of microRNAs encoded by myosin genes governs myosin expression and muscle performance. Dev Cell. 2009;17:662–73.
Bell ML, Buvoli M, Leinwand LA. Uncoupling of expression of an intronic microRNA and its myosin host gene by exon skipping. Mol Cell Biol. 2010;30:1937–45.
Li Q, Song XW, Zou J, Wang GK, Kremneva E, Li XQ, Zhu N, Sun T, Lappalainen P, Yuan WJ, Qin YW, Jing Q. Attenuation of microRNA-1 derepresses the cytoskeleton regulatory protein twinfilin-1 to provoke cardiac hypertrophy. J Cell Sci. 2010;123:2444–52.
Elia L, Contu R, Quintavalle M, Varrone F, Chimenti C, Russo MA, Cimino V, De Marinis L, Frustaci A, Catalucci D, Condorelli G. Reciprocal regulation of microRNA-1 and insulin-like growth factor-1 signal transduction cascade in cardiac and skeletal muscle in physiological and pathological conditions. Circulation. 2009;120:2377–85.
Ikeda S, He A, Kong SW, Lu J, Bejar R, Bodyak N, Lee KH, Ma Q, Kang PM, Golub TR, Pu WT. MicroRNA-1 negatively regulates expression of the hypertrophy-associated calmodulin and Mef2a genes. Mol Cell Biol. 2009;29:2193–204.
Kumarswamy R, Lyon AR, Volkmann I, Mills AM, Bretthauer J, Pahuja A, Geers-Knorr C, Kraft T, Hajjar RJ, Macleod KT, Harding SE, Thum T. SERCA2a gene therapy restores microRNA-1 expression in heart failure via an Akt/FoxO3A-dependent pathway. Eur Heart J. 2012;33:1067–75.
Care A, Catalucci D, Felicetti F, Bonci D, Addario A, Gallo P, Bang ML, Segnalini P, Gu Y, Dalton ND, Elia L, Latronico MV, Hoydal M, Autore C, Russo MA, Dorn 2nd GW, Ellingsen O, Ruiz-Lozano P, Peterson KL, Croce CM, Peschle C, Condorelli G. MicroRNA-133 controls cardiac hypertrophy. Nat Med. 2007;13:613–8.
Matkovich SJ, Wang W, Tu Y, Eschenbacher WH, Dorn LE, Condorelli G, Diwan A, Nerbonne JM, Dorn 2nd GW. MicroRNA-133a protects against myocardial fibrosis and modulates electrical repolarization without affecting hypertrophy in pressure-overloaded adult hearts. Circ Res. 2010;106:166–75.
Ren XP, Wu J, Wang X, Sartor MA, Qian J, Jones K, Nicolaou P, Pritchard TJ, Fan GC. MicroRNA-320 is involved in the regulation of cardiac ischemia/reperfusion injury by targeting heat-shock protein 20. Circulation. 2009;119:2357–66.
Hermeking H. p53 enters the microRNA world. Cancer Cell. 2007;12:414–8.
Bernardo BC, Gao XM, Winbanks CE, Boey EJ, Tham YK, Kiriazis H, Gregorevic P, Obad S, Kauppinen S, Du XJ, Lin RC, McMullen JR. Therapeutic inhibition of the miR-34 family attenuates pathological cardiac remodeling and improves heart function. Proc Natl Acad Sci U S A. 2012;109:17615–20.
Boon RA, Iekushi K, Lechner S, Seeger T, Fischer A, Heydt S, Kaluza D, Treguer K, Carmona G, Bonauer A, Horrevoets AJ, Didier N, Girmatsion Z, Biliczki P, Ehrlich JR, Katus HA, Muller OJ, Potente M, Zeiher AM, Hermeking H, Dimmeler S. MicroRNA-34a regulates cardiac ageing and function. Nature. 2013;495:107–10.
Dong S, Cheng Y, Yang J, Li J, Liu X, Wang X, Wang D, Krall TJ, Delphin ES, Zhang C. MicroRNA expression signature and the role of microRNA-21 in the early phase of acute myocardial infarction. J Biol Chem. 2009;284(43):29514–25.
Qian L, Van Laake LW, Huang Y, Liu S, Wendland MF, Srivastava D. miR-24 inhibits apoptosis and represses Bim in mouse cardiomyocytes. J Exp Med. 2011;208:549–60.
Hu S, Huang M, Nguyen PK, Gong Y, Li Z, Jia F, Lan F, Liu J, Nag D, Robbins RC, Wu JC. Novel microRNA prosurvival cocktail for improving engraftment and function of cardiac progenitor cell transplantation. Circulation. 2011;124:S27–34.
Wang JX, Jiao JQ, Li Q, Long B, Wang K, Liu JP, Li YR, Li PF. miR-499 regulates mitochondrial dynamics by targeting calcineurin and dynamin-related protein-1. Nat Med. 2011;17:71–8.
Li J, Donath S, Li Y, Qin D, Prabhakar BS, Li P. miR-30 regulates mitochondrial fission through targeting p53 and the dynamin-related protein-1 pathway. PLoS Genet. 2010;6, e1000795.
Aurora AB, Mahmoud AI, Luo X, Johnson BA, van Rooij E, Matsuzaki S, Humphries KM, Hill JA, Bassel-Duby R, Sadek HA, Olson EN. MicroRNA-214 protects the mouse heart from ischemic injury by controlling Ca(2)(+) overload and cell death. J Clin Invest. 2012;122:1222–32.
Cha MJ, Jang JK, Ham O, Song BW, Lee SY, Lee CY, Park JH, Lee J, Seo HH, Choi E, Jeon WM, Hwang HJ, Shin HT, Hwang KC. MicroRNA-145 suppresses ROS-induced Ca2+ overload of cardiomyocytes by targeting CaMKIIdelta. Biochem Biophys Res Commun. 2013;435:720–6.
Li R, Yan G, Li Q, Sun H, Hu Y, Sun J, Xu B. MicroRNA-145 protects cardiomyocytes against hydrogen peroxide (H(2)O(2))-induced apoptosis through targeting the mitochondria apoptotic pathway. PLoS One. 2012;7, e44907.
Valadi H, Ekstrom K, Bossios A, Sjostrand M, Lee JJ, Lotvall JO. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol. 2007;9:654–9.
Hunter MP, Ismail N, Zhang X, Aguda BD, Lee EJ, Yu L, Xiao T, Schafer J, Lee ML, Schmittgen TD, Nana-Sinkam SP, Jarjoura D, Marsh CB. Detection of microRNA expression in human peripheral blood microvesicles. PLoS One. 2008;3, e3694.
Mitchell PS, Parkin RK, Kroh EM, Fritz BR, Wyman SK, Pogosova-Agadjanyan EL, Peterson A, Noteboom J, O’Briant KC, Allen A, Lin DW, Urban N, Drescher CW, Knudsen BS, Stirewalt DL, Gentleman R, Vessella RL, Nelson PS, Martin DB, Tewari M. Circulating microRNAs as stable blood-based markers for cancer detection. Proc Natl Acad Sci U S A. 2008;105:10513–8.
Tijsen AJ, Creemers EE, Moerland PD, de Windt LJ, van der Wal AC, Kok WE, Pinto YM. MiR423-5p as a circulating biomarker for heart failure. Circ Res. 2010;106:1035–9.
Wang GK, Zhu JQ, Zhang JT, Li Q, Li Y, He J, Qin YW, Jing Q. Circulating microRNA: a novel potential biomarker for early diagnosis of acute myocardial infarction in humans. Eur Heart J. 2010;31:659–66.
Kuwabara Y, Ono K, Horie T, Nishi H, Nagao K, Kinoshita M, Watanabe S, Baba O, Kojima Y, Shizuta S, Imai M, Tamura T, Kita T, Kimura T. Increased microRNA-1 and microRNA-133a levels in serum of patients with cardiovascular disease indicate myocardial damage. Circ Cardiovasc Genet. 2011;4:446–54.
Fichtlscherer S, De Rosa S, Fox H, Schwietz T, Fischer A, Liebetrau C, Weber M, Hamm CW, Roxe T, Muller-Ardogan M, Bonauer A, Zeiher AM, Dimmeler S. Circulating microRNAs in patients with coronary artery disease. Circ Res. 2010;107:677–84.
Rossi MA. Pathologic fibrosis and connective tissue matrix in left ventricular hypertrophy due to chronic arterial hypertension in humans. J Hypertens. 1998;16:1031–41.
Swynghedauw B. Molecular mechanisms of myocardial remodeling. Physiol Rev. 1999;79:215–62.
Manabe I, Shindo T, Nagai R. Gene expression in fibroblasts and fibrosis: involvement in cardiac hypertrophy. Circ Res. 2002;91:1103–13.
Brown RD, Ambler SK, Mitchell MD, Long CS. The cardiac fibroblast: therapeutic target in myocardial remodeling and failure. Annu Rev Pharmacol Toxicol. 2005;45:657–87.
Khan R, Sheppard R. Fibrosis in heart disease: understanding the role of transforming growth factor-beta in cardiomyopathy, valvular disease and arrhythmia. Immunology. 2006;118:10–24.
Martos R, Baugh J, Ledwidge M, O’Loughlin C, Conlon C, Patle A, Donnelly SC, McDonald K. Diastolic heart failure: evidence of increased myocardial collagen turnover linked to diastolic dysfunction. Circulation. 2007;115:888–95.
van Rooij E, Sutherland LB, Thatcher JE, DiMaio JM, Naseem RH, Marshall WS, Hill JA, Olson EN. Dysregulation of microRNAs after myocardial infarction reveals a role of miR-29 in cardiac fibrosis. Proc Natl Acad Sci U S A. 2008;105:13027–32.
Ichimura A. miRNAs and regulation of cell signaling. FEBS J. 2011;278(10):1610–8.
Thum T, Gross C, Fiedler J, Fischer T, Kissler S, Bussen M, Galuppo P, Just S, Rottbauer W, Frantz S, Castoldi M, Soutschek J, Koteliansky V, Rosenwald A, Basson MA, Licht JD, Pena JT, Rouhanifard SH, Muckenthaler MU, Tuschl T, Martin GR, Bauersachs J, Engelhardt S. MicroRNA-21 contributes to myocardial disease by stimulating MAP kinase signalling in fibroblasts. Nature. 2008;456:980–4.
Bronnum H, Andersen DC, Schneider M, Sandberg MB, Eskildsen T, Nielsen SB, Kalluri R, Sheikh SP. miR-21 promotes fibrogenic epithelial-to-mesenchymal transition of epicardial mesothelial cells involving programmed cell death 4 and sprouty-1. PLoS One. 2013;8, e56280.
Roy S, Khanna S, Hussain SR, Biswas S, Azad A, Rink C, Gnyawali S, Shilo S, Nuovo GJ, Sen CK. MicroRNA expression in response to murine myocardial infarction: miR-21 regulates fibroblast metalloprotease-2 via phosphatase and tensin homologue. Cardiovasc Res. 2009;82:21–9.
Park MJ, Kim MS, Park IC, Kang HS, Yoo H, Park SH, Rhee CH, Hong SI, Lee SH. PTEN suppresses hyaluronic acid-induced matrix metalloproteinase-9 expression in U87MG glioblastoma cells through focal adhesion kinase dephosphorylation. Cancer Res. 2002;62:6318–22.
Zheng H, Takahashi H, Murai Y, Cui Z, Nomoto K, Niwa H, Tsuneyama K, Takano Y. Expressions of MMP-2, MMP-9 and VEGF are closely linked to growth, invasion, metastasis and angiogenesis of gastric carcinoma. Anticancer Res. 2006;26:3579–83.
Duisters RF, Tijsen AJ, Schroen B, Leenders JJ, Lentink V, van der Made I, Herias V, van Leeuwen RE, Schellings MW, Barenbrug P, Maessen JG, Heymans S, Pinto YM, Creemers EE. miR-133 and miR-30 regulate connective tissue growth factor: implications for a role of microRNAs in myocardial matrix remodeling. Circ Res. 2009;104:170–8. 6p following 8.
Zhao Y, Ransom JF, Li A, Vedantham V, von Drehle M, Muth AN, Tsuchihashi T, McManus MT, Schwartz RJ, Srivastava D. Dysregulation of cardiogenesis, cardiac conduction, and cell cycle in mice lacking miRNA-1-2. Cell. 2007;129:303–17.
Yang B, Lin H, Xiao J, Lu Y, Luo X, Li B, Zhang Y, Xu C, Bai Y, Wang H, Chen G, Wang Z. The muscle-specific microRNA miR-1 regulates cardiac arrhythmogenic potential by targeting GJA1 and KCNJ2. Nat Med. 2007;13:486–91.
Jongsma HJ. Diversity of gap junctional proteins: does it play a role in cardiac excitation? J Cardiovasc Electrophysiol. 2000;11:228–30.
Saffitz JE, Laing JG, Yamada KA. Connexin expression and turnover: implications for cardiac excitability. Circ Res. 2000;86:723–8.
Lerner DL, Yamada KA, Schuessler RB, Saffitz JE. Accelerated onset and increased incidence of ventricular arrhythmias induced by ischemia in Cx43-deficient mice. Circulation. 2000;101:547–52.
Diaz RJ, Zobel C, Cho HC, Batthish M, Hinek A, Backx PH, Wilson GJ. Selective inhibition of inward rectifier K+ channels (Kir2.1 or Kir2.2) abolishes protection by ischemic preconditioning in rabbit ventricular cardiomyocytes. Circ Res. 2004;95:325–32.
Wang Z, Yue L, White M, Pelletier G, Nattel S. Differential distribution of inward rectifier potassium channel transcripts in human atrium versus ventricle. Circulation. 1998;98:2422–8.
Xiao J, Luo X, Lin H, Zhang Y, Lu Y, Wang N, Yang B, Wang Z. MicroRNA miR-133 represses HERG K+ channel expression contributing to QT prolongation in diabetic hearts. J Biol Chem. 2007;282:12363–7.
Luo X, Xiao J, Lin H, Li B, Lu Y, Yang B, Wang Z. Transcriptional activation by stimulating protein 1 and post-transcriptional repression by muscle-specific microRNAs of IKs-encoding genes and potential implications in regional heterogeneity of their expressions. J Cell Physiol. 2007;212:358–67.
Goldoni D, Yarham JM, McGahon MK, O’Connor A, Guduric-Fuchs J, Edgar K, McDonald DM, Simpson DA, Collins A. A novel dual-fluorescence strategy for functionally validating microRNA targets in 3′ untranslated regions: regulation of the inward rectifier potassium channel K(ir)2.1 by miR-212. Biochem J. 2012;448:103–13.
Lu Y, Zhang Y, Wang N, Pan Z, Gao X, Zhang F, Shan H, Luo X, Bai Y, Sun L, Song W, Xu C, Wang Z, Yang B. MicroRNA-328 contributes to adverse electrical remodeling in atrial fibrillation. Circulation. 2010;122:2378–87.
Liu Z, Zhou C, Liu Y, Wang S, Ye P, Miao X, Xia J. The expression levels of plasma micoRNAs in atrial fibrillation patients. PLoS One. 2012;7, e44906.
Yang WJ, Yang DD, Na S, Sandusky GE, Zhang Q, Zhao G. Dicer is required for embryonic angiogenesis during mouse development. J Biol Chem. 2005;280:9330–5.
Wang S, Olson EN. AngiomiRs—key regulators of angiogenesis. Curr Opin Genet Dev. 2009;19:205–11.
Mendell JT. miRiad roles for the miR-17-92 cluster in development and disease. Cell. 2008;133:217–22.
Dews M, Homayouni A, Yu D, Murphy D, Sevignani C, Wentzel E, Furth EE, Lee WM, Enders GH, Mendell JT, Thomas-Tikhonenko A. Augmentation of tumor angiogenesis by a Myc-activated microRNA cluster. Nat Genet. 2006;38:1060–5.
Bonauer A, Carmona G, Iwasaki M, Mione M, Koyanagi M, Fischer A, Burchfield J, Fox H, Doebele C, Ohtani K, Chavakis E, Potente M, Tjwa M, Urbich C, Zeiher AM, Dimmeler S. MicroRNA-92a controls angiogenesis and functional recovery of ischemic tissues in mice. Science. 2009;324:1710–3.
Nicoli S, Standley C, Walker P, Hurlstone A, Fogarty KE, Lawson ND. MicroRNA-mediated integration of haemodynamics and Vegf signalling during angiogenesis. Nature. 2010;464:1196–200.
Harris TA, Yamakuchi M, Ferlito M, Mendell JT, Lowenstein CJ. MicroRNA-126 regulates endothelial expression of vascular cell adhesion molecule 1. Proc Natl Acad Sci U S A. 2008;105:1516–21.
Endo-Takahashi Y, Negishi Y, Nakamura A, Ukai S, Ooaku K, Oda Y, Sugimoto K, Moriyasu F, Takagi N, Suzuki R, Maruyama K, Aramaki Y. Systemic delivery of miR-126 by miRNA-loaded Bubble liposomes for the treatment of hindlimb ischemia. Sci Rep. 2014;4:3883.
Kuehbacher A, Urbich C, Zeiher AM, Dimmeler S. Role of Dicer and Drosha for endothelial microRNA expression and angiogenesis. Circ Res. 2007;101:59–68.
Wang S, Aurora AB, Johnson BA, Qi X, McAnally J, Hill JA, Richardson JA, Bassel-Duby R, Olson EN. The endothelial-specific microRNA miR-126 governs vascular integrity and angiogenesis. Dev Cell. 2008;15:261–71.
Fish JE, Santoro MM, Morton SU, Yu S, Yeh RF, Wythe JD, Ivey KN, Bruneau BG, Stainier DY, Srivastava D. miR-126 regulates angiogenic signaling and vascular integrity. Dev Cell. 2008;15:272–84.
Chen Y, Gorski DH. Regulation of angiogenesis through a microRNA (miR-130a) that down-regulates antiangiogenic homeobox genes GAX and HOXA5. Blood. 2008;111:1217–26.
Fasanaro P, D’Alessandra Y, Di Stefano V, Melchionna R, Romani S, Pompilio G, Capogrossi MC, Martelli F. MicroRNA-210 modulates endothelial cell response to hypoxia and inhibits the receptor tyrosine kinase ligand Ephrin-A3. J Biol Chem. 2008;283:15878–83.
Lee DY, Deng Z, Wang CH, Yang BB. MicroRNA-378 promotes cell survival, tumor growth, and angiogenesis by targeting SuFu and Fus-1 expression. Proc Natl Acad Sci U S A. 2007;104:20350–5.
Hua Z, Lv Q, Ye W, Wong CK, Cai G, Gu D, Ji Y, Zhao C, Wang J, Yang BB, Zhang Y. MiRNA-directed regulation of VEGF and other angiogenic factors under hypoxia. PLoS One. 2006;1, e116.
Cimmino A, Calin GA, Fabbri M, Iorio MV, Ferracin M, Shimizu M, Wojcik SE, Aqeilan RI, Zupo S, Dono M, Rassenti L, Alder H, Volinia S, Liu CG, Kipps TJ, Negrini M, Croce CM. miR-15 and miR-16 induce apoptosis by targeting BCL2. Proc Natl Acad Sci U S A. 2005;102:13944–9.
Suarez Y, Fernandez-Hernando C, Pober JS, Sessa WC. Dicer dependent microRNAs regulate gene expression and functions in human endothelial cells. Circ Res. 2007;100:1164–73.
Poliseno L, Tuccoli A, Mariani L, Evangelista M, Citti L, Woods K, Mercatanti A, Hammond S, Rainaldi G. MicroRNAs modulate the angiogenic properties of HUVECs. Blood. 2006;108:3068–71.
Ji R, Cheng Y, Yue J, Yang J, Liu X, Chen H, Dean DB, Zhang C. MicroRNA expression signature and antisense-mediated depletion reveal an essential role of MicroRNA in vascular neointimal lesion formation. Circ Res. 2007;100:1579–88.
Liu X, Cheng Y, Zhang S, Lin Y, Yang J, Zhang C. A necessary role of miR-221 and miR-T2009;104:476–87.
Maegdefessel L, Azuma J, Toh R, Merk DR, Deng A, Chin JT, Raaz U, Schoelmerich AM, Raiesdana A, Leeper NJ, McConnell MV, Dalman RL, Spin JM, Tsao PS. Inhibition of microRNA-29b reduces murine abdominal aortic aneurysm development. J Clin Invest. 2012;122:497–506.
Merk DR, Chin JT, Dake BA, Maegdefessel L, Miller MO, Kimura N, Tsao PS, Iosef C, Berry GJ, Mohr FW, Spin JM, Alvira CM, Robbins RC, Fischbein MP. miR-29b participates in early aneurysm development in Marfan syndrome. Circ Res. 2012;110:312–24.
Maegdefessel L, Azuma J, Toh R, Deng A, Merk DR, Raiesdana A, Leeper NJ, Raaz U, Schoelmerich AM, McConnell MV, Dalman RL, Spin JM, Tsao PS. MicroRNA-21 blocks abdominal aortic aneurysm development and nicotine-augmented expansion. Sci Transl Med. 2012;4:122ra22.
Bagga S, Bracht J, Hunter S, Massirer K, Holtz J, Eachus R, Pasquinelli AE. Regulation by let-7 and lin-4 miRNAs results in target mRNA degradation. Cell. 2005;122:553–63.
Goren Y, Kushnir M, Zafrir B, Tabak S, Lewis BS, Amir O. Serum levels of microRNAs in patients with heart failure. Eur J Heart Fail. 2012;14:147–54.
Horie T, Ono K, Nishi H, Nagao K, Kinoshita M, Watanabe S, Kuwabara Y, Nakashima Y, Takanabe-Mori R, Nishi E, Hasegawa K, Kita T, Kimura T. Acute doxorubicin cardiotoxicity is associated with miR-146a-induced inhibition of the neuregulin-ErbB pathway. Cardiovasc Res. 2010;87:656–64.
Kuwabara Y, Horie T, Baba O, Watanabe S, Nishiga M, Usami S, Izuhara M, Nakao T, Nishino T, Otsu K, Kita T, Kimura T, Ono K. MicroRNA-451 exacerbates lipotoxicity in cardiac myocytes and high-fat diet-induced cardiac hypertrophy in mice through suppression of the LKB1/AMPK pathway. Circ Res. 2015;116(2):279–88.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Ono, K. (2015). microRNAs and Cardiovascular Remodeling. In: Santulli, G. (eds) microRNA: Medical Evidence. Advances in Experimental Medicine and Biology, vol 888. Springer, Cham. https://doi.org/10.1007/978-3-319-22671-2_10
Download citation
DOI: https://doi.org/10.1007/978-3-319-22671-2_10
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-22670-5
Online ISBN: 978-3-319-22671-2
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)