
Abstract
Neuromyelitis optica spectrum disorders (NMOSD) are most often characterized by severe involvement of the optic nerves and/or spinal cord in a mono- or multiphasic manner and associated with anti-aquaporin 4 (AQP4) antibody (Ab) positivity. Optical coherence tomography (OCT) showed moderate to severe retinal axonal loss in neuromyelitis optica spectrum disorders (NMOSD) after optic neuritis (ON) episode. Peripapillary retinal nerve fiber layer (RNFL) and macular thinning seem more marked in NMOSD than in multiple sclerosis (MS) and sometimes (8.7–25.6 %) accompanied by a nonspecific macular change: microcystic macular edema (MME). NMOSD patients without ON might present subclinical retinal axonal loss, but it remains a matter of debate and seems clearly to occur in a lower intensity than in MS. The mechanism of this subclinical involvement remains unclear but could be due to anti-AQP4 antibodies and AQP4 expression on astrocytes inside the optic nerve and RNFL and on retinal Müller cells. OCT may help to differentiate NMOSD from MS by rigorous analysis of eyes with multiple sclerosis associated ON (MSON) and without MSON. In NMOSD, correlations between visual disability and OCT parameters are strong, whereas correlations between clinical disability and OCT parameters remain to be discussed. A few brain magnetic resonance imaging (MRI) studies coupled to OCT investigated optic ways imaging and found interesting correlation between retinal axonal loss and brain white and gray matter parameters. OCT is a wonderful tool enabling us to quantify retinal axonal loss in NMOSD. The main limitations of OCT studies in NMOSD are the small size of NMOSD cohort and sometimes a severe residual visual acuity enabling not to perform OCT.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Neuromyelitis optica spectrum disorders
- Multiple sclerosis
- Optic neuritis
- Optical coherence tomography
- Microcystic macular edema
- Anti-aquaporin 4 antibody
- Retinal nerve fiber layer
- Retinal atrophy
Introduction
Neuromyelitis optica (NMO) is an inflammatory demyelinating autoimmune disease of the central nervous system (CNS), classically affecting optic nerve(s) uni- or bilaterally and the spinal cord with an extensive involvement. It can be monophasic, but most of the time relapsing. Progressive form seems very rare (<1 %) and questionable. For a long time, NMO has been thought to be a variant of multiple sclerosis (MS), but clinical, neuroimaging, immunological, and pathological findings enable us nowadays to strictly distinguish both of them [1]. NMO affects older people than MS (mean age of 40 years old)—most often female patients (sex ratio F/H = 9). Clinical relapse of optic neuritis and myelitis is considered more severe than in MS because of usually incomplete recovery [2, 3]. All patients suffering from neuromyelitis optica (NMO) had a past history of clinical optic neuritis [4]. In the last decade, knowledge about NMO physiopathology has much improved, notably with the discovery of an NMO-specific autoantibody directed against aquaporin 4 (AQP4) protein, which is the main channel regulating water homeostasis in CNS [5, 6]. New rare phenotypes have also been described, involving the brain in an extensive manner [7, 8] or involving more specifically locations with high expression of AQP4 (periaqueductal, periventricular; [7, 9–11]). More sensitive tests for anti-AQP4 antibody (Ab) detection have been developed [12]. It has been demonstrated that patients presenting only 1 episode of transverse myelitis [13] or 2 or more episodes of isolated optic neuritis [14] being positive for anti-AQP4 Ab have a high risk to develop other severe relapses and NMO. In the same way, up to 5–6 % of patients suffering from chronic relapsing ON or single isolated ON or relapsing isolated ON presented anti-AQP4 Ab [15]. Thus, a concept of high-risk syndrome for clinical NMO conversion has emerged and allowed early therapeutic intervention in a disease where sequelae are the consequence of relapse and not the consequence of progressive lesion accumulation. For all these reasons, NMO criteria have to evolve, and recently the Mayo Clinic Research Group with other international experts proposed modified criteria for neuromyelitis optica spectrum disorders (NMOSD) [16]. This classification should replace the historical notion of Devic disease. In these new proposed criteria, anti-AQP4 Ab detection takes a major place. Thus, patients without ON can be diagnosed as NMOSD.
Except in Asian countries, NMOSD remains a rare disease and more rare than MS. While the first optical coherence tomography study (OCT) in MS was published in 1999 [17], the first OCT studies in NMO were published a decade later [18, 19]. As NMOSD is not yet considered as a variant of MS, OCT findings in NMOSD are also quite different from MS. OCT may help to distinguish NMOSD patients from MS patients and appears as a wonderful outcome measure to evaluate retinal axonal loss and efficacy of potential aggressive therapies in NMOSD.
In this review, we will firstly focus on eyes with past history of optic neuritis (ON eyes) in NMOSD and then develop the arguments in favor or against a subclinical retinal axonal loss in non-ON eyes (NON eyes) of NMOSD patients. We will also consider the potential correlations between OCT values and disability and the added value of optic ways magnetic resonance imaging (MRI).
Optic Neuritis Eyes of NMOSD Patients
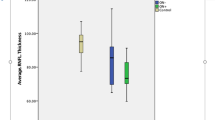
Most of the published NMOSD studies investigating anterior visual pathways damage by OCT in comparison to healthy controls (HC) or MS patients reported a severe retinal axonal loss versus HC [18–26] and a more severe retinal axonal loss in NMOSD than in MS [19–22, 24, 25, 27–31] as illustrated in Fig. 6.1. One study was interested only in NMOSD patients [32]. Up to now, all OCT studies in NMOSD involved adult patients. No study has been interested in OCT findings of pediatric NMOSD patients.

Peripapillary and macular OCT scan from a healthy control (HC; female 30 years old), a multiple sclerosis associated optic neuritis eye (MSON eye; female 32 years old), and a neuromyelitis optica spectrum disorder associated optic neuritis eye (NMOSD-ON eye; 28 years old). MS and NMOSD-ON eyes suffered from 1 episode of clinical ON. Patients’ ON eyes presented peripapillary retinal nerve fiber layer (pRNFL), macular ganglion cells layer (mGCL) atrophy, and macular inner nuclear layer (mINL) thickening versus HC. In MSON eye, pRNFL atrophy predominates on temporal quadrant with an increased nasal/temporal (N/T) ratio. In NMOSD-ON eye, all pRNFL quadrants are involved by atrophic process and N/T ratio is normal
NMOSD Versus HC
All studies including NMOSD patients and HC demonstrated that peripapillary retinal nerve fiber layer (pRNFL) and total macular volume (TMV)/macular thickness were significantly reduced after 1 or more clinical episodes of ON versus HC. Ratchford et al. reported that a single episode of ON in NMOSD causes an estimated decrease of 31 μm in pRNFL thickness compared to an estimated decrease of 10 μm in RRMS [20]. All studies reporting subanalysis of pRNFL quadrants reported significant atrophy in all quadrants for comparison of NMOSD and HC [18, 19, 24–26].
NMOSD Versus MS
Most of the studies including NMOSD and MS patients demonstrated that global pRNFL and TMV/macular thickness were significantly reduced after ON in both diseases but with a significant greater extent in NMOSD [19–22, 24, 25, 27, 29, 33], which is in agreement with consideration that ON is clinically more severe in NMOSD than in MS [2, 3]. Moreover visual disability of NMOSD-ON eyes is worse than in MSON eyes [3, 20, 24, 25, 27, 29, 31]. One additional study highlighted no significance but only a trend toward significance about a decreased VA in NMOSD-ON eyes versus MSON eyes [28]. Most of the previous OCT studies in NMOSD did not adjust for the number of clinical ON [19–22, 24, 25, 28, 29, 31] and/or did not perform statistical analysis with generalized estimation equation (GEE) models. The number of ON episodes increases the retinal axonal loss [29] and GEE is the best adapted statistical test for data with hierarchical structure including clusters of eyes on an individual level. Nevertheless, 1 study involving NMOSD and MS patients with only 1 MSON episode showed a greater pRNFL decrease in NMOSD [20] and another one with adjustment to the number of ON episodes showed more global pRNFL in NMOSD [27].
If post-ON retinal axonal loss seems to be higher in NMOSD than in MS, it seems not to involve all pRNFL quadrants with the same magnitude or as in MS. Compared to MS patients, NMOSD patients present frequently an inferior [19, 22, 24, 25, 27, 29] and a superior [22, 24, 27, 29] pRNFL atrophy after ON. Since temporal pRNFL atrophy is predominant in MSON eyes, comparison of NMOSD-ON eyes and MSON eyes rarely found more marked temporal pRNFL atrophy in NMOSD-ON eyes [19, 24, 25]. Merle et al. found temporal pRNFL atrophy in NMOSD eyes versus MS eyes but ON and NON eyes were mixed for both populations [19]. Interestingly nasal/temporal (N/T) pRNFL ratio in NMOSD remains in normal range [24, 26] arguing in favor of a more overall atrophy in NMOSD than in RRMS.
Only three studies did not show significant difference in retinal atrophy between NMOSD and MS [23, 26, 34]. In the first study, the authors compared ON eyes affected by only 1 episode of ON between NMOSD and MS and showed no difference in macular RNFL and GCIP thicknesses. However, INL thickness of NMOSD population was significantly thicker than in the MS population, and the NMOSD population closed to that already published in another paper [22] presented significantly more pRNFL atrophy than MS. The second study [34] did not highlight significant pRNFL differences between ON eyes of NMOSD and MS (p = 0.46). Indeed, it seems that patients with NMOSD presented more ON episodes than MS patients. The third study [26] found pRNFL thinning in some nasal quadrants with a TMV decreasing in NMOSD versus MS, but after adjustment, notably including the number of ON episodes in GEE analysis, differences did not persist any longer.
It remains quite difficult to summarize all previously referenced NMOSD-OCT studies because all of them have not included MS patients or HC and because comparison sometimes included mixed MSON and NON eyes or specifically investigated MSON eyes or NON eyes or both.
Microcystic Macular Edema
For highlighting microcystic macular edema (MME), it is mandatory to perform OCT with a 4th-generation OCT (spectral-domain OCT). In MS, MME might occur in the absence of any clinical MSON episode [35–37]. However, these three studies did not report optic nerve imaging of the MME eye without MSON, and it is difficult to consider that MME and severe pRNFL thinning could be due to only retrograde transsynaptic degeneration. Five studies investigated specifically or reported MME in NMOSD. All these studies showed that NMOSD patients with MME have always presented at least 1 clinical episode of ON [24, 26, 30, 32, 36]. MME is most often located in the inner nuclear layer (INL) and sometimes the inner plexiform layer (IPL). It is not specific of neuroinflammatory diseases [38], but it is a marker of severe optic neuropathy as illustrated in an NMOSD patient in Fig. 6.2a–e.

Peripapillary and macular OCT scan from an NMOSD patient who alternatively experienced multiple optic neuritis on both sides. Clinical episodes were more severe on the left side. (a) Right peripapillary OCT scan showing slight pRNFL atrophy and normal N/T pRNFL ratio. (b) Left peripapillary OCT scan showing profound pRNFL atrophy and normal N/T pRNFL ratio. (c) Ocular fundus OCT image centered on fovea showing macular changes. (d) Ocular fundus OCT image centered on fovea (red arrow) showing macular changes (circled in yellow) and papilla (white arrow). (e) Vertical OCT macular scans showing microcystic macular edema (MME) in the inner nuclear layer (white arrows) and in the inner plexiform layer (yellow arrow). Red arrows indicated areas without any individualized cystic lesions typical of MME at a location with macular changes on ocular fundus (possible INL thickening)
The proportion of NMOSD patients presenting MME is ranged between 8.7 and 25.6 % and the proportion of NMOSD-ON eyes with MME between 6.3 and 29.8 % [24, 26, 30, 32, 36] (Table 6.1) [19, 21, 24, 26, 30, 32, 35–39]. MME seems more frequently observed in NMOSD than in MS [32, 36]. NMOSD patients with MME presented thinner pRNFL than NMOSD patients without MME [32]. In agreement with this last finding, it has been shown that patients with MME have presented more episodes of ON than those without, but these latter were younger [30]. All pathologies included MS, CRION, and Whatever the cause of ON (MS, NMOSD, Chronic Relapsing Inflammatory Optic Neuropathy) is, MME is associated with a thinner pRNFL than the contralateral eye not affected by ON, but not with a lower total macular volume [36]—probably because of the slight INL thickening observed on ON eyes in this cohort. In MS, INL thickening is observed in MME eyes but also in MSON eyes without MME compared to contralateral NON eyes [35]. Fernandes et al. did not report MME in their cohort but interestingly reported the same INL thickening in NMOSD eyes with only 1 episode of ON versus HC but also versus MS patients with only 1 episode of MSON [23]. INL thickening seems to be of greater magnitude in NMOSD versus MS. MME is not always highlighted on an OCT scan, whereas ocular fundus may show macular changes (Fig. 6.2c–e). Inner nuclear layer thickening may be the ultimate step before microcyst formation.
In MS, the “outer” retinal layers (ORL; INL to photo receptor layer) seem not to be involved in the retinal atrophic process [21, 26, 40]. Balk and coworkers discussed a potential retinal neuroplasticity [40, 41]. In NMOSD, the same external retinal conservation has been found [26] whereas the thickness of ONL, which takes part of ORL, has been reported significantly increased in NMOSD-ON eyes presenting MME [24]. A recent study coupling OCT and optic nerve imaging in a MS/NMOSD/idiopathic ON cohort suggested that optic nerve with clinical or subclinical T2 lesion might be associated with an INL and ONL thickening whatever the presence of MME [42]. If ORL in NMOSD-ON eyes did not suffer from a retinal atrophic process, a thickening process may be discussed.
Non-optic Neuritis Eyes of NMOSD Patients: Is There Any Subclinical Retinal Involvement in NMOSD?
If a large majority of OCT studies considered that NMOSD patients affected by ON presented a more severe retinal atrophy than MS patients, subclinical retinal axonal loss in NMOSD is more discussed. Only studies including HC [18–26] enable us to really discuss whether or not a subclinical retinal axonal loss in NMOSD exists. However, Park et al. [25] did not show interest in NMOSD-NON eyes, and in two other studies [22, 23], all NMOSD eyes presented past history of ON. Regardless of the very few patients without ON in the NMOSD group, the French collaborative work was not able to do a statistical comparison, but pRNFL values between HC and NMOSD-NON eyes were very comparable [18]. Global peripapillary RNFL [24, 34] and total macular volume [24] of NMOSD-NON eyes appear in some studies similar to HC eyes. In other studies, macular involvement in NMOSD-NON eyes is highlighted [21, 26, 30]. GCIP [21, 30], average macular thicknesses/TMV [21, 30], and GCL volume [26] are reduced in NMOSD-NON eyes versus HC. Merle et al. [19] showed that global pRNFL value was significantly reduced in NMOSD-NON eyes versus HC, and a recent work [26] only found significant atrophy in temporal and naso-inferior quadrants of pRNFL.
If subclinical retinal involvement may be discussed in NMOSD, it seems that it is probably of a lesser extent than in MS since it has been reported that there are lower global pRNFL [27] and temporal pRNFL [26] values in MS-NON eyes than in NMOSD-NON eyes.
Astroglial cells and Müller cells are AQP4-expressing cells. Müller cells are located inside the retina and astroglial cells inside the RNFL and the optic nerve [43, 44] but also in the chiasma, optic tract, and brain. Without any clinical ON episode, anti-AQP4 Ab may induce their pathological process directly on these cells inside the different retinal layers (astrocytes and Müller cells) and on the rest of the optic ways (astrocytes). Some NMOSD studies have already described extensive [8] or more located brain abnormalities [7, 10], particularly in the brainstem and near the lateral ventricules where the lateral geniculate nucleus and the posterior part of optic radiations (OR) are located, respectively. As it has been demonstrated in MS [45], a retrograde transsynaptic degenerative process cannot be excluded in NMOSD.
How Could OCT Help to Distinguish NMOSD and MS?
Firstly, we propose to focus on ON eyes and what could be in favor of NMOSD (versus MS) is the presence of more severe pRNFL atrophy in global analysis but more particularly in another quadrant than temporal quadrant and a more frequent maculopathy (more atrophy, more MMO) associated with worse visual disability. Secondly, we propose to focus on NON eyes and an absence of temporal pRNFL atrophy would be in favor of NMOSD. In Table 6.1, the main results of OCT studies in NMOSD and MS are summarized.
Kim recently proposed that after a first episode of ON, global pRNFL less than 78.9 μm associated with a high-contrast visual acuity of less than 0.4 logMar leads to a specificity of 100 % in favor of NMOSD diagnostic [33].
Further multicentric collaborative studies with spectral-domain OCT and larger cohorts would be able to examine the question in more detail and to confirm or not the results of all previous exploratory OCT studies in NMOSD.
OCT and Magnetic Resonance Imaging in NMOSD
Another imaging biomarker that may help to distinguish these two diseases is optic nerve magnetic resonance imaging (MRI). In NMOSD, demyelinating lesion seen on MRI may involve more frequently the posterior part of the optic nerve and the chiasma [46, 47] with a greater extent [47] than in MS. To date no study has coupled optic nerve MRI and OCT assessments in NMOSD. One could expect that T2 lesion length in NMOSD may be higher in NMOSD than in MS, but these 2 latter studies with semiquantitative measurements remain contradictory. It was recently shown that 3D Double Inversion Recovery (DIR) sequence was superior to coronal 2D Short Tau Inversion Recovery - Fluid Attenuated Inversion Recovery (STIR-FLAIR) sequence in the detection of T2 optic nerve lesion [48]. This 3D sequence allows a direct T2 hypersignal length measurement, strongly associated with retinal axonal loss in a MS/NMOSD/idiopathic ON cohort [42] as illustrated in an NMOSD patient in Fig. 6.3a–e.

Brain 3D double inversion recovery (DIR) MRI sequence and peripapillary OCT scans of an NMOSD patient with past history of severe bilateral optic neuritis. (a) Coronal reconstruction centered on optic nerves showing bilateral T2 hyperintensities (yellow and red arrows for the right and left optic nerves, respectively) in retrobulbar (top image) and orbital (bottom image) portions. (b) Axial reconstruction showing bilateral extended T2 hyperintensities on optic nerves. The length of T2 hyperintense lesion measured between both colored dotted lines is 38 mm for both sides. (c) Oblique reconstruction along the right (yellow arrow) and the left (red arrow) optic nerves showing extended T2 hyperintensities. (d) Right profound global pRNFL atrophy (40 μm) with nasal/temporal pRNFL ratio in normal ranges according to Heidelberg Spectralis Healthy Controls database. (e) Left profound global pRNFL atrophy (37 μm) with nasal/temporal pRNFL ratio in normal ranges according to Heidelberg Spectralis Healthy Controls database
If similar metabolic measurement by proton(H+)-magnetic resonance spectroscopy (H-MRS) in NMOSD and HC argued in favor of brain white matter (WM) sparing in NMOSD [49], many diffusion tensor imaging (DTI) studies [50] showed altered DTI parameters in NMOSD versus HC in many tracts and notably optic radiations [50–53]. In addition, voxel-based morphometry studies [50, 54–56] showed atrophic process in WM and grey matter of NMOSD patients. Recently, a Brazilian study group showed for the first time a high correlation between average pRNFL thickness and pericalcarine cortical thickness [56]. In this study, many other cortical areas were reduced in NMOSD versus HC. In MS associated ON, a relationship between retinal thicknesses and occipital cortex thickness seems to be weaker [40, 41].
Disability in NMOSD and OCT Correlations
In NMOSD, mild to very good correlations between retinal thicknesses (peripapillary RNFL and/or macular thicknesses) and visual disability, scored by high-contrast visual acuity or low-contrast visual acuity or visual field or P100 latency, have been found [18–22, 24, 26, 29]. These correlations were stronger and more likely to be observed specifically in the ON eye group.
Correlation studies between OCT values and EDSS remain contradictory. Very few studies investigated this issue [18, 19, 26, 34, 56]. No [19] or moderate correlations with trend toward significance (pRNFL in ON eyes [34]) or moderate significant correlations between EDSS (visual FS included) and few OCT parameters (TMV and GCC [26]) but also excellent correlation (global pRNFL/EDSS without visual FS [18], global pRNFL/EDSS [56]) have been reported. No correlations were observed by focusing on NON eyes [26, 34]. Furthermore, NMOSD patients with MME did not present higher clinical disability [30]. Since there are an obvious link between disease duration and the age of the patient but also the probability for the patient to present with an ON episode, lower RNFL values were reported significantly associated with longer disease duration [34]. Expanded disability status scale is a disability scale established for MS patients [57]. It includes visual, brainstem, pyramidal, sensory, cerebellar, bladder/bowel, and cognitive functional scores. It does not seem to be the best adapted disability scale for NMOSD, whereas it is the most widely used. Neurologists are used to performing EDSS scores during their consultation and other disability scales require more time to be performed. There is a need for the development of a more specific disability scale in NMOSD [58], A more specific disability scale may be validated in the future but, OCT will probably remain a better biomarker of brain axonal loss in MS than in NMOSD.
Follow-Up Study of NMOSD Population
There is only one study reporting longitudinal OCT follow-up in NMOSD population [59]. With 3rd-generation OCT (time domain), the authors found significant retinal axonal loss in global pRNFL and all 4 classical pRNFL quadrants by comparing 2 different OCT assessments of a NMOSD cohort (n = 30). The 2 OCT assessments were spaced 18 months in mean. Retinal axonal loss was only observed on eyes affected by ON previous to the first assessment and may occur independently of relapses (ON or not) during the follow-up. This pRNFL decreasing is difficult to explain but we can make different hypotheses. Peripapillary RNFL decreasing may be secondary to ON that occurred previous to the first assessment (<3 months after relapse) because pRNFL decrease in MSON eyes begins at the third month and seems maximal at 6 months, with some data in favor of a continuing axonal loss in the affected eye for at least 12 months [60, 61]. The authors argued also that new ON occurring in ON eyes have potentially been missed by both patient and neurologist and that few patients presented ON during the follow-up. Up to now, no OCT study with SD-OCT and longitudinal NMOSD follow-up has been published. Because of its higher definition (<5 μm) and higher reproducibility [62, 63], SD-OCT will be better than TD-OCT to appreciate retinal axonal loss in a follow-up study.
Difficulties of OCT Studies in NMOSD
Optical coherence tomography studies in NMOSD remain more difficult to perform than in MS. NMOSD is a rare disease and most of the monocentric studies already published included small-sized population (n < 30). Multicentric OCT studies have to emerge in NMOSD in order to increase the power of statistical analyses, but an additional difficulty in a multicenter study will be to perform OCT scans with the same 4th-generation OCT device. However, the Baltimore OCT study group recently showed a good concordance between CIRRUS (Zeiss) and Spectralis (Heidelberg) OCT scan for retinal segmentation using an open source segmentation algorithm [64].
Optic neuritis in NMOSD are more severe than MS and, in spite of aggressive therapeutic management, it is not unusual to observe severe sequelae and some cases of blindness. It has been previously reported that up to 54 % of NMOSD-ON eyes with monophasic disease course were totally blind [2] and that bilateral severe visual loss (<1/10) appeared in a median time of 13 years [3]. Weak residual visual function after ON leads to difficulty in the acquisition of OCT and to an OCT of lesser quality.
Another difficulty is to know if we have to consider anti-AQP4 Ab-negative and Ab-positive patients together or if they are two different types of NMOSD disease. Recently, it has been discussed that NMOSD patients negative for anti-AQP4 Ab could have anti-myelin-oligodendrocytes-glycoprotein Ab [65] or that NMOSD patients could have both anti-AQP4 and anti-KIR4.1 Ab [66]. Kir4.1 is a ubiquitous potassium channel notably coexpressed with AQP4 by Müller cells.
Indeed, NMOSD patients are older than MS patients. Older patients have greater chance to have other confounding ocular pathologies and naturally thinner retinal thickness [67]. Obviously, complete ophthalmological evaluations may exclude these confounding factors.
NMOSD is more frequent in Asian countries and Afro-Caribbean populations [1]. Peripapillary RNFL thickness and macular volume are known to vary according to ethnic origin [68–70]. As an example, Asian, Hispanic, and Indian people have thicker pRNFL than European people. Thus, cautiousness is required for comparing OCT studies with Caucasian patients and OCT studies with Asian patients. It has to be taken into account when matching to HC or MS patients is performed—notably for future multicentric studies.
Advantages of OCT Studies in NMOSD
Benign MS are very rare in NMOSD. Thus it seems unlikely to miss (physician) or to forget (the patient) any ON episode. It is very important in OCT study to know how many ON episodes occurred because the more ON episodes occur, the more atrophy you observed.
Subclinical retinal involvement (directly within the retina or as a result of optic ways involvement) remains discussed in NMOSD and very probably of a lesser extent in NMOSD than in MS. In MS, it is difficult to know if retinal atrophy occurring without any clinical episode of ON is due to subclinical MSON or to axonal retrograde transsynaptic degeneration. For testing potential neuroprotective drugs at the acute phase of ON, this could be an advantage of studies in NMOSD versus studies in MS.
Conclusion
OCT is a wonderful tool for evaluating retinal axonal secondary to ON in neuroinflammatory diseases affecting the optic nerve—notably NMOSD. OCT may help to differentiate NMOSD and MS. In future NMOSD therapeutic trials, OCT has to be an evaluation criterion. Multimodal studies (OCT, optic nerve and brain MRI, visual evoked potentials) are still lacking in NMOSD and are necessary to better understand the optic ways involvement in NMOSD. Multicentric follow-up studies will be able to clearly confirm or not a subclinical axonal loss in NMOSD, particularly in macula.
References
Wingerchuk DM, Lennon VA, Luchinetti CF, et al. The spectrum of neuromyelitis optica. Lancet Neurol. 2007;6:805–15.
Wingerchuk DM, Hogancamp WF, O’Brien PC, et al. The clinical course of neuromyelitis optica (Devic’s syndrome). Neurology. 1999;53:1107–14.
Merle H, Olindo S, Bonnan M, et al. Natural history of the visual impairment of relapsing neuromyelitis optica. Ophthalmology. 2007;114:810–5.
Wingerchuck DM, Lennon VA, Pittock SJ. Revised diagnostic criteria for neuromyelitis optica. Neurology. 2006;66:1485–9.
Lennon VA, Wingerchuck DM, Kryzer TJ, et al. A serum autoantibody marker of neuromyelitis optica: distinction from multiple sclerosis. Lancet. 2004;364:2106–12.
Lennon VA, Kryzer TJ, Pittock SJ, et al. IgG marker of spinal multiple sclerosis binds to the aquaporin-4 water channel. J Exp Med. 2005;202:473–7.
Pittock SJ, Weinshenker BG, Luchinetti CF, et al. Neuromyelitis optica brain lesions localized at sites of high aquaporin 4 expression. Arch Neurol. 2006;63:964–8.
Ayrignac X, Daliere CC, Nerrant E, et al. Extensive cerebral white matter involvement in a patient with NMO spectrum disorder. Mult Scler. 2014;20:1401–3.
Nakashima I, Fujihara K, Miyazawa I, et al. Clinical and MRI features of Japanese patients with multiple sclerosis positive for NMO-IgG. J Neurol Neurosurg Psychiatry. 2006;77:1073–5.
Banker P, Sonni S, Kister I, et al. Pencil-thin ependymal enhancement in neuromyelitis optica spectrum disorders. Mult Scler. 2012;18:1050–3.
Appiwattanakul M, Popescu BF, Matiello M, et al. Intractable vomiting as the initial presentation of neuromyelitis optica. Ann Neurol. 2010;68:757–61.
Höfteberger R, Sabater L, Marignier R, et al. An optimized immunohistochemistry technique improves NMO-IgG detection: study comparison with cell-based assays. PLoS One. 2013;8, e79083.
Weinshenker BG, Wingerchuck DM, Vukusic S, et al. Neuromyelitis optica IgG predicts relapse after longitudinally extensive transverse myelitis. Ann Neurol. 2006;59:566–9.
Matiello M, Lennon VA, Jacob A, et al. NMO-IgG predicts the outcome of recurrent optic neuritis. Neurology. 2008;70:2197–200.
Petzold A, Pittock S, Lennon V, Maggiore C, Weinshenker BG, Plant GT. Neuromyelitis optica-IgG (aquaporin-4) autoantibodies in immune mediated optic neuritis. J Neurol Neurosurg Psychiatry. 2010;81:109–11.
Wingerchuk DM, Banwell B, Bennett JL, et al. International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology. 2015 Jun 19. pii: 10.1212/WNL.0000000000001729. [Epub ahead of print]..
Parisi V, Manni G, Spadaro M, Colacino G, Restuccia R, Marchi S, et al. Correlation between morphological and functional retinal impairment in multiple sclerosis patients. Invest Ophthalmol Vis Sci. 1999;40(11):2520–7.
de Seze J, Blanc F, Jeanjean L, Zéphir H, Labauge P, Bouyon M, et al. Optical coherence tomography in neuromyelitis optica. Arch Neurol. 2008;65(7):920–3.
Merle H, Olindo S, Donnio A, et al. Retinal nerve fiber layer thickness in neuromyelitis optica. Invest Ophthalmol Vis Sci. 2008;49:4412–7.
Ratchford JN, Quigg ME, Conger A, et al. Optical coherence tomography helps differentiate neuromyelitis optica and MS optic neuropathies. Neurology. 2009;73:302–8.
Syc SB, Saidha S, Newsome SD, et al. Optical coherence tomography segmentation reveals ganglion cell layer pathology after optic neuritis. Brain. 2012;135:521–33.
Monteiro ML, Fernandes DB, Apóstolos-Pereira SL, Callegaro D. Quantification of retinal neural loss in patients with neuromyelitis optica and multiple sclerosis with or without optic neuritis using Fourier-domain optical coherence tomography. Invest Ophthalmol Vis Sci. 2012;53(7):3959–66.
Fernandes DB, Raza AS, Nogueira RGF, et al. Evaluation of inner retinal layers in patients with multiple sclerosis of neuromyelitis optica using optical coherence tomography. Ophthalmology. 2013;120:387–94.
Schneider E, Zimmermann H, Oberwahrenbrock T, et al. Optical coherence tomography reveals distinct patterns of retinal damage in neuromyelitis optica and multiple sclerosis. PLoS One. 2013;8(6):e66151.
Park KA, Kim J, Oh YS. Analysis of spectral domain optical coherence tomography measurements in optic neuritis: differences in neuromyelitis optica, multiple sclerosis, isolated optic neuritis and normal healthy controls. Acta Ophthalmol. 2014;92:e57–65.
Outteryck O, Majed B, Defoort-Dhellemmes S, Vermersch P, Zéphir H. A comparative optical coherence tomography study in neuromyelitis optica spectrum disorder and multiple sclerosis. Mult Scler. 2015 Mar 31. pii: 1352458515578888. [Epub ahead of print]
Naismith RT, Tutlam NT, Xu J, et al. Optical coherence tomography differs in neuromyelitis optica compared with multiple sclerosis. Neurology. 2009;72:1077–82.
Green AJ, Cree BA. Distinctive retinal fibre layer and vascular changes in neuromyelitis optica following optic neuritis. J Neurol Neurosurg Psychiatry. 2009;80:1002–5.
Nakamura M, Nakazawa T, Doi H, et al. Early high dose intravenous methylprednisolone is effective in preserving retinal nerve fiber layer thickness in patients with neuromyelitis optica. Graefes Arch Clin Exp Ophthalmol. 2010;248:1777–85.
Sortichos ES, Saidha S, Byraiah G, et al. In vivo identification of morphologic retinal abnormalities in neuromyelitis optica. Neurology. 2013;80:1406–14.
Bichuetti DB, de Camargo AS, Falcao AB, et al. The retinal nerve fiber layer of patients with neuromyelitis optica and chronic relapsing optic neuritis is more severely damaged than patients with multiple sclerosis. J Neuroophthalmol. 2013;33:220–4.
Gelfand JM, Cree BA, Nolan R, et al. Microcystic inner nuclear layer abnormalities and neuromyelitis optica. JAMA Neurol. 2013;70:629–33.
Kim NH, Shin YJ, Jeong KS, Cho JY and Kim HJ. Optical coherence tomography after first optic neuritis for the differentiation between neuromyelitis optica and multiple sclerosis. Poster 677, ECTRIMS/ACTRIMS 2014 at Boston.
Lange AP, Sadjadi R, Zhu F, et al. Spectral-domain optical coherence tomography of retinal nerve fiber layer thickness in NMO patients. J Neuroophthalmol. 2013;33:213–9.
Saidha S, Sortichos ES, Ibrahim MA, et al. Microcystic macular oedema, thickness of the inner nuclear layer of the retina, and disease characteristics in multiple sclerosis: a retrospective study. Lancet Neurol. 2012;11:963–72.
Kaufhold F, Zimmermann H, Schneider E, et al. Optic neuritis is associated with inner nuclear layer thickening and microcystic macular edema independently of multiple sclerosis. PLoS One. 2013;8, e71145.
Burggraaff MC, Trieu J, de Vries-Knoppert WA, Balk L, Petzold A. The clinical spectrum of microcystic macular edema. Invest Ophthalmol Vis Sci. 2014;55:952–61.
Abegg M, Dysli M, Wolf S, et al. Microcystic macular edema. Retrograde maculopathy caused by optic neuropathy. Ophthalmology. 2014;121:142–9.
Gelfand JM, Nolan R, Schwartz DM, et al. Microcystic macular oedema in multiple sclerosis is associated with disease severity. Brain. 2012;135:1786–93.
Balk LJ, Steenwijk MD, Tewarie P, Daams M, Killestein J, Wattjes MP, Vrenkern H, Barkhof F, Polman CH, Uitdehaag BMJ, Petzold A. Bidirectional trans-synaptic axonal degeneration in the visual pathway in multiple sclerosis. J Neurol Neurosurg Psychiatry. 2015;86(4):419–24.
Balk LJ, Twisk JW, Steenwijk MD, Daams M, Tewarie P, Killestein J, et al. A dam for retrograde axonal degeneration in multiple sclerosis? J Neurol Neurosurg Psychiatry. 2014;85(7):782–9.
Hadhoum N, Hodel J, Defoort-Dhellemmes S, et al. Length of optic nerve double inversion recovery hypersignal: a new imaging biomarker for measuring axonal loss in neuroinflammatory diseases. Mult Scler. 2015. [Epub ahead of print].
Hamann S, Zeuthen T, La Cour M, et al. Aquaporins in complex tissues: distribution of aquaporins 1–5 in human and rat eye. Am J Physiol. 1998;274:1332–45.
Nagelhus EA, Veruki ML, Torp L, et al. Aquaporin 4 water channel protein in the rat retina and optic nerve: polarized expression in Müller cells and fibrous astrocytes. J Neurosci. 1998;18:2506–19.
Gabilondo I, Martinez-Lapiscina EH, Martinez-Heras E, et al. Trans-synaptic axonal degeneration in the visual pathway in multiple sclerosis. Ann Neurol. 2014;75:98–107.
Khanna S, Sharma A, Huecker J, Gordon M, Naismith RT, Van Stavern GP. Magnetic resonance imaging of optic neuritis in patients with neuromyelitis optica versus multiple sclerosis. J Neuroophthalmol. 2012;32:216–20.
Storoni M, Davagnanam I, Radon M, Siddiqui A, Plant GT. Distinguishing optic neuritis in neuromyelitis optica spectrum disease from multiple sclerosis: a novel magnetic resonance imaging scoring system. J Neuroophthalmol. 2013;33:123–7.
Hodel J, Outteryck O, Bocher AL, Zéphir H, Lambert O, Benadjaoud MA, Chechin D, Pruvo JP, Leclerc X. Comparison of 3D double inversion recovery and 2D STIR FLAIR MR sequences for the imaging of optic neuritis: pilot study. Eur Radiol. 2014;24(12):3069–75.
de Seze J, Blanc F, Kremer S, Collongues N, Fleury M, Marcel C, Namer IJ. Magnetic resonances spectroscopy evaluation in patients with neuromyelitis optica. J Neurol Neurosurg Psychiatry. 2010;81:409–11.
Pichiecchio A, Tavazzi E, Poloni G, Ponzio M, Palesi F, Pasin M, Piccolo L, Tosello D, Romani A, Bergamaschi R, Piccolo G, Bastianello S. Advanced magnetic resonance imaging of neuromyelitis optica: a multiparametric approach. Mult Scler. 2012;18:817–24.
Rocca MA, Agosta F, Mezzapesa DM, Martinelli V, Salvi F, Ghezzi A, Bergamaschi R, Comi G, Filippi M. Magnetization transfer and diffusion tensor MRI show gray matter damage in neuromyelitis optica. Neurology. 2004;62:476–8.
Yu C, Lin F, Li K, Jiang T, Qin W, Sun H, Chan P. Pathogenesis of normal-appearing white matter damage in neuromyelitis optica: diffusion-tensor MR imaging. Radiology. 2008;246:222–8.
Liu Y, Duan Y, Yu C, Wang J, Huang J, Ye J, Butzkueven H, Li K, Shu N. A tract-based diffusion study of cerebral white matter in neuromyelitis optica reveals widespread pathological alterations. Mult Scler. 2012;18:1013–21.
Rueda Lopes FC, Doring T, Martins C, Cabral FC, Malfetano FR, Pereira VCSR, Alves-Leon S, Gasparetto EL. The role of demyelination in neuromyelitis optica damage: diffusion-tensor MR imaging study. Radiology. 2012;263:235–42.
Chanson JB, Lamy J, Rousseau F, et al. White matter volume is decreased in the brain of patients with neuromyelitis optica. Eur J Neurol. 2013;20:361–7.
von Glehn F, Jarius S, Cavalcanti Lira RP, Alves Ferreira MC, von Glehn FH, Costa E, Castro SM, Beltramini GC, Bergo FP, Farias AS, et al. Structural brain abnormalities are related to retinal nerve fiber layer thinning and disease duration in neuromyelitis optica spectrum disorders. Mult Scler. 2014;20:1189–97.
Kurtzke JF. Rating neurological impairment in multiple sclerosis: an expanded disability status scale (EDSS). Neurology. 1983;33:1444–52.
Bennett JL, de Seze J, Lana-Peixoto M, et al. Neuromyelitis optica and multiple sclerosis: seeing differences through optical coherence tomography. Mult Scler. 2015;21:678–88.
Bouyon M, Collongues N, Zéphir H, et al. Longitudinal follow-up of vision in a neuromyelitis optica cohort. Mult Scler. 2013;19:1320–2.
Costello F, Coupland S, Hodge W, et al. Quantifying axonal loss after optic neuritis with optical coherence tomography. Ann Neurol. 2006;59:963–9.
Costello F, Hodge W, Pan YI, et al. Tracking retinal nerve fiber layer loss after optic neuritis: a prospective study using optical coherence tomography. Mult Scler. 2008;14:893–905.
Ho J, Sull AC, Vuong LN, Chen Y, Liu J, Fujimoto JG, Schuman JS, Duker JS. Assessment of artifacts and reproducibility across spectral- and time-domain optical coherence tomography. Ophthalmology. 2009;116:1960–70.
Huang J, Liu X, Wu Z, Xiao H, Dustin L, Sadda S. Macular thickness measurements in normal eyes with time-domain and Fourier-domain optical coherence tomography. Retina. 2009;29:980–7.
Bhargava P, Lang A, Al-Louzi O, et al. Applying an open source segmentation algorithm to different OCT devices in multiple sclerosis patients and healthy controls: Implications for clinical trials. Mult Scler Int. 2015 May 18. doi: 10.1155/2015/136295. [Epub ahead of print].
Sato DK, Callegaro D, Lana-Peixoto MA, et al. Distinction between MOG antibody-positive and AQP4 antibody-positive NMO spectrum disorders. Neurology. 2014;82:474–81.
Marignier R, Ruiz A, Cavagna S, Benetollo C, Durand-Dubief F, Vukusic S, Giraudon P. Potassium channel Kir4.1: a novel target for neuromyelitis optica autoantibodies ? Platform session PS7.6; ACTRIMS ECTRIMS 2014 at Boston.
Alamouti B, Funk J. Retinal thickness decreases with age: an OCT study. Br J Ophthalmol. 2003;87:899–901.
Budenz DL, Anderson RA, Varma R, et al. Determinants of normal retinal nerve fiber layer thickness measured by Stratus OCT. Ophthalmology. 2007;114:1046–52.
Girkin CA, McGwin G, Sinai MJ, et al. Variation in optic nerve and macular structure with age and race with spectral-domain optical coherence tomography. Ophthalmology. 2011;118:2403–8.
Knight OJ, Girkin GA, Budenz DL, et al. Effect of race, age, and axial lenght on optic nerve head parameters and retinal nerve fiber layer thickness measured by Cirrus HD-OCT. Arch Ophthalmol. 2012;130:312–8.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Outteryck, O., Vermersch, P. (2016). OCT Findings in Neuromyelitis Optica Spectrum Disorders. In: Petzold, A. (eds) Optical Coherence Tomography in Multiple Sclerosis. Springer, Cham. https://doi.org/10.1007/978-3-319-20970-8_6
Download citation
DOI: https://doi.org/10.1007/978-3-319-20970-8_6
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-20969-2
Online ISBN: 978-3-319-20970-8
eBook Packages: MedicineMedicine (R0)