
Abstract
Anthrax toxin is one of two major virulence factors secreted by pathogenic Bacillus anthracis, the etiologic agent of anthrax. Because inhalational anthrax is highly fatal, the agent has been weaponized for biowarfare and bioterrorism. Anthrax toxin is comprised of three individually nontoxic proteins, protective antigen (PA), lethal factor (LF), and edema factor (EF). But, to physiologically function, these individual subunits assemble into potent cytotoxins containing PA plus LF and/or EF. The PA component oligomerizes into a heptamer or octamer, which can insert into a host cell membrane to form a protein translocase channel. Under a transmembrane proton gradient driving force, LF and EF translocate through the narrow PA channel into the cytosol of the host cell. The narrowness of the channel necessitates that LF and EF unfold during translocation. This channel is unusual in this respect, because it contains its own unfoldase and translocase machinery. Highly nonspecific and dynamic clamp sites in the PA channel catalyze these activities. Anthrax toxin has been used extensively as a biophysical model to interrogate the molecular basis of translocation-coupled unfolding and translocation. It is being actively targeted by therapeutics to inhibit its function. New biotechnological adaptations use the toxin as a cancer therapy and generalized protein delivery vehicle.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Anthrax
B. anthracis is the only pathogen in its genus known to cause epidemic disease in mammals, including humans. Attributed to its global reach are its names: Greek, άνθραξ (“anthrax” meaning “coal”); German, Milzbrand (“spleen fire”) and Hadernkrankheit (“rag disease”); European, “Black Bane”; French, charbon (“charcoal”); Siberian Plague; Lodiana fever; and Indian, Pali Plague (Turnbull 1996). Anthrax was the first infectious disease to be linked to a specific bacterium, definitively establishing the Henle-Koch Postulates (Evans 1976). The disease is zoonotic and transmitted from animals to humans through three canonical infectious routes: cutaneous (~95 % of cases), inhalational (~5 %), and gastrointestinal (<1 %) (Mock and Fouet 2001; Koehler 2009).
1.1 Pathogenesis
Inhalation anthrax is the most pathogenic form of the disease. Inhaled B. anthracis spores germinate following phagocytosis, disseminating to nearby regional lymph nodes and the red pulp of the spleen. Lymphatic dissemination precedes hematogenous spread and terminal haemorrhagic septicemia. The high bacterial load of terminal anthrax (~109 bacilli ml–1) is favorable to its life cycle, especially given that it increases the probability that extremely stable infectious spores will be deposited into the soils surrounding an animal carcass for the next victim to contact, inhale, or ingest (Beyer and Turnbull 2009). Endowing B. anthracis with its extreme pathogenicity are its virulence plasmids, pXO1 and pXO2, which produce two major virulence factors, anthrax toxin and a poly-γ-d-glutamate (γDPGA) capsule, respectively.
1.2 Vaccine
To thwart anthrax’s march at the start of the twentieth century timely vaccination programs were instituted. Modernity and industrial-scale agricultural processing and global and railway transportation of animals, hides, and furs led to the heightened severity of worldwide epidemics (Turnbull 1991). Pasteur’s prototypic bacterial vaccine worked as the encapsulation virulence plasmid, pXO2, was heat-cured, attenuating the bacillus’ virulence (Mikesell et al. 1983). Thus the anthrax toxin protective antigen (PA) was likely present in the earliest toxigenic cellular vaccine. More readily deployable and practical livestock vaccinations were required, and Max Sterne later isolated the toxigenic but “uncapsulated” 34 F2 strain (Sterne 1939), which today is the live-spore vaccine used for livestock (Turnbull 1991).
1.3 Biowarfare and Bioterrorism
Bioweaponization has again made anthrax an existential threat to civilization. Humankind’s tragic attraction to chemical and biological weapons is rooted in the Greek, τόξον (toxon, meaning bow or arrow) (Mayor 2008). Anthrax weaponization is not hard to imagine given its infamous history as a ravager of civilizations, its ability to manifest itself asymptomatically in the early-stages of infection, its rapid progression to acute disease, and its ability to be widely disseminated in the war theater as a highly infectious airborne aerosol of resilient spores. Correspondingly, the eminent threat of biowarfare intensified anthrax vaccinology research. Live-spore vaccines used in the former Soviet Union and China for military were not without risk. In Britain, an acellular culture filtrate of B. anthracis 34 F2 containing anthrax toxin PA, was prepared and combined with the adjuvant alum to make Anthrax Vaccine Precipitated (Belton and Strange 1954; Strange and Belton 1954; Wright et al. 1954). The American acellular vaccine, Anthrax Vaccine Adsorbed, now BioThrax (Pittman et al. 2002; Wasserman et al. 2003), is safe and is the best pre-exposure defense against inhalational anthrax (Friedlander et al. 1999).
Anthrax biowarfare and bioterrorism incidents throughout the twentieth century included clandestine German and highly organized Japanese (Williams and Wallace 1989) efforts in World War I and II, respectively. These incidents and subsequent bioweapons buildups during the Cold War led to the signing of the 1972 Biological Weapons Convention. Nonetheless, rogue nations, terrorist groups, and disturbed individuals unfortunately either maintained underground bioweapons efforts or carried out bioterrorist attacks. During the Amerithrax bioterrorism attack of 2001, spore-laden letters were sent in two separate mailings via the US Postal system to two US senators in Washington D.C., television and print media centers in New York City, NY, and a publisher in Boca Raton, FL, leading all told to 11 inhalation and 11 cutaneous infections, which ultimately caused 5 fatalities (Jernigan et al. 2002). Cleanup costs were $1 billion due to the tenacity of the spores in the environment. Cutting-edge genomic evidence (Rasko et al. 2011) identified the strain and flask from which the spores were derived. After the prime suspect committed suicide, the FBI closed its case.
2 Anthrax Virulence Factors
B. anthracis would be a benign soil bacillus were it not for its two large virulence plasmids, pXO1 (Mikesell et al. 1983) and pXO2 (Green et al. 1985; Uchida et al. 1985). These plasmids express its two principle virulence factors: the tripartite toxin, anthrax toxin, encoded on pXO1; and a unique peptidoglycan-anchored γDPGA capsule encoded on pXO2. The toxin dampens the innate and adaptive immune response in myriad of cell types, induces hypotension, assists in dissemination of the bacillus, and causes edema and a shock-like death (Baldari et al. 2006; Frankel et al. 2009; Moayeri and Leppla 2009). The capsule also functions to evade immune cell activities by thwarting phagocytosis.
2.1 Anthrax Toxin
Anthrax toxin, originally discovered by Harry Smith in the 1950s (Smith and Keppie 1954), is a binary toxin (Gill 1978; Barth et al. 2004) comprised of three ~80–90 kDa proteins, called lethal factor (LF), edema factor (EF), and protective antigen (PA) (Frankel et al. 2009; Collier 2009). LF and EF are the two enzymatically active A moieties; PA is the cell-binding and channel-forming B moiety, making anthrax toxin an A2B binary toxin. LF is a 90-kDa Zn2+ protease that cleaves numerous mitogen-activated protein kinase kinases (Duesbery et al. 1998; Vitale et al. 2000) and the innate immunity inflammasome sensor, NLRP1 (Levinsohn et al. 2012; Chavarría-Smith and Vance 2013), thus interfering with many host cell signaling pathways. EF is a 89-kDa Ca2+ and calmodulin-dependent adenylyl cyclase (Leppla 1982), which increases the concentration of cyclic-AMP, causing edema and facilitating dissemination (Dumetz et al. 2011). Crystal structures of LF and EF (Pannifer et al. 2001; Drum et al. 2002) and PA (Petosa et al. 1997) are known. PA is an 83-kDa Ca2+-binding proprotein that contains four domains, D1, D2, D3 and D4 (Fig. 9.1a). The D1′ domain is what remains of the D1 prodomain upon proteolytic cleavage. Cleaved PA oligomerizes, assembles with LF and EF, and ultimately transforms into a translocase channel that delivers LF and EF into the cytosol of host cells. Hence, while PA, LF, and EF are individually nontoxic, the binary combinations, PA + LF and PA + EF, are toxic and known as anthrax lethal toxin (LeTx) and edema toxin (EdTx), respectively.


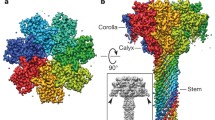
Monomeric and oligomeric PA structures. (a) Ribbons depiction of the 83-kDa monomeric PA proprotein (Feld et al. 2012b) colored by domain: the prodomain (D1, gray); LF/EF binding domain (D1′, green); channel-forming domain (D2, blue); oligomerization domain (D3, magenta); and receptor-binding domain (D4, gold). (b) Left, axial view of the PA8 prechannel (gray space-fill) co-complex with four LFN (blue) (Feld et al. 2010). LFN α1 (blue ribbon) is docked into the α clamp (green space-fill). Right, sagittal section of the PA8 prechannel with one LFN shown for clarity. (c) PA7 channel EM structure (blue surface) with indicated dimensions (Katayama et al. 2008). (d) Left, PA channel cartoon (gray) illustrating α- (green), ϕ- (green), and charge-clamp sites (Feld et al. 2012a). Right, clamp-site details and putative modes of action based on the structure/function of the α clamp-LFN α1 interaction (Feld et al. 2010), functional and spectroscopic measurements of the ϕ clamp (Krantz et al. 2005), or functional analysis of an electrostatic model of the β-barrel charge clamp (Wynia-Smith et al. 2012)
Holotoxin Assembly
To function, LeTx or EdTx holotoxin complexes must first self-assemble. Two proposed assembly pathways are known: on cell-surfaces (Collier and Young 2003; Collier 2009) or in serum (Ezzell and Abshire 1992; Panchal et al. 2005; Moayeri et al. 2007; Ezzell et al. 2009; Kintzer et al. 2009, 2010a, b). In the former, PA binds to a host-cell-surface receptors, called the anthrax toxin receptors (ANTXR1 and ANTXR2) (Bradley et al. 2001; Scobie et al. 2003). PA is a proprotein that must be cleaved between D1 and D1' (Fig. 9.1a) by a furin or furin-like protease (Klimpel et al. 1992; Molloy et al. 1992) to produce the 20-kDa (PA20) and 63-kDa (PA63) fragments. Similar fragments are also observed with trypsin nicking (Blaustein et al. 1989; Christensen et al. 2005). PA63 remains bound to the cell-surface receptor, allowing it to assemble at the cell surface to make ring-shaped heptameric (PA7) (Milne et al. 1994; Petosa et al. 1997; Lacy et al. 2004a) and octameric (PA8) (Kintzer et al. 2009, 2010a; Feld et al. 2010) prechannel oligomers (Fig. 9.1b). Prechannel oligomers then bind LF and EF to form holotoxin complexes.
In the serum of rabbits and guinea pigs, a protease resides there that can nick PA to form PA63 (Ezzell and Abshire 1992; Panchal et al. 2005). In bovine serum, this cleavage can allow PA63 to self-assemble with LF or EF to make assembled PA7- and PA8-prechannel LeTx or EdTx complexes (Kintzer et al. 2009, 2010a). The putative mammalian serum protease activity varies according to species (rats versus mice) (Moayeri et al. 2007), however, suggesting this mechanism may not be generalizable (Bann 2012). Nevertheless, several other highly active proteases are part of the B. anthracis secretome, and they can activate PA (Pflughoeft et al. 2014).
Host-Cell Receptor Binding
Following assembly, LeTx or EdTx complexes bind to cells via an extremely tight, sub-picomolar interaction with the anthrax toxin receptor (ANTXR) (Wigelsworth et al. 2004), as recently reviewed (van der Goot and Young 2009). Two ANTXR have been described, tumor endothelial marker 8 (TEM8) (Bradley et al. 2001) and capillary morphogenesis protein 2 (CMG2) (Scobie et al. 2003), but these have been renamed to ANTXR1 and ANTXR2, respectively (Young and Collier 2007). Another potential receptor, heterodimeric integrin β1, was also recently described (Martchenko et al. 2010). An ANTXR1/2 co-receptor was also described (Wei et al. 2006), albeit knockouts of the receptor show that it is not required in a mouse infection model (Young et al. 2007; Ryan and Young 2008). The widely studied receptors, ANTXR1 and ANTXR2, typically function as binding sites for the extracellular matrix, i.e., collagen type IV (Nanda et al. 2004) and basal lamina (Bell et al. 2001), respectively. Mutations in ANTXR2, for example, can lead to the rare genetic disorders juvenile hyaline fibromatosis and infantile systemic hyalinosis (Hanks et al. 2003). In vivo ANTXR2 is the major receptor mediating not only the pathology of toxemia but also the susceptibility to spore infection (Liu et al. 2009). The affinity of PA for either of these receptors is much greater than the physiological substrates, allowing PA to compete with natural ligands. The structure of ANTXR2 is known both in isolation (Lacy et al. 2004b) and in complex with PA monomer (Santelli et al. 2004) and the PA7 prechannel (Lacy et al. 2004a). ANTXR interactions are key to assembling (Kintzer et al. 2010b) and stabilizing holotoxin complexes (Miller et al. 1999; Lacy et al. 2004a; Kintzer et al. 2010b, 2012). A crystal structure of ANTXR1 (Fu et al. 2010) has also been reported.
Channel Formation
Upon endocytosis, the toxin-bearing vesicles traffic to the lysosome. Subsequent acidification of the endosomal compartment is essential to toxin action (Friedlander 1986). Lower pH conditions drive a conformational change in the PA oligomer, transforming it from the prechannel to the channel state (Blaustein et al. 1989; Miller et al. 1999; Kintzer et al. 2012). Overall, the putative channel is mushroom-shaped in its architecture (Fig. 9.1c), sharing much in common structurally with the known prechannel structures, notably the pore-facing loops localize similarly (Krantz et al. 2005). However, there are two major differences: first, the phenylalanine-clamp (ϕ-clamp) pore-facing loop, most critical to facilitating protein translocation, converges from a wider 25–30 Å diameter opening in the prechannel to a much narrower <10 Å diameter opening in the channel (Fig. 9.1d) (Krantz et al. 2005); and second, additional loops in a Greek-key motif unfurl and refold into a large ~100-Å-long, 14- or 16-stranded β barrel (Petosa et al. 1997; Nassi et al. 2002). Models of the β barrel (Nguyen 2004) are consistent with it being 12–15 Å in diameter, or about as wide as an α helix (Krantz et al. 2004), as seen in an autotransporter structure (Oomen et al. 2004).
LF/EF Unfolding and Translocation
Due to the aforementioned steric constraints, translocation of folded full-length LF and EF necessitates unfolding (Krantz et al. 2004, 2005). Endosomal acidification creates a destabilizing environment for LF and EF’s amino-terminal domains (LFN and EFN), facilitating their unfolding (Krantz et al. 2004). Furthermore, endosomal acidification establishes a proton motive force (PMF), which is comprised of the proton gradient chemical potential (ΔpH) and an electrical potential (Δψ) (Thoren et al. 2009; Brown et al. 2011). The ΔpH can drive the translocation of LF and EF (Krantz et al. 2006). Largely, in the endosomal context, where the latter Δψ is estimated to be minimal, it is the former ΔpH chemical gradient that drives translocation, and it was shown that the ΔpH on its own is sufficient (Brown et al. 2011). The ΔpH is critical to unfolding LF and EF as well as translocating their unfolded chains (Thoren et al. 2009). The mechanism of force transduction is largely consistent with a charge-state Brownian ratchet (Krantz et al. 2006; Basilio et al. 2009; Thoren et al. 2009; Pentelute et al. 2010; Pentelute et al. 2011; Brown et al. 2011; Wynia-Smith et al. 2012), albeit there are features of that model that do not explain all aspects of translocation, notably the high proton dependence of substrate unfolding (Thoren et al. 2009; Brown et al. 2011; Feld et al. 2012a). Following translocation into the cytosol, LF and EF refold and carry out their enzymatic functions, which serve to disrupt the host cell’s normal physiology.
2.2 γDPGA Capsule
Pathogenic strains of B. anthracis also secrete a γDPGA virulence factor (Candela and Fouet 2006), which consists of long, linear polypeptides (50–200 kDa) of d-Glu linked via amide linkages between the γ-carboxylate and α-amino group of adjacent monomers (Bruckner et al. 1953). Several gene products, Cap A-E, on the pXO2 virulence plasmid produce the thick capsule layer of γDPGA that surrounds the bacillus. While γDPGA was first isolated from B. anthracis, the genes responsible for its synthesis were determined by homology to B. subtilis (Ashiuchi et al. 2001). Cap A, Cap B, Cap C, and Cap E form the membrane-embedded molecular machinery that produces and secretes γDPGA (Candela and Fouet 2005; Candela et al. 2005). Cap D, anchors γDPGA to the peptidoglycan cell wall, and it also cleaves γDPGA into dissociable fragments; these function to greatly augment virulence and allow the bacillus to evade host innate immune defenses (Uchida et al. 1993; Candela and Fouet 2005). During infection, serum levels of γDPGA polymers reach up to 1–2 mg ml−1 in vivo (Boyer et al. 2009). The individual anthrax toxin components are secreted through the hydrogel layer of γDPGA capsule surrounding the bacillus. The free capsule polymers can interact with PA in vivo (Ezzell et al. 2009), and PA, LF and EF in vitro, and these interactions can facilitate holotoxin assembly and stability (Kintzer et al. 2012). γDPGA polymers can be either toxin-activating (Jang et al. 2011) or toxin-deactivating (Kintzer et al. 2012) depending on the molecular weight of the γDPGA and its preparation procedures.
3 PA Oligomerization
Assembled anthrax toxin complexes have been studied for many years with some controversy surrounding their stoichiometry and heterogeneity. Nevertheless, recent research has determined two possible PA oligomer stoichiometries and demonstrated new biochemical methods to isolate and detect particular complexes for physiological and biophysical studies.
3.1 PA Heptamer
The earliest work showed that the 83-kDa PA component can be cleaved by trypsin to make nicked PA (nPA); and nicking yields 63-kDa and 20-kDa fragments, called PA63 and PA20, respectively (Blaustein et al. 1989). PA63 was isolated from PA20 via mono Q anion-exchange chromatography; and PA63, but not PA20, formed cation-conducting channels in planar lipid bilayers (Blaustein et al. 1989). Anion-exchange purified PA63 also yields a stable oligomer, which was homoheptameric (PA7) by electron microscopy (Milne et al. 1994). Furthermore, PA oligomers were also observed in cell extracts, although the stoichiometry of the cell-surface-assembled complexes was not characterized (Milne et al. 1994). The oligomer was subsequently crystallized initially to low resolution (Petosa et al. 1997) and then to 3.6-Å resolution (Lacy et al. 2004a) and shown in either case to be a homoheptamer. The crystal structure reveals a large surface comprised of the D1′ domain and the twin Ca2+-ion-binding sites, where LF and EF were known to bind. There is extensively buried surface between D1′, D2, and parts of D3 between PA monomers. The inside of the oligomeric ring is defined by D2 and key pore loops shown to be critical for protein translocation via the PA channel. The D4 domains are located on the outside of the oligomer, and the D4 metal ion-dependent adhesion site, which is key for binding to the anthrax toxin receptor, is located on the bottom face opposite of the D1′ LF/EF-binding face (Lacy et al. 2004a).
To determine the PA:LF holotoxin co-complex stoichiometry, PA7 oligomers were extensively characterized by radioisotope-labeling, light-scattering, and ultracentrifugation studies (Mogridge et al. 2002a; Wigelsworth et al. 2004; Phillips et al. 2013). To simplify matters, the PA-binding, amino-terminal domain of LF (LFN) was used. When PA7 oligomers form co-complexes with LFN or LF the stoichiometry is consistent with 7:3 and the PA7LF3 complex (Mogridge et al. 2002a). Mutagenesis studies on PA revealed that binding interactions between PA and LFN occurred at PA2 dimer interfaces (Cunningham et al. 2002; Mogridge et al. 2002b), hence explaining the observed 7:3 stoichiometry. A prior study using native polyacrylamide gel electrophoresis, however, suggested that nPA and LF form co-complexes with a 1:1 stoichiometry (Singh et al. 1999).
3.2 PA Oligomer Heterogeneity
Electrospray ionization mass spectrometry (ESI-MS) determined that when nPA co-assembles with LFN it forms two distinct oligomers, the well-known heptamer (PA7) and a unique octamer (PA8), at 7:3 and 8:4 PA:LFN stoichiometries, respectively (Kintzer et al. 2009). Two even-numbered intermediates, PA2LFN and PA4(LFN)2, were also observed by ESI-MS. The unique PA8 species was not observed in prior radioisotope-labeling, light-scattering, crystallographic, and EM studies, because Q anion-exchange purified PA oligomers (the dominant form studied) are >95 % PA7 and contain only a small percentage (~5 %) of PA8 oligomers (Kintzer et al. 2009).
The PA8 oligomer assembly mechanism was characterized further by EM and electrophysiology. EM was used to estimate the ratio of PA7:PA8 oligomers, which was ~2:1 when nPA was co-assembled with LFN, EFN, or dimeric anthrax toxin receptor (Kintzer et al. 2009). Moreover, when carboxy-terminal His6-tagged PA was assembled upon Chinese hamster ovary (CHO) cells and subsequently purified by His6-affinity chromatography, it was also found that it formed a similar 2:1 ratio of PA7:PA8. Planar lipid bilayer electrophysiology studies showed that PA oligomer samples enriched in the PA8 species revealed a 20–30 % subpopulation of channels that had ~10 % greater conductance (Kintzer et al. 2009). These larger channels translocate LFN, EFN, LF and EF at similar rates to the PA7 oligomer, revealing PA8 oligomers are physiologically active translocases.
3.3 PA Octamer
The PA8 crystal structure was solved to 3.2 Å using a membrane-insertion loop deleted construct (PAΔMIL) (Kintzer et al. 2009). PAΔMIL unlike wild-type PA enabled purification of a high population (28 %) of free PA8 oligomers by Q anion-exchange. A unique property of the PA8 complex is that is it more pH and thermostable than the PA7 complex, and hence (PAΔMIL)7 complexes were removed from (PAΔMIL)8 by precipitating them away from a heterogenous mixture by treatment with ~10 % ethanol and mildly acidic conditions (pH 5.7). This procedure yielded homogeneous (PAΔMIL)8 complexes that crystallized in a square-planar architecture. (PAΔMIL)8 contains more extensively buried interface between adjacent PA subunits than the PA7 structure, when accounting for the difference in the MIL between the two constructs. Based on the known PA7 oligomer structure, it was suspected that the interaction of the MIL with neighboring PA subunits at the D4-D2 inter-PA interface could alter the oligomeric stoichiometry. This hypothesis was tested by chemically cross-linking the D4-D2 intra-PA interface. Depending upon the cross-link length, different oligomeric ratios could be produced, which favored the PA8 architecture (Feld et al. 2012b). An innovative synthetic octamerization strategy was devised by mutating the known D2-D3 oligomerization interface (Mogridge et al. 2001) in two separate PA monomer constructs, which could form a mutant, but functional, PA-PA interface (Phillips et al. 2013). When these two mutant PA are combined then only the even-numbered PA8 oligomers can form. This method works well and supports the physiological activity previously reported for WT PA8 complexes (Kintzer et al. 2010a).
3.4 Unique Serum Stability of PA8
Currently, WT PA8 oligomers can only be isolated as co-complexes with LFN, EFN, LF and EF. For example, a heterogenous 2:1 mixture of PA7(LFN)3 and PA8(LFN)4 is first produced by co-assembling nPA with LFN at pH 8, 0 °C. Then the mixture is pH- and temperature-challenged at pH 7, 37 °C, causing the PA7(LFN)3 complexes to precipitate as prematurely formed, insoluble channels (Kintzer et al. 2010a). EFN, LF and EF co-complexes with PA8 can be produced by ligand exchange using purified PA8(LFN)4 and an excess of the former (Kintzer et al. 2010a, 2012). Due to this stability difference, PA8LF4 predominated over PA7LF3 complexes in defibrinated bovine serum at physiological pH and temperature (37 °C) (Kintzer et al. 2010a). Moreover, the macrophage cytolysis activities of the PA7LF3 and PA8LF4 complexes were compared after incubations in defibrinated bovine serum finding that the PA8LF4 complex exhibited a 30-min half life, whereas the PA7LF3 complex was significantly less stable with a <5 min half life (Kintzer et al. 2010a). Hence, PA8 toxin complexes are more thermostable than the PA7 toxin complexes under conditions encountered in mammalian serum.
On cell surfaces, by contrast, PA oligomer complexes contact the soluble integrin-like domain of the anthrax toxin receptor (Lacy et al. 2004b). This interaction occurs via ANTXR2 and D2 and D4 of PA (Lacy et al. 2004a; Santelli et al. 2004). Since D2 and D4 are effectively stapled together by the ANTXR2 interaction, PA is unable to form the channel state readily unless the pH is dropped to about 5.5 (Lacy et al. 2004a). Receptor stabilization is roughly equivalent for PA7 and PA8 complexes; therefore, the stabilization advantage of the PA8 architecture over the PA7 architecture resides only within the milieu of the serum (Kintzer et al. 2010b). Since all of the observed PA in the serum of animal infection models late in infection is proteolytically activated as PA63 (Ezzell and Abshire 1992; Panchal et al. 2005; Ezzell et al. 2009), then significant serum-based assembly of anthrax toxin is possible, and octamer formation in the serum is another potential route of anthrax toxin assembly (Kintzer et al. 2009, 2010b). Detailed examination of serum-based assembly and the oligomeric architecture of toxin complexes in anthrax infection models are future areas of research.
4 Holotoxin Assembly
LF and EF bind to PA through a homologous ~250-residue PA-binding domain (referred to as LFN and EFN, respectively), and these domains share 35 % sequence identity and 55 % similarity (Quinn et al. 1991). The stoichiometry of assembled co-complexes with LF, EF, LFN, or EFN depends on the identity of the PA oligomer, where PA7 binds three LF and/or EF (Mogridge et al. 2002a; Wigelsworth et al. 2004; Kintzer et al. 2009, 2010a) and PA8 binds four LF and/or EF (Fig. 9.1b) (Kintzer et al. 2009, 2010a; Feld et al. 2010). Heterologous fusions to the C-terminal end of the PA-binding domain, LFN, was sufficient to deliver proteins into host cells in a PA-dependent manner, suggesting that the domain is necessary and sufficient for translocation (Arora and Leppla 1993; Milne et al. 1995). However, later studies, determined that amino-terminal His6, poly-lysine, and poly-arginine tags were also sufficient to deliver heterologous proteins into cells in a PA-dependent manner, albeit higher concentrations of heterologous substrate were required than with the LFN fusions (Milne et al. 1995; Blanke et al. 1996). While not understood at the time, these seemingly opposing sets of results revealed that PA oligomers contain two distinct binding subsites for LF and EF. One subsite was the tight-affinity site specific for the homologous folded domains of EFN and LFN, and the other subsite was a lower-affinity site that was far less specific and could bind unstructured peptides (like His6 tags).
LeTx Prechannel Core Complex Structure
Many subsequent mutagenesis studies confirmed that the binding sites comprised of specific contacts between PA and LFN and EFN (Cunningham et al. 2002; Lacy et al. 2002; Mogridge et al. 2002b; Lacy et al. 2005; Melnyk et al. 2006; Feld et al. 2010). Hydrogen exchange mass spectrometry (Melnyk et al. 2006) and rigid-body computational docking algorithms (Schueler-Furman et al. 2005; Lacy et al. 2005) were used to deduce the structure of a co-complex between PA oligomers and LFN. These models did not resolve why poly-cationic sequences (by themselves) were sufficient to localize heterologous proteins to PA oligomers. Also the computationally-docked model (Lacy et al. 2005) did not reconcile why PA point mutations (notably R178A) disrupted LFN binding (Cunningham et al. 2002).
The PA8 oligomer was exploited as a structural platform to answer these questions due to its enhanced thermostability and C4 symmetry. The structure (Fig. 9.1b) solved to 3.1 Å was representative of the PA8(LFN)4 co-complex (Feld et al. 2010). Unlike the isolated structure of LFN (Pannifer et al. 2001), the LFN subunits in the PA8(LFN)4 co-complex are in a partially unfolded conformation. This co-complex is one of the few examples, where the unfolding process has been visualized. The first α helix (α1) and β strand (β1) of LFN are in a partly unfolded conformation that is dissociated about 40 Å from the remainder of the folded structure (Feld et al. 2010). Hence, there are two distinct binding subsites for each LFN on the surface of the PA8 oligomer. In one subsite, the α1/β1 portion of LFN docks into a deep cleft between PA subunits, called the “α clamp”; and in the other subsite, the carboxy-terminal portion of of LFN binds in a separate subsite contained within a single PA subunit (Feld et al. 2010). While the latter subsite was well defined in mutagenesis and computer modeling studies (Lacy et al. 2005), the former α-clamp site was not. This discrepancy was due to the fact the modeling approach employed (Schueler-Furman et al. 2005) did not allow for conformational changes, such as partial unfolding.
The walls of the α-clamp cleft are comprised of the twin Ca2+ binding sites, which are fully-conserved within all known PA homologs (in other Clostridium and Bacillus binary toxins). The twin Ca2+ binding sites are such that their side chains are pointed inward toward the Ca2+ ions, leaving mostly backbone surface area contacts with the α1 helix. By virtue of the larger than average backbone exposure, the interactions with the α clamp are very nonspecific (Feld et al. 2010). The proposed mechanism of nonspecific helix recognition is consistent with the broad substrate specificity of calmodulin, a well known adapter, which uses a pair of twin Ca2+ sites to recognize α helices (Meador et al. 1992, 1993; Shen et al. 2005). Hence nonspecific and nonhomologous poly-cationic sequences also bind to the α-clamp cleft remarkably well (Feld et al. 2010), explaining prior observations that poly-cationic tracks are necessary and sufficient for heterologous protein delivery (Blanke et al. 1996).
5 PA Channel
Recent biophysical efforts have elucidated the basic structural features of the PA channel (Fig. 9.1c, d). The suggested resemblance of the PA channel to staphylococcal α hemolysin (Song et al. 1996; Benson et al. 1998) was based on morphological similarities not homology. The PA channel also possesses a ring-shaped oligomeric architecture, is SDS-resistent, and utilizes a hollow β-barrel domain to penetrate the membrane (Benson et al. 1998; Nassi et al. 2002). From modeling studies (Nguyen 2004), the β-barrel stem is extended, and it likely can accommodate structures as wide as an α helix (10–15 Å-wide) (Krantz et al. 2004; Oomen et al. 2004). The inward-facing pore loops within the soluble prechannel oligomer are also inward-facing within the cap of the channel (Krantz et al. 2005). By EM, the PA7 channel is indeed mushroom-shaped and approximately 170 Å tall × 125 Å wide at its maximum dimensions (Fig. 9.1c) (Katayama et al. 2008). The wider, cap is about 70 Å long, and like the prechannel, the topmost surface contains the LF/EF binding sites. The 100 Å long stem beneath the cap inserts into the membrane bilayer (Fig. 9.1d) (Katayama et al. 2010).
5.1 Peptide Clamps as Unfoldase and Translocase Active Sites
Functional electrophysiology studies laid the early groundwork in mapping the major active sites in the PA translocase, referred to collectively as “peptide clamps” (Fig. 9.1d) (Thoren and Krantz 2011; Feld et al. 2012a). These peptide-clamp sites have been empirically shown to catalyze the translocation of LF and EF into cells (Krantz et al. 2005; Feld et al. 2010; Wynia-Smith et al. 2012). Based on the locations of these clamps, the PA translocase channel can be divided into three sections: (i) the substrate docking surface on the top of the cap, which contains the “α clamp” (Feld et al. 2010); (ii) a critical hydrophobic constriction point with at opening near the base of the cap containing a ring of F427 residues, called the “ϕ clamp” (Krantz et al. 2005); and (iii) the highly anionic charged region in the solvophilic β-barrel stem portion, called the “charge clamp” (Wynia-Smith et al. 2012). Nonspecific substrate recognition is a key feature of a peptide translocase, because as the protein translocates, the segment of polypeptide sequence within the transporter will change continuously during translocation (Krantz et al. 2005; Thoren and Krantz 2011; Brown et al. 2011; Feld et al. 2012a; Wynia-Smith et al. 2012). In general, these clamps allow the PA channel to interact with a translocating polypeptide substrate via broad chemical or morphological features, imparting a high degree of nonspecificity (Thoren and Krantz 2011; Feld et al. 2012a). It is no surprise, therefore, that functional clamp sites are observed throughout the length of the channel.
Peptide-Clamp Paradox
The observed peptide clamp sites (Fig. 9.1d) may on their surface appear paradoxical: on one hand tight binding to clamp sites may impede protein translocation; on the other, when clamp sites are functionally disrupted, protein unfolding and translocation are kinetically inaccessible at physiological driving forces (Krantz et al. 2005; Feld et al. 2010; Thoren and Krantz 2011; Brown et al. 2011; Wynia-Smith et al. 2012). Because functioning clamp sites lower the activation energy of protein translocation, then these sites likely bind and release the translocating chain in a dynamic manner, much like an enzyme binds its substrate and releases product during catalysis. A protonation and deprotonation dynamic of the substrate, for example, could modulate interaction between the channel and a charge-clamp site. Under these conditions, a clamp site, therefore, is not a static binding site found in a more simplistic antibody-epitope interaction. Rather, a clamp site more closely resembles a dynamic binding site for polypeptide, where, for example, a clamp site can be modulated from a higher-affinity binding mode to a lower-affinity binding mode. Considering the structural plasticity of polypeptides and molecular machines, these putative dynamics are reasonable.
5.2 Transition to the Channel State
As the details of the structure of the PA channel have become apparent, much interest has been given to the acidic pH-induced conformational change from the prechannel to the channel (Bann 2012). Mutagenesis studies established a series of dominant-negative inhibitor (DNI) point mutations in PA that could allow for PA assembly into prechannel oligomers but would prevent formation of the channel state (Sellman et al. 2001b; Mourez et al. 2003). These possess therapeutic potential as a single copy of a DNI PA in an otherwise wild-type PA oligomer rendered the toxin harmless in vitro (Janowiak et al. 2009) and in a toxemia animal model (Sellman et al. 2001a). Hence DNI mutations provided the insight that certain molecular interactions occur in the channel that are less critical to prechannel oligomerization. 2-fluorohistidine substitution, however, did not effect the pH dependence of PA channel formation (Wimalasena et al. 2007), contrary to prior expectations that His residues sensed endosomal acidification to trigger the channel transition (Blaustein et al. 1989; Petosa et al. 1997).
Among the DNIs, the more notable D425K, F427A, and K397D mutations localize within the known PA7 and PA8 prechannel structures in adjacent pore-facing loops: the ϕ-clamp loop (2β10-2β11 loop) and the 2β7-2β8 loop. Biophysical characterizations of these sites demonstrate considerable inward narrowing and convergence of these loops as the channel forms. Electron paramagnetic resonance spectroscopy showed Cys-spin-labeled ϕ-clamp sites collapsed to a narrow pore of dimensions <10 Å (Krantz et al. 2005). Furthermore, bilayer-electrophysiology (Krantz et al. 2005), circular-dichroism (CD) spectroscopy (Kintzer et al. 2012), and liposome-pore-formation (Sun et al. 2008) assays revealed many mutations (such as F427A) slowed, but did not prevent, channel formation, whereas others (F427G, F427D, and F427R) blocked pore formation (Sun et al. 2008). Phylogenetic comparisons to the Clostridium binary toxins revealed covariance at two positions (D426Q and K397Q). PA K397Q channels were less functional translocases, but they could be complemented with the D426Q mutation, suggesting that the ϕ-clamp site and the 2β7-2β8 loop engaged each other through inter-PA contacts (Melnyk and Collier 2006). In summary, a major step in channel formation is the collapse and assembly of the ϕ clamp and adjacent 2β7-2β8 loop.
ϕ-clamp assembly, however, does not appear to be the only rate-limiting aspect of the mechanism of channel formation. The interfacial D2-D4 contacts on the periphery also must be disrupted not only within each individual PA subunit (intra-PA) but also between the membrane-insertion loop and a D4 in an adjacent PA subunit (inter-PA) (Kintzer et al. 2012). The intra-PA D2-D4 contacts occur in a water-coordinated interface between the two domains, where mutations in the interface can increase the pH threshold for channel formation in bilayers and by CD (Kintzer et al. 2012). Consistent with this view ANTXR2 (Lacy et al. 2004a; Sun et al. 2007; Kintzer et al. 2010b, 2012) or γ-DPGA (Kintzer et al. 2012) binding to PA oligomers stabilizes the intra-PA D2-D4 interface, and either of these ligands can significantly limit the rate of channel formation. Not surprisingly the loss of structure in intra- and inter-D2-D4 contacts is needed, because the membrane-insertion loops (2β2–2β3) situated between D2 and D4 must detach, unfold, and refold into the membrane-spanning β-barrel. The final step of PA channel formation involves the penetration of the β-barrel across the hydrophobic lipid bilayer as deduced from the D315A point mutation, which does not block channel formation in solution by CD, but does block insertion into planar lipid bilayers (Kintzer et al. 2012).
Thus while other pathways have been suggested (Bann 2012), a possible mechanism of prechannel-to-channel conversion (Kintzer et al. 2012) first requires the detachment of the various intra- and inter-D2-D4 interfaces in the oligomer; this dissociation or unfolding is followed by the collapse and proper organization of the ϕ-clamp site; an organized ϕ-clamp site then initiates the folding of the β-barrel; lastly, the barrel must penetrate the bilayer to form a functional translocase channel. The logic of the proposed scheme is not only based on functional mutagenesis studies but also on the idea that the prechannel must partially unfold before it can refold into the channel.
6 Translocation
Anthrax toxin is an excellent model system to study transmembrane protein translocation in part because the three subunits of the toxin can be expressed recombinantly and worked on in isolation. PA channels can be studied using whole-cell (Wolfe et al. 2005) and planar lipid bilayer electrophysiology (Finkelstein 2009) using a painted-membrane technique (Mueller et al. 1963). Single PA channels, when inserted into planar bilayers, form an open and ungated cation-conducting channel at modest potentials (Blaustein et al. 1989, 1990; Krantz et al. 2005). But when small molecule blocking ions (Blaustein and Finkelstein 1990; Blaustein et al. 1990; Orlik et al. 2005; Krantz et al. 2005) or protein substrates, such as LFN, are added, the conductance is blocked (Zhang et al. 2004a, b; Krantz et al. 2006; Thoren et al. 2009; Basilio et al. 2011b). Upon raising the driving force, substrate translocation may be monitored as the time-dependent increase in conductance. In an LFN translocation experiment under symmetrical pH 5.6, PA7 oligomer is added to the cis side to form channels; upon stabilization of the current, the cis compartment is perfused; LFN is added to block conductance and the cis compartment is prefused a second time, removing excess LFN; and finally the voltage is raised, driving LFN unfolding and translocation (Fig. 9.2a). Translocation is observed as PA channel conductance is restored and can be replotted to show a kinetic time course (Fig. 9.2b). With this method, the applied driving force can be externally controlled and continuously adjusted to identify key barriers in the translocation pathway (Zhang et al. 2004b; Krantz et al. 2006; Thoren et al. 2009; Thoren and Krantz 2011; Brown et al. 2011; Wynia-Smith et al. 2012).


Electrophysiological studies of translocation. (a) Ensemble planar lipid bilayer translocation experiment is shown for a bilayer formed between 1 mL chamber volumes and bathed in symmetric universal bilayer buffer (UBB) at pH 5.6 (Thoren et al. 2009). PA7 channel formation was recorded as the increase in conductance at a Δψ of 20 mV (cis positive). Upon stabilization, the cis side was perfused (perf.) with 10 mL of fresh UBB. Then LFN was added to the cis side, causing an exponential decrease in current as LFN docks inside the PA channels. Following a second perfusion of the cis side, the voltage was raised, driving the translocation of LFN. (b) To better observe the S-shaped translocation kinetics, the translocation phase from panel (a) is replotted. Time zero is set to the time when Δψ was raised to 70 mV. Fraction translocated is given as the level of current observed divided by the expected current, assuming a linear current-voltage relationship. (c) A single-channel translocation experiment under a 2-unit ΔpH and Δψ of 0 mV in asymmetrical KCl and pH conditions: cis buffer was 100 mM KCl, pH 5.6; trans buffer was pH 7.6 with no added KCl. A synthetic peptide (LF residues 1–50) was added to a single PA channel, and two-state opened (O) and closed (C) channel conductances were observed where a closed dwell time (τC) is highlighted. (d) τC from the recording in panel (c) were statistically analyzed as the CDF, P(τC ≤ t), or probability a τC is less than or equal to time, t
A second and perhaps more powerful technique has emerged to study protein translocation at the single-channel level (Kintzer et al. 2009; Thoren et al. 2009; Basilio et al. 2011b). With this method, a single PA channel can be inserted into a planar lipid bilayer, and peptide or protein substrate translocations can be inferred from the step-like openings and closings of the channel (Fig. 9.2c). Dwell times (τ) can be determined for when the channel is closed (τC) or open (τO). The τC values report on unblocking processes, including translocation and dissociation of the protein from the channel, whereas τO values report on the binding kinetics of the substrate to the channel. While individual measured τ values are stochastic, they can be analyzed statistically using the cumulative distribution function (CDF) (Fig. 9.2d). When combined with a driving-force analysis, translocation can be studied independently of dissociation. Single-channel assays have been used to determine pore size of the PA channel to deduce relative populations of PA7 and PA8 oligomers and determine their ability to translocate proteins (Kintzer et al. 2009); they have been used to observe transient intermediates during docking (Brown et al. 2011) and translocation (Thoren et al. 2009); and they have been employed to dissect the unusual S-shaped kinetics of translocation (Basilio et al. 2011b). The only disadvantage of single-channel analyses of translocation is the time investment of the investigator in the assays. Otherwise, there are tremendous advantages: single-channel assays, for example, can elucidate more complex chemical kinetic mechanisms, observe intermediate conductance states, and determine the order of transient kinetic intermediates.
Topologically, LF and EF initiate translocation starting from their unstructured 20–30 residue-long amino-terminal ends (Zhang et al. 2004a). Albeit when poly-cationic tracks are added to the carboxy-terminal end of LFN or heterologous fusions (Milne et al. 1995), the protein can also enter the PA channel and translocate into cells at 10- to 100-fold reduced efficiency (Sharma and Collier 2014). Nevertheless, because the amino-terminal ends are unstructured, it is unlikely LF and EF translocate to any significant extent from their carboxy-terminal ends, since the latter would require their unfolding to produce significant unstructured carboxy-terminal peptide to initiate translocation. Initiation (or docking) is charge and sequence (Pentelute et al. 2010, 2011; Brown et al. 2011; Wynia-Smith et al. 2012); length (Zhang et al. 2004a); pH (Brown et al. 2011); and PA-channel dependent at both the ϕ- (Krantz et al. 2005) and α-clamp sites (Feld et al. 2010). Empirically, LF, EF, LFN and EFN translocation kinetics are highly complex and cannot be fit to simple single-exponential functions; rather these kinetics are S-shaped (Fig. 9.2b) and indicative of many consecutive steps (Zhang et al. 2004b; Krantz et al. 2005, 2006; Thoren et al. 2009; Brown et al. 2011; Wynia-Smith et al. 2012). The relative lifetime of these complex kinetic records has been quantified by measuring the time for half of the substrate to translocate (t½) (Krantz et al. 2006).
6.1 Driving Forces and Barriers
For anthrax toxin, protein unfolding and translocation are driven by the PMF comprised of a Δψ and a ΔpH (Thoren et al. 2009). The electrical potential, Δψ, relates to the activation energy of translocation, ΔG‡(Δψ)
ΔG‡° is the ΔG‡ when Δψ is 0 mV; F is Faraday’s constant; and z is the charge required to cross the limiting barrier. Similarly, a chemical potential-modulated energy barrier, ΔG‡(ΔpH), relates to ΔpH
ΔG‡° is the ΔG‡ when ΔpH is 0 units; n is the number of protons used to cross the limiting barrier; R is the gas constant; and T is the temperature. ΔG‡(Δψ, ΔpH) values can be estimated by computing t½ values (in seconds) extracted from primary kinetic records under different applied potentials, Δψ or ΔpH, where c is a one-second constant
In principle and in practice, either of these driving forces can affect the activation energy of the two major barrier-crossing steps in the mechanism of translocation (Thoren et al. 2009). A pure Δψ can drive the translocation of the small substrate, LFN (Zhang et al. 2004b; Krantz et al. 2005, 2006; Thoren and Krantz 2011). However, EFN, LF, and EF translocate poorly under a pure Δψ (Krantz et al. 2006; Wynia-Smith et al. 2012), and efficient translocation of these substrates also requires a ΔpH (Krantz et al. 2006; Kintzer et al. 2009; Feld et al. 2010). A diode-like rectifying blockade of PA channels with LF was reported under a pure Δψ, even at high driving potentials, demonstrating inefficient translocation under that condition (Halverson et al. 2005). The ΔpH by itself, on the other hand, is sufficient for translocation, and mutagenesis of LFN suggests protonation and deprotonation of Asp and Glu residues play a key role in harnessing the ΔpH (Brown et al. 2011). Hence the PA channel is a proton/protein symporter (Krantz et al. 2006; Brown et al. 2011; Wynia-Smith et al. 2012).
Overall, in the translocation mechanism, there are two major limiting barriers (Thoren et al. 2009). In ΔG‡(Δψ, ΔpH) versus driving force (Δψ or ΔpH) plots, the observed curves are not linear as would be expected for a simple single-barrier crossing kinetic mechanism. (Of course, a single-barrier model cannot be expected to fully describe translocation records that are S-shaped.) Instead, ΔG‡ versus Δψ or ΔpH plots are boomerang-shaped with two asymptotic linear extremes, indicating that there are two major limiting barriers in the translocation mechanism (Thoren et al. 2009). One barrier has a high driving-force dependence, i.e., a high z value or n value for Δψ-dependent and ΔpH-dependent translocation, respectively. The second has a ~10-fold lower diving-force dependence. A two-major-limiting-barriers model was fit using mean-transit-time theory for two sequential first-order reactions (Fersht 1998; Thoren et al. 2009). From a series of destabilizing point mutations in LFN, the high driving-force-dependent barrier was shown to be limited by protein unfolding, but the lower-force-dependent barrier was not dependent on unfolding. This two-barrier model pertains only to the steps following substrate docking and initiation as deduced from single-channel experiments (Thoren et al. 2009). The oversimplification of the two-major-limiting-barriers model is that the folding and translocation processes could have multiple similar height barriers in succession, which is likely the case for the latter lower-force unfolding-independent translocation barrier. (See arguments below).
Barrier-less Drift-diffusion Model of Translocation
An alternative barrier-less model of translocation, the drift-diffusion model, has been applied to LFN translocation (Basilio et al. 2011b). Based on electrodiffusion, the model represents LFN as a rigid and positively charged rod dominated by Brownian motion and an applied Δψ. Over a narrow Δψ range (45−60 mV) the model fits the S-shaped single-channel translocation records remarkably well. Yet, fits to the semi-natural-log t½ versus Δψ plot are crudely linear. By comparison, when a larger dynamic range of Δψ records (from 30 to 150 mV) are analyzed, however, the empirical data are not linear on a semi-log plot (RT ln t½ versus Δψ), but rather the empirical relation is boomerang shaped with two linear extrema (Krantz et al. 2006; Thoren et al. 2009; Brown et al. 2011; Wynia-Smith et al. 2012). The experimental results of Basilio (Basilio et al. 2011b) are important in that they imply the post-unfolding step(s) contain many small barriers. But, the barrier-less drift-diffusion model, while a nice addition, is overly simplified, since it ignores unfolding, a process shown to limit translocation (Wesche et al. 1998; Zhang et al. 2004b; Thoren et al. 2009).
Toward a Unifying Barrier Model
A unifying interpretation is that translocation is universally barrier limited. This hypothesis is cogent with myriad of biochemical processes: protein folding and unfolding, peptide binding and dissociation, and enzyme catalysis (Fersht 1998). At low potentials <50 mV, the major limiting barrier(s) is (are) dominated by unfolding (Thoren et al. 2009; Brown et al. 2011). This notion is evident in that destabilizing point mutations shift the observed ΔG‡ versus Δψ curves at voltages <50 mV but not at higher potentials (Thoren et al. 2009). At higher potentials, translocation is not limited by unfolding, but rather it is logically hypothesized to be limited by the movement of the unfolded chain through the channel (Thoren et al. 2009). To maintain S-shaped kinetics, i.e., when the latter translocation barrier is limiting at higher potentials, a unifying picture may be that translocation requires the crossing of many equivalent height barriers, which must be traversed in serial/consecutive order. Kinetic schemes comprised of a series of irreversible steps with similar lifetimes may be analyzed using a gamma-function (Feller 2008), and these models yield lag-phase S-shaped kinetics (Floyd et al. 2008). A multi-barrier-limited interpretation also explains the use of an extremely slow diffusion constant (D) of 10−11 cm s−1 in the drift-diffusion model (Basilio et al. 2011b) (much slower than the D of 10−8 cm s−1 expected for free diffusion through a narrow pore (Simon et al. 1992; Chauwin et al. 1998)). The slower D estimated by the drift-diffusion model for PA suggests the channel binds and releases the peptide chain (likely at the peptide-clamp sites) in a repetitious manner along the length of the protein substrate (Krantz et al. 2005; Thoren and Krantz 2011; Feld et al. 2012a). Consistently, other slow D constants of 10−15 cm s−1 have been estimated for mitochondrial protein import and were rationalized as being due to peptide-pore interactions (Chauwin et al. 1998). In summary, binding and release steps are barrier-limited events immediately analogous to peptide-protein binding and dissociation reactions.
A unifying model of unfolding and translocation will be broadly relevant to understanding transmembrane protein translocation in the cell and mitochondrion (Simon et al. 1992; Chauwin et al. 1998; Huang et al. 1999) and the unfolding and translocation by ring-shaped oligomeric ATP-dependent disaggregase, unfoldase, and degradase molecular machines (Burton et al. 2001; Kenniston et al. 2003). At the very highest potentials, the observed rate of PA translocation begins to plateau in a way consistent with the maximal rates observed in many other ATP-dependent molecular machines that unfold and translocate proteins in the cell (Thoren et al. 2009). Of particular note, single-molecule translocation studies, which measure displacements and forces, show step-wise displacement, consistent with nm-length movements during translocation (Aubin-Tam et al. 2011; Maillard et al. 2011). Step-wise displacements in the anthrax toxin translocation mechanism would manifest in a series of consecutive barrier crossings and hence the empirically observed S-shaped kinetics.
6.2 Translocation-Coupled Unfolding
The earliest indication that protein unfolding limited protein translocation came from cellular studies of anthrax toxin translocation using heterologous LFN fusions (Wesche et al. 1998). Addition of fold-stabilizing ligands and disulfide bonds further supported this view that unfolding was required prior to translocation (Wesche et al. 1998; Zhang et al. 2004b; Juris et al. 2007). Protons, on the other hand, are likely a key physiological ligand that serves to assist in unfolding LFN and EFN through the destabilization of their native states (Krantz et al. 2004). Correspondingly, ΔG‡ values for translocation at low driving forces (~50 mV) correlate with equilibrium changes in stability (Krantz et al. 2006). Because the unfolding-limited barrier coincides with the force-dependent barrier, the unfolding transition state (TS) was mapped onto the structure of LFN using 21 different destabilizing point mutations (Thoren et al. 2009). Upon applying protein folding ϕ analysis (Fersht 1998), it was found that the unfolding TS is asymmetrically localized in the β-sheet subdomain of LFN; hence, the β-sheet subdomain is the “mechanical breakpoint” (Thoren et al. 2009) (according to nomenclature used in mechanical unfolding studies (Crampton and Brockwell 2010)). Once the rate-limiting β-sheet-subdomain structure is ruptured the rest of the LFN unfolds (Thoren et al. 2009).
The aforementioned translocation-coupled unfolding mechanism deduced from the study of the destabilized LFN mutants (Thoren et al. 2009) is also supported well by the crystal structure of the PA8(LFN)4 prechannel core complex (Feld et al. 2010). The α1 helix and β1 strand of LFN are unfolded in that prechannel structure, and this partially unfolded starting state was verified in the channel-LFN co-complex using planar bilayer electrophysiology (Feld et al. 2010). One interesting destabilizing mutation in LFN (M40A), the only outlier in that study, exhibited an unusually negative translocation-coupled unfolding ϕ value (Thoren et al. 2009). The negative ϕ value was indicative of slow translocation despite the fact that LFN M40A is destabilized. This residue is in the center of the α1 helix, which is situated in the α-clamp site on the surface of the PA8(LFN)4 structure. Mutations in this site affect the stability of the co-complex, where M40A caused LFN to bind more tightly to the PA channel (Feld et al. 2010). Tight binding correlated with slower translocation for multiple substitutions at M40. Furthermore, B-factor analysis and fluorescence anisotropy studies corroborated that the partial unfolding of the α1/β1 structure leads to increased disorder in the β-sheet subdomain (Feld et al. 2010). Therefore, prior to translocation, these interactions with the α clamp initiate some destabilization to the key rate-limiting structure in the LFN substrate.
6.3 Brownian-Ratchet Mechanism
A Brownian-ratchet (Astumian 1997), or charge-state-ratchet (Krantz et al. 2006), model of translocation is one possible explanation for how the ΔpH may be harnessed by the PA channel to produce physical force on the substrate protein (Fig. 9.3a). In this mechanism, acidic residues in the substrate protonate on the lower pH side upon entering the PA channel. Protonation is required because the channel is cation-selective (Blaustein et al. 1989) and hence anion-repulsive to deprotonated Asp/Glu residues (Krantz et al. 2006). Brownian motion then dominates the chain within the channel allowing it to move past the major cation-selective site. As protonated acidic groups emerge beyond the major cation-selective (i.e., anion-repulsive) site in the channel via Brownian motion, they then deprotonate down gradient to the higher pH side, thus reforming a net anion-anion repulsion between the chain and the channel. Therefore, the asymmetric ΔpH condition effectively enforces the direction of protonation and deprotonation, providing directionality to translocation.


ΔpH force-transduction models. (a) Charge-state Brownian ratchet (Krantz et al. 2006) as described in the text. The ϕ clamp constriction, charge clamp, proton binding (H+), and Brownian motion (↔) are indicated. Peptide chain is solely in the extended-chain configuration (Basilio et al. 2011a). (b) Helix-compaction model as described in the text (Feld et al. 2012a). The α clamp is also indicated. Peptide chain is either α-helical (coiled loops) or extended chain. The displacement of the “load” (black sphere) in either model is illustrative and meant only to indicate application of a productive force that could unfold or translocate a protein substrate
Empirical evidence for the ΔpH-driven charge-state ratchet has come from 2-sulfonato-ethyl-methanethiosulfonate (MTSES) modification of Cys residues or semisynthetic chemistry, either of which added an SO3− modification to LFN (Basilio et al. 2009; Pentelute et al. 2010). These modifications are effectively non-protonatable anionic sites at physiological pH. Many, but not all, of these SO3−-modified LFN showed significant decreases in the rate of translocation and suggested anionic sites were blocked by the strong anion-repulsive features of the channel. The positional effects of these SO3− modifications were complex, appearing to combine sterics and local electrostatics (Basilio et al. 2009). Additional semisynthetic peptide ligation studies showed positive and negative charges were required to utilize the ΔpH (Pentelute et al. 2011).
Detailed mutagenesis of LFN showed replacement of all anionic residues on the amino terminus up to residue 85 slowed the relative rate of ΔpH-driven translocation 100–1000 fold (Brown et al. 2011). An LFN with an uncharged amino terminus (i.e., no D, E, H, K, or R residues) was similarly deficient, but when only Asp/Glu residues were reintroduced at various positions, especially between residues 20 and 30 (the “20s cassette”), then translocation could be restored. Reintroduction of positively charged residues on their own did not stimulate ΔpH-driven translocation. The position of the 20 cassette is critical to driving the unfolding of LFN. When an uncharged spacer was inserted between the 20s cassette and the folded portion of LFN, then the rate of translocation was greatly impeded. However, this uncharged-spacer defect could be reversed with the LFN L145A (Thoren et al. 2009) destabilizing point mutation (Brown et al. 2011). A chimera study was conducted, which recombined the amino-terminal ends of LFN and EFN, with the goal to understand why EFN translocated 100-fold slower than LFN (Wynia-Smith et al. 2012). The major difference in the ΔpH-driven translocation of EFN is a general lack of negatively charged residues in EFN’s effective “20s cassette” (using LFN residue numbering). When too many Asp/Glu residues were added to EFN’s effective “20s cassette”, however, translocation slowed; therefore, an appropriate balance of charges is needed in the force-generation cassette (Wynia-Smith et al. 2012). Thus the highly charged 20 cassette is the ΔpH force-transduction cassette, and its position and Asp/Glu charge composition are required for ΔpH-driven translocation.
A charge-clamp site was electrostatically modeled to the upper portion of the β barrel; and when the strongly anionic clamp was mutated to neutral residues, cation-selectivity was reduced, and Δψ- and ΔpH-driven translocation was impeded (Wynia-Smith et al. 2012). While the major electrostatic sites both in the substrate and the channel have been identified, there are some curious deficiencies with the Brownian-ratchet mechanism when applied to anthrax toxin translocation. First, the location of the 20s cassette relative to the folded domain greatly affected ΔpH-driven translocation, suggesting mechanical force generation has significant positional requirements (Brown et al. 2011). A positional requirement is not anticipated in a Brownian-ratchet model, because this ratchet does not generate large enough force to unfold proteins, and the channel would need to wait for the protein to unfold prior to translocation (Glick 1995). However, if the channel were to wait for unfolding to equilibrate prior to translocating (as suggested (Basilio et al. 2011b)), then the location of the 20s cassette would not greatly influence the translocation kinetics (Brown et al. 2011). The next section considers these perceived deficiencies and provides an update to the overall mechanism.
6.4 Molecular Basis of Force Transduction
A key aim in the investigation of ΔpH-driven protein translocation is to understand the molecular basis of force transduction. The charge-state Brownian ratchet (Krantz et al. 2006) is compelling, but the energies garnered from a Brownian ratchet are near RT. These energies are lower than what may be needed to unfold LF and EF on a reasonable timescale. The protein unfolding step is more highly proton dependent than the subsequent translocation step; moreover, unfolding possesses a strong positional requirement for anionic, proton-binding residues in the 20s cassette of LFN. A complementary view is that there are two phases to this force-transduction mechanism (Brown et al. 2011; Feld et al. 2012b; Wynia-Smith et al. 2012). The first phase is a proton-binding step that manifests in a force-generating power stroke required for unfolding, and the latter phase contains the salient features of the charge-state Brownian ratchet.
The mechanics of force transduction requires some consideration of the conformation of the translocating polypeptide chain. There are two different views. One view, the “extended-chain model” (Basilio et al. 2011a), is that the translocating chain is acted upon in the extended-chain configuration, where ϕ/ψ angles would be 180°/180°. In that model for significant force to be applied to the load (the folded protein) the chain would need to be fully extended. Another view, the “helix-compaction model” or “proton-helix engine model” (Feld et al. 2012a) considers that the translocating chain could bind protons and convert from a more extended conformation to a more helical one (Fig. 9.3b). This interconversion would substantially contract the end-to-end length of a polypeptide about 1.5-2 Å per residue, allowing for force to be applied to the load. Alternatively a formed helix may be electrostatically pulled into the channel via interactions with the positive end of its helical dipole and other residue charges induced upon protonation. Either mechanism would yield a similar net favorable displacement on the load. While the former extended-chain model considers the chain to remain in a single conformation, the latter model considers that the translocating chain cycles between contracted α-helical and extended conformations in myriad serial steps along the length of the substrate chain.
The helix-compaction model (Fig. 9.3b) has emerged from recent data showing helical structure in the α clamp of the prechannel co-complex with LFN (Feld et al. 2010). Also surprisingly slow kinetics (orders of magnitude slower than LFN translocation) are observed in streptavidin-trapping experiments of a highly extended 33-residue portion of LFN in the PA channel (Basilio et al. 2011a). In the current picture, the various peptide-clamp sites in the PA channel coordinate their activities to promote the cyclic turnover of translocating chain from the extended-chain state to the collapsed and compacted helical state. Proton binding would either lead to helical compaction of a small portion of the translocating chain into the PA channel or the net attraction of a preformed helix deeper into the channel. For either possibility, proton-driven compaction or attraction may then be the major force generation step or “power stroke” (Glick 1995) in this mechanism, explaining the positional effects of the 20s cassette observed in the unfolding of LFN (Brown et al. 2011). Subsequently, the now helical segment would then have to convert to a more extended state. This transition could be driven by a putative conversion of the ϕ-clamp site to a more constricted state (only accommodating to extended chain). This conversion is suggested by work done on the ϕ-clamp-loop network in the PA channel (Melnyk and Collier 2006), and also by the fact that the ϕ-clamp loop has been solved in two different structures in the two different PA8 complexes (3HVD (Kintzer et al. 2009) and 3KWV (Feld et al. 2010)) (Feld et al. 2012a). Once in the extended-chain state, the charge-state Brownian-ratchet phase of the mechanism ensues. Interaction at the the α-clamp and ϕ-clamp sites may serve as backstops to prevent retrotranslocation during chain extension past the charge clamp, thus favoring that the polypeptide release its protons down gradient instead (Fig. 9.3b). The now anionic peptide will be trapped as the charge-clamp will repel the peptide, preventing its retrotranslocation (Krantz et al. 2006; Brown et al. 2011; Wynia-Smith et al. 2012). The transport cycle can then repeat on the next segment of polypeptide.
Allosteric associations between proton binding and the α, ϕ, and charge clamps may be likely pivotal to the latter mechanism (Feld et al. 2012a). For example, the ϕ clamp may be triggered to change state when the α clamp is either occupied or vacated; and/or the degree of protonation of the substrate or clamp site may trigger a change in state. If proton binding directly drives clamp transitions, then uncharged sequences could be moved through the channel, albeit without the benefit of the charge clamp acting to prevent backsliding. Overall, this conceptualization should serve as a framework for continued experimentation. Future work should address the allostery and mechanics of the ϕ-, α- and charge-clamp sites both structurally through EM, NMR and crystallography and functionally through single-channel electrophysiology.
7 Co-translocation Factors from the Host
The PA channel is sufficient to translocate LF and EF in a fully reconstituted planar bilayer electrophysiology assay at room temperature (Zhang et al. 2004b; Krantz et al. 2005, 2006; Thoren et al. 2009; Feld et al. 2010; Brown et al. 2011; Wynia-Smith et al. 2012). However, ATP-dependent and -independent cytosolic factors (including heat shock protein 90, thioredoxin reductase and the β subunit of the coat protein complex) can expedite diphtheria toxin A domain (DTA) translocation (Ratts et al. 2003, 2005) and the heterologous LFN-DTA fusion substrate (Tamayo et al. 2008). Yet the role of these chaperones in anthrax toxin LF and EF translocation is less clear; the enhancement of DTA enzyme activity may be peculiar to DTA; it could either indicate that chaperones assisted unfolding and translocation or chaperones functioned post-translocationally to refold the heterologous substrate. For instance, in cellular assays, drug compounds, which inhibit the chaperones, heat shock protein 90, and cyclophilin, block LFN-DTA but not LF translocation (Dmochewitz et al. 2011). If host-cell chaperones significantly assist LF and EF translocation, it is unlikely that cellular chaperones could operate in pure isolation because a portion of LF or EF must translocate via the PA channel in order to then allow a host chaperone to engage and complete translocation.
8 Therapeutics and Biotechnologies
There are many technologies poised to either inhibit or exploit the PA channel as a new protein-delivery platform. Since the studies of tetraalkylammonium ion blockade (Blaustein and Finkelstein 1990), a variety of other tetra-substituted ammonium and phosphoniums and other cationic small molecules have been tested as potential inhibitors of the channel (Orlik et al. 2005; Krantz et al. 2005). Derivatives of β-cyclodextrin have been successfully utilized as potent inhibitors of the staphylococcal α-toxin pore-forming channel (Gu et al. 1999). Numerous similar β-cyclodextrin derivatives (Nestorovich and Bezrukov 2014) were synthesized with multiple cationic sites to complement the anionic charge of the PA channel; and channel blockade appears to be a primary mode of action (Karginov et al. 2005). The β-cyclodextrin approach is applicable to other homologous Clostridium binary toxin channels (Bezrukov et al. 2012). The PA channel is also being exploited: to treat cancer through targeted cytotoxicity (Wein et al. 2013; Phillips et al. 2013; McCluskey and Collier 2013), to deliver antigens and active proteins into T cells (Shaw and Starnbach 2008), and to dissect innate immune sensing pathways (Kofoed and Vance 2011; Zhao et al. 2011; von Moltke et al. 2012). New protein engineering approaches with native peptide ligation will allow this protein-delivery science to scale to highly flexible screening platforms (Liao et al. 2014). These are just some of the many new and exciting PA channel therapeutics and biotechnologies.
References
Arora N, Leppla SH (1993) Residues 1-254 of anthrax toxin lethal factor are sufficient to cause cellular uptake of fused polypeptides. J Biol Chem 268:3334–3341
Ashiuchi M, Nawa C, Kamei T et al (2001) Physiological and biochemical characteristics of poly gamma-glutamate synthetase complex of Bacillus subtilis. Eur J Biochem FEBS 268:5321–5328
Astumian RD (1997) Thermodynamics and kinetics of a Brownian motor. Science 276:917–922
Aubin-Tam ME, Olivares AO, Sauer RT et al (2011) Single-molecule protein unfolding and translocation by an ATP-fueled proteolytic machine. Cell 145:257–267. doi:10.1016/j.cell.2011.03.036, S0092-8674(11)00313-8 [pii]
Baldari CT, Tonello F, Paccani SR, Montecucco C (2006) Anthrax toxins: a paradigm of bacterial immune suppression. Trends Immunol 27:434–440. doi:10.1016/j.it.2006.07.002
Bann JG (2012) Anthrax toxin protective antigen-Insights into molecular switching from prepore to pore. Protein Sci 21:1–12. doi:10.1002/pro.752
Barth H, Aktories K, Popoff MR, Stiles BG (2004) Binary bacterial toxins: biochemistry, biology, and applications of common Clostridium and Bacillus proteins. Microbiol Mol Biol Rev 68:373–402
Basilio D, Jennings-Antipov LD, Jakes KS, Finkelstein A (2011a) Trapping a translocating protein within the anthrax toxin channel: implications for the secondary structure of permeating proteins. J Gen Physiol 137:343–356. doi:10.1085/jgp.201010578, jgp.201010578 [pii]
Basilio D, Juris SJ, Collier RJ, Finkelstein A (2009) Evidence for a proton-protein symport mechanism in the anthrax toxin channel. J Gen Physiol 133:307–314
Basilio D, Kienker PK, Briggs SW, Finkelstein A (2011b) A kinetic analysis of protein transport through the anthrax toxin channel. J Gen Physiol 137:521–531. doi:10.1085/jgp.201110627, jgp.201110627 [pii]
Bell SE, Mavila A, Salazar R et al (2001) Differential gene expression during capillary morphogenesis in 3D collagen matrices: regulated expression of genes involved in basement membrane matrix assembly, cell cycle progression, cellular differentiation and G-protein signaling. J Cell Sci 114:2755–2773
Belton FC, Strange RE (1954) Studies on a protective antigen produced in vitro from Bacillus anthracis: medium and methods of production. Br J Exp Pathol 35:144–152
Benson EL, Huynh PD, Finkelstein A, Collier RJ (1998) Identification of residues lining the anthrax protective antigen channel. Biochemistry 37:3941–3948
Beyer W, Turnbull PCB (2009) Anthrax in animals. Mol Aspects Med 30:481–489. doi:10.1016/j.mam.2009.08.004
Bezrukov SM, Liu X, Karginov VA et al (2012) Interactions of high-affinity cationic blockers with the translocation pores of B. anthracis, C. botulinum, and C. perfringens binary toxins. Biophys J 103:1208–1217. doi:10.1016/j.bpj.2012.07.050
Blanke SR, Milne JC, Benson EL, Collier RJ (1996) Fused polycationic peptide mediates delivery of diphtheria toxin A chain to the cytosol in the presence of anthrax protective antigen. Proc Natl Acad Sci U S A 93:8437–8442
Blaustein RO, Finkelstein A (1990) Voltage-dependent block of anthrax toxin channels in planar phospholipid bilayer membranes by symmetric tetraalkylammonium ions. Effects on macroscopic conductance. J Gen Physiol 96:905–919
Blaustein RO, Koehler TM, Collier RJ, Finkelstein A (1989) Anthrax toxin: channel-forming activity of protective antigen in planar phospholipid bilayers. Proc Natl Acad Sci U S A 86:2209–2213
Blaustein RO, Lea EJ, Finkelstein A (1990) Voltage-dependent block of anthrax toxin channels in planar phospholipid bilayer membranes by symmetric tetraalkylammonium ions. Single-channel analysis. J Gen Physiol 96:921–942
Boyer AE, Quinn CP, Hoffmaster AR et al (2009) Kinetics of lethal factor and poly-d-glutamic acid antigenemia during inhalation anthrax in rhesus macaques. Infect Immun 77:3432–3441. doi:10.1128/IAI.00346-09, IAI.00346-09 [pii]
Bradley KA, Mogridge J, Mourez M et al (2001) Identification of the cellular receptor for anthrax toxin. Nature 414:225–229
Brown MJ, Thoren KL, Krantz BA (2011) Charge requirements for proton gradient-driven translocation of anthrax toxin. J Biol Chem 286:23189–23199. doi:10.1074/jbc.M111.231167, M111.231167 [pii]
Bruckner V, Kovacs J, Denes G (1953) Structure of poly-d-glutamic acid isolated from capsulated strains of B. anthracis. Nature 172:508
Burton RE, Siddiqui SM, Kim YI et al (2001) Effects of protein stability and structure on substrate processing by the ClpXP unfolding and degradation machine. EMBO J 20:3092–3100
Candela T, Fouet A (2005) Bacillus anthracis CapD, belonging to the γ-glutamyltranspeptidase family, is required for the covalent anchoring of capsule to peptidoglycan. Mol Microbiol 57:717–726
Candela T, Fouet A (2006) Poly-gamma-glutamate in bacteria. Mol Microbiol 60:1091–1098. doi:10.1111/j.1365-2958.2006.05179.x
Candela T, Mock M, Fouet A (2005) CapE, a 47-amino-acid peptide, is necessary for Bacillus anthracis polyglutamate capsule synthesis. J Bacteriol 187:7765–7772. doi:10.1128/JB.187.22.7765-7772.2005
Chauwin JF, Oster G, Glick BS (1998) Strong precursor-pore interactions constrain models for mitochondrial protein import. Biophys J 74:1732–1743
Chavarría-Smith J, Vance RE (2013) Direct proteolytic cleavage of NLRP1B is necessary and sufficient for inflammasome activation by anthrax lethal factor. PLoS Pathog 9, e1003452. doi:10.1371/journal.ppat.1003452
Christensen KA, Krantz BA, Melnyk RA, Collier RJ (2005) Interaction of the 20 kDa and 63 kDa fragments of anthrax protective antigen: kinetics and thermodynamics. Biochemistry 44:1047–1053. doi:10.1021/bi047791s
Collier RJ (2009) Membrane translocation by anthrax toxin. Mol Aspects Med 30:413–422. doi:10.1016/j.mam.2009.06.003, S0098-2997(09)00035-1 [pii]
Collier RJ, Young JA (2003) Anthrax toxin. Annu Rev Cell Dev Biol 19:45–70
Crampton N, Brockwell DJ (2010) Unravelling the design principles for single protein mechanical strength. Curr Opin Struct Biol 20:508–517. doi:10.1016/j.sbi.2010.05.005, S0959-440X(10)00075-8 [pii]
Cunningham K, Lacy DB, Mogridge J, Collier RJ (2002) Mapping the lethal factor and edema factor binding sites on oligomeric anthrax protective antigen. Proc Natl Acad Sci U S A 99:7049–7053
Gill DM (1978) Seven toxic peptides that cross cell membranes. In: Jeljaszewicz J, Wadstrom T (eds) Bacterial Toxins and Cell Membranes. Academic, New York, pp 291–332
Dmochewitz L, Lillich M, Kaiser E et al (2011) Role of CypA and Hsp90 in membrane translocation mediated by anthrax protective antigen. Cell Microbiol 13:359–373. doi:10.1111/j.1462-5822.2010.01539.x
Drum CL, Yan SZ, Bard J et al (2002) Structural basis for the activation of anthrax adenylyl cyclase exotoxin by calmodulin. Nature 415:396–402
Duesbery NS, Webb CP, Leppla SH et al (1998) Proteolytic inactivation of MAP-kinase-kinase by anthrax lethal factor. Science 280:734–737
Dumetz F, Jouvion G, Khun H et al (2011) Noninvasive imaging technologies reveal edema toxin as a key virulence factor in anthrax. Am J Pathol 178:2523–2535. doi:10.1016/j.ajpath.2011.02.027
Evans AS (1976) Causation and disease: the Henle-Koch postulates revisited. Yale J Biol Med 49:175–195
Ezzell JW, Abshire TG (1992) Serum protease cleavage of Bacillus anthracis protective antigen. J Gen Microbiol 138:543–549
Ezzell JW, Abshire TG, Panchal R et al (2009) Association of Bacillus anthracis capsule with lethal toxin during experimental infection. Infect Immun 77:749–755. doi:10.1128/IAI.00764-08, IAI.00764-08 [pii]
Feld GK, Brown MJ, Krantz BA (2012a) Ratcheting up protein translocation with anthrax toxin. Protein Sci 21:606–624. doi:10.1002/pro.2052
Feld GK, Kintzer AF, Tang II et al (2012b) Domain flexibility modulates the heterogeneous assembly mechanism of anthrax toxin protective antigen. J Mol Biol 415:159–174. doi:10.1016/j.jmb.2011.10.035, S0022-2836(11)01172-7 [pii]
Feld GK, Thoren KL, Kintzer AF et al (2010) Structural basis for the unfolding of anthrax lethal factor by protective antigen oligomers. Nat Struct Mol Biol 17:1383–1390. doi:10.1038/nsmb.1923, nsmb.1923 [pii]
Feller W (2008) An introduction to probability theory and its applications. Wiley, Hoboken
Fersht AR (1998) Structure and mechanism in protein science: a guide to enzyme catalysis and protein folding. Freeman, New York
Finkelstein A (2009) Proton-coupled protein transport through the anthrax toxin channel. Philos Trans R Soc Lond B Biol Sci 364:209–215. doi:10.1098/rstb.2008.0126, 74702356P024100L [pii]
Floyd DL, Ragains JR, Skehel JJ et al (2008) Single-particle kinetics of influenza virus membrane fusion. Proc Natl Acad Sci U S A 105:15382–15387. doi:10.1073/pnas.0807771105
Frankel AE, Kuo S-R, Dostal D et al (2009) Pathophysiology of anthrax. Front Biosci Landmark Ed 14:4516–4524
Friedlander AM (1986) Macrophages are sensitive to anthrax lethal toxin through an acid-dependent process. J Biol Chem 261:7123–7126
Friedlander AM, Pittman PR, Parker GW (1999) Anthrax vaccine: evidence for safety and efficacy against inhalational anthrax. JAMA 282:2104–2106
Fu S, Tong X, Cai C et al (2010) The structure of tumor endothelial marker 8 (TEM8) extracellular domain and implications for its receptor function for recognizing anthrax toxin. PLoS One 5, e11203. doi:10.1371/journal.pone.0011203
Glick BS (1995) Can Hsp70 proteins act as force-generating motors? Cell 80:11–14
Green BD, Battisti L, Koehler TM et al (1985) Demonstration of a capsule plasmid in Bacillus anthracis. Infect Immun 49:291–297
Gu LQ, Braha O, Conlan S et al (1999) Stochastic sensing of organic analytes by a pore-forming protein containing a molecular adapter. Nature 398:686–690
Halverson KM, Panchal RG, Nguyen TL et al (2005) Anthrax biosensor: protective antigen ion channel asymmetric blockade. J Biol Chem 280:34056–34062
Hanks S, Adams S, Douglas J et al (2003) Mutations in the gene encoding capillary morphogenesis protein 2 cause juvenile hyaline fibromatosis and infantile systemic hyalinosis. Am J Hum Genet 73:791–800
Huang S, Ratliff KS, Schwartz MP et al (1999) Mitochondria unfold precursor proteins by unraveling them from their N-termini. Nat Struct Biol 6:1132–1138
Jang J, Cho M, Chun JH et al (2011) The poly-γ-d-glutamic acid capsule of Bacillus anthracis enhances lethal toxin activity. Infect Immun 79:3846–3854. doi:10.1128/IAI.01145-10, IAI.01145-10 [pii]
Janowiak BE, Finkelstein A, Collier RJ (2009) An approach to characterizing single-subunit mutations in multimeric prepores and pores of anthrax protective antigen. Protein Sci 18:348–358. doi:10.1002/pro.35
Jernigan DB, Raghunathan PL, Bell BP et al (2002) Investigation of bioterrorism-related anthrax, United States, 2001: epidemiologic findings. Emerg Infect Dis 8:1019–1028. doi:10.3201/eid0810.020353
Juris SJ, Melnyk RA, Bolcome RE et al (2007) Cross-linked forms of the isolated N-terminal domain of the lethal factor are potent inhibitors of anthrax toxin. Infect Immun 75:5052–5058. doi:10.1128/IAI.00490-07, IAI.00490-07 [pii]
Karginov VA, Nestorovich EM, Moayeri M et al (2005) Blocking anthrax lethal toxin at the protective antigen channel by using structure-inspired drug design. Proc Natl Acad Sci U S A 102:15075–15080. doi:10.1073/pnas.0507488102
Katayama H, Janowiak BE, Brzozowski M et al (2008) GroEL as a molecular scaffold for structural analysis of the anthrax toxin pore. Nat Struct Mol Biol 15:754–760. doi:10.1038/nsmb.1442, nsmb.1442 [pii]
Katayama H, Wang J, Tama F et al (2010) Three-dimensional structure of the anthrax toxin pore inserted into lipid nanodiscs and lipid vesicles. Proc Natl Acad Sci U S A 107:3453–3457
Kenniston JA, Baker TA, Fernandez JM, Sauer RT (2003) Linkage between ATP consumption and mechanical unfolding during the protein processing reactions of an AAA+ degradation machine. Cell 114:511–520
Kintzer AF, Sterling HJ, Tang II et al (2010a) Anthrax toxin receptor drives protective antigen oligomerization and stabilizes the heptameric and octameric oligomer by a similar mechanism. PLoS One 5, e13888
Kintzer AF, Sterling HJ, Tang II et al (2010b) Role of the protective antigen octamer in the molecular mechanism of anthrax lethal toxin stabilization in plasma. J Mol Biol 399:741–758. doi:10.1016/j.jmb.2010.04.041, S0022-2836(10)00429-8 [pii]
Kintzer AF, Tang II, Schawel AK et al (2012) Anthrax toxin protective antigen integrates poly-γ-d-glutamate and pH signals to sense the optimal environment for channel formation. Proc Natl Acad Sci U S A 109:18378–18383. doi:10.1073/pnas.1208280109, 1208280109 [pii]
Kintzer AF, Thoren KL, Sterling HJ et al (2009) The protective antigen component of anthrax toxin forms functional octameric complexes. J Mol Biol 392:614–629. doi:10.1016/j.jmb.2009.07.037, S0022-2836(09)00876-6 [pii]
Klimpel KR, Molloy SS, Thomas G, Leppla SH (1992) Anthrax toxin protective antigen is activated by a cell surface protease with the sequence specificity and catalytic properties of furin. Proc Natl Acad Sci U S A 89:10277–10281
Koehler TM (2009) Bacillus anthracis physiology and genetics. Mol Aspects Med 30:386–396. doi:10.1016/j.mam.2009.07.004, S0098-2997(09)00052-1 [pii]
Kofoed EM, Vance RE (2011) Innate immune recognition of bacterial ligands by NAIPs determines inflammasome specificity. Nature 477:592–595. doi:10.1038/nature10394, nature10394 [pii]
Krantz BA, Finkelstein A, Collier RJ (2006) Protein translocation through the anthrax toxin transmembrane pore is driven by a proton gradient. J Mol Biol 355:968–979
Krantz BA, Melnyk RA, Zhang S et al (2005) A phenylalanine clamp catalyzes protein translocation through the anthrax toxin pore. Science 309:777–781
Krantz BA, Trivedi AD, Cunningham K et al (2004) Acid-induced unfolding of the amino-terminal domains of the lethal and edema factors of anthrax toxin. J Mol Biol 344:739–756
Lacy DB, Lin HC, Melnyk RA et al (2005) A model of anthrax toxin lethal factor bound to protective antigen. Proc Natl Acad Sci U S A 102:16409–16414
Lacy DB, Mourez M, Fouassier A, Collier RJ (2002) Mapping the anthrax protective antigen binding site on the lethal and edema factors. J Biol Chem 277:3006–3010
Lacy DB, Wigelsworth DJ, Melnyk RA et al (2004a) Structure of heptameric protective antigen bound to an anthrax toxin receptor: a role for receptor in pH-dependent pore formation. Proc Natl Acad Sci U S A 101:13147–13151
Lacy DB, Wigelsworth DJ, Scobie HM et al (2004b) Crystal structure of the von Willebrand factor A domain of human capillary morphogenesis protein 2: an anthrax toxin receptor. Proc Natl Acad Sci U S A 101:6367–6372. doi:10.1073/pnas.0401506101 [doi] 0401506101 [pii]
Leppla SH (1982) Anthrax toxin edema factor: a bacterial adenylate cyclase that increases cyclic AMP concentrations of eukaryotic cells. Proc Natl Acad Sci U S A 79:3162–3166
Levinsohn JL, Newman ZL, Hellmich KA et al (2012) Anthrax lethal factor cleavage of Nlrp1 is required for activation of the inflammasome. PLoS Pathog 8:e1002638. doi:10.1371/journal.ppat.1002638, PPATHOGENS-D-12-00046 [pii]
Liao X, Rabideau AE, Pentelute BL (2014) Delivery of antibody mimics into mammalian cells via anthrax toxin protective antigen. Chembiochem 15:2458–2466. doi:10.1002/cbic.201402290
Liu S, Crown D, Miller-Randolph S et al (2009) Capillary morphogenesis protein-2 is the major receptor mediating lethality of anthrax toxin in vivo. Proc Natl Acad Sci U S A 106:12424–12429. doi:10.1073/pnas.0905409106
Maillard RA, Chistol G, Sen M et al (2011) ClpX(P) generates mechanical force to unfold and translocate its protein substrates. Cell 145:459–469. doi:10.1016/j.cell.2011.04.010, S0092-8674(11)00429-6 [pii]
Martchenko M, Jeong S-Y, Cohen SN (2010) Heterodimeric integrin complexes containing beta1-integrin promote internalization and lethality of anthrax toxin. Proc Natl Acad Sci U S A 107:15583–15588. doi:10.1073/pnas.1010145107
Mayor A (2008) Greek fire, poison arrows, and scorpion bombs: biological & chemical warfare in the ancient world. Overlook Press, New York
McCluskey AJ, Collier RJ (2013) Receptor-directed chimeric toxins created by sortase-mediated protein fusion. Mol Cancer Ther 12:2273–2281. doi:10.1158/1535-7163.MCT-13-0358
Meador WE, Means AR, Quiocho FA (1992) Target enzyme recognition by calmodulin: 2.4 a structure of a calmodulin-peptide complex. Science 257:1251–1255
Meador WE, Means AR, Quiocho FA (1993) Modulation of calmodulin plasticity in molecular recognition on the basis of x-ray structures. Science 262:1718–1721
Melnyk RA, Collier RJ (2006) A loop network within the anthrax toxin pore positions the phenylalanine clamp in an active conformation. Proc Natl Acad Sci U S A 103:9802–9807. doi:10.1073/pnas.0604000103, 0604000103 [pii]
Melnyk RA, Hewitt KM, Lacy DB et al (2006) Structural determinants for the binding of anthrax lethal factor to oligomeric protective antigen. J Biol Chem 281:1630–1635
Mikesell P, Ivins BE, Ristroph JD, Dreier TM (1983) Evidence for plasmid-mediated toxin production in Bacillus anthracis. Infect Immun 39:371–376
Miller CJ, Elliott JL, Collier RJ (1999) Anthrax protective antigen: prepore-to-pore conversion. Biochemistry 38:10432–10441
Milne JC, Blanke SR, Hanna PC, Collier RJ (1995) Protective antigen-binding domain of anthrax lethal factor mediates translocation of a heterologous protein fused to its amino- or carboxy-terminus. Mol Microbiol 15:661–666
Milne JC, Furlong D, Hanna PC et al (1994) Anthrax protective antigen forms oligomers during intoxication of mammalian cells. J Biol Chem 269:20607–20612
Moayeri M, Leppla SH (2009) Cellular and systemic effects of anthrax lethal toxin and edema toxin. Mol Aspects Med 30:439–455. doi:10.1016/j.mam.2009.07.003, S0098-2997(09)00051-X [pii]
Moayeri M, Wiggins JF, Leppla SH (2007) Anthrax protective antigen cleavage and clearance from the blood of mice and rats. Infect Immun 75:5175–5184. doi:10.1128/IAI.00719-07, IAI.00719-07 [pii]
Mock M, Fouet A (2001) Anthrax. Annu Rev Microbiol 55:647–671
Mogridge J, Cunningham K, Collier RJ (2002a) Stoichiometry of anthrax toxin complexes. Biochemistry 41:1079–1082
Mogridge J, Cunningham K, Lacy DB et al (2002b) The lethal and edema factors of anthrax toxin bind only to oligomeric forms of the protective antigen. Proc Natl Acad Sci U S A 99:7045–7048
Mogridge J, Mourez M, Collier RJ (2001) Involvement of domain 3 in oligomerization by the protective antigen moiety of anthrax toxin. J Bacteriol 183:2111–2116
Molloy SS, Bresnahan PA, Leppla SH et al (1992) Human furin is a calcium-dependent serine endoprotease that recognizes the sequence Arg-X-X-Arg and efficiently cleaves anthrax toxin protective antigen. J Biol Chem 267:16396–16402
Mourez M, Yan M, Lacy DB et al (2003) Mapping dominant-negative mutations of anthrax protective antigen by scanning mutagenesis. Proc Natl Acad Sci U S A 100:13803–13808
Mueller P, Rudin DO, Tien HT, Westcott WC (1963) Methods for the formation of single bimolecular lipid membranes in aqueous solution. J Phys Chem 67:534–535
Nanda A, Carson-Walter EB, Seaman S et al (2004) TEM8 interacts with the cleaved C5 domain of collagen alpha 3(VI). Cancer Res 64:817–820
Nassi S, Collier RJ, Finkelstein A (2002) PA63 channel of anthrax toxin: an extended β-barrel. Biochemistry 41:1445–1450
Nestorovich EM, Bezrukov SM (2014) Designing inhibitors of anthrax toxin. Expert Opin Drug Discov 9:299–318. doi:10.1517/17460441.2014.877884
Nguyen TL (2004) Three-dimensional model of the pore form of anthrax protective antigen. Structure and biological implications. J Biomol Struct Dyn 22:253–265
Oomen CJ, Van Ulsen P, Van Gelder P et al (2004) Structure of the translocator domain of a bacterial autotransporter. EMBO J 23:1257–1266
Orlik F, Schiffler B, Benz R (2005) Anthrax toxin protective antigen: inhibition of channel function by chloroquine and related compounds and study of binding kinetics using the current noise analysis. Biophys J 88:1715–1724
Panchal RG, Halverson KM, Ribot W et al (2005) Purified Bacillus anthracis lethal toxin complex formed in vitro and during infection exhibits functional and biological activity. J Biol Chem 280:10834–10839. doi:10.1074/jbc.M412210200
Pannifer AD, Wong TY, Schwarzenbacher R et al (2001) Crystal structure of the anthrax lethal factor. Nature 414:229–233
Pentelute BL, Barker AP, Janowiak BE et al (2010) A semisynthesis platform for investigating structure-function relationships in the N-terminal domain of the anthrax Lethal Factor. ACS Chem Biol 5:359–364. doi:10.1021/cb100003r
Pentelute BL, Sharma O, Collier RJ (2011) Chemical dissection of protein translocation through the anthrax toxin pore. Angew Chem Int Ed Engl 50:2294–2296. doi:10.1002/anie.201006460
Petosa C, Collier RJ, Klimpel KR et al (1997) Crystal structure of the anthrax toxin protective antigen. Nature 385:833–838
Pflughoeft KJ, Swick MC, Engler DA et al (2014) Modulation of the Bacillus anthracis secretome by the immune inhibitor A1 protease. J Bacteriol 196:424–435. doi:10.1128/JB.00690-13
Phillips DD, Fattah RJ, Crown D et al (2013) Engineering anthrax toxin variants that exclusively form octamers and their application to targeting tumors. J Biol Chem 288:9058–9065. doi:10.1074/jbc.M113.452110
Pittman PR, Kim-Ahn G, Pifat DY et al (2002) Anthrax vaccine: immunogenicity and safety of a dose-reduction, route-change comparison study in humans. Vaccine 20:1412–1420
Quinn CP, Singh Y, Klimpel KR, Leppla SH (1991) Functional mapping of anthrax toxin lethal factor by in-frame insertion mutagenesis. J Biol Chem 266:20124–20130
Rasko DA, Worsham PL, Abshire TG et al (2011) Bacillus anthracis comparative genome analysis in support of the Amerithrax investigation. Proc Natl Acad Sci U S A 108:5027–5032. doi:10.1073/pnas.1016657108, 1016657108 [pii]
Ratts R, Trujillo C, Bharti A et al (2005) A conserved motif in transmembrane helix 1 of diphtheria toxin mediates catalytic domain delivery to the cytosol. Proc Natl Acad Sci U S A 102:15635–15640. doi:10.1073/pnas.0504937102, 0504937102 [pii]
Ratts R, Zeng H, Berg EA et al (2003) The cytosolic entry of diphtheria toxin catalytic domain requires a host cell cytosolic translocation factor complex. J Cell Biol 160:1139–1150. doi:10.1083/jcb.200210028, jcb.200210028 [pii]
Ryan PL, Young JA (2008) Evidence against a human cell-specific role for LRP6 in anthrax toxin entry. PLoS One 3:e1817. doi:10.1371/journal.pone.0001817
Santelli E, Bankston LA, Leppla SH, Liddington RC (2004) Crystal structure of a complex between anthrax toxin and its host cell receptor. Nature 430:905–908
Schueler-Furman O, Wang C, Baker D (2005) Progress in protein-protein docking: atomic resolution predictions in the CAPRI experiment using RosettaDock with an improved treatment of side-chain flexibility. Proteins 60:187–194. doi:10.1002/prot.20556
Scobie HM, Rainey GJ, Bradley KA, Young JA (2003) Human capillary morphogenesis protein 2 functions as an anthrax toxin receptor. Proc Natl Acad Sci U S A 100:5170–5174. doi:10.1073/pnas.0431098100, 0431098100 [pii]
Sellman BR, Mourez M, Collier RJ (2001a) Dominant-negative mutants of a toxin subunit: an approach to therapy of anthrax. Science 292:695–697
Sellman BR, Nassi S, Collier RJ (2001b) Point mutations in anthrax protective antigen that block translocation. J Biol Chem 276:8371–8376
Sharma O, Collier RJ (2014) Polylysine-mediated translocation of the diphtheria toxin catalytic domain through the anthrax protective antigen pore. Biochemistry 53:6934–6940. doi:10.1021/bi500985v
Shaw CA, Starnbach MN (2008) Antigen delivered by anthrax lethal toxin induces the development of memory CD8+ T cells that can be rapidly boosted and display effector functions. Infect Immun 76:1214–1222. doi:10.1128/IAI.01208-07
Shen Y, Zhukovskaya NL, Guo Q et al (2005) Calcium-independent calmodulin binding and two-metal-ion catalytic mechanism of anthrax edema factor. EMBO J 24:929–941
Simon SM, Peskin CS, Oster GF (1992) What drives the translocation of proteins? Proc Natl Acad Sci U S A 89:3770–3774
Singh Y, Klimpel KR, Goel S et al (1999) Oligomerization of anthrax toxin protective antigen and binding of lethal factor during endocytic uptake into mammalian cells. Infect Immun 67:1853–1859
Smith H, Keppie J (1954) Observations on experimental anthrax: demonstration of a specific lethal factor produced in vivo by Bacillus anthracis. Nature 173:689
Song L, Hobaugh MR, Shustak C et al (1996) Structure of staphylococcal α-hemolysin, a heptameric transmembrane pore. Science 274:1859–1866
Sterne M (1939) The use of anthrax vaccines prepared from avirulent (uncapsulated) variants of Bacillus anthracis. Ondersteport J Vet Sci Anim Indust 13:307–312
Strange RE, Belton FC (1954) Studies on a protective antigen produced in vitro from Bacillus anthracis: purification and chemistry of the antigen. Br J Exp Pathol 35:153–165
Sun J, Lang AE, Aktories K, Collier RJ (2008) Phenylalanine-427 of anthrax protective antigen functions in both pore formation and protein translocation. Proc Natl Acad Sci U S A 105:4346–4351. doi:10.1073/pnas.0800701105, 0800701105 [pii]
Sun J, Vernier G, Wigelsworth DJ, Collier RJ (2007) Insertion of anthrax protective antigen into liposomal membranes: effects of a receptor. J Biol Chem 282:1059–1065
Tamayo AG, Bharti A, Trujillo C et al (2008) COPI coatomer complex proteins facilitate the translocation of anthrax lethal factor across vesicular membranes in vitro. Proc Natl Acad Sci U S A 105:5254–5259. doi:10.1073/pnas.0710100105, 0710100105 [pii]
Thoren KL, Krantz BA (2011) The unfolding story of anthrax toxin translocation. Mol Microbiol 80:588–595. doi:10.1111/j.1365-2958.2011.07614.x
Thoren KL, Worden EJ, Yassif JM, Krantz BA (2009) Lethal factor unfolding is the most force-dependent step of anthrax toxin translocation. Proc Natl Acad Sci U S A 106:21555–21560. doi:10.1073/pnas.0905880106, 0905880106 [pii]
Turnbull PC (1991) Anthrax vaccines: past, present and future. Vaccine 9:533–539
Turnbull PCB (1996) Bacillus Medical Microbiology. University of Texas Medical Branch at Galveston, Galveston
Uchida I, Makino S, Sasakawa C et al (1993) Identification of a novel gene, dep, associated with depolymerization of the capsular polymer in Bacillus anthracis. Mol Microbiol 9:487–496
Uchida I, Sekizaki T, Hashimoto K, Terakado N (1985) Association of the encapsulation of Bacillus anthracis with a 60 megadalton plasmid. J Gen Microbiol 131:363–367
Van der Goot G, Young JA (2009) Receptors of anthrax toxin and cell entry. Mol Aspects Med 30:406–412. doi:10.1016/j.mam.2009.08.007, S0098-2997(09)00059-4 [pii]
Vitale G, Bernardi L, Napolitani G et al (2000) Susceptibility of mitogen-activated protein kinase kinase family members to proteolysis by anthrax lethal factor. Biochem J 352:739–745
Von Moltke J, Trinidad NJ, Moayeri M et al (2012) Rapid induction of inflammatory lipid mediators by the inflammasome in vivo. Nature 490:107–111. doi:10.1038/nature11351
Wasserman GM, Grabenstein JD, Pittman PR et al (2003) Analysis of adverse events after anthrax immunization in US Army medical personnel. J Occup Environ Med Am Coll Occup Environ Med 45:222–233
Wein AN, Liu S, Zhang Y et al (2013) Tumor therapy with a urokinase plasminogen activator-activated anthrax lethal toxin alone and in combination with paclitaxel. Invest New Drugs 31:206–212. doi:10.1007/s10637-012-9847-1
Wei W, Lu Q, Chaudry GJ et al (2006) The LDL receptor-related protein LRP6 mediates internalization and lethality of anthrax toxin. Cell 124:1141–1154. doi:10.1016/j.cell.2005.12.045, S0092-8674(06)00199-1 [pii]
Wesche J, Elliott JL, Falnes PO et al (1998) Characterization of membrane translocation by anthrax protective antigen. Biochemistry 37:15737–15746
Wigelsworth DJ, Krantz BA, Christensen KA et al (2004) Binding stoichiometry and kinetics of the interaction of a human anthrax toxin receptor, CMG2, with protective antigen. J Biol Chem 279:23349–23356
Williams P, Wallace D (1989) Unit 731: Japan’s secret biological warfare in World War II. Free Press, New York
Wimalasena DS, Cramer JC, Janowiak BE et al (2007) Effect of 2-fluorohistidine labeling of the anthrax protective antigen on stability, pore formation, and translocation. Biochemistry 46:14928–14936. doi:10.1021/bi701763z
Wolfe JT, Krantz BA, Rainey GJ et al (2005) Whole-cell voltage clamp measurements of anthrax toxin pore current. J Biol Chem 280:39417–39422
Wright GG, Hedberg MA, Slein JB (1954) Studies on immunity in anthrax. III. Elaboration of protective antigen in a chemically defined, non-protein medium. J Immunol 72:263–269
Wynia-Smith SL, Brown MJ, Chirichella G et al (2012) Electrostatic ratchet in the protective antigen channel promotes anthrax toxin translocation. J Biol Chem 287:43753–43764. doi:10.1074/jbc.M112.419598, M112.419598 [pii]
Young JA, Collier RJ (2007) Anthrax toxin: receptor binding, internalization, pore formation, and translocation. Annu Rev Biochem 76:243–265. doi:10.1146/annurev.biochem.75.103004.142728
Young JJ, Bromberg-White JL, Zylstra C et al (2007) LRP5 and LRP6 are not required for protective antigen-mediated internalization or lethality of anthrax lethal toxin. PLoS Pathog 3, e27. doi:10.1371/journal.ppat.0030027, 07-PLPA-RA-0017 [pii]
Zhang S, Finkelstein A, Collier RJ (2004a) Evidence that translocation of anthrax toxin’s lethal factor is initiated by entry of its N terminus into the protective antigen channel. Proc Natl Acad Sci U S A 101:16756–16761
Zhang S, Udho E, Wu Z et al (2004b) Protein translocation through anthrax toxin channels formed in planar lipid bilayers. Biophys J 87:3842–3849
Zhao Y, Yang J, Shi J et al (2011) The NLRC4 inflammasome receptors for bacterial flagellin and type III secretion apparatus. Nature 477:596–600. doi:10.1038/nature10510
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Krantz, B.A. (2015). Anthrax Toxin Protective Antigen Forms an Unusual Channel That Unfolds and Translocates Proteins Across Membranes. In: Delcour, A.H. (eds) Electrophysiology of Unconventional Channels and Pores. Springer Series in Biophysics, vol 18. Springer, Cham. https://doi.org/10.1007/978-3-319-20149-8_9
Download citation
DOI: https://doi.org/10.1007/978-3-319-20149-8_9
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-20148-1
Online ISBN: 978-3-319-20149-8
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)