
Abstract
Chromosomal fragile sites are specific loci that show gaps, breaks, or rearrangements in metaphase chromosomes when cells are cultured under conditions that partially inhibit DNA synthesis, inducing replication stress. The common fragile sites (CFS) are numerous in the human genome, and are essentially observed in all individuals. Although the molecular basis for chromosome fragility of CFSs remains incompletely understood, there is now general agreement that CFS sequences have a distinct replication programme that combines late replication with failure to activate origins in the core regions of the CFSs during replication stress, resulting in the failure to complete replication. The CFSs exhibit several features characteristic of highly unstable or recombinogenic regions of the genome, and CFSs have been shown to mediate genetic instability in cancers, including during the early stages of tumourigenesis. In this chapter, we review the molecular features of CFSs, as well as the relationship of CFSs to genomic alterations in cancer cells.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Chromosomal fragile sites
- Common fragile sites
- DNA replication
- Genetic instability
- Mechanism of instability
1 Historical Aspects and Characteristics of Fragile Sites
Chromosomal fragile sites are specific loci that show gaps, breaks, or rearrangements in metaphase chromosomes when cells are cultured under particular conditions that partially inhibit DNA synthesis [1, 2]. Fragile sites are grouped into two major classes based upon their frequency in the population, as well as the culture conditions required for their expression. The “rare fragile sites” number at least 30, and are found in less than 5 % of the population, in many instances, in only one or a few families. A number of rare fragile sites, including the FRAXA, FRAXE, FRAXF, FRA16A, FRA16B, and FRA11B, have been cloned or mapped at the molecular level (reviewed in [1]). With the exception of the FRA16B, the mutation leading to the expression of fragility is the expansion and methylation of a CGG trinucleotide repeat, and chromosome breakage occurs within this small segment of DNA. FRA16B also involves the expansion of a repeat – 33 bp AT-rich minisatellite. Expansion of these repeats can give rise to genetic disease by modifying the expression of genes in which they are located, as in the case of the FRAXA and FRAXF, or by mediating chromosomal deletions, as seen in some cases of Jacobsen Syndrome [1].
In contrast, numerous “common fragile sites” (CFSs) have been recognized – 87 CFSs are listed in the NCBI database (http://www.ncbi.nlm.nih.gov/gene/?term=%28common+fragile+site%20%29+AND+%22Homo+sapiens%22[porgn%3A__txid9606]). However, the precise number depends on the inducing conditions, cell type, and analytical methods; a recent study reported 230 CFSs, although most of these sites were expressed very infrequently [3]. As described below, the greater the impairment of replication, the more CFSs observed, until the cessation of replication. The expression of CFSs varies in different cell types, but the CFSs are essentially observed in all individuals [4, 5]. At least 45 common fragile sites have now been mapped at the sequence level, including the most frequently expressed sites: FRA3B , FRA6F, FRA7G , FRA7F, FRA16D , FRAXB (reviewed in [6]). Molecular analysis has provided evidence that the CFSs differ from the rare fragile sites in several ways. First, the CFSs span large genomic regions, ranging from 160 kb to greater than 10 Mb, and genomic breakage and instability occurs over a large region (reviewed in [2, 6, 7]). Second, despite extensive analysis of several CFSs, no specific sequence elements or repeat motifs, such as the trinucleotide repeats characteristic of rare fragile sites, have been identified to be required for their expression (reviewed in [2, 7]). Common fragile sites are conserved, and have also been observed in many other mammalian species, such as mouse, hamster, primates, dogs, cattle, and deer mouse. Furthermore, at least eight mouse CFSs have human CFS orthologs: Fra14A2 (FRA3B), Fra8E (FRA16D), Fra6C1 (FRA4F), Fra12C1 (FRA7K), Fra2D (FRA2G ), Fra6A3.1 (FRA7G), Fra6C1 (FRA7H ), and Fra4C2 (FRA9E), and regions orthologous to the human FRA3B/FHIT and FRA16D/WWOX are conserved in Mus musculus [8]. In yeast, chromosome breaks at specific sites called “replication slow zones” have been proposed to be analogous to CFSs [9]. Thus, fragile sites appear to be maintained across species, although their function is unknown.
The common fragile sites exhibit several features characteristic of highly unstable or recombinogenic regions of the genome. In addition to forming breaks and gaps on metaphase chromosomes, they are preferred sites for sister chromatid exchanges (SCEs), chromosomal deletions and rearrangements, the integration of transfected plasmid DNA or viruses, e.g., HPV, and the initiation of breakage-fusion-bridge (BFB) cycles, leading to gene amplification [2, 6, 7]. CFSs have also been shown to be preferred sites for structural variation in stem cells [10], and copy number variants in the human germline [11]. Recent studies have shed light on the role of CFSs in genetic instability in cancer cells. For example, Bignell et al. demonstrated that a substantial proportion of homozygous and hemizygous deletions in cancer cells cluster in CFSs [12]. The compendium of CFSs principally consists of large regions containing genes >300 kb in length, and over half of the recurrent molecular deletions in cancer cells originate in CFSs that are associated with large genes.
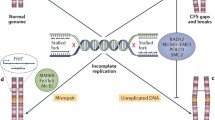
At present, the molecular basis for chromosome fragility of CFSs remains incompletely understood (Fig. 5.1). Local genomic features, including G-negative chromosomal bands distal to centromeres, enrichment for ALU repeats, high DNA flexibility, CpG island density, transcription start site density, H3K4me1 coverage, and mononucleotide microsatellite coverage are significant predictors [6, 7, 13]. We first demonstrated that CFSs replicate late in S-phase, and sometimes remain incompletely replicated in metaphase cells, [14] and there is now general agreement that CFSs remain incompletely replicated at the onset of mitosis following replication stress, making them prone to breakage. Moreover, CFS instability is dependent on ATR signalling and is associated with other DNA damage response factors [2]. For the past decade, several nonexclusive models have existed for CFS instability. The first model posits that CFSs contain sequences that are difficult to replicate, leading to stalled replication forks and, ultimately, replication fork collapse. The second model suggests that CFSs contain a paucity of replication origins, resulting in incomplete replication under replication stress. Recent studies of eight of the major CFSs have resulted in a convergence of both models revealing that a distinct replication programme combining late replication with failure to activate origins in the core regions of the CFSs following replication stress is responsible for the failure to complete replication. Because replication programmes differ in various cell types, different repertoires of expressed CFSs are found in human cells [6]. In this chapter, we review the features of DNA replication of common fragile sites, and the role of replication in the genetic instability characteristic of these sequences, as well as the relationship of CFSs to genomic alterations in cancer cells.

Model for the induction of common fragile sites. The model predicts that CFSs have a distinct replication programme that combines late replication with failure to activate origins in the core regions of the CFSs following replication stress, ultimately leading to long stretches of ssDNA. (a) Left panel. Common fragile sites could represent slow replicating regions as a result of an unusual chromatin structure, the presence of bulky DNA-protein complexes hindering replication fork progression, or persistence of post-replicative structures in the presence of APH. In this event, origins may initiate replication in early- to mid-S phase, but replication continues into late S phase. Right panel. CFSs have been shown to have an unusual distribution of primary and secondary origins – a lower density of primary origins at fragile sites may prevent completion of replication in the presence of APH within the S phase. A lower density of secondary origins, or lack of initiation at the secondary origins, may prevent rescue of replication by these inefficient origins when the primary origin is stalled (Green bar) or slowed in the presence of APH leading to Fig. 5.1 (continued) unreplicated regions within fragile sites. (b) Left panel. The ssDNA binding protein, RPA, coats the resultant unreplicated ssDNA and recruits the DNA damage response checkpoint proteins, including ATR, which activate S-phase or G2/M checkpoints. Right panel. Repair of these regions mediated by RAD51 and PRKDC (DNA-PKcs) and other proteins promotes replication fork progression. Some CFS sequences may escape checkpoint activation or are left unrepaired, resulting in an unreplicated region in G2/M. c MUS81-EME1 is recruited to such sites in prophase or early metaphase, and cleaves any remaining replication forks at CFSs (red circles represent FANCD2 foci at CFSs) to permit the sister chromatids to be disjoined in anaphase, giving rise to the characteristic cytological appearance of chromosome breaks/gaps at metaphase. Thereafter, the unreplicated DNA is repaired in the subsequent S phase. Repair of DNA breaks can result in molecular deletions or structural chromosomal rearrangements involving CFSs, which have been identified in cancers (Figure modified from references, [2, 38, 77])
2 Mechanisms of Fragile Site Expression
2.1 Brief Overview of DNA Replication
DNA replication in eukaryotes initiates at specific sites called origins of replication [15]. In Saccharomyces cerevisiae, origins of replication, known as Autonomously Replicating Sequence (ARS) share an A/T rich, 11 bp ARS consensus sequence that is recognized by the origin recognition complex (ORC) proteins. In contrast, the identification of metazoan origins of replication has proven to be much more difficult. Over the past two decades, a number of approaches have been undertaken to define metazoan origins, including low-throughput methods, e.g., two-dimensional gel electrophoresis techniques or nascent strand abundance assays and, more recently, genome-scale approaches that are combined with microarray or sequencing technologies (reviewed in [16, 17]). There is a notable low level of reproducibility between laboratories and across methods, especially in the case of the genome-wide techniques, suggesting that both cell intrinsic, i.e., only a subset of the active origins of any particular cell population have been mapped, and cell extrinsic, i.e., the subset of origins identified is method-dependent, factors are involved (reviewed in [16, 17]). Nevertheless, these methods have demonstrated that, despite the evolutionary conservation of the replication machinery, metazoan origins do not have the sequence specificity observed in S. cerevisiae – rather, they may be defined by DNA structure, such as G-quadruplex-forming DNA motifs [18].
The molecular mechanism of replication initiation is a highly conserved and tightly regulated process in all eukaryotes (reviewed in [19]). The first step involves licensing of origins in the late M or early G1 phase by the assembly of pre-replicative complexes (pre-RCs) at non-active origins as well as at the active origins, which includes ORC1-6, CDT1, CDC6, and minichromosome maintenance (MCM) 2-7 complex. Using genome-wide origin mapping approaches, metazoan genomes were found to have a very large number of origins, up to one every 11 kb, only a subset of which are activated in any given cell within a population [16, 18].
The second step corresponds to the loading of CDC45, which is triggered by two kinases, cyclin-CDK and CDC7-DBF4. The cyclin-CDK complex leads to progression of cells into S phase, and CDC7-DBF4 leads to activation of origin-firing by phosphorylation of the MCM proteins. Origin activation is followed by initiation of DNA replication by loading of the single-stranded DNA (ssDNA) binding protein, Replication Protein A [20], and the primase-DNA polymerase complex. The bidirectional replication fork is now active and can move into the elongation phase. In higher eukaryotes, the origins are not synchronously activated at the onset of the S phase; rather, they follow a precise and reproducible sequence of initiation throughout S phase (reviewed in [21]). Although not completely understood, this temporal replication programme has been linked to multiple biological factors: GC content, LINE (Long Interspersed Nuclear Elements) density, gene density, transcriptional activity, chromatin structure and, more recently, with large-scale chromatin folding (reviewed in [21]). For instance, transcriptionally active, GC-rich euchromatin tends to replicate before the condensed, silent, and GC-poor heterochromatin. As described later, the integrity of DNA replication is monitored during S phase by checkpoint proteins [22]. If replication is stalled or the DNA template damaged, the checkpoint proteins arrest the cell in S phase, and prevent entry into G2 until the fork is restored or the damage repaired.
2.2 Replication Dynamics of the Common Fragile Sites
2.2.1 Characteristics of the Inducers of Fragile Site Expression
The majority of the CFSs are induced by aphidicolin (APH) and, less frequently, bromo-2′deoxyuridine (BrdU), 5-azacytidine (5-Aza-C), 5-fluorouracil and camptothecin (reviewed in [2]), chemicals that interfere with DNA replication. Moreover, fragile site expression requires induction during the preceding S phase [14]. APH is an antibiotic, which inhibits DNA polymerases α, δ, and ε by competing with the incorporation of dCTP and, to a lesser extent, dTTP. High doses of APH (from 15 to 300 μM) block DNA elongation very rapidly and trigger an intra-S checkpoint, blocking cell cycle progression in early S phase and preventing initiation at late replicating domains [23]. At the lower doses of APH used for CFS induction (0.2–0.8 μM), cells still progress through S phase, but do so much more slowly than in an unperturbed S phase [24]. BrdU is incorporated into DNA in place of thymidine; at high concentrations of BrdU, S-phase progression is blocked [25]. 5-Aza-C, an inhibitor of DNA methyltransferases, inhibits chromatin condensation within G bands and heterochromatin (late-replicating) and, perhaps as a direct consequence, advances the replication timing of late-replicating chromosomal regions [26].
As mentioned above, the nature of the fragile site-inducing agents suggested that fragile site expression was likely to involve DNA synthesis. DNA repair mechanisms may also play a role, since caffeine, an inhibitor of the G2 checkpoint, increases the number of cells expressing CFSs. These observations, together with the high frequency of SCEs [27] and chromosome rearrangements at CFSs, led investigators to propose a number of years ago that fragile sites were associated with unreplicated DNA or DNA strand breaks.
2.2.2 Replication Dynamics of FRA3B and FRA16D
The FRA3B , at 3p14.2, lies within the Fragile Histidine Triad (FHIT) gene, and is the most highly expressed CFS in lymphoblastoid cells when DNA replication is perturbed by APH [4, 28]. The FHIT gene spans 1.6 Mb, but encodes only a 1.1 kb transcript. Large intragenic deletions within the FRA3B sequences, have been identified in a variety of tumour cells [4, 28]. By analyzing the replication timing of FRA3B in peripheral blood lymphocytes and human Epstein-Barr virus (EBV)-transformed lymphoblastoid cell lines, we and others showed that FRA3B alleles replicate in late S phase in untreated cells [14, 24, 29]. Exposure to APH resulted in a small, but significant, delay in the timing of replication of the FRA3B alleles, and some cells entered mitosis without completing the replication of these sequences [14].
To elucidate the link between DNA replication and CFS expression, our laboratory mapped active origins in the FHIT/FRA3B locus in non-malignant lymphoblastoid cells, using two independent methods, a nascent strand DNA assay combined with microarray analysis developed in our laboratory [30] and chromatin immunoprecipitation targeting ORC and MCM proteins [24] and mapped 100 ± 22 origins within the 1.6 Mb region. Several of the origins that mapped within the FRA3B core were also identified in an independent analysis of another lymphoblastoid cell line using the bubble-trapping method combined with deep sequencing analysis [31]. We found that FRA3B had significantly fewer, smaller, and more widely-dispersed origins as compared to its flanking non-CFS sequences (Lucas et al. unpublished results). Using a DNA combing and FISH method, the Debatisse laboratory did not detect initiation events within the FRA3B core, suggesting that the FRA3B region is replicated by long-travelling forks coming from origins located outside of the FRA3B [32]. Nonetheless, the approach used did not exclude the presence of “low efficiency” origins within the FRA3B in comparison to surrounding non-fragile regions. Indeed, we observed significantly less newly-replicated DNA in untreated cells at origins located within FRA3B, as compared to those located in flanking, non-fragile regions, suggesting that CFS origins are less efficient and/or have a faster fork speed (Lucas et al. unpublished results) [24]. Furthermore, Letessier et al. demonstrated a direct correlation between DNA replication and expression of breakage at FRA3B in cells with differential levels of breakage. That is, low origin density and late completion of DNA replication in untreated cells were linked to high levels of CFS expression in APH-treated cells, whereas higher origin density and earlier replication were linked to low levels of breakage [32].
Taken together, these results suggest that, in lymphoblastoid cells under basal growth conditions, the FRA3B is characterized by a low density of weak origins in comparison to its flanking non-CFS sequences (Fig. 5.1) [24, 32, 33]. In the presence of APH, dormant origins fail to fire in the FRA3B region (Lucas et al. unpublished results), [32] strongly suggesting that FRA3B does not respond properly to replication stress [24, 32].
The FRA16D , at 16q23, is the second most highly-expressed CFS in human lymphoblastoid cells [4]. The boundaries of the genetically unstable sequences comprising the FRA16D span ~2.5 Mb, and include the WWOX gene, which spans ~1 Mb (reviewed in [34]). Large intragenic deletions within WWOX have been identified in a variety of tumours, including breast, esophageal, lung, ovarian, colon, and prostate carcinomas [34, 35]. Furthermore, it has been suggested that WWOX may function as a suppressor of tumour growth. Several laboratories have demonstrated that the FRA16D sequences replicate late in S phase and that, the entire FRA16D is contained within one or more late-replicating domains [24, 32, 34, 35].
2.2.3 Replication Dynamics of Other Common Fragile Sites
The FRA7H , at 7q32.3, spans a 161 kb region of intergenic DNA, that is 58 % AT-rich, and predicted to contain four regions of high flexibility [36]. Using FISH analysis of asynchronous human lymphoma cells, Hellman et al. showed that the FRA7H alleles initiate replication in the mid-S phase in an asynchronous manner with one allele replicating earlier than the other, without allelic specificity [37]. Furthermore, the FRA7H exhibited a bipolar gradient of replication, where replication initiates and occurs earlier at the centre of the 160 kb region than the adjacent regions on either side. APH delays replication at FRA7H and enhances the replication timing difference within the 160 kb region. Overall, these results suggest that the FRA7H region has intrinsic features that may delay replication.
The FRA7G , at 7q31.2, corresponds to an AT-rich (61 % AT-rich), 800 kb region that encompasses several genes (TES, CAV1, CAV2, and MET), and shows loss of heterozygosity (LOH) in several human malignancies [38]. TES (TESTIN) may represent a candidate tumour suppressor gene, and MET is amplified in many tumours. Hellman et al. showed that breaks at FRA7G in a gastric carcinoma cell line led to amplification of the MET gene by a BFB mechanism, providing further evidence for a role for CFSs in the amplification of oncogenes [39]. Although the absolute replication timing of FRA7G within S phase is unknown, Hellman et al., demonstrated that the FRA7G has a biallelic replication pattern, with one allele replicating late and the other one earlier, and that the replication fork(s) progress unusually slowly within the fragile site [39]. At present, the effect of APH on the replication dynamics of FRA7G is unknown.
In two other CFSs, FRA1H and FRA2G , replication initiates during early to mid S-phase, but there is an intrinsic delay in replication progression and, by late S phase, approximately half of the CFS sequences remain unreplicated [40]. Using DNA combing techniques and FISH, Ozeri-Galai et al. determined that the FRA16C – which shares the same AT-rich genomic region as the FRA16B rare fragile site – is characterized by slow fork progression, and fork stalling at AT-rich sequences under basal conditions. Under replication stress, the frequency of fork stalling is exacerbated, and there is a failure to activate additional origins [41]. Finally, FRA6E – which contains the large 1.3 Mb PARK2 gene – contains long AT-rich repeats across which replication is slowed [42]. Thus, CFS expression combines late and slow replication, increased replication fork arrest, and an apparent paucity of active origins leading to replication stress and instability.
2.2.4 Possible Mechanism(s) Linking DNA Replication and Fragile Site Expression
2.2.4.1 Slow Replication Domains and Replication Transition Zones
As described earlier for the FRA3B , CFSs may represent sequences that replicate very slowly under normal growth conditions, potentially due to a low density of less efficient origins, and that are unable to recover from a further delay in DNA synthesis following replication stress. The link between origin density/efficiency, slow completion of replication, and DNA breaks at CFSs was confirmed by comparing the replication dynamics and the frequency of breaks for several CFSs that show differential expression in two cell types – fibroblasts and lymphoblasts [32, 34]. Furthermore, CFS regions seem to represent transition zones between early and late replicating domains [43]. Interestingly, genome-wide mapping of the replication dynamics of the long arms of human chromosomes 11 and 21 by PCR amplification of flow sorted BrdU-labelled cells has shown that genes implicated in cancer and other diseases are significantly over-represented in the transition regions between early and late replication domains [44].
2.2.4.2 Stalled Replication Forks
APH inhibits replication fork elongation, leading to stalled forks. In this event, a convergent replication fork extending from a distant origin may complete replication, resulting in a delay in the process. Another potential consequence is the uncoupling of the DNA unwinding by the replicative helicase from the replication machinery, as observed in Xenopus egg extracts treated with APH during both the initiation and elongation steps, leading to the accumulation of ssDNA regions, and triggering the formation of abnormal structures [45]. Another consequence of the replication machinery dissociation is that replication may not be able to resume, since some components (MCM2-7) can only be loaded onto the chromatin in the G1 phase. Interestingly, these effects are only observed in cells with a mutation in the S phase checkpoint proteins Mec1p (ATM /ATR ortholog) or Rad53p (CHEK2), or RecQ helicase Sgs1p (BLM homolog).
In addition, CFSs could be more prone to form secondary DNA structures that are difficult to replicate, such as hairpins, or could lead to even more aberrant structures, when located near a stalled fork. Indeed, DNA sequence analysis of the FRA3B , FRA7G , FRA7H , and FRA16D revealed that the CFSs contain multiple regions that have the potential to form unusual DNA structures, including high flexibility, low stability, and non-B-forming sequences [36]. Similarly, as suggested by Cha and Kleckner, some regions of the genome could be preferential sites for the formation of DNA-protein complexes, which could hinder the passage of the replication fork [9].
Stalled replication forks or the presence of unreplicated DNA, may be converted to DSBs, and prolonged replication inhibition results in the accumulation of DSBs. Non-homologous end joining (NHEJ) and single strand annealing are employed by cells to process DSBs in the early cellular response [46]. These two pathways would not be expected to result in a visible fragile site lesion in the ensuing mitosis, but rather in deletion of fragile site sequences in one or both of the daughter cells. As DSBs accumulate, RAD51-mediated homologous recombination (HR ) becomes the predominant mechanism of repair [47], a process that can result in formation of SCEs, as has been observed at CFSs. DSBs may also be repaired by ligation with homologous sequences from another chromosome, resulting in gross rearrangements, such as an unbalanced translocation, or they may be sites for ligation of exogenous DNA, e.g., viral sequences, as discussed later in Sect. 5.3.
2.2.4.3 Replication Defects at Fragile Sites and Checkpoints
In eukaryotic cells, the duplication of the genome during S phase and its transmission during G2-M phase is monitored at multiple levels (reviewed in [19, 48]). Normal checkpoint mechanisms ensure that DNA replication occurs once, and only once, per cell cycle, and that mitosis does not begin until DNA replication is complete. The ssDNA present at stalled replication forks leads to recruitment of the ATR (Ataxia Telangiectasia Mutated- and Rad3-related) kinase, which, in turn, activates a variety of proteins, including the CHEK1 protein kinase. Phosphorylation by CHEK1 leads to sequestration of the CDC25C phosphatase in the cytoplasm, thereby abrogating activation of the mitotic CDK1 by dephosphorylation, and leading to cell cycle arrest in the S phase. Response to DSBs is mediated similarly by another checkpoint kinase, ATM (Ataxia Telangiectasia Mutated), leading to activation of CHEK2, and resulting in cell cycle arrest and DNA damage repair. However, a threshold level of unreplicated DNA may be required to activate the checkpoint(s), and very low levels of DNA replication very late in the cell cycle may not be sufficient to delay mitotic entry. Sequences with impaired replication progression, or that replicate very late, would have a shorter period of time for DNA repair before the onset of mitosis. Unreplicated regions of DNA could affect localized chromatin structure, and manifest the recombinogenic properties of CFSs. In cultured cells challenged with APH or other CFS inducers, a fraction of cells escape the ATR replication checkpoint via a poorly understood mechanism, despite sustaining replication defects (stalled forks, aberrant replication structures, unreplicated DNA regions, etc.) at fragile site sequences. Moreover, fragile site induction is exacerbated in human cells in the absence of ATR or downstream targets, such as BRCA1, the Fanconi anaemia proteins, SMC1A/B, and CHEK1, indicating that the fragile site sequences are monitored by checkpoints, but sometimes escape [2, 9].
2.2.4.4 Transcription and Replication at CFSs
In bacteria and yeast, collisions of transcription complexes with moving replication forks cause genetic instability. To avoid this phenomenon, replication and transcription are spatially and temporally coordinated in eukaryotic cells. Helmrich et al. analyzed five CFSs associated with large genes, and found that the time required to transcribe genes >800 kb spans more than a single cell cycle, and that the long genes replicate late, regardless of their transcriptional activity. Regions of concomitant transcription and replication in late S phase lead to collisions of transcriptional machinery with replication forks, creating R-loops (RNA:DNA hybrids) resulting in breakage at CFSs, such as the FRA3B embedded within the >1.6 Mb FHIT gene, and the FRA16D within the 1.1 Mb WWOX gene [49]. In contrast, the results of other reports are not consistent with these findings. Le Tallec et al., observed plasticity in the location of the breaks within CFSs in different cell types, suggesting that transcription units per se do not set the borders of CFSs [34], and Jiang et al. found that the level of expression of the FRA3B was unrelated to the expression of FHIT in several lymphoid cell lines [50]. Moreover, this mechanism is unlikely to explain the fragility of all CFSs, since a large fraction of CFSs are not associated with large genes. Additional studies will be needed to clarify this relationship.
2.2.4.5 Chromatin Structure at CFSs
Epigenetically defined chromatin structure plays a critical role in the regulation of DNA replication and gene transcription. For example, open chromatin, characterized by the enrichment of active histone H3 acetylation marks, can facilitate origin firing during replication and lead to early replication during S phase [51]. Given that CFSs are late-replicating and manifest replication stress, Jiang et al. investigated whether chromatin conformation at CFSs plays a role in impaired DNA replication [50]. By using chromatin immunoprecipitation coupled with microarray analysis (ChIP-CHIP), the investigators mapped histone H3K9/14 acetylation (H3K9/14Ac) levels at the six most commonly expressed CFSs in EBV-transformed lymphoblastoid cells, and noted that the chromatin at CFSs was characterized by hypoacetylation as compared to the surrounding, non-fragile DNA sequences. In addition, chromatin at the FRA3B was more resistant to micrococcal nuclease treatment, suggesting that CFS chromatin assumed a more condensed conformation. In this regard, treatment of the cells with the histone deacetylase inhibitor, Trichostatin A (TSA), reduced breakage at these CFSs, which was accompanied by an increase of H3K9/14Ac at these sites. Thus, this study linked chromatin conformation to genomic instability at CFSs, and established hypoacetylation as a characteristic epigenetic pattern of CFSs that may contribute to their defective response to replication stress.
2.3 Other Classes of Fragile Sites (Early-Replicating AID -Independent)
Recently, a different class of fragile sites was identified using genome-wide approaches [52]. Barlow et al. mapped early activating replication origins by Repli-Seq and RPA-associated ssDNA at stalled replication forks by ChIP-seq in synchronized early S phase B lymphocytes treated with hydroxyurea (HU), an inhibitor of ribonucleotide reductase that induces replication stress by the depletion of deoxynucleotide pools. Surprisingly, they observed a substantial overlap between the two sets of loci (nearly 80 %). Moreover, the majority of the RPA-bound sites were also marked with the DNA damage marker γ-H2AX and fork-repairing complex components, BRCA1 and SMC5, further confirming that these RPA-bound loci at early replication origins were sites of stalled and collapsed replication forks. In contrast, they did not detect similar DNA damage sites at known CFSs. To distinguish these sites of replication failure from canonical CFS that replicate in late-S phase, the authors designated these regions as Early Replicating Fragile Sites (ERFSs). The authors further demonstrated that DNA damage at ERFSs is ATR-dependent, but not activation-induced cytidine deaminase (AID )-dependent, suggesting that similar defects of DNA repair mechanisms may be involved in both ERFS and CFS expression. Moreover, oncogenic stress, such as MYC overexpression, triggers fragility at both ERFSs and CFSs and, like CFSs, ERFSs are often embedded within genomic regions that are deleted or amplified in cancers. Despite many similarities between ERFSs and CFSs, these two classes of fragile sites differ in several ways. First, ERFSs are associated with early firing replication origins, whereas CFSs typically replicate late. Second, ERFS sequences are enriched for CpG dinucleotides, whereas CFS sequences are AT-rich. Third, ERFS loci contain a high density of activated origins, whereas those CFSs that have been mapped at high resolution have a low density of activated origins [24, 30, 32]. Fourth, ERFSs are often associated with promoters of highly transcribed genes that are characterized by open chromatin conformation; CFSs are embedded in introns of large genes with more condensed chromatin conformation [50]. Further studies are needed to elucidate the different mechanisms through which genomic instability arises from these two classes of fragile sites.
3 Relationship of Fragile Sites to Cancer
More than 30 years ago, fragile sites were implicated in the recurring chromosomal abnormalities in cancer. In 1984, Le Beau and Rowley reported an association between the chromosomal location of fragile site and the breakpoints of the recurring chromosomal abnormalities, including translocations, inversions, deletions, and amplification in leukaemias and lymphomas [53]. Many of these abnormalities target oncogenes, such as MYB, MOS, MYC , and HRAS, suggesting that fragile sites may act as predisposing factors for chromosomal rearrangements, particularly those involving genes known to induce malignant transformation. During the past few decades, new evidence has revealed that CFSs play a much broader role in inducing genetic instability in cancers. Chromosomal abnormalities involving CFSs have been shown to inactivate tumour suppressor genes, enhance oncogene expression, and facilitate the integration of viral sequences, which may result in further genotoxic stress and lead to selection of clones that eventually develop into a malignant disease. Herein, we discuss the potential mechanisms that lead to CFS expression in cancers, and their molecular consequences.
3.1 Mechanisms Leading to Common Fragile Site Expression in Cancer
3.1.1 Oncogene-Induced DNA Replication Stress
CFSs are induced experimentally in vitro by low doses of APH, a DNA polymerase inhibitor. Recently, Arlt et al. demonstrated that treatment with low doses of HU leads to the formation of de novo copy number variants (CNVs) in cultured fibroblasts, and that these CNVs resembled the characteristics of CFSs induced by APH [11]. As described earlier, HU induces replication stress through a different mechanism than APH, via the depletion of deoxyribonucleotide pools, thereby impeding replication fork progression [54]. Results from this study suggest that regardless of the source, replication stress is a causal factor of deleterious CNVs, especially within CFSs.
In cancers, oncogene activation can lead to DNA replication stress, increased CFS expression, and the subsequent induction of genomic aberrations in several ways (reviewed in Hills and Diffley [55]). First, deregulation of the TP53 and RB1/E2F pathways and overexpression of MYC or HPV E7 leads to a reduction in licensing of replication origins. Given that some CFSs are either inherently origin-deficient or fail to activate secondary origins following replication fork stalling [24, 30, 41], reduced origin licensing could further enhance these deficiencies and lead to increased fork collapse and accumulation of unreplicated ssDNA within CFSs. Second, once replication initiates, overexpression of oncogenes, such as CCNE, HPV E6 and E7, MYC, and RAS family genes can increase origin firing. This is particular harmful to CFSs that are embedded within large genes, and could be more susceptible to replication interference by the transcriptional machinery, leading to collisions between replication forks and transcription complexes and, eventually, the formation of DSBs. Increased origin firing within these CFSs may increase the chance that such collisions occur within CFSs. Third, many prereplicative complexes (pre-RC) components, such as CDT1 and CDC6 can act as oncogenes, and are often upregulated in response to RAS gene and CCNE overexpression. These activated pre-RC components lead to origin re-licensing, and the subsequent depletion of deoxyribonucleotide pools, a form of replication stress that is similar to HU treatment, which is known to induce CFSs. Taken together, it is possible that the increased genomic alterations of CFS loci seen in cancer cells are due, in part, to the replication stress induced by overexpression of oncogenes.
3.1.2 Mutations in Checkpoint and DNA Repair Pathways
DNA replication checkpoints and DNA repair pathways play important roles in the surveillance of the DNA damage associated with CFS expression. Unreplicated ssDNA and DSBs induced from collapsed stalled replication forks at CFSs are recognized by checkpoint proteins, and DNA damage sensing enzymes, such as ATR and ATM , which in turn activates repair pathways, including NHEJ. CFS expression is elevated when components of these pathways are mutated or downregulated, including ATR, ATM, CHEK1, BRCA1, FANCD2, PRKDC (DNA-PK), WRN, and BLM (reviewed in [7]), that are frequently mutated in cancer. For example, a survey of mutations and copy number alterations of ATR in cBioPortal, an online database for Cancer Genomics (http://www.cbioportal.org/public-portal/), reveals that ATR is targeted by missense and nonsense mutations, and frame-shift indels in a number of cancers, including bladder, breast, colorectal, head and neck, lung, ovarian, pancreas, melanoma, stomach, thyroid, and uterine cancers [56, 57]. Moreover, the aggregate frequency of mutations within select genes encoding components of the DNA damage checkpoint and repair pathways (ATR, ATM, BRCA1, CHEK1, FANCD2, RAD51, PRKDC, WRN, BLM) ranges from 10 to 40 % in cancer, with the higher frequency in solid tumours. Therefore, defects of DNA damage checkpoints and DNA repair due to frequent mutations in cancer may facilitate the expression of CFSs and lead to the pronounced genomic instability seen in cancer cells.
3.1.3 Aberrant Epigenetic States
In addition to the genetic features of CFS, a potential link between the epigenetic chromatin structure and CFS expression has been established recently. Jiang et al. demonstrated that several of the most frequent CFSs, including FRA3B and FRA16D , are characterized by a more condensed chromatin conformation than their surrounding, non-fragile regions, due to the lack of active histone acetylation marks [50]. Treatment with TSA and/or 5-Aza-C reduced chromosomal breakage at CFSs. Recently, mutations targeting epigenetic regulators have been identified in many types of cancers. For example, the majority of non-Hodgkin lymphomas carry mutations within the genes encoding KMT2D (an H3K4 methyltransferase), CREBBP and EP300 (histone and non-histone acetyltransferases), and EZH2 (H3K27 methyltransferase) (reviewed in [58]). In myeloid malignancies, enzymes that regulate DNA methylation (DNMT3A), and hydroxymethylation (IDH1, IDH2, TET2) are frequently mutated as well [59]. Similar phenomena are also observed in solid tumours (reviewed in [60]). Although most studies have focused on elucidating the consequence(s) of these epigenetic modifier mutations in the regulation of gene promoters, it is reasonable to predict that these mutations may target broader genomic regions, including CFS sequences, to establish an aberrant epigenetic landscape in cancers. For example, mutations in CREBBP or EP300 may further exacerbate hypoacetylation of CFSs, resulting in increased breakage. Further studies on the epigenetic mechanisms of CFS expression, particularly in cancers, are needed to shed light on the role of epigenetic marks and genomic instability involving CFSs.
3.2 Role of Fragile Sites in Chromosomal Alterations in Cancer
3.2.1 Inactivation of Tumour Suppressor Genes by Deletion
CFS expression has long been associated with genomic instability in cancers, including the gain or loss of genetic material spanning CFS loci, and translocations involving CFSs [61]. These genetic alterations can lead to inactivation of tumour suppressor genes or ectopic overexpression of oncogenes. For example, the FRA3B is embedded within a large tumour suppressor gene, FHIT, that is frequently deleted in lung and breast cancer, as well as other carcinomas [62]. Although Fhit –/– KO mice exhibited only a marginal increase of tumourigenesis in response to various carcinogens, crossing these mice with other disease models, such as Vhl –/– KO or Nit1 –/– KO animals, rendered full penetrance of tumour development (reviewed in [63]), suggesting a cooperative role for FHIT during tumourigenesis. Recently, Saldivar et al. showed that loss of Fhit expression in precancerous lesions initiates genomic instability that may eventually facilitate malignant transformation, linking alterations at CFSs to the origin of cancer genomic instability [64]. Other examples of tumour suppressor gene loss involving CFSs, include WWOX within the FRA16D , PARK2 within the FRA6E, and CAV1 and TES within the FRA7K [63].
3.2.2 Overexpression of Oncogenes by Amplification
In addition to the loss of genetic material involving CFSs, genomic amplification of the MET oncogene with boundaries within FRA7G sequences was observed in a gastric carcinoma cell line [39] and primary esophageal adenocarcinoma [65]. Amplification of the MET locus leads to overexpression of MET, resulting in a poor prognosis. By applying dual-colour FISH, Hellman et al. mapped the centromeric boundary of the amplified region within the FRA7G, and demonstrated that amplification of the MET locus via FRA7G breakage was organized in an inverted repeat fashion, as predicted by the BFB model [39]. They proposed that an initial break occurred at the telomeric end, and led to end-fusion of the sister chromatids; thereafter, ongoing replication stress might induce persistent FRA7G expression resulting in successive amplification and cycles of BFB [39]. In addition to FRA7G, FRA7I has also been implicated in duplication of the PIP gene via BFB cycles in human breast cancer [66]. However, an oncogenic role for PIP has yet to be established.
3.2.3 Deregulation of Genes via Chromosomal Translocations
In addition to the aberrations described above, CFSs have also been linked to the formation of chromosomal translocations in cancer. It is notable that FRA3B , the most commonly expressed CFS, was cloned by mapping the genomic sequences involved in the t(3;8)(p14.2;q24.1) noted in a family with hereditary renal cell carcinoma [67, 68]. This translocation disrupts FHIT, resulting in its inactivation. A similar phenomena was also observed for FRA16D , which was found to be involved in the recurring t(14;16) (q32.3;q23) in multiple myeloma (MM) [69]. This translocation not only results in a truncated allele of the tumour suppressor gene, WWOX, but also places the MAF oncogene near the IGH locus, resulting in enhanced MAF expression [69]. Exactly how genomic instability at CFSs mediates the formation of translocations is not fully understood. The t(14;16) may be mediated by the RAG1, RAG2, and AID (activation-induced cytidine deaminase) proteins, which normally participate in rearranging the B-cell immunoglobulin genes and T-cell receptor genes to increase the diversification of antibodies [70]. Indeed, by using a novel Translocation Capture Sequencing method, Klein et al. mapped chromosomal rearrangements in B lymphocytes and demonstrated that AID was responsible for many translocations involving MYC and IGH in B-cell lymphomas [71]. Determining whether CFSs, such as FRA16D, contain DNA sequences or chromatin structures that can be recognized by RAGs and AID requires further investigation. It has also been proposed that BFB cycles and NHEJ can induce chromosomal fusions [70]. Finally, DSBs resulting from collapsed replication forks within CFSs may be another potential source of translocations.
3.2.4 Integration of Viral DNA Sequences
Due to the high frequency of DSBs at CFS, they were predicted to be the preferred sites for the integration of foreign DNA. Indeed, Rassool et al. utilized this feature to clone the FRA3B by transfecting exogenous marker DNA into cells in which FRA3B expression was induced by APH, and observed preferential integration of the marker DNA at the FRA3B locus [72]. In cancers, CFSs have been found to be the integration sites for viral DNA sequences. For example, human papillomavirus (HPV), the most important cancer-related virus, is preferentially integrated into CFSs in cervical cancer cells [73, 74]. Recent studies demonstrated that expression of the HPV16 E6/E7 genes leads to replication stress by significantly decreasing the cellular nucleotide pools, raising the possibility that CFSs may be prone to increased expression in HPV infected cells, facilitating successive (and preferential) integration of viral sequences [75, 76].
3.2.5 New Potential Cancer-Specific Fragile Sites
The recent expanded efforts to map copy number alterations (CNAs) in a large cohort of tumours and the development of sophisticated bioinformatics analyses has led to new insights into the genomic alterations involving CFSs in cancer. Bignell et al. profiled the genotype status and CNAs in 746 publicly available cancer cell lines across multiple tissue types by using Affymetrix SNP6.0 arrays [12]. They detected large homozygous deletion (HD) clusters preferentially targeting recessive cancer genes (tumour suppressor genes) and CFS loci. In addition, they observed different structural signatures of HD clusters targeting recessive cancer genes and CFSs. That is, there was a threefold increase in homozygous deletions at known recessive cancer genes than hemizygous deletions, whereas there were 66 % more hemizygous deletions occurring at known CFSs than homozygous deletions. This suggests that there is a higher rate of DNA breakage within CFSs affecting one allele, some of which subsequently acquired other deletions in the remaining allele. Moreover, using this structural signature, the authors showed that the majority of the unclassified HD clusters had structural features of CFS loci, suggesting that there are potentially more CFSs that have not been identified or mapped precisely. In this regard, CFSs have largely been examined in lymphocytes. A recent study combining Repli-Seq with cytogenetic analysis found the distribution of CFSs in fibroblasts is quite different from that of lymphocytes [34]. This study further showed that over 50 % of recurrent cancer deletions originate from CFSs associated with large genes in different tissue types. Therefore, it is reasonable to predict that these unclassified HD clusters span CFSs that are specific to certain tissues, and have yet to be mapped.
4 Future Directions and Unanswered Questions
The application of new technologies has led to substantial advances in our understanding of the genomic characteristics of CFSs, and DNA replication patterns in these regions of the genome. Elucidating the molecular basis of CFSs and their inherent instability is important in that they provide a unique opportunity to examine the molecular events that follow certain types of replication stress, and how such replication stress leads to genetic instability within the replication-sensitive CFS sequences, ultimately leading to deletions, translocations, and other genomic aberrations in cancer. In addition, their instability in the earliest stages of tumour development provides an opportunity to examine their link to cell cycle checkpoints and DNA repair pathways. However, a number of questions remain, and we outline a few of these here. For example, what is the full spectrum of replication patterns at CFSs, and its relationship to DNA repair and cell cycle checkpoints? Does interference between transcription and replication play a mechanistic role in the expression of some CFSs? In vivo, what cellular processes/pathways lead to replication stress and genomic instability in premalignant cells and in cancer cells? Are there additional genomic aberrations in cancer cells that are mediated by genomic instability at CFSs? Are there mechanistic parallels between genomic instability at ERFSs and CFSs? Do CFSs have a biological function, or conserved function?
With respect to the last point, whether CFSs have a biological role has been the subject of considerable speculation. The evolutionary conservation of CFSs in widespread phyla argues for a conserved function. Nonetheless, such conservation is counterintuitive, given the likelihood that genetically unstable sequences might be detrimental to survival and, thus, selected against during evolution. Durkin and Glover proposed that the inherent fragility of these regions might in and of itself serve a valuable biological function [2]. They posited that CFSs may be among the last sequences to replicate, thereby serving to signal to the cell that replication is complete. Cell cycle checkpoints would monitor these sites, blocking entry into mitosis until their replication was complete. Intriguing data from the Hickson laboratory challenge this view, and suggest that breakage at CFSs actually promotes genomic stability [77]. These investigators observed that the DNA structure-specific nuclease MUS81-EME1 localizes to CFS loci in early mitotic cells. In contrast to the prevailing view that CFSs result from chromatin breaks during chromosome condensation, they found that cleavage of replication forks at CFSs (presumably unreplicated DNA) is an active MUS81-EME1 process, that promotes faithful sister chromatid disjunction at anaphase – replication would then be completed in the daughter cells in the subsequent S phase, thereby preserving the integrity of the genome. Further studies are needed to evaluate this intriguing model, as well as to unravel the complexity of CFS instability, and it’s relevance to the development and progression of cancer.
References
Sutherland GR (2003) Rare fragile sites. Cytogenet Genome Res 100:77–84
Durkin SG, Glover TW (2007) Chromosome fragile sites. Annu Rev Genet 41:169–192
Mrasek K, Schoder C, Teichmann AC et al (2010) Global screening and extended nomenclature for 230 aphidicolin-inducible fragile sites, including 61 yet unreported ones. Int J Oncol 36:929–940
Glover TW, Berger C, Coyle J, Echo B (1984) DNA polymerase alpha inhibition by aphidicolin induces gaps and breaks at common fragile sites in human chromosomes. Hum Genet 67:136–142
Yunis JJ, Soreng AL, Bowe AE (1987) Fragile sites are targets of diverse mutagens and carcinogens. Oncogene 1:59–69
Debatisse M, Le Tallec B, Letessier A, Dutrillaux B, Brison O (2012) Common fragile sites: mechanisms of instability revisited. Trends Genet TIG 28:22–32
Ozeri-Galai E, Bester AC, Kerem B (2012) The complex basis underlying common fragile site instability in cancer. Trends Genet TIG 28:295–302
McAllister BF, Greenbaum IF (1997) How common are common fragile sites: variation of aphidicolin-induced chromosomal fragile sites in a population of the deer mouse (Peromyscus maniculatus). Hum Genet 100:182–188
Cha RS, Kleckner N (2002) ATR homolog Mec1 promotes fork progression, thus averting breaks in replication slow zones. Science 297:602–606
Hussein SM, Batada NN, Vuoristo S et al (2011) Copy number variation and selection during reprogramming to pluripotency. Nature 471:58–62
Arlt MF, Ozdemir AC, Birkeland SR, Wilson TE, Glover TW (2011) Hydroxyurea induces de novo copy number variants in human cells. Proc Natl Acad Sci U S A 108:17360–17365
Bignell GR, Greenman CD, Davies H et al (2010) Signatures of mutation and selection in the cancer genome. Nature 463:893–898
Fungtammasan A, Walsh E, Chiaromonte F, Eckert KA, Makova KD (2012) A genome-wide analysis of common fragile sites: what features determine chromosomal instability in the human genome? Genome Res 22:993–1005
Le Beau MM, Rassool FV, Neilly ME et al (1998) Replication of a common fragile site, FRA3B, occurs late in S phase and is delayed further upon induction: implications for the mechanism of fragile site induction. Hum Mol Genet 7:755–761
Bell SP, Dutta A (2002) DNA replication in eukaryotic cells. Annu Rev Biochem 71:333–374
Gilbert DM (2012) Replication origins run (ultra) deep. Nat Struct Mol Biol 19:740–742
Mechali M (2010) Eukaryotic DNA, replication origins: many choices for appropriate answers. Nat Rev Mol Cell Biol 11:728–738
Besnard E, Babled A, Lapasset L et al (2012) Unraveling cell type-specific and reprogrammable human replication origin signatures associated with G-quadruplex consensus motifs. Nat Struct Mol Biol 19:837–844
Lucas I, Raghuraman MK (2003) The dynamics of chromosome replication in yeast. Curr Top Dev Biol 55:1–73
Achille A, Biasi MO, Zamboni G et al (1996) Chromosome 7q allelic losses in pancreatic carcinoma. Cancer Res 56:3808–3813
Rhind N, Gilbert DM (2013) DNA replication timing. Cold Spring Harb Perspect Med 3:1–26
Machida Y, Dutta A (2005) Cellular checkpoint mechanisms monitoring proper initiation of DNA replication. J Biol Chem 280:6253–6256
Dimitrova DS, Gilbert DM (2000) Temporally coordinated assembly and disassembly of replication factories in the absence of DNA synthesis. Nat Cell Biol 2:686–694
Palakodeti A, Lucas I, Jiang Y et al (2010) Impaired replication dynamics at the FRA3B common fragile site. Hum Mol Genet 19:99–110
Drouin R, Lemieux N, Richer CL (1990) Analysis of DNA replication during S-phase by means of dynamic chromosome banding at high resolution. Chromosoma 99:273–280
Collins N, Poot RA, Kukimoto I, Garcia-Jimenez C, Dellaire G, Varga-Weisz PD (2002) An ACF1-ISWI chromatin-remodeling complex is required for DNA replication through heterochromatin. Nat Genet 32:627–632
Glover TW, Stein CK (1987) Induction of sister chromatid exchanges at common fragile sites. Am J Hum Genet 41:882–890
Huebner K, Garrison PN, Barnes LD, Croce CM (1998) The role of the FHIT/FRA3B locus in cancer. Annu Rev Genet 32:7–31
Wang L, Darling J, Zhang JS, Huang H, Liu W, Smith DI (1999) Allele-specific late replication and fragility of the most active common fragile site, FRA3B. Hum Mol Genet 8:431–437
Lucas I, Palakodeti A, Jiang Y et al (2007) High-throughput mapping of origins of replication in human cells. EMBO Rep 8:770–777
Mesner LD, Valsakumar V, Cieslik M, Pickin R, Hamlin JL, Bekiranov S (2013) Bubble-seq analysis of the human genome reveals distinct chromatin-mediated mechanisms for regulating early- and late-firing origins. Genome Res 23:1774–1788
Letessier A, Millot GA, Koundrioukoff S et al (2011) Cell-type-specific replication initiation programs set fragility of the FRA3B fragile site. Nature 470:120–123
Hansen RS, Thomas S, Sandstrom R et al (2010) Sequencing newly replicated DNA reveals widespread plasticity in human replication timing. Proc Natl Acad Sci U S A 107:139–144
Le Tallec B, Millot GA, Blin ME, Brison O, Dutrillaux B, Debatisse M (2013) Common fragile site profiling in epithelial and erythroid cells reveals that most recurrent cancer deletions lie in fragile sites hosting large genes. Cell Rep 4:420–428
Palakodeti A, Han Y, Jiang Y, Le Beau MM (2004) The role of late/slow replication of the FRA16D in common fragile site induction. Genes Chromosomes Cancer 39:71–76
Mishmar D, Rahat A, Scherer SW et al (1998) Molecular characterization of a common fragile site (FRA7H) on human chromosome 7 by the cloning of a simian virus 40 integration site. Proc Natl Acad Sci U S A 95:8141–8146
Hellman A, Rahat A, Scherer SW, Darvasi A, Tsui LC, Kerem B (2000) Replication delay along FRA7H, a common fragile site on human chromosome 7, leads to chromosomal instability. Mol Cell Biol 20:4420–4427
Lucas I, Palakodeti A, Le Beau MM (2007) The implications of DNA replication in common fragile site expression. In: Arrieta I, Penagarikano O, Télez M (eds) Fragile sites: new discoveries and changing perspectives. Nova Science Publishers, Hauppauge, pp 67–98
Hellman A, Zlotorynski E, Scherer SW et al (2002) A role for common fragile site induction in amplification of human oncogenes. Cancer Cell 1:89–97
Pelliccia F, Bosco N, Curatolo A, Rocchi A (2008) Replication timing of two human common fragile sites: FRA1H and FRA2G. Cytogenet Genome Res 121:196–200
Ozeri-Galai E, Lebofsky R, Rahat A, Bester AC, Bensimon A, Kerem B (2011) Failure of origin activation in response to fork stalling leads to chromosomal instability at fragile sites. Mol Cell 43:122–131
Palumbo E, Matricardi L, Tosoni E, Bensimon A, Russo A (2010) Replication dynamics at common fragile site FRA6E. Chromosoma 119:575–587
Debatisse M, El Achkar E, Dutrillaux B (2006) Common fragile sites nested at the interfaces of early and late-replicating chromosome bands: cis acting components of the G2/M checkpoint? Cell Cycle 5:578–581
Watanabe Y, Fujiyama A, Ichiba Y et al (2002) Chromosome-wide assessment of replication timing for human chromosomes 11q and 21q: disease-related genes in timing-switch regions. Hum Mol Genet 11:13–21
Pacek M, Walter JC (2004) A requirement for MCM7 and Cdc45 in chromosome unwinding during eukaryotic DNA replication. Embo J 23:3667–3676
Musio A, Montagna C, Mariani T et al (2005) SMC1 involvement in fragile site expression. Hum Mol Genet 14:525–533
Saintigny Y, Delacote F, Vares G et al (2001) Characterization of homologous recombination induced by replication inhibition in mammalian cells. Embo J 20:3861–3870
Sancar A, Lindsey-Boltz LA, Unsal-Kacmaz K, Linn S (2004) Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu Rev Biochem 73:39–85
Helmrich ABM, Tora L (2011) Collisions between replication and transcription complexes cause common fragile site instability at the longest human genes. Mol Cell 44:966–977
Jiang Y, Lucas I, Young DJ et al (2009) Common fragile sites are characterized by histone hypoacetylation. Hum Mol Genet 18:4501–4512
Vogelauer M, Rubbi L, Lucas I, Brewer BJ, Grunstein M (2002) Histone acetylation regulates the time of replication origin firing. Mol Cell 10:1223–1233
Barlow JH, Faryabi RB, Callen E et al (2013) Identification of early replicating fragile sites that contribute to genome instability. Cell 152:620–632
LeBeau MM, Rowley JD (1984) Heritable fragile sites in cancer. Nature 308:607–608
Yarbro JW (1992) Mechanism of action of hydroxyurea. Semin Oncol 19:1–10
Hills SA, Diffley JF (2014) DNA replication and oncogene-induced replicative stress. Curr Biol CB 24:R435–R444
Cerami E, Gao J, Dogrusoz U et al (2012) The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2:401–404
Gao J, Aksoy BA, Dogrusoz U et al (2013) Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 6:pl1
Jiang Y, Hatzi K, Shaknovich R (2013) Mechanisms of epigenetic deregulation in lymphoid neoplasms. Blood 121:4271–4279
Abdel-Wahab O, Levine RL (2013) Mutations in epigenetic modifiers in the pathogenesis and therapy of acute myeloid leukemia. Blood 121:3563–3572
Berdasco M, Esteller M (2010) Aberrant epigenetic landscape in cancer: how cellular identity goes awry. Dev Cell 19:698–711
Arlt MF, Durkin SG, Ragland RL, Glover TW (2006) Common fragile sites as targets for chromosome rearrangements. DNA Repair 5:1126–1135
Iliopoulos D, Guler G, Han SY et al (2006) Roles of FHIT and WWOX fragile genes in cancer. Cancer Lett 232:27–36
Drusco A, Pekarsky Y, Costinean S et al (2011) Common fragile site tumor suppressor genes and corresponding mouse models of cancer. J Biomed Biotechnol 2011:984505
Saldivar JC, Bene J, Hosseini SA et al (2013) Characterization of the role of Fhit in suppression of DNA damage. Adv Biol Regul 53:77–85
Miller CT, Lin L, Casper AM et al (2006) Genomic amplification of MET with boundaries within fragile site FRA7G and upregulation of MET pathways in esophageal adenocarcinoma. Oncogene 25:409–418
Ciullo M, Debily MA, Rozier L et al (2002) Initiation of the breakage-fusion-bridge mechanism through common fragile site activation in human breast cancer cells: the model of PIP gene duplication from a break at FRA7I. Hum Mol Genet 11:2887–2894
Wilke CM, Guo SW, Hall BK et al (1994) Multicolor FISH mapping of YAC clones in 3p14 and identification of a YAC spanning both FRA3B and the t(3;8) associated with hereditary renal cell carcinoma. Genomics 22:319–326
Yamakawa K, Takahashi E, Murata M, Okui K, Yokoyama S, Nakamura Y (1992) Detailed mapping around the breakpoint of (3;8) translocation in familial renal cell carcinoma and FRA3B. Genomics 14:412–416
Chesi M, Bergsagel PL, Shonukan OO et al (1998) Frequent dysregulation of the c-maf proto-oncogene at 16q23 by translocation to an Ig locus in multiple myeloma. Blood 91:4457–4463
Nambiar M, Raghavan SC (2011) How does DNA break during chromosomal translocations? Nucleic Acids Res 39:5813–5825
Klein IA, Resch W, Jankovic M et al (2011) Translocation-capture sequencing reveals the extent and nature of chromosomal rearrangements in B lymphocytes. Cell 147:95–106
Rassool FV, McKeithan TW, Neilly ME, van Melle E, Espinosa R 3rd, Le Beau MM (1991) Preferential integration of marker DNA into the chromosomal fragile site at 3p14: an approach to cloning fragile sites. Proc Natl Acad Sci U S A 88:6657–6661
Wilke CM, Hall BK, Hoge A, Paradee W, Smith DI, Glover TW (1996) FRA3B extends over a broad region and contains a spontaneous HPV16 integration site: direct evidence for the coincidence of viral integration sites and fragile sites. Hum Mol Genet 5:187–195
Thorland EC, Myers SL, Gostout BS, Smith DI (2003) Common fragile sites are preferential targets for HPV16 integrations in cervical tumors. Oncogene 22:1225–1237
Bester AC, Roniger M, Oren YS et al (2011) Nucleotide deficiency promotes genomic instability in early stages of cancer development. Cell 145:435–446
Spardy N, Duensing A, Hoskins EE, Wells SI, Duensing S (2008) HPV-16 E7 reveals a link between DNA replication stress, fanconi anemia D2 protein, and alternative lengthening of telomere-associated promyelocytic leukemia bodies. Cancer Res 68:9954–9963
Ying S, Minocherhomji S, Chan KL et al (2013) MUS81 promotes common fragile site expression. Nat Cell Biol 15:1001–1007
Acknowledgements
The authors thank past and present members of the Le Beau laboratory for their support.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Jiang, Y., Lucas, I., Le Beau, M.M. (2015). Common Chromosomal Fragile Sites and Cancer. In: Rowley, J., Le Beau, M., Rabbitts, T. (eds) Chromosomal Translocations and Genome Rearrangements in Cancer. Springer, Cham. https://doi.org/10.1007/978-3-319-19983-2_5
Download citation
DOI: https://doi.org/10.1007/978-3-319-19983-2_5
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-19982-5
Online ISBN: 978-3-319-19983-2
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)