
Abstract
A detailed analysis of the plant cell division cycle is required to understand the changes in metabolic, developmental, and physiological processes. Determination of DNA synthesis phase (S-phase) of the cell division cycle is particularly important to assess the proliferative status of a given cell, tissue, or organ. Several methods exist to detect and quantify the S-phase of the cell division cycle. In comparison to frequently used detection by bromodeoxyuridine (BrdU) and tritiated thymidine incorporation, recently introduced ethynyl deoxyuridine method (EdU) described here, affords several advantages. EdU assay offers a simple, rapid, and nonradioactive method for proliferation analysis in plant cells while preserving morphological integrity. Here, we describe the 5-ethynyl-2′-deoxyuridine (EdU)-based S-phase assay for use in plant cells and tissues.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
Assaying cell proliferation is crucial in assessing cell health, characterizing cellular responses to various treatments or genetic modifications. It is also essential to determine the degree of synchronicity in cell division cycle synchronization experiments. Most cell proliferation assays estimate the number of cells by either incorporating a modified nucleotide during cell proliferation or by measuring the total nucleic acid content of lysed or isolated cells. The most accurate method is direct measurement of new DNA synthesis , which traditionally was achieved by the incorporation of tritium-labeled thymidine and detection by autoradiography [1, 2]. This radioactive method has been replaced by others, such as the incorporation of the thymidine analog, 5-bromo-2-deoxyuridine (BrdU) , into DNA followed by immunodetection with a specific antibody raised against BrdU [3, 4]. Although effective and sensitive, this method requires DNA denaturation or digestion (using hydrochloric acid, heat, or DNase) to expose BrdU to the antibody [5, 6]. The use of acid or heat often destroys cell morphology and damages the epitope of many proteins. This hinders their immunocytochemical detection with fluorescence-labeled antibodies. Using DNase, however, poses difficulty in obtaining reproducible levels of DNA digestion while analyzing different cell types or samples [7].
The antibody-based detection method of BrdU assay also necessitates cell wall digestion in experiments carried out on plant cells. Therefore, protoplasts or partially cell wall-digested cells/organs/tissue sections are often used for BrdU-based detection of proliferative activity in plants [8]. However, a significant wounding and osmotic stress is imposed on live plant cells due to treatment with cell wall-digesting enzymes. Moreover, specific optimization of the digestion medium, of the type and concentration of the enzymes, and their osmolarity is required for each plant species, organ, and cell type under investigation [9]. Partial cell wall digestion or release of protoplasts not only prolongs the experimental duration but also causes substantial reorganization of cytoskeleton and activation of stress- and defense-related genes. It is also possible to first chemically fix the cells and then partially digest their cell wall to diminish stress-related artifacts. However, this approach requires highly pure and expensive cell wall digestion enzymes, as crude enzyme preparations contain impurities such as proteases and nucleases that can significantly compromise cellular integrity [10].
Recently, a new agent 5-ethynyl-2′-deoxyuridine (EdU) based assay has emerged as a remarkable alternative to the abovementioned methods. EdU is a terminal alkyne containing nucleoside analog of thymidine and is incorporated into DNA during active DNA synthesis [11]. The EdU detection method is based on click chemistry. “Click” chemistry (Huisgen 1, 3-dipolar cycloaddition) is a type of chemical reaction that occurs at room temperature and is catalyzed by copper Cu(I), resulting in the formation of a strong covalent bond between an azide (present in the structure of the fluorochrome) and an alkyne group (present in EdU) [12, 13] (Fig. 18.1).

Copper-catalyzed azide-alkyne cycloaddition reaction. EdU incorporated into DNA during the S-phase of the cell cycle is detected by copper(I)-catalyzed click coupling to an azide-derivatized fluorophore. The reaction of EdU with Alexa Fluor 488 azide is shown as an example
The alkyne group is quite unreactive in biological systems and thus allows use in living cells [14, 15]. The small molecular size of the detection fluorochrome, compared to that of antibodies required for BrdU-based immunodetection, enables efficient penetration into plant cells, without the need for cell wall digestion or harsh DNA denaturation treatments [16]. This considerably simplifies the procedure and reduces the duration of the assay. In addition, the mild non-caustic EdU proliferation assay components keep the proteins intact, allowing their parallel immunocytochemical detection with fluorescence-labeled antibodies. Although initially developed for application in cultured mammalian cells, the EdU assay has been successfully applied in a wide variety of species including bacteria [17], yeast [18, 19], and a broad spectrum of animals [20–23] and plants [16].
The EdU-based assay has already been applied in several plant tissues and organs such as Arabidopsis root tips [24–29], leaf/petiole junction [30], and inflorescence [31]. The assay was also used to differentiate cells in early and late S-phase in root tips of Arabidopsis seedlings [32]. The assay was successfully applied in alfalfa root tips [33] and suspension-cultured cells [34], tomato root tips [35, 36], field bean root tips [37], asparagus cladodes [38], tobacco suspension-cultured cells [39], rice suspension-cultured cells [40], and maize root tips [41]. Further, this assay was used to determine the proliferation activity of different cell types of the anther locule of maize [42]. Here, we describe detailed cell synchronization and EdU-detection protocols for both monocot (rice)- and dicot (Arabidopsis)-cultured cells and roots.
2 EdU Labeling of Suspension-Cultured Cells
2.1 Materials
-
1.
Arabidopsis thaliana ecotype Landsberg erecta (cell line MM1) culture medium: Murashige and Skoog (MS) medium with 0.5 mg/L naphthalene acetic acid (NAA) and 0.05 mg/L kinetin.
-
2.
Oryza sativa L. ssp japonica cv. “Unggi-9” cell culture medium: G1 medium with 1 mg/L 2,4-dichlorophenoxy acetic acid (2,4-D) [16] .
-
3.
5-ethynyl-2′-deoxyuridine (EdU) stock solution: 10 mM EdU is prepared in dimethyl sulfoxide (DMSO) as a 1000× concentrated stock solution (see Note 1).
-
4.
Hydroxyurea (HU): HU prepared as a 1 M solution in water and sterilized with a filter such as 0.22 µm pore-sized Millipore mixed cellulose ester membrane. HU should be freshly prepared for each experiment as prolonged storage in aqueous medium may cause decomposition (see Note 2).
-
5.
Formaldehyde stock solution (8 % w/v): Dissolve 8 g paraformaldehyde powder in 100 mL water by heating to 60–70 °C in a fume hood. Add drops of 5 M KOH to the warm milky solution until it becomes completely clear. Heating at alkaline pH depolymerizes paraformaldehyde. After cooling to room temperature, adjust pH to between 6.5 and 7.5 with 5 % (v/v) H2SO4. Aliquots of this stock solution can be stored frozen for 6 months (see Note 3).
-
6.
Fixation solution (4 % formaldehyde in PBS with Triton X-100): Mix 8 % formaldehyde stock solution with equal volume of 2× PBS (2× PBS: 5.4 mM KCl, 2.94 mM KH2PO4, 274 mM NaCl, and 16 mM Na2HPO4, pH 7.4) and add Triton X-100 to a final concentration of 0.05 % (see Note 5).
-
7.
EdU detection cocktail: 1× PBS, 40 mM sodium ascorbate, 0.5 % Triton X-100, 4 mM CuSO4, 2.5–20 µM Alexa Fluor 488 azide (Invitrogen). To prevent oxidation of the formed Cu (I) to non-catalytic Cu (II) species, prepare the detection cocktail freshly and use within 15 min. The sequence of adding the components is important. Follow the sequence given above.
-
8.
DNA staining (DAPI) solution: Prepare 1 mg/mL DAPI (4,6-diamidino-2-phenylindole) solution in DMSO (10,000× stock) and dilute to 100 ng/mL in 1× PBS (see Note 4).
-
9.
Mounting solution: For short-term mounting of samples, use 1× PBS which prevents cell shrinkage. For long-term preservation of samples, use an anti-fade mounting solution such as Fluoromount-G (Southern Biotech; see Note 7).
-
10.
Consumables: 1.5 mL microfuge tubes, 4 mL cylindrical polypropylene tubes with cap, microscope slides, and circular coverslips.
-
11.
Equipment: Laminar flow hood, fume hood, roller/rocker plate, desktop centrifuge with swing-out rotor, fluorescence microscope, or laser scanning fluorescence confocal microscope with appropriate filter sets.
2.2 Method
2.2.1 Synchronization of Rice Suspension-Cultured Cells
-
1.
Seven-day-old suspension culture of rice is kept in the same medium for two more days to induce partial nutrient starvation and subcultured in a 1:3 ratio of conditioned medium to fresh medium ratio on the 9th day following previous subculturing .
-
2.
Freshly prepared, filter sterilized HU at a final concentration of 20 mM is immediately added to the cells and incubated for a period of 36 h to block them in the S-phase of cell cycle.
-
3.
HU block is removed by washing the cells 3 × 10 min with the sterile supernatant of a 1-day-old rice suspension culture (see Note 8).
-
4.
Samples are taken before wash, after wash and at 2 h intervals following the wash.
2.2.2 Synchronization of Arabidopsis Suspension-Cultured Cells
-
1.
Seven-day-old suspension culture of Arabidopsis is kept in the same culture medium for one more day (8th day) to induce partial nutrient starvation and subcultured by diluting 20× with fresh medium.
-
2.
Freshly prepared, filter sterilized HU at a final concentration of 5 mM is immediately added to the cells and incubated for a period of 36 h to block them in the S-phase of cell cycle .
-
3.
HU block is removed by washing the cells 3 × 10 min with the sterile supernatant of a 36-h-old Arabidopsis suspension culture (see Note 8).
-
4.
Samples are taken before wash, after wash and at 2 h intervals following the wash.
2.2.3 EdU Labeling
-
1.
Incubate 1 mL of cells in a 1.5 mL microfuge tube on a roller with 10 µM final concentration of EdU for 30 min in its own culture medium at culture growth temperature.
-
2.
Settle cells by centrifuging for 5 min at 400g. Wash the pellet in 1.5 mL 1× PBS for 5 min (in a roller) and transfer to 4 mL tube, recentrifuge and discard the supernatant.
-
3.
Fix the cell pellet for 45 min (rice) or 30 min (Arabidopsis) in 4 mL fixation solution on a roller. Centrifuge and replace the fixative with the same volume of 1× PBS. At this stage, the samples can be kept in the refrigerator for several weeks until further processing.
-
4.
Wash cells 3 × 5 min with 1× PBS. Collect 200 µL (1/20th of total fixed cells) of cells containing the smallest clusters (see Note 9). Collect cells by centrifugation, discard the supernatant and incubate the cells in 150 µL EdU detection cocktail by rotating for 30 min at room temperature in 0.2 mL microfuge tube (see Note 6).
-
5.
After 2 × 5 min washes with 1× PBS (0.2 mL each), label the nucleus with 1× PBS containing DAPI (100 ng/mL final concentration) for 5 min. Mount 20 µL aliquot of the cells onto a microscope slide with a coverslip and gently press with a tissue paper to flatten the clusters. The rest of the labeled cells can be kept in the fridge, in a dark container for several days.
-
6.
Using a fluorescence microscope, count the number of EdU-positive cells among 500–1000 DAPI-labeled nuclei and calculate the percentage of S-phase nuclei. Note that some cells which are at the very beginning or at the very end of DNA synthesis may display spotty or patchy EdU signal. As an additional indicator of cell cycle status, mitotic index of the samples can also be calculated in parallel using the same sample (Fig. 18.2a, b).
Fig. 18.2 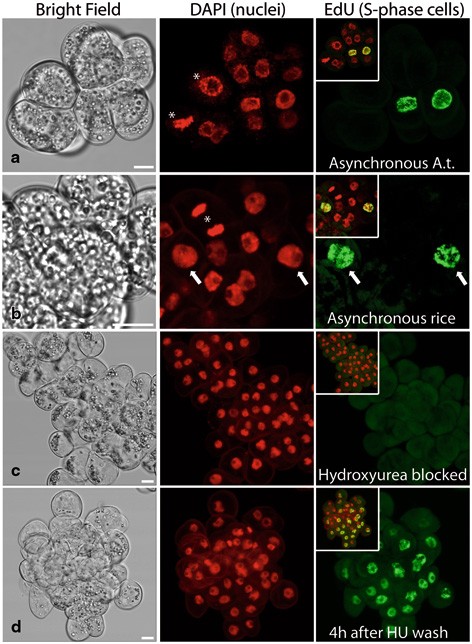
EdU labeling of S-phase nuclei on suspension-cultured cells of Arabidopsis (cell line “MM1,” Panel a) and rice (cv. “Unggi-9,” Panels b–d). Asynchronously dividing (a, b), hydroxyurea (HU)-blocked (c) and HU-released (4 h after HU wash) cells were treated with 10 mM EdU for 30 min. Nuclei were labeled with DAPI (middle column) and mitotic cells of asynchronously dividing cultures were highlighted with asterisks. Arrows indicate EdU-incorporated nuclei. Scale bars = 10 µm

3 EdU Labeling of Root Tissue
3.1 Materials
-
1.
Sterile half-strength MS agar plates containing 0.7 % agar with no hormones.
-
2.
Oryza sativa L. ssp japonica cv. “Nipponbare” and Arabidopsis thaliana ecotype Columbia-0 seeds.
-
3.
Sterilization solutions: 70 % ethanol and 4 % (v/v) commercial bleach (e.g., domestos containing 4.8 g sodium hypochlorite per 100 g) in water.
-
4.
5-ethynyl-2′-deoxyuridine (EdU) stock solution: see Sect. 18.2.1
-
5.
Formaldehyde stock solution (8 % w/v): see Sect. 18.2.1
-
6.
Fixation solution (4 % formaldehyde in PBS with TritonX-100): see Sect. 18.2.1
-
7.
EdU detection cocktail: see Sect. 18.2.1
-
8.
DNA staining (DAPI) solution: see Sect. 18.2.1
-
9.
Mounting solution: see Sect. 18.2.1
-
10.
Consumables: see Sect. 18.2.1
-
11.
Equipment: see Sect. 18.2.1
3.2 Method
-
1.
Dehusk rice seeds before sterilization. Place rice or Arabidopsis seeds in a 1.5 mL microfuge tube containing 70 % ethanol and roll for 1 min.
-
2.
Replace ethanol with 4 % (v/v) commercial bleach and roll for 15 min.
-
3.
Wash seeds 5 × 5 min in sterile distilled water and place them on sterile half strength MS agar plates. Seeds grown at 22 °C in long day conditions (16 h light/8 h dark cycle).
-
4.
Seven-day-old seedlings were incubated in half strength liquid MS medium with 10 µM EdU for a period of 30 min to 2 h at room temperature.
-
5.
Roots of rice seedlings were cut directly under fixation solution in a petri dish. Arabidopsis seedlings were fixed as whole. Both rice roots and Arabidopsis seedlings were fixed for 30 min at room temperature (see Note 10).
-
6.
Replace the fixer with 1× PBS. At this stage, the samples can be stored in the refrigerator for several days.
-
7.
Wash the root tips/seedlings 3 × 5 min in 1× PBS. Cut Arabidopsis root tips under 1× PBS after washes. Incubate root tips in 200 µL detection cocktail by rotating for 30 min at room temperature in 0.2 mL microfuge tube (see Note 6).
-
8.
After 3 × 5 min washes with 1× PBS, counterstain the nucleus with 1× PBS containing DAPI (500 ng/mL) at room temperature for 20 min.
-
9.
Mount root tips onto a microscope slide using a coverslip and mounting solution (e.g., Fluoromount-G) and gently press with a tissue paper to flatten the root tips and to blot the excess mounting solution (see Note 7).
-
10.
For quantification of labeling, individual labeled nuclei in a given root region (root tip, stele, etc., Fig. 18.3) can be counted (see Sect. 18.2.2.3). Alternatively, total fluorescence intensity of the EdU-linked fluorochrome can be measured in a predetermined region of interest [16].
Fig. 18.3 
EdU labeling of S-phase cells on various root tissues of Arabidopsis (Ecotype “Columbia-0”) and rice (cv. “Nipponbare”). Roots of seedlings were immersed in 10 µM EdU for 30 min (rice) or 2 h (A.t: Arabidopsis). EdU-incorporated S-phase cells at root tips (a, b), stele (c) and lateral root meristem (d) are shown. Nuclei were labeled with DAPI (middle column). Scale bars = 50 µm

4 Notes
-
1.
Caution: EdU is toxic, use appropriate precautions. EdU can interfere with cell cycle progression in long-term experiments. For long-term cell cycle studies (i.e., EdU incubation during complete cell division cycle) , a less toxic derivative, F-ara-EdU can be used [43–45].
-
2.
Caution: HU is toxic and carcinogenic; use appropriate precautions. More efficient but significantly more expensive inhibitor, aphidicolin can also be used instead of HU.
-
3.
Paraformaldehyde is very hazardous in case of skin contact, eye contact, or inhalation (irritant/corrosive). Work in a fume hood and wear protective equipment. Avoid repeated freeze-thaw cycles of frozen aliquots. Thawed aliquots may require reheating to 60–70 °C for complete dissolution. Discard the aliquot if reheating does not clear up the precipitate. A fixation solution prepared from powdered formaldehyde is a better fixative than commercially available liquid formaldehyde solution [10].
-
4.
Caution: DAPI which is used as a nuclear counterstain is a known mutagen; use appropriate precautions. Other nucleic acid dyes such as Hoechst (DNA specific) or Propidium iodide (DNA/RNA stain) can also be used to locate the nuclei and chromosomes. All nucleic acid intercalating dyes should be handled with extreme care due to health hazards.
-
5.
The addition of Triton X-100 to the fixation solution provides uniform fixation with reduced cell shrinkage.
-
6.
Fluorochrome-containing solutions should not be exposed to strong light; therefore, incubations should be performed at dark or under dim light. The simplest solution is to wrap the samples in aluminum foil during incubations.
-
7.
Glycerol-based (or high osmolarity) mounting media may cause cell shrinkage but they better suit imaging with high numerical aperture oil immersion objectives. Mounting the samples in PBS or water-based anti-fade solutions prevents cell shrinkage; however, care must be taken not to dry out the sample. Sealing the coverslip or occasional PBS loading may be necessary for prolonged observations to prevent sample drying.
-
8.
One-day-old (rice) or 36-h-old (Arabidopsis) conditioned culture mediums were prepared by subculturing separate cultures 1 day (or 36 h in case of Arabidopsis) before the time of HU wash. We have found that using such conditioned mediums for HU washes is far more efficient than using fresh culture mediums for both rice and Arabidopsis.
-
9.
A brief settling on bench settles the largest and heaviest clusters leaving the smaller clusters in suspension. Nylon mesh can also be used to collect a population of finer clusters as long as the smaller size of the cluster is not due to genetic variation. Selecting smaller clusters makes S-phase and mitotic index counting easier.
-
10.
Since the fixer contains Triton X-100, plants with thin and fragile seedlings (like Arabidopsis) can also be fixed as a whole without cutting the roots (as shown in Fig. 18.3b–d). However, we have found that roots have to be cut before incubation with the EdU detection cocktail for fast and efficient penetration of the azide containing fluorochrome.
5 Interpretation and Conclusions (Troubleshooting)
Faint/no EdU labeling can be caused by:
-
1.
The sequence of adding the components of the EdU detection cocktail is important. If the detection cocktail turns milky or develops a precipitate after the addition of all the components, it will not work efficiently.
-
2.
Sodium ascorbate may have degraded. Stock solutions must be stored at ≤ − 20 °C. Thus stored, the solution is stable for several months. If the solution has turned brown, it has degraded. Discard and prepare new stock solution.
-
3.
EdU may have degraded due to improper storage. Aliquots of EdU stock solution must be stored at ≤ − 20 °C. At this storage condition, the solution is stable for at least a year.
-
4.
At the time of EdU addition, the cell culture or the tissue under consideration may not contain DNA synthesizing cells due to suboptimal growth conditions, stress, inhibitory chemicals, or altered genetic makeup. For asynchronously dividing cultures and young tissues, DAPI labeling of fixed samples and detection of mitosis can help to quickly assess the proliferative status of the culture.
References
Taylor JH, Woods PS, Hughes WL (1957) The organization and duplication of chromosomes as revealed by autoradiographic studies using tritium labeled thymidine. Proc Natl Acad Sci U S A 43:122–128
Mendelsohn ML (1962) Autoradiographic analysis of cell proliferation in spontaneous breast cancer of C3H mouse. The growth fraction. J Natl Cancer Inst 28:1015–1029
Gratzner HG (1982) Monoclonal antibody to 5-bromo- and 5-iododeoxyuridine: a new reagent for detection of DNA replication. Science 218:474–475
Leif RC, Stein JH, Zucker RM (2004) A short history of the initial application of anti-5-BrdU to the detection and measurement of S phase. Cytometry A 58:45–52
Dolbeare F, Gratzner H, Pallavicini MG, Gray JW (1983) Flow cytometric measurement of total DNA content and incorporated bromodeoxyuridine. Proc Natl Acad SciU SA 80:5573–5577
Moran R, Darzynkiewicz Z, Staiano-Coico L, Melamed MR (1985) Detection of BrdUrd incorporation by monoclonal antibodies: role of the DNA denaturation step. J Histochem Cytochem 33:821–827
Holm M, Thomsen M, Hoyer M, Hokland P (1998) Optimization of a flow cytometric method for the simultaneous measurement of cell surface antigen, DNA content and in vivo BrdUrd incorporation into normal and malignant hematopoietic cells. Cytometry A 32:28–36
Stroobants C, Sossountzov L, Miginiac E (1990) DNA Synthesis in excised tobacco leaves after Bromodeoxyuridine incorporation: immunohistochemical detection in semi-thin spurr sections. J Histochem Cytochem 38:641–647
Fowke LC, Cutler AJ (1994) Plant protoplast techniques. In: Harris N, Oparka KJ (eds) Plant cell biology: a practical approach. IRL, Oxford, pp 177–196
Goodbody KC, Lloyd CW (1994) Immunofluorescence techniques for the analysis of the cytoskeleton. In: Harris N, Oparka KJ (eds) Plant cell biology: a practical approach. IRL, Oxford, pp 221–243
Salic A, Mitchison TJ (2008) A chemical method for fast and sensitive detection of DNA synthesis in vivo. Proc Natl Acad Sci U S A 105:2415–2420
Kolb HC, Sharpless KB (2003) The growing impact of click chemistry on drug discovery. Drug Discov Today 8:1128–1137
Darzynkiewicz Z, Traganos F, Zhao H, Halicka HD, Li J (2011) Cytometry of DNA replication and RNA synthesis: historical perspective and recent advances based on “click chemistry”. Cytometry A 79:328–337
Best MD (2009) Click chemistry and bioorthogonal reactions: unprecedented selectivity in the labeling of biological molecules. BioChemistry 48:6571–6584
Agard NJ, Baskin JM, Prescher JA, Lo A, Bertozzi CR (2006) A comparative study of bioorthogonal reactions with azides. ACS Chem Biol 1:644–648
Kotogány E, Dudits D, Horváth GV, Ayaydin F (2010) A rapid and robust assay for detection of S-phase cell cycle progression in plant cells and tissues by using ethynyl deoxyuridine. Plant Meth 6:5
Ferullo DJ, Cooper DL, Moore HR, Lovett S (2010) Cell cycle synchronization of E. coli using the stringent response, with fluorescence labeling assays for DNA content and replication. Methods 48:8–13
Hua H, Kearsey SE (2011) Monitoring DNA replication in fission yeast by incorporation of 5-ethynyl-2′-deoxyuridine. Nucl Acids Res 39:e60
Lopez-Serra L, Lengronne A, Borges V, Kelly G, Uhlmann F (2013) Budding yeast Wapl controls sister chromatid cohesion maintenance and chromosome condensation. Curr Biol 23:64–69
Kaiser CL, Kamien AJ, Shah PA, Chapman BJ, Cotanche DA (2009) 5-ethynyl-2′-deoxyuridine labeling detects proliferating cells in the regenerating avian cochlea. Laryngoscope 119:1770–1775
Warren M, Puskarczyk K, Chapman SC (2009) Chick embryo proliferation studies using EdU labeling. Dev Dyn 238:944–949
Diermeier-Daucher S, Clarke ST, Hill D, Vollmann-Zwerenz A, Bradford JA, Brockhoff G (2009) Cell type specific applicability of 5-ethynyl-2′-deoxyuridine (EdU) for dynamic proliferation assessment in flow cytometry. Cytometry A 75:535–546
Limsirichaikul S, Niimi A, Fawcett H, Lehmann A, Yamashita S, Ogi T (2009) A rapid non-radioactive technique for measurement of repair synthesis in primary human fibroblasts by incorporation of ethynyl deoxyuridine (EdU). Nucl Acids Res 37:e31
Notaguchi M, Wolf S, Lucas WJ (2012) Phloem-mobile Aux/IAA transcripts target to the root tip and modify root architecture. J Integr Plant Biol 54:760–772
Kakar K, Zhang H, Scheres B, Dhonukshe P (2013) CLASP-mediated cortical microtubule organization guides PIN polarization axis. Nature 495:529–533
Perilli S, Perez-Perez JM, Di Mambro R, Peris CL, Diaz-Trivino S, Del Bianco M, Pierdonati E, Moubayidin L, Cruz-Ramirez A, Constantino P, Scheres B, Sabatini S (2013) RETINOBLASTOMA-RELATED protein stimulates cell differentiation in the Arabidopsis root meristem by interacting with cytokinin signaling. Plant Cell 25:4469–4478
Zhu Y, Weng M, Yang Y, Zhang C, Li Z, Shen WH, Dong A (2011) Arabidopsis homologues of the histone chaperone ASF1 are crucial for chromatin replication and cell proliferation in plant development. Plant J 66:443–455
Xu D, Huang W, Li Y, Wang H, Hunag H, Cui X (2012) Elongator complex is critical for cell cycle progression and leaf patterning in Arabidopsis. Plant J 69:792–808
Xiong Y, McCormack M, Li L, Hall Q, Xiang C, Sheen J (2013) Glucose–TOR signalling reprograms the transcriptome and activates meristems. Nature 496:181–186
Ichihashi Y, Kawade K, Usami T, Horiguchi G, Takahashi T, Tsukaya H (2011) Key proliferative activity in the junction between the leaf blade and leaf petiole of Arabidopsis. Plant Physiol 157:1151–1162
She W, Grimanelli D, Rutowicz K, Whitehead MW, Puzio M, Kotlinski M, Jerzmanowski A, Baroux C (2013) Chromatin reprogramming during the somatic-to-reproductive cell fate transition in plants. Development 140:4008–4019
Hayashi K, Hasegawa J, Matasunaga S (2013) The boundary of the meristematic and elongation zones in roots: endoreduplication precedes rapid cell expansion. Sci Rep 3:2723
Bazin J, Khan GA, Combier JP, Bustos-Sanmamed P, Debernardi JM, Rodriguez R, Sorin C, Palatnik J, Hartmann C, Crespi M, Lelandais-Briere C (2013) miR396 affects mycorrhization and root meristem activity in the legume Medicago truncatula. Plant J 74:920–934
Ayaydin F, Kotogány E, Abraham E, Horváth GV (2011) Synchronization of Medicago sativa cell suspension culture. Meth Mol Biol 761:227–238
Ron M, Dorrity MW, de Lucas M, Toal T, Hernandez RJ, Little SA, Maloof JN, Kliebenstein DJ, Brady SM (2013) Identification of novel loci regulating interspecific variation in root morphology and cellular development in tomato. Plant Physiol 162:755–768
Kuznetsova MA, Sheval EV (2013) Detection of replication sites in plant cell nuclei by using semithin sections. Cell Tissue Biol 7:375–378
Schubert I, Schubert V, Fuchs J (2011) No evidence for “break-induced replication” in a higher plant—but break-induced conversion may occur. Front Plant Sci 2:8
Nakayama H, Yamaguchi T, Tsukaya H (2012) Acquisition and diversification of cladodes: leaf-like organs in the genus Asparagus. Plant Cell 24:929–940
Tresch S, Schmotz J, Grossmann K (2011) Probing mode of action in plant cell cycle by the herbicide endothall, a protein phosphatase inhibitor. Pestic Biochem Physiol 99:86–95
Dudits D, Abraham E, Miskolczi P, Ayaydin F, Bilgin M, Horváth GV (2011) Cell-cycle control as a target for calcium, hormonal and developmental signals: the role of phosphorylation in the retinoblastoma-centred pathway. Ann Bot 107:1193–1202
Bass HW, Wear EE, Lee TJ, Hoffman GG, Gumber HK, Allen GC, Thompson WF, Hanley-Bowdoin L (2014) A maize root tip system to study DNA replication programmes in somatic and endocycling nuclei during plant development. J Exp Bot 65:2747–2756
Kelliher T, Walbot V (2011) Emergence and patterning of the five cell types of the Zea mays anther locule. Dev Biol 350:32–49
Neef AB, Luedtkel NW (2011) Dynamic metabolic labeling of DNA in vivo with arabinosyl nucleosides. Proc Natl Acad Sci U S A 108:20404–20409
Cruz-Ramirez A, Diaz-Trivino S, Wachsman G, Du Y, Arteaga-Vazquez M, Zhang H, Benjamins R, Blilou I, Neef AB, Chandler V, Scheres B (2013) A SCARECROW-RETINOBLASTOMA protein network controls protective quiescence in the Arabidopsis root stem cell organizer. PLOS Biol 11:e1001724
Qu D, Wang G, Wang Z, Zhou L, Chi W, Cong S, Ren X, Liang P, Zhang B (2011) 5-ethynyl-2′-deoxycytidine as a new agent for DNA labeling: detection of proliferating cells. Anal Biochem 417:112–121
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Kuntam, S., Ayaydin, F. (2015). Detection of S-Phase of Cell Division Cycle in Plant Cells and Tissues by Using 5-Ethynyl-2′-Deoxyuridine (EdU). In: Yeung, E., Stasolla, C., Sumner, M., Huang, B. (eds) Plant Microtechniques and Protocols. Springer, Cham. https://doi.org/10.1007/978-3-319-19944-3_18
Download citation
DOI: https://doi.org/10.1007/978-3-319-19944-3_18
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-19943-6
Online ISBN: 978-3-319-19944-3
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)