
Abstract
Heart failure (HF) is a clinical diagnosis combining characteristic symptoms with physical or instrumental findings. No single tests can absolutely establish its presence or absence. An episode of acute heart failure is a complex clinical condition defined as a rapid or gradual onset of symptoms and signs of heart failure requiring immediate medical attention. Despite therapeutic advances in the care of chronic HF, the prognosis of patients with acute heart failure syndromes (AHFS) remains poor, with substantial morbidity and mortality and high health care cost. Management of AHFS remains a major challenge in current clinical practice given the heterogeneity of underlying pathophysiological mechanisms and clinical presentations. No single treatment algorithm may be feasible for all patients. Identification of the acute precipitating factors as well as noninvasive characterization of cardiac filling pressures and output is necessary in order to align therapeutic strategies to patient clinical profile. Improving hemodynamics and minimizing adverse events are the key goals of early in-hospital therapies traditionally including oxygen, noninvasive ventilation (NIV), diuretics, vasodilators and inotropic agents. However, still today we lack robust evidence linking hospital management with post-discharge outcomes, therefore the use available pharmacological agents for AHFS are largely empirical. In this chapter currently available data on diagnosis, clinical assessment and management of patients presenting with AHFS will be discussed.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Pulmonary Capillary Wedge Pressure
- Filling Pressure
- Loop Diuretic
- Acute Decompensated Heart Failure
- Inotropic Agent
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Case Report
A 64-year-old man was admitted to the emergency room for acute dyspnea. The patient reported fatigue and dyspnea for minimum efforts during the days before. He reported no recent pathological findings.
1.1 Medical History and Cardiovascular Risk Factors
-
Type 2 diabetes mellitus.
-
2013: access to the emergency room for asthenia and dizziness; on that occasion, high levels of glucose were found.
1.2 Allergies
None
1.3 Social History
He used to smoke about 30 cigarettes/day some years ago.
1.4 Home Medications
Rosuvastatin 20 mg at 9.00 p.m., metformin 850 mg at 12.00 a.m and at 8.00 p.m.
1.5 Vital Signs
-
Temperature: 36.5 °C
-
Heart rate: 126 beats per minutes
-
Blood pressure: 170/100 mmHg
-
Respiratory rate: 22/min
-
Oxygen saturation while breathing in ambient air: 92 %
1.6 Physical Examination
-
General: alert, awake, and oriented but slightly agitated
-
Neck: slight jugular venous distention, no lymphadenopathy, and no carotid murmur
-
Cardiovascular: early diastolic gallop with S3 and, systolic murmur 2/6 at the mesocardium without radiation to the armpit, neck, or carotid vessels
-
Lungs: breath sounds diffusely decreased, in particular at the lung bases, and rales up to medium shots bilaterally
-
Abdomen: plain, no pulsatile masses, normal bowel sounds in all four quadrants, no high-pitched or tinkling sounds, resonant to percussion, soft, nondistended/nontender, no rebound or guarding, no costovertebral angle tenderness, hepatomegaly up to 2 cm from the costal margin, no splenomegaly, and Giordano and Murphy signs negative
-
Neurological: negative cerebellar test, cranial nerve intact, no focal deficit, and reflections normoexcitable
-
Psychiatric: normal
-
Skin: pale, cold, and sweaty with cyanosis of the extremities
1.7 Routine Laboratory Tests
-
Complete blood count: leukocytosis with neutrophilia (WBC 10.760/mmc, 91.20 % neutrophils), hemoglobin 13.5 g/dl, and platelets 248,000/mmc
-
Inflammatory markers: ESR 29 mm/h and CRP 0.6 mg/dl
-
Hepatic function: GOT normal, GPT with slight increase (61 U/l), γ-GT 121 U/l, and ALP, total bilirubin (direct and indirect), and coagulation normal
-
Normal renal function (creatinine 0.82 mg/dl, BUN 38 mg/dl, eGFR 92.8 ml/min/1.73 m2)
-
Electrolytes (Na+, K+, Ca++, Mg++, Cl−): normal
-
Fasting blood glucose: 179 mg/dl
-
Myocardial necrosis markers: normal CK-MB and Hs-TnI 0.059 ng/ml (n.v. 0–0.055)
-
BNP: 744 pg/ml
-
Thyroid function: normal TSH and fT4 and fT3 2.10 pg/ml (n.v. 2.2–4.2 pg/ml)
The blood gas analysis performed in ambient air showed pH = 7.41, pO2 = 58 mmHg, pCO2 = 40 mmHg, and p/F = 276. ECG showed a sinus tachycardia (heart rate was 126 beats per minutes), normal atrioventricular and intraventricular conduction, and nonspecific alterations of ventricular repolarization.
1.8 Chest X-Ray
X-ray showed signs compatible with acute pulmonary edema.
1.9 What Are the Possible Causes of Worsening Acute Dyspnea and Orthopnea in This Patient?
There are several causes that may acutely unbalance the left ventricle function. There may be cardiac, extracardiac, or iatrogenic triggers; however, dyspnea can also be related to diseases affecting primarily the lungs. These are the possible causes in this patient:
-
Acute myocardial infarction
-
Hypertensive crisis
-
Arrhythmias
-
Acute myopericarditis
-
Lung diseases (bacterial pneumonia)
-
ARDS
-
Pulmonary embolism
-
Valvular disease (acute mitral regurgitation)
The patient was apyretic and CRP was negative, although there was a mild leukocytosis with neutrophilia.
1.10 EKG
EKG was negative for ischemic alterations, and hs-troponin I was minimally altered with normal CK-MB.
According to these data, acute myocardial infarction, bacterial pneumonia, arrhythmias, and acute myopericarditis were initially excluded as possible causes of dyspnea. The patient was then treated with furosemide bolus and infusion of nitroglycerin to reduce high blood pressure initially encountered. A CPAP (continuous positive airway pressure) was positioned and was set a FiO2 of 50 % and PEEP (positive end-expiratory pressure) of 10 cmH2O. The patient showed marked improvement in dyspnea, and blood gas analysis showed a significant increase in pO2 (pO2 = 58 mmHg → 139 mmHg). This favorable response to treatment could make us exclude a noncardiogenic acute pulmonary edema (ARDS), which is characterized by severe hypoxemia refractory to increased FiO2 and reduced lung compliance.
1.11 Echocardiography
An echocardiography was also recorded: “tricuspid aortic valve with normal valve opening; standard size of the aortic root and ascending aorta with mild ectasia of the aortic arch. Mild left atrial enlargement (44 ml/m2). Normal right ventricle size and systolic function (TAPSE 22 mm). Slightly dilated left ventricle with severe reduction of systolic global function (EF 25 %) and diffuse hypokinesia; modest pericardial effusion more evident close to the right sections and conditioning initial atrial collapse. No significant gradients. Mild mitral insufficiency, mild tricuspid regurgitation with high pulmonary arterial pressure (60 mmHg). Inferior vena cava dilated and hypo-collapsing.” Echocardiogram ruled out the presence of significant valvular disease and dysfunction and dilatation of the right sections but showed severe left ventricular dysfunction associated with mild pericardial effusion. At this diagnostic–therapeutic point, an underlying ischemic heart disease or a myopericarditis could not be excluded.
1.12 Coronary Angiography
An invasive coronary angiography documented the absence of hemodynamically significant stenosis, and an eco-color-Doppler of the lower limbs excluded the presence of a deep vein thrombosis (to rule out thromboembolic pulmonary disease).
1.13 Therapy
After resolution of the acute phase, a specific therapy for heart failure was given to the patient (ACEI, beta-blockers, potassium sparing), and in a few days, a good cardiovascular compensation was restored (demonstrated also by the gradual reduction of BNP: 744 pg/ml → 250 pg/ml). The absence of a compatible clinical history, the slightest movement of hs-troponin (0.059 ng/ml → 0.061 ng/ml → 0.033 ng/ml), and the constant negativity of inflammatory markers made the myopericarditis an unlikely cause of the acute pulmonary edema. We thought the hypertensive crisis was the cause of acute heart failure.
1.14 Final Diagnosis
The final diagnosis was “hypertensive crisis complicated by acute pulmonary edema in patientswith hypokinetic-dilated cardiomyopathy without hemodynamically significant stenosis” After 2 months, the patient was asymptomatic and in good hemodynamic compensation. A new echocardiogram showed the absence of pericardial effusion and an improvement in ejection fraction (EF: 25 % → 39 %).
2 Definition and Clinical Classification of Acute Heart Failure Syndromes (AHFSs)
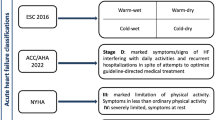
According to the latest European Society of Cardiology (ESC) guidelines, heart failure (HF) can be defined as “an anomaly of cardiac structure or function impairing heart’s ability to deliver oxygen at a rate commensurate with the requirements of the metabolizing tissues despite normal filling pressures or at the expense of increased filling pressures” [1]. The clinical manifestations of heart failure result from the impaired forward cardiac output (forward failure) and/or elevated venous pressure related (backward failure) to the failing heart. The clinical syndrome of HF may result from disorders of any aspect of cardiac function including pericardial disease, myocardial disease, endocardial disease, valvular heart disease, arrhythmias and conduction disorders, congenital heart disease, high output state, or volume overload state. These patients may present with reduced or preserved left ventricular (LV) systolic function. LV ejection fraction (LVEF) is considered important in classification of patients with HF because of differing patient prognosis and response to therapies and because most clinical trials selected patients based on LV ejection fraction (LVEF). For this reason, patients with HF are broadly categorized (with some difference among major international guidelines) in HF with preserved EF (normal or mildly reduced LVEF and LV not dilated) and HF with reduced EF (LVEF usually ≤35–40 %). An episode of acute heart failure or acute heart failure syndrome (AHFS) is usually defined as a rapid or gradual onset (or change) of symptoms and signs of heart failure (HF) requiring immediate medical attention (unplanned hospitalization or office room visit). Patients with AHFS are generally classified into those presenting with HF for the first time (de novo AHF) and those with worsening chronic HF.
2.1 Pathophysiology of AHFS
Regardless of the underlying cause or superimposed precipitating factor, pulmonary and systemic congestion (due to increased left- and/or right-side heart filling pressures), with or without low cardiac output, is the unifying finding in the broad spectrum of hemodynamic models in AHFS [2, 3]. Congestion and not low cardiac output is the main cause for AHFS [4–7].
High left-side filling pressure results in pulmonary hypertension (pulmonary congestion) with increased pulmonary capillary wedge pressure (PCWP) that preceded the subsequent clinical congestion with pulmonary interstitial and alveolar edema.
High right-side filling pressure results in systemic venous hypertension (systemic congestion) with increased central venous pressure (CVP) leading to jugular vein distension and often subsequent peripheral edema and gradual body weight gain [8]. Volume overload is only one of the possible hemodynamic perturbations that may explain the elevated filling pressure. Additional pathophysiologic mechanisms are afterload mismatch (increased afterload) and abnormal end-ventricular diastolic pressure (related to ventricular diastolic dysfunction/abnormal compliance and valvular regurgitation). Pulmonary congestion, with or without associated systemic signs, may be the results of two different pathways. The cardiac (central) pathway is the mechanism by which a low cardiac output (usually an acute decrease induced by a variety of precipitant mechanisms including ischemia, arrhythmia, inflammatory activation or progression of underlying HF process induced by progressive myocardial dysfunction) leads to a further neurohormonal activation, lower renal perfusion (cardiorenal syndrome), and fluid accumulation with systemic congestion (overload fluid retention) [9, 10]. The vascular (peripheral) pathway is related to increased vascular stiffness/resistance with acute afterload mismatch impairing systolic performance and resulting in redistribution of fluid from systemic to pulmonary circulation rather than in general fluid retention [2, 9, 10]. Venous volume mobilization of the splanchnic circulation has also been proposed as complementary mechanism [11]. Although in most cases both pathways are active during an AHF event, the magnitude of one pathway may predominate in each patient and is usually suspected according to AHF initial clinical presentation.
2.2 Precipitants of Acute Heart Failure
Approximately 80 % of acute decompensated heart failure (ADHF) patients have a worsening of chronic heart failure. In such patients with preexisting HF, one or more identifiable exacerbating factors not necessarily related to the evolution of the underlying HF disease can be often identified (up to 70 %) (Table 7.1). Detection and treatment of precipitating factors is necessary both for acute management of an episode of AHF and prevention of its recurrence.
2.3 Clinical Profiles at Presentation
The two major classes of symptoms in HF are those due to volume overload (dyspnea, orthopnea, paroxysmal nocturnal dyspnea, cough, gastrointestinal symptoms) and those due to a reduction in CO (fatigue and weakness). The most common are dyspnea and fatigue. Dyspnea (at exertion or at rest) is related to complex physiological mechanisms involving both pulmonary venous congestion and a buildup of lactic acid by working muscle increasing the ventilatory response to exercise. On the other hand, low cardiac output state often results in fatigue and weakness due to reduced skeletal muscle perfusion or atrophy. Elevated systemic venous pressures like those occurring in volume overload or right ventricular dysfunction states are responsible for abdominal discomfort (liver congestion and abdominal ascites), anorexia, and peripheral edema. Common physical findings are summarized in Table 7.2. The most common clinical findings are dyspnea (approximately 90 %), rales, and peripheral edema (65 %).
On the basis of typical clinical and hemodynamic characteristics, AHF patients may present with one of several distinct clinical profiles considering that some overlap between groups may exist [8]. The main clinical profiles and relative features are summarized in Table 7.3.
Another classification scheme has been previously proposed (Forrester classification) and is based on the severity of disease at presentation rather than on the cause of HF [12, 13]. It is a simple strategy to classify patients into specific hemodynamic profiles that may be helpful to guide the initial management strategy. Accordingly, a patient presenting with AHFS may be classified into one of the four specific hemodynamic profiles based on the absence or presence of signs of congestion (wet or dry) and the adequacy of peripheral perfusion (warm or cold): warm and dry, warm and wet, cold and dry, and cold and wet.
2.4 Clinical Assessment and Diagnosis of AHF
Traditionally, the diagnosis of HF is a clinical diagnosis combining characteristic symptoms with physical findings, and still today no single tests can absolutely establish its presence or absence. Unfortunately, signs and symptoms of HF often overlap with those of other common medical conditions (especially with chronic lung disease), and those more specific are also less common (like orthopnea and paroxysmal nocturnal dyspnea) or less reproducible (third heart sound and jugular venous distension) so that several ancillary tests, also contributing to determine mechanisms underlying the AHF, are usually needed to support the clinical diagnosis of AHF.
A chest radiography should be performed initially because it may aid in diagnosis of HF as well as in ruling out other differential diagnoses (e.g., pneumonia). Findings suggestive of HF include cardiomegaly (cardiac-to-thoracic width ratio above 50 %), upper zone vascular redistribution (cephalization), interstitial edema with Kerley B-lines, alveolar edema, and pleural effusions. Radiographic evidence of signs of pulmonary congestion in a patient with dyspnea makes the diagnosis of heart failure more likely; however, the absence of radiographic pulmonary congestion does not exclude diagnosis of AHF. Patients with chronic heart failure, despite AHF symptoms and elevated PCWP, may have few radiographic signs because of enhanced lymphatic drainage. Electrocardiography (ECG) is not useful for diagnosis but offers possible clues to identify both specific treatable precipitating factors of AHF (acute myocardial ischemia and arrhythmias) and also possible etiology of HF (e.g., Q wave in ischemic cardiomyopathy).
Laboratory tests (blood chemistry and hematological tests) are useful to guide initial therapy, to detect reversible cause of HF (e.g., hypocalcemia, thyroid dysfunction) and comorbidities (anemia), and to obtain prognostic information. Serial monitoring of myocardial necrosis biomarkers (troponin) is recommended initially for diagnostic (exclude acute coronary syndrome) and prognostic purpose. Troponin elevation in acute HF does not necessarily indicate the presence of an acute coronary syndrome. A significant number of patients with AHFS have increased levels of troponin as a result of myocardial injury during AHF episode resulting from ischemic injury and myocyte apoptosis. Such troponin elevation is associated, however, with poor long-term prognosis.
Measurement of natriuretic peptide (NP) levels is helpful especially when the diagnosis is in question. Natriuretic peptides (BNP and NT-proBNP) are a family of hormones released in increased amounts from myocytes (especially ventricular) secondary to myocardial stretch and elevated end-diastolic filling pressure as occurs in AHFS. Increased NP levels are indicators of both the presence and severity of illness. Accordingly, European guidelines recommend measurement of NP levels both to exclude alternative causes of dyspnea and to obtain prognostic information. Patient presenting with acute onset or worsening of symptoms suggestive of HF with a plasma BNP level <100 pg/ml or NT-proBNP <300 pg/ml is unlikely to have AHFS. For patients presenting in nonacute way (slow onset of symptoms), a lower exclusion NP cutoff point should be used to avoid “false-negative” diagnosis (35 pg/ml for BNP and 125 pg/ml for NT-proBNP). Results of NP tests should be always interpreted in the context of all available clinical data and should not be used in isolation to diagnose HF. A variety of conditions associated with myocardial stretch even in the absence of AHF can still be associated with NP elevation (e.g., atrial fibrillation, pulmonary hypertension, and pulmonary embolism). In addition, NP levels are falsely increased in renal failure and tend to be lower in obese patients.
An initial bedside transthoracic echocardiography is recommended both to support the diagnosis of AHFS and to determine its etiology through an assessment of cardiac anatomy and function (left and right ventricular systolic function and wall motion, diastolic function, valvular function, and pulmonary artery pressure). The TD-derived E/Ea parameter is being used to noninvasively estimate LV filling pressures. In addition, especially for those with hypotensive AHFS, echocardiographic assessment of inferior vena cava (IVC) diameter and its respiratory variation aid to determine the patient volume status.
2.5 AHFS Management
The main goal of short-term therapy (hours to days) for AHFS has been to achieve the lowest left ventricular filling pressure possible without decreasing cardiac output (especially renal perfusion), increasing heart rate, or further activating neurohormones because these factors have been associated with a worse prognosis [2]. The physician’s challenge is that many of the current medications that improve hemodynamics and symptoms may have potential deleterious effect on such variables [8].
Currently, the use of available pharmacological agents for the acute management of AHFS is largely empirical. None of the employed agents would meet today’s standards for approval based on evidence for clinical efficacy and safety. However matter, no major clinical practice guidelines include any therapeutic class I, level-of-evidence A recommendations for the pharmacological treatment of AHFS [1].
Evaluation and management of AHFS include three main phases: the initial or early phase (stabilization phase), the in-hospital phase, and the discharge phase. The main goals of each phase are summarized in Table 7.4.
2.6 Initial Management Strategy
After treatment of life-threatening conditions, improving hemodynamics and correlated symptoms are the key goals in early management. This requires a basic understanding of pathophysiologic mechanisms underlying an episode of acute HF and how potential overt precipitants adversely affect the cardiovascular system. These conditions and all HF precipitants should be targeted and treated for optimal results. Aligning treatment decision to initial patient clinical profile can yield to treat specific subgroups of patients with more tolerable therapies.
Taking the above consideration in mind, according to recommendations from ESC 2012 Guidelines [1], early management of acute pulmonary edema/congestion includes an initial intravenous bolus of loop diuretics at time of presentation (usually furosemide 40 mg i.v. or 2.5 time the total outpatient oral loop diuretic dose) and eventually an i.v. vasodilator if SBP >110 mmHg (class of recommendation II, level B) or an i.v. inotropic agent if SBP <85 mmHg (class of recommendation II, level of evidence C) as adjunctive therapy. Of note, in the American Guidelines (AHA 2013) on heart failure, no specific cutoff values exclude the use of a vasodilator, but its use is advocated generally in the absence of symptomatic hypotension. Subsequently, patients should be reevaluated (within 1 h) for adequate response. Response to treatment includes reduction in dyspnea and adequate diuresis (>100 ml/h urine production in first 2 h), accompanied by an increase in oxygen saturation and usually reduction in respiratory rate and heart rate. In the absence of adequate response, all clinical-laboratory parameters should be reassessed (ECG, echocardiogram with hemodynamic measures, and principle laboratory tests) and several options considered. The most common cause of inadequate response is, however, poor response to the diuretic regimen utilized. Strategies to enhance diuretic efficacy will be discussed below in this chapter. AHF patients unresponsive to diuretic pharmacological therapy may be eventually considered for transient venovenous ultrafiltration (UF) that allows mechanical extracorporeal removal of plasma water. In patients with persistent hypotension (low CO) despite initial vasoactive therapy, other conditions like acute ischemic mechanical complications, severe valve dysfunction (particularly aortic stenosis), or alternative diagnoses (e.g., pulmonary embolism) requiring primary intervention rather than palliation of consequences should be reconsidered. Pulmonary artery catheterization may be sometimes useful in such unresponsive patients especially to ensure that hypotension is not due to inadequate LV filling pressure enabling more tailored vasoactive therapy (both inotropes and vasopressors). Finally, in unresponsive patients with persistent hypotension or cardiogenic shock with a rapid deterioration, a short-term mechanical circulatory support (including intra-aortic balloon pump and ECMO) may be considered as a “bridge to decision therapy.”
Approximately 80 % of patients are hospitalized with worsening of HF. For those with new-onset HF who stabilize after initial management, a chronic HF should be considered, and they should be treated according to recommendation of current guidelines. Initiation or implementation of evidence-based pharmacological therapies for chronic heart failure such as beta- blockers, ACE inhibitors, aldosterone-blocking agents, ARB, and electrical device should occur soon during this phase after stabilization. This topic will be extensively addressed in the chapter on chronic heart failure. For the acute setting, it is important to underline that the outpatient oral HF medications should be always carefully reviewed at admission. Generally, HF therapy should be continued at same doses during an AHFS episode unless the patient has hypotension or contraindications (such as hyperkalemia and severe renal failure for ACE inhibitors, angiotensin receptor blockers, aldosterone antagonists) that may require dose reduction or complete withholding. Several reports have shown that continuation of HF medical therapy with ACE inhibitors (or angiotensin receptor antagonists) and with beta- blockers for most patients is usually well tolerated and results in better outcomes [14, 15].
2.7 Ventilation
Oxygen supplementation should be titrated in order to keep the patient comfortable achieving arterial oxygen saturation above 90 %; caution is required in patients at risk of CO2 retention. In the presence of significant respiratory distress, noninvasive positive-pressure ventilation (CPAP or BiPAP) may be immediately considered to relieve dyspnea and to improve hypoxia, metabolic disturbance, and hemodynamic parameters (reduced LV wall stress and cardiac work) in the absence of contraindications (hypotension, vomiting, depressed consciousness, pneumothorax).
Previous studies and meta-analysis support the use of noninvasive ventilation (NIV) in cardiogenic pulmonary edema showing that besides respiratory and metabolic improvement, its early use can also prevent the need for endotracheal intubation. However, in the recent Cardiogenic Pulmonary Oedema trial (3CPO), no differences other than an improvement in dyspnea were seen in the rates of death or intubation between patients treated with NIV compared to standard oxygen therapy [16]. Currently, according to ESC guidelines, NIV should be considered in such dyspneic patients generally when SBP is not below 85 mmHg (class of recommendation IIa, level of evidence B).
2.8 Opiates
Opiates such as morphine sulfate may be beneficial in pulmonary edema because it is thought to induce mild venodilatation (thereby reducing preload) and reduce anxiety and distress associated with dyspnea.
Despite wide empiric use, small previous trial raised concern about the safety of morphine, because its use has been associated with increased need for invasive ventilation and greater in-hospital mortality [17, 18]. At present, according to the latest European guidelines, an i.v. opiate, along with antiemetic medication, may be considered in particularly restless and distressed patients to relieve anxiety and improve breathlessness. Patients should be carefully monitored because opiates can induce respiratory depression.
2.9 Diuretic Therapy
Diuretics are the mainstay of therapy in managing congestion in ADHS. Although safety and efficacy of diuretics have not been established in randomized controlled trials, long observational experience has shown their efficacy in relieving congestive symptoms. By reducing intravascular volume, diuretic therapy in ADHF lowers CVP and PCWP reducing pulmonary and peripheral edema often increasing forward-stroke volume and CO. In addition, when given intravenously, loop diuretics may act as vasodilators (principally venodilators) with additional benefit on renal and pulmonary congestion. Despite the demonstrated efficacy in managing congestion, the use of aggressive diuretic regimens may be associated with neurohormonal activation, worsening renal function, electrolyte abnormalities, and arrhythmias and so with adverse clinical outcomes [19, 20].
Current guidelines recommend administering the lowest dosage in order to achieve and maintain euvolemia avoiding volume depletion and dehydration.
Loop diuretics have the most rapid onset and most powerful effect. In acute setting, like AHFS, intravenous rather than oral administration is recommended because of greater drug bioavailability and more rapid onset of action. Diuretic dosing should be individualized and titrated according to patient status and initial response. Considering the bolus therapy, usually the diuretic effect begins within 30 min with a peak at 1–2 h [21]. Common suggested initial doses of intravenous loop diuretics are 40 mg for furosemide, 0.5–1 mg for bumetanide, and 5–10 mg for torsemide in patients who are not receiving loop diuretics [22]. In patients who have been already taking a loop diuretic, the dose should be almost equal or greater (i.e., 2.5 times) than the maintenance oral dose (in ESC guidelines, the greater dose is advocated) [1]. As discussed above, poor response to the diuretic is a common cause of inadequate response to initial therapeutic approach in AHFS. Now a widely accepted definition of diuretic resistance in HF is still lacking. In HF patients, the diuretic dose–response curve may be shifted downward and to the right because of a reduction in both renal drug delivery and natriuretic response. So higher doses are required to achieve a given diuretic response, and the maximal effect may be bunted [23]. Once a single effective dose has been determined, it should be administered multiple times per day (two or three times) according to the magnitude of diuresis needed. If there is little or no response, experts recommend dose doubling at 2 h intervals as needed (until effective diuresis is demonstrated) up to the maximum effective doses (ceiling doses over which no further diuresis will be achieved). Suggested maximum effective doses of loop diuretics in heart failure and renal insufficiency have been previously described [22]. Regarding the use of furosemide, in patients with HF and normal renal function, suggested maximal intravenous doses are 40–80 mg, but in the presence of renal insufficiency, larger staring doses may be required up to 160 or 200 mg in moderate and severe renal impairment, respectively. Doses of 250 mg or above should be given by infusion over 4 h.
From a pharmacokinetic and pharmacodynamic perspective, there are potential benefits of continuous infusion versus intermittent bolus. Continuous infusion results in more constant delivery of diuretics to the tubule with increased diuresis probably minimizing intermittent periods of the known postsodium retention effect. After a starting loading dose, suggested starting infusion rate of loop diuretics varies with the level of renal function (GFR or creatinine clearance). For furosemide, suggested doses are 10 mg/h with a GFR > 75 ml/min, 10–20 mg/h with a GFR 25–75 ml/min, and 20–40 mg if GFR < 25 ml/min. If an adequate response has not occurred within 1h, a loading dose should be repeated and then the infusion rate uptitrated [22].
However, existing data still does not allow definitive recommendations for clinical practice because even in a recent trial, no clear benefit or harms with intermittent bolus versus continuous infusion strategy have been demonstrated [24].
Several strategies can be tried to overcome such diuretic resistance especially during in-hospital phase. A common method for treating diuretic resistance is the sequential nephron blockade by adding a thiazidic or thiazide-like diuretic (DCT diuretics) [25, 26]. Metolazone and hydrochlorothiazide are the most common molecules used in combination with loop diuretics. Many clinicians prefer metolazone, a thiazide-like diuretic, because it has a longer half-life and a preserved efficacy in advanced renal failure (GFR below 20 ml/min) [27]. However, such combination therapy is associated with significant increase in adverse effect especially when high doses of DCT diuretics are used compared to either therapy alone [28]. it is advisable to start with lower dose of DCT diuretic (hydrochlorothiazide 12.5–25 mg or metolazone 2.5–5 mg) and maintain a daily regimen for a short period with careful monitoring of electrolyte balance and fluid depletion [29].
2.10 Additional Strategy to Enhance Diuresis
Dopamine infusion at low doses (≤3 μg/kg/min) may selectively activate dopamine receptors (DA1 and DA2) resulting in renal vasodilation and increasing renal blood flow. However, a significant benefit of standard use of dopamine has not been confirmed in recent randomized studies [30]. Although there is uncertainty, in the latest 2012 ESC guidelines, it is advocated to start infusion with low dose of dopamine (at 2.5 mcg/kg/min) in patients with poor response to diuretic regimen.
Salt and fluid restriction is another strategy that has also been commonly used during initial management of AHF patients, although as noted in recent European guidelines no firm evidence exists to support this practice. By reducing sodium load at the nephron, postdiuretic sodium retention may be reduced, especially when sodium intake is high. Generally, it is common to restrict sodium intake <2 g/day and fluid intake <1.5–2.0 L/day [1].
2.11 Ultrafiltration
AHF patients unresponsive to diuretic therapy may be considered for venovenous ultrafiltration (UF). UF allows mechanical extracorporeal removal of plasma water across a semipermeable membrane in response to a transmembrane pressure gradient (convective transfer). Venovenous UF is performed at bedside via a central or peripheral vascular access using a transportable UF console. With slow continuous UF usually performed in HF patients, the amount of ultrafiltrate created is small (2–4 ml/min) and does not require replacement with substitution fluid. Compared to hypotonic urine output achieved with loop diuretics, the ultrafiltrate (or volume removed) is isotonic to plasma and therefore removed more sodium (and less potassium). In addition, UF allows a better control of plasma water removal rate that can be tuned to match the putative refilling rate from the interstitium (approximately 15 ml/min) avoiding intravascular depletion and the vicious cycle of further neurohormonal activation [31].
2.12 Intravenous Vasodilators
By reducing both preload and afterload and therefore cardiac filling pressure, vasodilators may have beneficial hemodynamic effects in ADHS by reducing pulmonary congestion and usually increasing CO. The mechanisms for increased CO include left and right ventricular afterload reduction, improved diastolic ventricular properties, reduced mitral regurgitation, and eventually reduction in myocardial ischemia.
Currently approved intravenous vasodilators in clinical practice are organic nitrates such as nitroglycerin (NTG) and isosorbide dinitrate (ISDN), inorganic nitrates such as sodium nitroprusside (SNP), and nesiritide (currently not available in many European countries).
Despite that nitrates have been used to relieve symptoms and improve hemodynamic acute HF for many years, their use is still based on limited evidence primarily from small, single-center studies [32]. Currently, the use of intravenous vasodilators is recommended (class II and level of evidence B) to relieve symptoms and to reduce pulmonary congestion in patients in AHFS with intact blood pressure. In the latest 2012 ESC guidelines [1], their use is advocated only in patients with a BP greater than 110 mmHg, compared to ACCF/AHA guidelines where their use is limited only in the presence of symptomatic hypotension. Suggested intravenous doses of vasodilators in AHFS are indicated in Table 7.5.
Among organic nitrates, the most widely used is NTG. At low modest doses, intravenous NTG acts primarily through venodilatation, while at higher doses (>40 mcg/min) the effect of arteriolar dilatation begins to be apparent [33]. However, despite a graded dose–response curve, a variable interindividual response exists also related to baseline levels of systemic vascular resistance. A process of careful uptitration is always needed to avoid sudden BP reduction or hypotension. A significant drawback for intravenous nitrate particularly with NTG is the phenomenon of tachyphylaxis that may occur in 15–30 % of patients within 24 h probably related to strong counter-regulatory neurohormonal activation that leads to sodium and water retention [34].
Sodium nitroprusside (SNP) is the sodium salt of a complex molecule that breaks down in the blood interacting with oxyhemoglobin and directly releasing NO and cyanide into circulation. SNP is a potent vasodilator with the faster onset of action (within 60–90 s). Its short half-life (approximately 2 min) facilitates early establishment in the intensive care unit of an individual patient’s optimal level of vasodilation. Even low doses produce an equivalent venous and arteriolar dilatation resulting in balanced vasodilation of both sides of the circulation [35]. Because SNP can cause significant hypotension, it is usually used in intensive care settings even with invasive arterial monitoring. It is postulated that SNP may potentially increase the risk of a coronary steel phenomenon: as opposed to nitroglycerin’s preferential effect on larger conductance vessels, SNP dilates smaller resistance vessels creating a low-pressure system distal to occluded vessels that diverts critical pressure-dependent flow from ischemic areas [36]. The clinical significance of these observations is uncertain, and the true incidence of clinically significant coronary steal remains unknown.
2.13 Inotropes and Vasopressors
Despite the hemodynamic benefits in the short-term management, positive inotropic agents have not demonstrated improved outcomes in patients with HF in both hospital and outpatient settings. Rather, data from some registries and post hoc analyses of RCT suggest an increased morbidity and mortality with inotrope use in HF. In fact, as opposed to hemodynamic benefits, inotropes may cause sinus tachycardia and precipitate myocardial ischemia and arrhythmias [37–39].
In AHFS, the use of intravenous inotropic agents may be helpful to improve CO in patients with severe LV dysfunction with hypotension (PAS < 85 mmHg) and/or low-output syndrome. In such patients, the marginal systemic perfusion may limit institution and adequate response to the other pharmacological treatment like diuretics. On the other hand, at the cost of increasing afterload and decreasing cardiac output, drugs with arterial vasoconstriction action (e.g., norepinephrine or dopamine at high doses) may be a temporizing measure to redistribute CO from extremities to vital organs, and their use is restricted to patients with persistent hypoperfusion despite optimization of cardiac filling pressure and the concomitant use of inotropes. The use of inotropic/vasopressor agents will be addressed in the chapter on the management shock. In the 2102 ESC guidelines, the use of inotropes is in class IIa of recommendation, level of evidence C, and the use of vasopressor in class IIb, level of evidence C [1].
Calcium sensitizers such as levosimendan are a new category of inotropic agents that exert positive inotropic effects by increasing the affinity of troponin C for calcium. Levosimendan exertion also has a vasodilatory effect by blocking adenosine triphosphate-dependent potassium channels in the vascular smooth muscle cells. Such inotropic and vasodilator effects may result in increased CO and reduced filling pressures in AHF patients. Compared to the classic inotropic agents, calcium sensitizers have two major pharmacodynamic advantages: first, increase in contractile force occurs without increasing calcium loading that is associated with enhanced myocardial oxygen consumption, increased heart rate, and arrhythmias; second, the inotropic effect is not attenuated by concomitant treatment with beta-blockers. At present, the real risk/benefit ratio of levosimendan in AHFS is still debated in light of less favorable outcomes observed in a recent study compared to placebo [40]. Currently, levosimendan is approved in Europe as a second-line agent for severe low-output HF refractory standard therapy, and according to ESC guidelines its use may be considered (recommendation class IIb, level of evidence C) especially in patients with chronic beta-blocker therapy to overcome the beta-blockade effect.
2.14 Thromboembolism Prophylaxis
Several mechanisms like increased systemic venous pressure, low cardiac output, and procoagulant blood changes may increase the risk of venous thromboembolism in patients with HF. For this reason, in the absence of contraindication to anticoagulation, thromboembolism prophylaxis (e.g., LMWH) is currently recommended when HF patients are hospitalized (if not already anticoagulated) to reduce the risk of deep vein thrombosis and pulmonary embolism (recommendation class I, level of evidence A) [1].
References
McMurray JJ, Adamopoulos S, Anker SD, Auricchio A, Böhm M, Dickstein K, Falk V, Filippatos G, Fonseca C, Gomez-Sanchez MA, Jaarsma T, Køber L, Lip GY, Maggioni AP, Parkhomenko A, Pieske BM, Popescu BA, Rønnevik PK, Rutten FH, Schwitter J, Seferovic P, Stepinska J, Trindade PT, Voors AA, Zannad F, Zeiher A, ESC Committee for Practice Guidelines (2012) ESC guidelines for the diagnosis and treatment of acute and chronic heart failure 2012: The Task Force for the Diagnosis and Treatment of Acute and Chronic Heart Failure 2012 of the European Society of Cardiology. Developed in collaboration with the Heart Failure Association (HFA) of the ESC. Eur Heart J 33(14):1787–1847
Gheorghiade M, De Luca L, Fonarow GC, Filippatos G, Metra M, Francis GS (2005) Pathophysiologic targets in the early phase of acute heart failure syndromes. Am J Cardiol 96(6A):11G–17G
Yancy CW (2008) Vasodilator therapy for decompensated heart failure. J Am Coll Cardiol 52(3):208–210
Gheorghiade M, Vaduganathan M, Fonarow GC, Bonow RO (2013) Rehospitalization for heart failure: problems and perspectives. J Am Coll Cardiol 61(4):391–403
Cleland JG, Swedberg K, Follath F et al (2003) The EuroHeart Failure survey programme—a survey on the quality of care among patients with heart failure in Europe. Part 1: patient characteristics and diagnosis. Eur Heart J 24:442–463
Adams KF Jr, Fonarow GC, Emerman CL et al, for the ADHERE Scientific Advisory Committee and Investigators (2005) Characteristics and outcomes of patients hospitalized for heart failure in the United States: rationale, design, and preliminary observations from the first 100,000 cases in the Acute Decompensated Failure National Registry (ADHERE). Am Heart J 149:209–216
Fonarow GC, Abraham WT, Albert NM et al (2004) Organized program to initiate lifesaving treatment in hospitalized patients with heart failure (OPTIMIZE-HF): rationale and design. Am Heart J 148:43–51
Gheorghiade M, Pang PS (2009) Acute heart failure syndromes. J Am Coll Cardiol 53(7):557–573
Cotter G, Felker GM, Adams KF, Milo-Cotter O, O’Connor CM (2008) The pathophysiology of acute heart failure–is it all about fluid accumulation? Am Heart J 155(1):9–18
Metra M, Felker GM, Zacà V, Bugatti S, Lombardi C, Bettari L, Voors AA, Gheorghiade M, Dei CL (2010) Acute heart failure: multiple clinical profiles and mechanisms require tailored therapy. Int J Cardiol 144(2):175–179
Fallick C, Sobotka PA, Dunlap ME (2011) Sympathetically mediated changes in capacitance: redistribution of the venous reservoir as a cause of decompensation. Circ Heart Fail 4:669e75
Nohria A, Tsang SW, Fang JC, Lewis EF, Jarcho JA, Mudge GH, Stevenson LW (2003) Clinical assessment identifies hemodynamic profiles that predict outcomes in patients admitted with heart failure. J Am Coll Cardiol 41(10):1797–1804
Forrester JS, Diamond G, Chatterjee K, Swan HJ (1976) Medical therapy of acute myocardial infarction by application of hemodynamic subsets (first of two parts). N Engl J Med 295(24):1356–1362
Metra M, Torp-Pedersen C, Cleland JG et al (2007) Should beta-blocker therapy be reduced or withdrawn after an episode of decompensated heart failure? Results from COMET. Eur J Heart Fail 9:901
Fonarow GC, Abraham WT, Albert NM et al (2008) Influence of beta-blocker continuation or withdrawal on outcomes in patients hospitalized with heart failure: findings from the OPTIMIZE-HF program. J Am Coll Cardiol 52:190
Gray A, Goodacre S, Newby DE, Masson M, Sampson F, Nicholl J, 3CPO Trialists (2008) Noninvasive ventilation in acute cardiogenic pulmonary edema. N Engl J Med 359(2):142–151
Hoffman JR, Reynolds S (1987) Comparison of nitroglycerin, morphine and furosemide in treatment of presumed pre-hospital pulmonary edema. Chest 92:586–593
Peacock WF, Hollander JE, Diercks DB et al (2008) Morphine and outcomes in acute decompensated heart failure: an ADHERE analysis. Emerg Med J 25:205–209
Domanski M, Norman J, Pitt B, Haigney M, Hanlon S, Peyster E (2003) Diuretic use, progressive heart failure, and death in patients in the Studies Of Left Ventricular Dysfunction (SOLVD). J Am Coll Cardiol 42:705–708
Hasselblad V, Gattis Stough W, Shah MR et al (2007) Relation between dose of loop diuretics and outcomes in a heart failure population: results of the ESCAPE trial. Eur J Heart Fail 9:1064–1069
Sagar S, Sharma BK, Sharma PL, Wahi PL (1984) A comparative randomized double-blind clinical trial of bumetanide and furosemide in congestive cardiac failure and other edema states. Int J Clin Pharmacol Ther Toxicol 22:473–478
Brater DC (1998) Diuretic therapy. N Engl J Med 339:387–395
Ellison DH (2001) Diuretic therapy and resistance in congestive heart failure. Cardiology 96(3–4):132–143
Felker GM, Lee KL, Bull DA, Redfield MM, Stevenson LW, Goldsmith SR, LeWinter MM, Deswal A, Rouleau JL, Ofili EO, Anstrom KJ, Hernandez AF, McNulty SE, Velazquez EJ, Kfoury AG, Chen HH, Givertz MM, Semigran MJ, Bart BA, Mascette AM, Braunwald E, O’Connor CM, NHLBI Heart Failure Clinical Research Network (2011) Diuretic strategies in patients with acute decompensated heart failure. N Engl J Med 364(9):797–805
Ghose RR, Gupta SK (1981) Synergistic action of metolazone with loop diuretics. Br Med J 282:1432–1433
Fliser D, Schroter M, Neubeck M et al (1994) Coadministration of thiazides increases the efficacy of loop diuretics even in patients with renal failure. Kidney Int 46:482–488
Dargie HJ, Allison ME, Kennedy AC et al (1972) High dosage metolazone in chronic renal failure. Br Med J 4:196–198
Oster JR, Epstein M, Smoller S (1983) Combined therapy with thiazide-type and loop diuretic agents for resistant sodium retention. Ann Intern Med 99:405–406
Ernst ME, Moser M (2009) Use of diuretics in patients with hypertension. N Engl J Med 361(22):2153–2164
Chen HH, Anstrom KJ, Givertz MM, Stevenson LW, Semigran MJ, Goldsmith SR, Bart BA, Bull DA, Stehlik J, LeWinter MM, Konstam MA, Huggins GS, Rouleau JL, O’Meara E, Tang WH, Starling RC, Butler J, Deswal A, Felker GM, O’Connor CM, Bonita RE, Margulies KB, Cappola TP, Ofili EO, Mann DL, Dávila-Román VG, McNulty SE, Borlaug BA, Velazquez EJ, Lee KL, Shah MR, Hernandez AF, Braunwald E, Redfield MM, NHLBI Heart Failure Clinical Research Network (2013) Low-dose dopamine or low-dose nesiritide in acute heart failure with renal dysfunction: the ROSE acute heart failure randomized trial. JAMA 310(23):2533–2543
Felker GM, Mentz RJ (2012) Diuretics and ultrafiltration in acute decompensated heart failure. J Am Coll Cardiol 59(24):2145–2153
Piper S, McDonagh T (2014) The role of intravenous vasodilators in acute heart failure management. Eur J Heart Fail 16(8):827–834
Bayley S, Valentine H, Bennett ED (1984) The haemodynamic responses to incremental doses of intravenous nitroglycerin in left ventricular failure. Intensive Care Med 10:139–145
Gupta D, Georgiopoulou VV, Kalogeropoulos AP, Marti CN, Yancy CW, Gheorghiade M, Fonarow GC, Konstam MA, Butler J (2013) Nitrate therapy for heart failure: benefits and strategies to overcome tolerance. JACC Heart Fail 1(3):183–191
Elkayam U, Janmohamed M, Habib M, Hatamizadeh P (2008) Vasodilators in the management of acute heart failure. Crit Care Med 36(1 Suppl):S95–S105
Mann T, Cohn PF, Holman LB et al (1978) Effect of nitroprusside on regional myocardial blood flow in coronary artery disease: Results in 25 patients and comparison with NTG. Circulation 57:732–738
Elkayam U, Tasissa G, Binanay C et al (2007) Use and impact of inotropes and vasodilator therapy in hospitalized patients with severe heart failure. Am Heart J 153:98–104
Abraham WT, Adams KF, Fonarow GC, et al; ADHERE Scientific Advisory Committee and Investigators; ADHERE Study Group (2005) In- hospital mortality in patients with acute decompensated heart failure requiring intravenous vasoactive medications: an analysis from the acute decompensated heart failure national registry (adhere). J Am Coll Cardiol 46:57–64
O’Connor CM, Gattis WA, Uretsky BF et al (1999) Continuous intravenous dobutamine is associated with an increased risk of death in patients with advanced heart failure: insights from the Flolan International Randomized Survival Trial (first). Am Heart J 138:78–86
Packer M, Colucci W, Fisher L, Massie BM, Teerlink JR, Young J, Padley RJ, Thakkar R, Delgado-Herrera L, Salon J, Garratt C, Huang B, Sarapohja T, REVIVE Heart Failure Study Group (2013) Effect of levosimendan on the short-term clinical course of patients with acutely decompensated heart failure. JACC Heart Fail 1(2):103–111. doi:10.1016/j.jchf.2012.12.004. Epub 2013 Apr 1
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Romandini, A., Maffei, S. (2015). Acute Heart Failure and Pulmonary Edema. In: Capucci, A. (eds) Clinical Cases in Cardiology. Springer, Cham. https://doi.org/10.1007/978-3-319-19926-9_7
Download citation
DOI: https://doi.org/10.1007/978-3-319-19926-9_7
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-19925-2
Online ISBN: 978-3-319-19926-9
eBook Packages: MedicineMedicine (R0)