
Abstract
Aniridia is characterized by congenital hypoplasia of the iris and alterations of other structures of the eye, including cornea, crystalline lens, optic nerve, and retina. Patients suffer from early onset of nystagmus, photophobia, amblyopia, and severely decreased visual acuity. In 70 % of cases, aniridia is inherited in an autosomal dominant fashion, while it is sporadic in about 30 % of cases. In the great majority of patients, this disease is caused by heterozygous mutations in the PAX6 gene, which encodes for a transcription factor, very well conserved along phylogeny and critical for eye morphogenesis. Aniridia-causing mutations can be of various types, from single base substitution to large chromosomal deletions. All of them determine a loss of function of the gene. When chromosomal deletions are large and involve the WT1 gene, subjects suffer from the WAGR (Wilm’s tumor, Aniridia, Genitourinary abnormalities, mental Retardation) syndrome. Both prenatal or postnatal genetic test is available. It is indicated when isolated or WAGR is present, as well as other eye disorders potentially associated with PAX6 mutations. Genetic testing is useful for differentiating aniridia caused by mutations only in the PAX6 gene from those forms associated with the deletion of contiguous genes.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others
Keywords
Introduction
Aniridia (from Greek, meaning “without” [an-] and “iris” [-iridia]) is an extremely rare eye condition. Its prevalence in Norway and Sweden is estimated to be 1:76,000 population and 1:70,000 population, respectively [1]. The estimated point prevalence is 1 in 40,000 live births in Denmark [2] and 0.42 in 100,000 live births in Spain [3]. It affects males and females equally.
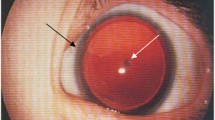
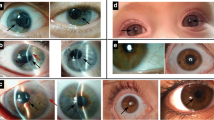
Aniridia (Online Mendelian Inheritance in Man [OMIM] 106210) is characterized by congenital hypoplasia of the iris which can vary considerably from milder forms to complete bilateral aplasia.
Besides the lack of iris tissue, aniridia also shows alterations of other structures of the eye: cornea, crystalline lens, optic nerve, and retina. The fovea, the central part of the retina which enables detailed vision, and the optic nerve are often not fully developed (hypoplasia/dysplasia). This is associated with the early onset of nystagmus, photophobia, amblyopia, and severely decreased visual acuity.
In adolescents and adults aniridia can manifest itself with keratopathies, including central epithelial defects, corneal opacities, peripheral vascular pannus, and limbal stem cell deficiency. A further decrease in vision occurs with the development of cataracts, lens displacement and glaucoma [4].
In 70 % of cases, aniridia is inherited in an autosomal dominant fashion, while it is sporadic in about 30 % of cases [5]. It is caused by mutations in the PAX6 gene (located on chromosome 11p), which plays an important role in cell differentiation and embryonic development, as it is involved in the morphogenesis of the eye, the olfactory bulb, the neural tube, the brain, and non-central nervous system organs such as the pancreas and the intestines [2]. In the majority of persons with aniridia, there is a loss of function of one copy of the gene PAX6 intragenic mutations are observed in two-thirds of cases, whereas chromosomal rearrangements are found in about one third (deletions, translocations, and inversions). The mutations can affect the structural gene or the regions of other genes that regulate development (e.g., SOX2), adhesion cells, and structural proteins of the cornea and lens.
Clinically, aniridia may manifest itself as an isolated eye abnormality without apparent systemic involvement or as part of a more complex constellation of conditions. Large alterations in chromosome 11p, comprising PAX6 and the adjacent WT1 gene, lead to a contiguous gene syndrome, the WAGR syndrome (Wilms tumor, Aniridia, Genitourinary abnormalities, and mental Retardation) [6].
The Gillespie syndrome (OMIM 206700), another extremely rare congenital condition, is characterized by aplasia of the pupil border, cerebellar ataxia, and delayed psychomotor development. Gillespie syndrome is genetically distinct from aniridia, although PAX6 mutations have been described in two persons with a phenotype similar to the Gillespie syndrome.
Aniridia is registered in Orphanet, the reference portal for information on rare diseases and orphan drugs, under the number ORPHA77.
The PAX6 Gene
Aniridia is caused by mutations of the PAX6 gene that encodes a highly conserved transcription regulator involved in the ocular development of animals from the fruit fly (Drosophila melanogaster) to humans [7–9]. The PAX6 gene was cloned in 1991 [10] and in 1992 a cDNA homologue was isolated from mouse embryo [11]. The human and mouse proteins show nearly complete sequence homology and both proteins are members of the PAX protein family, comprising nine members that share a paired domain. Each of the genes encoding PAX proteins has a tissue-specific expression; each PAX protein is involved in the development and function of one or more organs. The paired domain is about 120 amino acids long and is responsible for specific interactions with DNA sequences. The PAX6 protein interacts with the DNA sequences through the homeodomain which extends for about another 60 amino acids at the C terminal of the paired domain [12].
The PAX6 gene is highly conserved phylogenetically. Nearly all animals have at least one gene very similar to human PAX6. For example, the fruit fly has a gene that encodes the paired domain and the homeodomain which has extended sequence homology with the human PAX6 gene; it is called eyeless (ey) because some of its mutant allelic variants are associated with ocular structure anomalies [13].
In humans, the PAX6 gene is located on the short arm of chromosome 11 (11p13), about 22.4 kb long and comprising 14 exons [9]. The mature transcript of PAX6 is about 2.7 kb long [10]. PAX6 transcription is regulated by two promoters, P0 and P1, which are differently regulated by elements in cis and activated in tissue-specific fashion [14, 15].
The protein encoded by the PAX6 gene, in addition to the domains for interaction with DNA (paired and homeodomain), has a domain at the C terminal (PST), rich in proline, serine, and threonine.
Preceding the PST region is a linker region, 78 amino acids long, which contains a high percentage of glycine (16.7 %) and glutamine (12.8 %) residues [9].
The paired domain is subdivided into an N-terminal subdomain (residues 1–60) containing a beta short motif and three alpha-helices arranged in a helix-turn-helix motif, and a C-terminal subdomain (residues 77–133) containing three alpha-helices. There do not appear to be direct protein-protein interactions between the two subdomains.
The homeodomain is a protein domain with about 60 amino acids and is characterized by three alpha-helical-like structures (helix I, II and III) folded into a compact globular structure [16, 17]. The tissue-specific expression of the PAX6 gene is identical in the mouse and humans. It is expressed in various tissues during embryonic development and in the adult organism.
PAX6 plays a centrally important role in the complete development of the eye lens and the transcriptional activation of its structural genes, such as the zeta-crystallins [18, 19]. It also plays a determinant role in the differentiation of pluripotent progenitors of the retinal cells and in maintaining their tissue-specific expression [20, 21]. The presence of the isoform containing exon 5a ensures for correct eye growth [21].
The PAX6 gene is expressed during the earliest stage of embryonic development of the pancreas and continues to be expressed in adult endocrine cells. Mutant mice homozygous for PAX6 lack cells able to produce pancreatic glucagon, suggesting that the gene is essential for the differentiation of pancreatic alpha cells [22]. In addition, PAX6, by binding to common elements in the promoters of genes for insulin, glucagon and somatostatin, activates the gene promoters that encode these hormones [23]. Reports have described cases of patients presenting aniridia and diabetes associated with PAX6 mutations, suggesting that the two conditions share a common regulating mechanism [24].
In the nervous system, PAX6 controls the migration and differentiation of several specific progenitors of neural brain cells. The presence of PAX6 in association with Emx2 factor regulates the formation of cortical areas and confers area identity to diverse cells [25, 26]. Analysis of its genetic expression in mutant mice has shown that PAX6 regulates the expression of Neurog2 in the spinal cord and differentially controls distinct enhancers along the dorsoventral axis [27]. Radial glial cells, precursors of astrocytes, are ubiquitous in the central nervous system during its development.
Experimental studies have shown that cells isolated from the cortex of mice mutant for PAX6, have less neurogenic potential, suggesting the importance of PAX6 in the differentiation of the central nervous system [28].
PAX6 is also involved in the development of Rathke’s pouch and the anterior pituitary gland; its expression is essential for the differentiation of various types of cells (somatotropic, lactotropic, thyrotropic) along the dorsoventral axis of the adenohypophysis [29].
A study on the molecular basis for hypophyseal dysfunctions in the mouse and humans identified 12 transcription factors considered critical for hypophyseal development and function, including the PAX6 gene [30].
Genetic Basis of Aniridia
Aniridia is transmitted in autosomal dominant fashion. Each gene in every cell is present in two copies (alleles) one each from both parents. A disorder is referred to as dominant when it is expressed in a heterozygous person (i.e., a person with only one mutant allele). The affected person transmits the mutation on average to 50 % of his or her children, irrespective of the sex of the child. Most persons with aniridia (about 70 %) have a parent with the condition (familial aniridia), whereas the remaining 30 % do not (sporadic aniridia) [5]. Sporadic aniridia arises from a new mutation during gametogenesis. The rate of pathogenetic mutation of the PAX6 gene is about 10−5 to 10−6, meaning that each healthy individual has a probability between 1:100,000 and 1:1,000,000 of having a child with aniridia caused by a new mutation.
Aniridia may manifest itself clinically as an isolated ocular anomaly caused by point mutations in PAX6 or by deletions of the structural gene or the regions regulating its expression. In 15 % of cases, aniridia is the clinical expression of the WAGR syndrome (Wilms tumor, a rare kidney cancer; Aniridia; Genitourinary abnormalities; and mental Retardation) which is caused by a cytogenetically visible deletion in the 11p13 band or by a submicroscopic deletion involving the PAX6 gene and the adjacent WT1 gene [6].
An interactive database for the analysis of PAX6 mutations is available at http://lsdb.hgu.mrc.ac.uk/home.php?select_db=PAX6. The database currently contains information on 359 mutations, 92 % of which are associated with congenital eye defects and 8 % apparently neutral polymorphisms [31, 32]. The pathogenic mutations comprise: nonsense mutations (36 % of the total); frameshift deletions or insertions (24 %); missense mutations (17 %); splicing mutations (12 %); in frame deletions or insertions (6 %); and run on mutations (5 %). Nonsense mutations introduce a premature stop codon; in splicing mutations and frameshift deletions or insertions, the protein following the mutation is strongly altered and therefore nonfunctional.
These three categories of mutations constitute over 72 % of all pathogenic mutations identified to date [6, 31–34]. Of the pathogenic mutations present in the database, about 90 % are associated with aniridia and about 10 % with other phenotypes such as a foveal hypoplasia, microophthalmia, and optic nerve defects [31, 32]. Among the mutations responsible for aniridia, few missense 2 % mutations encode proteins with a likely loss of function [32, 35–38]. Of the 29 mutations known to be associated with eye defects (without aniridia), 69 % are missense mutations [32].
This means that aniridia is more often associated with mutations that lead to complete inactivation of the protein (nonsense mutations, frameshift, splicing, deletion of the entire gene or a significant part of it), whereas other ocular phenotypes are associated with missense mutations. This is probably because missense mutations lead to changes in a single amino acid. This class of mutations does not completely inactivate protein function but rather modifies it, resulting in a phenotype different from aniridia.
Missense mutations are generally located in the paired domain (exons 5, 5a, 6, and 7) and are associated with phenotypes that affect the tissues involved in aniridia, such as the fovea, the optic nerve, and the iris [39, 40].
The mutations that introduce a premature stop codon have presumably a negative dominant effect in that the PAX6 protein trunk containing only the domains for DNA binding could acquire a major capacity for binding the target sequences without activating the genes downstream and thus interfere with normal protein function [41, 42].
It could be expected that the mutations that truncate the normal protein sequence of PAX6 are associated with a less severe form of the condition (or do not lead to its development) if the mutation alters only the C-terminal of the proteins while sparing the functional domains. Actually, however, genotype-phenotype correlations of mutations in the database suggest that the position of the truncating mutation does not have a precise role, hence the phenotype consequences in vivo. The truncating mutations associated with aniridia are not correlated with their location [32]. It is possible that nonsense-mediated decay is the pathogenically responsible molecular mechanism. Nonsense- mediated decay is the mechanism through which mRNAs containing a premature stop codon are decayed before they can produce large amounts of protein trunks [43]. The available data suggest the hypothesis that aniridia is due to haploinsufficiency because of the loss of allele function.
This does not appear to be due to premature termination of the protein but rather to the nonsense-mediated delay mechanism [31, 32].
The majority of patients (80–90 %) with aniridia are heterozygous for PAX6 mutations [44] (see also the database mentioned above). In humans, homozygous mutations (i.e., when both alleles carry the mutation) are lethal and cause a phenotype similar to that seen in the mouse, characterized by anophthalmia and central nervous system defects [45]. Also other organisms with homozygous PAX6 mutations present anomalous phenotypes, for example, small eye mice, eyeless Drosophila, and Caenorhabditis elegans [13, 46–48]. Homozygous small eye mice die shortly after birth, have no eyes or nasal cavities and present brain defects [7].
Genetic Analysis
Point mutations of the PAX6 gene are identified by DNA sequencing. The deletions (small and large) are identified with molecular (multiple ligation-dependent probe amplification [MLPA]) or cytogenetic techniques (fluorescent in situ hybridization [FISH]). In these cases the possible deletion of the WT1 gene, associated with the risk of Wilms tumor in the WAGR syndrome, is evaluated.
The sensitivity of genetic testing (i.e., a test’s ability to identify a mutation) is less than 100 %. In the WAGR syndrome, cytogenetic screening has a sensitivity of about 70 %. In isolated aniridia, the complete panel of molecular tests has a sensitivity of about 65 %.
When a pathogenic mutation is detected in a person with aniridia, screening can be extended to other family members. To pregnant women may be offered prenatal genetic testing (chorionic villous sampling CVS or amniocentesis).
Theoretically, preconceptional genetic testing is another possibility, analyzing the first polar globule of an affected mother.
In cases of de novo mutation, the neonate should be tested for the possible involvement of the WT1 gene, due to the higher risk of developing Wilms tumor.
Genetic analysis of PAX6 is indicated when isolated or syndromic aniridia (WAGR) is present, as well as other disorders potentially associated with PAX6 mutations (Peters anomaly, papillary ectopia, foveal hypoplasia, coloboma, optic nerve hypoplasia).
Medically, genetic testing is useful for differentiating aniridia caused by mutations only in the PAX6 gene from those forms associated with the deletion of contiguous genes.
References
Edén U, Iggman D et al (2008) Epidemiology of aniridia in Sweden and Norway. Acta Ophthalmol 86:727–729
Grindley JC, Davidson DR, Hill RE (1995) The role of Pax-6 in eye and nasal development. Development 121:1433–1442
Bermejo E, Martínez-Frías ML (2002) Defectos congénitos oculares:algunos aspectos clínicos y epidemiológicos. Bol ECEMC: Rev Dismor Epidemiol 1:43–48
Tornqvist K (2008) Aniridia: sight-threatening and hard to cure. Acta Ophthalmol 86:704–705
Valenzuela A, Cline RA (2004) Ocular and nonocular findings in patients with aniridia. Can J Ophthalmol 39:632–638
Crolla JA, van Heyningen V (2002) Frequent chromosome aberrations revealed by molecular cytogenetic studies in patients with aniridia. Am J Hum Genet 71:1138–1149
Hill RE, Favor J et al (1991) Mouse Small eye results from mutation in a paired-like omeobox containing gene. Nature 354:522–525
Jordan T, Hanson I et al (1992) The human PAX6 gene is mutated in two patients with aniridia. Nat Genet 90:41–54
Glaser T, Walton DS, Maas RL (1992) Genomic structure, evolutionary conservation and aniridia mutations in the human PAX6 gene. Nat Genet 2:232–238
Ton CC, Hirvonen H et al (1991) Positional cloning and characterization of a paired box -and homeobox – containing gene from the aniridia region. Cell 67(6):1059–1074
Ton CC, Miwa H, Saunders GF (1992) Small eye (Sey): cloning and characterization of the murine homolog of the human aniridia gene. Genomics 13(2):251–256
Stuart ET, Kioussi C, Gruss P (1993) Mammalian Pax genes. Annu Rev Genet 27:219–236
Quiring R, Walldorf U et al (1994) Homology of the eyeless gene of Drosophila to the Small eye gene in mice and aniridia in humans. Science 265:785–789
Plaza S, Dozier C et al (1995) Quail Pax-6 (Pax- QNR) mRNAs are expressed from two promoters used differentially during retina development and neuronal differentiation. Mol Cell Biol 15:3344–3353
Plaza S, Saule S, Dozier C (1999) High conservation of cis-regulatory elements between quail and human for the Pax-6 gene. Dev Genes Evol 209:165–173
Tsao DH, Gruschus JM et al (1995) The three dimensional solution structure of the NK-2 homeodomain from the Drosophila. J Mol Biol 251:297–307
Esposito G, Fogolari F et al (1996) Analysis of the solution structure of the homeodomain of rat thyroid transcription factor 1 by 1H-NMR spectroscopy and restrained molecular mechanics. Eur J Biochem 241:101–113
Ashery-Padan R, Marquardt T et al (2000) Pax6 activity in the lens primordium is required for lens formation and for correct placement of a single retina in the eye. Genes Dev 14:2701–2711
Richardson J, Cvekl A, Wistow G (1995) Pax-6 is essential for lens-specific expression of z-crystallin. Proc Natl Acad Sci U S A 92:4676–4680
Marquardt T, Ashery-Padan R et al (2001) Pax6 is required for the multipotent state of retinal progenitor cells. Cell 105:43–55
Mann RS (2004) Two Pax are better than one. Nat Genet 36:10–11
St-Onge L, Sosa-Pineda B et al (1997) Pax6 is required for differentiation of glucagon-producing alpha-cells in mouse pancreas. Nature 387:406–409
Sander M, Neubuser A et al (1997) Genetic analysis reveals that PAX6 is required for normal transcription of pancreatic hormone genes and islet development. Genes Dev 11:1662–1673
Yasuda T, Kajimoto Y et al (2002) PAX6 mutation as a genetic factor common to aniridia and glucose intolerance. Diabetes 51:224–230
Glaser T, Jepeal L et al (1994) PAX6 gene dosage effect in a family with congenital cataracts, aniridia, anophthalmia and central nervous system defects. Nat Genet 7:463–471
Bishop KM, Goudreau G, O’ Leary DD (2000) Regulation of area identity in the mammalian neocortex by Emx2 and Pax6. Science 288:344–349
Scardigli R, Schuurmans C et al (2001) Crossregulation between neurogenin2 and pathways specifying neuronal identity in the spinal cord. Neuron 31:203–217
Heins N, Malatesta P et al (2002) Glial cells generate neurons: the role of the transcription factor Pax6. Nat Neurosci 5:308–315
Kioussi C, O’Connell S et al (1999) Pax6 is essential for establishing ventral-dorsal cell boundaries in pituitary gland development. Proc Natl Acad Sci 96:14378–14382
Cushman LJ, Camper SA (2001) Molecular basis of pituitary dysfunction in mouse and human. Mamm Genome 12:485–494
Prosser J, van Heyningen V (1998) PAX6 mutation reviewed. Hum Mutat 11:93–108
Tzoulaki I, White IM, Hanson IM (2005) PAX6 mutations: genotype-phenotype correlations. BMC Genet 6:27–39
Fisher E, Scambler P (1994) Human haploinsufficiency – one for sorrow, two for joy. Nat Genet 7:5–7
Fantes J, Redeker B et al (1995) Aniridia-associated cytogenetic rearrangements suggest that a position effect may cause the mutant phenotype. Hum Mol Genet 4:415–422
Tang HK, Chao LY, Saunders GF (1997) Functional analysis of paired box missense mutations in the PAX6 gene. Hum Mol Genet 6:381–386
Azuma N, Hotta Y et al (1998) Missense mutations in the PAX6 gene in aniridia. Invest Ophthalmol Vis Sci 39:2524–2528
Vincent MC, Pujo AL et al (2003) Screening for PAX6 gene mutations is consistent with haploinsufficiency as the main mechanism leading to various ocular defects. Eur J Hum Genet 11:163–169
Chauhan BK, Yang Y et al (2004) Functional interactions between alternatively spliced forms of Pax6 in crystallin gene regulation and in haploinsufficiency. Nucleic Acid Res 32(5):1696–1709
Azuma N, Yamaguchi Y et al (2003) Mutations of the PAX6 gene detected in patients with a variety of optic-nerve malformations. Am J Hum Genet 72:1565–1570
Hanson I, Churchill A et al (1999) Missense mutations in the most ancient residues of the PAX6 paired domain underlie a spectrum of human congenital eye malformations. Hum Mol Genet 8:165–172
Singh S, Tang HK et al (1998) Truncation mutations in the transactivating region of PAX6 result in dominant-negative mutants. J Biol Chem 273(34):21531–21541
Duncan MK, Cvekl A et al (2000) Truncated forms of Pax6 disrupt lens morphology in transgenic mice. Invest Ophthalmol Vis Sci 41:464–473
Byers PH (2002) Killing the messenger: new insights into nonsense-mediated mRNA decay. J Clin Invest 109:3–6
van Heyningen V, Williamson KA (2002) PAX6 in sensory development. Hum Mol Genet 11(10):1161–1167
Hodgson SV, Saunders KE (1980) A probable case of the homozygous condition of the aniridia gene. J Med Genet 17:478–480
Matsuo T, Osumi-Yamashita N et al (1993) A mutation in the Pax-6 gene in rat small eye is associated with impaired migration of midbrain crest cells. Nat Genet 3:299–304
Chisholm AD, Horvitz HR (1995) Patterning of the Caenorhabditis elegans head region by the Pax-6 family member vab-3. Nature 377:52–55
Zhang Y, Emmons SW (1995) Specification of senseorgan identity by Caenorhabditis elegans Pax-6 homologue. Nature 377:55–59
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Damante, G., D’Elia, A.V. (2015). Introduction – What Is Aniridia: Epidemiology, Clinical Features and Genetic Implications. In: Parekh, M., Poli, B., Ferrari, S., Teofili, C., Ponzin, D. (eds) Aniridia. Springer, Cham. https://doi.org/10.1007/978-3-319-19779-1_1
Download citation
DOI: https://doi.org/10.1007/978-3-319-19779-1_1
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-19778-4
Online ISBN: 978-3-319-19779-1
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)