
Abstract
Plasma cell dyscrasias can induce a variety of kidney diseases either with organized deposits - AL amyloidosis, type-1 cryoglobulin, and immunotactoid glomerulonephritis, or with non-organized deposits of light or heavy chains characteristic of LCDD and HCDD, respectively. In addition to these well-defined diseases, a recently described entity has emerged, characterized by non-Randall-type, non-organized glomerular deposition of complete IgG. The term proliferative glomerulonephritis with monoclonal IgG deposits (PGNMID) was proposed, as most of the cases reported in the initial series shared pathologic features with membranoproliferative glomerulonephritis (MPGN). Nevertheless, cellular proliferation is sometimes minimal and the glomerulopathy can then be categorized as atypical membranous nephropathy (MN). Diagnosis relies on immunofluorescence examination demonstrating light chain isotype restriction, mostly kappa (70–80 %), associated with gamma heavy chain subclass restriction, and on electron microscopy showing lack of organization of the granular electron-dense deposits. Interestingly, the IgG subclass seems to influence the glomerular pattern as most MPGN are associated with IgG3, while the majority of MN is associated with IgG1. Less than a third of all patients have evidence of dysproteinemia by serum and urine examination, while hypocomplementemia is observed in 30 % of them, despite negative testing for cryoglobulin. Renal prognosis is poor, leading to dialysis in 20 % of cases, although it can be improved by treatment of the underlying hemopathy which should be carefully searched for. Several reports suggest that rituximab, a B-cell depleting agent, could be a promising treatment in patients without overt malignancy, including those with post-kidney transplant recurrence of the disease. Several cases of GN with isolated renal C3 deposits and circulating monoclonal gammopathy have been recently reported. These cases might represent an unusual complication of plasma cell dyscrasia, related to complement activation through an autoantibody activity of the monoclonal Ig against a complement alternative pathway regulator protein such as complement factor H.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Membranoproliferative glomerulonephritis
- Monoclonal component
- Membranous nephropathy
- Anti-CD20 antibody
- Ig subclass
In the past 10 years, the spectrum of glomerular diseases associated with multiple myeloma and myeloma-related disorders has expanded with the use of appropriate reagents including highly specific anti-light chain (LC) and anti-heavy chain (HC) subclass antibodies, and electron microscopy. For a long time, since the identification of the variable region of a circulating Ig LC in amyloid fibrils by Glenner and associates [1], AL amyloid was considered the only cause of glomerular involvement in myeloma and related disorders. Then, the description of non-amyloid light chain deposition disease (LCDD) by Randall and associates [2] opened up new perspectives in plasma cell-related glomerular pathology, although the nodular glomerulosclerosis characteristic of the disease was constantly associated with LC deposits along renal tubules. Together with myeloma cast nephropathy, AL amyloidosis and LCDD are the most common complications of plasma cell-related disorders [3], thus indicating that the majority of these disorders are caused by Ig LC deposition in renal parenchyma.
Deposition of monoclonal Ig containing both heavy and light chains is far less common and may manifest as type-I cryoglobulinemic GN [4], Randall-type light and heavy chain deposition disease [5], immunotactoid GN [6–8], and light and heavy chain amyloidosis [9, 10] (Table 11.1).
In type 1 cryoglobulinemia, a membranoproliferative glomerulonephritis (MPGN) with macrophage infiltration is the most characteristic histologic pattern and the deposits are typically, but not invariably, organized into fibrillary or microtubular structures at the ultrastructural level. The hallmark of immunotactoid glomerulonephritis is the presence of highly organized non-amyloidotic microtubular deposits, usually of >30 nm in diameter, with hollow cores and parallel stacking, although thinner tubules can be observed [6]. Light and heavy chain amyloidosis is extremely rare and, similar to AL amyloidosis, is characterized by the presence of Congo red-positive deposits composed of haphazardly oriented fibrils that measure 8–14 nm in diameter. Among diseases with non-organized deposits, LHCDD is characterized by the presence of nodular sclerosing glomerulopathy by light microscopy, linear staining of glomerular and tubular basement membranes for a single heavy and light chain by immunofluorescence, and non-fibrillar, granular electron-dense deposits involving glomerular and tubular basement membranes by electron microscopy. Recently, a second entity has emerged, which is characterized by non-Randall-type and non-organized glomerular Ig deposition that does not conform to any of the previous categories [11–14]. In most cases reviewed by Nasr in 2004 [15] and 2009 [16], lesions were those of MPGN. The authors coined the term proliferative glomerulonephritis with monoclonal IgG deposits (PGNMID) to call this new entity. In other rarer cases, lesions were those of atypical membranous nephropathy (MN) [13, 15, 17–19]. Although the clinicopathological presentation of these patients is shared with common cases of MPGN and MN, specificity is provided by the monoclonal Ig deposits which should lead to adapted diagnostic procedure and therapeutic strategy.
In this chapter, we revisit the spectrum of non-organized monoclonal Ig deposits. We will discuss important diagnostic issues including demonstration of monoclonality of the deposits and search for underlying lymphocyte and/or plasma cell proliferation, as well as the treatment options in the light of recent pathophysiologic and therapeutic advances.
Proliferative GN with Non-Organized Monoclonal Ig Deposits
Alpers et al. [11] first described six patients with an MPGN pattern of GN, including mesangial hypercellularity, increased mesangial matrix and mesangial interposition, and monoclonal IgGκ and C3 staining. Granular subendothelial and mesangial deposits were seen by electron microscopy. None of these six patients had detectable serum or urine monoclonal Ig, and bone marrow examination in four patients was normal. The authors pointed out the female predominance (five of the six patients), the young age of onset (31 years old or less in three patients), and the absence of overt plasma cell dyscrasia.
Bridoux et al. [13] reported the cases of five patients manifesting glomerulopathy with non-organized, non-Randall-type monoclonal Ig deposits; two of these patients being described in detail by Touchard [12]. The mean age was 54 ± 17 years. All patients presented with microhematuria and renal failure; four of five had a nephrotic syndrome. Kidney biopsy revealed atypical membranous, endocapillary proliferative, and membranoproliferative patterns. By immunofluorescence, the glomerular capillary wall deposits consisted of IgG3κ in two patients, IgG3λ in one, IgG2κ in one, and isolated λLC in one. Corresponding monoclonal proteins were detected in serum or urine in three patients.
In 2004, Nasr et al. [15] reported the first extensive description of PGNMID in a series of 10 patients, and recently enlarged this series to 37 cases [16], thus allowing a thorough description of the disease
Epidemiology
Nasr et al. [15] reported a biopsy incidence of 0.21 % of a total of 4650 native biopsies referred to the Renal Pathology Laboratory of Columbia University College of Physicians and Surgeons from January 2000 to February 2003. By comparison, the biopsy incidences of AL amyloidosis and Randall-type MIDD were 1.66 and 0.52 % over the same time period, respectively. In Japan, Masai et al. [20] identified four patients with PGNMID after reviewing 5443 kidney biopsies (biopsy incidence of 0.07 %).
Clinical Features
In Nasr et al.’s largest series, the majority of patients were white (81 %) and female (62 %). All patients were adults and had a mean age of 55 years (range 20–81). At presentation, all patients had proteinuria. Proteinuria was in the nephrotic range in 69 % of patients, and 49 % developed full nephrotic syndrome. Microhematuria was documented in 77 % of patients. Two-thirds of patients had renal insufficiency, including three who were on hemodialysis. None of the patients had significant extra-renal symptoms. A case where crescentic glomerulonephritis was superimposed to PGNMID was recently reported with autoimmune hemolytic anemia, thus widening the spectrum of PGNMID [21]. Two cases with a rapidly favorable outcome were associated with Parvovirus B19 infection, suggesting that virus infection-associated immune disorders could be implicated in the pathogenesis of PGNMID [22].
Pathologic Findings
Four histologic patterns were observed. The most common seen in 57 % of cases was MPGN, often associated with endocapillary hypercellularity including focal macrophage infiltration. The second most common pattern, seen in 35 %, was predominantly endocapillary proliferative GN. The third histologic pattern, seen in 5 % of cases, was predominantly membranous GN but with focal endocapillary hypercellularity and segmental membranoproliferative features. The fourth and rarest pattern was pure mesangial proliferative GN. Crescents were present in 32 % of cases. Interstitial inflammation was predominantly focal and associated with a variable degree of tubular atrophy and arteriosclerosis.
Results of immunofluorescence staining with anti-LC isotype and anti-Ig subclass antibodies obtained in three different series [13, 16, 20] are shown in Table 11.2. Deposits were identified exclusively in the glomeruli. They were mostly granular and localized to the glomerular capillary wall and mesangium. IgG was the only Ig deposited, with the exception of a case where only λLC was detected [13] and another case with exclusive IgM κ deposits [23]. All cases showed LC isotype restriction, including 30 cases (76.9 %) with sole positivity for κ. Ig subclass analysis showed a huge predominance of IgG3 (69.2 % of cases), whereas IgG3 represents a minor subclass in healthy subjects (8 %) and myeloma patients (4 %) [24]. No case showed positivity for IgG4. On statistical analysis, IgG3 subtype correlated with the absence of M-spike, with only 2 of 21 patients with IgG3 deposits having a positive M-spike in Nasr et al.’s series [16]. Immunofluorescence studies using antibodies specific for γ-heavy chain, CH1, Ch2, and CH3 domains, and γ3 hinge did not show apparent deletion [16, 20].
In all cases, granular electron-dense deposits were confined to the glomerular compartment, while they were both glomerular and tubular in LHCDD. They were primarily subendothelial and mesangial, but subepithelial deposits were also seen. In Nasr et al.’s series [16], some patients showed rare ill-defined fibrils with focal lattice-like arrays although the deposits never formed well-organized structures as seen in fibrillary or immunotactoid GN.
Immunologic Data.
Only 14 of 52 (27 %) patients had evidence of dysproteinemia by serum and/ or urine electrophoresis and immunofixation [11, 13, 16, 20]. Of the 26 of 37 patients reported by Nasr et al. [16] who had no detectable monoclonal component in serum or urine, four were tested with the serum free LC assay; of these, three were found to have normal κ:λ ratio, and one (who had glomerular monoclonal IgG3κ deposits) had an elevated κ:λ ratio.
Bone marrow examination, performed in 30 patients [11, 16, 20], showed marrow plasmacytosis in two patients and clear signs of myeloma in one patient. None of the patients had lymphadenopathy, hepatomegaly, or lymphoma.
Search for cryoglobulinemia was negative in all patients (performed repeatedly in many patients), and none of the patients had any systemic manifestations of cryoglobulinemia. Serum complement was decreased in 11 of 41 (27 %) patients [16, 20]. Of the 11 patients with hypocomplementemia, 8 had IgG3 glomerular deposits and 3 had IgG1 glomerular deposits.
Treatment Outcome
In the largest series reported so far [16], 18 of 37 patients received immunosuppressive agents either with or without concurrent renin–angiotensin system (RAS) blockade. It is remarkable that 12 patients (37.5 %) developed complete (n = 4) or partial (n = 8) remission, whereas only two reached ESRD (Table 11.3).
Transplantation
Because 20–25 % of PGNMID progress to ESRD [16], potential recurrence of the disease in the allograft is an important issue. Nasr et al. [25] reported recurrence of PGNMID in four Caucasians (three women and one man), although no patient had a detectable circulating monoclonal component or hematologic malignancy. Recurrence was first documented by biopsy performed at a mean of 3.8 months posttransplant because of renal insufficiency (four patients), proteinuria (three patients), and microhematuria (three patients). Histologic patterns in the allograft were endocapillary or mesangial GN.
Monoclonal IgG deposits (three IgG3κ and one IgG3λ) in the transplants had identical heavy and light chain isotypes as in the native kidneys. Recurrence was treated with combined high-dose prednisone plus rituximab (n = 3) or plus cyclosporine (n = 1). After a mean posttransplant follow-up of 43 months, all four patients achieved reduction in proteinuria and three had reduction in creatinine. Repeat biopsies showed reduced histologic activity after treatment.
Posttransplant PGNMID has also been reported by other authors [26–28], either as a recurrence of a pre-transplant PGNMID or as a de novo glomerulopathy, in patients having reached ESRD for other reasons, such as polycystic disease or type-1 diabetes mellitus. These observations confirm the severity of the disease and the poor renal outcome despite non-rituximab immunosuppressive regimens.
Nonproliferative GN with Non-Organized Monoclonal Ig Deposits
Next to PGNMID, isolated case reports and small series suggested that some patients developed GNMID with no or minimal glomerular cell proliferation. One of the patients reported by Bridoux et al. [13] and described in detail by Touchard [12] had nephrotic syndrome related to thickened glomerular capillary walls with IgG3λ and complement deposits. Immunoblotting revealed the presence of monoclonal IgG3λ. Evans et al. [18] described a patient with follicular B-cell lymphoma who developed nephrotic syndrome related to subepithelial granular IgG1κ deposits. One patient in Nasr et al.’s series [15] of PGNMID had a pattern of MN, however, with segmental membranoproliferative features and IgG1κ deposits.
Komatsuda et al. [19] reviewed 5,443 kidney biopsies from their own department in Akita (Japan) and identified three patients with monoclonal immunoglobulin deposition disease associated with membranous features. All patients had proteinuria, and one patient developed nephrotic syndrome. Renal insufficiency was not observed. Cryoglobulin or monoclonal protein in serum and urine was not detected. A renal biopsy showed thickening of the glomerular capillary walls and spike formation. Tubulointerstitial and vascular alterations were mild or absent. Immunofluorescence studies revealed granular IgG3κ deposits in two patients and IgG1κ deposits in one patient, along the glomerular capillary walls. Significant deposition along the tubular basement membranes was not observed in any patient. Immunofluorescence studies using antibodies specific for γ-heavy chain Fab containing CH1 domain, CH2 domain, and CH3 domain did not show any apparent deletion. On confocal microscopy, glomerular colocalization of light and heavy chains was observed. Electron microscopy showed predominant subepithelial granular deposits without distinct ultrastructural organization. All patients were treated with steroids, and good effects were observed. A follow-up renal biopsy performed in one patient showed histological improvement. No patient developed myeloma or other hematological malignancy during the course of follow-up (mean 44 months).
Revisiting the Disease Spectrum of GN with Monoclonal Ig Deposits
To get further insight into the glomerulopathies with monoclonal Ig deposits, we recently reviewed the cases of 26 patients with non-cryoglobulinemic GN and monoclonal Ig deposits referred to three nephrology departments in Paris between 1980 and 2008 [17]. We found that there were more patients with MN (n = 14) than with MPGN (n = 12) (Fig. 11.1). In five of the MN patients, the glomerular lesions were, however, atypical with mesangial hypertrophy and increased mesangial cellularity (Fig. 11.1a). Overall, extracapillary proliferation with crescents was observed in 13 cases (4 of 14 MN, 9 of 12 MPGN), whereas glomerular necrotic lesions were present in only six biopsies. Interstitial inflammation with infiltration by neutrophils and nonmalignant lymphocytes was noted in 17 patients (65 %). Interstitial fibrosis with tubular atrophy ranged from absent or mild (57 %) to moderate (27 %) and severe (16 %). Vascular lesions were frequent, mainly arteriolar hyalinosis (15/26) and arteriosclerosis (19/26).

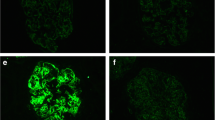
Pathological findings in glomerulonephritis with monoclonal Ig deposits. Light microscopy findings in MN membranous nephropathy, showing immune deposits on the external side of the glomerular basement membrane, with frequent mesangial hypertrophy (a, Masson’s trichrome stain); the deposits have irregular size (inset a) and are embedded in basement membrane expansions (b, JMS stain). In patients with membranoproliferative pattern, light microscopy shows proliferation of mesangial cells (c, Masson’s trichrome stain) and double contours (d, JMS stain). By immunofluorescence, parietal granular IgG deposits in MN (e) are different from the more diffuse pattern seen in the MPGN (f). Ultrastuctural studies found that most patients have granular, non-organized deposits in the subepithelial (g) or subendothelial spaces, whereas in some cases, the deposits show microtubular substructure (h), as previously described in immunotactoid glomerulonephritis. From Guiard et al. [17], with permission
Demographic, clinical, and biological characteristics of these patients are shown in Table 11.4. At presentation, all patients had glomerular proteinuria >1 g/24 h and most (85 %) of the patients presented with nephrotic syndrome. Mean serum creatinine level at presentation was 211 μmol/l (eGFR: 49.3 ± 34.6 ml/min/1.73 m2), and 14 of 26 (54 %) patients initially had significant renal dysfunction, including three patients who needed temporary hemodialysis. In eight cases (31 %), a circulating monoclonal IgG was detected by standard methods (serum and urine protein electrophoresis with immunofixation). In all of these cases, the serum monoclonal IgG had the same light and heavy chain isotype as the monoclonal compound identified in the glomerular deposits, on the renal biopsy. Hypocomplementemia was observed in 8 of 22 patients (36 %) with available data, showing either isolated C4 or combined C3 and C4 consumption. Low serum complement concentration was equally observed among patients with either MPGN or MN and independently of the monoclonal IgG isotype. Serum cryoglobulin, hepatitis C, hepatitis B, and HIV serology were negative in all patients.
Bone marrow examination and blood lymphocyte phenotype were performed in 22 of 26 patients and a hematological malignancy was identified in nine of them: two had multiple myeloma (MM), four had chronic lymphocytic leukemia (CLL), and three non-Hodgkin’s lymphoma (NHL). Five of the patients with malignancy had detectable serum monoclonal IgG. The hematological disease was revealed by the nephropathy in four of nine patients, whereas four patients had a long-standing history of hemopathy when GN was detected (mean delay was 32 months [3–89]). One patient, who was initially diagnosed with monoclonal gammopathy of undetermined significance, converted to overt MM 81 months after the onset of renal disease. A positron emission tomography (PET) scan was performed in three patients with no proven hematological malignancy and found no tumoral mass.
Although a circulating monoclonal IgG was detected in less than a third of the patients even by sensitive techniques, all patients did have monoclonal Ig glomerular deposits. Light chain isotype restriction was found in all patients with positivity for κ light chain in 80 % of patients. The subclasses of IgG deposits were determined for 21 patients: deposits stained for γ1 in eight patients (6 IgG1κ and 2 IgG1λ), γ2in two patients (IgG2κ), γ3 in ten patients (9 IgG3κ and 1 IgG3λ), and γ4 in one patient (IgG4κ). IgG subclass distribution was different according to the observed glomerular pattern: IgG3 deposits were identified in 80 % of cases in MPGN (seven of eight, IgG3k; one of eight, IgG3λ), whereas only 18 % of MN had IgG3 deposits (p = 0.0021). On the other hand, IgG1 deposits were present in 64 % of MN (four of seven, IgG1k; three of seven, IgGλ, whereas only 10 % of MPGN had IgG1 deposits (p = 0.014). In most of the examined patients (11 of 14), ultrastructural study showed that immune deposits were not organized. EM demonstrated large, granular deposits that were subepithelial in eight patients (with associated mesangial deposits in two of them) and subendothelial in three patients (Fig. 11.1g, h). Three patients had immunotactoid GN, with organized subepithelial deposits with microtubular substructure (Fig. 11.1h). The diameter of the microtubular structures was 25–40 nm. Of note, these three patients had CLL.
MPGN was also reported with IgM-secreting monoclonal proliferations in the absence of cryoglobulinemia [10].
Pathophysiological Considerations
One of the important points shown by the immunofluorescence studies is the striking correspondence between the localization of the IgG deposits, defining either MPGN or MN histological patterns, and the subclass of the monoclonal IgG found in the deposits. IgG3 is the predominant subclass in proliferative GN with monoclonal IgG deposits, as it is in type-1 cryoglobulinemia [4, 9, 16, 17]. Classic MPGN is triggered by deposition of immune complexes in the mesangium and the glomerular capillaries, activating the complement cascade and recruiting inflammatory cells such as macrophages and lymphocytes. In monoclonal IgG3-associated MPGN, there is no evidence for an antigen–antibody immune complex, either circulating or formed in situ. This rather uncommon serum subclass of human IgG (mean normal adult level, 0.42 mg/ml; range 0.18–0.80 mg/ml) is the most nephritogenic because of its ability to aggregate in the glomerular capillary via a specific Fc–Fc interaction. IgG3 is also the most positively charged human IgG, favoring its affinity towards the anionic sites of the glomerular membrane [29, 30]. This high avidity of IgG3 for the glomeruli may explain the fact that monoclonal components can remain undetectable in the serum of patients with proven monoclonal IgG3 kidney deposits. Last but not least, IgG3 has the greatest complement-fixing capacity, which in turn could activate downstream inflammatory mediators that promote glomerular leukocyte infiltration and proliferation. Interestingly, IgG3 is the predominant Ig subclass in monoclonal components observed in immunodeficiency states, including aging and treatment with anti-calcineurin inhibitors [31]. This observation suggests that PGNMID occurs in an unusual immunological setting that requires further investigation.
On the other hand, most (64 %) cases of monoclonal MN are due to IgG1 deposits, while IgG3 is rarely observed [17, 19, 32, 33]. These data confirm the observations of Bridoux et al. who found that five of ten patients with atypical MN due to monotypic Ig deposits had IgG1 subclass deposited in their glomeruli [6]. The one patient from Nasr et al.’s series with membranous features also had IgG1 deposits. Interestingly, IgG4 was not found in our series, although this subclass is the most prominent in idiopathic MN [34]. However, it is difficult to draw definitive conclusions about the propensity of IgG1 subclass for membranous deposits, because it is the most frequent Ig subclass found in monoclonal gammopathies [35]. Nevertheless, one of our previous reports supports the hypothesis that, in contrast to classic MN [36], the deposited immunoglobulin may, in some cases, not be directed against a local antigen, but rather precipitates, because of peculiar physicochemical properties [33]. In a patient with a membranous pattern of GN, the circulating monoclonal IgG1λ showed unusual in vitro aggregation properties, including dependence on low ionic strength and neutral pH, suggesting that electrostatic interactions had a role in the precipitation process. We speculate that in vivo precipitation is facilitated by the local concentration of the protein in glomerular basement membrane and the ionic properties of the negatively charged local milieu. Interestingly, the IgG precipitated from serum had a non-organized ultrastructure similar to that of kidney deposits [33]. On the other hand, we recently reported a very particular case of recurrent MN, occurring 13 days after kidney transplantation. The graft biopsy specimen showed granular staining for complement and monoclonal IgG3k and electron microscopy revealed subepithelial non-organized deposits. A search for hematologic disorders was negative. Retrospective evaluation of a biopsy sample from the native kidney revealed a similar pattern: monotypic IgG3k deposits together with C3, C1q, and C5b-9. Glomerular deposits contained PLA2R in both the graft and the native kidney, suggesting that the recurrence was the result of circulating monoclonal anti-PLA2R antibodies binding to PLA2R antigen expressed on donor podocytes. Confocal analysis of anti-PLA2R and anti-human IgG3 showed colocalization, and the patient had IgG3k-restricted circulating anti-PLA2R antibodies [37]. This case reveals that the occurrence of monoclonal MN should lead to systematic testing of anti-PLA2R antibodies.
A unique case of hypocomplementemic MPGN associated with monoclonal λ light chain dimers, isolated from the serum and urine, has also been reported [14]. The dimers formed a miniautoantibody against complement factor H and thus activated the alternative pathway of complement. Several cases of GN with isolated renal C3 deposits and circulating monoclonal gammopathy have been recently reported [38, 39]. These cases might represent an unusual complication of plasma cell dyscrasia, related to complement activation through an autoantibody activity of the monoclonal Ig against a complement alternative pathway regulator protein such as complement factor H, as it has been shown for one patient reported by Bridoux et al. [38]. Whether this can occur also in GN with monoclonal Ig deposits remains to be established, but several patients, both in Nasr’s series [16] and in our study [17], showed isolated C3 consumption and deposition in glomeruli, without involvement of C1q or low peripheral C4 levels, suggesting that complement activation in this setting can probably be mediated by the alternative pathway.
Diagnostic Considerations
Monoclonal gammopathy should be considered as an important and common cause of MPGN. The Mayo Clinic recently reviewed the case of 68 patients with MPGN who were negative for hepatitis B and C and were evaluated for gammopathies, during the period of 2001 through 2006 [40]. Twenty-eight (41.1 %) had serum and/or urine electrophoresis studies positive for monoclonal gammopathy. Sixteen patients had so-called MGUS (this term is usually not employed in the presence of visceral complications), while 12 patients had various lymphoplasmacytic cell proliferations including multiple myeloma (six patients), low-grade B-cell lymphoma (three patients), CLL (two patients), and lymphoplasmacytic lymphoma (LPL)/Waldenström’s macroglobulinemia (one patient). Ten of 28 patients had circulating monoclonal IgMk. Data of immunofluorescence microscopy of kidney biopsies correlated with immunofixation results. Therefore, all patients with a diagnosis of MPGN should be evaluated for an underlying monoclonal gammopathy. A careful analysis of the biopsy with anti-LC isotype antibodies is the first step of the workup. If light chain isotype restriction is found, then the biopsy should be analyzed with anti-γHC subclass antibodies to confirm monoclonality. The same analysis should be done for the patients with MN.
Irrespective of the histological pattern, MPGN or MN, the finding of monoclonal deposits should lead to analyze the organization of deposits by EM, and to search for cryoglobulinemia, circulating monoclonal component by highly sensitive techniques, and signs of a lymphoplasmacytic cell proliferation by bone marrow examination, blood lymphocyte phenotyping, and CT or PET scan.
In cases of endocapillary proliferative or MPGN in which the deposits stain for a single γHC subclass and a single LC, diagnostic considerations would include PGNMID, type-1 cryoglobulinemia GN, and immunotactoid GN (Table 11.5). Two points should be emphasized. First, the distinction with type-1 cryoglobulinemia may be difficult because the characteristic feature of thrombi with annular structure of deposits by EM is not always found. Second, EM may not be available or the results delayed and, therefore, the distinction with immunotactoid GN may be impossible for some time. However, from a therapeutic point of view, the key point is the presence of monoclonal Ig deposits which should lead to a detailed workup for a lymphoplasmacytic cell disorder and to appropriate treatment against the overt or low-grade incipience proliferation.
Therapeutic Considerations
All patients who present with a well-defined hematological malignancy, such as multiple myeloma and high-grade NHL associated with a monoclonal compound, must be treated according to the standard chemotherapy protocols, including newly introduced drugs such as thalidomide, bortezomib, or rituximab in addition to inhibitors of the RAS. If the treatment permits sustained hematological remission and suppression of the circulating monoclonal IgG, then the renal disease can disappear. In nonmalignant cases with a low-tumoral mass plasma cell dyscrasia or a low-grade lymphoproliferative disease, nephrologists have to convince hematologists that, as in AL amyloidosis, the treatment of the otherwise “benign” neoplasm is mandatory to hamper the renal disease. These last years, the term of Monoclonal Gammopathy of Renal Significance (MGRS) has emerged to describe those patients with monoclonal Ig-related renal disease, such as AL amyloidosis, MICDD, or PGNMID, and an hematological disorder which is more consistent with MGUS than with multiple myeloma [41]. In contrast with typical MGUS, for which treatment is not recommended, the identification of a MGRS must lead to a rapid therapeutic intervention, in order to preserve or restore kidney function but also to avoid recurrence of renal disease after kidney transplantation [42].
In our recent series [17], complete remission of the nephrotic syndrome was obtained in 13 patients (54 %). In all of these patients, remission of nephropathy was reached only after the disappearance of the circulating M-spike. Absence of renal remission was mainly observed among patients who were diagnosed at a late stage of chronic kidney disease, with elevated serum creatinine levels and presence of extensive fibrosis on the renal biopsy. The presence of an identified hematological malignancy was not associated with a worse renal outcome, and complete remission of nephropathy could be obtained in five of nine patients with myeloma, CLL, or lymphoma. Patients with MPGN or MN had the same prognosis. Complete or partial remission was obtained in 6 of 12 patients with MPGN and 8 of 14 patients with MN. Response to treatment was not associated with any clinical or laboratory feature, such as age, presence of malignancy, and level of proteinuria. The only nonstatistically significant differences between responders and nonresponders were the initial glomerular filtration rate.
For patients without overt malignancy, rituximab may be the optimal therapeutic choice. Indeed, our study confirms previously published data, showing that treatment with RAS inhibitors or corticosteroids alone is not sufficient to achieve long-term remission [17]. Rituximab has a very favorable benefit-to-tolerance ratio in this subgroup of patients. In the series reported by Nasr et al. [16], two of four patients with MPGN and monoclonal IgG deposits who received this B-cell depleting drug experienced partial remission. Three other reports on monoclonal MPGN or immunotactoid GN [40, 43, 44] also suggest that rituximab can be beneficial in this setting. In our series, five of seven patients with either MPGN or MN showed complete remission and two experienced a good-quality partial remission, with no major side effects. In the case of the monoclonal MN associated with IgG3-restricted antiPLA2R antibodies, rituximab permitted remission of the nephrotic syndrome and stabilization of serum creatinine [37]. Further studies are necessary to define which patients should be treated with this drug and what should be the best therapeutic scheme.
In conclusion, GNs with non-Randall-type, non-organized monoclonal Ig deposits are a new evolving entity whose diagnosis relies on a careful examination of the kidney biopsy with specific anti-LC isotype and anti-IgG subclass antibodies. Therefore, recognition of this entity mainly relies on the pathologist. The diagnosis of these diseases has two main consequences. The first is a detailed workup in search of a lymphoplasmacytic disorder. The second regards therapeutic strategy aimed at annihilating the underlying B-cell proliferation and improving the associated kidney disease.
References
Glenner GG, Terry W, Harada M, et al. Amyloid fibril proteins: proof of homology with immunoglobulin light chains by sequence analyses. Science. 1971;172:1150–1.
Randall RE, Williamson Jr WC, Mullinax F, et al. Manifestations of systemic light chain deposition. Am J Med. 1976;60:293–9.
Ivanyi B. Frequency of light chain deposition nephropathy relative to renal amyloidosis and Bence Jones cast nephropathy in a necropsy study of patients with myeloma. Arch Pathol Lab Med. 1990;114:986–7.
Karras A, Noel LH, Droz D, et al. Renal involvement in monoclonal (type I) cryoglobulinemia: two cases associated with IgG3 kappa cryoglobulin. Am J Kidney Dis. 2002;40:1091–6.
Lin J, Markowitz GS, Valeri AM, et al. Renal monoclonal immunoglobulin deposition disease: the disease spectrum. J Am Soc Nephrol. 2001;12:1482–92.
Bridoux F, Hugue V, Coldefy O, et al. Fibrillary glomerulonephritis and immunotactoid (microtubular) glomerulopathy are associated with distinct immunologic features. Kidney Int. 2002;62:1764–75.
Rosenstock JL, Markowitz GS, Valeri AM, et al. Fibrillary and immunotactoid glomerulonephritis: distinct entities with different clinical and pathologic features. Kidney Int. 2003;63:1450–61.
Nasr SH, Valeri AM, Cornell LD, et al. Fibrillary glomerulonephritis: a report of 66 cases from a single institution. Clin J Am Soc Nephrol. 2011;6:775–84.
Nasr SH, Colvin R, Markowitz GS. IgG1 lambda light and heavy chain renal amyloidosis. Kidney Int. 2006;70:7.
Audard V, Georges B, Vanhille P, et al. Renal lesions associated with IgM-secreting monoclonal proliferations: revisiting the disease spectrum. Clin J Am Soc Nephrol. 2008;3:1339–49.
Alpers CE, Tu WH, Hopper Jr J, Biava CG. Single light chain subclass (kappa chain) immunoglobulin deposition in glomerulonephritis. Hum Pathol. 1985;16:294–304.
Touchard G. Ultrastructural pattern and classification of renal monoclonal immunoglobulin deposits. In: Touchard G, Aucouturier P, Hermine O, Ronco P, editors. Monoclonal gammopathies and the kidney. Dordrecht: Kluwer; 2003. p. 95–117.
Bridoux F, Zanetta G, Vanhille P, Goujon JM, Vanhille P, Bauwens M, Chevet D, Ronco P, Preud’homme JL, Touchard G. Glomerulopathy with non-organized and non-Randall type monoclonal immunoglobulin deposits: a rare entity [abstract]. J Am Soc Nephrol. 2001;12:94A.
Jokiranta TS, Solomon A, Pangburn MK, et al. Nephritogenic lambda light chain dimer: a unique human miniautoantibody against complement factor H. J Immunol. 1999;15(163):4590–6.
Nasr SH, Markowitz GS, Stokes MB, et al. Proliferative glomerulonephritis with monoclonal IgG deposits: a distinct entity mimicking immune-complex glomerulonephritis. Kidney Int. 2004;65:85–96.
Nasr SH, Satoskar A, Markowitz GS, et al. Proliferative glomerulonephritis with monoclonal IgG deposits. J Am Soc Nephrol. 2009;20:2055–64.
Guiard E, Karras A, Plaisier E, et al. Patterns of non-cryoglobulinemic glomerulonephritis with monoclonal Ig deposits: correlation with IgG subclass and response to rituximab. Clin J Am Soc Nephrol. 2011;6:1609–16.
Evans DJ, Macanovic M, Dunn MJ, et al. Membranous glomerulonephritis associated with follicular B-cell lymphoma and subepithelial deposition of IgG1-kappa paraprotein. Nephron Clin Pract. 2003;93:c112.
Komatsuda A, Masai R, Ohtani H, et al. Monoclonal immunoglobulin deposition disease associated with membranous features. Nephrol Dial Transplant. 2008;23:3888–94.
Masai R, Wakui H, Komatsuda A, Togashi M, Maki N, Ohtani H, Oyama Y, Sawada K. Characteristics of proliferative glomerulonephritis with monoclonal IgG deposits associated with membranoproliferative features. Clin Nephrol. 2009;72:46–54.
Fujiwara T, Komatsuda A, Ohtani H, Togashi M, Sawada K, Wakui H. Proliferative glomerulonephritis with monoclonal IgG deposits in a patient with autoimmune hemolytic anemia. Clin Nephrol. 2013;79(6):494–8.
Fujita E, Shimizu A, Kaneko T, Masuda Y, Ishihara C, Mii A, Higo S, Kajimoto Y, Kanzaki G, Nagasaka S, Iino Y, Katayama Y, Fukuda Y. Proliferative glomerulonephritis with monoclonal immunoglobulin G3κ deposits in association with parvovirus B19 infection. Hum Pathol. 2012;43(12):2326–33.
Yahata M, Nakaya I, Takahashi S, Sakuma T, Sato H, Soma J. Proliferative glomerulonephritis with monoclonal IgM deposits without Waldenström’s macroglobulinemia: case report and review of the literature. Clin Nephrol. 2012;77(3):254–60.
Aucouturier P, Preud’Homme JL. Subclass distribution of human myeloma proteins as determined with monoclonal antibodies. Immunol Lett. 1987;16:55–7.
Nasr SH, Sethi S, Cornell LD, Fidler ME, Boelkins M, Fervenza FC, Cosio FG, D’Agati VD. Proliferative glomerulonephritis with monoclonal IgG deposits recurs in the allograft. Clin J Am Soc Nephrol. 2011;6(1):122–32.
Albawardi A, Satoskar A, Von Visger J, Brodsky S, Nadasdy G, Nadasdy T. Proliferative glomerulonephritis with monoclonal IgG deposits recurs or may develop de novo in kidney allografts. Am J Kidney Dis. 2011;58(2):276–81.
Sumida K, Ubara Y, Marui Y, Nakamura M, Takaichi K, Tomikawa S, Fujii T, Ohashi K. Recurrent proliferative glomerulonephritis with monoclonal IgG deposits of IgG2lambda subtype in a transplanted kidney: a case report. Am J Kidney Dis. 2013;62(3):587–90.
Batal I, Bijol V, Schlossman RL, Rennke HG. Proliferative glomerulonephritis with monoclonal immunoglobulin deposits in a kidney allograft. Am J Kidney Dis. 2014;63(2):318–23.
Capra JD, Kunkel HG. Aggregation of gamma-G3 proteins: relevance to the hyperviscosity syndrome. J Clin Invest. 1970;49:610–21.
Abdelmoula M, Spertini F, Shibata T, Gyotoku Y, Luzuy S, Lambert PH, Izui S. IgG3 is the major source of cryoglobulins in mice. J Immunol. 1989;143:526–32.
Aucouturier P, Bremard-Oury C, Clauvel JP, Debré M, Griscelli C, Seligmann M, Preud’homme JL. Serum IgG subclass levels in primary and acquired immunodeficiency. Monogr Allergy. 1986;20:62–74.
Moulin B, Ronco PM, Mougenot B, Francois A, Fillastre JP, Mignon F. Glomerulonephritis in chronic lymphocytic leukemia and related B-cell lymphomas. Kidney Int. 1992;42:127–35.
de Seigneux S, Bindi P, Debiec H, Alyanakian MA, Aymard B, Callard P, Ronco P, Aucouturier P. Immunoglobulin deposition disease with a membranous pattern and a circulating monoclonal immunoglobulin G with charge-dependent aggregation properties. Am J Kidney Dis. 2010;56:117–21.
Oliveira DB. Membranous nephropathy: an IgG4-mediated disease. Lancet. 1998;351:670–1.
Aucouturier P, Mounir S, Preud’homme JL. Distribution of IgG subclass levels in normal adult sera as determined by a competitive enzyme immunoassay using monoclonal antibodies. Diagn Immunol. 1985;3:191–6.
Ronco P, Debiec H. Advances in membranous nephropathy: success stories of a long journey. Clin Exp Pharmacol Physiol. 2011;38:410–6.
Debiec H, Hanoy M, Francois A, Guerrot D, Ferlicot S, Johanet C, Aucouturier P, Godin M, Ronco P. Recurrent membranous nephropathy in an allograft caused by IgG3k targeting the PLA2 receptor. J Am Soc Nephrol. 2012;23(12):1949–54.
Bridoux F, Desport E, Frémeaux-Bacchi V, Chong CF, Gombert JM, Lacombe C, Quellard N, Touchard G. Glomerulonephritis with isolated C3 deposits and monoclonal gammopathy: a fortuitous association? Clin J Am Soc Nephrol. 2011;6:2165–74.
Sethi S, Sukov WR, Zhang Y, Fervenza FC, Lager DJ, Miller DV, Cornell LD, Krishnan SG, Smith RJ. Dense deposit disease associated with monoclonal gammopathy of undetermined significance. Am J Kidney Dis. 2010;56:977–82.
Sethi S, Zand L, Leung N, Smith RJ, Jevremonic D, Herrmann SS, Fervenza FC. Membranoproliferative glomerulonephritis secondary to monoclonal gammopathy. Clin J Am Soc Nephrol. 2010;5:770–82.
Leung N, Bridoux F, Hutchison CA, Nasr SH, Cockwell P, Fermand JP, Dispenzieri A, Song KW, Kyle RA, International Kidney and Monoclonal Gammopathy Research Group. Monoclonal gammopathy of renal significance: when MGUS is no longer undetermined or insignificant. Blood. 2012;120(22):4292–5.
Fermand JP, Bridoux F, Kyle RA, Kastritis E, Weiss BM, Cook MA, Drayson MT, Dispenzieri A, Leung N, International Kidney and Monoclonal Gammopathy Research Group. How I treat monoclonal gammopathy of renal significance (MGRS). Blood. 2013;122(22):3583–90.
Bhat P, Weiss S, Appel GB, Radhakrishnan J. Rituximab treatment of dysproteinemias affecting the kidney: a review of three cases. Am J Kidney Dis. 2007;50:641–4.
Vilayur E, Trevillian P, Walsh M. Monoclonal gammopathy and glomerulopathy associated with chronic lymphocytic leukemia. Nat Clin Pract Nephrol. 2009;5:54–8.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Ronco, P., Karras, A., Plaisier, E. (2015). Non-Randall Glomerulonephritis with Non-Organized Monoclonal Ig Deposits. In: Picken, M., Herrera, G., Dogan, A. (eds) Amyloid and Related Disorders. Current Clinical Pathology. Humana Press, Cham. https://doi.org/10.1007/978-3-319-19294-9_11
Download citation
DOI: https://doi.org/10.1007/978-3-319-19294-9_11
Publisher Name: Humana Press, Cham
Print ISBN: 978-3-319-19293-2
Online ISBN: 978-3-319-19294-9
eBook Packages: MedicineMedicine (R0)