
Abstract
Lupus erythematosus (LE) can affect the skin, the internal organs, or both. In this chapter we review the cutaneous findings specific to LE as well as the nonspecific skin findings that may be associated with systemic LE. Cutaneous disease represents the second most common presentation in systemic lupus erythematosus (SLE), and as such, dermatologists play an important role in the evaluation and diagnosis for these patients by correlating clinical findings with those demonstrated on skin biopsy if needed, and by undertaking initial risk assessment for systemic disease.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Acute cutaneous LE
- Subacute cutaneous LE
- Chronic cutaneous LE
- Discoid LE
- Chilblain LE
- Tumid LE
- Lupus profundus
- Antimalarials
- Hydroxychloroquine
-
Cutaneous disease in systemic lupus erythematosus (SLE) is common. In contrast, the likelihood of systemic disease in patients with cutaneous lupus is variable and depends on cutaneous lupus subtype.
-
Cutaneous lupus subtypes may have significant overlap, both clinically and histologically. Clinicopathologic correlation and careful observation of lesion morphology is essential in establishing subtype.
-
In the setting of new onset subacute cutaneous lupus, a thorough evaluation of prescription and over-the-counter medication history is important to rule out iatrogenic disease.
-
Patients with cutaneous lupus should be evaluated for the presence of systemic disease through a complete history, review of systems, physical examination, and serologic testing as appropriate.
-
The goal of treating cutaneous lupus is to prevent progression of existing lesions and formation of new ones. Aggressive treatment is warranted to prevent disfigurement in scarring subtypes.
Interdisciplinary Introduction
Lupus erythematosus (LE) can affect the skin, the internal organs, or both. In this chapter we review the cutaneous findings specific to LE as well as the nonspecific skin findings that may be associated with systemic LE. Cutaneous disease represents the second most common presentation in systemic lupus erythematosus (SLE), and as such, dermatologists play an important role in the evaluation and diagnosis for these patients by correlating clinical findings with those demonstrated on skin biopsy if needed, and by undertaking initial risk assessment for systemic disease.
In patients with significant systemic disease, including central nervous system (CNS), renal, or other internal organ involvement, co-management in an interdisciplinary fashion is important, and the managing team should include a rheumatologist, nephrologist, neurologist, or other relevant specialists. An interdisciplinary approach may also be beneficial in patients with skin-limited disease who are managed with systemic medications, which can result in multi-system morbidity, including infection, osteoporosis, and metabolic or cardiovascular effects.
Epidemiology & Classification
Epidemiology
The incidence of cutaneous lupus erythematosus (CLE) is similar to that of SLE, but CLE is more common than SLE in males and in older adults [1, 2]. The female to male ratio in CLE is closer to 3 or 4:1, as opposed to the much higher ratio seen in SLE [2]. In CLE, smoking is a risk factor for refractoriness to therapy [3, 4].
Classification of Cutaneous Lupus Erythematosus
Gilliam Classification (Table 3.1)
Classification of lupus erythematosus (LE)-specific skin changes is based on lesion morphology. According to the widely used classification scheme suggested by Gilliam and Sontheimer [5], CLE may be divided as follows: acute cutaneous LE (ACLE); subacute cutaneous LE (SCLE); and chronic cutaneous LE (CCLE), with the last category including discoid LE (DLE), chilblain LE, tumid LE, and lupus profundus [5] (Table 3.1). We review the clinical features of each subtype in detail below.
Of note, there can be significant overlap between CLE subtypes, both clinically and histologically, and it is typically not possible to classify subtypes based solely on histology. Clinicopathologic correlation is extremely important, and careful observation of lesion morphology is paramount. It may not always be possible to make a definitive diagnosis of CLE subtype in every patient.
Other Approaches to CLE Subgrouping
Variants of the Gilliam classification have been proposed. There is a lack of international agreement on the proper classification of CLE, and there has been a recent proliferation of the potential ways to group subtypes of CLE [6, 7]. An international effort to develop definitions and groupings for subtypes of CLE is ongoing [8, 9].
LE-Nonspecific Skin Changes (Table 3.2)
There are numerous skin findings that can be seen in patients with LE but are not specific for LE, including vasculitis, vasculopathy, and nonscarring alopecia, among others. These findings, reviewed in detail in Table 3.2, are more frequently associated with SLE than is CLE alone [10]. The presence of these findings in patients with established disease should also prompt evaluation for an underlying flare of SLE.
CLE Association with SLE
The likelihood of SLE in patients with CLE is variable and depends largely on CLE subtype. In localized DLE, extracutaneous involvement is relatively uncommon. Patients with generalized DLE or papulosquamous SCLE are more likely to meet criteria for SLE, although they may have lower likelihood of CNS or renal involvement than other groups with SLE.
By contrast, about 80% of SLE patients have specific skin changes. The commonly used ACR-97 criteria for SLE diagnosis [11, 12] place great importance on skin manifestations, including both LE-specific skin changes (butterfly rash, discoid lesions), and relatively nonspecific skin changes (oral and nasal mucosal ulcers, and photosensitivity). In many patients with isolated cutaneous LE, skin signs and symptoms alone may thus fulfill the required four ACR criteria for SLE [13, 14] (Table 3.3).
New criteria for SLE were developed by the Systemic Lupus International Collaborating Clinics (SLICC) in 2012 [15], including 11 clinical and six immunologic criteria. Fulfillment of four or more criteria (including at least one clinical and one immunologic item) is required for a diagnosis of SLE. Among other changes, the SLICC criteria reduce the relative weighting of skin findings as compared to ACR-97 by consolidating the skin findings in LE into fewer categories, eliminating the redundancy in ACR-97 that allows patients to meet SLE criteria with skin-limited disease. Of note, alopecia is added as a criterion in the SLICC criteria, though this presents challenges, as SLE patients can have alopecia for many different reasons, not all of which are related to SLE. Differentiating alopecia related to SLE from alopecia due to other causes (e.g., medication-related telogen effluvium, androgenic alopecia, and alopecia areata, among others) is frequently challenging. For many patients the etiology of hair loss is multifactorial (Table 3.4).
Not surprisingly based on the changes reviewed, studies suggest greater sensitivity for the SLICC criteria as compared with the ACR-97, but poorer specificity [16]. Further testing and validation will be needed to determine the optimal criteria for SLE [15, 17].
Pathogenesis of Cutaneous Lupus Erythematosus
The pathogenesis of CLE is incompletely understood but involves the interaction of genetic and environmental factors to promote the development of a complex inflammatory cascade. Identified pathogenic factors include ultraviolet irradiation compounded by the accumulation of apoptotic cells due to decreased clearing or impaired macrophage phagocytic capacity, B cell defects, dysregulation of T cells, activation of dendritic cells (DCs), and chemokine and cytokine imbalances, particularly in type 1 interferon (IFN) [18]. Autoantibody-mediated antibody-dependent cellular cytotoxicity (ADCC) is a potential mechanism of tissue injury, particularly as certain autoantibodies are associated with certain phenotypes, as reviewed below [19].
Genetic Factors
Major genetic associations with CLE include the human leukocyte antigen (HLA) A1, B8, DR3, B7 and DR2 haplotypes [20]. In the SCLE subtype in particular, genetic studies have identified HLA types A1, B8, DR3, DQ2, DRw52 and C4null as susceptibility haplotypes [21]. SCLE is closely associated with the HLA haplotype DRB1*0301-B*08.6, which includes the 308A TNFa promoter polymorphism. This polymorphism has been associated with increased UV-induced TNFa production in keratinocytes [20, 22].
Additionally, polymorphisms affecting many genetic regions outside the major histocompatibility complex (MHC) regions increase susceptibility to CLE. These include genes encoding cytokines (IL-1 locus 2q13; IL-10 locus 1q31), cytokine receptors (gamma receptor II Fc RII locus 1q23; T cell receptor TCR locus 7q35), adhesion molecules (ICAM-1 locus 19p13.3–p13.2; E-selectin locus 1q23–25), antioxidant enzymes (glutathione-S-transferase M1 GST M1 locus 1p13), and apoptosis genes (Fas locus 10q24.1;TRIM39), ITGAM, TYK2, and CTLA4 [23,24,25,26,27]. Genes in the IFN pathway (e.g. IRF5) are associated with SLE and may also play a role in CLE [28].
Inherited deficiencies of complement components have also been strongly linked to CLE. The likely mechanism is accumulation of DNA and/or RNA inside cells, leading to IRF-3 dependent production and release of IFNa. Patients with C1q deficiency frequently develop LE-like photosensitive skin eruptions. C1q binds apoptotic cells and appears to play a role in the clearance of apoptotic keratinocytes [29]. C1 inhibitor, C1q, C2, and partial C4b deficiencies have been described in CLE.
Certain complement deficiencies have been associated with specific CLE subtypes: C2/C4 deficiencies are associated with SCLE, while C4 deficiency has been associated with LE profundus [30].
Missense mutations in the TREX1 gene, an exonuclease that digests single-stranded or mis-paired double-stranded DNA, underlies familial chilblain LE [31].
Ultraviolet Light and Apoptosis
Both Ultraviolet A (UVA) and Ultraviolet B (UVB) exposure can trigger CLE, although irradiation of a large spot size on a normally photo-exposed area is required to see induction of lesions in about half of CLE patients [32,33,34]. The exact role of UVR in CLE induction is unclear. UVB irradiation induces changes in keratinocyte membrane expression of autoantigens [35, 36]. It also is known that UVR induces DNA damage and that there are increased apoptotic cells in the epidermis in CLE. These increased apoptotic cells are seen in more than half of CLE biopsies after irradiation, and CLE patients may have defects in clearance of these cells [37]. As reviewed, patients deficient in C1q develop a photosensitive form of LE [38].
Innate Immunity
Antimicrobial peptides, including LL-37, are expressed in inflammatory and epithelial cells. These are upregulated in CLE skin [39, 40]. Antimicrobial peptides and other molecules present in CLE skin, including HMGB1, hyaluronic acid, self-nucleic acids, and nucleic acid-containing immune complexes, upregulate DCs through toll-like receptors and pathogen recognition receptors [41, 42]. Type I interferons are upregulated in CLE and activate the JAK/STAT1/2 signaling pathway, causing expression of IFN-stimulated genes that activate the adaptive immune system.
Inflammatory Cells
The interface dermatitis that is the hallmark of most subtypes of CLE, as reviewed below, includes an inflammatory infiltrate of DCs, as well as CCR5+, CD4+ T cells. CD8+ T cells can predominate in long-standing DLE. Th17, CD4+ T cells are important in SLE pathogenesis, but activation of type I IFN and IFN-g are more characteristic of DLE than activation of the IL-17 pathway [43]. Regulatory T cells are locally decreased in CLE, potentially contributing to autoimmunity [44].
Inflammatory Cytokines and Chemokines
A distinctive IFN signature is observed in the skin and blood of certain CLE patients [45, 46]. Specifically, this signature can be found only in subsets of CLE characterized by an interface dermatitis (DLE, SCLE); it correlates with CLE disease activity, but there is no difference in IFN signature in those meeting criteria for SLE relative to CLE alone [46].
The expression pattern of IFN-inducible proteins in CLE reflects the characteristic histological distribution of infiltrating immune cells in each subset [47]. Studies have demonstrated the CXCR3 ligands CXCL9 (interferon-gamma [IFNg]-induced monokine), CXCL10 (IFNg-inducible protein 10), and CXCL11 (IFN-inducible T cell alpha chemoattractant) as the most abundantly expressed chemokine family members in cutaneous LE [48]. In addition, IFN-lambda, recently demonstrated in the epidermis of CLE, is produced by keratinocytes and induces expression of CXCL9 [49]. Within cutaneous LE lesions, plasmacytoid and myeloid DCs accumulate in the dermis and are activated to produce type I IFN, as detected by the expression of IRF7 and MxA [50]. Type I INF induce proinflammatory cytokines and chemokines that support the cellular immune response.
The proinflammatory cytokines TNFa and IL-1 are upregulated by UVR and therefore may be important in CLE [51]. In addition, increased TNFa produced by peripheral blood mononuclear cells (PBMCs) in CLE patients correlates with increased disease activity [52].
Clinical Features
Each subtype of CLE has distinctive clinical features and differing frequency of association with SLE. However, the histology can overlap in some subtypes. Subtypes with scarring (e.g., DLE and lupus panniculitis) have the deepest and most dense inflammatory infiltrate on histology, while clinically transient subtypes (e.g., ACLE) are characterized by the most superficial infiltrate.
Acute Cutaneous Lupus Erythematosus (ACLE)
ACLE is an acute, non-scarring, photosensitive eruption that occurs in patients who frequently meet criteria for SLE. It is transient, and its appearance tends to mirror increased systemic activity. ACLE is associated with a younger age of SLE onset. Co-occurrence with other subtypes of CLE, especially SCLE, can occur.
Classically, ACLE presents as erythematous patches with fine scale and/or edema. Patients may initially mistake this rash for sunburn and only seek medical attention after it persists for several days. There are both localized and generalized forms of ACLE.
Localized ACLE is more common than generalized; it may present as either the classic “butterfly rash,” involving the malar cheeks and nasal bridge with sparing of the nasolabial folds, or between joints on the dorsal fingers. Involvement of other photodistributed sites (forehead, periorbital, sides of neck) can occur.
The generalized form of ACLE is less common and can be non-bullous or bullous. Non-bullous ACLE may appear as symmetric, discrete or coalescing macules and/or papules. It can also mimic dermatomyositis. Bullous ACLE can mimic bullous fixed drug or Stevens-Johnson syndrome (SJS)/toxic epidermolytic necrolysis (TEN), presenting with flaccid bullae and epidermal detachment. (See section below entitled “ Vesicobullous disease occurring as severe variants of ACLE, SCLE, and rarely, DLE.”)
Not all patients with SLE develop ACLE, however presence of ACLE is typically a sign of SLE. Malar ACLE was reported in up to 52% of SLE patients at the time of diagnosis in one study [53], and large U.S. lupus cohorts reported malar ACLE in 20–60% of patients. Rosacea can be mistaken for malar ACLE. Persistence of the eruption, involvement past the nasolabial fold onto the lip, presence of papules, and dynamic flushing would go against a diagnosis of ACLE. A biopsy may be needed to confirm the diagnosis.
ACLE tends to wax and wane with systemic activity. It does not scar but can result in post-inflammatory hyper- or hypopigmentation. Treatment of ACLE often requires treatment of underlying SLE .
Subacute Cutaneous Lupus Erythematosus (SCLE)
SCLE is a highly photosensitive, non-scarring CLE subtype. An estimated 35–40% of patients with SCLE meet criteria for SLE, although many patients do so by fulfilling 4 or more of the ACR-97 criteria involving skin lesions, photosensitivity, and serologies [2, 54, 55]. SCLE patients with SLE typically have only mild systemic symptoms, most commonly arthritis and myalgias; in the original series, no SCLE patients had serious CNS or renal disease [56].
SCLE most commonly involves sun-exposed areas, including the upper chest and back in a ‘V’ distribution, the extensor aspect of arms, and, occasionally, the sides of the face. The mid-face, scalp, and skin below the waist are usually spared.
Clinically, there are two forms of SCLE: annular and papulosquamous. Annular SCLE is characterized by scaly, erythematous, thin, coin-shaped plaques with raised red borders and a central clearing. The annular plaques tend to coalesce, producing a polycyclic array. In contrast, papulosquamous SCLE (Fig. 3.1) tends to have a psoriasis or eczema-like appearance in a sun-exposed distribution. Lesions may begin as small erythematous papules or plaques with fine scale. Although most patients are asymptomatic, mild pruritus may occur. Most patients have chronically active disease with intermittent sun-induced exacerbations, and although SCLE does not scar, it can result in significant hyperpigmentation or hypopigmentation.

Papulosquamous SCLE . Erythematous scaly patches and plaques on back
Up to 30% of SCLE cases are induced or exacerbated by a medication [54]. Widespread involvement may favor a drug-induced etiology. Presence of eosinophils on histopathology does not appear to reliably distinguish drug-induced from idiopathic disease. A 2012 population-based case-control study found terbinafine, TNF-alpha antagonists, antiepileptics, and proton pump inhibitors to be the most frequent culprits [54]. However, over 100 different agents have been implicated, with additional culprits including anti-hypertensive medications (calcium channel blockers and ACE inhibitors), nonsteroidal anti-inflammatory drugs, other antifungal agents, and chemotherapy agents. There have been reports of radiation therapy-induced SCLE and paraneoplastic SCLE [57].
In suspected cases of SCLE, consider testing for the anti-SSA/Ro antibody, the titers for which may be positive in up to 5% of cases when the ANA test is negative. Women with SCLE and certain autoantibodies (typically anti-SSA but rarely anti-RNP) have an increased risk of giving birth to infants with neonatal lupus (NL), due to transplacental passage of the autoantibody. Presence of Ro/SSA Antobody is also associated with risk of congenital heart block in the fetus or newborn. Women with anti-SSA/Ro antibody who become pregnant should be evaluated by Maternal-Fetal Medicine.
Discoid Lupus Erythematosus (DLE)
DLE is the most common type of CLE. Active DLE lesions often present as erythematous, scaly plaques. These may cause scarring, alopecia, and dyspigmentation, manifestations that become more pronounced over time. Patients classically develop atrophic plaques with central hypopigmentation and peripheral hyperpigmentation. Vitiligo-like hypopigmentation may also be seen (Fig. 3.2). Although typically asymptomatic, lesions may be tender. Erythema, tenderness, and/or scale are all signs of disease activity that can fluctuate and should be treated. Disfiguring scarring, burning pain and alopecia cause significant morbidity. Interestingly, a recent survey study noted that patients are bothered more by signs of activity (e.g. redness) than damage (scarring and dyspigmentation) [58, 59].

DLE with post-inflammatory hypopigmentation. Note the presence of activity (pink) and damage (hypopigmentation)
DLE can occur in a localized or a generalized distribution (Fig. 3.3). Localized DLE, which is more common, presents with lesions limited to the head and neck, with a propensity for the scalp and conchal bowl of the ear. Generalized DLE, the less common form, presents with lesions below the neck, typically on the extensor forearms and hands; these patients are at higher risk for developing SLE than those with localized disease [60, 61]. DLE lesions occur most commonly in sun-exposed areas but can also occur in non-sun-exposed regions, including, rarely, the palms and soles (<2%) as well as the mucosa (lips, oral cavity, genitalia) (Fig. 3.4a, b) [62].

Generalized DLE . Erythematous, scaly atrophic plaques on back and arms, with central hypopigmentation and hyperpigmentation at the periphery

Non-sun-exposed DLE (a) DLE affecting soles of feet. (b) Intraoral DLE
Hypertrophic DLE is a variant of DLE in which thick, keratotic plaques occur on the arms, hands, and face. Hypertrophic DLE can be confused clinically and histologically with warts, keratoacanthomas, squamous cell carcinomas, and hypertrophic lichen planus; a skin biopsy is often needed to confirm the diagnosis. Although hypertrophic DLE can mimic squamous cell carcinoma (SCC), SCC can also rarely develop within a DLE lesion, and a high index of suspicion is necessary for diagnosis [63].
Although most patients with DLE have skin-limited disease, recent data suggest that the risk of progression to SLE may be higher than previously thought. In 1975, Prystowsky et al. reported that <5–10% of adults with DLE progress to SLE [64], while in 2011, Grohhagen et al. reported a 16.7% risk of progression within 3 years of diagnosis [54]. Children with DLE are believed to have a greater likelihood of developing SLE than adults, with reported risk ranging from 23.5–26% [60, 65, 66]. In one retrospective study looking at 34 children <16 years old over a nine-year period, an association between DLE and SLE was seen in 23.5% of patients, with disseminated DLE lesions more frequent in those meeting SLE criteria (87.5% vs. 34%).
Cutaneous Lupus: Additional LE-Specific Skin Variants
Lupus Erythematosus Tumidus (Tumid Lupus)
Tumid lupus is a relatively uncommon variant of cutaneous lupus that is generally considered to be a skin-limited condition. Clinically, lesions appear as erythematous, edematous papules or plaques, sometimes annular, without overlying epidermal change or scarring. Tumid LE is characterized by extreme photosensitivity, with lesions occurring most commonly on the face, V of the neckline, upper back, and extensor upper extremities.
Histologically, there is no vacuolar interface dermatitis at the dermal-epidermal junction (DEJ), and direct immunofluorescence (DIF) is negative. Patients are typically ANA negative and do not have underlying SLE.
Lupus Erythematosus Panniculitis/Lupus Profundus
Lupus erythematous (LE) panniculitis is a scarring subtype of CLE characterized by intense inflammation in the fat lobules. It typically presents with tender, erythematous plaques or subcutaneous nodules without epidermal change. It occurs most commonly on the face, proximal extremities, upper trunk, and buttocks, but also scalp, breasts, and thighs. Lupus panniculitis involving the breast, also known as “lupus mastitis”, may present similarly to inflammatory breast cancer, and biopsy should readily rule out a malignancy. Lesions of LE panniculitis frequently occur in sun-protected areas, are often painful, and evolve into disfiguring, depressed areas of focal lipoatrophy (Fig. 3.5a, b). One third of cases of LE panniculitis may present with overlying DLE, in a phenomenon clinically termed “lupus profundus.”

(a) Lupus panniculitis of the face in a patient with SLE. Firm, violaceous plaques on the cheeks, rather than patches of erythema seen with ACLE. (b) Lupus profundus . Lupus panniculitis with overlying DLE involving the face
Clinically and histologically, lupus panniculitis can closely resemble subcutaneous panniculitis-like T-cell lymphoma [67]. Biopsy specimens should be reviewed by a dermatopathologist, and T-cell markers and gene rearrangement studies may be necessary to help differentiate the two entities .
Chilblain Lupus
Chilblain lupus is a rare form of CLE that resembles frostbite and occurs most commonly in children and young to middle-aged women. In a series of 33 patients with chilblains lasting more than one month, 24% were found to meet classification criteria for SLE at the time of diagnosis [68]. Patients present with single or multiple, erythematous to violaceous, painful and/or pruritic nodules, most commonly located on the dorsolateral aspect of fingers and toes, and rarely on the ears and nose. Lesions arise 12–24 hours after exposure to a cold and wet environment. Unlike classic chilblains , lesions of chilblain lupus often persist beyond cold months.
Neonatal Lupus
Neonatal lupus (NL) is a self-limited syndrome that occurs in infants whose mothers have anti-SSA/Ro antibodies, or less commonly anti-SSB/La or anti-RNP antibodies, due to transplacental passage of these antibodies. The cutaneous eruption in NL occurs in 1–2% of infants born to mothers with SLE or Sjögren Syndrome with positive anti-SSA/Ro but can occur in asymptomatic mothers as well. In one series, 44% of NL mothers were asymptomatic without a history of connective tissue disease, with 50% of these mothers subsequently developing SLE or Sjögren syndrome within 10 years [69].
Unlike most CLE subtypes, NL occurs equally in male and female infants [70]. It can include one or more of the following features: an SCLE-like eruption (15–95%); congenital heart block (10%); hepatobiliary disease (9–25%); cytopenias, including leukopenia, neutropenia, or thrombocytopenia (10–15%); varying neurologic findings, including hydrocephalus , non-specific white matter changes, and calcification of the basal ganglia; vasculopathy; and, rarely, stippling of the epiphyses (chondrodysplasia punctata) on x-ray [70, 71]. Complete heart block, which is permanent, is the most feared complication of NL; the other findings are transient and self-limited, typically self-resolving within 6–8 months as the maternal antibodies are cleared.
Skin
The skin eruption of NL can be present at birth but is most commonly detected between 4–6 weeks of age, often following the first sun exposure. Skin lesions morphologically resemble the annular lesions of SCLE and have the same histologic findings; further similarities between NL and adult SCLE include photosensitivity and a strong association with anti-SSA/Ro. Unlike SCLE, however, NL has a predilection for the periorbital face (resulting in the finding known as “raccoon eyes”) and scalp, though it can also present elsewhere on the body.
The skin lesions of NL typically self-resolve in the first year of life but can result in dyspigmentation and residual telangiectasias in 10–20% of patients. Corticosteroids can hasten resolution of NL, but there is no evidence that sequelae are prevented with treatment. Photoprotection is important, and pulsed dye laser can be used to treat residual telangiectasias.
Cardiac
Autoantibody-induced cardiac conduction abnormalities occur in the setting of a normal heart in NL, which is the most common cause of congenital heart block diagnosed in utero or during the neonatal period (See Disease Assessment section below for screening recommendations). Of note, anti-SSA/Ro is responsible for cardiac manifestations; NL induced by anti-RNP does not involve the heart. Cardiac NL has a mortality rate of approximately 20%, and approximately two-thirds of affected children require pacemakers [72].
Hematologic
Any hematological lineage can be affected in NL, but thrombocytopenia is the most common manifestation, generally occurring within the 1st week of life and self-resolving by age 2–4 weeks. Neutropenia occurs later, at 4–8 weeks (10–15%). Rare cases of hemolytic anemia, pancytopenia, or aplastic anemia have been reported [70].
Hepatobiliary
Hepatobiliary disease has been reported in 9–25% of infants with NL. The severity of involvement can vary widely, ranging from asymptomatic elevations in liver function tests , to mild hepatosplenomegaly, cholestasis, and hepatitis, to, rarely, death [70, 71].
Lupus-specific Vesiculobullous Disease
Bullous SLE (BSLE)
BSLE, also referred to as “bullous lupus,” is a neutrophilic, subepidermal, antibody-mediated, vesiculobullous condition that occurs as a clinical manifestation of SLE. It is occasionally the presenting sign of SLE. Unlike ACLE, blistering activity does not necessarily correlate with systemic disease activity, although parallel exacerbations, often between BSLE and lupus nephritis, have been described.
Clinically and histologically, BSLE can resemble neutrophil-rich bullous pemphigoid, epidermolysis bullosa acquisita, linear IgA dermatosis, or dermatitis herpetiformis, presenting with tense, fluid filled vesicles or bullae. It often scars and may lead to the development of milia. The bullous eruption has a rapid onset and is typically widespread and symmetric, favoring the upper trunk, proximal upper extremities, neck, and face. However, it can occur on both sun-exposed and unexposed skin, as well as nasal, oral, and genital mucous membranes. Skin lesions are typically pruritic, with symptoms ranging from mild to severe. Mucosal lesions are typically painful.
Elements required for diagnosis of BSLE include (1) meeting criteria for SLE, (2) having an acquired bullous eruption, (3) a skin biopsy showing a neutrophilic subepidermal blister, and (4) direct immunofluorescence (DIF) demonstrating IgG (typically linear deposition at the DEJ), with or without IgM or IgA. Lastly, evidence of antibodies to type VII collagen may be demonstrated by DIF, indirect IF, or ELISA [73, 74]. The cutaneous lesions in BSLE typically respond well to treatment with dapsone .
Vesiculobullous Disease Occurring as Severe Variants of ACLE, SCLE, and Rarely, DLE.
The lupus-specific vesiculobullous eruptions are distinctly different from BSLE in that they present with flaccid rather than tense bullae, typically have a positive Nikolsky sign, often involve the mucosa, can occur in the setting of any CLE subtype, and may occur in patients without SLE. The frequency of these vesiculobullous eruptions is unclear due to the differing presentations and nomenclature reported in the literature.
In TEN-like ACLE, also known as apoptotic pan-epidermolysis (ASAP), patients with SLE present with diffuse or patchy erythema, often photodistributed, that evolves rapidly into flaccid bullae [75]. Unlike drug-induced TEN, there is typically no or limited mucosal involvement, no clear drug culprit, and a better prognosis [76]. TEN-like SCLE, by contrast, is described as widespread, flaccid bullae in the context of preexisting, photodistributed SCLE lesions and positive anti-SSA/Ro or anti-SSB/La.
Erythema multiforme (EM)-like ACLE, SCLE, or DLE, also known as Rowell syndrome , is characterized by EM-like lesions (targetoid, erythematous plaques with central flaccid bullae and erosions) in the context of lupus erythematosus (Fig. 3.6) [76, 77]. In the original case series from 1963, Rowell et al. described four adult women with longstanding DLE, chilblain LE, and skin lesions resembling EM [78]. Zeitouni et al. redefined the diagnostic criteria in 2000 to require all of three major criteria (including the presence of lupus erythematosus, EM-like lesions, and a speckled pattern of ANA), as well as one of the minor criteria (including chilblains, positive anti-SSA/Ro or anti-SSB/La, or a positive rheumatoid factor) [79].

Bullous DLE/EM-like DLE. Bullae with surrounding violaceous erythema on the foot
A review compiling 142 cases from the international literature noted that EM-like CLE differed from classic EM in that it did not preferentially affect the distal extremities, infrequently involved the mucous membranes, and was only rarely associated with an identifiable trigger [76]. It is difficult to differentiate the EM-like lesions of Rowell syndrome from classic EM on routine skin biopsy. However, positive DIF has been reported in more than 50% of cases; this frequency is similar to that seen in classic SCLE/ACLE, suggesting that EM-like lesions represent morphologic variants of CLE [76]. Nonetheless, whether or not Rowell syndrome truly represents a variant of CLE or an entity in its own right is controversial.
Non-specific Cutaneous Lesions of LE
Vascular findings have been reported to occur in approximately 50% of patients with lupus. These include: Raynaud phenomenon, livedo reticularis, palmar erythema, subtle periungual telangiectasias, leukocytoclastic or urticarial vasculitis, antiphospholipid vasculopathy, and atrophy blanche.
Non-scarring alopecia related to SLE may be telogen effluvium (caused by the underlying condition or by medications such as methotrexate or glucocorticoids) or “lupus hair.” Lupus hair is characterized by thin, unruly terminal hairs that fracture easily, usually along the frontal hairline, and typically during exacerbations of SLE. The hair grows back when the disease activity subsides [80].
Other nonspecific cutaneous manifestations of LE can include photosensitivity, reticular erythematous mucinosis (REM), erythromelalgia, and anetoderma.
Diagnostic Considerations
A biopsy of lesional skin is the cornerstone of CLE diagnosis. DIF may be a useful adjunct, while lupus serologies are often less helpful. In the setting of new onset SCLE, a careful evaluation of prescription and over-the-counter medication history is important to rule out drug-induced disease.
Diagnosis of Cutaneous Lupus
Histopathology
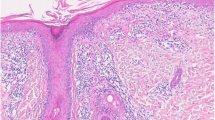
CLE histology classically shows an interface dermatitis (vacuolar degeneration of the DEJ) with perivascular lymphocytic inflammation and increased dermal mucin (Fig. 3.7). Of note, CLE and dermatomyositis may look histologically identical. However, the two conditions can be differentiated clinically.

Hematoxylin and eosin stained section of CLE skin lesion. Discoid LE showing focal interface dermatitis and dense perivascular and periadnexal lymphoid infiltrates
The CLE subtypes differ in the amount and depth of inflammation, though there may be overlapping histologic findings between the clinical phenotypes. ACLE, SCLE, and DLE all demonstrate vacuolar interface dermatitis but vary in the degree of dermal inflammation. ACLE has the sparsest and most superficial inflammation of the subtypes, while the superficial lymphocytic infiltrate in SCLE is slightly more robust. DLE demonstrates the greatest and deepest dermal inflammation, including periadnexal involvement with follicular plugging and scarring of the epidermis.
Both tumid lupus and lupus panniculitis lack epidermal changes. The findings seen in tumid lupus include prominent dermal mucin and a variable degree of perivascular and periadnexal lymphocytic infiltrate. Lupus panniculitis is characterized by a lobular lymphocytic infiltrate, hyalinizing fat necrosis, periseptal lymphoid follicles, and occasionally calcium deposition or overlying changes of DLE.
NL , if biopsied, looks identical to SCLE, while chilblain LE is characterized by a lymphocytic vasculitis with dermal edema. BSLE demonstrates a subepidermal, neutrophilic-rich blister, and the diagnosis can be confirmed by a positive DIF and ANA. The bullous variants of ACLE, SCLE, and DLE can look identical to drug-related TEN, SJS, or EM on histology, making a thorough history and a high index of suspicion important for accurate diagnosis.
Antibody Deposits in the Skin (DIF)
Skin biopsy to examine immunoreactant deposition by direct immunofluorescent (DIF) testing can be useful in either CLE or SLE. DIF may be performed on lesional skin in active lesions of CLE. When a DIF is performed on nonlesional, non-sun exposed skin, it is referred to as a “lupus band test.” Either test is considered positive when a continuous band of immunoreactants along the DEJ is observed. Antibody deposition at the DEJ is the most characteristic immunohistologic finding in lesions of cutaneous lupus and normal skin of patients with SLE. Although the lesional DIF can be helpful in establishing the diagnosis of CLE if the routine biopsy findings are non-specific, it does not replace routine histology as the method of choice for establishing a diagnosis of CLE.
In patients with known SLE, a lupus band test sampled from sun-exposed skin will be positive in 75% [81], and unexposed skin 50% of the time [82]. However 20% of the general population will have a positive lupus band test if sun-exposed skin is biopsied.
Uniquely, SCLE can demonstrate intraepidermal deposits by DIF, thought to be due to anti-Ro/SSA autoantibodies depositing directly in the epidermis rather than at the DEJ.
Evaluation for Systemic Disease
Both the ACR and SLICC criteria for SLE rely heavily on cutaneous manifestations for classification of SLE. Thus, as reviewed, according to the ACR-97 criteria, a patient with a positive ANA, photosensitivity, a malar rash and discoid lesions will fulfill criteria for SLE despite the absence of internal organ involvement. Despite this caveat, CLE can frequently accompany serious systemic involvement. In such cases, the rheumatologist can assist the dermatologist in co-management.
To date, there is no definitive way to predict if a lupus patient with solely cutaneous disease will develop involvement of other organs (Fig. 3.8). Once the diagnosis of CLE is confirmed by histology, a review of systems and a physical examination should be performed to evaluate for mucosal ulcers, fatigue, pleurisy, photosensitivity, joint pain, Raynaud’s syndrome, alopecia, history of miscarriages, or thrombotic events.

Evaluation/Management. 1. Mucosal ulcers, cytopenias, arthritis, miscarriages, or thrombosis. 2. Suggested laboratory work-up: CBC with differential, serum BUN and creatinine, urinalysis with microscopy, C3, C4, anti-dsDNA, anti-SSA, anti-SSB, anti-Smith, lupus anticoagulant, anti-cardiolipin, and beta-2-glycoprotein 1-antibodies
Initial serology screening should include the ANA measured by the immunofluorescence method. If the patient is being evaluated in the outpatient setting, it is prudent to wait for the results of the ANA test prior to ordering additional serologies. However, if the patient is acutely ill or meets criteria for SLE, additional initial serologies can include anti-dsDNA, anti-Smith, anti-SSA and -SSB antibodies, and C3 and C4 levels. If there is a history of thrombocytopenia, miscarriages, or thrombotic events, evaluation for the antiphospholipid syndrome with dilute Russell viper venom time (DRVVT), anticardiolipin antibodies, and anti-β2-glycoprotein-1 antibodies should be performed.
Principles of Management
The aim of treating CLE is to prevent progression of existing skin lesions and formation of new ones, with aggressive treatment warranted to prevent disfigurement in scarring subtypes. Management strategies include patient education and behavior modification, topical, and systemic therapies, often in combination (Table 3.5). Many systemic therapies have a delayed onset of action in CLE, and initial treatment with topical and intralesional corticosteroids can be important.
Prevention/Patient Education
Sun Protection
UVA and UVB exposure have been shown to induce CLE lesions [85], non-specific cutaneous eruptions, and even systemic symptoms in patients with and without SLE. A vehicle-controlled, randomized, double-blind trial demonstrated that the use of a broad-spectrum sunscreen by those with photosensitive CLE can prevent development of skin lesions [86]. Thus, minimizing UV exposure is a critical component of therapy, even in patients who do not report photosensitivity or worsening of skin lesions following sun exposure.
Strict sunscreen use is recommended. It should be applied 20–30 minutes prior to expected exposure in sufficient amount (approximately one ounce is required to cover the body of most adults), and with reapplication every 2 hours if sun exposure continues. The sunscreen should be labeled “broad spectrum” (indicating that it provides protection against both UVA and UVB), with a sun protection factor (SPF) of at least 50. Sun-protective clothing is important, including tight-weave fabrics, dark garments and wide-brimmed hats. Sunscreen-impregnated clothing can also be helpful.
UVB-specific protection techniques include avoiding extended outdoor exposure during peak UVB times (10 A.M. to 2 P.M.). Consideration may also be given to using fluorescent light bulbs with the lowest irradiance and/or applying UV-blocking shields to indoor lighting [87, 88], as indoor fluorescent lighting can emit UVB and exacerbate CLE.
UVA is harder to block, as it varies minimally by time of day or by season and can penetrate window glass. However, UV-blocking films can be applied to glass windows in cars, offices, and homes. Sunscreens providing UVA protection (such as those containing titanium dioxide, zinc oxide, mexoryl XL, and others) can also be helpful.
Evaluation for vitamin D deficiency is important in sun-avoiding patients, as sunlight is required for vitamin synthesis. Daily supplementation with at least 400 IU of vitamin D3 and periodic monitoring of 25-hydroxyvitamin D levels for deficiency are recommended.
Smoking Cessation
Multiple studies have reported that cigarette smokers with CLE have more severe disease than nonsmokers and that a subset of these patients are more refractory to therapies [3, 4, 89, 90]. Based on this, and the increased risk of cardiovascular disease in SLE, patients should therefore be counseled on smoking cessation [3].
Camouflage
Makeup products such as Dermablend, Covermark, or Bare Minerals can be helpful in improving cosmesis for patients with active CLE disease or residual pigmentary alterations.
Local Therapy
Topical corticosteroids, calcineurin inhibitors, and intralesional corticosteroids are first-line therapies for CLE.
Topical Steroids
High-potency topical corticosteroids have long been the mainstay for treatment of CLE, including for scarring subtypes of CLE on the face. However, there is only one randomized, controlled trial examining the efficacy of high-potency topical steroids in CLE. In a 12-week cross-over study of 78 DLE patients, excellent improvement or resolution of lesions was seen in 27% of patients treated with fluocinonide 0.05% cream at 6 weeks, as compared to 10% of patients treated with hydrocortisone 1% cream [91, 92].
Calcineurin Inhibitors
Topical calcineurin inhibitors are a good alternative for patients with persistent facial lesions despite therapy with topical corticosteroids, in whom the risk of continued potent topical steroid use outweighs the benefit. In a randomized, vehicle-controlled, multicenter trial, 20 patients with CLE treated with tacrolimus 0.1% ointment showed significantly more improvement after 28 and 56 days as compared to those treated with vehicle, though the difference was not significant at 84 days [93]. A double-blind, randomized, controlled trial compared tacrolimus 0.1% ointment to clobetasol propionate 0.05% ointment in 20 patients, using a split-face design. The two ointments showed equal efficacy, however, 61% of patients developed telangiectasias on the clobetasol side, as early as 3 weeks into therapy [94]. Although calcineurin inhibitors do not carry a risk of skin thinning, telangiectasias, cataracts, or glaucoma, patients may develop lentigines localized to the treatment site. In addition, calcineurin inhibitors carry a black box warning for a heightened risk of malignancy, specifically lymphoma, although there is no evidence to suggest a causal relationship [95].
Intralesional corticosteroids
Intralesional triamcinolone, given in concentrations ranging from 2.5–20 mg/cc depending on the thickness and location of the lesion being treated, can be effective in DLE. The injections may be repeated monthly while the lesions are active.
Laser
Although typically not used as first line therapy, pulsed-dye laser (PDL) has been demonstrated in several case reports and series to be a safe and effective treatment for DLE. An open prospective study of 12 DLE patients treated with PDL demonstrated efficacy after 6 weeks of treatment [96].
Systemic Therapy
Presently, there are no FDA-approved medications approved specifically for the treatment of CLE. Systemic therapies are indicated for CLE when disease is widespread, when a scarring subtype such as DLE or LE panniculitis affects a cosmetically disfiguring location, or in cases that are refractory to topical or intralesional therapy.
Many of the same systemic medications used to treat SLE are frequently employed in CLE. Exceptions include systemic corticosteroids, which are frequently used in SLE but reserved for severe, rapid-onset CLE; leflutamide, which is used in SLE but not CLE, and thalidomide, which is used for CLE but typically not for SLE.
Antimalarials
Oral antimalarials are considered first-line systemic therapy for all CLE subtypes that are not completely responsive to topical modalities. In addition, it has been suggested that the initiation of hydroxychloroquine (HCQ) for CLE may prevent development of SLE. Specifically, James et al. reported the treatment of HCQ resulted in a significant delay in the time from onset of the first symptom to SLE classification [97].
Antimalarials are immunomodulatory drugs with a mechanism of action that is incompletely understood but thought to involve inhibition of TLR signaling and subsequent inhibition of pro-inflammatory cytokines, as well as antithrombotic properties. It takes 2–3 months to obtain steady-state concentrations, which may account for the slow onset of therapeutic benefit. Because antimalarials can take up to 3–6 months to reach maximum efficacy, bridging with topical and intralesional therapy is important in CLE.
The three antimalarials currently used include HCQ (200–400 mg/day, ≤6.5 mg/kg/day), chloroquine (125–250 mg/day, ≤3.5–4 mg/kg/day), and quinacrine (100 mg/day). Quinacrine is currently only available at compounding companies and may not be covered by insurance.
In practice, HCQ is the antimalarial of choice due to the lower risk for retinopathy as compared to chloroquine (CQ). If there is no response or an incomplete response to HCQ therapy after 2 months, quinacrine may be added. Chang et al. demonstrated a 67% improvement rate of cutaneous disease with the addition of quinacrine to HCQ in patients who had previously failed HCQ monotherapy [98]. Interestingly, Frances et al. recently reported an association between complete remission and higher blood concentrations of HCQ, suggesting that it may be useful to consider checking serum HCQ concentrations in patients with refractory CLE [99]. Finally, if no response is seen, switching from HCQ to CQ can be therapeutically beneficial. Due to weight-based CQ dosing recommendations, patients may be advised not to take the medication on a certain number of days per week.
Antimalarials most commonly cause ocular and cutaneous side effects, most of which are reversible. All ocular side effects are more common with CQ than HCQ, and combined CQ and HCQ use is contraindicated because of additive eye toxicity. Quinacrine does not appear to cause eye toxicity. Corneal drug deposition may cause reversible ocular side effects that are not a contraindication to continued antimalarial therapy, including halos, blurred vision, photophobia, and reduction in accommodation.
True, irreversible retinopathy is uncommon with HCQ and preventable with screening. Premaculopathy (retinal pigment deposition resulting in paracentral and pericentral scotoma, usually without vision change), is reversible with cessation of antimalarials. However, continued administration can result in true retinopathy (“bull’s eye” pigment deposition, central scotoma, and visual acuity changes). In a 10-year retrospective study, eye toxicity was shown to be quite rare below 6.5 mg/kg ideal body weight per day of HCQ [100]. Recent CQ/HCQ retinopathy screening guidelines published by the American Academy of Ophthalmology suggest that the risk for retinopathy for patients treated with HCQ at 400 mg/day and CQ at 250 mg/day in the first 5 years of therapy is negligible; at 5 years, the risk increases to 1% [101]. However, the authors’ current practice is to monitor yearly with HCQ and at least twice a year with CQ. There has been a movement in the ophthalmology community to decrease HCQ dosing to 5 mg/kg/day. Our observation however is that many cutaneous lupus patients require higher dosing to control skin disease. When necessary, dosing options may be reviewed collaboratively with a retina specialist.
Cutaneous side effects of antimalarials include reversible blue-grey hyperpigmentation (10–30% of patients), progressive bleaching of skin or hair roots (10% of CQ), and yellow discoloration (quinacrine), among others.
Other side effects of antimalarials include gastrointestinal effects (CQ > HCQ, up to 10% intolerable), infrequent CNS effects (restlessness, headache, seizures, toxic psychosis), and rare hematologic effects (aplastic anemia caused by quinacrine and agranulocytosis caused by CQ) [102, 103]. There are also rare reports of ototoxicity, neuromyotoxicity, cardiomyopathy, and rhabdomyolysis [104, 105].
It is well known that HCQ has a positive effect on glucose and lipid levels [106,107,108,109]. Thus, CLE patients treated with HCQ benefit from decreased disease activity but possibly also improved glycemic and lipid control.
Antimalarial-resistant Disease
In patients with CLE who fail antimalarial therapy, a wide range of therapeutic options are available. Medication choice should be guided by comorbidities and the presence or absence of systemic involvement. Unfortunately, patients who fail antimalarial combination therapy are often also refractory to other systemic treatments. Antimalarials are typically continued while additional agents are added, specifically either immunosuppressants or immunomodulators.
Immunosuppressant Agents
Only about 50% of patient with CLE refractory to antimalarials respond to immunosuppressant therapy [58, 110]. Agents utilized include systemic corticosteroids, methotrexate, mycophenolate mofetil (MMF), and azathioprine (AZA). Choice of systemic agent for CLE should include consideration of other systemic manifestations of disease. For example, patients with concurrent arthritis may benefit from methotrexate, while presence of some types of nephritis may improve with MMF or AZA. Collaborative decision making for patients with active integumentary and systemic disease is necessary. It should be noted that all immunosuppressant medications confer increased risk of malignancy, particularly lymphoproliferative and skin cancers, in the group of patients with systemic disease.
Methotrexate
In patients who fail antimalarials, methotrexate at doses of 7.5–25 mg orally or subcutaneously once weekly has been noted to be effective in in retrospective studies and case reports [111,112,113,114,115]. A retrospective analysis of 43 treatment-refractory CLE patients who were started on oral or subcutaneous methotrexate found improvement in 98% of cases. Seven out of 43 patients developed severe side effects necessitating discontinuation of therapy [114].
Potential side effects of methotrexate include gastrointestinal toxicity, bone marrow suppression, supratherapeutic dosing in the setting of renal insufficiency, hepatotoxicity, pulmonary interstitial pneumonitis or fibrosis, and phototoxicity [111]. Co-administration of folic acid 1–5 mg daily, as well as rigorous evaluation for drug interactions prior to prescription, can be helpful in preventing bone marrow suppression. Folic acid is helpful for treating oral ulcerations and preventing or treating gastrointestinal upset. Switching to subcutaneous administration can also be helpful to prevent or ameliorate GI upset, and to avoid absorptive limitations at higher doses. Dividing the weekly methotrexate dose and administering 12 hours apart may increase bioavailability and therefore efficacy. Importantly, methotrexate is teratogenic (pregnancy class X), and pregnancy should be prevented during treatment and for 3 months after discontinuation.
Mycophenolate Mofetil (MMF) or Mycophenolate Sodium
Mycophenolate mofetil (MMF, 1–3 g/day) and mycophenolate sodium (720–2160 mg/day) have been shown to be effective in treating SCLE, DLE, and chilblain lupus in multiple case reports and small studies [116,117,118,119,120,121,122]. In some of these cases, patients were also being treated with HCQ. In an open pilot study, 10 patients with SCLE resistant to antimalarials and topical steroids achieved statistically significant reductions in the Cutaneous Lupus Erythematosus Disease Area and Severity Index (CLASI) (from 10.8 ± 6 to 2.9 ± 2.6) following 3-month treatment with MMF (1440 mg/day) [119]. A large multicenter trial of 370 patients compared MMF to cyclophosphamide for treatment of the non-renal aspects of lupus, including skin lesions. At 24 weeks, mucocutaneous LE had improved to mild or non-detectable disease in 84% of patients on MMF vs. 93% of patients on cyclophosphamide [123]. One small study demonstrated failure to MMF in 5 of 7 patients [124].
Gastrointestinal toxicity is common with MMF and can occur in up to 50% of patients. This can be prevented by taking the medication on a full stomach or changing to enteric-coated mycophenolate sodium. Hematologic abnormalities due to bone marrow suppression can occur in 2–11% of patients, including agranulocytosis, neutropenia, anemia, and thrombocytopenia.
The malignancy risk conferred by MMF is controversial. In the transplant population, <1% of patients treated with 2–3 g daily of MMF in combined immunosuppressive regimens develop lymphoma or lymphoproliferative disorders. In the dermatologic literature, there have been case reports of lymphoma, solid tumors, and Kaposi sarcoma developing in patients treated with MMF. The literature also includes conflicting data regarding the risk of non-melanoma skin cancer, with some studies reporting an increased risk of basal cell carcinomas in MMF, and some showing no association. MMF is teratogenic (pregnancy class D), and measures to prevent pregnancy are needed during treatment and 6 weeks following discontinuation of medication.
Azathioprine
Azathioprine (AZA) is used more commonly to treat SLE than CLE [125]. However, several small case series from the 1980s demonstrated successful treatment of DLE with AZA, dosed up to 2–2.5 mg/kg/day [126,127,128]. As with MMF, GI intolerance is the most common adverse effect of AZA, though dividing the dose to three times daily and taking the medication with meals can help.
AZA may carry an increased risk of lymphoproliferative malignancies and cutaneous SCC. However, one retrospective study (n = 358) comparing the incidence of lymphoma and other malignancies in patients with SLE treated with AZA versus those who had not received the drug found no significant difference between the two groups [129].
Other side effects of AZA include bone marrow suppression, with excess risk found in patients (up to 10%) who are deficient in thiopurine methyltransferase (TPMT); screening is important if planning to start at higher doses. Rarely, hepatitis (<1%) or a systemic hypersensitivity reaction such as drug reaction with systemic symptoms/drug-induced hypersensitivity syndrome (DRESS/DIHS) can occur. AZA is pregnancy category D, although some authors have suggested it is relatively safe in pregnancy.
Systemic Corticosteroids
Systemic corticosteroids are generally avoided as therapy for CLE due to the well-known side effects with chronic use; LE patients are at increased risk for developing avascular necrosis (AVN) at baseline. Systemic corticosteroids may, however, be beneficial for short courses in patients with severe or disfiguring CLE, when quick onset of action is needed. In such instances, prednisone may be initiated at 0.5–1 mg/kg/day and tapered over 2–4 weeks.
Important adverse effects of systemic corticosteroids include AVN and osteoporosis [130], particularly with long-term use. To date, there are no specific guidelines to assess and manage osteoporosis in lupus patients. In 2010, the American College of Rheumatology published recommendations for the prevention and treatment of glucocorticoid-induced osteoporosis [131], which were updated in 2017, providing a risk stratification scheme to determine which patients would benefit from bone mineral density testing and bisphosphonate therapy, among other interventions. It is important to encourage bone health and minimize fracture risk by encouraging patients to take vitamin D and calcium supplementation, engage in weight-bearing activities, stop smoking, and reduce alcohol intake. Fall risk assessment is also important.
AVN results from compromise of the bone vasculature with resultant death of the bone marrow and trabecular bone. Several pathologic processes may cause ischemia to bone, and in some instances the cause is not easily identifiable. The most common clinical presentation is pain, most commonly in the anterolateral femoral head. Plain film radiography may be helpful in the initial assessment of AVN, but the plain radiograph can remain normal for months after symptoms appear. Magnetic resonance imaging (MRI) is the most sensitive imaging modality to assess for AVN.
Immunomodulators
Dapsone
Dapsone (25–150 mg/day) has been employed in the treatment of BSLE, lupus panniculitis, SCLE, and DLE. The combined results of three case series including 55 CLE patients treated with dapsone showed a 55% improvement rate [132, 133]. Despite this report, dapsone is widely viewed as less effective in the treatment of CLE, with the exception of BSLE. Because dapsone targets neutrophils, it has been found to be exceptionally useful in treating this subtype, with dramatic response to doses as low as 50 mg/day [73].
Anticipated, dose-related side effects of dapsone include hemolytic anemia (presenting with fatigue and dark urine) and methemoglobinemia (shortness of breath, fatigue, headache, and blue lips). Although hemolytic anemia is expected, with an anticipated average decrease in hemoglobin by 2 g/dL, screening for G6PD deficiency is necessary to avoid severe hemolysis. Patients with diabetes should be counseled that dapsone-induced hemolysis can result in falsely low levels of HgbA1c. In addition, pulse oximetry readings can be spuriously low in all patients and should not necessarily be interpreted as a sign of respiratory decompensation in the absence of other findings. Methemoglobinemia can potentially be prevented with vitamin E (800 IU daily) or cimetidine (400 mg 3 times a day) [134, 135].
Idiosyncratic adverse effects of dapsone therapy include agranulocytosis (rare, presenting with fevers and signs of infection within the first 12 weeks of therapy), reversible peripheral neuropathy (predominantly motor, +/− sensory), GI upset, dapsone hypersensitivity syndrome (typically 3–6 weeks into therapy, and equivalent to DRESS/DIHS).
Thalidomide
Thalidomide (50–100 mg/day) has been shown to be highly efficacious in the treatment of DLE, SCLE, and tumid lupus [136,137,138], with studies showing 60–80% of patients achieving complete response. CLE typically responds quickly, beginning at 2–4 weeks, and doses can often be tapered after improvement [136, 139]. Hence, thalidomide works well as a rescue medication or for maintenance at low or intermittent dosing (e.g. 25 mg every 2–3 days) in an effort to minimize toxicity.
Common adverse effects of thalidomide include drowsiness, constipation, peripheral edema, and irregular menses. Use of thalidomide is limited by its more serious adverse effects, which include teratogenicity, idiosyncratic peripheral sensory neuropathy, venous thrombosis, and rare leukopenia. Only clinicians registered with the Risk Evaluation and Management Strategy (REMS) program can prescribe thalidomide. Pregnancy prevention is critical, and pregnancy tests are monitored via REMS. CBC should be done at baseline and after starting the drug. Neurologic examinations with sensory nerve action potential (SNAP) amplitudes should be done every 6 months.
Lenalidomide
Lenalidomide is a thalidomide derivative with a better side effect profile. It is a potential alternative to thalidomide in CLE, given promising results in a case series and two small open-label trials [46, 136, 140]. Like thalidomide, lenalidomide is helpful in treating refractory CLE; however, it carries less risk of sedation, constipation, peripheral neuropathy, and thrombophilic effects. Monitoring should include CBC (to evaluate for thrombocytopenia, neutropenia, leukopenia, or anemia), thyroid function tests (TFTs, as patients can develop hypothyroidism), and nerve conduction tests (for peripheral neuropathy). Of note, studies in transplant patients on lenalidamide have noted an increased risk for NMSC, and thus patients should have full body skin exams every 6–12 months.
Oral Retinoids
Oral retinoids are another option for CLE patients who fail antimalarial therapy. Multiple case reports support the efficacy of isotretinoin in this condition, while a randomized, controlled trial found acitretin to be effective in 50% of CLE patients [141,142,143]. Systemic retinoids are also strongly linked with teratogenicity, and pregnancy prevention is essential. Patients should also be monitored for leukopenia, pseudotumor cerebri, triglyceridemia, and rare hepatitis. Increased myalgias and muscle breakdown with elevated CPK are noted with isotretinoin in the absence of rhabdomyolysis, and bexarotene can be associated with central hypothyroidism.
Other Therapies
Other agents reported in the literature as treatments for CLE include clofazamine, rituximab, intravenous immunoglobulin (IVIG) and belimumab. Rituximab has recently been shown to have limited efficacy [144]. IVIG tends have a short-lived response in CLE, with mixed efficacy reported [145,146,147]. In general, IVIG can be used as a bridge while waiting for another systemic medication to take effect. Belimumab is a B-cell activating factor inhibitor that is FDA-approved for treatment of SLE; further evaluation for efficacy in treating CLE is warranted [148].
Disease and Comorbidity Assessment (Table 3.6)
Systemic Screening
For patients with CLE only, yearly screening with a CBC with differential, serum albumin, serum creatinine, urinalysis with microscopy, and spot urine for protein/creatinine ratio when significant proteinuria is detected on urinalysis is recommended and is the current practice of the authors. In addition to laboratory evaluation, careful review of systems and physical exam are needed at each visit to evaluate for signs or symptoms such as mucosal ulcers, pleurisy, and joint pain.
Malignancy Risk
The most significant systemic comorbidities seen in CLE affect patients with SLE. However, there is literature to suggest that SCLE in particular is associated with malignancy. There are about 15 reported cases associating SCLE with cancers, including adenocarcinoma of the breast, uterus, esophagus, lung and stomach, Hodgkin’s lymphoma, and hepatocellular carcinoma [57, 149,150,151]. The emergence of new SCLE in an older individual with otherwise negative serologic work-up for systemic or drug-induced lupus should prompt consideration for an underlying malignancy [149]. All patients with CLE should remain up to date with age-appropriate cancer screening.
Pregnancy
Pregnant women with CLE should be checked for ANA, anti-SSA/Ro, anti-SSB/La, and anti-U1-RNP if status is unknown, in order to risk stratify for development of NL in the infant. If maternal positivity of these autoantibodies is found, in utero frequent pulsed Doppler fetal echocardiography starting at 18 weeks gestational age is recommended. If second degree heart block is detected, treatment with fluorinated corticosteroids is recommended.
Infants born to mothers with positive ANA, anti-SSA/Ro, anti-SSB/La, or anti-U1-RNP should be evaluated at birth for hematologic and hepatic involvement (via CBC-D and liver function tests [LFTs]), regardless of whether they have rash. Neonates should also undergo electrocardiogram and possibly echocardiogram, in order to identify first-degree heart block, which may be clinically silent but puts them at risk for cardiac progression.
Neonates with no heart block at birth and a normal electrocardiogram typically do not develop heart block at a later date. It is reasonable to check CBC and LFTs periodically during the first year of life, although there is no universally accepted frequency of screening.
For women who give birth to an infant with NL, the risk for NL in subsequent pregnancies is approximately 25%. Subsequent pregnancies should be considered high risk and monitored closely. In addition, preemptive HCQ can be considered.
Summary
Patients with skin manifestations of lupus erythematosus must be systematically evaluated for SLE, as well as associated comorbidities, such as malignancy. Treatment is guided by the organ systems involved and the severity of the cutaneous disease. All patients should be counseled on sun avoidance, sunscreens, and sun protective clothing; those with vitamin D deficiency should receive replacement therapy.
Steroids and steroid-sparing agents can both be employed as topical therapies. There are few rigorous studies on the efficacy of systemic therapy in CLE, but antimalarials play an important role in the management of many patients with cutaneous lupus and may prevent progression to systemic disease. If HCQ alone is ineffective, combination therapy with HCQ and quinacrine is recommended. For aggressive or unresponsive skin disease, the addition of immunosuppressive agents or thalidomide (or its derivatives), often with oral steroids as bridge treatment, may be required.
Ongoing surveillance for flares or progression to systemic disease is required, but recommendations should be tailored to the severity of the underlying systemic disease. At least yearly systemic monitoring of urinalysis and CBC is recommended for patients with stable skin disease.
Patients with significant systemic disease or medication complications are often best managed through an interdisciplinary approach, with specialists including dermatologists, rheumatologists, and potentially nephrologists and neurologists, depending on the manifestations of the disease. In addition, patients should be monitored for co-existent autoimmune diseases and co-morbidities related to disease and therapies.
References
Jarukitsopa S, Hoganson DD, Crowson CS. Epidemiology of systemic lupus erythematosus and cutaneous lupus in a predominantly white population in the United States. Arthritis Care Res Hoboken; 2015.
Durosaro O, et al. Incidence of cutaneous lupus erythematosus, : a population-based study. Arch Dermatol. 2009;145:249–53.
Piette EW, Foering KP, Chang AY. Impact of smoking in cutaneous lupus erythematosus. Arch Dermatol. 2012;148:317–22.
Moghadam-Kia S, Chilek K, Gaines E. Cross-sectional analysis of a collaborative Web-based database for lupus erythematosus-associated skin lesions: prospective enrollment of 114 patients. Arch Dermatol. 2009;145:255–60.
Gilliam JN, Sontheimer RD. Distinctive cutaneous subsets in the spectrum of lupus erythematosus. J Am Acad Dermatol. 1981;4(4):471–5.
Kuhn A, et al. Clinical manifestations of cutaneous lupus erythematosus. Dermatol Ges. 2007;5:1124–37.
Lipsker D. The need to revisit the nosology of cutaneous lupus erythematosus: the current terminology and morphologic classification of cutaneous LE: difficult, incomplete and not always applicable. Lupus. 2010;19:1047–9.
Schultz HY, et al. From pathogenesis, epidemiology, and genetics to definitions, diagnosis, and treatments of cutaneous lupus erythematosus and dermatomyositis: a report from the 3rd International Conference on Cutaneous Lupus Erythematosus (ICCLE). Dermatology. 2015;135:7–12.
Merola JF, Nyberg F, Furukawa F. Redefining cutaneous lupus erythematosus: a proposed international consensus approach and results of a preliminary questionnaire. Lupus Sci Med. 2015;2:e000085.
Zecevic RD, et al. Skin lesions-an indicator of disease activity in systemic lupus erythematosus? Lupus. 2001;10:364.
Tan EM, Cohen AS, Fries JF. The revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1982;25:1271.
Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1997;40:1725.
Tan EM. Criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1984;25:53.
Albrecht JA, Braverman IM, Callen JP. Dermatology position paper on the revision of the ACR criteria for SLE. Lupus. 2004;13:839.
Petri M, Orbai AM, Alarcon GS. Derivation and validation of systemic lupus international collaborating clinics classification criteria for systemic lupus erythematosus. Arthritis Rheum. 2012;64(8):2677–86.
Ighe A, et al. Application of the systemic lupus interna tional collaborating clinics classification criteria on a Regional Swedish systemic lupus erythematosus register. Arthritis Res Ther. 2015;17:3.
Ines L, Silva C, Galindo M. Classification of systemic lupus erythematosus: systemic lupus international collaborating clinics versus american college of rheumatology criteria. Arthritis Care Res Hoboken; 2015.
Dey-Rao R, Sinha AA. Genome-wide transcriptional profiling of chronic cutaneous lupus erythematosus (CCLE) peripheral blood identifies systemic alterations relevant to the skin manifestation. Genomics. 2015;105:90–100.
Lee LA, et al. Autoantibodies of neonatal lupus erythematosus. Dermatol Antibodydependent cellular cytotoxicity and skin disease. J Invest Dermatol. 1985;85(suppl 165):963–6.
Millard TP, Kondeatis E, Cox A. A candidate gene analysis of three related photosensitivity disorders: cutaneous lupus erythematosus, polymorphic light eruption and actinic prurigo. Br J Dermatol. 2001;145:229.
Fischer GF, et al. Association between chronic cutaneous lupus erythematosus and HLA class II alleles. Hum Immunol. 1994;41:280–4.
Werth VP, et al. Association of a promoter polymorphism of TNFalpha with subacute cutaneous lupus erythematosus and distinct photoregulation of transcription. J Invest Dermatol. 2000;115:726–30.
Osmola A, et al. Genetic background of cutaneous forms of lupus erythematosus: update on current evidence. J Appl Genet. 2004;45:77–86.
Jarvinen TM, et al. Polymorphisms of the ITGAM gene confer higher risk of discoid cutaneous than of systemic lupus erythematosus. PLoS One. 2010;5:14212.
Kim-Howard X, Maiti AK, Anaya JM. ITGAM coding variant (rs1143679) influences the risk of renal disease, discoid rash and immunological manifestations in patients with systemic lupus erythematosus with European ancestry. Ann Rheum Dis. 2010;69:1329–32.
Sanchez E, Nadig A, Richardson BC. Phenotypic associations of genetic susceptibility loci in systemic lupus erythematosus. Ann Rheum Dis. 2011;70:1752–7.
Kunz M, Konig IR, Schillert A. Genome-wide association study identifies new susceptibility loci for cutaneous lupus erythematosus. Exp Dermatol. 2015;24(7):510–5.
Jarvinen TM, Hellquist A, Koskenmies S. Tyrosine kinase 2 and interferon regulatory factor 5 polymorphisms are associated with discoid and subacute cutaneous lupus erythematosus. Exp Dermatol. 2010;19:123–31.
Pickering MC, et al. Ultraviolet-radiation-induced keratinocyte apoptosis in C1q-deficient mice. J Invest Dermatol. 2001;117:52.
Nousari HC, et al. Generalized lupus panniculitis and antiphospholipid syndrome in a patient without complement deficiency. Ped Dermatol. 1999;16:273.
Lee-Kirsch MA, Gong M, Schulz H. Familial chilblain lupus, a monogenic form of cutaneous lupus erythematosus, maps to chromosome 3p. Am J Hum Genet. 2006;79:731.
Sanders CJ, et al. Photosensitivity in patients with lupus erythematosus:a clinical and photobiological study of 100 patients using a prolonged phototest protocol. Br J Dermatol. 2003;149:131–7.
Kuhn A, et al. Photoprovocation in cutaneous lupus erythematosus: study evaluating a standardized protocol. J Invest Dermatol. 2011;131:1622–30.
Rosenbaum M, Billet S, Patel P. Failure of physiologic doses of pure UVB or UVA to induce lesions in photosensitive lupus erythematosus: implications for phototesting and sunblocking strategies. Photodermatol Photoimmunol Photomed. 2006;22:290.
Furukawa F, et al. Binding of antibodies to the extractable nuclear antigens SS-A/Ro and SS-B/La is induced on the surface of human keratinocytes by ultraviolet light (UVL): implications for the pathogenesis of photosensitive cutaneous lupus. J Invest Dermatol. 1990;94:77.
Casciola-Rosen LA, Anhalt G, Rosen A. Autoantigens targets in systemic lupus erythematosus are clustered in two populations of surface structures on apoptotic keratinocytes. J Exp Med. 1994;179:1317.
Kuhn A, Herrmann M, Kleber S. Accumulation of apoptotic cells in the epidermis of patients with cutaneous lupus erythematosus after ultraviolet irradiation. see comment. Arthritis Rheum. 2007;54:939.
Botto M, Walport MJ. C1q, autoimmunity and apoptosis. Immunobiology. 2002;205:395–406.
Kreuter A, et al. Expression of antimicrobial peptides in different subtypes of cutaneous lupus erythematosus. Acad Dermatol. 2011;65:125–33.
Villanueva E, et al. Netting neutrophils induce endothelial damage, infiltrate tissues, and expose immunostimulatory molecules in systemic lupus erythematosus; 2011. p. 538–52.
Popovic K, Ek M, Espinosa A. Increased expression of the novel proinflammatory cytokine high mobility group box chromosomal protein 1 in skin lesions of patients with lupus erthematosus. Arthritis Rheum. 2005;52:3639.
Chang LM, Maheshwari P, Werth S. Identification and molecular analysis of glycosaminoglycans in cutaneous lupus erythematosus and dermatomyositis. J Histochem Cytochem. 2011;59:336–45.
Jabbari A, et al. Dominant Th1 and minimal Th17 skewing in discoid lupus revealed by transcriptomic comparison with psoriasis. J Invest Dermatol. 2014;134:87–95.
Franz B, Fritzsching B, Riehl A. Low number of regulatory T cells in skin lesions of patients with cutaneous lupus erythematosus. Arthritis Rheum. 2007;56:1910–20.
Wong D, et al. Interferon and biologic signatures in dermatomyositis skin: specificity and heterogeneity across diseases. PLoS One. 2012;7:29161.
Braunstein I, et al. The interferon-regulated gene signature is elevated in SCLE and DLE and correlates with CLASI score. Br J Dermatol. 2012;166:971–5.
Wenzel J, et al. The expression pattern of interferon-inducible proteins reflects the characteristic histological distribution of infiltrating immune cells in different cutaneous lupus erythematosus subsets. Br J Dermatol. 2007;157:752–7.
Gambichler T, et al. Cytokine and chemokine ligand expression in cutaneous lupus erythematosus. Eur J Dermatol. 2012;22:319–23.
Zahn S, et al. Evidence for a pathophysiological role of keratinocyte-derived type III interferon (IFNlambda) in cutaneous lupus erythematosus. J Invest Dermatol. 2011;131:133–40.
Meller S, Winterberg F, Gilliet M. Ultraviolet radiation-induced injury, chemokines, and leukocyte recruitment: an amplification cycle triggering cutaneous lupus erythematosus. Arthritis Rheum. 2005;52:1504–16.
Werth VP, Zhang W. Wavelength-specific synergy between ultraviolet radiation and interleukin-1 alpha in the regulation of matrix-related genes: mechanistic role for tumor necrosis factor-alpha. J Invest Dermatol. 1999;113:196–201.
Nabatian AS, et al. TNFα release in peripheral blood mononuclear cells of cutaneous lupus & dermatomyositis patients. Arthritis Res Ther. 2012;4:R1.
Rothfield N, Sontheimer RD, Bernstein M. Lupus erythematosus: systemic and cutaneous manifestations. Clin Dermatol. 2006;24:348–62.
Gronhagen CM, et al. Cutaneous lupus erythematosus and the association with systemic lupus erythematosus: a population-based cohort of 1088 patients in Sweden. Br J Dermatol. 2011;164:1335–41.
Cohen MR, Crosby D. Systemic disease in subacute cutaneous lupus erythematosus: a controlled comparison with systemic lupus erythematosus. J Rheumatol. 1994;21:1665–9.
Sontheimer RD, Thomas JR, Gilliam JN. Subacute cutaneous lupus erythematosus: a cutaneous marker for a distinct lupus erythematosus subset. Arch Dermatol. 1979;115(12):1409–15.
Evans KG, Heymann WR. Paraneoplastic subacute cutaneous lupus erythematosus: an underrecognized entity. Cutis. 2013;91:25–9.
Chang AY, et al. Quality of life differences between responders and nonresponders in the treatment of cutaneous lupus erythematosus. JAMA Dermatol. 2013;149:104–6.
Verma SM, et al. The impact of skin damage due to cutaneous lupus on quality of life. Br J Dermatol. 2014;170:315–21.
Sampaio MC, et al. dos Discoid lupus erythematosus in children-a retrospective study of 34 patients. Pediatr Dermatol. 2008;25:163–7.
Vera-Recabarren MA, et al. Comparative analysis of subacute cutaneous lupus erythematosus and chronic cutaneous lupus erythematosus: clinical and immunological study of 270 patients. Br J Dermatol. 2010;162:91–101.
Parish LC, Kennedy RJ, Hurley J. Palmar lesions in lupus erythematosus. Arch Dermatol. 1967;96:273–6.
Tao J, et al. Squamous cell carcinoma complicating discoid lupus erythematosus in Chinese patients: review of the literature. Acad Dermatol. 2012;66:695.
Prystowsky SD, Gilliam JN. Discoid lupus erythematosus as part of a larger disease spectrum. Correlation of clinical features with laboratory findings in lupus erythematosus. Arch Dermatol. 1975;111:1448–52.
Gysel D, et al. Van de Waard- Childhood discoid lupus erythematosus: report of five new cases and review of the literature. Acad Dermatol Venereol. 2002;16:143–7.
Moises-Alfaro C, et al. Discoid lupus erythematosus in children: clinical, histopathologic, and follow-up features in 27 cases. Pediatr Dermatol. 2003;20:103–7.
Magro CM, et al. Atypical lymphocytic lobular panniculitis: a clonal subcutaneous T-cell dyscrasia. J Cutan Pathol. 2008;35:947–54.
Viguier M, Pinquier L, Cavelier-Balloy B. Clinical and histopathologic features and immunologic variables in patients with severe chilblains. A study of the relationship to lupus erythematosus. Medicine (Baltimore). 2001;80:180–8.
Rivera TL, Izmirly PM, Birnbaum BK. Disease progression in mothers of children enrolled in the Research Registry for Neonatal Lupus. Ann Rheum Dis. 2009;68:828–35.
Buyon JP, et al. Neonatal lupus: basic research and clinical perspectives. Rheum Dis Clin North Am. 2010;31:299–313.
Lee LA, Sokol RJ, Buyon JP. Hepatobiliary disease in neonatal lupus: prevalence and clinical characteristics in cases enrolled in a national registry. Pediatrics. 2002;109:E11.
Buyon JP. Neonatal lupus and autoantibodies reactive with SSA/Ro-SSB/La. Scand J Rheumatol Suppl. 1998;107:23–30.
Hall RP, et al. Bullous eruption of systemic lupus erythematosus. Dramatic response to dapsone therapy. Ann Intern Med. 1982;97:165–70.
Gammon WR, et al. Bullous SLE: a phenotypically distinctive but immunologically heterogeneous bullous disorder. J Invest Dermatol. 1993;100:28S–34.
Ting W, et al. Toxic epidermal necrolysis-like acute cutaneous lupus erythematosus and the spectrum of the acute syndrome of apoptotic pan-epidermolysis (ASAP): a case report, concept review and proposal for new classification of lupus erythematosus vesiculobullous skin lesions. Lupus. 2004;13:941–50.
Torchia D, et al. Erythema multiforme and Stevens-Johnson syndrome/toxic epidermal necrolysis associated with lupus erythematosus. J Am Acad Dermatol. 2012;67:417–21.
Ziemer M, et al. Stevens-Johnson syndrome and toxic epidermal necrolysis in patients with lupus erythematosus: a descriptive study of 17 cases from a national registry and review of the literature. Br J Dermatol. 2012;166:575–600.
Rowell NR, et al. Lupus erythematosus and erythema multiforme-like lesions. A syndrome with characteristic immunological abnormalities. Arch Dermatol. 1963;83:176–80.
Zeitouni NC, et al. Redefining Rowell’s syndrome. Br J Dermatol. 2000;142:343–6.
Alarcon-Segovia D, Cetina JA. Lupus hair. Am J Med Sci. 1974;267:241–2.
Fabre VC, et al. Twenty percent of biopsy specimens from sun-exposed skin of normal young adults demonstrate positive immunofluorescence. Arch Dermatol. 1991;127:1006–11.
David-Bajar KM, Davis BM. Pathology, immunopathology, and immunohistochemistry in cutaneous lupus erythematosus. Lupus. 1997;6:145–57.
Li PH, Wong WH, Lee TL. Relationship between autoantibody clustering and clinical subsets in SLE: cluster and association analyses in Hong Kong Chinese. Rheumatology (Oxford). 2013;52:337–45.
Lee LA, et al. The autoantibody response to Ro/SSA in cutaneous lupus erythematosus. Arch Dermatol. 1994;130:1262.
Kuhn A, et al. Phototesting in lupus erythematosus: a 15-year experience. J Am Acad Dermatol. 2001;45:86–95.
Kuhn A, et al. Photoprotective effects of a broad-spectrum sunscreen in ultraviolet-induced cutaneous lupus erythematosus: a randomized, vehicle-controlled, double-blind study. J Am Acad Dermatol. 2010;64:37–48.
Klein RS, et al. Analysis of compact fluorescent lights for use by patients with photosensitive conditions. Photochem Photobiol. 2009;85(4):1004–10.
Sayre RM, Dowdy JC, Poh-Fitzpatrick M. Dermatological risk of indoor ultraviolet exposure from contemporary lighting sources. Photochem Photobiol. 2004;80:47–51.
Jewell ML, McCauliffe DP. Patients with cutaneous lupus erythematosus who smoke are less responsive to antimalarial treatment. J Am Acad Dermatol. 2000;42:983–7.
Wahie S, et al. Clinical and pharmacogenetic influences on response to hydroxychloroquine in discoid lupus erythematosus: a retrospective cohort study. J Invest Dermatol. 2011;131:1981–6.
Jessop S, Whitelaw DA, Delamere FM. Drugs for discoid lupus erythematosus. Cochrane Database Syst Rev. 2009;5:CD002954.
Roenigk HH, et al. Discoid lupus erythematosus. Diagnostic features and evaluation of topical corticosteroid therapy. Cutis. 1980;25:281–5.
Kuhn A, et al. Efficacy of tacrolimus 0.1 ointment in cutaneous lupus erythematosus a multicenter randomized doubleblind vehiclecontrolled trial. Acad Dermatol. 2011;65:54–64.
Tzung TY, et al. Tacrolimus vs. clobetasol propionate in the treatment of facial cutaneous lupus erythematosus a randomized doubleblind bilateral comparison study. Br J Dermatol. 2007;156:191–2.
Thaci D, Salgo R. Malignancy concerns of topical calcineurin inhibitors for atopic dermatitis: facts and controversies. Clin Dermatol. 2010;28:52–6.
Erceg A, et al. Efficacy and safety of pulsed dye laser treatment for cutaneous discoid lupus erythematosus. J Am Acad Dermatol. A randomized study of the effect of withdrawing hydroxychloroquine sulfate in systemic lupus erythematosus The Canadian Hydroxychloroquine Study Group 3241504. 1991;60:626–32.
James JA, Kim-Howard XR, Bruner BF. Hydroxychloroquine sulfate treatment is associated with later onset of systemic lupus erythematosus. Lupus. 2007;16:401–9.
Chang AY, et al. Response to antimalarial agents in cutaneous lupus erythematosus: a prospective analysis. Arch Dermatol. 2011;147:1261–7.
Frances C, Cosnes A, Duhaut P. Low blood concentration of hydroxychloroquine in patients with refractory cutaneous lupus erythematosus: a French multicenter prospective study. Arch Dermatol. 2012;148:479–84.
Levy GD, et al. Incidence of hydroxychloroquine retinopathy in 1,207 patients in a large multicenter outpatient practice. Arthritis Rheum. 1997;40:1482–6.
Marmor MF, et al. Revised recommendations on screening for chloroquine and hydroxychloroquine retinopathy. Ophthalmology. 2011;118:415–22.
Ward WQ, et al. Toxic psychosis: a complication of antimalarial therapy. J Am Acad Dermatol. 1985;12:863–5.
Schmid I, et al. Marrow transplantation for severe aplastic anemia associated with exposure to quinacrine. Blut. 1990;61:52–4.
Siddiqui AK, et al. Hydroxychloroquine-induced toxic myopathy causing respiratory failure. Chest. 2007;131:588–90.
Creel N, Werth VP. Rhabdomyolysis associated with quinacrine therapy in a patient with chronic cutaneous lupus erythematosus. J Drugs Dermatol. 2005;4:225.
Bili A, et al. Hydroxychloroquine use and decreased risk of diabetes in rheumatoid arthritis patients. J Clin Rheumatol. 2011;17(3):115–20.
Wasko MC, et al. Hydroxychloroquine and risk of diabetes in patients with rheumatoid arthritis. JAMA. 2007;298(2):187–93.
Chen YM, et al. Hydroxychloroquine reduces risk of incident diabetes mellitus in lupus patients in a dose-dependent manner: a population-based cohort study. Rheumatology (Oxford). 2015;54(7):1244–9.
Mercer E, et al. Hydroxychloroquine improves insulin sensitivity in obese non-diabetic individuals. Arthritis Res Ther. 2012;14(3):R135.
Wahie S, Meggitt SJ. Long-term response to hydroxychloroquine in patients with discoid lupus erythematosus. Br J Dermatol. 2013;169:653–9.
Kuhn A, et al. Methotrexate treatment for refractory subacute cutaneous lupus erythematosus. J Am Acad Dermatol. 2002;46:600–3.
Bohm L, Uerlich M, Bauer R. Rapid improvement of subacute cutaneous lupus erythematosus with low-dose methotrexate. Dermatology. 1997;194:307–8.
Malcangi G, et al. Bullous SLE: response to methotrexate and relationship with disease activity. Lupus. 2003;12:63–6.
Wenzel J, et al. Efficacy and safety of methotrexate in recalcitrant cutaneous lupus erythematosus: results of a retrospective study in 43 patients. Br J Dermatol. 2005;153:157–62.
Boehm IB, Boehm GA, Bauer R. Management of cutaneous lupus erythematosus with low-dose methotrexate: indication for modulation of inflammatory mechanisms. Rheumatol Int. 1998;18:59–62.
Cetkovska P, Pizinger K. Coexisting subacute and systemic lupus erythematosus after terbinafine administration: successful treatment with mycophenolate mofetil. Int J Dermatol. 2006;45:320–2.
Hanjani NM, Nousari CH. Mycophenolate mofetil for the treatment of cutaneous lupus erythematosus with smoldering systemic involvement. Arch Dermatol. 2002;138:1616–8.
Boehm I, Bieber T. Chilblain lupus erythematosus Hutchinson: successful treatment with mycophenolate mofetil. Arch Dermatol. 2001;137:235–6.
Kreuter A, et al. Mycophenolate sodium for subacute cutaneous lupus erythematosus resistant to standard therapy. Br J Dermatol. 2007;156:1321–7.
Goyal S, Nousari HC. Treatment of resistant discoid lupus erythematosus of the palms and soles with mycophenolate mofetil. J Am Acad Dermatol. 2001;45:142–4.
Schanz S, et al. Successful treatment of subacute cutaneous lupus erythematosus with mycophenolate mofetil. Br J Dermatol. 2002;147:174–8.
Gammon B, et al. Efficacy of mycophenolate mofetil in antimalarial-resistant cutaneous lupus erythematosus. Acad Dermatol. 2011;65:717–21.
Ginzler EM, et al. Nonrenal disease activity following mycophenolate mofetil or intravenous cyclophosphamide as induction treatment for lupus nephritis: findings in a multicenter, prospective, randomized, open-label, parallel-group clinical trial. Arthritis Rheum. 2010;62:211–21.
Pisoni CN, Obermoser G, Cuadrado MJ. Skin manifestations of systemic lupus erythematosus refractory to multiple treatment modalities: poor results with mycophenolate mofetil. Clin Exp Rheumatol. 2005;23:393–6.
Swaak AJ, et al. Statius Azathioprine in the treatment of systemic lupus erythematosus. A threeyear prospective study. Clin Rheumatol. 1984;3:285–91.
Ashinoff R, et al. Resistant discoid lupus erythematosus of palms and soles: successful treatment with azathioprine. J Am Acad Dermatol. 1988;19:961–5.
Tsokos GC, Caughman SW, Klippel JH. Successful treatment of generalized discoid skin lesions with azathioprine. Its use in a patient with systemic lupus erythematosus. Arch Dermatol. 1985;121:1323–5.
Shehade S. Successful treatment of generalized discoid skin lesions with azathioprine. Arch Dermatol. 1986;122:376–7.
Nero P, Rahman A, Isenberg DA. Does long term treatment with azathioprine predispose to malignancy and death in patients with systemic lupus erythematosus? Ann Rheum Dis. 2004;63:325–6.
Lakshminarayanan S, et al. Factors associated with low bone mineral density in female patients with systemic lupus erythematosus. J Rheumatol. 2001;28(1):102–8.
Cohen-Cline H, et al. Use of interactive voice response to improve colorectal cancer screening. Med Care. 2014;52(6):496–9.
Coburn PR, Shuster S. Dapsone and discoid lupus erythematosus. Br J Dermatol. 1982;106(1):105–6.
Lindskov R, Reymann F. Dapsone in the treatment of cutaneous lupus erythematosus. Dermatologica. 1986;172(4):214–7.
Prussick R, et al. The protective effect of vitamin E on the hemolysis associated with dapsone treatment in patients with dermatitis herpetiformis. Arch Dermatol. 1992;128(2):210–3.
Coleman MD, et al. The use of cimetidine to reduce dapsone-dependent methaemoglobinaemia in dermatitis herpetiformis patients. Br J Clin Pharmacol. 1992;34(3):244–9.
Cortés-Hernández J, et al. Thalidomide in the treatment of refractory cutaneous lupus erythematosus: prognostic factors of clinical outcome. Br J Dermatol. 2012;166(3):616–23.
Stevens RJ, et al. Thalidomide in the treatment of the cutaneous manifestations of lupus erythematosus: experience in sixteen consecutive patients. Br J Rheumatol. 1997;36(3):353–9.
Burrows NP, et al. Lupus erythematosus profundus with partial C4 deficiency responding to thalidomide. Br J Dermatol. 1991;125:62–7.
Pelle MT, Werth VP. Thalidomide in cutaneous lupus erythematosus. Am J Clin Dermatol. 2003;4(6):379–87.
Shah AA, Wigley FM, Hummers LK. Telangiectases in scleroderma: a potential clinical marker of pulmonary arterial hypertension. J Rheumatol. 2010;37(1):98–104.
Shornick JK, Formica N, Parke AL. Isotretinoin for refractory lupus erythematosus. J Am Acad Dermatol. 1991;24(1):49–52.
Ruzicka T, et al. Treatment of cutaneous lupus erythematosus with acitretin and hydroxychloroquine. Br J Dermatol. 1992;127(5):513–8.
Furner BB. Subacute cutaneous lupus erythematosus response to isotretinoin. Int J Dermatol. 1990;29(8):587–90.
Man A, et al. Changes in forced vital capacity over time in systemic sclerosis: application of group-based trajectory modelling. Rheumatology (Oxford). 2015;54(8):1464–71.
Ky C, et al. Efficacy of intravenous immunoglobulin monotherapy in patients with cutaneous lupus erythematosus: results of proof-of-concept study. Dermatol Reports. 2015;7(1):5804.
Strauss RM, et al. Good response of linear scleroderma in a child to ciclosporin. Br J Dermatol. 2004;150(4):790–2.
De Pità O, et al. Intravenous immunoglobulin therapy is not able to efficiently control cutaneous manifestations in patients with lupus erythematosus. Lupus. 1997;6(4):415–7.
Manzi S, et al. Effects of belimumab, a B lymphocyte stimulator-specific inhibitor, on disease activity across multiple organ domains in patients with systemic lupus erythematosus: combined results from two phase III trials. Ann Rheum Dis. 2012;71(11):1833–8.
Chaudhry SI, Murphy LA, White IR. Subacute cutaneous lupus erythematosus: a paraneoplastic dermatosis? Clin Exp Dermatol. 2005;30(6):655–8.
Jasim ZF, Walsh MY, Armstrong DK. Subacute lupus erythematosus-like rash associated with oesophageal adenocarcinoma in situ. Clin Exp Dermatol. 2007;32(4):443–5.
Shaheen B, Milne G, Shaffrali F. Subacute cutaneous lupus erythematosus associated with breast carcinoma. Clin Exp Dermatol. 2009;34(7):e480–1.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2022 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Pappas-Taffer, L., Gonzalez-Rivera, T.C., Werth, V.P. (2022). Cutaneous Lupus. In: Garg, A., Merola, J.F., Fitzpatrick, L. (eds) Interdisciplinary Approaches to Overlap Disorders in Dermatology & Rheumatology. Springer, Cham. https://doi.org/10.1007/978-3-319-18446-3_3
Download citation
DOI: https://doi.org/10.1007/978-3-319-18446-3_3
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-18445-6
Online ISBN: 978-3-319-18446-3
eBook Packages: MedicineMedicine (R0)