
Abstract
Autism spectrum disorders (ASD) are a heterogeneous and highly prevalent group of neurodevelopmental disorders. Genome analyses have revealed a large amount of candidate risk variants in ASD patients, of which most are rare and private. Currently available tools and techniques have not been sufficient to pinpoint genuine causative or risk variants for ASD among the multitude of variants discovered in each genome. Moreover, targeted drugs to ameliorate specific symptoms of ASD, such as language impairment, difficulties in social interaction, and repetitive behavior, are still unavailable. In vitro cellular and molecular analyses of stem cells to study this group of disorders is bringing new insights into this field. Induced pluripotent stem cells (iPSCs) have been successfully generated and used to model 6 syndromic, monogenic forms of ASD. The results have been very promising, confirming previous data obtained from studies using mouse models and human brain tissues. Disease modeling with stem cells from nonsyndromic ASD patients, albeit in its early stages, has revealed alterations in expression of cytoskeletal genes, compromised mTOR signaling, and reduced neuronal dendritic arborization in these patients. Finally, investigation of ASD iPSC-derived neurons has suggested IGF-1 as a candidate drug to ameliorate ASD symptoms in cases with different causative mutational mechanisms. Owing to the use of stem cells, we currently witness an amazing period of discoveries that have revolutionized our knowledge of ASD, with great expectation to deliver a better understanding of ASD pathophysiology.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others
Keywords
Introduction
Autism spectrum disorders (ASD) are a heterogeneous group of neurodevelopmental disorders characterized by severe developmental defects in social response and communication accompanied by inappropriate repetitive behavior [1]. ASD has an overall prevalence of ~1 % [2]. There is a strong male bias, with a ratio of about 4 males to 1 female, particularly among those with milder ASD forms [3]. ASD can be a clinical manifestation of several well-characterized monogenic disorders. The ASD penetrance in these disorders varies, and may reach values of ~60 % in Fragile X syndrome (FXS), tuberous sclerosis, and Timothy syndrome [4–6]. ASD is also a relevant phenotype in Rett, Rett-like or CDKL5-associated syndrome, Angelman and Phelan–McDermid syndromes (PMDS) [7–9]. The molecular genetic mutational mechanism is very well characterized in all of these conditions. For didactic purposes, we will refer to the group of monogenic disorders frequently associated with ASD as syndromic ASD forms, in contrast to ASD or nonsyndromic ASD, in which the major clinical manifestation is autism, associated or not with other clinical features, such as epilepsy and hyperactivity, and in which clinical features are not sufficient to define a syndrome.
A high heritability component in ASD has been estimated, varying from 50 to 90 % [3, 10]. The recurrence risk in families with only one ASD-affected individual is about 10 %, and male siblings of ASD individuals have a ~3-fold increase of the risk of being affected by ASD in comparison to female siblings [3, 10]. Recurrence risk also increases according to the number of ASD-affected individuals in the family, thus reinforcing the genetic basis of ASD. The disease model seems to be very complex, with a high level of heterogeneity. Based on several worldwide genome-wide association studies (GWAS), there is a consensus that common variants, each with a low predictive risk, play a role in ASD etiology; however, the actual contribution of these variants to ASD heritability is still unclear [3, 10, 11]. Genomic studies, in contrast, have revealed that rare CNVs or rare sequencing variants with moderate-to-high penetrance are associated with the etiology of ASD in at least 20 % of the cases [3, 10, 12, 13]. These studies have revealed a remarkable genetic heterogeneity, as less than 20 of the 100 candidate genes present recurrent putative pathogenic variants. Despite the high heterogeneity of ASD, a growing number of evidence has shown that candidate genes belong to convergent pathways, nowadays represented by regulation of transcription (chromatin remodeling genes and transcription factors), regulation of protein abundance (splicing, translation, and ubiquitination genes), synapse (cell adhesion, sodium and calcium channels, N-methyl-d-aspartate (NMDA) receptors, and synaptic scaffold genes), and intracellular signaling factors that regulate cell growth and proliferation (mTOR/PI3K and RAS genes) [3, 12, 13].
The majority of these candidate pathogenic variants have been classified as loss-of-function variants, which occur with a population frequency of about 5 % [14]. Therefore, one of the current challenges in the genomic analysis of ASD is pinpointing causative variants; moreover, it is necessary to define the penetrance associated with each one of them and how many pathogenic variants are sufficient for a complete ASD penetrance per individual. Finally, the functional effects of the identified variants must be demonstrated and be translated to the phenotypic effect. One approach to address the above-mentioned questions is to conduct functional studies in order to verify which of these candidate variants are sufficient to cause morphological and functional changes in neurons.
Stem cells have recently emerged as a promising alternative to conduct functional cellular studies, particularly in disorders where the tissue of interest is of limited or nearly no access. This is exactly the case for ASD and several other neurodevelopmental disorders, in which the ideal tissue of interest to conduct cellular and molecular studies is the developing brain. Another drawback is the impossibility to get an accurate diagnosis for these disorders before 1 year of age. Murine model systems provide genetic homogeneity and allow the study of behavioral phenotypes, but show limited applicability to understand how diseases affect human neocortical regions, and often do not recapitulate the complex human neurodevelopment [15, 16].
Stem cells from human exfoliated deciduous teeth (SHED) are an easily accessible cell type comprised by populations of mesodermal and neuroectodermal origin [17–19]. The identification of cellular pathways associated with diseases can be successfully achieved by studying SHED, particularly when they share the same embryological origin with cells of the main disease-affected tissue, as in the case of ASD [20–23]. However, the use of SHED is restricted by their limited potential to be differentiated into functional neurons. These difficulties have been overcome, thanks to the possibility of reprogramming somatic cells to a pluripotent state by overexpressing specific transcription factors [24, 25]. These cells, known as induced pluripotent stem cells (iPSCs), have opened a new world of possibilities, as they can be differentiated toward multiple cell lines, including neurons; therefore, the generation of disease-specific neurons by reprogramming somatic cells from ASD patients have empowered researchers to functionally characterize genetic alterations and determine how they lead to ASD neuronal phenotypes. In this regard, both glutamatergic and GABAergic neurons as well as astrocytes, which are of interest to study ASD-related phenotypes, can be obtained [26]. Further, cells from different sources, such as SHED and erythroblasts, in addition to fibroblasts, which were the first type of cells used to obtain iPSCs, can be reprogrammed by these transcription factors with similar efficiency [23, 27]. Even though human embryonic stem cells (hESCs) and neural stem cells (hNSCs) can also be employed to model diseases in a dish, they are in practice, more difficult to be obtained, and their use still represents an ethical issue. Also, the use of these cells is hampered by the impossibility to establish the diagnosis in the early developmental period, particularly for nonsyndromic forms of ASD (Fig. 8.1).

Schematic summary of different approaches to ASD modeling using human stem cells. Patient-derived embryonic stem cells (ESCs) and neural stem cells (NSCs) were used to model the syndromic ASD fragile-X syndrome. Although these patient-derived stem cells have the advantage of not requiring any genetic or epigenetic manipulations to be relevant for disease modeling, they are, in practice, difficult to be obtained. Stem cells from human exfoliated deciduous teeth (SHED) represent an accessible source of patient material and have recently been used to model nonsyndromic forms of ASD. Induced pluripotent stem cells (iPSCs) can be generated from adult cells from ASD patients and can be differentiated into the disease-affected cells. At least six monogenic disorders that include ASD features as part of the phenotype have been modeled by the use of iPSC methodology: Rett syndrome, CDKL5-associated ASD, Fragile-X syndrome, Prader–Willi/Angelman syndrome, Timothy syndrome, and Phelan–McDermid syndrome. iPSCs have also been used to model nonsyndromic forms of ASD, such as TRPC6-associated ASD. Finally, control-derived stem cells engineered to reduce (knockdown) a particular gene’s expression have been used to model the neurodevelopmental impact of Neurexin 1 (NRXN1) and Neuroligin 4X (NLGN4X) deletions, associated with nonsyndromic ASD and other neurodevelopmental disorders. Figure modified from Sterneckert et al. (2014)
We are currently at the dawn of human stem cell modeling of ASD and ASD-associated monogenic disorders. Although much effort still needs to be gathered to fully understand the etiology of these disorders, the results obtained so far have been encouraging and exciting, as they have unraveled new biological pathways and provided a causal relationship between pathogenic mutations and morphological and functional neuronal alterations. Importantly, in several situations, such findings recapitulate relevant cellular and/or molecular phenotypes previously reported in murine models or other approaches.
iPSCs: Modeling Monogenic Disorders Featuring ASD
To date, six genetically well-characterized monogenic disorders with ASD features have been modeled with the use of iPSC technology. In the following segment, we provide a short background and summarize the most significant findings and current state of the art for each disorder.
Fragile X Syndrome
FXS is the most commonly inherited form of intellectual disability, which may be accompanied by a characteristic appearance in affected males (large head, long face, prominent forehead and chin, protruding ears), connective tissue defects, and postpubescent macroorchidism [28, 29]. Autistic features may be present in up to 60 % of all cases, depending on the clinical criteria adopted [30–33]. FXS is caused by expansions of CGG trinucleotide repeats in the 5′ untranslated region of the FMR1 (fragile X mental retardation 1) gene in the X chromosome. The expansions lead to hypermethylation of the FMR1 promoter and consequent silencing of FMRP (fragile X mental retardation protein), an RNA-binding protein involved in mRNA localization and protein synthesis during synaptic plasticity [34, 35]. In hESCs derived from FXS-affected blastocyst-stage embryos, FMR1 expression is active and gene silencing takes place upon differentiation [36]. However, this is not observed in iPSCs derived from FXS patients, in which reprogramming adult cells to a pluripotent state does not reset the epigenetic marks associated with FMR1 silencing [37]. These findings illustrate how iPSCs and ESCs, albeit similar in many aspects, still exhibit differences that might be relevant to the disease under investigation and that should be considered when selecting cell types for disease modeling. In the case of FXS, since FMR1 is kept silenced during neuronal differentiation, FXS-derived iPSCs remain a suitable model to study neuronal changes caused by the expansion mutations. Additionally, one advantage of FXS-derived iPSCs is the ability to generate cells from clinically well-characterized patients and the possibility of investigating different-sized mutations present in the same individual (a consequence inherent to the dynamic nature of the expansions; [38, 39]). Such approach has shed light on the genotype–phenotype relationship in FXS, showing defective neurite formation and outgrowth occurring prior to synaptogenesis, during neuronal differentiation [38, 40]. Importantly, a previous study using an Fmr1-knockout mouse model and in vitro neural stem cells (NSCs) from postmortem brain of a fragile-X fetus also described reduced neurite outgrowth and branching and altered neuronal differentiation in FMRP-deficient NSCs [41].
Timothy Syndrome
Timothy syndrome (TS) is a rare, severe neurodevelopmental disorder accompanied by cardiac defects/arrhythmia and facial dysmorphisms, and one of the most penetrant monogenic forms of ASD. This disorder is caused by mutations in CACNA1C (calcium channel, voltage-dependent, L type, alpha 1C subunit), a gene encoding an alpha subunit of a voltage-dependent calcium channel (Cav1.2) [5]. Neurons derived from TS iPSCs showed action potentials and increased intracellular Ca++ concentration indicative of loss of calcium channel inactivation. TS neurons also showed abnormal expression of tyrosine hydroxylase, which could be rescued with the drug roscovitine, a compound that increases the inactivation of L-type calcium channels. Moreover, gene expression studies revealed that iPSC-derived neural progenitor cells (NPCs) and iPSC-derived neurons from TS patients show alterations in expression of genes previously associated with ASD, as well as similarities with gene expression patterns observed in postmortem idiopathic ASD brains [42, 43]. Also in TS-derived iPSCs, Krey et al. [44] observed that depolarization causes dendritic retraction in TS neurons, which occurs independently of the excessive Ca++ influx seen in these cells. In fact, their results suggested that the dendritic phenotype was caused by decreased binding between TS Cav1.2 and the GTPase Gem, leading to ectopic activation of RhoA. These findings directly link Cav1.2 channels to RhoA signaling in the brain and provide new grounds for studying ASD neuronal phenotypes in vitro.
Rett Syndrome
Rett Syndrome (RTT) is a severe progressive neurodevelopmental disorder mainly caused by mutations in the X-linked gene MECP2 (methyl CpG-binding protein 2) [45, 46]. RTT individuals undergo apparently normal development until 6–18 months of age, followed by impaired motor function, stagnation and regression of developmental skills, hypotonia, seizures, and autistic behavior [45, 47]. MeCP2, the encoded protein, is involved in transcriptional regulation by binding to methylated CpG dinucleotides, and recruiting proteins involved in chromatin remodeling [48]. Marchetto et al. [47] were the first group to model RTT in human cells. By generating iPSCs from RTT patient-derived fibroblasts, they found that deficiency of MECP2 in RTT neurons resulted in smaller soma, fewer dendritic spines and synapses, and impairment in calcium signaling and in excitatory synaptic transmission, by comparison to control, unaffected neurons. Most of these neuronal phenotypes, such as reduced soma size and dendritic arborization, have been subsequently corroborated by groups employing different techniques to generate MECP2-deficient neurons from pluripotent stem cells [49, 50]. Li et al. [50] used gene-editing techniques to generate MECP2-deficient hESCs, and showed that mutant neurons exhibit global reduction in translation and protein synthesis, and reduced AKT/mTOR activity. Studies in mouse models have suggested a role for astrocytes in RTT pathogenesis, which has been recently confirmed in human cells. Through differentiation of RTT iPSCs to astrocytes and employment of a series of co-culture experiments, Williams et al. [51] showed that MECP2-mutated astrocytes lead to reduction in neurite length and in the number of terminal ends of wild-type neurons, and this non-cell-autonomous influence was partially mediated by factors secreted by mutant astrocytes.
Modeling RTT cells in a dish has shown that neuronal disease-related phenotypes can be rescued in vitro. Treatment with insulin-like growth factor-1 (IGF-1) or gentamicin has been shown to improve synaptic density in RTT neurons [47], and addition of IGF-1 or BDNF (brain-derived neurotrophic factor) improved protein synthesis through activation of AKT/mTOR pathway. Importantly, these findings provide proof-of-principle evidence for the application of ASD iPSC-derived neurons in drug discovery.
CDKL5-Associated Syndrome
Dominant-negative mutations in CDKL5 (cyclin-dependent kinase-like 5), also located in the X chromosome, are responsible for a RTT-like phenotype [52, 53], herein referred to as CDKL5-associated syndrome. Patients mainly exhibit early-onset intractable seizures before 6 months of age, severe developmental delay, and autistic features [52–55]. CDKL5 encodes a serine/threonine kinase whose role in brain development is not fully understood. Cdkl5 silencing in a mouse model has shown the importance of this gene for dendritic spine morphogenesis and maintenance of synaptic contact, which occurs via interaction between the postsynaptic proteins NGL-1 and PSD-95, stabilized through phosphorylation of NGL-1 by CDKL5 [56]. The use of patient-derived iPSCs harboring loss-of-function mutations in CDKL5 further confirmed those findings, as patients’ iPSC-derived neurons exhibited a significantly reduced number of synaptic contacts and lacked presynaptic terminals [56].
Angelman Syndrome and Prader–Willi Syndrome
Angelman syndrome (AS) and Prader–Willi syndrome (PWS) were the first imprinting disorders described in humans, in which alterations in the chromosomal region 15q11-q13 lead to different phenotypes depending on which chromosome (paternal or maternal) is affected [57]. AS is characterized by significant intellectual disability, absent speech, frequent seizures, motor impairment, and a typical happy demeanor [58]. PWS is characterized by small stature, neonatal hypotonia, hypogonadism, mild-to-moderate intellectual disability, and compulsive hyperphagia [59]. AS is caused by loss of function of the maternally inherited allele of UBE3A (ubiquitin-protein ligase E3A), which undergoes tissue-specific genomic imprinting with silencing of the paternally inherited allele in brain tissues [60, 61]. When the paternal chromosome is deleted in the same chromosome region, individuals develop PWS due to loss of a cluster of several species of small nucleolar RNAs [62]. Although autistic features are reported in AS but not in PWS, the 15q11-q13 region has been systematically associated with ASD [63]. Chamberlain et al. [64] were the first to show that iPSCs derived from AS and PWS fibroblasts maintain the genomic imprinting at 15q11-q13. Moreover, they confirm that expression of the paternally inherited UBE3A is repressed upon neuronal differentiation in AS cells, recapitulating the main epigenetic characteristics of AS in vitro. Additionally, in neurons differentiated from PWS-derived iPSCs, Cruvinel et al. [65] showed that the zinc-finger protein ZNF274, in association with the histone methyltransferase SETDB1, might protect against methylation of the small nucleolar RNA cluster in the PWS region. Together these findings suggest that iPSC modeling of alterations at 15q11-q13 is a promising strategy to better understand PWS, AS and ASD.
Phelan–McDermid Syndrome
Another syndrome frequently associated with ASD is Phelan-McDermid syndrome (PMDS). This syndrome is caused by heterozygous deletions of variable sizes in chromosome 22 (region 22q13.3). Besides ASD, PMDS patients may exhibit hypotonia, normal to accelerated growth, and minor dysmorphic features [66]. Studies attempting to establish a critical region for the syndrome, in combination with analysis of rare ASD-related mutations and functional studies have appointed SHANK3 to be the most likely candidate responsible for the neurological phenotype in PMDS [7, 67]. Produced iPSC-derived neurons from PMDS patients and observed a reduced amplitude and frequency of spontaneous excitatory synaptic events. Such phenotype was caused by impaired AMPA- and NMDA-mediated transmission. Overexpression of SHANK3 rescued the electrophysiological alterations found, showing that this gene significantly contributes to the neuronal alterations in PMDS. By treating the PMDS neurons with IGF-1, the authors were also able to restore the excitatory synaptic defects. Intriguingly, treatment with IGF-1 decreased the expression of SHANK3 in control and PMDS neurons. They found that IGF-1 actually increases the number of synapses that lack SHANK3 but contains PSD-95, which have faster deactivation of excitatory currents, a kinetics that resemble that of neurons appearing later in development.
Stem Cells to Model Nonsyndromic ASD
Lymphocytes and fibroblasts have been used in several studies in order to dissect the cellular pathways altered in nonsyndromic forms of ASD [68, 69]. However, even though acquiring these cells is relatively simple and with minor ethical implications, they present limitations to study neurodevelopmental pathways, as they are mature, post-natal cell types, and possess non-neural embryological origins.
SHED are an alternative and interesting source of patient-derived cells to be studied, as they can be noninvasively isolated, they show the same early embryonic origin as neurons, and they express neural progenitor markers [70]. Therefore, SHED may bear genetic regulatory networks that resemble those found in neurons. As detailed below, in three recent studies, we show the applicability of SHED in dissecting the genetic regulatory circuitry in nonsyndromic ASD.
Griesi-Oliveira et al. [22] assessed the gene expression profile of SHED from seven nonsyndromic ASD individuals, with no defined genetic mechanism. By comparing cases and controls, the authors identified 683 differentially expressed genes (DEGs), of which a significant number is expressed in brain and is involved in mechanisms and molecular pathways previously associated with ASD, such as cytoskeleton regulation, axonal guidance, protein synthesis, and cellular adhesion. Among the identified DEGs, one of the upregulated genes was CHD8, which has been found to be mutated in about 0.3 % of ASD cases in more than one study [12, 71–74]. Interestingly, CHD8 is a co-regulator of androgen-responsive transcription [75] and androgen receptor, as well as a significant number of genes regulated by this receptor presented overexpression in the studied set of patients. The authors suggested that this might be a possible mechanism through which CHD8 can contribute to ASD, especially considering the skewed male-to-female prevalence in such disorders.
Suzuki et al. [76] used SHED to investigate the role of mTOR (mammalian target of rapamycin) signaling pathway in nonsyndromic ASD pathophysiology. mTOR-signaling pathway regulates several essential cellular processes including cell growth, proliferation, autophagy, and protein synthesis [77]. In the central nervous system, mTOR signaling is crucial from the early stages of neural development, controlling self-renewal and differentiation of hNSCs and, in neurons, mTOR signaling is involved in synapse formation and plasticity. Dysfunctional mTOR signaling and dysregulated protein synthesis in neuronal cells have been associated with several monogenic syndromes with high prevalence of autism, such as FXS, tuberous sclerosis, and PTEN-related syndromes, which are caused by mutations in, respectively, FMR1, TSC1/2, and PTEN, molecules known to be negative regulators of mTOR pathway [78]. Functional studies addressing mTOR-signaling activity in patients with nonsyndromic ASD were lacking. To examine this important question, Suzuki et al. have made use of cultured SHED derived from 13 patients with nonsyndromic ASD, who were negative for FRM1, TSC1/2, and PTEN mutations, and 11 age- and sex-matched controls. They observed that SHED derived from three patients (23 % of the patient sample) showed dysregulation of mTOR-signaling pathway in response to extracellular nutrient availability, enhanced proliferative capacity at higher cell densities, and reduced response to the antiproliferative effect of rapamycin, a specific mTOR inhibitor. Together, the results suggest that dysregulation of mTOR signaling plays an important role in the pathogenesis of a subgroup of nonsyndromic ASD, and that mTOR pathway components might be promising therapeutic targets for these patients. Interestingly, these results were further corroborated by two studies showing altered mTOR signaling in postmortem brain of patients with nonsyndromic ASD [79, 80].
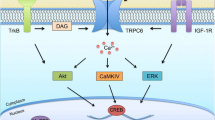
Griesi-Oliveira et al. [23], by molecularly characterizing the breakpoints of a balanced translocation between chromosomes 3 and 11 in a patient with nonsyndromic ASD, evidenced disruption of TRPC6, a gene that encodes a cation channel. They demonstrated that the transcriptome of SHED from this patient with haploinsufficiency of TRPC6 (TRPC6-mut) is dysregulated as compared to control SHED, with enrichment for genes important for cytoskeleton structure and regulation, such as SEMA3A, EPHA4, INA, and MAP2 [23]. Moreover, a significant number of differentially expressed genes between the TRPC6-mut patient’s SHED and control SHED are putative targets of CREB, a transcription factor known to be activated via calcium influx through TRPC6 [81]. Using hyperforin, a specific activator of the channel, the authors also confirmed that part of such genes could in fact be regulated by TRPC6 activation. In order to evaluate how representative these findings would be in neurons, Griesi-Oliveira et al. assessed the phenotype of neuronal cells derived from TRPC6-mut patient iPSC lines obtained from SHED, which represents the first work to evaluate a nonsyndromic case of ASD using such model. Calcium influx upon activation of TRPC6 was reduced in NPCs from the TRPC6-mut patient. Paralleling the results found in SHED, activation of TRPC6 in NPCs leads to expression regulation of some of CREB target genes. TRPC-mut neurons have shorter neurites, with a reduction in arborization complexity and lower density of dendritic spines and glutamatergic vesicles compared to control neurons. These results are consistent with previous and authors’ findings in rodent models [81, 82]. Moreover, using gain and loss of function models, authors demonstrated that such alterations could indeed be attributed to TRPC6 function. Interestingly, by taking advantage of a pair of isogenic iPSC line of a RTT patient, one with the mutated copy of MeCP2 inactivated by X-chromosome inactivation, and one with this copy activated, authors showed that MeCP2 is involved in TRPC6 expression regulation, pointing to a shared molecular pathway between a syndromic and nonsyndromic form of ASD. Although using cells in different developmental states, the work points to a mechanism in which haploinsufficiency of TRPC6 leads to reduced calcium influx and consequent dysregulation of the expression of neurodevelopmental genes, at least in part by CREB activity modulation. Such expression dysregulation then would lead to neuronal morphological and functional alterations (Fig. 8.2). Finally, Griesi-Oliveira and colleagues demonstrated that neuronal abnormalities in TRPC6-mut neurons could be rescued with hyperforin or IGF-1 treatment.

TRPC6 haploinsufficiency consequences in neuronal phenotype: TRPC6 disruption leads to a lower expression of these channels in cell membrane and consequent lower Ca++ influx into cells. Calcium signaling through TRPC6 leads to CREB activation, which is consequently diminished in TRPC6-mut cells. This leads to gene expression abnormalities, probably due to dysregulation of CREB activation. A significant number of such dysregulated genes are important for neuronal development and function, especially genes related to cytoskeleton dynamics. Indeed, TRPC6-mut neurons presented less and shorter neurites and a reduction on spine density and glutamatergic vesicles, when compared to controls
The use of control-derived stem cells engineered to reduce (knockdown) a particular gene’s expression can also be an approach to in vitro model molecular dysfunction associated with nonsyndromic ASD. In two recent studies, the neurodevelopmental impact of Neurexin 1 (NRXN1) and Neuroligin 4X (NLGN4X) deletions, known to be associated with nonsyndromic ASD and other neurodevelopmental disorders, were investigated using human iPSCs and hESCs as in vitro models [83, 84]. NRXN1 is a presynaptic neuronal adhesion molecule that interacts with postsynaptic neuroligins, such as NLGN4X, in excitatory and inhibitory synapses in the brain to form an inter-synaptic complex required for synapse formation and function. Zeng et al. [83] showed that reduction of NRXN1 expression in both iPSC-derived hNSCs and hESC-derived hNSCs leads to alterations in the expression levels of several genes involved in cell adhesion and neuron development during differentiation of the hNSCs into mature neurons. Additionally, NSCs with NRXN1 knockdown showed reduced astrocyte differentiation potential. These results suggest that NRXN1 deletion might impair nervous system development and synaptic adhesion and transmission. Using a similar approach, Shi et al. [84] knocked down NLGN4X expression in iPSC-derived NSCs and observed transcriptome alterations as well as morphological changes during differentiation of NSCs into mature neurons over a 6-week period. The authors observed that NLGN4X knockdown alters the expression patterns of several biological pathways including nervous system development and neuron differentiation, impairs the differentiation of the NSCs into neurons, and compromises neurite formation and inter-cell connections. In conclusion, these two studies combined in vitro stem cell models and targeted gene silencing to explore molecular, cellular, and neurodevelopmental effects of loss-of function mutations in ASD-associated genes.
Conclusions and Perspectives
Disease modeling in a dish with the use of stem cells has proven to be, so far, a very promising avenue to study ASD, both in its syndromic and nonsyndromic forms. In general, most of the alterations found in iPSC-derived neurons possessing different ASD pathogenic mutations are comparable to data obtained from animal models or brain-derived tissues or cells (Table 8.1). This remarkable concordance thus validates disease modeling with iPSCs, which, despite being an in vitro biological system, is able to reproduce in vivo observations. Nevertheless, in its current state, the use of reprogrammed cells should always be viewed as a complementary approach, as there are still limitations to translating how the morphological, functional or transcriptional changes observed in neural iPSC-derived cells lead to alterations in the human phenotype. In the near future, an extensive cellular characterization of a wide spectrum of ASD variability and mutational mechanisms is anticipated to unveil these relationships.
The monogenic ASD syndromic forms are caused by mutated genes belonging to different but related cellular pathways. The phenotypes can be caused by a variety of mutational mechanisms, including loss-of-function, gain-of-function, and dominant-negative mutations, associated with different functional effects at the cellular level. In spite of the genetic heterogeneity associated with the monogenic ASD syndromic forms, in general, the major pathophysiological consequences of the mutations in iPSC-derived neurons apparently are altered dendritic arborization and impaired synaptic function. It is also relevant to mention that comparable neuronal changes have been observed in iPSC modeling of nonsyndromic ASD. These observations raise some possibilities that deserve our attention: (a) impaired dendrite formation can be a feature shared by many neurodevelopmental disorders, as previously suggested [44]; (b) if impaired dendrite formation is such a common feature in these disorders, it will be impossible to assign this phenotype to specific neurodevelopmental phenotypes (e.g., ASD or cognitive deficit alone); (c) we still need to search for more specific synaptic changes or molecular markers at the cellular and molecular levels in order to establish precise correlations between neuronal phenotypes and clinical phenotypes, as suggested in CDKL5-associated syndrome. In CDKL5-mutated neurons, PSD-95, a protein that plays a significant role in learning and memory, is compromised as a consequence of the dominant-negative effect of the CDKL5 mutations, which would explain the severe mental impairment in these patients.
Mesenchymal stem cells can be easily accessed and manipulated. The few studies conducted on this type of cells have shown promising results. For example, transcriptome analysis conducted on SHED from idiopathic ASD patients and on one patient with haploinsufficiency of TRPC6 revealed cytoskeleton dynamic genes to be one of the most relevant dysregulated pathways [22, 23]. Such dysregulation would predictably result in abnormal dendritic development, which was found in TRPC6-mutated iPSC-derived neurons exhibiting less dendritic arborization and extension [23].
Finally, dysregulation of mTOR signaling has been found in about 25 % of ASD patients through analysis of SHED cultures. This is quite an unexpected proportion, as pathogenic variants in mTOR-related genes have been found in a much smaller proportion of ASD patients. Thus, the application of stem cells in the investigation of ASD etiology can not only be useful for dissecting the functional consequences of known mutations but also for aiding in the identification of common mechanisms involved in different ASD cases, even in those in which genetic alterations have not been identified.
Language impairment, difficulties in social interaction, and abnormal behavior with repetitive stereotyped movements are the main clinical hallmarks of ASD patients, and pharmacological treatments have yet to be elected to ameliorate these symptoms. Furthermore, due to the high genetic heterogeneity of nonsyndromic forms of ASD, personalized treatment for each patient has been expected to take place. However, the current studies in iPSC-derived neurons, both from syndromic and nonsyndromic patients, together with mouse models with different mutations, have revealed IGF-1 as a candidate molecule to rescue the phenotype in more than one situation. In this regard, in a phase one clinical trial using IGF-1 in PMDS, Kolevzon et al. [98] suggested in this pilot study that this drug was associated with improvement in both social impairment and restrictive behaviors in autistic children. The overall results indicate that IGF-1 might act in neuronal regulation in a very downstream manner, thus compensating any genetic alteration acting upstream. It will be important to investigate this hypothesis further, as understanding how IGF-1 regulates neuronal morphology and function can aid in finding a more universal drug for ASD treatment.
We stand in a very exciting period with great expectation to move toward a better understanding of ASD etiology and pathophysiology, and it seems that the use of stem cells will certainly change our knowledge in this field.
References
Ronemus M, Iossifov I, Levy D, Wigler M. The role of de novo mutations in the genetics of autism spectrum disorders. Nat Rev Genet. 2014;15(2):133–41.
Autism and Developmental Disabilities Monitoring Network Surveillance Year 2008 Principal Investigators, Centers for Disease Control and Prevention. Prevalence of autism spectrum disorders—Autism and Developmental Disabilities Monitoring Network, 14 sites, United States, 2008. MMWR Surveill Summ. 2012;61(3):1–19.
Brandler WM, Sebat J. From de novo mutations to personalized therapeutic interventions in autism. Annu Rev Med. 2015;66:487–507.
Curatolo P, Porfirio MC, Manzi B, Seri S. Autism in tuberous sclerosis. Eur J Paediatr Neurol. 2004;8(6):327–32.
Splawski I, Timothy KW, Sharpe LM, Decher N, Kumar P, Bloise R, Napolitano C, Schwartz PJ, Joseph RM, Condouris K, et al. Ca(V)1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell. 2004;119(1):19–31.
Lozano R, Rosero CA, Hagerman RJ. Fragile X spectrum disorders. Intractable Rare Dis Res. 2014;3(4):134–46.
Betancur C, Buxbaum JD. SHANK3 haploinsufficiency: a “common” but underdiagnosed highly penetrant monogenic cause of autism spectrum disorders. Mol Autism. 2013;4(1):17.
Prilutsky D, Palmer NP, Smedemark-Margulies N, Schlaeger TM, Margulies DM, Kohane IS. iPSC-derived neurons as a higher-throughput readout for autism: promises and pitfalls. Trends Mol Med. 2014;20(2):91–104.
Liu X, Takumi T. Genomic and genetic aspects of autism spectrum disorder. Biochem Biophys Res Commun. 2014;452(2):244–53.
Jeste SS, Geschwind DH. Disentangling the heterogeneity of autism spectrum disorder through genetic findings. Nat Rev Neurol. 2014;10(2):74–81.
Gaugler T, Klei L, Sanders SJ, Bodea CA, Goldberg AP, Lee AB, Mahajan M, Manaa D, Pawitan Y, Reichert J, et al. Most genetic risk for autism resides with common variation. Nat Genet. 2014;46(8):881–5.
De Rubeis S, He X, Goldberg AP, Poultney CS, Samocha K, Cicek AE, Kou Y, Liu L, Fromer M, Walker S, et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature. 2014;515(7526):209–15.
Yuen RK, Thiruvahindrapuram B, Merico D, Walker S, et al. Whole-genome sequencing of quartet families with autism spectrum disorder. Nat Med. 2015;21(2):185–91.
MacArthur DG, Balasubramanian S, Frankish A, Huang N, Morris J, Walter K, Jostins L, Habegger L, Pickrell JK, Montgomery SB, et al. A systematic survey of loss-of-function variants in human protein-coding genes. Science. 2012;335(6070):823–8.
Rakic P. Evolution of the neocortex: a perspective from developmental biology. Nat Rev Neurosci. 2009;10:724–35.
Chailangkarn T, Acab A, Muotri AR. Modeling neurodevelopmental disorders using human neurons. Curr Opin Neurobiol. 2012;22(5):785–90.
Cordero DR, Brugmann S, Chu Y, Bajpai R, Jame M, Helms JA. Cranial neural crest cells on the move: their roles in craniofacial development. Am J Med Genet A. 2011;155A(2):270–9.
Janebodin K, Horst OV, Ieronimakis N, Balasundaram G, Reesukumal K, Pratumvinit B, Reyes M. Isolation and characterization of neural crest-derived stem cells from dental pulp of neonatal mice. PLoS One. 2011;6(11):e27526.
Komada Y, Yamane T, Kadota D, Isono K, Takakura N, Hayashi S, Yamazaki H. Origins and properties of dental, thymic, and bone marrow mesenchymal cells and their stem cells. PLoS One. 2012;7(11):e46436.
Bueno DF, Sunaga DY, Kobayashi GS, Aguena M, Raposo-Amaral CE, Masotti C, Cruz LA, Pearson PL, Passos-Bueno MR. Human stem cell cultures from cleft lip/palate patients show enrichment of transcripts involved in extracellular matrix modeling by comparison to controls. Stem Cell Rev. 2010;7(2):446–57.
Kobayashi GS, Alvizi L, Sunaga DY, Francis-West P, Kuta A, Almada BV, Ferreira SG, de Andrade-Lima LC, Bueno DF, Raposo-Amaral CE, et al. Susceptibility to DNA damage as a molecular mechanism for non-syndromic cleft lip and palate. PLoS One. 2013;8(6):e65677.
Griesi-Oliveira K, Sunaga DY, Alvizi L, Vadasz E, Passos-Bueno MR. Stem cells as a good tool to investigate dysregulated biological systems in autism spectrum disorders. Autism Res. 2013;6(5):354–61.
Griesi-Oliveira K, Acab A, Gupta AR, Sunaga DY, Chailangkarn T, Nicol X, Nunez Y, Walker MF, Murdoch JD, Sanders SJ et al. Modeling non-syndromic autism and the impact of TRPC6 disruption in human neurons. Mol Psychiatry. 2014.
Yu J, Vodyanik MA, Smuga-Otto K, Antosiewicz-Bourget J, Frane JL, Tian S, Nie J, Jonsdottir GA, Ruotti V, Stewart R, et al. Induced pluripotent stem cell lines derived from human somatic cells. Science. 2007;318(5858):1917–20.
Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, Yamanaka S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131(5):861–72.
Kim DS, Ross PJ, Zaslavsky K, Ellis J. Optimizing neuronal differentiation from induced pluripotent stem cells to model ASD. Front Cell Neurosci. 2014;8:109.
Hubbard JJ, Sullivan SK, Mills JA, Hayes BJ, Torok-Storb BJ, Ramakrishnan A. Efficient iPS cell generation from blood using episomes and HDAC inhibitors. J Vis Exp. 2014;92:e52009. doi:10.3791/52009.
Bowen P, Biederman B, Swallow KA. The X-linked syndrome of macroorchidism and mental retardation: further observations. Am J Med Genet. 1978;2(4):409–14.
Garber KB, Visootsak J, Warren ST. Fragile X syndrome. Eur J Hum Genet. 2008;16(6):666–72.
Crawford DC, Acuna JM, Sherman SL. FMR1 and the fragile X syndrome: human genome epidemiology review. Genet Med. 2001;3(5):359–71.
Clifford S, Dissanayake C, Bui QM, Huggins R, Taylor AK, Loesch DZ. Autism spectrum phenotype in males and females with fragile X full mutation and premutation. J Autism Dev Disord. 2007;37(4):738–47.
Harris SW, Hessl D, Goodlin-Jones B, Ferranti J, Bacalman S, Barbato I, Tassone F, Hagerman PJ, Herman H, Hagerman RJ. Autism profiles of males with fragile X syndrome. Am J Ment Retard. 2008;113(6):427–38.
McDuffie A, Abbeduto L, Lewis P, Kover S, Kim JS, Weber A, Brown WT. Autism spectrum disorder in children and adolescents with fragile X syndrome: within-syndrome differences and age-related changes. Am J Intellect Dev Disabil. 2010;115(4):307–26.
Ashley Jr CT, Wilkinson KD, Reines D, Warren ST. FMR1 protein: conserved RNP family domains and selective RNA binding. Science. 1993;262(5133):563–6.
Sidorov MS, Auerbach BD, Bear MF. Fragile X mental retardation protein and synaptic plasticity. Mol Brain. 2013;6:15.
Eiges R, Urbach A, Malcov M, Frumkin T, Schwartz T, Amit A, Yaron Y, Eden A, Yanuka O, Benvenisty N, et al. Developmental study of fragile X syndrome using human embryonic stem cells derived from preimplantation genetically diagnosed embryos. Cell Stem Cell. 2007;1(5):568–77.
Urbach A, Bar-Nur O, Daley GQ, Benvenisty N. Differential modeling of fragile X syndrome by human embryonic stem cells and induced pluripotent stem cells. Cell Stem Cell. 2010;6(5):407–11.
Sheridan SD, Theriault KM, Reis SA, Zhou F, Madison JM, Daheron L, Loring JF, Haggarty SJ. Epigenetic characterization of the FMR1 gene and aberrant neurodevelopment in human induced pluripotent stem cell models of fragile X syndrome. PLoS One. 2011;6(10):e26203.
Liu J, Koscielska KA, Cao Z, Hulsizer S, Grace N, Mitchell G, Nacey C, Githinji J, McGee J, Garcia-Arocena D, et al. Signaling defects in iPSC-derived fragile X premutation neurons. Hum Mol Genet. 2012;21(17):3795–805.
Doers ME, Musser MT, Nichol R, Berndt ER, Baker M, Gomez TM, Zhang SC, Abbeduto L, Bhattacharyya A. iPSC-derived forebrain neurons from FXS individuals show defects in initial neurite outgrowth. Stem Cells Dev. 2014;23(15):1777–87.
Castren M, Tervonen T, Karkkainen V, Heinonen S, Castren E, Larsson K, Bakker CE, Oostra BA, Akerman K. Altered differentiation of neural stem cells in fragile X syndrome. Proc Natl Acad Sci U S A. 2005;102(49):17834–9.
Pasca SP, Portmann T, Voineagu I, Yazawa M, Shcheglovitov A, Pasca AM, Cord B, Palmer TD, Chikahisa S, Nishino S, et al. Using iPSC-derived neurons to uncover cellular phenotypes associated with Timothy syndrome. Nat Med. 2011;17(12):1657–62.
Tian Y, Voineagu I, Pasca SP, Won H, Chandran V, Horvath S, Dolmetsch RE, Geschwind DH. Alteration in basal and depolarization induced transcriptional network in iPSC derived neurons from Timothy syndrome. Genome Med. 2014;6(10):75.
Krey JF, Pasca SP, Shcheglovitov A, Yazawa M, Schwemberger R, Rasmusson R, Dolmetsch RE. Timothy syndrome is associated with activity-dependent dendritic retraction in rodent and human neurons. Nat Neurosci. 2013;16(2):201–9.
Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet. 1999;23(2):185–8.
Chahrour M, Zoghbi HY. The story of Rett syndrome: from clinic to neurobiology. Neuron. 2007;56(3):422–37.
Marchetto MC, Carromeu C, Acab A, Yu D, Yeo GW, Mu Y, Chen G, Gage FH, Muotri AR. A model for neural development and treatment of Rett syndrome using human induced pluripotent stem cells. Cell. 2010;143(4):527–39.
Chahrour M, Jung SY, Shaw C, Zhou X, Wong ST, Qin J, Zoghbi HY. MeCP2, a key contributor to neurological disease, activates and represses transcription. Science. 2008;320(5880):1224–9.
Cheung AY, Horvath LM, Grafodatskaya D, Pasceri P, Weksberg R, Hotta A, Carrel L, Ellis J. Isolation of MECP2-null Rett syndrome patient hiPS cells and isogenic controls through X-chromosome inactivation. Hum Mol Genet. 2011;20(11):2103–15.
Li Y, Wang H, Muffat J, Cheng AW, Orlando DA, Loven J, Kwok SM, Feldman DA, Bateup HS, Gao Q, et al. Global transcriptional and translational repression in human-embryonic-stem-cell-derived Rett syndrome neurons. Cell Stem Cell. 2013;13(4):446–58.
Williams EC, Zhong X, Mohamed A, Li R, Liu Y, Dong Q, Ananiev GE, Mok JC, Lin BR, Lu J, et al. Mutant astrocytes differentiated from Rett syndrome patients-specific iPSCs have adverse effects on wild-type neurons. Hum Mol Genet. 2014;23(11):2968–80.
Tao J, Van Esch H, Hagedorn-Greiwe M, Hoffmann K, Moser B, Raynaud M, Sperner J, Fryns JP, Schwinger E, Gecz J, et al. Mutations in the X-linked cyclin-dependent kinase-like 5 (CDKL5/STK9) gene are associated with severe neurodevelopmental retardation. Am J Hum Genet. 2004;75(6):1149–54.
Weaving LS, Christodoulou J, Williamson SL, Friend KL, McKenzie OL, Archer H, Evans J, Clarke A, Pelka GJ, Tam PP, et al. Mutations of CDKL5 cause a severe neurodevelopmental disorder with infantile spasms and mental retardation. Am J Hum Genet. 2004;75(6):1079–93.
Rademacher N, Hambrock M, Fischer U, Moser B, Ceulemans B, Lieb W, Boor R, Stefanova I, Gillessen-Kaesbach G, Runge C, et al. Identification of a novel CDKL5 exon and pathogenic mutations in patients with severe mental retardation, early-onset seizures and Rett-like features. Neurogenetics. 2011;12(2):165–7.
Archer HL, Evans J, Edwards S, Colley J, Newbury-Ecob R, O’Callaghan F, Huyton M, O’Regan M, Tolmie J, Sampson J, Clarke A, Osborne J. CDKL5 mutations cause infantile spasms, early onset seizures, and severe mental retardation in female patients. J Med Genet. 2006;43(9):729–34.
Ricciardi S, Ungaro F, Hambrock M, Rademacher N, Stefanelli G, Brambilla D, Sessa A, Magagnotti C, Bachi A, Giarda E, et al. CDKL5 ensures excitatory synapse stability by reinforcing NGL-1-PSD95 interaction in the postsynaptic compartment and is impaired in patient iPSC-derived neurons. Nat Cell Biol. 2012;14(9):911–23.
Buiting K. Prader-Willi syndrome and Angelman syndrome. Am J Med Genet C Semin Med Genet. 2010;154C(3):365–76.
Williams CA, Beaudet al, Clayton-Smith J, Knoll JH, Kyllerman M, Laan LA, Magenis RE, Moncla A, Schinzel AA, Summers JA, et al. Angelman syndrome 2005: updated consensus for diagnostic criteria. Am J Med Genet A. 2006;140(5):413–8.
Cassidy SB, Driscoll DJ. Prader-Willi syndrome. Eur J Hum Genet. 2009;17(1):3–13.
Rougeulle C, Glatt H, Lalande M. The Angelman syndrome candidate gene, UBE3A/E6-AP, is imprinted in brain. Nat Genet. 1997;17(1):14–5.
Vu TH, Hoffman AR. Imprinting of the Angelman syndrome gene, UBE3A, is restricted to brain. Nat Genet. 1997;17(1):12–3.
de Smith AJ, Purmann C, Walters RG, Ellis RJ, Holder SE, Van Haelst MM, Brady AF, Fairbrother UL, Dattani M, Keogh JM, et al. A deletion of the HBII-85 class of small nucleolar RNAs (snoRNAs) is associated with hyperphagia, obesity and hypogonadism. Hum Mol Genet. 2009;18(17):3257–65.
Moreira DP, Griesi-Oliveira K, Bossolani-Martins AL, Lourenco NC, Takahashi VN, da Rocha KM, Moreira ES, Vadasz E, Meira JG, Bertola D, et al. Investigation of 15q11-q13, 16p11.2 and 22q13 CNVs in autism spectrum disorder Brazilian individuals with and without epilepsy. PLoS One. 2014;9(9):e107705.
Chamberlain SJ, Chen PF, Ng KY, Bourgois-Rocha F, Lemtiri-Chlieh F, Levine ES, Lalande M. Induced pluripotent stem cell models of the genomic imprinting disorders Angelman and Prader-Willi syndromes. Proc Natl Acad Sci U S A. 2010;107(41):17668–73.
Cruvinel E, Budinetz T, Germain N, Chamberlain S, Lalande M, Martins-Taylor K. Reactivation of maternal SNORD116 cluster via SETDB1 knockdown in Prader-Willi syndrome iPSCs. Hum Mol Genet. 2014;23(17):4674–85.
Phelan K, McDermid HE. The 22q13.3 deletion syndrome (Phelan-McDermid syndrome). Mol Syndromol. 2012;2(3-5):186–201.
Shcheglovitov A, Shcheglovitova O, Yazawa M, Portmann T, Shu R, Sebastiano V, Krawisz A, Froehlich W, Bernstein JA, Hallmayer JF, Dolmetsch RE. SHANK3 and IGF1 restore synaptic deficits in neurons from 22q13 deletion syndrome patients. Nature. 2013;503(7475):267–71.
Hu VW, Nguyen A, Kim KS, Steinberg ME, Sarachana T, Scully MA, Soldin SJ, Luu T, Lee NH. Gene expression profiling of lymphoblasts from autistic and nonaffected sib pairs: altered pathways in neuronal development and steroid biosynthesis. PLoS One. 2009;4(6):e5775.
Chien WH, Gau SS, Chen CH, Tsai WC, Wu YY, Chen PH, Shang CY, Chen CH. Increased gene expression of FOXP1 in patients with autism spectrum disorders. Mol Autism. 2013;4(1):23.
Arthur A, Rychkov G, Shi S, Koblar SA, Gronthos S. Adult human dental pulp stem cells differentiate toward functionally active neurons under appropriate environmental cues. Stem Cells. 2008;26(7):1787–95.
Neale BM, Kou Y, Liu L, Ma’ayan A, Samocha KE, Sabo A, Lin CF, Stevens C, Wang LS, Makarov V, Polak P, Yoon S, Maguire J, Crawford EL, Campbell NG, Geller ET, Valladares O, Schafer C, Liu H, Zhao T, Cai G, Lihm J, Dannenfelser R, Jabado O, Peralta Z, Nagaswamy U, Muzny D, Reid JG, Newsham I, Wu Y, Lewis L, Han Y, Voight BF, Lim E, Rossin E, Kirby A, Flannick J, Fromer M, Shakir K, Fennell T, Garimella K, Banks E, Poplin R, Gabriel S, DePristo M, Wimbish JR, Boone BE, Levy SE, Betancur C, Sunyaev S, Boerwinkle E, Buxbaum JD, Cook Jr EH, Devlin B, Gibbs RA, Roeder K, Schellenberg GD, Sutcliffe JS, Daly MJ. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature. 2012;485(7397):242–5.
O’Roak BJ, Vives L, Fu W, Egertson JD, Stanaway IB, Phelps IG, Carvill G, Kumar A, Lee C, Ankenman K, Munson J, Hiatt JB, Turner EH, Levy R, O’Day DR, Krumm N, Coe BP, Martin BK, Borenstein E, Nickerson DA, Mefford HC, Doherty D, Akey JM, Bernier R, Eichler EE, Shendure J. Multiplex targeted sequencing identifies recurrently mutated genes in autism spectrum disorders. Science. 2012;338(6114):1619–22.
O’Roak BJ, Vives L, Girirajan S, Karakoc E, Krumm N, Coe BP, Levy R, Ko A, Lee C, Smith JD, Turner EH, Stanaway IB, Vernot B, Malig M, Baker C, Reilly B, Akey JM, Borenstein E, Rieder MJ, Nickerson DA, Bernier R, Shendure J, Eichler EE. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature. 2012;485(7397):246–50.
Sanders SJ, Murtha MT, Gupta AR, Murdoch JD, Raubeson MJ, Willsey AJ, Ercan-Sencicek AG, DiLullo NM, Parikshak NN, Stein JL, Walker MF, Ober GT, Teran NA, Song Y, El-Fishawy P, Murtha RC, Choi M, Overton JD, Bjornson RD, Carriero NJ, Meyer KA, Bilguvar K, Mane SM, Sestan N, Lifton RP, Günel M, Roeder K, Geschwind DH, Devlin B, State MW. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature. 2012;485(7397):237–41.
Menon T, Yates JA, Bochar DA. Regulation of androgen-responsive transcription by the chromatin remodeling factor CHD8. Mol Endocrinol. 2010;24(6):1165–74.
Suzuki AM, Griesi-Oliveira K, de Oliveira Freitas Machado C, Vadasz E, Zachi EC, Passos-Bueno MR, Sertie AL. Altered mTORC1 signaling in multipotent stem cells from nearly 25% of patients with nonsyndromic autism spectrum disorders. Mol Psychiatry. 2015. doi:10.1038/mp.2014.175.
Laplante M, Sabatini DM. mTOR signaling at a glance. J Cell Sci. 2009;122(Pt 20):3589–94.
Costa-Mattioli M, Monteggia LM. mTOR complexes in neurodevelopmental and neuropsychiatric disorders. Nat Neurosci. 2013;16(11):1537–43.
Tang G, Gudsnuk K, Kuo SH, Cotrina ML, Rosoklija G, Sosunov A, Sonders MS, Kanter E, Castagna C, Yamamoto A, Yue Z, Arancio O, Peterson BS, Champagne F, Dwork AJ, Goldman J, Sulzer D. Loss of mTOR-dependent macroautophagy causes autistic-like synaptic pruning deficits. Neuron. 2014;83(5):1131–43.
Oguro-Ando A, Rosensweig C, Herman E, Nishimura Y, Werling D, Bill BR, Berg JM, Gao F, Coppola G, Abrahams BS, Geschwind DH. Increased CYFIP1 dosage alters cellular and dendritic morphology and dysregulates mTOR. Mol Psychiatry. 2014.
Tai Y, Feng S, Ge R, Du W, Zhang X, He Z, Wang Y. TRPC6 channels promote dendritic growth via the CaMKIV-CREB pathway. J Cell Sci. 2008;121(Pt 14):2301–7.
Zhou J, Du W, Zhou K, Tai Y, Yao H, Jia Y, Ding Y, Wang Y. Critical role of TRPC6 channels in the formation of excitatory synapses. Nat Neurosci. 2008;11(7):741–3.
Zeng L, Zhang P, Shi L, Yamamoto V, Lu W, Wang K. Functional impacts of NRXN1 knockdown on neurodevelopment in stem cell models. PLoS One. 2013;8(3):e59685.
Shi L, Chang X, Zhang P, Coba MP, Lu W, Wang K. The functional genetic link of NLGN4X knockdown and neurodevelopment in neural stem cells. Hum Mol Genet. 2013;22(18):3749–60.
Chen RZ, Akbarian S, Tudor M, Jaenisch R. Deficiency of methyl-CpG binding protein-2 in CNS neurons results in a Rett-like phenotype in mice. Nat Genet. 2001;27:327–31.
Bauman ML, Kemper TL, Arin DM. Pervasive neuroanatomic abnormalities of the brain in three cases of Rett’s syndrome. Neurology. 1995;45:1581–6.
Ananiev G, Williams EC, Li H, Chang Q. Isogenic pairs of wild type and mutant induced pluripotent stem cell (iPSC) lines from Rett syndrome patients as in vitro disease model. PLoS One. 2011;6(9), e25255.
Mironov SL, Skorova E, Hartelt N, Mironova LA, Hasan MT, Kügler S. Remodelling of the respiratory network in a mouse model of Rett syndrome depends on brain-derived neurotrophic factor regulated slow calcium buffering. J Physiol. 2009;587:2473–85.
Chapleau CA, Calfa GD, Lane MC, Albertson AJ, Larimore JL, Kudo S, Armstrong DL, Percy AK, Pozzo-Miller L. Dendritic spine pathologies in hippocampal pyramidal neurons from Rett syndrome brain and after expression of Rett-associated MECP2 mutations. Neurobiol Dis. 2009;35:219–33.
Yazdani M, Deogracias R, Guy J, Poot RA, Bird A, Barde YA. Disease modeling using embryonic stem cells: MeCP2 regulates nuclear size and RNA synthesis in neurons. Stem Cells. 2012;30:2128–39.
Armstrong D, Dunn JK, Antalffy B, Trivedi R. Selective dendritic alterations in the cortex of Rett syndrome. J Neuropathol Exp Neurol. 1995;54:195–201.
Belichenko PV, Wright EE, Belichenko NP, Masliah E, Li HH, Mobley WC, Francke U. Widespread changes in dendritic and axonal morphology in Mecp2-mutant mouse models of Rett syndrome: evidence for disruption of neuronal networks. J Comp Neurol. 2009;514:240–58.
Djuric U, Cheung AY, Zhang W, Mok RS, Lai W, Piekna A, Hendry JA, Ross PJ, Pasceri P, Kim DS, Salter MW, Ellis J. MECP2e1 isoform mutation affects the form and function of neurons derived from Rett syndrome patient iPS cells. Neurobiol Dis. 2015;76:37–45.
Ballas N, Lioy DT, Grunseich C, Mandel G. Non-cell autonomous influence of MeCP2-deficient glia on neuronal dendritic morphology. Nat Neurosci. 2009;12:311–7.
Kishi N, Macklis JD. MeCP2 functions largely cell-autonomously, but also non-cell-autonomously, in neuronal maturation and dendritic arborization of cortical pyramidal neurons. Exp Neurol. 2010;222:51–8.
Braun K, Segal M. FMRP involvement in formation of synapses among cultured hippocampal neurons. Cereb Cortex. 2000;10:1045–52.
Bozdagi O, Sakurai T, Papapetrou D, Wang X, Dickstein DL, Takahashi N, Kajiwara Y, Yang M, Katz AM, Scattoni ML, Harris MJ, Saxena R, Silverman JL, Crawley JN, Zhou Q, Hof PR, Buxbaum JD. Haploinsufficiency of the autism-associated Shank3 gene leads to deficits in synaptic function, social interaction, and social communication. Mol Autism. 2010;1(1):15.
Kolevzon A, Bush L, Wang AT, Halpern D, Frank Y, Grodberg D, Rapaport R, Tavassoli T, Chaplin W, Soorya L, et al. A pilot controlled trial of insulin-like growth factor-1 in children with Phelan-McDermid syndrome. Mol Autism. 2014;5(1):54.
Castren M. Differentiation of neuronal cells in fragile X syndrome. Cell Cycle. 2006;5:1528–30.
Evans JC, Archer HL, Colley JP, Ravn K, Nielsen JB, Kerr A, Williams E, Christodoulou J, Gecz J, Jardine PE, et al. Early onset seizures and Rett-like features associated with mutations in CDKL5. Eur J Hum Genet. 2005;13(10):1113–20.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Passos-Bueno, M.R., Griesi-Oliveira, K., Sertié, A.L., Kobayashi, G.S. (2015). Stem Cells to Understand the Pathophysiology of Autism Spectrum Disorders. In: Zatz, M., Keith Okamoto, O. (eds) Stem Cells in Modeling Human Genetic Diseases. Stem Cell Biology and Regenerative Medicine. Springer, Cham. https://doi.org/10.1007/978-3-319-18314-5_8
Download citation
DOI: https://doi.org/10.1007/978-3-319-18314-5_8
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-18313-8
Online ISBN: 978-3-319-18314-5
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)