
Abstract
Uncommon causes of stroke represent up to 5 % of all ischemic strokes. Non-atherosclerotic vasculopathies (NAVs) account for a minority of strokes and are of particular importance in children and young adults, accounting for 14–25 % of strokes in patients under the age of 50. NAVs comprise a great variety of diseases with various underlying mechanisms including immunological, infective, collagen vascular, and hematological conditions. Increased availability of multimodal brain imaging and improved quality of noninvasive angiographic imaging has allowed more accurate and timely diagnoses, as well as better differentiation among such vasculopathies, resulting in appropriate management and enhanced outcomes. Arterial dissection, both traumatic and spontaneous, is the most common of the NAVs. Dissection is often interlinked with other arteriopathies including fibromuscular dysplasia and collagen vascular disorders. Other NAVs are being increasingly recognized, including reversible cerebral vasoconstriction syndrome, unilateral intracranial arteriopathy of childhood, moyamoya disease, post-radiation vasculopathies, and cerebral vasculitides. Mechanisms of stroke in NAVs vary from traumatic vessel injury to inflammation and infection, often in the setting of an underlying genetic predisposition. This chapter reviews the clinical manifestations, diagnosis, and management of the most common NAVs.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
FormalPara Case PresentationA 35-year-old woman developed a severe headache associated with nausea and vomiting. She had history of migraine with aura and generalized anxiety disorder, treated with propranolol and fluoxetine respectively. She took multiple doses of sumatriptan over a period of 48 h with minimal relief. On day 3, she presented to the emergency department with persistent intractable headache, altered mental status, and intermittent tingling of the right arm and face. She was disoriented to time and place, but had no focal neurologic deficit. Ancillary blood tests were normal. A CT scan of brain showed a cortical left frontal subarachnoid hemorrhage (SAH). Cerebrospinal fluid examination showed elevated red blood cell count with normal white blood cells count and slightly elevated protein level. A magnetic resonance angiogram (MRA) showed segmental narrowing in the middle cerebral arteries (MCAs), anterior cerebral arteries, and posterior cerebral arteries bilaterally, a finding confirmed by catheter cerebral angiogram. MR venogram was normal. Extensive vasculitis workup was non-contributory. The diagnosis or reversible vasoconstriction was made. She received intravenous magnesium sulfate, nimodipine, and topiramate. Despite overall improvement, she continued to complain of constant waxing and waning headaches for two consecutive weeks. On day 21, a CT scan of brain showed resolution of the SAH, with persistent vasoconstriction of the vessels of the circle of Willis bilaterally on CT angiogram (CTA). At 12 weeks, a repeat CTA showed resolution of vasoconstriction and normalization of vessels caliber in the MCAs, ACAs, and PCAs. At 12 months, she was asymptomatic except for occasional migraine attacks.
Introduction
Uncommon causes of stroke represent up to 5 % of all ischemic strokes [1]. Non-atherosclerotic vasculopathies (NAVs) account for a minority of strokes and are of particular importance in children and young adults, accounting for 14–25 % of strokes in patients under the age of 50. NAVs comprise a great variety of diseases with various underlying mechanisms including immunological, infective, collagen vascular, and hematological conditions. Increased availability of multimodal brain imaging and improved quality of noninvasive angiographic imaging has allowed more accurate and timely diagnoses, as well as better differentiation among such vasculopathies, resulting in appropriate management and enhanced outcomes. Arterial dissection, both traumatic and spontaneous, is the most common of the NAVs. Dissection is often interlinked with other arteriopathies including fibromuscular dysplasia and collagen vascular disorders. Other NAVs are being increasingly recognized, including reversible cerebral vasoconstriction syndrome, unilateral intracranial arteriopathy of childhood, moyamoya disease, post-radiation vasculopathies, and cerebral vasculitides. Mechanisms of stroke in NAVs vary from traumatic vessel injury to inflammation and infection, often in the setting of an underlying genetic predisposition. This chapter reviews the clinical manifestations, diagnosis, and management of the most common NAVs.
Cervicocephalic Arterial Dissection
Cervicocephalic arterial dissection (CCAD) is the most common of the NAVs accounting for 2.5 % of all ischemic strokes and for 15–20 % of cerebral infarctions in young adults [2–6]. CCAD usually involves the extracranial pharyngeal and distal segments of the internal carotid artery (ICA), at times extending to its petrous or supraclinoid segment [7, 8]. Dissection affecting the vertebral arteries accounts for one third of all CCADs [7, 8]. Vertebral artery dissections usually involve the distal third segment of the vessel, with intracranial extension occurring less frequently [5, 7, 8]. Intracranial dissections may follow closed head trauma or basilar skull fracture. A subintimal tear in a cervicocephalic carotid or vertebral artery occurs, followed by formation of an intramural hematoma within the layers of the tunica media [7, 9]. Longitudinal extension of the hematoma subsequently leads to tapering, luminal narrowing, focal stenosis, or occlusion of the affected vessel [7, 9]. When the intramural hematoma extends between the medial and adventitial layers, a false lumen or pseudoaneurysm may form [4, 5, 7]. Bilateral involvement is seen in 5–10 % of patients and usually raises the suspicion of underlying genetic component [10, 11]. The etiopathogenesis of CCAD remains unknown. Impaired endothelial-dependent vasodilatation occurs following a trivial trauma or when another precipitating event is present [9]. Hereditary, environmental, infective, and intrinsic factors may increase the risk of CCAD [11–14]. Abrupt cervical manipulation, in particular neck rotation and extension such as during contact sports or chiropractic manipulation, as well as minor neck extension (beauty parlor syndrome) may increase the risk of dissection (Table 16.1) [14–18]. Spontaneous dissection may occur, although patients often report a history of antecedent trivial trauma or strenuous effort such as during labor [7, 11, 12, 19, 20]. Patients with heritable connective tissue disorders and underlying arteriopathy such as FMD and collagen vascular disorders are in particular prone to arterial dissections [21–27]. Other conditions associated with increased risk of CCAD include migraine, preceding infections, arterial hypertension, MTHFR mutation, and homocysteinuria (Table 16.2) [28–32].
The clinical diagnosis of CCAD can be challenging. CCAD is often an asymptomatic incidental finding on MRI and CTA. Ipsilateral headache is the most common clinical presentation, with retro-orbital and retro-auricular pain often described in patients with carotid and vertebral artery dissections respectively [3, 5, 7, 33]. Other neurological symptoms resulting from direct compression by the dissecting aneurysm or vessel occlusion include partial Horner’s syndrome, cranial nerve palsies, pulsatile tinnitus, dysgeusia, and ocular symptoms. Focal neurologic deficits due to retinal and hemispheric ischemia may occur (Table 16.3) [5, 7, 33, 34]. In the Cervical Artery Dissection and Ischemic Stroke Patients (CADISP) Study Group, the presence of occlusive cervical artery dissection, multiple cervical artery dissections, and vertebral artery dissection were associated with an increased risk for delayed stroke [35]. Intracranial extension of arterial dissection may result in SAH. When CCAD is suspected, noninvasive studies such as Doppler ultrasonography, MRA or multisectional CTA head and neck are recommended. CTA and MRA are minimally invasive techniques that can provide high-resolution and high-contrast images of the arterial lumen and wall, with good sensitivity and specificity [36]. MRA and CTA have replaced conventional angiography, thereby facilitating early diagnosis and rapid treatment. Findings may include pseudoaneurysmal formation, intimal flap with double lumen (Fig. 16.1), vessel stenosis, or total arterial occlusion (Figs. 16.2 and 16.3). Fat suppression MRI techniques may reveal the presence of the intramural hematomas within the vessel wall [36] (Fig. 16.4). Conventional angiography has historically been the gold standard for the diagnosis of arterial dissection; its use however should be limited to selective cases where MRA or CTA is inconclusive. In patients with recurrent dissection, family history of dissection, or associated intracranial aneurysms, further workup to exclude FMD and collagen vascular disorders may be indicated [26].

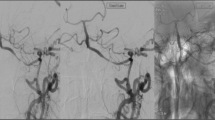
Cerebral angiogram showing cervical ICA dissection with (a) double lumen with intimal flap, and (b) pseudoaneurysmal formation

CTA of the neck showing distal vertebral artery dissection secondary to cervical fracture

(a) CTA neck showing a long segment of beaded focal stenosis in the left ICA suggestive of dissection extending into the petrous segment in a young woman with severe migraine following the use of triptans; (b) healing of the previously noted ICA dissection at 2 month follow-up

MRI brain with fat suppression shows right cervical ICA dissection with narrow eccentric flow-void surrounded by hyperintense crescent shaped intramural hematoma
In the absence of randomized clinical trials to compare various treatment options, the choice of stroke prevention therapy remains controversial. Treatment is usually aimed at preventing intramural extension, thrombus formation, and artery-to-artery embolization. Treatment options include intravenous or intra-arterial thrombolysis in patients eligible for alteplase. Optimal secondary prevention strategies in CCADS remain controversial. Options include antithrombotic therapies with either antiplatelet agents or anticoagulants; endovascular and surgical interventions are considered in selected patients with recurrent symptoms despite antithrombolytic therapies [37–43].
Intravenous thrombolysis with tissue plasminogen activator should be considered within the 4.5 h of the onset of symptoms when acute ischemic stroke is suspected [44, 45]. In the Cervical Artery Dissection and Ischemic Stroke (CADISP) registry, 68 of 616 patients received thrombolysis, majority of which in the intravenous route (55 patients) [46]. The use of thrombolysis was not associated with increased risk of bleeding [46]. Similar results were reported by Georgiadis et al. in 33 patients with acute ischemic stroke treated with intravenous thrombolysis without clinical deterioration, increased risk of SAH, pseudo-aneurysm formation, or arterial rupture [47].
Early preventive strategies should be initiated as the risk of recurrent ischemic events is highest within the first few weeks of the dissection. Anticoagulation with heparin followed by warfarin for 3–6 months has been empirically recommended except when intracranial extension is suspected [37, 47]. However, the value of anticoagulation in extracranial CCAD has not been established [43]. Data from a Cochrane review comparing antiplatelets with anticoagulants across 36 observational studies with 1,285 patients showed no differences in the odds of death or the occurrence of ischemic stroke between the two treatment modalities [38]. The results of the non-randomized arm of the Cervical Artery Dissection in Stroke Study (CADISS) which compared anticoagulation and antiplatelets for the prevention of recurrent stroke in carotid and vertebral dissection showed no difference between the two treatment arms [39]. The prospective multicenter randomized open label-controlled part of CADISS is ongoing [40]. Treatment should be customized based on acuteness of symptoms, clinical characteristics, symptom-recurrence, and imaging findings. Anticoagulation should be avoided when intracranial dissection is suspected due to increased risk of SAH [41]. Antiplatelets are often prescribed in patients with asymptomatic stenosis with subacute or late presentation. In contrast, the presence of thrombus in the dissected artery favors the use of anticoagulation [7, 37]. Surgical and endovascular interventions with angioplasty and stenting should be reserved to patients with recurrent symptoms who fail medical therapy [48–51]. The majority of CCADs heal spontaneously and outcome is usually favorable. Recurrence dissection in the involved vessel is very rare, often occurring within the first 2 months after the initial event [52, 53].
Fibromuscular Dysplasia
FMD is a non-atherosclerotic non-inflammatory segmental non-inflammatory vascular disease of unknown etiology affecting the medium and small sized arteries of virtually every arterial bed, predominantly the renal and extracranial segment of the ICA [54]. FMD may result in arterial stenosis, occlusion, aneurismal formation, or vessel dissection. While the prevalence of the disease is unknown, FMD is increasingly being diagnosed due to advances in neuroimaging. FMD is more common in young women between the ages of 30 and 50 years, especially in individuals with a history of migraines, thus hormonal factors have been postulated [54]. The disease is uncommon in children. Genetic susceptibility has been suggested in subsets of patients with autosomal mode of inheritance [55–57]. Histologically, medial fibroplasia accounts for 95–99 % of cases. Involvement of the intima and the adventitia is rare (<1 %) [58]. Angiographic characteristics observed in 80–90 % of cases of FMD include multifocal short segment of arterial stenoses with alternating mural dilatations and constriction giving the classic appearance of “string of beads”, predominantly in the mid- and distal portion of the internal carotid and vertebral arteries (medial fibroplasia or type 1) [54, 58] (Fig. 16.5). Less commonly, a unifocal concentric or band-like tubular stenosis may occur due to intimal fibroplasia (type 2). Rarely, adventitial involvement or medial hyperplasia may exit (type 3) [54, 58]. Clinical symptomatology is variable and nonspecific. Majority of cases are asymptomatic incidental findings on neuroangiographic studies. Symptoms may include headaches, pulsatile tinnitus, and blood pressure changes. Arterial dissections and cerebral aneurysms occur in 7–20 % of cases. Although often asymptomatic, these may be responsible for cerebral ischemia or subarachnoid hemorrhage [59, 60].

String of beads in the left vertebral artery and left ICA in a patient with fibromuscular dysplasia
When arterial dissection occurs, around 20 % of patients may develop transient ischemic symptoms or cerebral infarction [35]. FMD should be suspected in patients with bilateral CCADs, especially when intracranial aneurysms are present. Patients with hypertension, migraine, and history of cigarette smoking are more predisposed to FMD. The condition may coexist with other collagen vascular disorders such as cystic medial necrosis, Ehlers–Danlos syndrome (type IV), Marfan’s syndrome, Alport syndrome, and vasculitic conditions such as Takayasu’s disease [21, 60, 61]. Management of FMD is similar to that of CCAD. There are no randomized controlled trials of revascularization versus medical therapy in patients FMD. Medical management should always be the first choice of therapy, with percutaneous or surgical intervention reverted to patients with recurrent symptoms and cerebral aneurysms [62].
Moyamoya Disease
Moyamoya disease is an idiopathic progressive non-inflammatory intracranial occlusive arteriopathy of unknown etiology. It is characterized by vaso-occlusive changes involving the circle of Willis, typically the ICA terminus or proximal anterior cerebral arteries (ACA) and middle cerebral arteries (MCA), resulting in a complex network of collateral net-like tuft of vessels corresponding to the lenticulostriate and thalamoperforate arteries [63, 64]. Moyamoya disease predominantly affects children and young adults in the first or third decades of life with female preponderance. Moyamoya disease was first described by Suzuki and Takaku in 1969 in Japanese patients with abnormal net-like vessels in the base of brain appearing as “something hazy just like a puff of cigarette smoke drifting in the air” (moyamoya in Japanese) [63]. It has since been reported in every ethnic group. Histologically, there is excentric intimal hyperplasia with fibrosis of the cerebral arterial trunks, thinning of the media, and endothelial thickening leading to stenosis or occlusion of the lumen in the internal carotid terminus, ACAs and MCAs, without an obvious underlying inflammatory response [64–66]. The internal elastic lamina of the affected arteries is often tortuous; the adventitia is usually spared. Cerebral aneurysms are common [64]. Immuno-histochemical studies showed aberrant expression of IgG and S100A4 protein in vascular smooth muscle cells of the intracranial vascular wall, suggesting an underlying immune reaction [67, 68].
The pathogenesis of primary moyamoya disease is unclear, with genetic predisposition suggested. Genetic link to telomeric region of 17q25.3 was reported in Japanese family with autosomal dominant pattern [69]. Familial occurrence has also been reported in various ethnic groups in particular among identical twins [68, 70–77].
Clinical manifestations include headaches, cognitive impairment, mental retardation, encephalopathy, seizures, involuntary movements, transient neurological deficits, SAH, and focal neurological impairment secondary to ischemic or hemorrhagic strokes [64, 66, 78–81]. Moyamoya disease should be differentiated from secondary conditions associated with similar intracranial vascular stenotic pattern known as moyamoya syndrome (Table 16.4) [82].
In the absence of hematological, biochemical, and serologic findings, diagnosis is usually based on clinical presentation and neuroradiological and angiographic findings. Cerebral CT and MRI scans may reveal multiple infarctions or hemorrhages, often bilateral, in the distribution of the ACAs, MCAs, and watershed zones. Microbleeds are common. Cerebral atrophy is present in patients with recurrent symptoms due to progressive disease [83–87].
Angiographic findings include multiple mid-sized arterial irregularities and focal arterial stenoses or occlusion (Fig. 16.6) at the terminal portion of the ICAs bilaterally with distinct collateral channels formation at the base of the brain (Fig. 16.7). Except for the occlusion of the posterior cerebral arteries, the vertebrobasilar system is rarely involved. Six angiographic stages have been described: (1) bilateral suprasellar ICA narrowing, (2) collateral channels or moyamoya vessels at the base of the brain, (3) progressive ICA fork stenosis and prominent moyamoya vessels, (4) occlusion of the main arteries of the circle of Willis and extracranial collaterals, (5) further progression of stage 4 with prominent extracranial collaterals and disappearance of moyamoya vessels, and (6) complete absence of moyamoya vessels and major cerebral arteries with predominantly extracranial collaterals [63]. Intracranial aneurysms are common [80, 86]. The optimal treatment of moyamoya disease and timing of neurovascular intervention in symptomatic patients remain unclear. Medical therapy includes antiplatelet agents, vasodilators, and when seizures occur, antiepileptic agents. Patients with symptomatic progressive moyamoya disease are usually referred for neurovascular surgical intervention, with the goal to improve cerebral perfusion thereby halting disease progression, and thus reducing risk of stroke and clinical deterioration [87]. Surgical approaches include direct bypass with extracranial–intracranial anastomosis such as superficial temporal artery to middle cerebral artery (STA-MCA) bypass, indirect bypass such as encephalomyosynangiosis, encephaloduroarteriosynangiosis, encephalomyoarteriosynangiosis, omental pedicle transposition, durapexy, multiple cranial burr holes, multiple cranial burr holes with vessel synangiosis, or combined revascularization approaches [78, 82, 87–94]. Given the rarity of the disorder and of lack evidence-based guidelines, best surgical treatment options remain unknown. Intraoperative video angiography using indocyanine green is a promising technique to assess bypass graft patency in patients undergoing direct bypass with STA-MCA anastomosis [95].

MRA showing high-grade stenosis or occlusion of the supraclinoid ICA with subtle increased vascularity noted at the base of the skull adjacent to the cavernous sinus due to moyamoya collateral vessels

Bilateral supraclinoid ICA stenosis/MCA occlusion (arrow) on cerebral angiogram with extensive collaterals through lenticulostriates and thalamoperforates collaterals (arrowhead) consistent with moyamoya disease
Radiation Induced Vasculopathy
Radiation-induced vasculopathy is a common late complication of cranial radiation therapy. The condition is of particular importance in children treated with intracranial radiation for parasellar brain tumors and craniopharyngiomas [96–99]. Radiation-induced vasculopathy may develop months to years after radiotherapy, a risk persisting until adulthood (Fig. 16.8). The mechanism by which vasculopathy occurs after cranial irradiation remains unclear. Small and medium arteries are primarily affected, with progressive luminal narrowing due to endothelial thickening and medial fibrosis. Radiation injury to the large vessels is rare, usually occurring following radiation therapy for vascular malformations and pituitary tumors (Fig. 16.9). Head and neck radiation for the treatment of epithelial cancers or lymphomas is associated with delayed carotid atherosclerosis. Higher brain radiation with doses exceeding 50 Gy confers increased risk of radiation-induced vasculopathy leading to progressive cerebral arterial occlusive disease mimicking moyamoya syndrome [98, 100].

(a) Radiation induced basilar artery stenosis and PCA stenosis on MRA head in a patient with cranial radiation for pituitary macroadenoma. (b) Post-surgical changes in the pituitary fossa (arrow), with hypodensity in the basis pontis due to ischemic changes (arrowhead) on sagittal gadolinium enhanced MRI brain

(a) Radiation-induced vasculopathy changes on MRA head with irregular stenosis of the PCAs bilaterally in a 49-year-old woman with visual field defects and focal seizures. She had a medulloblastoma resected 25 years ago followed by cranial radiation therapy. (b) DWI shows area of restricted diffusion in the right occipital lobe. (c) Post-surgical changes are noted in left cerebellum on FLAIR sequence
Clinical manifestations include encephalopathy, seizures, and focal neurological deficits secondary to cerebral ischemia. Hemorrhages from radiation-induced vascular abnormalities are rare, often instead resulting from a chemotherapy effect on hemostatic system [98, 101, 102].
There is no effective treatment for radiation-induced vasculopathy. Physicians should focus on reducing radiation doses. The benefit of antiplatelet agents has not been established. Revascularization surgery may be considered in patients’ progressive arteriopathy (moyamoya syndrome) and recurrent neurological symptoms. In radiation-induced symptomatic carotid artery stenosis, surgical treatment with carotid endarterectomy may be equally effective as in non-irradiated carotid atherosclerosis [101, 102].
Reversible Cerebral Vasoconstriction Syndrome
Reversible cerebral vasoconstriction syndrome (RCVS) is characterized by thunderclap headaches, usually severe, with or without seizures or other neurologic symptoms, and segmental constriction of cerebral arteries, which resolves spontaneously within 3 months (Table 16.5) [103–105]. RCVS is also known as Call-Fleming syndrome, CNS pseudo-vasculitis, postpartum angiopathy, idiopathic thunderclap headache with reversible vasospasm, isolated benign cerebral vasculitis, and migraine angiitis. RCVS may occur in susceptible patients such as postpartum women, even without preeclampsia or eclampsia, due to transient failure of regulation of cerebral arterial tone with sympathetic overactivity [103, 106, 107]. Migraineurs with aura are more susceptible to the disease, especially when using vasoactive drug such as triptans or ergot-alkaloids. Other precipitants include nasal decongestants containing pseudoephedrine and ephedrine, illicit drugs including cannabis, cocaine, lysergic acid diethylamide, methamphetamine, selective serotonin reuptake inhibitors and selective noradrenaline reuptake inhibitors, catecholamine-secreting tumors, and intravenous immunoglobulin therapy (Table 16.6) [103, 107, 108]. RCVS is more common in women in the mid-40s, although it has been reported in children and in adults in every age group [104]. RCVS is often underdiagnosed with an unknown incidence. While relatively benign, serious ischemic and hemorrhagic events may occur in 5–10 % of patients, thus resulting in permanent neurological deficit [105, 108, 109]. Posterior reversible ischemic encephalopathy syndrome (PRES) may occur [103, 105–107].
The most common presentation includes recurrent severe rapidly escalating thunderclap headache (Table 16.7). Unlike in SAH, the headaches in RCVS are short-lived, lasting minutes to days, often recurrent, and gradually dissipating within 3 weeks of symptom-onset [103, 108, 110]. Patients typically endorse some form of exertional activity as a trigger prior to the onset of headaches. Seizures may occur especially when PRES develops. Transient neurological symptoms in particular visual disturbances mimicking migraine aura are common. When persistent focal deficit lasts beyond 1 h, stroke is suspected. In the absence of cerebral ischemia or ICH, neurological examination is usually normal [103, 108]. A surge in blood pressure may often occur due to the pain intensity.
Blood workup including ancillary laboratory tests with complete blood count (CBC), metabolic panel, and vasculitis panel is noncontributory. Cerebrospinal fluid (CSF) analysis is typically normal, sometimes with slightly elevated lymphocytic white blood cell (WBC) count.
MRI brain is usually normal. SAH, ICH, and cerebral infarction may occur. PRES-like findings are encountered in 10 % of patients (Fig. 16.10) [103, 104, 109]. Neuro-angiographic studies with MRA or CTA usually show diffuse vasoconstriction in the various arteries of the circle of Willis, both in the anterior and in the posterior circulation distribution (Fig. 16.11). Resolution of vasoconstrictive changes of the affected vessels and new constriction in previously normal vessels is not uncommon, with maximum changes seen within 2 weeks of initial onset of symptoms. Complete or substantial normalization of arteries is observed on follow-up angiographic studies within 12 weeks of clinical onset. Differential diagnosis includes aneurismal SAH, ICH, CCAD, meningitis, cerebral dural venous sinus thrombosis (CDVST), pituitary apoplexy, colloid cyst of the third ventricle, primary angiitis of the central nervous system (PACNS), and idiopathic primary thunderclap headache. The diagnosis of RCVS is made by exclusion when all other conditions are ruled out. RCVS is usually a uniphasic and self-limiting condition.

MRI FLAIR sequence demonstrates moderate signal intensity changes in the occipital lobes bilaterally typical of PRES

(a) MRA brain shows areas of severe vascular narrowing in the mid and distal basal artery (arrow) and the posterior cerebral arteries (arrowheads) in a 42-year-old woman with intractable migraine with aura and hyperemesis gravidarum. (b) Follow-up study at 3 months interval demonstrates near normalization of vessel caliber and contour
In the absence of randomized clinical trials, treatment for RCVS remains conservative, aiming at alleviating symptoms and preventing complications. Analgesics and antiepileptic agents are often administered to alleviate headaches and to prevent seizure recurrence. Prophylactic use of antiepileptic drugs is not advocated [103, 108]. Early administration of calcium channel blockers such as nimodipine or verapamil, and magnesium sulfate is often recommended. While short courses of glucocorticosteroids may help alleviating the cephalgic pain, they do not seem to prevent clinical deterioration in RCVS and thus should be avoided, in particular in patients with ischemic and hemorrhagic strokes [103, 107]. In refractory cases with rapidly deteriorating symptoms, intra-arterial administration of milrinone, verapamil or nimodipine may be considered [111, 112]. Intra-arterial balloon angioplasty should be restricted to patients with rapid clinical progression when all other treatment measures have failed [113, 114].
Unilateral Arteriopathy of Childhood
A vasculopathy unique to children is unilateral arteriopathy of childhood. Also known as transient cerebral arteriopathy (TCA), this condition is a non-progressive, often reversible, unilateral vasculopathy characterized by infarction in the lateral lenticulostriate artery territory due to non-progressive unilateral arterial disease affecting the supraclinoid ICA and its proximal branches. The hallmark of TCA is normalization or significant improvement of initial arterial stenotic changes on follow studies at 3–6 months interval. The pathophysiology of TCA is not well understood. A transient inflammatory process has been implicated, and a history of chickenpox preceding the ischemic event has been reported in 44 % of patients [115, 116]. The condition should be suspected in children with acute ischemic stroke especially with recurrent symptoms when other etiologies of cerebral infarctions are excluded [117, 118]. Vascular imaging reveals unilateral subcortical infarctions affecting the basal ganglia and internal capsule, with unilateral multifocal or segmental narrowing in the arterial wall of the distal ICA, proximal ACA or MCA [117, 118]. Transient worsening of the arterial lesions may occur up to 6 months from the initial symptom-onset in 20 % of patients, making differentiation from other intracranial causes of vasculopathies such as moyamoya disease and cerebral vasculitis a challenging process to the treating physician [119].
Despite its reversible course, children with TCA are left with focal neurological deficits due to the cerebral infarctions. Poor functional outcome tends to be more frequent in patients with initially progressive arteriopathy. Treatment includes antithrombotic agents, sometimes in combination with antiviral drugs [120].
Central Nervous System (CNS) Vasculitides
CNS vasculitides are uncommon cause of strokes in children and young adults leading to neuropsychiatric manifestations and devastating neurological deficits. They consist of a heterogenous group of systemic disorders, which include infection-related vasculitides, non-infectious inflammatory systemic vasculitides, autoimmune vasculitides, drug induced vasculitis, and the rare PACNS (Table 16.8).
Primary Angiitis of the Central Nervous System
PACNS is a rare vasculitic disorder involving the small and medium leptomeningeal arteries, with an annual incidence of 2.4 per one million persons per year. Since first described by Cravioto in 1959, the condition has been increasingly recognized as a devastating cause of recurrent stroke in young adults [121–123]. The disease is more common in men in the fourth decade of age, and solely affects the brain and spinal cord. Neurological manifestations include chronic nonspecific headaches, behavioral abnormalities, cognitive dysfunction, seizures, meningeal inflammation, multifocal neurological deficits, and recurrent strokes [121, 123–125].
Serological workup and lumbar puncture analysis are usually non-diagnostic. CSF analysis is necessary to exclude infective etiologies and other systemic conditions that may mimic PACNS. Findings are nonspecific, with mild pleocytosis in 80–90 % of patients, normal glucose and CSF protein. Diagnostic criteria include the presence of an unexplained neurologic deficit, in the absence of systemic vasculitides, and angiographic or histopathologic CNS arteritic process [121, 124].
Findings on MRI of brain are nonspecific with multiple infarctions of various ages in both cortical and subcortical distributions. Angiographic findings are also nonspecific, with a low sensitivity and specificity of less than 25 %. In the appropriate clinical scenario, the findings of multiple subcortical and cortical infarcts on CT or MRI brain, together with the presence of arterial beading, is suggestive of the condition (Fig. 16.12). Brain biopsy with sampling of the leptomeninges is the gold standard, but also carries a low yield. A negative brain biopsy does not preclude the diagnosis [125, 126].

Cerebral angiogram showing multiple irregularities in the MCA and ACA branches in PACNS
Differential diagnosis includes RCVS and secondary causes of cerebral vasculitis. Early recognition is crucial as treatment with corticosteroids with or without cytotoxic drugs can often prevent serious outcomes [126]. The disease if often progressive; if untreated, prognosis is poor.
Cerebral Amyloid Angiopathy
Cerebral amyloid angiopathy (CAA) is characterized by amyloid β deposition in the media and adventitia of leptomeningeal and cortical vessels [127, 128]. Amyloid β-related angiitis (ABRA) is a rare complication of CAA resulting from a granulomatous inflammatory response to beta amyloid (Aβ) deposition in the vessel walls [129–131]. The condition should be differentiated from PACNS, and from the perivascular non-destructive inflammatory infiltration or CAA-related inflammation (CAA-RI). Clinical symptomatology includes acute or subacute cognitive decline, headaches, seizures, uveo-meningitis, and focal neurological deficits [127, 132]. Unlike PACNS, ABRA often affects people of older age group usually in the seventh decade, without gender predilection [129]. Except for elevated erythrocyte sedimentation rate (ESR), serological markers of inflammation are usually normal. CSF abnormalities are common but nonspecific, including mild pleocytic lymphocytosis, elevated protein, and rarely oligoclonal bands. CSF tau and Aβ 41 are usually normal. APOE4 may be present in 70 % of patients [130, 131]. MRI characteristics include hyperintensities on T2-weighted image or fluid-attenuation inversion recovery (FLAIR) with minimal gadolinium-enhanced leptomeninges. The presence of microbleeds at the cortico-subcortical junction is often seen on susceptibility-weighted images. Cerebral infarcts may occur. Unlike non-inflammatory CAA, ICHs are uncommon [129, 133]. Brain biopsy is the gold standard with findings of transmural granulomatous vasculitis changes superimposed on CAA histological characteristics. Majority of patients with ABRA responds well to steroids and immunosuppressant agents such as cyclophosphamide [128, 129, 131].
Systemic Vasculitides
CNS vasculitis may occur secondary to idiopathic systemic and hypersensitivity vasculitis, autoimmune conditions, collagen vascular disorders, and various CNS infections. Clinical manifestations are diverse; fever, generalized malaise, weight loss, and fatigue are common. There is usually multi-organ involvement with renal, cardiac, arthropathic, dermatological, ocular, and pulmonary manifestations. The spectrum of neurological symptoms is broad and nonspecific. Headaches, cognitive disturbances, psychiatric manifestations, meningoencephalitis, myelopathy, myopathy, peripheral neuropathy, TIA-like symptoms, and recurrent ischemic or hemorrhagic strokes may occur. Large vessel arteritides are more commonly associated with cerebrovascular events. These include giant cell arteritis (GCA) and Takayasu’s disease.
GCA is a chronic granulomatous vasculitis mainly affecting the aorta and its branches, in particular the cranial arteries derived from the extracranial carotid arteries. It is the most common systemic vasculitis among women in the fifth decade of age, with an incidence of 3.5 per 100,000 per year [134, 135]. A dull temporal headache is reported in 90 % of patients, followed by visual symptoms, jaw claudication, weight loss, and fatigue. Other systemic manifestations may include fever, anorexia, night sweats, and muscle aches and stiffness due to polymyalgia rheumatica. Scalp tenderness and temporal artery swelling may occur. Visual loss, cerebral ischemia, and tongue infarction are the most feared complications. Cerebral infarctions occur in 3–4 % of patients with GCA. They are often due to occlusion of an extracranial segment of the vertebral or carotid arteries or their branches. Occlusion of the short posterior ciliary artery leading to choroidal ischemia results in anterior ischemic optic neuropathy (AION), the most common cause of GCA-related permanent visual loss [135, 136]. The diagnosis is straightforward in the presence of headache, visual loss, and elevated ESR. Temporal artery biopsy remains the gold standard for the diagnosis. Ultrasonography may play a role in selecting biopsy site [135, 137]. MRI and positron emission tomography may help detect any ischemic and active inflammatory changes. Despite the absence of good evidence from clinical trials, high doses corticosteroids therapy (40–60 mg of prednisone) is the treatment of choice and should be initiated even when ESR is normal or when biopsy results are inconclusive. Alternatively, steroids-sparing drugs such as methotrexate should be considered in patients intolerant to steroids or who require prolonged steroids therapy [134, 136]. Combination therapy with methotrexate and steroids is a safe alternative to steroids-only treatment [138].
Takayasu’s arteritis or pulseless disease is a chronic granulomatous large vessel panarteritis predominantly affecting the aortic arch or its branches [139]. Unlike GCA, the condition usually affects young women under the age of 40. Clinical symptoms include renovascular hypertension due to renal artery stenosis, intermittent claudication, decreased peripheral pulses, headaches, visual disturbances, focal neurologic deficits, cerebral ischemia, and rarely PRES-like clinical manifestations and imaging findings [139–141]. Laboratory workup is nonspecific, often revealing a normochromic or hypochromic anemia, leukocytosis, elevated ESR, and impaired renal function. Angiographic findings may include renal artery stenosis, as well as narrowing of and wall thickening within the aortic arch and its major branches. Medical treatment includes steroids and immunosuppressant therapies with methotrexate, mycophenolate mofetil, or azathioprine. Endovascular intervention should only be considered in patients with progressive symptoms refractory to conventional treatment. As a general rule, endovascular intervention should be avoided during the active phase of the disease due to a very high rate of arterial restenosis [142].
Kawasaki disease is a multisystemic vasculitis affecting the medium and small vessels, more commonly in infants and young children [143–148]. The disease manifests as an acute febrile mucocutaneous inflammation, with lymphadenitis, coronary artery inflammation, and widespread aneurismal formation. Cerebral infarctions are uncommon [149].
Polyarteritis nodosa is an uncommon systemic necrotizing pan-arteritis of small and medium-sized arteries, which may be associated with pseudo-aneurysmal formation. It affects the heart, kidneys, skin, and gastrointestinal tract. The cerebral vessels are rarely involved. Intraparenchymal and SAH may occur [143].
While neuropsychiatric symptoms and peripheral nervous system involvement are common in systemic lupus erythematosus (SLE), true immune complex-mediated CNS vasculitis and lupus cerebritis are uncommon. When cerebral ischemia occurs, it often results from cardiac embolism associated with Libman–Sacks endocarditis, or due to the presence of circulating antiphospholipid antibodies leading to thrombotic arterial occlusion [150–152]. Cerebral venous and dural sinus thrombosis may occur [149].
Small vessel vasculitides such as ANCA-associated small vessel vasculitis, microscopic polyangiitis, granulomatous polyangiitis, and Cogan’s syndrome rarely affect the CNS.
Sjogren syndrome is a chronic inflammatory autoimmune condition with multiorgan involvement including sicca symptoms, peripheral and cranial neuropathy, and myelopathy. CNS is rarely involved when cerebral dural venous sinus thrombosis occurs. Cerebral ischemia is rare [144].
Behçet’s disease is a multisystem inflammatory disease affecting the arteries and veins, characterized by relapsing oral and genital ulcerations, recurrent uveitis, iritis, and synovitis [153, 154]. Small vessel arteritis, thromboangiitis, cutaneous vasculitis, and cerebral aneurysms may occur [155, 156]. Clinical manifestations include headaches, cranial neuropathies, vestibulopathy aseptic meningitis, seizures, and cerebral venous thrombosis. Cerebral arterial vasculitic involvement and ischemic strokes are rare [153, 154, 157].
Sarcoidosis is a rare granulomatous disease of unknown etiology with multi-organs involvement. It primarily affects the eyes, skin, and lungs. Neurosarcoidosis with involvement of the brain parenchyma, hypothalamic–pituitary pathway, meninges, cranial nerves, and cerebral vasculature is not uncommon. Cerebral infarctions and transient ischemic attacks (TIAs) may rarely be the initial presenting manifestations of the disease [156]. ICH has been reported in <0.6 % of cases of neurosarcoidosis [158].
Kohlmeier–Degos, or malignant atrophic papulosis, is a rare systemic thrombo-obliterative vasculopathy of the medium and small size vessels, characterized by cutaneous, gastrointestinal, and neurological involvement [159]. Thrombosis of the cerebral arteries and intracerebral hemorrhage may occur due to coagulopathy or primary endothelial dysfunction [160]. Death usually occurs within 2–3 years from the onset of systemic involvement.
Other rare causes of CNS vasculitis include Henoch–Schönlein purpura, infectious etiologies and toxicity related to cancer treatment [161–163]. Chemotherapy agents associated with CNS vasculopathies and dural sinus thrombosis include, but are not limited, to l-aspariginase, methotrexate, BCNU, cisplatin, cyclophosphamide, cyclosporine, and tacrolimus [162, 164–166].
Various infectious etiologies may be associated with increased risk of ischemic and hemorrhagic strokes through various mechanisms including thrombophlebitis, vessel invasion, and cardioembolism [163, 167–175]. Of particular interest is syphilitic arteritis, an obliterative endarteritis involving the large and medium-sized vessels (Heubner arteritis), and less frequently the small cerebral arteries of the brain, meninges, and spinal cord (Nissl arteritis). Meningovascular syphilis occurs 5–10 years after the onset of untreated syphilis. Early manifestations are not uncommon in patients with human immunodeficiency virus (HIV) infections [35, 174]. Neurological symptoms include behavioral changes, seizures, and focal neurological deficits. When cerebral ischemia occurs, it most commonly affects the MCA and its branches. CSF pleocytosis and elevated protein, together with a reactive serology is suggestive. Cerebral angiography often demonstrates a diffuse angiopathy with concentric narrowing of the large vessels, and focal narrowing and dilatation of the small vessels. Aortic dissection may occur secondary to aortitis. Penicillin remains the drug of choice for neurosyphilis.
In summary, the differential diagnosis of CNS vasculitis is diverse. The diagnosis is based on clinical presentation with progressive neurological deficit, presence or absence of multisystem involvement, serological testing, neuroimaging studies with CT scans and MRI brains, and cerebral angiographic findings (Table 16.9). Neurological manifestations are broad and nonspecific. Serological findings are often nonspecific. CSF may be normal or may demonstrate nonspecific changes consistent with an inflammatory process, including mild lymphocytic pleocytosis and increased protein. CSF analysis and detailed serological and hypercoagulability testing are however necessary to exclude infective etiologies and secondary causes of vasculitis including systemic and connective tissue disorders. Accurate diagnosis is essential to prevent disease progression and to initiate management strategies and to secure precise treatment decisions. Advances in neuroimaging techniques have helped distinguish inflammatory from non-inflammatory vascular lesions. Digital subtraction angiography often shows segmental narrowing affecting multiple intracranial vessels. Tissue diagnosis with brain and leptomeningeal biopsy remains the gold standard.
Management of patients PACNS or multisystem secondary vasculitis is aimed at halting the inflammatory process and thus the disease progression. Combination of corticosteroids and immunosuppressant agents (mainly cyclophosphamide) has been shown to produce favorable clinical outcome. Other agents such as methotrexate, azathioprine, and rituximab have been used with variable results. While antiplatelet agents may be considered in patients with ischemic strokes, their use remains arbitrary in the absence of randomized clinical trials.
Anatomical Vascular Anomalies
Common anatomic variations in the configuration of the circle of Willis and its branches have been linked to the increased risk of ischemic or hemorrhagic strokes. These include but are not limited to vessel fenestration and duplication, hypoplasia and agenesis, coiling, tortuosity, elongation, and kinking. While asymptomatic in the majority, such anatomical variations may increase the risk of stroke thought different mechanisms including compression, dilatation of vascular channel, and intracranial aneurismal formation [175, 176].
Agenesis and hypoplasia of one or both internal carotid arteries is a rare, usually asymptomatic developmental anomaly that occurs during the early phase of embryonic development. Initially reported by Verbiest in 1954, ICA hypoplasia is detected in less than 0.01 % of the population [176, 177]. Flow to the anterior circulation is achieved through collateral pathways of the circle of Willis via dilated basilar artery and posterior communicating arteries, or through transcranial anastomosis between the extracranial and intracranial carotid systems, or through persistent embryonic vessels. Failure of collaterals, arterial compression, or dilated vascular channels may lead to cerebral ischemia. Associated intracranial aneurysms occur in 25–35 % of patients and are often responsible for intraparenchymal and subarachnoid hemorrhages [178].
Duplicates or fenestrations of the intracranial arteries are rare congenital anomalies. Unlike the anterior circulation vessels, fenestration is more common in the vertebrobasilar system, with basilar artery fenestration observed in around 5 % of the general population [179] (Fig. 16.13). Arterial fenestrations are usually asymptomatic. When saccular aneurysms form at the site of the fenestration or duplication, subarachnoid hemorrhage may occur [178].

MRA head showing (a) fenestrated ACA, and (b) basilar artery
Conclusion
Several non-atherosclerotic vasculopathies are responsible for ischemic strokes in particular in children and young adults. CCADs are among the most common non-atherosclerotic vasculopathies. Other conditions such as radiation-induced vasculopathy, moyamoya disease, and cerebral vasculitis are rare but potentially treatable conditions that should be considered in particular in young patients with recurrent cerebral ischemic events. Accurate diagnosis is necessary to initiate the appropriate treatment, to slow disease progression, and to improve outcome.
Abbreviations
- ABRA:
-
Amyloid β-related angiitis
- ACA:
-
Anterior cerebral arteries
- CNS:
-
Central nervous system
- CAA:
-
Cerebral amyloid angiopathy
- CSF:
-
Cerebrospinal fluid
- CCAD:
-
Cervicocephalic arterial dissection
- FMD:
-
Fibromuscular dysplasia
- GCA:
-
Giant cell arteritis
- ICA:
-
Internal carotid artery
- MCA:
-
Middle cerebral artery
- NAV:
-
Non-atherosclerotic vasculopathy
- PRES:
-
Posterior reversible ischemic encephalopathy syndrome
- PACNS:
-
Primary angiitis of the CNS
- RCVS:
-
Reversible cerebral vasoconstriction syndrome
- SAH:
-
Subarachnoid hemorrhage
References
Biller J, Adams Jr HP, Bruno A, Love BB, Marsh III EE. Mortality in acute cerebral infarction in young adults – a ten-year experience. Angiology. 1991;42(3):224–30.
Dafer RM, Biller J. Non-atherosclerotic cerebral vasculopathies. In: Gilman S, editor. Neurobiology of disease. San Diego, CA: Elsevier; 2006. p. 255–64.
Dafer RM, Biller J. Non-atherosclerotic vasculopathies. In: Biller J, editor. Stroke in Children and young adults. 2nd ed. Philadelphia, PA: Saunders Elsevier; 2009. p. 115–34.
Saver JL, Easton JDH. Dissection and trauma of cervicocerebral arteries. In: Barnett HJM, Mohr JP, Stein BM, Yatsu FM, editors. Stroke, pathophysiology, diagnosis and management. 2nd ed. New York, NY: Churchill Livingstone Inc; 1992. p. 671–88.
Mokri B. Cervicocephalic arterial dissections. In: Bogousslavsky J, Caplan LR, editors. Uncommon causes of stroke. Cambridge: Cambridge University Press; 2001. p. 211–29.
Schievink WI, Mokri B, Piepgras DG. Spontaneous dissections of cervicocephalic arteries in childhood and adolescence. Neurology. 1994;44(9):1607–12.
Schievink WI. Spontaneous dissection of the carotid and vertebral arteries. N Engl J Med. 2001;344(12):898–906.
Fisher CM, Ojemann RG, Roberson GH. Spontaneous dissection of cervico-cerebral arteries. Can J Neurol Sci. 1978;5(1):9–19.
Baumgartner RW, Lienhardt B, Mosso M, Gandjour J, Michael N, Georgiadis D. Spontaneous and endothelial-independent vasodilation are impaired in patients with spontaneous carotid dissection: a case-control study. Stroke. 2007;38(2):405–6.
Lucas C, Moulin T, Deplanque D, Tatu L, Chavot D. Stroke patterns of internal carotid artery dissection in 40 patients. Stroke. 1998;29(12):2646–8.
Treiman GS, Treiman RL, Foran RF, Levin PM, Cohen JL, Wagner WH, et al. Spontaneous dissection of the internal carotid artery: a nineteen-year clinical experience. J Vasc Surg. 1996;24(4):597–605.
Caso V, Paciaroni M, Bogousslavsky J. Environmental factors and cervical artery dissection. Front Neurol Neurosci. 2005;20:44–53.
Grau AJ, Brandt T, Buggle F, Orberk E, Mytilineos J, Werle E, et al. Association of cervical artery dissection with recent infection. Arch Neurol. 1999;56(7):851–6.
Schievink WI, Atkinson JL, Bartleson JD, Whisnant JP. Traumatic internal carotid artery dissections caused by blunt softball injuries. Am J Emerg Med. 1998;16(2):179–82.
Smith WS, Johnston SC, Skalabrin EJ, Weaver M, Azari P, Albers GW, et al. Spinal manipulative therapy is an independent risk factor for vertebral artery dissection. Neurology. 2003;60(9):1424–8.
Mourad JJ, Girerd X, Safar M. Carotid-artery dissection after a prolonged telephone call. N Engl J Med. 1997;336(7):516.
Schellhas KP, Latchaw RE, Wendling LR, Gold LH. Vertebrobasilar injuries following cervical manipulation. JAMA. 1980;244(13):1450–3.
Weintraub MI. Beauty parlor stroke syndrome: report of five cases. JAMA. 1993;269(16):2085–6.
Wiebers DO, Mokri B. Internal carotid artery dissection after childbirth. Stroke. 1985;16(6):956–9.
Rubinstein SM, Peerdeman SM, van Tulder MW, Riphagen I, Haldeman S. A systematic review of the risk factors for cervical artery dissection. Stroke. 2005;36(7):1575–80.
Schievink WI, Bjornsson J, Piepgras DG. Coexistence of fibromuscular dysplasia and cystic medial necrosis in a patient with Marfan’s syndrome and bilateral carotid artery dissections. Stroke. 1994;25(12):2492–6.
Vila N, Millan M, Ferrer X, Riutort N, Escudero D. Levels of alpha1-antitrypsin in plasma and risk of spontaneous cervical artery dissections: a case-control study. Stroke. 2003;34(9):E168–9.
Schievink WI, Michels VV, Piepgras DG. Neurovascular manifestations of heritable connective tissue disorders. A review. Stroke. 1994;25(4):889–903.
Hademenos GJ, Alberts MJ, Awad I, Mayberg M, Shepard T, Jagoda A, et al. Advances in the genetics of cerebrovascular disease and stroke. Neurology. 2001;56(8):997–1008.
Volker W, Ringelstein EB, Dittrich R, Maintz D, Nassenstein I, Heindel W, et al. Morphometric analysis of collagen fibrils in skin of patients with spontaneous cervical artery dissection. J Neurol Neurosurg Psychiatry. 2008;79(9):1007–12.
Schievink WI, Wijdicks EF, Michels VV, Vockley J, Godfrey M. Heritable connective tissue disorders in cervical artery dissections: a prospective study. Neurology. 1998;50(4):1166–9.
Brandt T, Morcher M, Hausser I. Association of cervical artery dissection with connective tissue abnormalities in skin and arteries. Front Neurol Neurosci. 2005;20:16–29.
Guillon B, Berthet K, Benslamia L, Bertrand M, Bousser MG, Tzourio C. Infection and the risk of spontaneous cervical artery dissection: a case-control study. Stroke. 2003;34(7):e79–81.
Pezzini A, Del ZE, Archetti S, Negrini R, Bani P, Albertini A, et al. Plasma homocysteine concentration, C677T MTHFR genotype, and 844ins68bp CBS genotype in young adults with spontaneous cervical artery dissection and atherothrombotic stroke. Stroke. 2002;33(3):664–9.
Pezzini A, Caso V, Zanferrari C, Del ZE, Paciaroni M, Bertolino C, et al. Arterial hypertension as risk factor for spontaneous cervical artery dissection. A case-control study. J Neurol Neurosurg Psychiatry. 2006;77(1):95–7.
Tzourio C, Benslamia L, Guillon B, Aidi S, Bertrand M, Berthet K, et al. Migraine and the risk of cervical artery dissection: a case-control study. Neurology. 2002;59(3):435–7.
Pezzini A, Granella F, Grassi M, Bertolino C, Del ZE, Immovilli P, et al. History of migraine and the risk of spontaneous cervical artery dissection. Cephalalgia. 2005;25(8):575–80.
Mokri B, Sundt Jr TM, Houser OW. Spontaneous internal carotid dissection, hemicrania, and Horner’s syndrome. Arch Neurol. 1979;36(11):677–80.
Mokri B, Silbert PL, Schievink WI, Piepgras DG. Cranial nerve palsy in spontaneous dissection of the extracranial internal carotid artery. Neurology. 1996;46(2):356–9.
Lichy C, Metso A, Pezzini A, Leys D, Metso T, Lyrer P, et al. Predictors of delayed stroke in patients with cervical artery dissection. Int J Stroke. 2012;11:10–4949.
Pelkonen O, Tikkakoski T, Pyhtinen J, Sotaniemi K. Cerebral CT and MRI findings in cervicocephalic artery dissection. Acta Radiol. 2004;45(3):259–65.
Menon R, Kerry S, Norris JW, Markus HS. Treatment of cervical artery dissection: a systematic review and meta-analysis. J Neurol Neurosurg Psychiatry. 2008;79(10):1122–7.
Lyrer P, Engelter S. Antithrombotic drugs for carotid artery dissection. Cochrane Database Syst Rev 2003;(3):CD000255.
Kennedy F, Lanfranconi S, Hicks C, Reid J, Gompertz P, Price C, et al. Antiplatelets vs anticoagulation for dissection: CADISS nonrandomized arm and meta-analysis. Neurology. 2012;79(7):686–9.
Antiplatelet therapy vs. anticoagulation in cervical artery dissection: rationale and design of the Cervical Artery Dissection in Stroke Study (CADISS). Int J Stroke 2007;2(4):292–6.
Metso TM, Metso AJ, Helenius J, Haapaniemi E, Salonen O, Porras M, et al. Prognosis and safety of anticoagulation in intracranial artery dissections in adults. Stroke. 2007;38(6):1837–42.
Furie KL, Kasner SE, Adams RJ, Albers GW, Bush RL, Fagan SC, et al. Guidelines for the prevention of stroke in patients with stroke or transient ischemic attack: a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. 2011;42(1):227–76.
Kernan WN, Ovbiagele B, Black HR, Bravata DM, Chimowitz MI, Ezekowitz MD, et al. American Heart Association Stroke Council, Council on Cardiovascular and Stroke Nursing, Council on Clinical Cardiology, and Council on Peripheral Vascular Disease. Guidelines for the prevention of stroke in patients with stroke and transient ischemic attack: a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. 2014;45(7):2160–236.
Derex L, Nighoghossian N, Turjman F, Hermier M, Honnorat J, Neuschwander P, et al. Intravenous tPA in acute ischemic stroke related to internal carotid artery dissection. Neurology. 2000;54(11):2159–61.
Georgiadis D, Lanczik O, Schwab S, Engelter S, Sztajzel R, Arnold M, et al. IV thrombolysis in patients with acute stroke due to spontaneous carotid dissection. Neurology. 2005;64(9):1612–4.
Engelter ST, Dallongeville J, Kloss M, Metso TM, Leys D, Brandt T, et al. Thrombolysis in cervical artery dissection – data from the Cervical Artery Dissection and Ischaemic Stroke Patients (CADISP) database. Eur J Neurol. 2012;19(9):1199–206.
Georgiadis D, Arnold M, von Buedingen HC, Valko P, Sarikaya H, Rousson V, et al. Aspirin vs anticoagulation in carotid artery dissection: a study of 298 patients. Neurology. 2009;72(21):1810–5.
DeOcampo J, Brillman J, Levy DI. Stenting: a new approach to carotid dissection. J Neuroimaging. 1997;7(3):187–90.
Finsterer J, Strassegger J, Haymerle A, Hagmuller G. Bilateral stenting of symptomatic and asymptomatic internal carotid artery stenosis due to fibromuscular dysplasia. J Neurol Neurosurg Psychiatry. 2000;69(5):683–6.
Edwards NM, Fabian TC, Claridge JA, Timmons SD, Fischer PE, Croce MA. Antithrombotic therapy and endovascular stents are effective treatment for blunt carotid injuries: results from longterm followup. J Am Coll Surg. 2007;204(5):1007–13.
Chiche L, Praquin B, Koskas F, Kieffer E. Spontaneous dissection of the extracranial vertebral artery: indications and long-term outcome of surgical treatment. Ann Vasc Surg. 2005;19(1):5–10.
Engelter ST, Lyrer PA, Kirsch EC, Steck AJ. Long-term follow-up after extracranial internal carotid artery dissection. Eur Neurol. 2000;44(4):199–204.
Touze E, Gauvrit JY, Moulin T, Meder JF, Bracard S, Mas JL. Risk of stroke and recurrent dissection after a cervical artery dissection: a multicenter study. Neurology. 2003;61(10):1347–51.
Slovut DP, Olin JW. Fibromuscular dysplasia. N Engl J Med. 2004;350(18):1862–71.
Bigazzi R, Bianchi S, Quilici N, Salvadori R, Baldari G. Bilateral fibromuscular dysplasia in identical twins. Am J Kidney Dis. 1998;32(6):E4.
Halpern MM, Sanford HS, Viamonte Jr M. Renal-artery abnormalities in three hypertensive sisters. Probable familial fibromuscular hyperplasia. JAMA. 1965;194(5):512–3.
Rushton AR. The genetics of fibromuscular dysplasia. Arch Intern Med. 1980;140(2):233–6.
Olin JW, Gornik HL, Bacharach JM, Biller J, Fine LJ, Gray BH, et al. Fibromuscular dysplasia: state of the science and critical unanswered questions: a scientific statement from the american heart association. Circulation. 2014;129(9):1048–78.
Cloft HJ, Kallmes DF, Kallmes MH, Goldstein JH, Jensen ME, Dion JE. Prevalence of cerebral aneurysms in patients with fibromuscular dysplasia: a reassessment. J Neurosurg. 1998;88(3):436–40.
Hudgins LB, Limbacher JP. Fibromuscular dysplasia in Alport’s syndrome. J Tenn Med Assoc. 1982;75(11):733–5.
Schievink WI, Limburg M. Angiographic abnormalities mimicking fibromuscular dysplasia in a patient with Ehlers-Danlos syndrome, type IV. Neurosurgery. 1989;25(3):482–3.
Persu A, Touze E, Mousseaux E, Barral X, Joffre F, Plouin PF. Diagnosis and management of fibromuscular dysplasia: an expert consensus. Eur J Clin Invest. 2012;42(3):338–47.
Suzuki J, Takaku A. Cerebrovascular “moyamoya” disease. Disease showing abnormal net-like vessels in base of brain. Arch Neurol. 1969;20(3):288–99.
Suzuki J, Kodama N. Moyamoya disease – a review. Stroke. 1983;14(1):104–9.
Yamashiro Y, Takahashi H, Takahashi K. Cerebrovascular Moyamoya disease. Eur J Pediatr. 1984;142(1):44–50.
Yilmaz EY, Pritz MB, Bruno A, Lopez-Yunez A, Biller J. Moyamoya: Indiana University Medical Center experience. Arch Neurol. 2001;58(8):1274–8.
Lin R, Xie Z, Zhang J, Xu H, Su H, Tan X, et al. Clinical and immunopathological features of Moyamoya disease. PLoS One. 2012;7(4):e36386.
Weinberg DG, Arnaout OM, Rahme RJ, Aoun SG, Batjer HH, Bendok BR. Moyamoya disease: a review of histopathology, biochemistry, and genetics. Neurosurg Focus. 2011;30(6):E20.
Yamauchi T, Tada M, Houkin K, Tanaka T, Nakamura Y, Kuroda S, et al. Linkage of familial moyamoya disease (spontaneous occlusion of the circle of Willis) to chromosome 17q25. Stroke. 2000;31(4):930–5.
Kaneko Y, Imamoto N, Mannoji H, Fukui M. Familial occurrence of moyamoya disease in the mother and four daughters including identical twins. Neurol Med Chir (Tokyo). 1998;38(6):349–54.
Kang HS, Kim SK, Cho BK, Kim YY, Hwang YS, Wang KC. Single nucleotide polymorphisms of tissue inhibitor of metalloproteinase genes in familial moyamoya disease. Neurosurgery. 2006;58(6):1074–80.
Mineharu Y, Takenaka K, Yamakawa H, Inoue K, Ikeda H, Kikuta KI, et al. Inheritance pattern of familial moyamoya disease: autosomal dominant mode and genomic imprinting. J Neurol Neurosurg Psychiatry. 2006;77(9):1025–9.
Nanba R, Kuroda S, Tada M, Ishikawa T, Houkin K, Iwasaki Y. Clinical features of familial moyamoya disease. Childs Nerv Syst. 2006;22(3):258–62.
Shetty-Alva N, Alva S. Familial moyamoya disease in Caucasians. Pediatr Neurol. 2000;23(5):445–7.
Zafeiriou DI, Ikeda H, Anastasiou A, Vargiami E, Vougiouklis N, Katzos G, et al. Familial moyamoya disease in a Greek family. Brain Dev. 2003;25(4):288–90.
Linfante I, Ciarmiello A, Fusco C, Ronga B, Andreone V. Similar TIAs and corresponding alterations in regional cerebral perfusion in Caucasian monozygotic twins with moyamoya disease. Clin Imaging. 2002;26(6):378–81.
Andreone V, Ciarmiello A, Fusco C, Ambrosanio G, Florio C, Linfante I. Moyamoya disease in Italian monozygotic twins. Neurology. 1999;53(6):1332–5.
Han DH, Nam DH, Oh CW. Moyamoya disease in adults: characteristics of clinical presentation and outcome after encephalo-duro-arterio-synangiosis. Clin Neurol Neurosurg. 1997;99 Suppl 2:S151–5.
Serdaru M, Gray F, Merland JJ, Escourolle R, Grumbach R. Moyamoya disease and intracerebral hematoma. Clinical pathological report. Neuroradiology. 1979;18(1):47–52.
Rao M, Zhang H, Liu Q, Zhang S, Hu L, Deng F. Clinical and experimental pathology of Moyamoya disease. Chin Med J (Engl). 2003;116(12):1845–9.
Oppenheim JS, Gennuso R, Sacher M, Hollis P. Acute atraumatic subdural hematoma associated with moyamoya disease in an African-American. Neurosurgery. 1991;28(4):616–8.
Khan N, Schuknecht B, Boltshauser E, Capone A, Buck A, Imhof HG, et al. Moyamoya disease and Moyamoya syndrome: experience in Europe; choice of revascularisation procedures. Acta Neurochir (Wien). 2003;145(12):1061–71.
Houkin K, Aoki T, Takahashi A, Abe H. Diagnosis of moyamoya disease with magnetic resonance angiography. Stroke. 1994;25(11):2159–64.
Tsuchiya K, Makita K, Furui S. Moyamoya disease: diagnosis with three-dimensional CT angiography. Neuroradiology. 1994;36(6):432–4.
Yamada I, Nakagawa T, Matsushima Y, Shibuya H. High-resolution turbo magnetic resonance angiography for diagnosis of Moyamoya disease. Stroke. 2001;32(8):1825–31.
Yamada I, Matsushima Y, Suzuki S. Moyamoya disease: diagnosis with three-dimensional time-of-flight MR angiography. Radiology. 1992;184(3):773–8.
Smith ER, Scott RM. Surgical management of moyamoya syndrome. Skull Base. 2005;15(1):15–26.
Kim SK, Seol HJ, Cho BK, Hwang YS, Lee DS, Wang KC. Moyamoya disease among young patients: its aggressive clinical course and the role of active surgical treatment. Neurosurgery. 2004;54(4):840–4.
Matsushima T, Inoue TK, Suzuki SO, Inoue T, Ikezaki K, Fukui M, et al. Surgical techniques and the results of a fronto-temporo-parietal combined indirect bypass procedure for children with moyamoya disease: a comparison with the results of encephalo-duro-arterio-synangiosis alone. Clin Neurol Neurosurg. 1997;99 Suppl 2:S123–7.
Isono M, Ishii K, Kamida T, Inoue R, Fujiki M, Kobayashi H. Long-term outcomes of pediatric moyamoya disease treated by encephalo-duro-arterio-synangiosis. Pediatr Neurosurg. 2002;36(1):14–21.
Touho H. A simple surgical technique of direct anastomosis for treatment of moyamoya disease: technical note. Surg Neurol. 2004;62(4):366–8.
Kim DS, Kang SG, Yoo DS, Huh PW, Cho KS, Park CK. Surgical results in pediatric moyamoya disease: angiographic revascularization and the clinical results. Clin Neurol Neurosurg. 2007;109(2):125–31.
Houkin K, Kuroda S, Nakayama N. Cerebral revascularization for moyamoya disease in children. Neurosurg Clin N Am. 2001;12(3):575–84. ix.
Dauser RC, Tuite GF, McCluggage CW. Dural inversion procedure for moyamoya disease. Technical note. J Neurosurg. 1997;86(4):719–23.
Woitzik J, Horn P, Vajkoczy P, Schmiedek P. Intraoperative control of extracranial-intracranial bypass patency by near-infrared indocyanine green videoangiography. J Neurosurg. 2005;102(4):692–8.
Grenier Y, Tomita T, Marymont MH, Byrd S, Burrowes DM. Late postirradiation occlusive vasculopathy in childhood medulloblastoma. Report of two cases. J Neurosurg. 1998;89(3):460–4.
Ferroir JP, Marro B, Belkacemi Y, Stilhart B, Schlienger M. Cerebral infarction related to intracranial radiation arteritis twenty-four years after encephalic radiation therapy. Rev Neurol (Paris). 2007;163(1):96–8.
Bitzer M, Topka H. Progressive cerebral occlusive disease after radiation therapy. Stroke. 1995;26(1):131–6.
Aoki S, Hayashi N, Abe O, Shirouzu I, Ishigame K, Okubo T, et al. Radiation-induced arteritis: thickened wall with prominent enhancement on cranial MR images report of five cases and comparison with 18 cases of Moyamoya disease. Radiology. 2002;223(3):683–8.
Ishikawa N, Tajima G, Yofune N, Nishimura S, Kobayashi M. Moyamoya syndrome after cranial irradiation for bone marrow transplantation in a patient with acute leukemia. Neuropediatrics. 2006;37(6):364–6.
Omura M, Aida N, Sekido K, Kakehi M, Matsubara S. Large intracranial vessel occlusive vasculopathy after radiation therapy in children: clinical features and usefulness of magnetic resonance imaging. Int J Radiat Oncol Biol Phys. 1997;38(2):241–9.
Mueller S, Fullerton HJ, Stratton K, Leisenring W, Weathers RE, Stovall M, et al. Radiation, atherosclerotic risk factors, and stroke risk in survivors of pediatric cancer: a report from the Childhood Cancer Survivor Study. Int J Radiat Oncol Biol Phys. 2013;86(4):649–55.
Ducros A. Reversible cerebral vasoconstriction syndrome. Lancet Neurol. 2012;11(10):906–17.
Headache Classification Committee of the International Headache Society (IHS). The international classification of headache disorders. 3rd edition (beta version). Cephalalgia. 2013;33(9):629–808.
Ducros A, Boukobza M, Porcher R, Sarov M, Valade D, Bousser MG. The clinical and radiological spectrum of reversible cerebral vasoconstriction syndrome. A prospective series of 67 patients. Brain. 2007;130(Pt 12):3091–101.
Calabrese LH, Dodick DW, Schwedt TJ, Singhal AB. Narrative review: reversible cerebral vasoconstriction syndromes. Ann Intern Med. 2007;146(1):34–44.
Ducros A, Bousser MG. Reversible cerebral vasoconstriction syndrome. Pract Neurol. 2009;9(5):256–67.
Whyte CA, Calabrese LH. Reversible cerebral vasoconstriction syndrome. Headache. 2009;49(4):597–8.
Moskowitz SI, Calabrese LH, Weil RJ. Benign angiopathy of the central nervous system presenting with intracerebral hemorrhage. Surg Neurol. 2007;67(5):522–7.
Ghosh PS, Rothner AD, Zahka KG, Friedman NR. Reversible cerebral vasoconstriction syndrome: a rare entity in children presenting with thunderclap headache. J Child Neurol. 2011;26(12):1580–4.
Bouchard M, Verreault S, Gariepy JL, Dupre N. Intra-arterial milrinone for reversible cerebral vasoconstriction syndrome. Headache. 2009;49(1):142–5.
French KF, Hoesch RE, Allred J, Wilder M, Smith AG, Digre KB, et al. Repetitive use of intra-arterial verapamil in the treatment of reversible cerebral vasoconstriction syndrome. J Clin Neurosci. 2012;19(1):174–6.
Ioannidis I, Nasis N, Agianniotaki A, Katsouda E, Andreou A. Reversible cerebral vasoconstriction syndrome: treatment with multiple sessions of intra-arterial nimodipine and angioplasty. Interv Neuroradiol. 2012;18(3):297–302.
Farid H, Tatum JK, Wong C, Halbach VV, Hetts SW. Reversible cerebral vasoconstriction syndrome: treatment with combined intra-arterial verapamil infusion and intracranial angioplasty. AJNR Am J Neuroradiol. 2011;32(10):E184–7.
Braun KP, Bulder MM, Chabrier S, Kirkham FJ, Uiterwaal CS, Tardieu M, et al. The course and outcome of unilateral intracranial arteriopathy in 79 children with ischaemic stroke. Brain. 2009;132(Pt 2):544–57.
Amlie-Lefond C, Bernard TJ, Sebire G, Friedman NR, Heyer GL, Lerner NB, et al. Predictors of cerebral arteriopathy in children with arterial ischemic stroke: results of the International Pediatric Stroke Study. Circulation. 2009;119(10):1417–23.
Sebire G. Transient cerebral arteriopathy in childhood. Lancet. 2006;368(9529):8–10.
Chabrier S, Rodesch G, Lasjaunias P, Tardieu M, Landrieu P, Sebire G. Transient cerebral arteriopathy: a disorder recognized by serial angiograms in children with stroke. J Child Neurol. 1998;13(1):27–32.
Bernard TJ, Manco-Johnson MJ, Lo W, MacKay MT, Ganesan V, DeVeber G, et al. Towards a consensus-based classification of childhood arterial ischemic stroke. Stroke. 2012;43(2):371–7.
Yeon JY, Shin HJ, Seol HJ, Kim JS, Hong SC. Unilateral intracranial arteriopathy in pediatric stroke: course, outcome, and prediction of reversible arteriopathy. Stroke. 2014;45(4):1173–6.
Salvarani C, Brown Jr RD, Hunder GG. Adult primary central nervous system vasculitis. Lancet. 2012;380(9843):767–77.
Cravioto H, Feigin I. Noninfectious granulomatous angiitis with a predilection for the nervous system. Neurology. 1959;9:599–609.
Calabrese LH, Mallek JA. Primary angiitis of the central nervous system. Report of 8 new cases, review of the literature, and proposal for diagnostic criteria. Medicine (Baltimore). 1988;67(1):20–39.
Hajj-Ali RA, Calabrese LH. Primary angiitis of the central nervous system. Autoimmun Rev. 2013;12(4):463–6.
Calabrese L. Primary angiitis of the central nervous system: the penumbra of vasculitis. J Rheumatol. 2001;28(3):465–6.
Calabrese LH. Diagnostic strategies in vasculitis affecting the central nervous system. Cleve Clin J Med. 2002;69 Suppl 2:SII105–8.
Scolding NJ, Joseph F, Kirby PA, Mazanti I, Gray F, Mikol J, et al. Abeta-related angiitis: primary angiitis of the central nervous system associated with cerebral amyloid angiopathy. Brain. 2005;128(Pt 3):500–15.
Danve A, Grafe M, Deodhar A. Amyloid beta-related angiitis - a case report and comprehensive review of literature of 94 cases. Semin Arthritis Rheum. 2014;14:10.
Salvarani C, Hunder GG, Morris JM, Brown Jr RD, Christianson T, Giannini C. Abeta-related angiitis: comparison with CAA without inflammation and primary CNS vasculitis. Neurology. 2013;81(18):1596–603.
Bogner S, Bernreuther C, Matschke J, Barrera-Ocampo A, Sepulveda-Falla D, Leypoldt F, et al. Immune activation in amyloid-beta-related angiitis correlates with decreased parenchymal amyloid-beta plaque load. Neurodegener Dis. 2014;13(1):38–44.
Kinnecom C, Lev MH, Wendell L, Smith EE, Rosand J, Frosch MP, et al. Course of cerebral amyloid angiopathy-related inflammation. Neurology. 2007;68(17):1411–6.
Child ND, Braksick SA, Flanagan EP, Keegan BM, Giannini C, Kantarci OH. Amyloid-beta-related angiitis presenting as a uveomeningeal syndrome. Neurology. 2013;81(20):1796–8.
Eng JA, Frosch MP, Choi K, Rebeck GW, Greenberg SM. Clinical manifestations of cerebral amyloid angiopathy-related inflammation. Ann Neurol. 2004;55(2):250–6.
Gonzalez-Gay MA, Martinez-Dubois C, Agudo M, Pompei O, Blanco R, Llorca J. Giant cell arteritis: epidemiology, diagnosis, and management. Curr Rheumatol Rep. 2010;12(6):436–42.
Borchers AT, Gershwin ME. Giant cell arteritis: a review of classification, pathophysiology, geoepidemiology and treatment. Autoimmun Rev. 2012;11(6-7):A544–54.
Fraser JA, Weyand CM, Newman NJ, Biousse V. The treatment of giant cell arteritis. Rev Neurol Dis. 2008;5(3):140–52.
Schmidt WA. Ultrasound in vasculitis. Clin Exp Rheumatol. 2014;32(1 Suppl 80):S71–7.
Hoffman GS, Cid MC, Hellmann DB, Guillevin L, Stone JH, Schousboe J, et al. A multicenter, randomized, double-blind, placebo-controlled trial of adjuvant methotrexate treatment for giant cell arteritis. Arthritis Rheum. 2002;46(5):1309–18.
Kohrman MH, Huttenlocher PR. Takayasu arteritis: a treatable cause of stroke in infancy. Pediatr Neurol. 1986;2(3):154–8.
Arnaud L, Haroche J, Mathian A, Gorochov G, Amoura Z. Pathogenesis of Takayasu’s arteritis: a 2011 update. Autoimmun Rev. 2011;11(1):61–7.
Camara-Lemarroy CR, Lara-Campos JG, Perez-Contreras E, Rodriguez-Gutierrez R, Galarza-Delgado DA. Takayasu’s arteritis and posterior reversible encephalopathy syndrome: a case-based review. Clin Rheumatol. 2013;32(3):409–15.
Liang P, Hoffman GS. Advances in the medical and surgical treatment of Takayasu arteritis. Curr Opin Rheumatol. 2005;17(1):16–24.
Bousser MG, Biousse V. Small vessel vasculopathies affecting the central nervous system. J Neuroophthalmol. 2004;24(1):56–61.
Benseler S, Schneider R. Central nervous system vasculitis in children. Curr Opin Rheumatol. 2004;16(1):43–50.
Fujiwara S, Yamano T, Hattori M, Fujiseki Y, Shimada M. Asymptomatic cerebral infarction in Kawasaki disease. Pediatr Neurol. 1992;8(3):235–6.
Lapointe JS, Nugent RA, Graeb DA, Robertson WD. Cerebral infarction and regression of widespread aneurysms in Kawasaki’s disease: case report. Pediatr Radiol. 1984;14(1):1–5.
Suda K, Matsumura M, Ohta S. Kawasaki disease complicated by cerebral infarction. Cardiol Young. 2003;13(1):103–5.
Templeton PA, Dunne MG. Kawasaki syndrome: cerebral and cardiovascular complications. J Clin Ultrasound. 1987;15(7):483–5.
Laversuch CJ, Brown MM, Clifton A, Bourke BE. Cerebral venous thrombosis and acquired protein S deficiency: an uncommon cause of headache in systemic lupus erythematosus. Br J Rheumatol. 1995;34(6):572–5.
Freire BF, da Silva RC, Fabro AT, dos Santos DC. Is systemic lupus erythematosus a new risk factor for atherosclerosis? Arq Bras Cardiol. 2006;87(3):300–6.
Chartash EK, Lans DM, Paget SA, Qamar T, Lockshin MD. Aortic insufficiency and mitral regurgitation in patients with systemic lupus erythematosus and the antiphospholipid syndrome. Am J Med. 1989;86(4):407–12.
Fluture A, Chaudhari S, Frishman WH. Valvular heart disease and systemic lupus erythematosus: therapeutic implications. Heart Dis. 2003;5(5):349–53.
Gan J, Zheng HB, Xi J, Zhou D, Shang HF, Lai XH. A case of neuro-vasculo-Behcet disease. Eur J Neurol. 2007;14(7):e16–7.
Mohammed RH, Nasef A, Kewan HH, Al SM. Vascular neurobehcet disease: correlation with current disease activity forum and systemic vascular involvement. Clin Rheumatol. 2012;31(7):1033–40.
Corse AM, Stern BJ. Neurosarcoidosis and stroke. Stroke. 1990;21(1):152–3.
Hodge MH, Williams RL, Fukui MB. Neurosarcoidosis presenting as acute infarction on diffusion-weighted MR imaging: summary of radiologic findings. AJNR Am J Neuroradiol. 2007;28(1):84–6.
Peno IC, De las Heras Revilla V, Carbonell BP, Di Capua SD, Ferrer ME, Garcia-Cobos R, et al. Neurobehcet disease: clinical and demographic characteristics. Eur J Neurol. 2012;19(9):1224–7.
O’Dwyer JP, Al-Moyeed BA, Farrell MA, Pidgeon CN, Collins DR, Fahy A, et al. Neurosarcoidosis-related intracranial haemorrhage: three new cases and a systematic review of the literature. Eur J Neurol. 2013;20(1):71–8.
Fernandez-Perez ER, Grabscheid E, Scheinfeld NS. A case of systemic malignant atrophic papulosis (Kohlmeier-Degos’ disease). J Natl Med Assoc. 2005;97(3):421–5.
Scheinfeld N. Malignant atrophic papulosis. Clin Exp Dermatol. 2007;32(5):483–7.
Sokol DK, McIntyre JA, Short RA, Gutt J, Wagenknecht DR, Biller J, et al. Henoch-Schonlein purpura and stroke: antiphosphatidylethanolamine antibody in CSF and serum. Neurology. 2000;55(9):1379–81.
Macdonald DR. Neurologic complications of chemotherapy. Neurol Clin. 1991;9(4):955–67.
Igarashi M, Gilmartin RC, Gerald B, Wilburn F, Jabbour JT. Cerebral arteritis and bacterial meningitis. Arch Neurol. 1984;41(5):531–5.
Reddy AT, Witek K. Neurologic complications of chemotherapy for children with cancer. Curr Neurol Neurosci Rep. 2003;3(2):137–42.
Armstrong T, Gilbert MR. Central nervous system toxicity from cancer treatment. Curr Oncol Rep. 2004;6(1):11–9.
Schiff D, Wen P. Central nervous system toxicity from cancer therapies. Hematol Oncol Clin North Am. 2006;20(6):1377–98.
Atalaia A, Ferro J, Antunes F. Stroke in an HIV-infected patient. J Neurol. 1992;239(6):356–7.
Menge T, Neumann-Haefelin T, von Giesen HJ, Arendt G. Progressive stroke in an HIV-1-positive patient under protease inhibitors. Eur Neurol. 2000;44(4):252–4.
Ortiz G, Koch S, Romano JG, Forteza AM, Rabinstein AA. Mechanisms of ischemic stroke in HIV-infected patients. Neurology. 2007;68(16):1257–61.
Poltera AA. Thrombogenic intracranial vasculitis in tuberculous meningitis. A 20 year “post mortem” survey. Acta Neurol Belg. 1977;77(1):12–24.
Leiguarda R, Berthier M, Starkstein S, Nogues M, Lylyk P. Ischemic infarction in 25 children with tuberculous meningitis. Stroke. 1988;19(2):200–4.
de Carvalho CA, Allen JN, Zafranis A, Yates AJ. Coccidioidal meningitis complicated by cerebral arteritis and infarction. Hum Pathol. 1980;11(3):293–6.
Bonawitz C, Castillo M, Mukherji SK. Comparison of CT and MR features with clinical outcome in patients with Rocky Mountain spotted fever. AJNR Am J Neuroradiol. 1997;18(3):459–64.
Olin JW, Froehlich J, Gu X, Bacharach JM, Eagle K, Gray BH, et al. The United States Registry for Fibromuscular Dysplasia: results in the first 447 patients. Circulation. 2012;125(25):3182–90.
Iqbal S. A comprehensive study of the anatomical variations of the circle of Willis in adult human brains. J Clin Diagn Res. 2013;7(11):2423–7.
Alpers BJ, Berry RG, Paddison RM. Anatomical studies of the circle of Willis in normal brain. AMA Arch Neurol Psychiatry. 1959;81(4):409–18.
Verbiest H. Radiological findings in a case with absence of the left internal carotid artery and compression of several cranial nerve roots in the posterior fossa by the basilar artery. Med Contemp. 1954;72(12):601–9.
Yoon SM, Chun YI, Kwon Y, Kwun BD. Vertebrobasilar junction aneurysms associated with fenestration: experience of five cases treated with Guglielmi detachable coils. Surg Neurol. 2004;61(3):248–54.
Kloska SP, Schlegel PM, Strater R, Niederstadt TU. Causality of pediatric brainstem infarction and basilar artery fenestration? Pediatr Neurol. 2006;35(6):436–8.
Acknowledgment
The author would like to thank Vimal Patel, PhD, medical and scientific writer at NorthShore Neurological Institute for assistance with preparing figures and copy-editing of the text.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Dafer, R.M. (2016). Secondary Prevention After Non-atherosclerotic Cerebral Vasculopathies. In: Ovbiagele, B. (eds) Ischemic Stroke Therapeutics. Springer, Cham. https://doi.org/10.1007/978-3-319-17750-2_16
Download citation
DOI: https://doi.org/10.1007/978-3-319-17750-2_16
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-17749-6
Online ISBN: 978-3-319-17750-2
eBook Packages: MedicineMedicine (R0)