
Abstract
Facioscapulohumeral MD (FSHD or FSH) is a complex, inheritable muscle disease. Although frequently cited as the third most common type of MD in older reports, many newer sources rank FSHD as the most prevalent type of MD, occurring at a rate of some 7 cases/1,000 persons, as compared with DMD/BMD (5 cases/1,000) and myotonic dystrophy (4.5 cases/1,000). The identification of FSHD as the most common type of MD has important ramifications, for example, when allocating future Federal (U.S.) funding for research, and in terms of the potential market size for future FSHD treatments. FSHD has only recently attracted attention from the pharmaceutical industry, largely due to major advances in the understanding of the gene/mechanism of disease, including over-expression of a protein called DUX4. Most individuals with FSHD inherit the mutation from a parent with the disease, with 10–33 % of all FSHD cases resulting from a de novo (or sporadic) mutation. The major symptom of FSHD is progressive weakening and loss of skeletal muscles. The usual location of these weaknesses at onset is the origin of the name: face (facio), shoulder girdle (scapulo), and upper arms (humeral). There is currently no disease modifying treatment or cure for FSHD. Most treatments proposed to “treat” FSHD have not yet been tested in randomized clinical trials. These may include: hormone supplementation, protein supplements (creatinine monohydrate), or drugs used to decrease inflammation (e.g., prednisone). To better understand and validate their use, many are now being properly investigated in clinical trials.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others
Keywords
- Facioscapulohumeral MD
- Hormone supplementation
- Protein supplements (creatinine monohydrate)
- Prednisone
- Lumbar lordosis
- Scoliosis
- Beevor’s sign
- DUX4 gene
- FSHD1
- FSHD2
- Infantile FSHD
- Creatine kinase
- Electromyograph
- Muscle biopsy
- Orthotics
- Mobility devices
- Counseling
Introduction
Facioscapulohumeral muscular dystrophy, a complex, inheritable muscle disease, was historically known as Landouzy–Déjérine disease—named after the French neurologists Dr. Joseph Jules Déjérine (1849–1917) and Dr. Louis Théophile Joseph Landouzy (1845–1917). It is more commonly known today as FSHD or FSH. Landouzy and Déjérine first named the disease FSHD in 1885 in order to distinguish the disease from the only other form of muscular dystrophy (MD) known at the time, DMD [1, 2].
Although frequently reported as the third most common type of MD in older reports and articles, many newer sources [3–5], including a May 2014 report by Orphanet, ranks FSHD as the most prevalent type of MD [6]. According to Orphanet, FSHD is the most prevalent MD with 7 cases/1,000 persons reported as compared with DMD/BMD (5 cases/1,000 persons) and Steinert myotonic dystrophy (4.5 cases/1,000 persons). Other prominent Websites, such as FSH Canada, also list FSHD as “the most prevalent of the nine primary types of MD affecting adults and children” [7]. Informal discussions with those afflicted with FSHD, as well as researchers and proponents of FSHD treatments, indicate that the incidence of FSHD is probably underreported. This may reflect the fact that some patients with FSHD—such as those with mild symptoms, or those with an onset late in life—may not ever be formally diagnosed and thus may not be reported or included in patient registries.
The identification of FSHD as the most common type of MD has important ramifications, for example, when allocating future Federal (U.S.) funding for research. Daniel P. Perez, CEO and Founder of the FSH Society, has testified nearly 50 times before Congress. Due to his leadership efforts and testimony, funding for FSHD has grown from $1.5m/year in 2003 (out of $39.1m for all types of MD) to $5–6m in 2009–2013, a significant increase, but not yet aligned, based on prevalence alone. According to the most recent testimony by Mr. Perez to the U.S. Senate Appropriations Committee on May 16, 2014, FSHD is one of the most common adult MDs with a prevalence of 1:15,000–1:20,000 [6, 7].
Another important factor related to prevalence of disease is the potential market size for future FSHD treatments. Quite simply, a larger prevalence means a larger potential market and this may have an influence on the interest levels of third party capital providers investing in FSHD clinical trials and treatments. This is a win–win situation for both those afflicted with MD and those wishing to invest in the healthcare marketplace.
Despite being the most common form of MD, FSHD has only recently attracted attention from big pharma (e.g., GSK), largely due to major advances in the understanding of the gene/mechanism of disease [8]. For example, recent advances in the understanding of the cause of FSHD point to over-expression of a protein called DUX4, which is normally suppressed in adult muscles, but is activated in FSHD. A more detailed explanation is provided below in the section titled, “The Proposed Cause of FSHD.”
Overview of Symptoms of FSHD
The major symptom of FSHD is progressive weakening and loss of skeletal muscles. The usual location of these weaknesses at onset is the origin of the name: face (facio), shoulder girdle (scapulo), and upper arms (humeral). Early weaknesses of the muscles of the eye (open and close) and mouth (smile, pucker, whistle) are distinctive. These symptoms, in combination with weaknesses in the muscles that stabilize the scapulae (shoulder blades), are often the basis of the physician’s clinical diagnosis of FSHD.
The progression of FSHD is quite variable, even among afflicted siblings [9]. For most patients, the progression of the disease is usually relatively slow; however, it usually worsens during adolescent years as the muscular framework fails to keep pace with the expanding and lengthening skeletal structure.
Other skeletal muscles invariably weaken. Involvement of muscles of the foot, hip girdle, and abdomen is common. With FSHD, most affected people develop unbalanced (side-to-side) weaknesses. The reason for this asymmetry is unknown [10].
In most cases, FSHD muscle involvement starts in the face and slowly progresses to the shoulder and upper-arm muscles and then down to the abdominal and foot-extensor muscles. Foot drop and foot weakness can be early manifestations and are generally accepted as part of the natural course of the disease [11, 12].
Initial signs of FSHD also include difficulty reaching above the shoulder level, scapular winging, and facial weakness. Weakness in the abdominal muscles can cause a protuberant abdomen and lumbar lordosis and scoliosis (curvatures of the lower spine). The inability to run and balance and loss of power may manifest in patients with FSHD and may be more noticeable in FSHD patients who play sports, take dance classes, or even school gymnastics classes, as FSHD patients compete with peers.
The lower abdominal muscles are usually weaker than the upper abdominal muscles. This distribution of weakness causes a positive Beevor’s sign—a characteristic weakness of the lower abdominal muscles, involving the movement of the navel towards the head on flexing the neck—and, according to the FSH Society’s Website and other sources [13], prognostic for FSHD.
Other symptoms in FSHD patients include chronic pain in the majority of patients (50–80%) with severe pain in up to 23% [14–16], vision abnormalities (due to vascular abnormalities of blood vessels in the back of the eye, which cause visual problems in only about 1% of the cases) [13], progressive hearing loss (correlated with the severity of genetic abnormalities and especially in severe infantile cases), cardiac arrhythmias (generally asymptomatic), and cognitive impairment, sometimes with epilepsy. The latter conditions are rare, but may be seen in severe, early-onset cases.
Autosomal Dominance
Most individuals with FSHD inherit the mutation from a parent with the disease. DNA is the means of transmission of inheritable traits from parent to child and occurs via chromosome transfer from one generation to the next. Each chromosome contains a strand of DNA. Human cells usually contain 46 chromosomes, 23 from each parent. Children inherit one member of each of the 23 pairs of chromosomes from each parent. Forty-four of the chromosomes, also called autosomes, are homologous pairs (numbered 1 through 22), with each strand of the pair having the same size, order, and arrangement with genes for the same traits in the same position on the chromosome. The remaining chromosome pair consists of the nonhomologous sex chromosomes X and Y. A mother donates an X chromosome, and a father donates either an X or Y chromosome. Therefore, males have one X chromosome and one Y chromosome, while females have two X chromosomes [17].
FSHD is the result of a DNA mutation on one member of the chromosome 4 pair. FSHD is highly penetrant. This means that when a person inherits a chromosome 4 with the FSHD mutation, there is a high probability that discernible muscle weaknesses will develop. Since weakness still occurs in the presence of the normal member of the chromosome 4 pair, the disease is considered dominant. FSHD is, therefore, a dominant inherited disease, meaning only one parent has to have the disease gene or deletion for his or her child to inherit FSHD. Since each parent donates only one member of each chromosome pair to a child, the probability of passing the disease to an offspring is 50%.
If one has a blood parent, sibling, or other relative who has the FSHD mutation, there may be a risk of carrying that mutation. Often, when a person is diagnosed with FSHD, the disease is discovered to be throughout the extended family tree and over many generations. It is important to be aware that there may be other family members who are affected but unaware that they may have FSHD or may be at risk for FSHD. Professionals with knowledge of genetics and inheritance of FSHD can advise them regarding that risk.
The Proposed Cause of FSHD
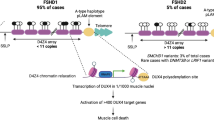
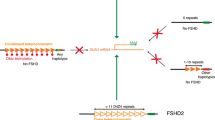
A DNA mutation causes FSHD. The gene that is linked to FSHD is unknown, but its approximate location is toward the end of the DNA of the long arm of chromosome 4. The specific genetic location of the FSHD deletion is 4q35 in the D4Z4 DNA region. Although the precise details are not yet known, the most probable cause of FSHD is inappropriate expression of protein called DUX4 by a “double homeobox protein 4 gene” [18, 19]. According to the FSH Society’s Website, approximately 2% of FSHD cases are not linked to the 4q35 deletion on chromosome 4.
Researchers are investigating the molecular connection between deletion and FSHD. The size of the deletion has a relationship to the severity of the disease—patients with the fewest repeats (the largest deletion) typically have the most severe symptoms.
The DUX4 gene is normally expressed in germ line tissue (cells associated with reproduction, such as sperm and ovaries) and repressed in somatic cells (non-germ cells associated with forming other parts of the human body), but becomes over-expressed in FSHD patients and is toxic to muscle cells.
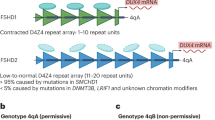
Two forms of the disease are recognized and reported in the literature: FSHD1 and FSHD2. About 95% of patients with FSHD have the FSHD1 form, where one allele (called D4Z4) is contracted and the other D4Z4 allele is normal [20, 21]. De novo, or sporadic contraction of D4Z4, account for 10–30% of FSHD1 cases [22, 23].
Less than 5% of FSHD patients have no contracted D4Z4 repeat arrays, but may still have abnormal DNA and are termed FSHD2 cases [24]. Patients with FSHD2 sometimes have another mutated gene, called SMCHD1, which appears to upregulate D4Z4 and be the cause of some, if not most, cases of FSHD2 [25]. There is no generally accepted estimate of its incidence, but this is unlikely to exceed 2% of all cases of FSHD.
Infantile FSHD (IFSHD) is characterized by onset in early childhood. There is no generally accepted estimate of its incidence, but it is rare. FSHD occurs in all racial groups and with equal frequency in both sexes.
One cannot clinically distinguish one type of FSHD from the other.
De Novo Cases of FSHD [19]
Studies report from 10% to as high as 33% of all FSHD cases result from a de novo (or sporadic) mutation. Approximately 20% of reported de novo cases are those inherited from a seemingly unaffected parent who is a “germline mosaic,” meaning that only the mother’s or father’s germ cells (the egg or sperm) are affected. When a germline mosaic is involved, the parent appears unaffected but the children are at risk.
In the remaining 80% of de novo cases, neither parent’s genes are affected; a new spontaneous mutation results in a chromosome 4 deletion that causes FSHD. When the 4q35 deletion fragment appears in a sporadic FSHD case, it is transmitted in an autosomal dominant (only one parent needs to be affected) manner to succeeding generations. The probability, then, of passing the disease to an offspring is 50%.
Onset of Symptoms
Although the FSHD gene is present at birth, weaknesses are generally not noticeable until the second decade. Sometimes, muscle weaknesses are slight throughout adulthood. A physician can usually clinically recognize and diagnose FSHD beyond the age of 20. However, it is important to realize that the onset of FSHD is highly variable and may require several physicians, with differing specialties, to diagnose younger patients or patients with milder symptoms.
In IFSHD, a young child or an infant develops symptoms. In IFSHD, there are facial weaknesses during the first two years of life in addition to other typical muscle weaknesses of FSHD. Some of these children also experience early hearing losses and retinal abnormalities.
Prognosis of FSHD
Predicting the exact course and outcome of FSHD is impossible because the rapidity and extent of muscle loss differs considerably among FSHD patients—even among siblings. Some report few difficulties throughout life, while others need orthotic devices (e.g., abdominal brace, leg or foot braces) or a wheelchair as walking becomes difficult or impossible. The degree of severity in an FSHD parent cannot accurately predict the extent of disability that may develop in that parent’s child.
Some reports suggest than men with FSHD are more severely affected than women [26, 27].
There is certainty that some skeletal muscles will weaken and waste throughout life and that this can, and often does, cause limitations on personal and occupational activities. The heart and internal (smooth) muscles seem spared and, with rare exceptions, those with FSHD have a normal life span.
Diagnosing FSHD
Physical examinations by clinicians familiar with the disease, such as neurologists associated with an Muscular Dystrophy Association (MDA) Center, are dependable when there are clinical symptoms that follow an expected location and pattern of weakening muscles.
Often the physician will supplement a physical examination with inquiries about a possible family history of FSHD and may wish to take blood samples to help make the diagnosis. Other diagnostic tools that a physician may employ to confirm a diagnosis of FSHD include:
-
Measurement of specific enzyme levels in the blood (e.g., creatine kinase or CK).
-
An electromyograph or EMG, which records abnormal electrical activity of a functioning skeletal muscle.
-
A muscle biopsy, where a small piece of muscle tissue is analyzed for visible abnormalities by a histopathologist.
-
A DNA test, especially for equivocal cases for patients at younger ages and some at-risk adults with mild or asymptomatic cases. This test is highly reliable for most cases.
-
The test detects the 4q35 DNA deletion described earlier. Although several factors may occasionally complicate the test, the FSH Society states that confirmation of the 4q35 deletion is 98% reliable as a presumptive diagnosis of FSHD. The test requires no more than a small amount of blood that one’s physician sends to a testing laboratory. The laboratory extracts sufficient DNA for the test from the cells present in the blood.
-
Currently, there is no DNA test available for those cases where there is no linkage between FSHD and chromosome 4.
-
-
Prenatal testing, which is available for those persons interested.
Many physicians will also refer a patient diagnosed with FSHD to undergo further testing that may include:
-
A visual exam, including a retinal examination
-
Pulmonary function tests
-
Cardiac tests
-
Orthopedic examination
-
Radiographic examination (especially for scoliosis and lordosis to determine a baseline and to monitor progression)
-
Referral for physical therapy
-
Orthotic (man-made support devices, usually made of cloth, plastic, or rubber) to support the abdomen (e.g., with lordosis) or extremities (e.g., foot drop)
-
Measurements for assisted mobility devices, such as a scooter, Segway-type of device, or electronic wheelchair
Neurologists are often the primary physicians in muscle disease clinics since muscles do their work through stimulation by nerves. Physiatrists are physicians who work with chronic neuromuscular conditions. Periodic visits with a neurologist or physiatrist are useful to monitor the progress of FSHD and to obtain referrals to other professionals and services. An orthopedist (a physician concerned with the skeletal system and associated muscles, joints, and ligaments) can offer advice about mobility issues and other functional problems of the muscular/skeletal system. Those with experience in MD/FSHD are generally the most helpful.
Physical therapy, including light exercise, helps preserve flexibility. Swimming is especially helpful in this regard because of buoyancy and may make movements easier. According to the FSH Society’s website, a patient with FSHD should stay as active as possible, with rest breaks as needed during exercise and activities.
Occupational therapists can help with suggestions for adaptations and physical aids that can often partially free an FSHD patient from some constrictions of the disease. Foot drop can sometimes be managed with ankle-foot orthotics (AFOs) and knee-ankle-foot orthotics (KAFOs). Occupational and ergonomic therapists may even visit a FSHD patient’s home and make recommendations to improve the way a patient with FSHD can navigate their house or room.
Speech and hearing therapists can help with limitations imposed by hearing loss and weakened facial musculature to improve speech and communication.
Psychiatry and Psychological Counseling
While it is beyond the scope of this chapter to delve into all of the facets of psychiatric and psychological counseling for patients afflicted with FSHD, suffice it to say that a diagnosis of FSHD can be devastating to the patient, the patient’s family, and the patient’s caregivers. A patient, if diagnosed with clinical depression by a physician, may benefit from medication (e.g., antidepressants) and regular counseling sessions. This is not inexpensive, but may be covered, at least in part, by insurance carriers, though there is usually a cost difference between in-network (physicians who may or may not be familiar with MD) and out-of-network physicians (who could be those who are most familiar with MD or those most sought after, but may or may not be in the insurance carrier’s plan).
There are many different types of experts who can provide counseling services for patients with FSHD; however, for simplicity, three are described below:
-
A psychiatrist, a medically trained physician trained to treat mental disorders, can prescribe medication, if needed, to a patient with FSHD.
-
A psychologist (e.g., Ph.D.), trained to understand mental disorders, can help a patient learn new skills required to cope with complex, life-altering diseases such as FSHD. These experts cannot prescribe medication, but typically cost less per hour than psychiatrists.
-
A Licensed Specialist Clinical Social Worker (LSCSW) or Licensed Clinical Social Worker (LCSW), with a Master’s degree in Social Work plus supervised experience and the required amount of continuing education, can counsel patients and families with FSHD and typically cost less than psychologists and psychiatrists.
Treatments for FSHD
There is currently no disease modifying treatment or cure for FSHD. Most treatments currently considered or proposed to “treat” FSHD have not yet been tested in randomized clinical trials. These may include: hormone supplementation (e.g., testosterone), protein supplements (creatinine monohydrate), or drugs used to decrease inflammation (e.g., prednisone). To better understand and validate their use, many are now being properly investigated in clinical trials. See paragraph below titled, “Clinical Trials for Patients with FSHD” for additional details.
Pain is part of FSHD in many patients. No specific treatments are available. Pain medication and mild physiotherapy are often prescribed with moderate results.
Sometimes an orthopedic surgeon may recommend attaching the scapulae (shoulder blades) to the back to improve motion of the arms. An individual who is considering such surgery should consult with their neurologist or physiatrist and an orthopedic surgeon. Discussion of this procedure with individuals who have undergone the surgery is highly recommended.
Sometimes an orthopedic surgeon may wish to fuse the spinal column of a FSHD patient in order to correct lordosis/scoliosis. As this surgery is rarely performed, an individual may wish to consult with their neurologist, physiatrist, or other orthopedic/pediatric surgeons to determine possible negative or untoward consequences. Because this surgery may decrease or eliminate compensatory mechanisms and decrease remaining ambulatory time for a patient with FSHD, it is advisable to obtain several professional opinions to understand the risks and benefits of such a surgery before deciding upon this type of surgery for an ambulatory patient with FSHD.
Further discussion of treatment for patients with MD, not exclusively for FSHD, is provided in Chapter 7.
Mobility Devices for Patients with FSHD
If a FSHD patient requires a mobility device, such as an electronic wheelchair (usually due to fatigue associated with walking or the inability to walk) or scooter, there is an extensive and sometimes protracted process that a patient must undergo to satisfy insurance requirements justifying their use and procurement. Based on the author’s experience, time limitations exist for physicians seeking to obtain a wheelchair for their FSHD patient (e.g., the physician recommending the procurement of a wheelchair must have seen the FSHD patient within the last six months) and may be limited to physicians practicing in the patient’s state of residence. This can be difficult for patients with FSHD who seek optimum treatment and may need to travel out of state to visit a physician or may have visited a physician within their state, but not within the six month “statute of limitations.” Therefore, the patient with FSHD and his/her caregiver should seek the advice of mobility provider to make sure that the initial application has the highest probability of success upon initial submission/request. NuMotion, a provider of wheelchairs in North Carolina (and a hub for the MDA “closet” where devices for persons afflicted with MD are offered for free), is one example of a mobility provider that not only understands the situation for families dealing with MD, but can act as an advocate for the entire family.
The MDA can be helpful in assisting patients with FSHD or even provide loaner wheelchairs—called “closet wheelchairs”—free of charge for those that qualify or for patients waiting for a customized wheelchair. There may be assistance regarding their upkeep and repair, and patients and caregivers should inquire about such services when procuring a wheelchair. It is beyond the scope of this chapter to discuss mobility transport, but it may include: wheelchair with ramps to place the wheelchair in a van/truck; exterior carriers that mount to a hitch on the back of a vehicle; and collapsible scooters that can be taken apart to carry in the trunk of a car.
Clinical Trials for Patients with FSHD
Clinicaltrials.gov lists 17 trials using the search term “FSHD”, but this may be a little misleading as the 17th trial (as accessed on June 2, 2014) actually lists the title of the trial as for DMD and not FSHD. Most of the trials posted are related to the study of antioxidants, protein supplementation, and physical therapy and may be out of date or abandoned for lack of efficacy or the inability to obtain additional funding. Some of the compounds under investigation, such as albuterol and prednisone, have been marketed for many years, but are not approved for FSHD, and therefore, have not been formally studied in the clinical trial setting for the treatment of FSHD. Some products, at first blush, appear to be unique, such as Wyeth’s (now Pfizer’s) MYO-029; however, the last update was in 2007 and the Website states that the trial is now closed. A review of ADIS reports (in June 2014) lists MYO-029 as “discontinued in Phase II.”
Additional discussion of treatments, including a discussion of clinical trials, is provided in Chapter 12.
FSHD Respiratory Insufficiency [10]
Respiratory involvement may be seen with FSHD. Evaluation of the symptoms and signs of respiratory insufficiency should be sought during routine clinic visits in patients with moderate to severe FSHD. Regular monitoring of respiratory function is suggested as individuals might experience insufficiency over a long period of time without presenting signs.
Symptomatic respiratory insufficiency can be initially managed with nighttime noninvasive pressure support, e.g., a BiPAP machine. In very severe cases, patients may require the use of a ventilator. For FSHD patients with respiratory insufficiency, in standard practice, trauma (ER, ICU), surgery, and anesthesiology settings, care should be taken not to suppress respiratory drive with narcotics unless it is a situation of palliative care. It is important to notify the doctors about FSHD and any respiratory problems the patient might have or be at risk for.
Oxygen supplementation can be detrimental to patients with hypercarbic (high CO2) respiratory failure and lead to worsening CO2 levels. Oxygen should generally not be administered unless BiPAP or similar ventilatory support is also being used. Consultation with the patient’s primary physician and a pulmonologist can enable periodic monitoring of CO2 levels in the office/school setting or pulmonary function lab in the hospital.
References
Landouzy L, Déjérine J. De la myopathie atrophique progressive. Rev Med. 1885;5:81–117, 253–366.
Upadhyaya M, Cooper DN, editors. FSHD, facioscapulohumeral muscular dystrophy: clinical medicine and molecular cell biology. London: BIOS Scientific Publishers; 2004; ISBN: 18599 62440.
Facts and Statistics about FSHD. University of Massachusetts Medical School. http://www.umassmed.edu/wellstone/aboutfshd/FSHDfacts/. Accessed 11 Jun 2014.
Flanigan KM, et al. Genetic characterization of a large, historically significant Utah kindred with facioscapulohumeral muscular dystrophy. Neuromuscul Disord. 2001;11:525–9.
Mostacciuolo ML, et al. Facioscapulohumeral muscular dystrophy: epidemiological molecular study in a north-east Italian population sample. Clin Genet. 2009;75:550–5.
Prevalence of rare diseases: bibliographic data, www.orpha.net, May 2014 Number 1, Orphanet Report Series. http://www.orpha.net/orphacom/cahiers/docs/GB/Prevalence_of_rare_diseases_by_alphabetical_list.pdf. Accessed 30 May 2014.
FSHD Canada Foundation website. http://www.fshd.ca/what_overview.html. Accessed 9 June 2014.
Fred Hutchinson Cancer Research Center partners with GlaxoSmithKline to develop muscular dystrophy therapeutics. Newswise website. http://www.newswise.com/articles/fred-hutchinson-cancer-research-center-partners-with-glaxosmithkline-to-develop-muscular-dystrophy-therapeutics (2012). Accessed 25 Oct 2013.
Lunt PW, Harper PS. Genetic counseling in facioscapulohumeral muscular dystrophy. J Med Genet. 1991;28:655–64.
FSH Society website. http://www.fshsociety.org/pages/about.html. Accessed 23 May 2014.
Tyler FH, Stephens FE. Studies in disorders of muscle: II: clinical manifestations and inheritance of facioscapulohumeral muscular dystrophy in a large family. Ann Int Med. 1950;32:640–60.
Padberg GW. Facioscapulohumeral disease. Thesis, Leiden; 1982.
Darras BT. UptoDate: facioscapulohumeral muscular dystrophy. Wolters Kluwer Health; 2014, p. 16.
Van der Kooi EL, Kalkman JS, Linderman E, et al. Effects of training and albuterol on pain and fatigue in facioscapulohumeral muscular dystrophy. J Neurol. 2007;254:931.
Jensen MP, Hoffman AJ, Stoelb BL, et al. Chronic pain in persons with myotonic dystrophy and fasioscapulohumeral muscular dystrophy. Arch Phys Med Rehabil. 2008;89:320.
Padua L, Aprile I, Frusciante R, et al. Quality of life and pain in patients with facioscapulohumeral muscular dystrophy. Muscle Nerve. 2009;40:200.
Facts about genetics and neuromuscular diseases, copyrighted by the Muscular Dystrophy Association (MDA); 2011, p. 15.
Gabriels J, Beckers MC, Ding H, et al. Nucleotide sequence of the partially deleted D4Z4 locus in a patient with FSHD identifies a putative gene within each 3.3kb element. Gene. 1999;236:25.
Richards M, Coppee F, Thomas N, et al. Facioscalpulohumeral muscular dystrophy (FSHD): an enigma unraveled? Hum Genet. 2012;131:325.
Lemmers RJ, Miller DG, van der Maarel SM. Facioscapulohumeral muscular dystrophy. Gene Rev. http://www.ncbi.nlm.nih.gov/books/NBK1443/. Accessed 28 Jun 2014.
Stratland JM, Tawi R. Facioscapulohumeral muscular dystrophy: molecular pathological advances and future directions. Curr Opin Neurol. 2001;24:423.
Kohler J, Rupilius B, Otto M, et al. Germline mosaicism in 4q35 facioscapulohumeral muscular dystrophy (FSHD1A) occurring predominantely in oogenesis. Hum Genet. 1996;98:485.
Bakker E, Van der Wielen MJ, Voorhoeve E, et al. Diagnostic, predictive, and prenatal testing for facioscapulohumeral muscular dystrophy: diagnostic approach for sporadic and familial cases. J Med Genet. 1996;33:29.
van Overveld PG, Lemmers RJ, Sandkuijl LA, et al. Hypomethylation of D4Z4 in 4q-linked and non-4q-linked facioscapulohumeral muscular dystrophy. Nat Genet. 2003;35:315.
Lemmers RJ, Tawul R, Petek LM, et al. Digenic inheritance of an SMCHD1 mutation and an FSHD-permissive D4Z4 allele causes facioscapulohumeral muscular dystrophy type 2. Nat Genet. 2012;44:1370.
Zatz M, Marie SK, Cerqueira A, et al. The facioscapulohumeral muscular dystrophy (FSHD1) gene affects males more severely and more frequently than females. Am J Med Genet. 1998;77:155.
Tonini MM, Passos-Bueno MR, Cerqueira A, et al. Asymptomatic carriers and gender differences in facioscapulohumeral muscular dystrophy (FSHD). Neuromuscul Disord. 2004;14:33–8.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Huml, R.A., Perez, D.P. (2015). FSHD: The Most Common Type of Muscular Dystrophy?. In: Huml, R. (eds) Muscular Dystrophy. Springer, Cham. https://doi.org/10.1007/978-3-319-17362-7_3
Download citation
DOI: https://doi.org/10.1007/978-3-319-17362-7_3
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-17361-0
Online ISBN: 978-3-319-17362-7
eBook Packages: MedicineMedicine (R0)