
Abstract
This chapter describes computational approaches to study amyloid formation. The first part addresses identification of potential amyloidogenic regions in the amino acid sequences of proteins and peptides. Next, we discuss nucleation and aggregation sites in protein folding and misfolding. The last part describes up-to-date kinetic models of amyloid fibrils formation. Numerous studies show that protein misfolding is initiated by specific amino acid segments with high amyloid-forming propensity. The ability to identify and, ultimately, block such segments is very important. To this end, many prediction algorithms have been developed which vary greatly in their effectiveness. We compared the predictions for 30 proteins by using different methods and found that, at best, only 50 % of residues in amyloidogenic segments were predicted correctly. The best results were obtained by using the meta-servers that combine several independent approaches, and by the method PASTA2. Thus, correct prediction of amyloidogenic segments remains a difficult task. Additional data and new algorithms that are becoming available are expected to improve the accuracy of the prediction methods, particularly if they use 3D structural information on the target proteins. At the same time, our understanding of the kinetics of fibril formation is more advanced. The current kinetic models outlined in this chapter adequately describe the key features of amyloid nucleation and growth. However, the underlying structural details are less clear, not least because of the apparently different mechanisms of amyloid fibril formation which are discussed. Ultimately, the detailed understanding of the structural basis for amyloidogenesis should help develop rational therapies to block this pathogenic process.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Amyloidogenic segments
- Amyloid prediction methods
- Linear and exponential aggregation kinetics
- Domain swapping
- Nucleation of protein folding and misfolding
9.1 Introduction
More than 40 human diseases are currently known to critically involve protein misfolding and deposition as amyloid fibrils in organs and tissues (Chiti and Dobson 2006). These diseases, collectively called amyloidoses, differ in their etiology and clinical presentation and can be classified as primary vs. secondary, acquired vs. hereditary, and systemic vs. focal diseases. Primary amyloidosis is caused by the deposition of a specific protein. An example is AL amyloidosis caused by deposition of immunoglobulin light chains that are overproduced in plasma cells (Hayman et al. 2001). Secondary amyloidosis occurs as a consequence of another underlying disorder. For example, AA amyloidosis, which is a common complication of chronic inflammation, involves deposition of a proteolytic fragment of serum amyloid A (SAA) that is overproduced in inflammation. In contrast to AL and AA that are acquired diseases, most other amyloidoses have a genetic origin and involve autosomal dominant mutations that make a normally soluble globular protein amyloidogenic. Examples include mutations in proteins such as transthyretin, apolipoproteins A-I and A-II, gelsolin, lysozyme, cystatin, fibrinogen, etc. (Benson 2003). These types of amyloidosis are usually systemic diseases affecting multiple tissues and organs (kidney, liver, heart, etc.). In contrast, focal diseases are localized and affect a single organ where amyloid fibers are deposited, such as brain in neurodegenerative diseases. The best known of such disorders involve depositions of amyloid-beta (Aβ) peptide in Alzheimer’s disease and of prion proteins in Creutzfeld-Jakob and Mad Cow diseases. To modulate and, ultimately, block the pathologic transition from the native functional protein conformation into amyloid, it is essential to unravel the properties of the protein sequence and structure underlying this transition.
To form amyloid, a protein molecule must undergo major conformational changes. In fact, the fibril core always consists of β-sheets in which individual β-strands are oriented perpendicular to the main axis of the fibril (Jiménez et al. 1999), whereas the native protein may or may not contain the β-sheet structure. Conversion of amyloidogenic proteins into fibrils is often associated with cytotoxicity (Bucciantini et al. 2004). Notably, immature water-soluble fibrils, pre-fibrillar aggregates and oligomers are typically more toxic to cells than mature insoluble amyloid fibrils (Bucciantini et al. 2004). The structures of fibril precursors, which are rich in β-sheet, and the mechanisms of their toxic action were proposed to be similar for different proteins. This idea is based on the observation that specific antibodies that bind to toxic protofibrils of the Aβ peptide can also bind to fibril precursors formed by other proteins with unrelated amino acid sequences, suggesting structural similarity of these precursors (Kayed et al. 2003).
Importantly, numerous experimental studies show that the ability to form fibrils is not limited to amyloidogenic proteins associated with diseases, but is an inherent property of various structurally unrelated proteins (Chiti et al. 1999; Fändrich et al. 2001). Moreover, an increasing number of proteins are found to form functional amyloid in vivo (Fowler et al. 2007). These findings raise a question: what factors trigger protein misfolding into amyloid? Is it a particular amino acid sequence of a protein, its structural properties, interactions with ligands (such as lipids and heparan sulfate proteoglycans, which are ubiquitous components of amyloid deposits in vivo), or the lack thereof? What mechanisms can protect normal globular proteins from becoming amyloidogenic?
Evolutionary selection against aggregation resulted in an increased content of amino acids that inhibit protein aggregation (Tartaglia et al. 2005), such as Pro that disrupts the β-sheet structure, Gly that confers mobility to the polypeptide chain and thereby increases the entropic cost for ordering (Rauscher et al. 2006), as well as the increased content of charged residues (Kovacs et al. 2010) that confer protein solubility. However, the demands for protein folding (described in part 4 of this chapter) as well as the functional requirements can make it difficult to fully eliminate protein misfolding. In this chapter, we address the basic properties of the primary, secondary and tertiary protein structure that confer amyloidogenic properties, and describe the computational methods that enable one to predict amyloidogenic regions in proteins and peptides and to understand the kinetic steps and the underlying physical processes involved in amyloid fibril formation.
9.2 Identification of Protein Sites Responsible for Amyloid Formation
9.2.1 Structural Determinants of Amyloidogenic Propensity of Proteins and Peptides
Although it is currently accepted that most if not all proteins can form amyloid fibers under certain conditions in vitro (Chiti and Dobson 2006), it remains a major challenge to predict whether or not a given protein or peptide actually forms amyloid at near-physiologic conditions. The difficulty stems, in part, from a wide range of external and internal factors that can influence protein misfolding and amyloid formation in vivo and in vitro. External factors such as the local pH and the interactions (or lack thereof) with various ligands can critically modulate protein folding and shift the balance towards misfolding and aggregation (Fändrich et al. 2001). Protein mutations and post-translational modifications can also importantly influence amyloid formation. Another crucial determinant is the protein concentration determined by the balance between the generation of a potentially amyloidogenic protein or peptide and its proteolysis and clearance. Among the major internal determinants are the overall stability of the native protein conformation (addressed below) and the presence of local protein regions with high propensity to initiate the misfolding. The latter aspect is addressed later in parts 2 and 3 of this chapter.
In vivo and in vitro studies consistently show that reduced structural stability of globular proteins tends to promote amyloid fibril formation (Chiti and Dobson 2006). In vitro studies demonstrate that mildly denaturing conditions leading to partial protein unfolding promote fibril growth (Fändrich et al. 2003), perhaps due to increased solvent accessibility of the aggregation-prone regions (Dobson 1999). This idea is supported by the observation that many naturally occurring mutations and variations associated with amyloid diseases reduce protein stability (Gertz and Rajkumar 2010). However, exceptions from this general trend have also been observed in various proteins, such as immunoglobulin light chains or apolipoprotein A-I (Sánchez et al. 2006; Klimtchuk et al. 2010; Das et al. 2014), suggesting that fibril formation by a mutant protein does not always correlate with its reduced stability. Thus, although partial protein destabilization is necessary for amyloid fiber formation, it is apparently not sufficient.
Notably, many amyloid diseases involve natively unfolded proteins or peptides. Examples described in this volume include Aβ peptide in Alzheimer’s disease, α-synuclein in Parkinson’s disease, amylin in type 2 diabetes, serum amyloid A in inflammation-linked amyloidosis, and several small apolipoproteins such as apoA-II or apoC-II that readily form amyloid fibrils in vivo and/or in vitro. Partial folding of these intrinsically disordered proteins is believed to be prerequisite for their β-aggregation (see Chap. 2 by Uversky in this volume). Thus, the current paradigm in the field is that amyloid formation is initiated by the partially folded structural intermediates. However, such partial folding alone is often insufficient to form amyloid, suggesting that additional factors are involved.
Extensive experimental evidence accumulated since 1990s suggests that protein misfolding is usually initiated by specific amino acid sequence motives that, when exposed to solvent, are more liable to aggregation than the rest of the polypeptide sequence (Tenidis et al. 2000; von Bergen et al. 2000; Ivanova et al. 2004). A protein can become non-amyloidogenic upon deletion of such motifs (Ivanova et al. 2004); in addition, certain mutations in these sensitive motives can either diminish or amplify the amyloidogenic propensity of a protein (Ivanova et al. 2004). Furthermore, synthetic peptide fragments corresponding to these amyloidogenic regions, which are sometimes as short as five residues (López de la Paz and Serrano 2004), can form amyloid fibrils similar to those formed by the full-length proteins (Thompson et al. 2000). Identification of such regions in proteins and peptides is crucial for understanding the mechanism of amyloid formation and, ultimately, for developing targeted therapies to modulate or block amyloidosis.
9.2.2 Development of Prediction Methods for Amyloidogenic Regions
One of the earlier prediction methods of amyloidogenic regions is based on the idea that protein misfolding is initiated by the packing defects in the tertiary structure, which lead to increased solvent accessibility of the backbone hydrogen bonds, termed “insufficient wrapping” (Fernández et al. 2003). The authors reported that 10 % of protein structures deposited to the Protein Data Bank have such packing defects, and proposed an algorithm to search for such potentially labile structural regions. Obviously, this method requires detailed knowledge of the 3D structure of the target protein, which is not always available.
Most other prediction methods do not rely on the 3D structural information of the target protein and use the primary structure as an input. Since fibril formation involves conformational changes leading to an increased β-sheet content (Jiménez et al. 1999; Yoon and Welsh 2004), a computational algorithm was proposed to search for polypeptide chain segments with high propensity to form β-sheet. This approach can identify short segments with β-sheet propensity, yet it cannot predict whether or not a given protein is likely to form amyloid.
Another method to predict possible amyloidogenic regions in the primary sequence is based on the experimental studies of amyloidogenic properties of six-residue synthetic peptides with various amino acid sequences (López de la Paz and Serrano 2004). The authors determined the hexapeptide sequence (STVIIE) that has the highest propensity to form amyloid fibrils in vitro, and used this motif to search for amyloidogenic regions in other proteins. Interestingly, amyloid fiber formation was observed upon insertion of the hexapeptide STVIIE at the N-terminus of the SH3-domain of α-spectrin, a water-soluble protein that normally does not form fibrils (Esteras-Chopo et al. 2005). Thus, the combined computational and experimental studies supported the idea that the amyloidogenic propensity of a protein can be localized in short residue segments.
This idea was further supported in studies of murine β2-microglobulin that normally does not form amyloid. The variable segment in residues 83–89 was substituted for a seven-residue sequence (NHVTLSQ) from human β2-microglobulin that forms amyloid. The substitution induced amyloid formation by the murine protein in vitro (Ivanova et al. 2004). Importantly, this synthetic amyloidogenic heptapeptide (NHVTLSQ) forms amyloid in solution, whereas the peptide with a similar amino acid composition but scrambled sequence (QVLHTSN) does not. Studies such as this clearly show that not only the composition of the amino acids but also their order determines the amyloidogenic properties. Furthermore, on the basis of their experimental studies the authors proposed a structural model of β2-microglobulin amyloid in which a β-zipper spine is decorated with the remaining part of the protein molecule that partially retains its native fold (Ivanova et al. 2004).
Many other studies addressed the link between the amino acid sequence of a protein and its ability to form amyloid (Idicula-Thomas and Balaji 2005). The authors reported that proteins with low thermal stability and increased life time have increased propensity to form amyloid fibrils in vivo, and determined characteristics of the amino acid sequence which correlate with the fibril formation. Their results showed that a seven-residue peptide with a high β-sheet propensity, which was inserted into an α-helix, increased the propensity of this helix to convert into a β-sheet under mildly denaturing conditions. These studies support the idea that a particular composition and sequence of short amino acid stretches is crucial for amyloid formation. The authors proposed a function for predicting amyloidogenic properties of proteins on the basis of their amino acid sequences, and tested it by using the SwissProtein data base. The results suggested that 32 % of all proteins in the database were amyloidogenic; further, of the 54 proteins that readily formed fibrils, 75 % were correctly identified as amyloidogenic.
9.2.3 FoldAmyloid Algorithm
Several research groups have developed methods for identifying regions within polypeptide chains that are responsible for amyloid formation. One of such methods proposed by our team is FoldAmyloid (Galzitskaya et al. 2006; Garbuzynskiy et al. 2010). FoldAmyloid is based on the well-known concept of enthalpy-entropy compensation stating that a sufficient number of contacts between residues, which provide favorable enthalpy contribution to the free energy of protein stability, is required to compensate for the loss of conformational entropy upon protein arrangement into a more organized compact state (Galzitskaya et al. 2000). Since the enthalpy is determined by the combined strength of the short-range packing interactions, we hypothesized that if the mean expected packing density, which determines the average number of residue contacts within a given distance, is lower than the threshold (i. e. the normal packing density for globular proteins), the protein will remain unfolded. Alternatively, if the mean expected packing density exceeds the threshold, resulting in an increased number of residue contacts, this will favor amyloid formation. In fact, since amyloid fibrils are thermostable, insensitive to proteases, and rich in β-sheet (Kajava et al. 2004), they are expected to contain such densely packed regions.
We tested this hypothesis computationally and demonstrated that the ability of proteins to fold and form such densely packed regions is often responsible for amyloid formation (Garbuzynskiy et al. 2010). In addition to the packing density for individual amino acids (contact scale), we obtained the probability scales inferred from the statistical analyses of protein structures, such as the scale for the main chain hydrogen bond formation (donor scale). These scales were incorporated into server FoldAmyloid (http://bioinfo.protres.ru/FoldAmyloid) for predicting amyloidogenic regions in a protein sequence.
The server was tested on a database (http://bioinfo.protres.ru/fold-amyloid/amyloid_base.html) containing 144 peptides that readily form amyloid as well as 263 peptides that do not. The contact scale correctly identified 75 % of amyloid-forming peptides and 74 % of peptides that did not form amyloid. The donor scale correctly determined 69 % of peptides that formed amyloid and 78 % of those that did not. Using these scales with an equal weight, we created a hybrid scale that predicted correctly 80 % of amyloid-forming peptides and 72 % of peptides that did not form amyloid (Garbuzynskiy et al. 2010).
9.2.4 Other Prediction Methods
During the last decade, several algorithms for predicting amyloidogenic segments have been developed and improved. One of such algorithms, Zyggregator (Tartaglia and Vendruscolo 2008), uses side chain hydrophobicity, the tendency to form α-helices and β-sheets, and the protein net charge to determine the aggregation profile of a protein and predict its folding rate (Chiti et al. 2003). This method is available online at http://www-vendruscolo.ch.cam.ac.uk/ggt23.html.
The method Tango (Fernandez-Escamilla et al. 2004), preceded by Agadir (Muñoz and Serrano 1994), predicts the protein’s probability to form a particular secondary structure. Agadir uses a statistical analysis of the empirical properties of amino acids based on 3D protein structures to calculate the relative probability of amino acid stretches to fold into a helical or globular conformation. Tango employs a similar approach but considers four possible structural states: α-helix, β-turn, α-helical and β-sheet aggregates, as well as the unfolded (random coil) state (Fernandez-Escamilla et al. 2004). This method is available online at http://tango.crg.es/protected/academic/calculation.jsp.
Several more recent methods such as Waltz employ machine-learning algorithms and are trained on the data sets of peptides with empirically determined amyloidogenic properties (Maurer-Stroh et al. 2010). Waltz was trained on the database of hexapeptides about one half of which tends to form amyloid at neutral pH. In contrast to black-box methods where the weights assigned to individual amino acids have no physical meaning, the weights is Waltz generally represent the amyloid-forming propensity of amino acids. To obtain the weight values, the authors aligned the training set onto itself and created a position-specific scoring matrix, which reflects the probability of a given type of amino acid to be found in a particular position in the amyloidogenic hexapeptide. Interestingly, the results showed that hydrophobic and aromatic residues are favored in the middle of the hexapeptide; this contrasts with the overall tolerance for placement of charged and polar amino acids. The resultant algorithm is based on a combination of the position-specific scoring matrices and additional empirical information on the amyloid-forming propensity obtained from the physicochemical analyses of the designed set of hexapeptides. In addition, the authors proposed to distinguish the aggregates by their morphology as either fibril-like or amorphous, as the properties of peptides that tend to form such aggregates distinctly differ. In general, Waltz was reported to achieve better prediction results than its predecessor, Tango (Maurer-Stroh et al. 2010).
The SecStr method (Hamodrakas et al. 2007) is based on the hypothesis that regions with a high predisposition to form α-helices as well as β-sheets, as determined by at least three methods for secondary structure prediction, can act as conformation switches that tend to promote amyloid formation. The program is available online at http://biophysics.biol.uoa.gr.
Among the amyloid prediction methods, a special place belongs to AmylPred2, a meta-server for consensus analysis (Tsolis et al. 2013). The server combines 11 methods including FoldAmyloid, Tango, and Waltz. The authors show that, judging from the Matthews correlation coefficient that measures the quality of binary classifications in machine learning, AmylPred2 outperforms its composite methods. However, the results suggest that the overall sensitivity of all methods is relatively low (~50 % for the best cases, ~40 % for AmylPred2) due to significant overprediction of amyloidogenic regions. Other problems emerging from the analysis of certain target proteins where that the predictions by various methods vary greatly. Nevertheless, with inclusion of more experimental data and additional prediction methods, the server can become very useful in finding consensus among various algorithms and predicting amyloidogenic segments with greater reliability. This server is available at http://biophysics.biol.uoa.gr/onlinetools.html.
PASTA2 is a good example of a support vector machine method, which is based on the regression analysis to build a model that divides a given dataset into classes on the basis of the training examples. This method shows an excellent potential for identifying amyloidogenic regions. PASTA2 is an extension of the PASTA method that calculates the propensity of a polypeptide segment to form a cross-β structure on the basis of hydrogen-bonding statistics found in the β-strands. The authors used the training set consisting of ~2,500 protein domains, each under 100 a. a. long, with known high-resolution structures (<1.8 Å resolution). In addition to the energetic parameters, PASTA2 predicts structural features such as the secondary structure (e. g. parallel or antiparallel β-sheet) and intrinsic disorder. According to the authors’ data, this approach can outperform AmylPred2; however, overprediction of amyloidogenic segments remains an issue with PASTA (Trovato et al. 2006; Walsh et al. 2014). The PASTA 2.0 server can be accessed at http://protein.bio.unipd.it/pasta2/.
One of the recently proposed methods, termed GAP, is a result of advances in machine learning (Thangakani et al. 2014). The method is based on the idea of “paired frequencies” postulating that, since the N and N + 2 side chains are similarly oriented in any β-strand, amyloid-forming sequences are expected to have position-specific amino acid propensities similar to those found in the secondary structures of globular proteins. GAP has been tested on 310 amyloid-forming peptides. In spite of its relatively poor performance on amyloidogenic proteins (described in the next section), the method yielded several interesting results. For example, the propensities for β-structure formation in globular proteins differed from those in amyloid-forming peptides (Thangakani et al. 2014). The authors also found that, in spite of the partial overlap, there was a distinct difference in the pairing propensities of the amino acids for the peptides forming amyloid fibrils versus amorphous β-structured aggregates.
The growing volume of experimental data on amyloid formation by proteins and peptides, combined with the development of new computational approaches, leads to continuous improvement in the accuracy of the predictions. Still, these predictions have inherent limitations that are discussed below.
9.3 Experimental Verification of Theoretical Amyloid Predictions
9.3.1 Two Types of Amyloidogenic Segments
The locations of amyloidogenic regions have been determined by experimental methods, such as mass spectrometry, for a number of globular proteins and peptides that readily form fibers in vivo or in vitro. Many of these proteins and peptides are listed in Table 9.1. In addition, several hundred artificial peptides have been shown to form fibrillar aggregates in vitro; most of these peptides were derived from the amyloidogenic segments of natural proteins. Our analysis of the available experimental data from these proteins and peptides revealed that most amyloidogenic segments have one common property: they have an elevated content of hydrophobic residues (Galzitskaya et al. 2006).
The only notable exception are prion domains of yeast proteins, such as Sup35 and Ure2, which have a high content of polar residues, particularly asparagine (N) and glutamine (Q) (Nelson et al. 2005). The X-ray crystal structure of the GNNQQNY peptide responsible for Sup35 aggregation was determined by (Nelson et al. 2005). The results revealed that hydrogen bonds formed by the protein backbone and the side chains are important for structural stabilization. Aggregates of a similar type can be formed in numerous disease-related proteins containing long polyglutamine tracts (up to several dozen Gln) whose length varies among the patients; particularly long polyQ tracts lead to protein aggregation (Yang et al. 2014) in disorders such as Huntington’s disease.
In sum, there are two distinct types of amyloidogenic regions found in naturally occurring proteins. The first type is rich in hydrophobic residues and stabilizes the fibril via the hydrophobic interactions. The second type contributes to fibril formation via the hydrogen bonds formed by polar residues such as Gln and Asn in prion-like domains and in polyQ proteins. Most amyloid prediction methods, including FoldAmyloid (Galzitskaya et al. 2006; Garbuzynskiy et al. 2010), are designed to search for the regions of the first type, which have been subjects of extensive experimental studies. Although FoldAmyloid potentially allows for identification of amyloidogenic regions of the second type, the existing experimental data are insufficient to test this aspect of its performance.
9.3.2 Performance of Prediction Algorithms Using 30 Amyloidogenic Proteins
To test the performance of various sequence-based prediction methods, we used experimentally defined amyloidogenic regions in proteins which were originally compiled and tested by (Walsh et al. 2014). Two methods, Waltz and FoldAmyloid, which were not included in the original work, were added with some modifications. For MetAmyl, which is a meta-server that predicts amyloidogenic regions by using a combination of four methods, we inherited the results from the original analysis that showed good performance by this method (Emily et al. 2013). We excluded the FISH-Amyloid (Gasior and Kotulska 2014) and FoldAmyloid hybrid methods whose predictions were less accurate. Further, we excluded four prion proteins from the test set since their amyloidogenic regions have a distinct amino acid composition, which necessitates specialized prediction algorithms such as that proposed by (Alberti et al. 2009). The final test set consisted of 30 amyloidogenic proteins and peptides listed in Table 9.1.
Our analysis has two limitations. First, for practical reasons, only a subset of the peptide fragments of the test proteins has been studied experimentally. Therefore, in many cases it is not known whether (i) all predicted non-amyloidogenic residues are truly non-amyloidogenic (except for the experimentally verified amyloidogenic regions predicted as non-amyloidogenic, i. e. false negatives); (ii) all regions predicted to form amyloid actually do so. However, one should keep in mind that proteins are not expected to have many amyloidogenic regions. Second, the average length of the experimentally verified amyloidogenic regions in the test proteins is much larger than the polypeptide segments actually forming the amyloid core (usually 5–11 amino acids) (Gilead and Gazit 2005), which increases the probability of predicting correct amyloidogenic regions.
With these caveats, our analysis clearly showed that, despite high prediction performance stated in many publications, the actual performance is not very good as judged by the number of total true positives. As expected, the best predictors are meta-servers that combine several independent approaches. The top predictor, MetAmyl, yielded more than half correctly predicted amyloidogenic residues (661 out of 986). However, it also had the highest overprediction rate: almost four times more regions have been predicted to be amyloidogenic than experimentally confirmed. Notably, MetAmyl, which is based on a consensus of four methods, predicts more true positives than another meta-server, AmylPred2, which uses 11 methods. Among the non-meta-servers, PASTA2 and FoldAmyloid yielded the best results; notably, the results for PASTA2 were close to those for the meta-servers. This relatively good performance may be due to the fact that both PASTA2 and FoldAmyloid use averaged structural information on globular proteins to optimize the prediction performance, although the protein datasets for these methods are different.
Several methods such as Waltz, Tango and GAP were trained on the database of peptides that form amyloid fibrils in vitro. The description of GAP states that ~90 % true positives were obtained by using the peptides database (Thangakani et al. 2014). However, our test of the proteins listed in Table 9.1 suggests that GAP significantly overpredicts the amyloidogenic regions; for example, GAP predicted ~150 amyloidogenic regions for the first target protein, prolactin, which is unrealistically high. Because of this high overprediction rate, GAP was not included in our final analysis.
Waltz was tested in two regimes, the best performance and the maximal specificity regime. In the regime of maximal specificity, Waltz produced fewer overpredictions than other methods. The obvious drawback of such high specificity is missed amyloidogenic regions, i.e. relatively few true positives and many false negatives. Moreover, comparison of the prediction results by Waltz and its ancestor, Tango, showed that the performance of these two methods is not significantly different (Table 9.1).
The general reason for the limited accuracy of various prediction methods (~50 % correctly predicted residues in amyloidogenic regions by our estimate) is that these methods do not take into account the actual 3D structural information on individual proteins. As a result, the prediction methods are expected to underestimate the influence of long-range interactions among residues that are distant in the primary sequence but close in 3D space. These and other structural features may be crucial, at least for some proteins (for example, see the lipid surface-binding proteins apoA-I and apoA-IV described in Chap. 8 by Das and Gursky in this volume). The effect is further exacerbated when the training dataset for the machine learning algorithms contains only polypeptides but no globular proteins, as was the case with GAP. Another reason for the limited performance may be attributed to particular amino acid sequences giving rise to various types of aggregates, e.g. β-sheet rich amorphous aggregates versus fibrils, as suggested by the peptide studies (Thangakani et al. 2014).
9.3.3 Peptide Test Case: Huntingtin-Based 17-Residue Sequences
To improve the performance of the prediction methods, their results should be compared with the experimental data obtained from a wide range of proteins and peptides. Such a comparison is reported in many recent studies. One example is the study using the amino acid sequence of a 17-residue N-terminal peptide of huntingtin, a polyQ-containing protein that forms amyloid in Huntington’s disease (Roland et al. 2013). The N-terminal 17-mer peptide of huntingtin forms highly α-helical aggregates that do not spontaneously convert into β-sheet-rich fibers under near-physiologic conditions. The authors generated 15 peptides with scrambled sequences and analyzed the aggregation properties of these peptides as well as their ability to form β-sheet-rich fibrils. The experimentally determined amyloidogenic properties were compared with the properties of the scrambled sequences predicted with Zyggregator, Waltz, Zipper, and Tango. Although the peptide that was predicted to be particularly amyloidogenic readily formed amyloid fibrils in vitro, the general quality of the predictions was not very high. Individual methods varied in the number of the predicted amyloidogenic sequences and in their ability to correctly identify the fibril-forming peptides. Most methods overpredicted the peptide amyloidogenicity. The authors proposed that this discrepancy between the theory and the experiment may be due, in part, to experimental limitations (e. g. low micromolar peptide concentrations used in these experiments might have been insufficient for fiber formation by some peptides), as well as to the limited fundamental understanding of the link between the primary peptide structure and the process of amyloid formation. At the same time, underprediction of two out of five amyloid-forming peptides by all four methods demonstrated that, evidently, there are certain amino acid sequences whose propensity to form amyloid is not sufficiently accounted for by these prediction methods.
9.3.4 Protein Test Case: Glucan Transferase Bgl2p
Glucan transferase Bgl2p (230 amino acids) is the major thermostable protein in the yeast cell wall. Amyloidogenic regions in Bgl2p were initially predicted by using FoldAmyloid (Galzitskaya et al. 2006; Garbuzynskiy et al. 2010), followed by the experimental demonstration that this protein readily forms amyloid fibrils in vitro (Kalebina et al. 2008). The protein was predicted to have a strong amyloid-forming potential (seven amyloidogenic segments); this explains why the mutational analysis of this protein by using several amino acid substitutions could not reduce its amyloidogenic potential in vitro.
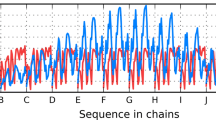
Next, we used a consensus analysis to predict amyloidogenic peptides in Bgl2p. To do so, we used six programs: PASTA, Tango, Waltz, Aggrescan (de Groot et al. 2012), DHPred (Zimmermann and Hansmann 2006), and FoldAmyloid. Residue segments that were predicted to be amyloidogenic by at least four out of six methods were chosen for the peptide synthesis; another peptide, which was predicted by three methods (Aggrescan, DHPred, and FoldAmyloid), has also been synthesized; finally, an additional synthetic peptide fragment was used as a non-amyloidogenic control. Thus, four 10-residue peptides have been synthesized (Fig. 9.1, gray highlight): (1) AEGFTIFVGV (residues 80–89, predicted by up to 5 methods), (2) VDSWNVLVAG (residues 166–175, up to 3 methods), (3) VMANAFSYWQ (residues 187–196, up to 4 methods), and (4) NDVRSVVADI (residues 141–150, 0 methods, non-amyloidogenic) (Bezsonov et al. 2013). Aggregation properties of these peptides and of the full-length protein were studied in the pH range 4.4–7.5. Fluorescence spectroscopy (ThT binding) and fluorescence microscopy clearly showed that peptide 4, which was predicted to be non-amyloidogenic, actually did not form fibrils under any experimental conditions explored. Peptides 1 and 3, which were predicted by up to five methods, readily formed amyloid fibrils in all experiments. Peptide 2 predicted by up to three methods formed amyloid at acidic pH but not at pH 7.5. Full-length protein Bgl2p also readily formed amyloid at pH 4.7 but not at pH 7.5, suggesting a positive correlation in the aggregation behavior of Bgl2p and peptide 2 (Bezsonov et al. 2013). Notably, peptide fragments corresponding to all amyloidogenic regions that were predicted by the consensus methods formed amyloid fibrils, demonstrating the utility of the consensus methods for predicting amyloidogenic properties in vitro.

Potential amyloidogenic determinants in glucan transferase Bgl2p from Saccharomyces cerevisiae cell wall (UniProtKB/TrEMBL entry number P15703). Amino acids that were predicted by individual algorithms to be located in amyloidogenic segments (*); amino acids that were predicted by four or more methods to be amyloidogenic are boxed. The N-terminal signal sequence is in light gray letters. Generally, the signal sequences which bind lipid are largely hydrophobic and are predicted to be amyloidogenic. Sequences of the four peptides that have been synthesized and experimentally investigated are highlighted in gray. “aa” indicates residue numbers; “seq” shows protein amino acid sequence. The number in brackets relates to the serial number of synthesized peptide mentioned in the text (Figure is modified from (Bezsonov et al. 2013))
A more important and, arguably, more challenging task is to identify amyloidogenic segments that are critical to amyloid formation in a biological context, and to predict the effects of protein mutations and modifications on amyloid-forming propensity in vivo. To this end, (Belli et al. 2011) compared the aggregation propensity predicted by different methods with the limited experimental data available on the aggregation propensities of several wild-type and mutant proteins from E. coli. The data were based on the quantitative measurements of the levels of mutant proteins found in inclusion bodies relative to the wild type (Carrió et al. 2005; Wang et al. 2008). The authors concluded that, despite limited ability to predict amyloid formation in a biological context, “algorithms that have been developed to predict amyloid formation in vitro also offer a considerable degree of accuracy for predicting amyloid propensity in vivo.”
9.4 Nucleation and Aggregation Sites in Protein Folding and Misfolding
Since polypeptide chain can either fold into a native structure or misfold and form aggregates or amyloid fibrils, these processes compete, and the outcome can depend on the rate-limiting transition states as well as the folding and misfolding intermediates. For some proteins such as human acylphosphatase, the transition states in folding and aggregation are structurally unrelated (Chiti et al. 2002), suggesting structurally distinct pathways for folding and aggregation. In other proteins such as β2-microglobulin, partially unfolded species that resemble folding intermediates have been implicated in amyloid formation (Jahn et al. 2006), suggesting that the free-energy landscapes for folding and misfolding of these proteins may be related. Identification of a folding intermediate as the key precursor of β2-microglobulin fibril elongation under physiological conditions provided direct experimental evidence that the folding and aggregation landscapes for this protein coincide, at least initially, and diverge only at the level of a native-like folding intermediate that resembles the immunoglobulin fold (Jahn et al. 2006). Thus, there is no single rule describing the relationship between the regions important for folding of the native structure and for amyloid formation.
A crucial rate-limiting event in protein folding is the formation of a folding nucleus, which is a structured part of the polypeptide chain in the high-energy transition state. A detailed analysis of the formation and evolution of the folding nucleus in amyloidogenic proteins may help understand what properties make these proteins amyloidogenic. However, experimental data delineating both the folding nucleus and the amyloidogenic regions in the same protein are often lacking. Since the folding nucleus is unstable, it is difficult to investigate it experimentally. An elaborate experimental approach, called Φ-analysis, has been developed to indirectly assess the structure of the folding nuclei (Matouschek et al. 1989). By introducing point mutations into a protein structure, it is possible to find residues whose mutations have a similar destabilizing effect on the transition state and on the native state. The Φ-value for a mutation in residue r is defined as: Φ = Δ r [F(T) − F(U)] / Δ r [F(N) − F(U)] (9.1)Here Δ r [F(N) − F(U)] is the mutation-induced change in the free energy difference between the native (N) and the unfolded (U) state, and Δ r [F(T) − F(U)] is the mutation-induced change in the free energy difference between the transition (T) state (which is the high-energy rate-limiting state in protein unfolding) and the unfolded (U) state. Most Φ-values vary from 0 to 1; Φ = 1 indicates that the mutated residue is in the folding nucleus. The values of Φ < 0 or Φ > 1 are rare and indicate non-native contacts in the transition state.
Since the Φ-analysis is very labor-intensive, there is general paucity of experimental data identifying folding nuclei in amyloidogenic proteins. To overcome this problem, we used the available data to compare: (i) the experimentally identified amyloidogenic regions with the predicted folding nuclei (Galzitskaya and Finkelstein 1999; Garbuzynskiy et al. 2004) (for proteins with experimentally identified amyloidogenic regions), and (ii) the experimentally identified folding nuclei with the predicted amyloidogenic regions (for proteins with experimentally identified folding nuclei). The results revealed that most experimentally determined amyloidogenic segments (12 regions, Table 9.2) overlap the predicted folding nuclei (Fig. 9.2), and most predicted amyloidogenic segments overlap the experimentally determined folding nuclei (Galzitskaya and Garbuzynskiy 2008; Galzitskaya 2009, 2011a). On average, Φ-values for residues in amyloidogenic regions were significantly greater than those outside these regions. This implies that the amyloidogenic regions tend to overlap the folding nucleus of a native protein structure. Consequently, amyloidogenic regions can nucleate either the normal protein folding or the misfolding into amyloid fibrils, thus playing a key role in the competition between these processes.

Distribution of the predicted Φ-values (Galzitskaya and Finkelstein 1999; Garbuzynskiy et al. 2004) in protein regions that have been demonstrated experimentally to be amyloidogenic (black bars) or non-amyloidogenic (gray bars). The regions from seven proteins have been used for this analysis: acylphosphatase (1aps), β2-microglobulin (1im9), gelsolin (1kcq), transthyretin (1bm7), lysozyme (193l), myoglobin (1wla), and human prion (1qm0)
Furthermore, our sequence and structural analysis suggests that these regions usually contain clusters of large apolar side chains, leading to restricted motions of the polypeptide backbone and thereby helping nucleate the ordered structure. Since protein folding nuclei are determined based on the Φ-value analysis, while the amyloidogenic regions are predicted from the analysis of the primary structure, the overlap between the two regions observed in our studies enables us to predict the nucleation sites for protein folding on the basis of the primary structure analysis (Galzitskaya and Garbuzynskiy 2008; Galzitskaya 2009, 2011a).
As first proposed by Eisenberg’s group, amyloid formation can involve domain swapping whereby two or more polypeptide chains swap identical structural elements to form oligomers (Bennett et al. 1994, 1995). Proteins with a wide range of unrelated amino acid sequences and structures can oligomerize via the domain swapping (Galzitskaya 2011b). The residues from such swapped regions acquire their stable conformation early in the folding process, suggesting that these regions are important for correct protein folding as well as misfolding. We compiled a data base of proteins that contain swapped domains as well as the proteins that have been crystallized in the monomeric form. The folding nuclei were determined based on the monomeric protein structures with the experimental error for Ф-value of ±0.1, and the amyloidogenic segments were predicted using the amino acid sequence analysis by FoldAmyloid. Together, the results showed that, in 11 out of 17 proteins, the regions with Ф > 0.5 that are probably responsible for folding overlapped with the swapped regions of the polypeptide chain. Furthermore, in 11 out of 17 proteins, the swapped regions overlapped with the predicted amyloidogenic regions (Galzitskaya 2011b). These results support the idea that protein regions undergoing domain swapping are often critical for correct protein folding as well as in misfolding.
9.5 Possible Mechanisms and Kinetic Models of Amyloid Growth
9.5.1 Linear Nucleation-Elongation Model
A commonly used method to study amyloid formation is to monitor the time course of amyloid growth by tracking the binding of diagnostic dyes Thioflavin T (ThT) or Congo Red (CR). Binding to amyloid-like aggregates increases fluorescence intensity of these dyes, which can be used to track the increase in concentration of amyloid-like aggregates in real time (Buxbaum and Linke 2012). Such kinetic experiments using fluorescence can be performed relatively easily (e. g. see the Chap. 4 by Singh et al. on amylin in this volume) and can help dissect the complex multistep pathways of amyloid fiber formation.
Amyloid formation can be considered as a polymerization reaction. Most quantitative models for linear polymerization stem from the work performed more than half a century ago (Oosawa et al. 1959) proposing a kinetic model for actin polymerization. The experimental data showed that actin polymerization is akin to a condensation reaction that takes place only if the concentration of the initial reactant (actin) exceeds the critical threshold. The highly cooperative character of the reaction was supported by two observations: (i) increase in actin concentration resulted in a higher reaction rate at early stages; (ii) addition of a nucleus with a pre-formed aggregate led to rapid polymerization of free actin.
The linear polymerization (i. e. nucleation – consecutive elongation) model was used to explain protofibril formation by hemoglobin in sickle-cell anemia (Hofrichter et al. 1974). The reaction had a high free energy barrier for nucleation. The authors defined a nucleus as the least thermodynamically stable oligomer that can initiate further growth of protofibrils.
Frieden and Goddette (1983) developed additional aspects of the linear model of actin polymerization. They noted that each event of monomer attachment to the growing polymer chain has its own rate constant, and that the reaction begins with the monomer activation, which in the case of actin involved Mg2+-induced conformational changes.
Goldstein and Stryer (1986) further explored the linear model of protein polymerization. They defined the nucleus as a “primer” of a certain size whose formation led to changes in kinetic constants. The goal was to explore numerical methods for optimal fitting of various experimental data to the model; important improvements in the experimental approach have also been proposed.
9.5.2 Exponential Growth Model
Even though the nucleation-consecutive elongation is a common mechanism of protein polymerization, many experimental data on protein polymerization and fibril formation cannot be adequately described by this simple model (Foderà et al. 2008; Xue et al. 2008; Cohen et al. 2013). To describe these data, an “exponential growth” mechanism was proposed. This mechanism reflects an increased number of sites for monomer attachment upon fibril growth in processes such as fibril fragmentation, secondary nucleation, branching, etc. (Fig. 9.3).

General scheme for amyloid formation depicting linear and exponential growth models. M monomer, O oligomer, O n* oligomer of critical size n* that forms the nucleus that is the transient species with high free energy, P amyloid aggregate, k m+ rate constant of monomer attachment to the oligomer, k m− rate constant of monomer dissociation from the oligomer, k n+ rate constant of amyloid seed formation (the least stable specie on the reaction pathway), k exp rate constant of the exponential growth event (bifurcation, fragmentation etc.), k p rate constant of monomer attachment to the growing amyloid aggregate, α non-cross-beta aggregate, β cross-beta aggregate
The first experiments that were aimed to test the exponential growth model addressed the kinetics of actin polymerization. Reagents such as Ca2+ and Mg2+ were known to disassemble actin filaments, but the mechanism of their action was unclear. Wegner and Savko demonstrated that actin filaments can undergo spontaneous fragmentation during polymerization reaction. This explained why the nucleation-consecutive elongation model failed to adequately fit the data (Wegner and Savko 1982). Only when fibril fragmentation was accounted for, the model could adequately approximate the kinetic data.
Ferrone et al. (1980) developed a model of heterogeneous nucleation to explain the effect of “extreme autocatalysis” and the strong concentration-dependence observed in aggregation of sickle-cell hemoglobin. This two-step model assumed that, first, regular nucleation leads to the formation of a protofibril; next, additional protofibrils are formed on its surface. This model included two equations describing homogeneous nucleation at the first stage and heterogeneous nucleation at the second stage; the system of equations was solved numerically. The results suggested that the surface available for addition of monomers increases mainly due to the increase in the size of the aggregate, as the protofibril surface can provide new nucleation sites. The concept of heterogeneous nucleation was novel and has importantly contributed to the theory of protein aggregation.
More recently, Miranker and colleagues explored amyloid fibril formation by amylin, a small polypeptide hormone that deposits in type 2 diabetes (also see Chap. 4 by Singh et al. in this volume). Kinetic studies showed that nucleation can proceed via two pathways: protofibril-independent (primary) and protofibril-dependent (secondary). The balance between the two depends on the external interface. In the presence of such an interface, the primary mechanism is dominant; alternatively, the secondary mechanism dominates (Ruschak and Miranker 2007).
Normally, amyloid fibrils are unbranched linear polymers. Interestingly, fibril branching (i. e. growth in a tree-like structure) was observed during amyloid formation by a small hormone glucagon; in these studies, single fibril growth was monitored in real time by using TIRF microscopy (Andersen et al. 2009). Clearly, such branching can lead to exponential growth kinetics.
9.5.3 Mixed Models
Exponential growth model predicts a longer lag phase (nucleation) and/or a faster propagation phase (growth) as compared to the nucleation-consecutive elongation model. Notably, in the latter model, the number of fibers during linear growth is roughly proportional to the number of nuclei formed during the lag phase, because fiber growth is energetically more favorable than the nucleation. Therefore, after a certain time, the number of fibrils becomes constant. In contrast, the number of fibrils continues to increase upon fragmentation (which commonly occurs in amyloid fibrils), branching (which is uncommon in amyloid) and other exponential growth scenarios. This difference is the key distinction between the nucleation-consecutive elongation and the exponential growth models. Since certain experimental data cannot be adequately described by either model alone (Wegner and Savko 1982), several mixed models for protein polymerization have been proposed based on a combination of the nucleation at the first stage and exponential growth at the second stage. In case of amyloid, the most probable mechanism of exponential growth in the second stage is fibril fragmentation (Serio et al. 2000; Xue et al. 2008).
Radford and colleagues approximated the aggregation kinetics of β2-microglobulin with a modular system of kinetic equations (Xue et al. 2008). A set of modules describing various steps in the aggregation mechanism was selected, and various combinations of these modules were used to fit the experimental data obtained by ThT fluorescence. The best fit was obtained by using a model that included a module for polymerization with a consecutive monomer attachment, and another module for fragmentation.
Morris, Finke and colleagues proposed a simple model to describe the process of amyloid aggregation where exponential growth model incorporated a linear relationship between the “ends” of the growing aggregate and its mass (Morris et al. 2009). Although the non-quadratic mass accumulation during early stages of growth can be described by this model, the analytical solution represents a sigmoid curve. Hence, the model did not apply to proteins displaying non-sigmoid reaction kinetics (Giehm and Otzen 2010).
Knowles and colleagues analytically solved equations describing fiber formation with fragmentation (Knowles et al. 2009). Their model included the nucleation stage and the exponential growth stage with fragmentation; the latter was essential for the accurate approximation of the experimental data. The authors reported that nearly all proteins showed linear scaling in the logarithmic coordinates of relative concentration versus relative lag time, with the constant exponential coefficient (i.e. the dependence lnTlag ~ const + γln[C], with a constant γ = −0.5). The nature of such “scaling” was addressed by Cohen et al. (2011) who expanded the model by adding secondary nucleation, i.e. nucleus formation on the surface of growing fibrils, and solved the equations analytically. The authors report that γ depends on the size n of the secondary nucleus, γ = −(n + 1)/2. In the exponential growth model in the absence of bifurcation, n = 0 and hence, γ = −0.5, which is the “scaling” constant (Knowles et al. 2009). Thus γ reflects the specific mechanism of the amyloid formation and can potentially be used to assess the exponential growth mechanism on the basis of the kinetic data.
Recently, we reported a detailed analysis of various kinetic mechanisms of amyloid growth (Dovidchenko et al. 2014). A useful parameter in this analysis is Lrel which describes the ratio between the duration of the lag phase and the time required to include all monomers into the growing polymer (Fig. 9.4). We found that: (i) the linear growth corresponds to a very narrow range of Lrel ≤ 0.2 and occurs only if Lrel is independent of the initial monomer concentration; (ii) these limitations do not apply to the exponential growth. Further, we showed that Lrel is determined by the size of the primary nucleus (n*), which is the smallest least stable aggregate on the reaction pathway, and of the secondary nucleus (n 2), which mediates branching on the surface of the growing fibril. We determined the dependence of Lrel on the initial monomer concentration and used it to calculate n* and n 2. Notably, we found that the scaling effect described by (Knowles et al. 2009) is a general feature of the polymerization reaction which reflects both the nucleus size and the specific scenario for amyloid growth (illustrated in Fig. 9.4).

Alternative scenarios for amyloid growth and the corresponding kinetic parameters. T2 is the time of inclusion of all monomers into the aggregate, Lrel is the ratio between the duration of the lag phase and the time of inclusion of all monomers into the growing polymer, [M∑] is the total monomer concentration, n* is the size of the primary nucleus, and n 2 is the size of the secondary nucleus
To determine the sizes of the primary and secondary nuclei and the mechanism of amyloid growth, we used this model to approximate the experimental kinetic data recorded from eight proteins: insulin, β2-microglobulin, yeast prion Sup35, yeast prion Ure2p, murine WW domain, Aβ42, TI I27, and apolipoprotein C-II. The results are summarized in Table 9.3.
Interestingly, in most cases of exponential growth, the size of the primary nucleus, n*, was close to 1, suggesting that the protein monomer is sufficient to initiate amyloid formation. Alternatively, n* ≅ 1 may relate to a group of protein molecules that act as a single entity in solution; another alternative is that a protein monomer initiates fibril growth from an aggregated state. Thus, kinetic data alone are insufficient to unambiguously determine the fibrillation mechanisms. Since molecular mechanisms of amyloid formation can vary from protein to protein, one ought to use additional experimental techniques (e.g. various types of microscopy, ultracentrifugation, etc.) to carefully rule out alternative scenarios before deciding on the precise mechanism of fibrillation. An example of a combined kinetic and structural approach that utilizes atomic force microscopy to determine the detailed mechanism of amylin fibrillation is described by Jeremic and his team in Chap. 4 of this volume.
In sum, substantial progress has been made in our understanding of the kinetic aspects of amyloid formation. In some cases (such as Aβ42 or apolipoprotein C-II, Table 9.3) it is possible to determine the mechanism of aggregation solely on the basis of the kinetic data, while in many other cases additional structural information is required. By combining computational and experimental approaches, one can determine the size of the primary nucleus, which is the critical state in the fibrillation pathway.
9.6 Concluding Remarks
Despite recent advances in the development and improvement of the sequence-based amyloid prediction algorithms, much remains to be done in this area. The results of our comparative analysis, expanded and averaged in Table 9.4, show that, despite current improvements, individual algorithms have limited accuracy and specificity.
The recent improvements are illustrated by comparing two best-performing individual methods, FoldAmyloid (which is a relatively old and simple approach) and PASTA2 (a new more sophisticated approach that performs best among the non-meta-servers). As evident from Table 9.4, the accuracy and sensitivity of PASTA2 predictions are clearly better than those of FoldAmyloid. At the same time, comparison of the overall prediction quality shows only modest improvement, which is due to low sensitivity partly caused by overprediction and partly by the coarseness of the methods. Even the incorporation of several methods into the meta-servers MetAmyl and AmylPred2 does not drastically improve the results. Apparently, the methods that are entirely sequence-based are approaching their limit.
The problem may perhaps be partially overcome by using the training sets containing small proteins and their domains rather than short peptides forming the amyloid core. However, a greater problem is that the existing prediction methods do not incorporate 3D structural information of the target proteins. Because sequence-based prediction of the 3D structure of globular proteins is still unattainable, accurate prediction of the native environment of the peptides forming the amyloid core is also unattainable. The situation is somewhat analogous to the prediction of the antibody-binding protein epitopes on the basis of the primary structure: in both instances, the properties of interest depend on the 3D protein structure and hence, cannot be accurately predicted based on the amino acid sequence alone. We believe that, to qualitatively improve the amyloid prediction methods, it is perhaps necessary to incorporate additional information that critically influences proteins’ propensity to form amyloid, such as the native 3D structure. In fact, a prediction test carried out on natively-unfolded amyloidogenic proteins and peptides (Ahmed and Kajava 2013) showed much better performance than the analysis of folded proteins reported in this chapter. Ultimately, in addition to 3D structure, other important factors such as the protein dynamics and the environmental conditions (e. g. the presence of lipid membranes) should also be considered.
Significant progress has been achieved in our understanding of the link between the normal folding and the misfolding of proteins, and in elucidating the kinetic features of protein misfolding and aggregation. Sophisticated kinetic models have been proposed to accurately describe the complex pathways of protein fibrillation, which include nucleation, branching, fragmentation, and growth from the surface. However, detailed structural understanding of these reaction steps is still lacking for most proteins. Such a detailed structural understanding may possibly provide a key element to improve the accuracy of amyloid prediction.
Prediction of protein fibrillation in vivo remains a major challenge, since a wide array of environmental factors can influence fiber nucleation and growth in the biological context. For example, most amyloid deposits found in vivo contain additional components (such as lipids, cell membrane components such as heparan sulfate proteoglycans, and apolipoproteins), and the complex role of these components in amyloidogenesis is far from clear. Moreover, some proteins form so-called functional amyloids in living cells, wherein the assembly and disassembly of fibrils occurs in response to biological clues (Chiti and Dobson 2006). Understanding the mechanisms of fibril assembly and disassembly in vivo and in vitro is not only of fundamental scientific importance, but may also help develop new therapeutic targets against amyloidosis.
References
Ahmed AB, Kajava AV (2013) Breaking the amyloidogenicity code: methods to predict amyloids from amino acid sequence. FEBS Lett 587:1089–1095
Alberti S, Halfmann R, King O, Kapila A, Lindquist S (2009) A systematic survey identifies prions and illuminates sequence features of prionogenic proteins. Cell 137:146–158
Andersen CB, Yagi H, Manno M, Martorana V, Ban T, Christiansen G, Otzen DE, Goto Y, Rischel C (2009) Branching in amyloid fibril growth. Biophys J 96:1529–1536
Belli M, Ramazzotti M, Chiti F (2011) Prediction of amyloid aggregation in vivo. EMBO Rep 12:657–663
Bennett MJ, Choe S, Eisenberg D (1994) Domain swapping: entangling alliances between proteins. Proc Natl Acad Sci U S A 91:3127–3131
Bennett MJ, Schlunegger MP, Eisenberg D (1995) 3D domain swapping: a mechanism for oligomer assembly. Protein Sci 4:2455–2468
Benson MD (2003) The hereditary amyloidoses. Best Pract Res Clin Rheumatol 17:909–927
Bezsonov EE, Groenning M, Galzitskaya OV, Gorkovskii AA, Semisotnov GV, Selyakh IO, Ziganshin RH, Rekstina VV, Kudryashova IB, Kuznetsov SA, Kulaev IS, Kalebina TS (2013) Amyloidogenic peptides of yeast cell wall glucantransferase Bgl2p as a model for the investigation of its pH-dependent fibril formation. Prion 7:175–184
Binger KJ, Pham CLL, Wilson LM, Bailey MF, Lawrence LJ, Schuck P, Howlett GJ (2008) Apolipoprotein C-II amyloid fibrils assemble via a reversible pathway that includes fibril breaking and rejoining. J Mol Biol 376:1116–1129
Bucciantini M, Calloni G, Chiti F, Formigli L, Nosi D, Dobson CM, Stefani M (2004) Prefibrillar amyloid protein aggregates share common features of cytotoxicity. J Biol Chem 279:31374–31382
Buxbaum JN, Linke RP (2012) A molecular history of the amyloidoses. J Mol Biol 421:142–159
Carrió M, González-Montalbán N, Vera A, Villaverde A, Ventura S (2005) Amyloid-like properties of bacterial inclusion bodies. J Mol Biol 347:1025–1037
Chamberlain AK, MacPhee CE, Zurdo J, Morozova-Roche LA, Hill HA, Dobson CM, Davis JJ (2000) Ultrastructural organization of amyloid fibrils by atomic force microscopy. Biophys J 79:3282–3293
Chiti F, Dobson CM (2006) Protein misfolding, functional amyloid, and human disease. Annu Rev Biochem 75:333–366
Chiti F, Webster P, Taddei N, Clark A, Stefani M, Ramponi G, Dobson CM (1999) Designing conditions for in vitro formation of amyloid protofilaments and fibrils. Proc Natl Acad Sci U S A 96:3590–3594
Chiti F, Taddei N, Baroni F, Capanni C, Stefani M, Ramponi G, Dobson CM (2002) Kinetic partitioning of protein folding and aggregation. Nat Struct Biol 9:137–143
Chiti F, Stefani M, Taddei N, Ramponi G, Dobson CM (2003) Rationalization of the effects of mutations on peptide and protein aggregation rates. Nature 424:805–808
Cohen SIA, Vendruscolo M, Welland ME, Dobson CM, Terentjev EM, Knowles TPJ (2011) Nucleated polymerization with secondary pathways. I. Time evolution of the principal moments. J Chem Phys 135:065105
Cohen SIA, Linse S, Luheshi LM, Hellstrand E, White DA, Rajah L, Otzen DE, Vendruscolo M, Dobson CM, Knowles TPJ (2013) Proliferation of amyloid-β42 aggregates occurs through a secondary nucleation mechanism. Proc Natl Acad Sci U S A 110:9758–9763
Collins SR, Douglass A, Vale RD, Weissman JS (2004) Mechanism of prion propagation: amyloid growth occurs by monomer addition. PLoS Biol 2:e321
Das M, Mei X, Jayaraman S, Atkinson D, Gursky O (2014) Amyloidogenic mutations in human apolipoprotein A-I are not necessarily destabilizing – a common mechanism of apolipoprotein A-I misfolding in familial amyloidosis and atherosclerosis. FEBS J 281:2525–2542
De Groot NS, Castillo V, Graña-Montes R, Ventura S (2012) AGGRESCAN: method, application, and perspectives for drug design. Methods Mol Biol 819:199–220
Dobson CM (1999) Protein misfolding, evolution and disease. Trends Biochem Sci 24:329–332
Dovidchenko NV, Finkelstein AV, Galzitskaya OV (2014) How to determine the size of folding nuclei of protofibrils from the concentration dependence of the rate and lag-time of aggregation. I. Modeling the amyloid protofibril formation. J Phys Chem B 118:1189–1197
Emily M, Talvas A, Delamarche C (2013) MetAmyl: a METa-predictor for AMYLoid proteins. PLoS One 8:e79722
Esteras-Chopo A, Serrano L, López de la Paz M (2005) The amyloid stretch hypothesis: recruiting proteins toward the dark side. Proc Natl Acad Sci U S A 102:16672–16677
Fändrich M, Fletcher MA, Dobson CM (2001) Amyloid fibrils from muscle myoglobin. Nature 410:165–166
Fändrich M, Forge V, Buder K, Kittler M, Dobson CM, Diekmann S (2003) Myoglobin forms amyloid fibrils by association of unfolded polypeptide segments. Proc Natl Acad Sci U S A 100:15463–15468
Ferguson N, Berriman J, Petrovich M, Sharpe TD, Finch JT, Fersht AR (2003) Rapid amyloid fiber formation from the fast-folding WW domain FBP28. Proc Natl Acad Sci U S A 100:9814–9819
Fernández A, Kardos J, Scott LR, Goto Y, Berry RS (2003) Structural defects and the diagnosis of amyloidogenic propensity. Proc Natl Acad Sci U S A 100:6446–6451
Fernandez-Escamilla A-M, Rousseau F, Schymkowitz J, Serrano L (2004) Prediction of sequence-dependent and mutational effects on the aggregation of peptides and proteins. Nat Biotechnol 22:1302–1306
Ferrone FA, Hofrichter J, Sunshine HR, Eaton WA (1980) Kinetic studies on photolysis-induced gelation of sickle cell hemoglobin suggest a new mechanism. Biophys J 32:361–380
Foderà V, Librizzi F, Groenning M, van de Weert M, Leone M (2008) Secondary nucleation and accessible surface in insulin amyloid fibril formation. J Phys Chem B 112:3853–3858
Fowler DM, Koulov AV, Balch WE, Kelly JW (2007) Functional amyloid – from bacteria to humans. Trends Biochem Sci 32:217–224
Frieden C, Goddette DW (1983) Polymerization of actin and actin-like systems: evaluation of the time course of polymerization in relation to the mechanism. Biochemistry (Mosc) 22:5836–5843
Galzitskaya OV (2009) Are the same or different amino acid residues responsible for correct and incorrect protein folding? Biochem Biokhimii͡a 74:186–193
Galzitskaya OV (2011a) Misfolded species involved regions which are involved in an early folding nucleus. In: Haggerty LM (ed) Protein struct. Nova Science Publishers, Hauppauge, pp 1–30
Galzitskaya OV (2011b) Regions which are responsible for swapping are also responsible for folding and misfolding. Open Biochem J 5:27–36
Galzitskaya OV, Finkelstein AV (1999) A theoretical search for folding/unfolding nuclei in three-dimensional protein structures. Proc Natl Acad Sci U S A 96:11299–11304
Galzitskaya OV, Garbuzynskiy SO (2008) Folding and aggregation features of proteins. In: O’Doherty CB, Byrne AC (eds) Protein misfolding. Nova Science Publishers, New York, pp 99–112
Galzitskaya OV, Surin AK, Nakamura H (2000) Optimal region of average side-chain entropy for fast protein folding. Protein Sci 9:580–586
Galzitskaya OV, Garbuzynskiy SO, Lobanov MY (2006) FoldUnfold: web server for the prediction of disordered regions in protein chain. Bioinformatics 22:2948–2949
Garbuzynskiy SO, Finkelstein AV, Galzitskaya OV (2004) Outlining folding nuclei in globular proteins. J Mol Biol 336:509–525
Garbuzynskiy SO, Lobanov MY, Galzitskaya OV (2010) FoldAmyloid: a method of prediction of amyloidogenic regions from protein sequence. Bioinformatics 26:326–332
Gasior P, Kotulska M (2014) FISH Amyloid – a new method for finding amyloidogenic segments in proteins based on site specific co-occurence of aminoacids. BMC Bioinformatics 15:54
Gertz MA, Rajkumar SV (eds) (2010) Amyloidosis: diagnosis and treatment. Humana Press, New York
Giehm L, Otzen DE (2010) Strategies to increase the reproducibility of protein fibrillization in plate reader assays. Anal Biochem 400:270–281
Gilead S, Gazit E (2005) Self-organization of short peptide fragments: from amyloid fibrils to nanoscale supramolecular assemblies. Supramol Chem 17:87–92
Goldstein RF, Stryer L (1986) Cooperative polymerization reactions. Analytical approximations, numerical examples, and experimental strategy. Biophys J 50:583–599
Hamodrakas SJ, Liappa C, Iconomidou VA (2007) Consensus prediction of amyloidogenic determinants in amyloid fibril-forming proteins. Int J Biol Macromol 41:295–300
Hayman SR, Bailey RJ, Jalal SM, Ahmann GJ, Dispenzieri A, Gertz MA, Greipp PR, Kyle RA, Lacy MQ, Rajkumar SV, Witzig TE, Lust JA, Fonseca R (2001) Translocations involving the immunoglobulin heavy-chain locus are possible early genetic events in patients with primary systemic amyloidosis. Blood 98:2266–2268
Hofrichter J, Ross PD, Eaton WA (1974) Kinetics and mechanism of deoxyhemoglobin S gelation: a new approach to understanding sickle cell disease. Proc Natl Acad Sci U S A 71:4864–4868
Idicula-Thomas S, Balaji PV (2005) Understanding the relationship between the primary structure of proteins and their amyloidogenic propensity: clues from inclusion body formation. Protein Eng Des Sel PEDS 18:175–180
Ivanova MI, Sawaya MR, Gingery M, Attinger A, Eisenberg D (2004) An amyloid-forming segment of beta2-microglobulin suggests a molecular model for the fibril. Proc Natl Acad Sci U S A 101:10584–10589
Jahn TR, Parker MJ, Homans SW, Radford SE (2006) Amyloid formation under physiological conditions proceeds via a native-like folding intermediate. Nat Struct Mol Biol 13:195–201
Jaroniec CP, MacPhee CE, Astrof NS, Dobson CM, Griffin RG (2002) Molecular conformation of a peptide fragment of transthyretin in an amyloid fibril. Proc Natl Acad Sci U S A 99:16748–16753
Jiménez JL, Guijarro JI, Orlova E, Zurdo J, Dobson CM, Sunde M, Saibil HR (1999) Cryo-electron microscopy structure of an SH3 amyloid fibril and model of the molecular packing. EMBO J 18:815–821
Jones S, Manning J, Kad NM, Radford SE (2003) Amyloid-forming peptides from beta2-microglobulin-Insights into the mechanism of fibril formation in vitro. J Mol Biol 325:249–257
Kajava AV, Baxa U, Wickner RB, Steven AC (2004) A model for Ure2p prion filaments and other amyloids: the parallel superpleated beta-structure. Proc Natl Acad Sci U S A 101:7885–7890
Kalebina TS, Plotnikova TA, Gorkovskii AA, Selyakh IO, Galzitskaya OV, Bezsonov EE, Gellissen G, Kulaev IS (2008) Amyloid-like properties of Saccharomyces cerevisiae cell wall glucantransferase Bgl2p: prediction and experimental evidences. Prion 2:91–96
Kayed R, Head E, Thompson JL, McIntire TM, Milton SC, Cotman CW, Glabe CG (2003) Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science 300:486–489
Klimtchuk ES, Gursky O, Patel RS, Laporte KL, Connors LH, Skinner M, Seldin DC (2010) The critical role of the constant region in thermal stability and aggregation of amyloidogenic immunoglobulin light chain. Biochemistry (Mosc) 49:9848–9857
Knowles TPJ, Waudby CA, Devlin GL, Cohen SIA, Aguzzi A, Vendruscolo M, Terentjev EM, Welland ME, Dobson CM (2009) An analytical solution to the kinetics of breakable filament assembly. Science 326:1533–1537
Kovacs E, Tompa P, Liliom K, Kalmar L (2010) Dual coding in alternative reading frames correlates with intrinsic protein disorder. Proc Natl Acad Sci U S A 107:5429–5434
Kozhukh GV, Hagihara Y, Kawakami T, Hasegawa K, Naiki H, Goto Y (2002) Investigation of a peptide responsible for amyloid fibril formation of beta 2-microglobulin by achromobacter protease I. J Biol Chem 277:1310–1315
Krebs MR, Wilkins DK, Chung EW, Pitkeathly MC, Chamberlain AK, Zurdo J, Robinson CV, Dobson CM (2000) Formation and seeding of amyloid fibrils from wild-type hen lysozyme and a peptide fragment from the beta-domain. J Mol Biol 300:541–549
López de la Paz M, Serrano L (2004) Sequence determinants of amyloid fibril formation. Proc Natl Acad Sci U S A 101:87–92
Lu X, Wintrode PL, Surewicz WK (2007) Beta-sheet core of human prion protein amyloid fibrils as determined by hydrogen/deuterium exchange. Proc Natl Acad Sci U S A 104:1510–1515
Matouschek A, Kellis JT, Serrano L, Fersht AR (1989) Mapping the transition state and pathway of protein folding by protein engineering. Nature 340:122–126
Maurer-Stroh S, Debulpaep M, Kuemmerer N, Lopez de la Paz M, Martins IC, Reumers J, Morris KL, Copland A, Serpell L, Serrano L, Schymkowitz JWH, Rousseau F (2010) Exploring the sequence determinants of amyloid structure using position-specific scoring matrices. Nat Methods 7:237–242
Maury CP, Nurmiaho-Lassila EL (1992) Creation of amyloid fibrils from mutant Asn187 gelsolin peptides. Biochem Biophys Res Commun 183:227–231
Morris AM, Watzky MA, Finke RG (2009) Protein aggregation kinetics, mechanism, and curve-fitting: a review of the literature. Biochim Biophys Acta 1794:375–397
Muñoz V, Serrano L (1994) Elucidating the folding problem of helical peptides using empirical parameters. Nat Struct Biol 1:399–409
Nelson R, Sawaya MR, Balbirnie M, Madsen A, Riekel C, Grothe R, Eisenberg D (2005) Structure of the cross-beta spine of amyloid-like fibrils. Nature 435:773–778
Oosawa F, Asakura S, Hotta K, Imai N, Ooi T (1959) G-F transformation of actin as a fibrous condensation. J Polym Sci 37:323–336
Picotti P, De Franceschi G, Frare E, Spolaore B, Zambonin M, Chiti F, de Laureto PP, Fontana A (2007) Amyloid fibril formation and disaggregation of fragment 1–29 of apomyoglobin: insights into the effect of pH on protein fibrillogenesis. J Mol Biol 367:1237–1245
Rauscher S, Baud S, Miao M, Keeley FW, Pomès R (2006) Proline and glycine control protein self-organization into elastomeric or amyloid fibrils. Structure 14:1667–1676
Roland BP, Kodali R, Mishra R, Wetzel R (2013) A serendipitous survey of prediction algorithms for amyloidogenicity. Biopolymers 100:780–789
Ruschak AM, Miranker AD (2007) Fiber-dependent amyloid formation as catalysis of an existing reaction pathway. Proc Natl Acad Sci U S A 104:12341–12346
Sánchez IE, Tejero J, Gómez-Moreno C, Medina M, Serrano L (2006) Point mutations in protein globular domains: contributions from function, stability and misfolding. J Mol Biol 363:422–432
Selivanova OM, Suvorina MY, Dovidchenko NV, Eliseeva IA, Surin AK, Finkelstein AV, Schmatchenko VV, Galzitskaya OV (2014) How to determine the size of folding nuclei of protofibrils from the concentration dependence of the rate and lag-time of aggregation. II. Experimental application for insulin and LysPro insulin: aggregation morphology, kinetics, and sizes of nuclei. J Phys Chem B 118:1198–1206
Serio TR, Cashikar AG, Kowal AS, Sawicki GJ, Moslehi JJ, Serpell L, Arnsdorf MF, Lindquist SL (2000) Nucleated conformational conversion and the replication of conformational information by a prion determinant. Science 289:1317–1321
Tartaglia GG, Vendruscolo M (2008) The Zyggregator method for predicting protein aggregation propensities. Chem Soc Rev 37:1395–1401
Tartaglia GG, Pellarin R, Cavalli A, Caflisch A (2005) Organism complexity anti-correlates with proteomic beta-aggregation propensity. Protein Sci 14:2735–2740
Tenidis K, Waldner M, Bernhagen J, Fischle W, Bergmann M, Weber M, Merkle ML, Voelter W, Brunner H, Kapurniotu A (2000) Identification of a penta- and hexapeptide of islet amyloid polypeptide (IAPP) with amyloidogenic and cytotoxic properties. J Mol Biol 295:1055–1071
Thangakani AM, Kumar S, Nagarajan R, Velmurugan D, Gromiha MM (2014) GAP: towards almost 100 percent prediction for β-strand-mediated aggregating peptides with distinct morphologies. Bioinformatics 30:1983–1990
Thompson A, White AR, McLean C, Masters CL, Cappai R, Barrow CJ (2000) Amyloidogenicity and neurotoxicity of peptides corresponding to the helical regions of PrP(C). J Neurosci Res 62:293–301
Trovato A, Chiti F, Maritan A, Seno F (2006) Insight into the structure of amyloid fibrils from the analysis of globular proteins. PLoS Comput Biol 2:e170
Tsolis AC, Papandreou NC, Iconomidou VA, Hamodrakas SJ (2013) A consensus method for the prediction of “Aggregation-Prone” peptides in globular proteins. PLoS One 8:e54175
Von Bergen M, Friedhoff P, Biernat J, Heberle J, Mandelkow EM, Mandelkow E (2000) Assembly of tau protein into Alzheimer paired helical filaments depends on a local sequence motif ((306)VQIVYK(311)) forming beta structure. Proc Natl Acad Sci U S A 97:5129–5134
Walsh I, Seno F, Tosatto SCE, Trovato A (2014) PASTA 2.0: an improved server for protein aggregation prediction. Nucleic Acids Res 42:W301–W307
Wang Q, Johnson JL, Agar NYR, Agar JN (2008) Protein aggregation and protein instability govern familial amyotrophic lateral sclerosis patient survival. PLoS Biol 6:e170
Wegner A, Savko P (1982) Fragmentation of actin filaments. Biochemistry 21:1909–1913
Wright CF, Teichmann SA, Clarke J, Dobson CM (2005) The importance of sequence diversity in the aggregation and evolution of proteins. Nature 438:878–881
Xue W-F, Homans SW, Radford SE (2008) Systematic analysis of nucleation-dependent polymerization reveals new insights into the mechanism of amyloid self-assembly. Proc Natl Acad Sci U S A 105:8926–8931
Yang H, Li J-J, Liu S, Zhao J, Jiang Y-J, Song A-X, Hu H-Y (2014) Aggregation of polyglutamine-expanded ataxin-3 sequesters its specific interacting partners into inclusions: implication in a loss-of-function pathology. Sci Rep 4:6410
Yoon S, Welsh WJ (2004) Detecting hidden sequence propensity for amyloid fibril formation. Protein Sci 13:2149–2160
Zhu L, Zhang X-J, Wang L-Y, Zhou J-M, Perrett S (2003) Relationship between stability of folding intermediates and amyloid formation for the yeast prion Ure2p: a quantitative analysis of the effects of pH and buffer system. J Mol Biol 328:235–254
Zimmermann O, Hansmann UHE (2006) Support vector machines for prediction of dihedral angle regions. Bioinformatics 22:3009–3015
Acknowledgement
We are grateful to Dr. Olga Gursky for her valuable comments and editorial help. This study was supported by the Russian Science Foundation grant №14-14-00536.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Dovidchenko, N.V., Galzitskaya, O.V. (2015). Computational Approaches to Identification of Aggregation Sites and the Mechanism of Amyloid Growth. In: Gursky, O. (eds) Lipids in Protein Misfolding. Advances in Experimental Medicine and Biology, vol 855. Springer, Cham. https://doi.org/10.1007/978-3-319-17344-3_9
Download citation
DOI: https://doi.org/10.1007/978-3-319-17344-3_9
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-17343-6
Online ISBN: 978-3-319-17344-3
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)