
Abstract
Development of the epidermis is marked by terminal differentiation of keratinocytes. The mechanisms that drive this process at the chromatin level are poorly understood. However, recent genomics approaches, including comparative genomic studies, ENCODE data, and biochemical studies to elucidate the 3-dimensional genome architecture with next-generation sequencing technology, have greatly advanced our understanding of the molecular mechanisms that govern epidermal differentiation. Specifically, the identification of enhancers, a class of regulatory elements, and their role in driving key transcriptional events has revealed novel molecular mechanisms for epidermal development. Here we discuss the history of the “enhancer” concept and our current understanding of enhancer-promoter interactions that drive transcriptional activation for epidermal differentiation.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others

10.1 Introduction
The epidermis at the surface of the skin provides a tractable and spatially hierarchical model to investigate the development of committed cells. The architecture of the epidermis provides the key structure for the physical barrier of the skin. Epidermal cells, or keratinocytes, in the most internal basal layer of the epidermis must strike a critical balance between self-renewal and differentiation in order to build a functional barrier across the entire body [1]. Keratinocyte self-renewal is characterized by parallel cell division within the basal layer. Expression of Keratin 5 (K5) and Keratin 14 (K14) marks these basal proliferating keratinocytes.
During differentiation, basal keratinocytes divide asymmetrically, giving rise to suprabasal keratinocytes that migrate outwards, entering the spinous layer [1,2,3]. Spinous and granular keratinocytes activate Keratin 1 (K1) and Keratin 10 (K10) expression concomitant with K5/K14 downregulation. During late terminal differentiation, the keratinocytes coordinately express many Epidermal Differentiation Complex (EDC) genes, including filaggrin (FLG) and FLG-like, late cornified envelope (LCE) , small proline-rich region (SPRR) , and S100 [4,5,6,7]. As the keratinocytes reach the outermost stratum corneum, they enucleate and are surrounded by a cornified envelope, the basic structural unit of the skin barrier, which is formed by transglutaminase-1-mediated cross-linking of scaffold proteins. The cornified envelopes are sealed together by keratinocyte-derived extruded lipids to form a semi-permeable barrier [2]. In mice, the pattern of functional barrier acquisition corresponds with maturation of the cornified envelopes, and proceeds from specific dorsal initiation sites at embryonic day (E)16, spreading to converge at the dorsal and ventral midline so that the whole embryo is impermeable by E17 [8].
Epidermal differentiation can be recapitulated in vitro by exposure of proliferating keratinocytes to high calcium levels [9, 10]. This process, called calcium switching, stimulates calcium receptor (CaR) and downstream phosphokinase C (PKC) signaling, which activates the Fos/Jun family of transcription factors that play important roles in keratinocyte differentiation (reviewed in [11]). Fos and Jun proteins form homo- or heterodimers that compose the AP-1 transcription factor complex [12]. In normal epidermis as well as in organotypic epidermal cultures, the expression pattern of AP-1 proteins is tightly regulated even within the differentiated layers [13]. Fos proteins are found in the nuclei of both basal and suprabasal keratinocytes. JunB and JunD are expressed in all layers of normal epidermis. Interestingly, c-Jun is expressed in the spinous layer, then disappears and reemerges in the outermost granular layer directly at the transition zone to the stratum corneum. As will be discussed in this chapter, many of the genes expressed in keratinocytes, in either proliferative or differentiated layers of the epidermis, contain AP-1 binding sites in their promoter regions. This suggests that specific combinations of AP-1 protein complexes bind to the enhancers for genes expressed at successive stages of epidermal differentiation.
Transcriptional regulation clearly plays a major role in development of the epidermis. So how does the keratinocyte decipher the genome to activate a transcriptional program specific to epidermal differentiation? Early studies of gene regulation focused on single gene promoters and the transcription factor binding sites contained within [14]. However, not all transcriptional activation is attributable to biochemical activity at the gene promoter, thus suggesting the contribution of other loci. Complete sequencing of the genomes of humans and model organisms has revolutionized our ability to further define, and ascertain the functions of, noncoding sequences in the regulation of gene expression [14,15,16]. Here we discuss the history of the conceptual advances in our understanding of the roles of enhancer elements in epidermal development. We discuss known functions for enhancer regulation of key genes that define the stages of epidermal development; the genetic, genomic and epigenetic features that allow us to identify enhancers; and the approaches that will enable a thorough understanding of the dynamic role of enhancers in gene activation.
10.2 What Is an Enhancer?
The concept of an “enhancer ” emerged in 1981. In that year, Pierre Chambon and George Khoury independently discovered a non-coding 72 bp tandem repeat sequence, upstream of the SV40 early gene promoters that was required for transcription [17, 18]. A subsequent study from Walter Schaffner identified the ability of the SV40 DNA sequence to “enhance” the expression of rabbit β-globin even when the SV40 sequence was placed thousands of base pairs away from the β-globin gene promoter in an expression vector [19]. Chambon observed similar results using the gene for conalbumin [20]. Further experiments performed by Paul Berg and Michael Fromm showed that the SV40 sequence was able to “enhance” transcription independent of its location (upstream or downstream of its target gene) and orientation (forward or reverse) [21]. This established the SV40 sequence as the prototype of a novel genetic element, an enhancer, and established the definition of a classical enhancer as a non-coding sequence that can modulate gene expression in a position- and orientation-independent manner.
The discovery of the SV40 enhancer paved the way for the identification of enhancers in other tissue types [15]. Often, searches for enhancers were prioritized and interrogated in the sequences surrounding the target genes that included upstream or downstream sequences, including 5′ and 3′ untranslated regions (UTRs), introns, and intergenic regions (reviewed in [22]).
In vivo studies of putative enhancers identified the spatiotemporal specificity of these sequences to ensure biologically relevant cell- and tissue-specific gene expression (reviewed in [23, 24]). Biochemical studies further identified clusters or arrays of transcription factor binding sites that act as “building blocks” of cis-regulatory modules that also show enhancer activity (reviewed in [15, 16, 25]). The enrichment of multiple transcription factor binding sites within an enhancer facilitates cell-specific expression largely attributable to combinatorial and differential binding of transcription factor family members in the context of different microenvironments. Based on these later studies, we more loosely define an enhancer as a non-coding sequence, containing clusters of transcription factor binding sites, that drives cell-, tissue-, or developmental stage-specific gene expression. Subsequent findings, described below, paved the way for the application of comparative genomics to enhancer discovery.
10.3 Transcriptional Regulation in Epidermal Development by Non-Promoter Sequences
Prior to the availability of whole genome sequences, biological insights into the transcriptional activation of key epidermal differentiation target genes initially focused on sequences immediately upstream of the transcription start site, relying on conservation of these sequences between mouse and human to identify putative regulatory regions [14]. These regions were cloned into reporter constructs and tested in vitro and in vivo to determine their ability to drive gene expression in the expected spatio-temporal pattern. Subsequent genetic deletion studies enabled the discovery of minimal promoter and regulatory core elements for activity and tissue specificity within segments of the cloned fragments. While the discovery of these promoter sequences identified key molecular players in gene activation and facilitated in vivo epidermal-specific genetic studies, it also highlighted the paucity of data to explain every nuance in gene expression for epidermal terminal differentiation.
10.3.1 Transcriptional Regulation of the Basal Keratin Genes (K5 and K14)
K5 and K14 are specifically expressed in mitotically active basal keratinocytes in most types of stratified epithelia. K5 and K14 proteins form stable heterodimers that are further cross-linked to provide structural support for cells [26, 27]. Their co-expression suggests that similar transcriptional mechanisms permit coordinated regulation of the two genes [28, 29].
Earlier studies identified marked similarities between the upstream sequences of K14 to viral and immunoglobulin enhancer elements [30]. Although 2.5 kb of 5′ upstream and 70 bp of 3′ downstream noncoding sequences of a cloned spliced human K14 gene are capable of directing gene expression in various cell lines, in vivo these regions drive epidermal-specific expression, demonstrated by reporter gene activity in transgenic mice that coincides with endogenous K14 expression [31]. An additional study of a 2 kb upstream sequence of K14 in transgenic mice also identified epidermal and outer root sheath hair follicle-specific reporter gene expression, conferred by a 700 bp sequence conserved between mouse and human, that exhibited keratinocyte-specific open chromatin as measured by DNaseI hypersensitivity [32]. The upstream cis-regulatory elements and promoter region of K14 contained binding sites for the transcription factor ETS, as well as AP-2, AP-1, and SP1 sites shared by the K5 regulatory region [32,33,34,35,36] (see below).
A similar study of the human K5 upstream sequence identified a 6 kb region that controls cell type-specific transcriptional activity [37]. A 90 bp sequence within this region is sufficient to activate expression specifically in the epidermis, hair follicles, and tongue. However, the pattern of expression was aberrantly switched from the basal layer to differentiated suprabasal cells. This suggested minimal promoter activity in the 90 bp upstream sequence for keratinocyte-specific expression and a requirement for other enhancers within the 6 kb upstream region for directing expression to the correct layers within stratified epithelia. The link between AP-2, SP1 and other unknown transcription factors for K5-tissue-specific activation was later determined based on the discovery of these transcription factor binding sites in the 6 kb sequence upstream of the human K5 transcription start site and corroborated with comparative studies of the bovine K5 promoter sequence [35, 37].
The mechanisms controlling coordinate expression of K5 and K14 were further elucidated by the discovery that mice lacking the transcription factor p63 fail to express K5 or K14 and do not develop a stratified epidermis [38, 39]. Biochemical studies revealed the presence of p63-responsive regulatory elements in an epithelial-specific enhancer 1.4 kb upstream of the K14 promoter [40,41,42,43] and in a transcriptionally active region upstream of K5 [44]. The shared and unique transcription factor binding sites harbored by the K5 and K14 enhancers suggest that both common and unique molecular mechanisms are involved in regulating their expression.
10.3.2 Transcriptional Regulation of the Suprabasal Genes
10.3.2.1 Early Differentiation: Regulation of K1 and K10 Expression
K1 and K10 are expressed in post-mitotic differentiating keratinocytes, and like K14 and K5, form heterodimers that are important for tissue integrity [45]. A 10.8 kb region surrounding the human K1 gene is sufficient to direct tissue-specific expression in transgenic mice during development, and is responsive to calcium induced differentiation in vitro [46]. Two calcium responsive elements were identified within a 1.7 kb enhancer in the 3′ flanking sequences of K1 [47, 48] including one element at the proximal 5′ end of the enhancer that is activated by AP-1 binding, and another that either suppresses or promotes the calcium response depending on the presence of vitamin D or retinoic acid, respectively [48]. Epidermal specific expression of K1 is directed by 207 bp of sequence at the distal 3′ end of this enhancer; however the transcription factor responsible for driving this expression has not been identified [47].
Insights into the transcriptional regulation of K10 expression were provided by analyses of mice deficient in the CCAAT/enhancer binding protein family member C/EBPβ, which exhibit decreased expression of K10 and K1 and mild epidermal hyperplasia [49]. This work was motivated by the observed increases in C/EBPα, C/EBPβ and C/EBPδ mRNA levels, as well as C/EBPα and C/EBPβ protein induction, upon calcium-induced differentiation of mouse primary keratinocytes [50]. DNaseI footprinting and gel-shift assays performed on the human K10 promoter, which had been identified via homology with the bovine sequence, revealed that C/EBP binding to three distinct elements was required for K10 expression [51]. While C/EBPα and C/EBPβ both activate K10, they are differentially expressed in stratified epidermis. C/EBPβ acts early in the basal and spinous layers of the epidermis and is later superseded by C/EBPα during keratinocyte differentiation and upward migration into the granular layer. Besides C/EBP binding to three intact C/EBP binding sites within the K10 promoter , full activation of K10 during differentiation also requires specific binding of AP-2 [51]. Consistent with co-expression of K1 and K10, consensus C/EBP binding sites are also observed in the upstream promoter regions of the K1 gene [52, 53].
10.3.2.2 Regulation of EDC Genes
Terminal differentiation in the epidermis is marked by the expression of four gene families that lie within the EDC and are coordinately activated at the transcriptional level: small proline-rich region (SPRR), late cornified envelope (LCE), filaggrin (FLG) and FLG-like (FLG-like), and S100 genes [4,5,6,7]. Pioneer studies to elucidate gene expression in relation to epidermal differentiation targeted the SPRR gene family members involucrin (IVL) and loricrin (LOR). These two important marker genes are distinctively expressed in terminally differentiated keratinocytes, and encode structural proteins that are cross-linked with many of the other proteins encoded by EDC genes to form the cornified envelope, the basic structural unit of the stratum corneum [54]. IVL is cross-linked early in the formation of the cornified envelope [55] and LOR is in turn cross-linked to the existing scaffolding containing IVL [56]. In the developing mouse embryo, Ivl and Lor transcripts are upregulated as early as E15.5 [57], and protein expression can be observed by E16.5, corresponding to the onset of skin barrier formation [6, 8]. The tight correlation of IVL and LOR expression with keratinocyte terminal differentiation makes them ideal candidates for studying the mechanisms that underlie the switch from a proliferating to a differentiating program.
10.3.2.3 Involucrin
A 3.7 kb upstream sequence of IVL directs keratinocyte-specific expression of a β-galactosidase reporter gene in transgenic mice [58]. Deletion analysis showed that this sequence comprises distal- and proximal-regulatory regions (DRR and PRR) or enhancers [59, 60], and contains an AP-1 binding site that is required for expression and synergizes with an adjacent SP1 binding site [61]. The DRR AP-1 site binds FRA-1, JunB, JunD, and p300, a histone acetyltransferase often associated with enhancers [62, 63], while the SP1 site interacts with SP1, SP3, and KLF4 transcription factors [59, 61, 64,65,66]. Because FRA-1 and KLF4 are both known to interact with p300 [65, 67], these data indicate that a complex of transcription factors forms on the DRR to drive IVL expression during keratinocyte differentiation.
10.3.2.4 Loricrin
Transcriptional activation of mouse loricrin expression was first localized to a 6.5 kb region spanning the loricrin gene [68]. Transgenic reporter mice in which LOR coding sequences were replaced by a β-galactosidase gene revealed that the remaining 1.5 kb of 5′-flanking sequence, a small noncoding exon, a 1.1 kb intron, a single coding exon, and 2.2 kb of 3′-flanking sequence from the mouse loricrin gene drive epidermal-specific, but not differentiation-specific expression. Minimal promoter activity, dependent on an AP1 site conserved between mouse and human, was mapped to a 60 bp sequence upstream of the transcription start site. In the case of the human LOR gene, enhancers located within 1.5 kb of 5′-flanking sequence and 9 kb of 3′-sequence are responsible for tissue- and differentiation-specific expression of a human LOR transgene in transgenic mice [69]. As few as 154 bp of 5′ sequence upstream from the cap site can direct expression specifically in cultured NHEK and HaCAT keratinocytes, in an SP1/c-Jun and p300/CREB-dependent manner [70]. Differential occupation of this site by SP3/CREB-1/CREMα/ATF-1/Jun B, and an AP-2-like protein (named keratinocyte-specific repressor-1 (KSR-1)) in a repressive state, and SP1/c-Jun/p300/CBP in an active state during differentiation, enabled LOR transcriptional resolution in stratified layers [70].
10.4 Identification of Enhancers by Comparative Genomics in the Post-Human Genome Era
The early studies described above identified the transcription factors responsible for transcriptional regulation of major epidermal differentiation genes. However, the molecular mechanisms underlying transcription factor-driven activation of these genes were less clear. Furthermore, these studies were limited to the analysis of proximal promoter and enhancer regions. The availability of complete genome sequences for a wide range of model organisms and animal species greatly facilitated the identification of putative enhancer and other regulatory sequences. Specifically, it shifted the discovery of enhancers toward a more systematic procedure – high-throughput and on a genome-wide scale. The discovery of putative enhancers was predicated on the identification of noncoding sequence conservation across multiple species and facilitated by multiple-sequence alignments [71]. Indeed, a study of conserved noncoding sequences present between human and pufferfish or ultraconserved between human and mouse found that many of these conserved noncoding sequences exhibit enhancer activities that are developmental- and tissue-specific in transgenic mouse assays [72]. Below we discuss the discovery of enhancers for relevant skin biology genes using comparative genomics.
10.4.1 SPRR Genes
The availability of whole genome data enabled the discovery of conserved noncoding sequences (CNSs and based on sequence alignments) in the SPRR locus that is clustered within the EDC [73]. Small proline-rich (SPRR) proteins are the primary constituents of the cornified envelope [74]. Many of the SPRRs are coordinately upregulated under stress conditions to rapidly build a temporary barrier [73]. Of the DNaseI hypersensitive sites (HSs) residing within CNSs, one demonstrated enhancer activity under conditions when the SPRR genes are coordinately upregulated, suggesting its potential as an enhancer region that coordinates SPRR gene expression.
10.4.2 PADI3
The family of peptidylarginine deaminases (PADs) is encoded by a cluster of 5 PADI genes on human chromosome 1p35–36 [75]. Peptidylarginine deaminase 3 (PADI3) encodes PAD3, which is involved in filaggrin metabolism, releasing individual filaggrin monomers that contribute to the natural moisturizing functions of the skin barrier. Using a comparative genomic approach, an enhancer located 86 kb from the PADI3 gene promoter was identified and determined to be calcium sensitive [76]. This enhancer was found to trigger expression of PADI3 upon epidermal keratinocyte differentiation, and links PADI3 expression to AP-1 transcription factors through chromatin opening and looping (see Sect. 10.5).
10.4.3 The Role of the CNE 923 Enhancer in Coordinate Regulation of the EDC
The coordinate regulation of EDC genes, as well as the synteny and linearity of the EDC locus across a wide range of mammalian species, suggest a molecular mechanism originating at the proximal genomic level. To delineate this, investigators screened for enhancer elements within the EDC [7]. In 2010, 48 conserved noncoding elements (CNEs) within the human EDC were identified from sequence alignments of orthologous EDC loci across eutherian (human, chimpanzee, macaque, mouse, rat, dog) and metatherian (opossum) mammals. Approximately 50% of these CNEs exhibited dynamic regulatory activity, and were thus identified as potential cis-regulatory elements or enhancers that might synergistically or independently coordinate EDC gene expression during skin barrier formation. Among these, human CNE 923, located approximately 923 kb from the transcriptional start site of the most 5’ EDC gene, S100A10, induced the highest luciferase reporter activity in proliferating and differentiated keratinocytes. CNE 923 exhibited DNaseI hypersensitivity in primary human keratinocytes, and was sufficient to drive reporter gene expression in the developing E16.5 epidermis in transgenic reporter mice. The activity of CNE 923 was monitored in an independent transgenic mouse line and was sufficient to drive β-galactosidase activity in the same dorsal to ventral pattern of barrier acquisition that coincided with EDC gene activation [57]. These studies provided compelling evidence for CNE 923 as an epidermal-specific enhancer and a potential LCR. CNE 923 was also noted to form dynamic chromatin interactions with a number of EDC genes, and this was sensitive to keratinocyte differentiation and dependent on the AP-1 transcription factor. The role of CNE 923 for mediating EDC chromatin architecture is discussed further in Sect. 10.7.
10.4.4 p63
A long-range cis-regulatory enhancer of p63 (p63LRE) spanning a 12 kb region in mice was recently identified by comparative genomics [77]. p63LRE comprises two evolutionarily conserved modules acting in concert to control tissue- and layer-specific expression of the p63 gene. Both modules are in an accessible and active chromatin state in human and mouse keratinocytes and in embryonic epidermis, and are strongly bound by p63. p63LRE activity is dependent on p63 expression in embryonic skin and also in the commitment of human induced pluripotent stem cells toward an epithelial cell fate. C/EBPα, C/EBPβ, and the POU domain-containing protein Pou3f1 repress p63 expression during keratinocyte differentiation by binding the p63LRE enhancer. The availability of these transcription factors in the outermost layers of the epidermis accounts for increased repression of p63, thereby relieving p63-mediated repression of EDC and keratinocyte differentiation genes in these layers, and limiting p63 activity to the basal layers. We discuss p63-bound sites and enhancer regions on a genome-wide scale in Sect. 10.7.4.
10.5 Epigenetics and Chromatin Remodeling
So far, we have discussed studies that make compelling arguments for transcriptional regulation in which the key determinants are transcription factor binding to individual nucleotide motifs. However, this view has been challenged by our increased understanding of the non-random packaging of linear DNA into histones, which alters the accessibility of DNA segments to transcription factor binding, and by the discovery of specific post-translational modifications to histone components that dynamically regulate DNA accessibility. Specifically, the identification of histone deacetylase (HDAC) and histone acetyltransferase (HAT) enzymes and their targets furnished the first direct evidence linking histone modification states to transcriptional regulation [78, 79]. These findings paved the way for investigations into higher order chromatin structure and genome compartmentalization. HATs catalyze the transfer of an acetyl group from acetyl CoA to the ε-amino group of lysine residues on histones. This mark is generally associated with active genes. Conversely, HDACs remove the acetyl group from acetyl-lysine (Ac-Lys) to regenerate the free ε-amino group, causing chromatin compaction and a transcriptionally repressive environment (reviewed in [80, 81]). The identification and characterization of other classes of histone modifying enzymes soon followed, implicating kinases [82, 83], lysine and arginine-specific methyltransferases [84,85,86], arginine deiminases [87, 88], ubiquitinases [89], deubiquitinases [90,91,92], and lysine- and arginine-specific demethylases (HDMs) in transcription regulation [93,94,95]. Collectively, these discoveries highlighted the role of chromatin modifications in governing eukaryotic gene expression and other DNA-dependent functions and ushered in a new era of chromatin-based epigenetic studies (reviewed in [96, 97]).
Within the context of the skin, p300 and CBP, two enhancer-associated HATs, have been implicated in the regulation of IVL and LOR (discussed earlier) [65, 70]. Epidermal-specific deletion of Actl6a, an essential component of HATs, resulted in de-repression of KLF4 and Brg1/Brm binding, thereby aberrantly activating epidermal differentiation genes and abolishing epidermal progenitor function [98]. HDACs also play important roles in epidermal development, specifically in preventing senescence of basal progenitor cells [99, 100]. Epidermal-specific deletion of both HDAC1 and 2 in embryonic epidermis resulted in a phenotype resembling the effects of loss of p63. This phenotype was associated with de-repression of ΔNp63-repressed target genes including the senescence gene p16 [101]. HDAC1/2 localizes to the promoter regions of ΔNp63-repressed targets in cultured human keratinocytes, and histones in these regions are hyper-acetylated following HDAC inhibition, indicating a requirement for HDAC1/2 in ΔNp63-mediated repression. Together, these data reveal essential roles for histone modifying enzymes in controlling the activities of key regulators of epidermal development (reviewed in [102]).
Ezh2, an essential component of the polycomb repressor complex 2 (PRC2) is also important for epidermal development [103]. Epidermal-specific loss of Ezh2 resulted in early epidermal differentiation owing to precocious recruitment of AP-1 transcription factor to the EDC. This indicated a role for Ezh2 in gene repression in proliferating keratinocytes by promoting histone H3K27 trimethylation (H3K27me3). However, loss of Ezh2, while decreasing H3K27me3 marks, was not sufficient to alter transcriptional status unless AP-1 was also recruited to the affected region, emphasizing an important and direct role for AP-1 in keratinocyte differentiation.
Recent cell biology studies have also highlighted the importance of chromatin remodeling in permitting efficient and coordinate regulation of clusters of genes in epidermal development. During epidermal development, the EDC locus moves away from the nuclear periphery and towards the nuclear interior prior to activation of EDC gene expression [104]. Ablation of either p63, a master regulator of epidermal development [38, 39, 105], or Satb1, a higher-order genome organizer that binds to the EDC in epidermal progenitor cells, caused altered chromatin conformation of the EDC, and loss of expression of genes that lie in the central domain of the EDC [106]. These findings identified Satb1 as an important downstream target of p63 required for proper establishment of higher-order EDC chromatin structure and coordinated gene expression [106]. Similarly, p63 and its direct target Brg1 are essential in remodeling the higher-order chromatin structure of the EDC and positioning the locus within the 3D chromatin landscape to allow efficient expression of EDC genes in epidermal progenitor cells during skin development [107, 108].
10.6 Methods to Identify Enhancers by ENCODE in the Post-Human Genome Era
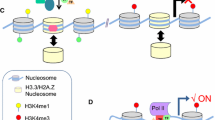
Increased understanding of the chromatin state of the genome has forced us to re-examine initial models for the control of gene expression that focused entirely on the role of cis-regulatory elements (Sect. 10.3). Following completion of the human genome sequence, the National Human Genome Research Institute, recognizing the need to more fully understand the regulation of gene expression, launched the Encyclopedia of Non-Coding Elements (ENCODE), a collaborative public research project to identify and characterize the function of noncoding elements in the genome, and develop the tools and technology to achieve this goal [63]. Next-generation sequencing was instrumental in producing these genome datasets in a cost-effective manner. The ENCODE studies as well as work by others have greatly facilitated our ability to identify enhancers on a genome-wide scale based on chromatin modifications that are unique to these regulatory elements, such as DNaseI hypersensitivity (open chromatin), histone modification epigenetic marks (H3K27Ac, H3K4me1), and transcription factor binding (p300, activating TFs) associated with functional enhancers (reviewed in [15, 25, 109]). High-throughput chromatin immunoprecipitation linked with deep sequencing (ChIP-seq) for specific histone modifications has enabled the discovery of new enhancers and paved the way for further downstream functional analyses (reviewed in [15]). Table 10.1 lists signature genomic marks and methods to identify enhancers.
Chromatin looping and tracking have been proposed as models to explain how distant enhancers are able to regulate their target genes [143, 144]. The first experimental demonstration of direct interactions between distantly located enhancers and target genes was made possible by the development of chromosome conformation capture (3C) techniques [125]. 3C was used to demonstrate that loop formation between the β-globin LCR enhancer and gene accompanied transcriptional activation [113,114,147], and established a paradigm that was later validated for numerous other loci, including the α-globin gene cluster, TH2, IFNG, MHC class II and IgH loci [148]. Transcription factor ChIP-chip studies also revealed that enhancers could be located even further from their target genes than previously thought, as far as 10–20 kbs to several Mbs away [149]. Often, these proximal and distal enhancers interact to co-regulate a target gene.
Recent improvements in chromosome conformation capture methods have allowed us to examine the chromatin interactions of genomic regions at varying levels of depth and resolution. 4C (circular chromosome conformation capture) detects all interacting sequences with a sequence of interest using a bait such as an enhancer [126]. 5C (chromosome conformation capture carbon-copy) is designed to detect many known interactions with numerous baits and typically within a gene locus [127], while the Hi-C approach is aimed at detecting all chromatin interactions [128]. Methods such as ChIA-PET (Chromatin interaction analysis with paired-end tag sequencing ) combine 5C and Hi-C methods to simultaneously identify genome-wide chromatin interactions and the proteins that bind interacting sequences [150]. Evidence for epigenetic modifications, chromatin looping, and the interplay between the two has been obtained relatively recently, and has provided new insights to our understanding of the biochemical aspects of enhancer-mediated transcriptional regulation.
10.7 Emerging Concepts for Understanding Enhancer-Promoter Interactions
10.7.1 Mechanisms Underlying Enhancer-Promoter Interactions and Topological Association of Chromatin Domains in the Regulation of Gene Expression
Apart from the aforementioned roles of p63, Satb1, Brg1, and AP-1 in transcriptional regulation during epidermal differentiation, additional molecular mechanisms underlying transcriptional regulation by enhancers are less clear. Here we discuss studies that have elucidated the mechanisms of enhancer-promoter interactions in epidermal biology, and lessons we can learn from other tissue models.
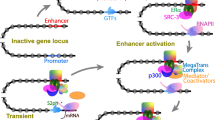
10.7.1.1 The Formation and Biology of Enhancer-Promoter Chromatin Loops
Formation of enhancer-promoter chromatin loops as a mechanism to drive gene activation has emerged as a major concept in the exploration of enhancer-promoter interactions. One of the best studied loci is the evolutionarily conserved β-globin locus that plays key roles in hematopoiesis (reviewed in [151]). The 5 globin genes (ε, Gγ, Aγ, δ and β) form a cluster, and are expressed in a developmental-stage- and tissue-specific manner, controlled by a LCR. The GATA-1 transcription factor and Ldb1 are required to form a chromatin interaction between the β-globin LCR and the β-globin promoter for transcriptional activation in erythroid cells [152, 153]. The requirement for formation of a chromatin loop for gene transcription was demonstrated using artificial zinc fingers (ZF) [154] to force chromatin loop formation by tethering Ldb1 to the β-globin locus control region in GATA-1 null erythroblasts. This was found to be sufficient to activate β-globin gene expression. This work was the first to demonstrate the causality of chromatin spatial interactions in promoting gene transcription.
10.7.1.2 Regulation of the EDC by CNE 923 Via AP-1 Mediated Chromatin Interactions
The organization of the EDC as a conserved cluster of genes with related functions [7] is reminiscent of the organization of the β-globin locus [151]. Moreover, the identification of the EDC enhancer CNE 923 as an epidermal specific enhancer (described in Sect. 10.4.3), suggests its potential function as an LCR that drives coordinate and concomitant EDC gene expression in a manner similar to the β-globin LCR. In line with this, 3C experiments performed with respect to the mouse orthologous CNE 923 sequence in proliferating primary mouse keratinocytes demonstrated that this sequence interacts with nine EDC gene promoters (Sprr2a1, Sprr2d, Sprr2f, Sprr1b, Sprr3, Ivl, Lce1b, Lce1a2, and Crct1 gene promoters) that lie as far as 500 kb from CNE 923, despite the lack of EDC gene expression in these cells, suggesting a poised and enhancer-mediated chromatin state [57]. After calcium-induced differentiation, CNE 923 interacted with the promoters of 11 EDC gene, including Lce3b, S100a6, Sprr2a1, Sprr2b, Sprr3, Sprr4, Ivl, Lce6a, Lce1b, Lce1e, and Crct1. The interaction between CNE 923 and S100a6, located 2 Mb apart, suggested that the EDC chromatin domain is compacted during differentiation to bring more linearly distal genes into close proximity with the enhancer. These results highlight dynamic chromatin looping interactions with 923 that are associated with concomitant EDC gene expression.
Bioinformatics analysis of human CNE 923 identified two highly conserved sequence blocks (PhastCons) that are required for enhancer activity. A consensus AP-1 transcription factor binding site within the most 5′ block is required for maximal enhancer activity, and pharmacological inhibition of AP-1 binding in calcium-induced keratinocytes represses EDC gene expression, and causes loss of c-Jun/AP-1 binding to 923 and aberrant chromatin remodeling. These observations identify a link between an epidermal-specific EDC enhancer and c-Jun/AP-1 transcription factor binding, and together with other studies [104, 106, 107], suggest that further analysis of the 3D structure of chromatin would aid our understanding of EDC regulation.
10.7.2 Higher Level Chromatin Architecture : Topologically Associated Domains (TAD) and Chromosome Territories (CT)
Topologically associated domains (TADs) were first identified by Hi-C [155, 156], and are defined as distinct clusters of enhancer-promoter interactions [157, 158]. At the highest order of chromosome organization, spatially proximal TADs compose a chromosome territory (CT), a compartment within the nucleus that is often segregated in a chromosome-specific manner (reviewed in [159]). Actively transcribed gene-rich loci that are in an open conformation are more likely to loop out of their CTs, suggesting that the space between CTs is important for genomic loci to access the transcription machinery (reviewed in [159]).
The importance of long range enhancer-promoter interactions in the context of a CT was demonstrated in studies of the developing limb bud [160], where differential expression of the Sonic hedgehog (Shh) gene is mediated by specific interactions between the Shh promoter and a long-range enhancer MFCS1. In the intermediate portion of the limb bud, which lacks Shh expression, the long-range enhancer is spatially and linearly distant from the Shh coding region. In anterior limb bud cells, the long-range enhancer interacts with the Shh coding region, but the interactors remain in a poised state within their CT. However, in cells of the zone of polarizing activity (ZPA) where Shh is actively expressed, 3D-FISH showed that the interacting regions relocate outside the CT.
A similar mechanism has been observed in the control of EDC gene expression in differentiating keratinocytes, where Satb1 binds to several sites across the EDC locus, compacting the EDC chromatin architecture into a densely looped structure that, upon relocalization of the locus into the nuclear interior by Brg1, enables efficient and coordinate activation of EDC genes [106, 107]. Similar mechanisms may be employed to control genes such as K1 and K10 which are coordinately activated as keratinocytes transition from basal to suprabasal layers of the epidermis.
10.7.3 Involvement of Cohesin and CTCF in Forming Active Chromatin Hubs
Enhancer activity can also be modulated by insulators that function as physical barriers to optimal enhancer-promoter formation for transcriptional activation. Here we discuss newly recognized attributes of enhancers and new direct roles for CCCTC-binding factor (CTCF) bound insulators in enhancer–promoter interactions and in broadly configuring the genome (Reviewed in [143, 161]).
Cohesin is a complex of proteins that holds sister chromatids together after DNA replication, until the sister chromatids separate at anaphase (reviewed in [162]). Analysis of the effects of mutations in cohesin subunits identified a role for cohesin in regulating enhancer-promoter interactions and gene expression (reviewed in [162]). Subsequently, it was discovered that mammalian cohesin complexes can be recruited to DNaseI hypersensitive sites and conserved noncoding sequences by the CTCF DNA binding protein [134,135,165]. CTCF often binds at insulators and at boundary elements to demarcate active chromatin hubs and limit the effect of enhancers [166], and cohesin contributes to CTCF’s enhancer blocking activity [163, 165]. Studies of the apolipoprotein gene cluster [167], the globin locus [168], and the T-cell receptor (Tcra) locus [169] demonstrated cooperation of CTCF and cohesin to mediate insulators corresponding to TAD boundaries , thereby maintaining proper chromatin loop formation and localization of transcriptional apparatus at the gene promoters to control gene expression.
These mechanisms are relevant to human disease, as chromosomal rearrangements of the conserved TAD-spanning WNT6/IHH/EPHA4/PAX3 locus that disrupt a CTCF-associated boundary domain within a TAD, cause limb malformations in humans. Mice harboring the equivalent disease-relevant rearrangements were generated using CRISPR/Cas9-genome editing, and displayed ectopic limb expression of a gene that lies within the locus but is not normally expressed in limb development, due to misplacement of a cluster of limb enhancers relative to TAD boundaries [158]. This finding demonstrates the functional importance of TADs for orchestrating gene expression via genome architecture and suggests the utility of analyzing disease-associated large-scale chromosomal rearrangements in delineating TAD boundaries.
Genomic studies in other tissue types and loci have also greatly advanced our understanding of the molecular mechanisms of CTCF-mediated chromatin looping events. Chromatin conformation Hi-C capture data with parallel CTCF ChIP-seq identified a nonrandom pattern of forward and reverse orientation for a given pair of CTCF binding sites involved within a chromatin loop [141,142,172]. Subsequent studies later identified a functional role for the directionality of CTCF binding sites to influence chromatin topology and enhancer-promoter function [173, 174]. CRISPR/Cas9-generated inversion of a genomic region spanning CTCF boundary elements in the P-cadherin enhancer altered chromatin topology [173]. Deletions of individual CTCF binding sites by CRISPR/Cas9 led to loss of CTCF, reduced cohesin binding, and reduced or abolished chromatin looping, while inversions restored CTCF and cohesin binding but reduced chromatin looping [174]. However, the impact of the presence or directionality of CTCF binding sites on proximal gene expression varied, suggesting the influence of additional factors. This was further evidenced by deletions of 4 CTCF binding sites in the casein locus that did not affect gene expression but instead caused more distal de novo Sultd1 activation by way of other nearby enhancers [175]. These studies demonstrated a novel governing principle for chromatin architecture in controlling gene expression, and holds great potential for more accurate and contextual prediction of the functionality of enhancers in the skin and other tissues.
10.7.4 The Role of Mediator and Super-Enhancers
More recently, a new class of enhancers called “super-enhancers” has been identified [176]. Super-enhancers are marked by high levels of Mediator coactivator complex occupation as determined by ChIP-seq and span much larger distances than typical enhancers (approximately 8.7 kb versus 703 bp). Mediator is a major component of the transcription pre-initiation complex (PIC) machinery with RNA polymerase II (RNA pol II) and is required for activator-dependent transcription in vitro and in vivo (reviewed in [177]). Reduced levels of Mediator specifically affect gene expression near super-enhancers [176]. This was convincingly demonstrated by the loss of enhancer-promoter loops of select genes upon deletion of Mediator [178, 179]. Mediator-occupied super-enhancers also exhibited enriched binding of transcription factors that are master regulators involved in cell-identity in ESCs, pro-B cells, T helper cells, myotubes, and macrophages, among other cell types. However, one can argue that Mediator and even cohesin binding may not be entirely necessary for gene activation as it was recently determined that chromatin looping between the globin LCR and the β globin locus via Ldb1 was established in the absence of Mediator and cohesion binding [180]. More recent work identified an emergent paradigm for super-enhancers in the direct biogenesis of master regulator miRNAs for tissue specificity [181].
A role for super-enhancers was recently identified in epidermal stem cells [182]. The target genes associated with epidermal stem cell-specific super-enhancers identified by H3K27Ac and Mediator ChIP-seq methods contain a high frequency of binding motifs for the transcription factors Sox9, Lhx2, Nfatc1 and Nfib, that are important for maintaining hair follicle stem cells [154,155,156,186]. ChIP-seq experiments showed that these transcription factors bind at high frequency to super-enhancers relative to typical enhancers. Lineage tracing during mouse epidermal development, wound-healing, and in cell culture showed that super-enhancers are remodeled according to their cellular environment, supporting the idea that enhancers are activated or silenced in a lineage-specific fashion. The dynamic behavior of these regulatory elements in human keratinocyte progenitors as well as in their differentiating progeny was further supported by the discovery of genomic profiles for superenhancers (H3K27ac) and typical enhancers (H3K4me1, H3K27ac, H3K4me3) [187] that were different across these distinct keratinocyte states. Binding of the p63 master regulator for keratinocyte differentiation was observed in a core set of super-enhancers and enhancers (shared between mouse and human) associated with keratinocyte-specific gene expression, but also notably in mouse-specific regions that underlie species-specific transcriptional differences [188].
Analysis of sites bound by Mediator thus enables identification of key transcription factors and enhancer sequences in a variety of cell types and their sensitivity to changing conditions, highlighting the potential of this approach as a tool to pinpoint important regulatory sequences involved in cell and tissue homeostasis, even without prior knowledge of the transcription factors or genes involved.
10.7.5 Non-coding RNAs
As discussed above, the establishment and maintenance of chromosome territories or TADs bring enhancer and promoter interactions into close proximity to facilitate gene activation (reviewed in [157]). In addition to the roles of cohesin and CTCF occupation in enhancer-promoter interactions, long non-coding RNAs (or lncRNAs) may also serve as a scaffold for the assembly of transcription factors and chromatin remodeling enzymes at the promoter [189]. The lncRNA HOTAIR, first discovered in adult skin [190], targets members of the polycomb repressive complex 2 (PRC2) to specific genomic loci, including Ezh2 [189] which, as discussed earlier, regulates epidermal differentiation [103]. HOTAIR also directly interacts with LSD1 [189], a protein complex that is associated with maintenance of epidermal stem cells in an undifferentiated state [191]. Moreover, PRC2 and LSD1 interact in a HOTAIR dependent manner [189], suggesting a potential role for HOTAIR in epidermal development.
More recently, evidence of transcription of functional enhancer RNA (eRNA) from non-coding enhancer sequences has emerged [192]. eRNAs are considered a separate class of non-coding RNA. Unlike lncRNAs, which are marked by H3K4me3 at their promoters and are frequently spliced and polyadenylated, eRNAs are transcribed from enhancer regions marked by H3K4me1, an absence of H3K4me3 histone modifications, and to a lesser extent, post-transcriptional modifications. During transcriptional activation, enhancer RNAs participate in a bi-directional enhancer-promoter activation feedback loop, whereby chromatin looping between the enhancer and promoter brings the bi-directionally transcribed eRNA near the target gene to drive gene expression, at the same time allowing the eRNA to stabilize the enhancer-promoter loop with the help of the Mediator complex (reviewed in [87]). This mechanism for eRNA mediated gene activation has been identified in the expression of various lineage-specific genes, including macrophage-specific genes [193], estrogen-regulated genes in breast cancer cells [194], and p53-regulated genes that induce cell cycle arrest [195]. To date, a role for eRNAs in epidermal development has yet to be identified, but should be kept in mind as a possible mode of gene regulation.
10.8 Conclusion
In this Chapter, we have outlined the history of scientific discoveries related to the transcriptional regulation of key genes involved in skin biology leading up to the concept of enhancers and the molecular mechanisms that orchestrate this process at the chromatin level in the post-human genome and post-ENCODE eras. Our current knowledge of the mechanisms regulating expression of genes involved in determining the proliferative or differentiated state of keratinocytes identifies common patterns in modes of regulation. These observations are consistent with mechanisms identified in other tissue systems. However, regulation of keratinocyte-specific genes appears to heavily utilize p63, AP-1, SP1, and C/EBP transcription factors in particular.
Earlier studies of proximal regions of epidermal genes including keratin genes, IVL, and LOR, and functional genetic approaches to delineate the sequences that drive transcriptional activities, underscored the role of the complex interplay of AP-1 proteins, SP1 and C/EBP transcription factors in epidermal gene regulation. However, we have yet to understand the larger regulatory landscape of the keratin gene clusters.
By contrast, studies of the EDC locus have benefited from the availability of bioinformatics tools and high-throughput methods, revealing that proper control of gene expression is an intricate hierarchy of events that depends firstly on appropriate post-translational regulation of histones to designate chromatin regions as accessible or inaccessible, followed by further organization and remodeling of the EDC together with proper nuclear spatial positioning [106, 107]. The establishment of regulatory landscapes and chromosome territories or topologically associated domains (TADs) brings regulatory enhancers and promoters into close proximity, allowing AP-1, together with other as yet unknown factors, to be poised for efficient coordinate activation of the EDC genes upon induction of epidermal differentiation [57].
The varied roles of AP-1 transcription factors provide an example of how different combinations of common transcription factors are brought together to form complexes to modulate or alter gene expression under different conditions such as spatial context and developmental stage. Distinct complexes may be formed by altering the occupancy of DNA binding proteins at arrays of enhancer elements in close proximity to gene promoters. Complex assembly may also be driven in part by long-range chromatin interactions that bring distal enhancers close to their target gene promoters, and this can be mediated in a myriad of ways. Within the field of skin biology, we have only recently begun to define the mechanisms by which enhancer-promoter interactions initially occur.
10.9 Future Studies
Further dissection of the regulatory principles underlying the gene expression patterns that accompany and drive epidermal development will require a two-pronged approach. The availability of whole genome sequences from increasing numbers of species [196], high-throughput techniques such as ChIP-seq, RNA-seq, ATAC-seq, and the development of bioinformatics tools to allow the integration of such data [197], have made it possible to approach the analyses of gene regulation at the genomics level. Specifically, these advances have enabled researchers to identify enhancer landscapes across the genome and further elucidate the principles that govern enhancer-promoter interactions and genome organization relevant to gene expression. Such an approach identified a surprising role for the DNA methyltransferases, Dnmt3a and Dnmt3b, in occupying enhancers and functionally linked them to epidermal stem cell function [198]. Furthermore, the discovery of skin disease sequence variants enriched in superenhancers highlights the clinical impact of this research and future studies in this area [187].
While several key mechanisms, such as the roles of chromatin looping, 3D genomic architecture, and non-coding RNAs were first demonstrated in non-epidermal tissues and cell-types, they are crucial in constructing a framework for understanding epidermal-specific enhancer-promoter interactions. For example, cohesin and CTCF have been shown to play a role in maintaining enhancer-promoter interactions in a multitude of cell-types and tissues [138,139,169, 199]. This strongly suggests that they are also likely to be major players in mediating the formation of chromatin loops in differentiating keratinocytes. This could be tested by identifying changes in CTCF and cohesin occupation in the different epidermal layers, as well as through functional genetic studies.
In order to completely understand the mechanisms driving enhancer regulated gene expression, we must continue to incorporate multi-disciplinary approaches and novel methods to approach the problem from genetic, molecular and cellular perspectives. Drawing from the expertise of the evolutionary biology field, we are now able to identify candidate regions with regulatory potential faster than ever before [71, 72, 200]. Additional enhancers that are not evolutionarily conserved can be identified using sequencing technologies (see Table 10.1 and [201]). Current advances in genome editing (CRISPR/Cas9, TALENs) have also made it easier to test the functions of endogenous enhancers [158, 173,174,204] and provide an improvement over the use of artificial transgenes that remove enhancers from their appropriate genomic context. The precision of these genome-editing methods enables us to directly test hypotheses regarding enhancer functions at specific locus/loci within the regulatory landscape. For example, CRISPR/Cas9 genome editing of several enhancers has demonstrated their requirement for gene expression [158, 173,174,204]. A high-throughput genetic screen for a targeted set of regulatory elements in the POU5F1 locus using CRISPR/Cas9 editing has also confirmed the functional role of enhancers for POU5F1 expression, while simultaneously revealing a new class of “TEMP” enhancers that are characterized by temporary loss of gene expression and weak reporter activity [205]. CRISPR/Cas9-mediated recombination of orthologous yet divergent enhancer sequences in mice has also provided a comparative functional assay to further assess the importance of transcription factor binding sites during development [204]. For instance, in vivo replacement of a mouse Sonic hedgehog enhancer with the orthologous snake-specific Sonic hedgehog enhancer led to a limb defect that was rescued by introduction of an ETS binding site that had been lost in the snake. In addition to modern genetics and genomics approaches, molecular tools, such as live-imaging and high-resolution microscopy and biomechanics studies will extend our understanding of the dynamics of enhancer-promoter interactions.
Elucidation of the molecular biology and biochemistry of enhancer-promoter interactions has set the stage for a new era of investigation into the mechanisms of transcriptional regulation. Armed with new methodologies for genome sequencing and editing and protein engineering to both discover enhancers and to rapidly test their functions, we are well placed to achieve a more comprehensive understanding of the principles of genome architecture that modulate cellular transcriptomes.
Abbreviations
- 3C:
-
Chromosome conformation capture
- ChIP:
-
Chromatin immunoprecipitation
- CNE:
-
Conserved noncoding element
- CNS:
-
Conserved noncoding sequence
- CT:
-
Chromosome Territory
- CTCF:
-
CCCTC-binding factor
- DRR:
-
Distal regulatory region
- EDC:
-
Epidermal Differentiation Complex
- FLG :
-
Filaggrin
- HAT:
-
Histone acetyltransferase
- HDAC:
-
Histone deactylase
- IVL:
-
Involucrin
- LCE:
-
Late cornified envelope
- LCR:
-
Locus control region
- LOR:
-
Loricrin
- PAD:
-
Peptidylarginine deaminase
- SPRR:
-
Small proline-rich region
- TAD:
-
Topologically associated domains
References
Hsu YC, Li L, Fuchs E. Emerging interactions between skin stem cells and their niches. Nat Med. 2014;20(8):847–56.
Lechler T, Fuchs E. Asymmetric cell divisions promote stratification and differentiation of mammalian skin. Nature. 2005;437(7056):275–80.
Blanpain C, Fuchs E. Epidermal homeostasis: a balancing act of stem cells in the skin. Nat Rev Mol Cell Biol. 2009;10(3):207–17.
Mischke D, Korge BP, Marenholz I, Volz A, Ziegler A. Genes encoding structural proteins of epidermal cornification and S100 calcium-binding proteins form a gene complex (“epidermal differentiation complex”) on human chromosome 1q21. J Invest Dermatol. 1996;106(5):989–92.
Zhao XP, Elder JT. Positional cloning of novel skin-specific genes from the human epidermal differentiation complex. Genomics. 1997;45(2):250–8.
Marshall D, Hardman MJ, Nield KM, Byrne C. Differentially expressed late constituents of the epidermal cornified envelope. Proc Natl Acad Sci U S A. 2001;98(23):13031–6.
de Guzman Strong C, Conlan S, Deming CB, Cheng J, Sears KE, Segre JA. A milieu of regulatory elements in the epidermal differentiation complex syntenic block: implications for atopic dermatitis and psoriasis. Hum Mol Genet. 2010;19(8):1453–60.
Hardman MJ, Sisi P, Banbury DN, Byrne C. Patterned acquisition of skin barrier function during development. Development. 1998;125(8):1541–52.
Yuspa SH, Kilkenny AE, Steinert PM, Roop DR. Expression of murine epidermal differentiation markers is tightly regulated by restricted extracellular calcium concentrations in vitro. J Cell Biol. 1989;109(3):1207–17.
Pillai S, Bikle DD, Mancianti ML, Cline P, Hincenbergs M. Calcium regulation of growth and differentiation of normal human keratinocytes: modulation of differentiation competence by stages of growth and extracellular calcium. J Cell Physiol. 1990;143(2):294–302.
Bikle DD, Xie Z, Tu CL. Calcium regulation of keratinocyte differentiation. Expert Rev Endocrinol Metab. 2012;7(4):461–72.
Shaulian E, Karin M. AP-1 in cell proliferation and survival. Oncogene. 2001;20(19):2390–400.
Mehic D, Bakiri L, Ghannadan M, Wagner EF, Tschachler E. Fos and jun proteins are specifically expressed during differentiation of human keratinocytes. J Invest Dermatol. 2005;124(1):212–20.
Visel A, Bristow J, Pennacchio LA. Enhancer identification through comparative genomics. Semin Cell Dev Biol. 2007;18(1):140–52.
Shlyueva D, Stampfel G, Stark A. Transcriptional enhancers: from properties to genome-wide predictions. Nat Rev Genet. 2014;15(4):272–86.
Levo M, Segal E. In pursuit of design principles of regulatory sequences. Nat Rev Genet. 2014;15(7):453–68.
Benoist C, Chambon P. In vivo sequence requirements of the SV40 early promotor region. Nature. 1981;290(5804):304–10.
Gruss P, Dhar R, Khoury G. Simian virus 40 tandem repeated sequences as an element of the early promoter. Proc Natl Acad Sci U S A. 1981;78(2):943–7.
Banerji J, Rusconi S, Schaffner W. Expression of a beta-globin gene is enhanced by remote SV40 DNA sequences. Cell. 1981;27(2 Pt 1):299–308.
Moreau P, Hen R, Wasylyk B, Everett R, Gaub MP, Chambon P. The SV40 72 base repair repeat has a striking effect on gene expression both in SV40 and other chimeric recombinants. Nucleic Acids Res. 1981;9(22):6047–68.
Fromm M, Berg P. Simian virus 40 early- and late-region promoter functions are enhanced by the 72-base-pair repeat inserted at distant locations and inverted orientations. Mol Cell Biol. 1983;3(6):991–9.
Kleinjan DA, van Heyningen V. Long-range control of gene expression: emerging mechanisms and disruption in disease. Am J Hum Genet. 2005;76(1):8–32.
Levine M. Transcriptional enhancers in animal development and evolution. Curr Biol. 2010;20(17):R754–63.
Williamson I, Hill RE, Bickmore WA. Enhancers: from developmental genetics to the genetics of common human disease. Dev Cell. 2011;21(1):17–9.
Hardison RC, Taylor J. Genomic approaches towards finding cis-regulatory modules in animals. Nat Rev Genet. 2012;13(7):469–83.
Coulombe PA, Fuchs E. Elucidating the early stages of keratin filament assembly. J Cell Biol. 1990;111(1):153–69.
Lee CH, Coulombe PA. Self-organization of keratin intermediate filaments into cross-linked networks. J Cell Biol. 2009;186(3):409–21.
Moll R, Franke WW, Schiller DL, Geiger B, Krepler R. The catalog of human cytokeratins: patterns of expression in normal epithelia, tumors and cultured cells. Cell. 1982;31(1):11–24.
Lersch R, Stellmach V, Stocks C, Giudice G, Fuchs E. Isolation, sequence, and expression of a human keratin K5 gene: transcriptional regulation of keratins and insights into pairwise control. Mol Cell Biol. 1989;9(9):3685–97.
Marchuk D, McCrohon S, Fuchs E. Complete sequence of a gene encoding a human type I keratin: sequences homologous to enhancer elements in the regulatory region of the gene. Proc Natl Acad Sci U S A. 1985;82(6):1609–13.
Vassar R, Rosenberg M, Ross S, Tyner A, Fuchs E. Tissue-specific and differentiation-specific expression of a human K14 keratin gene in transgenic mice. Proc Natl Acad Sci U S A. 1989;86(5):1563–7.
Sinha S, Degenstein L, Copenhaver C, Fuchs E. Defining the regulatory factors required for epidermal gene expression. Mol Cell Biol. 2000;20(7):2543–55.
Leask A, Rosenberg M, Vassar R, Fuchs E. Regulation of a human epidermal keratin gene: sequences and nuclear factors involved in keratinocyte-specific transcription. Genes Dev. 1990;4(11):1985–98.
Leask A, Byrne C, Fuchs E. Transcription factor AP2 and its role in epidermal-specific gene expression. Proc Natl Acad Sci U S A. 1991;88(18):7948–52.
Ohtsuki M, Flanagan S, Freedberg IM, Blumenberg M. A cluster of five nuclear proteins regulates keratin gene transcription. Gene Expr. 1993;3(2):201–13.
Sinha S, Fuchs E. Identification and dissection of an enhancer controlling epithelial gene expression in skin. Proc Natl Acad Sci U S A. 2001;98(5):2455–60.
Byrne C, Fuchs E. Probing keratinocyte and differentiation specificity of the human K5 promoter in vitro and in transgenic mice. Mol Cell Biol. 1993;13(6):3176–90.
Mills AA, Zheng B, Wang XJ, Vogel H, Roop DR, Bradley A. p63 is a p53 homologue required for limb and epidermal morphogenesis. Nature. 1999;398(6729):708–13.
Yang A, Schweitzer R, Sun D, Kaghad M, Walker N, Bronson RT, Tabin C, Sharpe A, Caput D, Crum C, McKeon F. p63 is essential for regenerative proliferation in limb, craniofacial and epithelial development. Nature. 1999;398(6729):714–8.
Candi E, Rufini A, Terrinoni A, Dinsdale D, Ranalli M, Paradisi A, De Laurenzi V, Spagnoli LG, Catani MV, Ramadan S, Knight RA, Melino G. Differential roles of p63 isoforms in epidermal development: selective genetic complementation in p63 null mice. Cell Death Differ. 2006;13(6):1037–47.
Romano RA, Birkaya B, Sinha S. A functional enhancer of keratin14 is a direct transcriptional target of deltaNp63. J Invest Dermatol. 2007;127(5):1175–86.
Boldrup L, Coates PJ, Gu X, Nylander K. DeltaNp63 isoforms regulate CD44 and keratins 4, 6, 14 and 19 in squamous cell carcinoma of head and neck. J Pathol. 2007;213(4):384–91.
Medawar A, Virolle T, Rostagno P, de la Forest-Divonne S, Gambaro K, Rouleau M, Aberdam D. DeltaNp63 is essential for epidermal commitment of embryonic stem cells. PLoS One. 2008;3(10):e3441.
Romano RA, Ortt K, Birkaya B, Smalley K, Sinha S. An active role of the DeltaN isoform of p63 in regulating basal keratin genes K5 and K14 and directing epidermal cell fate. PLoS One. 2009;4(5):e5623.
Magin TM, Vijayaraj P, Leube RE. Structural and regulatory functions of keratins. Exp Cell Res. 2007;313(10):2021–32.
Rosenthal DS, Steinert PM, Chung S, Huff CA, Johnson J, Yuspa SH, Roop DR. A human epidermal differentiation-specific keratin gene is regulated by calcium but not negative modulators of differentiation in transgenic mouse keratinocytes. Cell Growth Differ. 1991;2(2):107–13.
Huff CA, Yuspa SH, Rosenthal D. Identification of control elements 3′ to the human keratin 1 gene that regulate cell type and differentiation-specific expression. J Biol Chem. 1993;268(1):377–84.
Lu B, Rothnagel JA, Longley MA, Tsai SY, Roop DR. Differentiation-specific expression of human keratin 1 is mediated by a composite AP-1/steroid hormone element. J Biol Chem. 1994;269(10):7443–9.
Zhu S, Oh HS, Shim M, Sterneck E, Johnson PF, Smart RC. C/EBPbeta modulates the early events of keratinocyte differentiation involving growth arrest and keratin 1 and keratin 10 expression. Mol Cell Biol. 1999;19(10):7181–90.
Oh HS, Smart RC. Expression of CCAAT/enhancer binding proteins (C/EBP) is associated with squamous differentiation in epidermis and isolated primary keratinocytes and is altered in skin neoplasms. J Invest Dermatol. 1998;110(6):939–45.
Maytin EV, Lin JC, Krishnamurthy R, Batchvarova N, Ron D, Mitchell PJ, Habener JF. Keratin 10 gene expression during differentiation of mouse epidermis requires transcription factors C/EBP and AP-2. Dev Biol. 1999;216(1):164–81.
Johnson LD, Idler WW, Zhou XM, Roop DR, Steinert PM. Structure of a gene for the human epidermal 67-kDa keratin. Proc Natl Acad Sci U S A. 1985;82(7):1896–900.
Rieger M, Franke WW. Identification of an orthologous mammalian cytokeratin gene. High degree of intron sequence conservation during evolution of human cytokeratin 10. J Mol Biol. 1988;204(4):841–56.
Segre JA. Epidermal barrier formation and recovery in skin disorders. J Clin Invest. 2006;116(5):1150–8.
Eckert RL, Crish JF, Efimova T, Dashti SR, Deucher A, Bone F, Adhikary G, Huang G, Gopalakrishnan R, Balasubramanian S. Regulation of involucrin gene expression. J Invest Dermatol. 2004;123(1):13–22.
Nithya S, Radhika T, Jeddy N. Loricrin – an overview. J Oral Maxillofac Pathol. 2015;19(1):64–8.
Oh IY, Albea DA, Goodwin ZA, Quiggle AM, Baker BP, Guggisberg AM, Geahlen JH, Kroner GM, de Guzman Strong C. Regulation of the dynamic chromatin architecture of the epidermal differentiation complex is mediated by a c-Jun/AP-1-modulated enhancer. J Invest Dermatol. 2014;134:2371–80.
Carroll JM, Taichman LB. Characterization of the human involucrin promoter using a transient beta-galactosidase assay. J Cell Sci. 1992;103(Pt 4):925–30.
Welter JF, Crish JF, Agarwal C, Eckert RL. Fos-related antigen (Fra-1), junB, and junD activate human involucrin promoter transcription by binding to proximal and distal AP1 sites to mediate phorbol ester effects on promoter activity. J Biol Chem. 1995;270(21):12614–22.
Banks EB, Crish JF, Eckert RL. Transcription factor Sp1 activates involucrin promoter activity in non-epithelial cell types. Biochem J. 1999;337(Pt 3):507–12.
Banks EB, Crish JF, Welter JF, Eckert RL. Characterization of human involucrin promoter distal regulatory region transcriptional activator elements-a role for Sp1 and AP1 binding sites. Biochem J. 1998;331(Pt 1):61–8.
Ogryzko VV, Schiltz RL, Russanova V, Howard BH, Nakatani Y. The transcriptional coactivators p300 and CBP are histone acetyltransferases. Cell. 1996;87(5):953–9.
ENCODE Project Consortium, Birney E, Stamatoyannopoulos JA, Dutta A, Guigó R, Gingeras TR, Margulies EH, Weng Z, Snyder M, Dermitzakis ET, Thurman RE, Kuehn MS, Taylor CM, Neph S, Koch CM, Asthana S, Malhotra A, Adzhubei I, Greenbaum JA, Andrews RM, Flicek P, Boyle PJ, Cao H, Carter NP, Clelland GK, Davis S, Day N, Dhami P, Dillon SC, Dorschner MO, Fiegler H, Giresi PG, Goldy J, Hawrylycz M, Haydock A, Humbert R, James KD, Johnson BE, Johnson EM, Frum TT, Rosenzweig ER, Karnani N, Lee K, Lefebvre GC, Navas PA, Neri F, Parker SC, Sabo PJ, Sandstrom R, Shafer A, Vetrie D, Weaver M, Wilcox S, Yu M, Collins FS, Dekker J, Lieb JD, Tullius TD, Crawford GE, Sunyaev S, Noble WS, Dunham I, Denoeud F, Reymond A, Kapranov P, Rozowsky J, Zheng D, Castelo R, Frankish A, Harrow J, Ghosh S, Sandelin A, Hofacker IL, Baertsch R, Keefe D, Dike S, Cheng J, Hirsch HA, Sekinger EA, Lagarde J, Abril JF, Shahab A, Flamm C, Fried C, Hackermüller J, Hertel J, Lindemeyer M, Missal K, Tanzer A, Washietl S, Korbel J, Emanuelsson O, Pedersen JS, Holroyd N, Taylor R, Swarbreck D, Matthews N, Dickson MC, Thomas DJ, Weirauch MT, Gilbert J, Drenkow J, Bell I, Zhao X, Srinivasan KG, Sung WK, Ooi HS, Chiu KP, Foissac S, Alioto T, Brent M, Pachter L, Tress ML, Valencia A, Choo SW, Choo CY, Ucla C, Manzano C, Wyss C, Cheung E, Clark TG, Brown JB, Ganesh M, Patel S, Tammana H, Chrast J, Henrichsen CN, Kai C, Kawai J, Nagalakshmi U, Wu J, Lian Z, Lian J, Newburger P, Zhang X, Bickel P, Mattick JS, Carninci P, Hayashizaki Y, Weissman S, Hubbard T, Myers RM, Rogers J, Stadler PF, Lowe TM, Wei CL, Ruan Y, Struhl K, Gerstein M, Antonarakis SE, Fu Y, Green ED, Karaöz U, Siepel A, Taylor J, Liefer LA, Wetterstrand KA, Good PJ, Feingold EA, Guyer MS, Cooper GM, Asimenos G, Dewey CN, Hou M, Nikolaev S, Montoya-Burgos JI, Löytynoja A, Whelan S, Pardi F, Massingham T, Huang H, Zhang NR, Holmes I, Mullikin JC, Ureta-Vidal A, Paten B, Seringhaus M, Church D, Rosenbloom K, Kent WJ, Stone EA; NISC Comparative Sequencing Program; Baylor College of Medicine Human Genome Sequencing Center; Washington University Genome Sequencing Center; Broad Institute; Children's Hospital Oakland Research Institute, Batzoglou S, Goldman N, Hardison RC, Haussler D, Miller W, Sidow A, Trinklein ND, Zhang ZD, Barrera L, Stuart R, King DC, Ameur A, Enroth S, Bieda MC, Kim J, Bhinge AA, Jiang N, Liu J, Yao F, Vega VB, Lee CW, Ng P, Shahab A, Yang A, Moqtaderi Z, Zhu Z, Xu X, Squazzo S, Oberley MJ, Inman D, Singer MA, Richmond TA, Munn KJ, Rada-Iglesias A, Wallerman O, Komorowski J, Fowler JC, Couttet P, Bruce AW, Dovey OM, Ellis PD, Langford CF, Nix DA, Euskirchen G, Hartman S, Urban AE, Kraus P, Van Calcar S, Heintzman N, Kim TH, Wang K, Qu C, Hon G, Luna R, Glass CK, Rosenfeld MG, Aldred SF, Cooper SJ, Halees A, Lin JM, Shulha HP, Zhang X, Xu M, Haidar JN, Yu Y, Ruan Y, Iyer VR, Green RD, Wadelius C, Farnham PJ, Ren B, Harte RA, Hinrichs AS, Trumbower H, Clawson H, Hillman-Jackson J, Zweig AS, Smith K, Thakkapallayil A, Barber G, Kuhn RM, Karolchik D, Armengol L, Bird CP, de Bakker PI, Kern AD, Lopez-Bigas N, Martin JD, Stranger BE, Woodroffe A, Davydov E, Dimas A, Eyras E, Hallgrímsdóttir IB, Huppert J, Zody MC, Abecasis GR, Estivill X, Bouffard GG, Guan X, Hansen NF, Idol JR, Maduro VV, Maskeri B, McDowell JC, Park M, Thomas PJ, Young AC, Blakesley RW, Muzny DM, Sodergren E, Wheeler DA, Worley KC, Jiang H, Weinstock GM, Gibbs RA, Graves T, Fulton R, Mardis ER, Wilson RK, Clamp M, Cuff J, Gnerre S, Jaffe DB, Chang JL, Lindblad-Toh K, Lander ES, Koriabine M, Nefedov M, Osoegawa K, Yoshinaga Y, Zhu B, de Jong PJ. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature. 2007;447(7146):799–816.
Balasubramanian S, Sturniolo MT, Dubyak GR, Eckert RL. Human epidermal keratinocytes undergo (−)-epigallocatechin-3-gallate-dependent differentiation but not apoptosis. Carcinogenesis. 2005;26(6):1100–8.
Crish JF, Eckert RL. Synergistic activation of human involucrin gene expression by Fra-1 and p300 – evidence for the presence of a multiprotein complex. J Invest Dermatol. 2008;128(3):530–41.
Chew YC, Adhikary G, Xu W, Wilson GM, Eckert RL. Protein kinase C delta increases Kruppel-like factor 4 protein, which drives involucrin gene transcription in differentiating keratinocytes. J Biol Chem. 2013;288(24):17759–68.
Kaczynski J, Cook T, Urrutia R. Sp1- and Kruppel-like transcription factors. Genome Biol. 2003;4(2):206.
DiSepio D, Jones A, Longley MA, Bundman D, Rothnagel JA, Roop DR. The proximal promoter of the mouse loricrin gene contains a functional AP-1 element and directs keratinocyte-specific but not differentiation-specific expression. J Biol Chem. 1995;270(18):10792–9.
Yoneda K, Steinert PM. Overexpression of human loricrin in transgenic mice produces a normal phenotype. Proc Natl Acad Sci U S A. 1993;90(22):10754–8.
Jang SI, Steinert PM. Loricrin expression in cultured human keratinocytes is controlled by a complex interplay between transcription factors of the Sp1, CREB, AP1, and AP2 families. J Biol Chem. 2002;277(44):42268–79.
Siepel A, Bejerano G, Pedersen JS, Hinrichs AS, Hou M, Rosenbloom K, Clawson H, Spieth J, Hillier LW, Richards S, Weinstock GM, Wilson RK, Gibbs RA, Kent WJ, Miller W, Haussler D. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 2005;15(8):1034–50.
Pennacchio LA, Ahituv N, Moses AM, Prabhakar S, Nobrega MA, Shoukry M, Minovitsky S, Dubchak I, Holt A, Lewis KD, Plajzer-Frick I, Akiyama J, De Val S, Afzal V, Black BL, Couronne O, Eisen MB, Visel A, Rubin EM. In vivo enhancer analysis of human conserved non-coding sequences. Nature. 2006;444(7118):499–502.
Martin N, Patel S, Segre JA. Long-range comparison of human and mouse Sprr loci to identify conserved noncoding sequences involved in coordinate regulation. Genome Res. 2004;14(12):2430–8.
Cabral A, Voskamp P, Cleton-Jansen AM, South A, Nizetic D, Backendorf C. Structural organization and regulation of the small proline-rich family of cornified envelope precursors suggest a role in adaptive barrier function. J Biol Chem. 2001;276(22):19231–7.
Chavanas S, Mechin MC, Takahara H, Kawada A, Nachat R, Serre G, Simon M. Comparative analysis of the mouse and human peptidylarginine deiminase gene clusters reveals highly conserved non-coding segments and a new human gene, PADI6. Gene. 2004;330:19–27.
Chavanas S, Adoue V, Mechin MC, Ying S, Dong S, Duplan H, Charveron M, Takahara H, Serre G, Simon M. Long-range enhancer associated with chromatin looping allows AP-1 regulation of the peptidylarginine deiminase 3 gene in differentiated keratinocyte. PLoS One. 2008;3(10):e3408.
Antonini D, Sirico A, Aberdam E, Ambrosio R, Campanile C, Fagoonee S, Altruda F, Aberdam D, Brissette JL, Missero C. A composite enhancer regulates p63 gene expression in epidermal morphogenesis and in keratinocyte differentiation by multiple mechanisms. Nucleic Acids Res. 2015;43(2):862–74.
Taunton J, Hassig CA, Schreiber SL. A mammalian histone deacetylase related to the yeast transcriptional regulator Rpd3p. Science. 1996;272(5260):408–11.
Brownell JE, Zhou J, Ranalli T, Kobayashi R, Edmondson DG, Roth SY, Allis CD. Tetrahymena histone acetyltransferase A: a homolog to yeast Gcn5p linking histone acetylation to gene activation. Cell. 1996;84(6):843–51.
Yang XJ, Seto E. HATs and HDACs: from structure, function and regulation to novel strategies for therapy and prevention. Oncogene. 2007;26(37):5310–8.
Wang Z, Zang C, Cui K, Schones DE, Barski A, Peng W, Zhao K. Genome-wide mapping of HATs and HDACs reveals distinct functions in active and inactive genes. Cell. 2009;138(5):1019–31.
Sassone-Corsi P, Mizzen CA, Cheung P, Crosio C, Monaco L, Jacquot S, Hanauer A, Allis CD. Requirement of Rsk-2 for epidermal growth factor-activated phosphorylation of histone H3. Science. 1999;285(5429):886–91.
Thomson S, Clayton AL, Hazzalin CA, Rose S, Barratt MJ, Mahadevan LC. The nucleosomal response associated with immediate-early gene induction is mediated via alternative MAP kinase cascades: MSK1 as a potential histone H3/HMG-14 kinase. EMBO J. 1999;18(17):4779–93.
Chen D, Ma H, Hong H, Koh SS, Huang SM, Schurter BT, Aswad DW, Stallcup MR. Regulation of transcription by a protein methyltransferase. Science. 1999;284(5423):2174–7.
Gary JD, Lin WJ, Yang MC, Herschman HR, Clarke S. The predominant protein-arginine methyltransferase from Saccharomyces cerevisiae. J Biol Chem. 1996;271(21):12585–94.
Rea S, Eisenhaber F, O'Carroll D, Strahl BD, Sun ZW, Schmid M, Opravil S, Mechtler K, Ponting CP, Allis CD, Jenuwein T. Regulation of chromatin structure by site-specific histone H3 methyltransferases. Nature. 2000;406(6796):593–9.
Wang Y, Wysocka J, Sayegh J, Lee YH, Perlin JR, Leonelli L, Sonbuchner LS, McDonald CH, Cook RG, Dou Y, Roeder RG, Clarke S, Stallcup MR, Allis CD, Coonrod SA. Human PAD4 regulates histone arginine methylation levels via demethylimination. Science. 2004;306(5694):279–83.
Cuthbert GL, Daujat S, Snowden AW, Erdjument-Bromage H, Hagiwara T, Yamada M, Schneider R, Gregory PD, Tempst P, Bannister AJ, Kouzarides T. Histone deimination antagonizes arginine methylation. Cell. 2004;118(5):545–53.
Robzyk K, Recht J, Osley MA. Rad6-dependent ubiquitination of histone H2B in yeast. Science. 2000;287(5452):501–4.
Emre NC, Ingvarsdottir K, Wyce A, Wood A, Krogan NJ, Henry KW, Li K, Marmorstein R, Greenblatt JF, Shilatifard A, Berger SL. Maintenance of low histone ubiquitylation by Ubp10 correlates with telomere-proximal Sir2 association and gene silencing. Mol Cell. 2005;17(4):585–94.
Gardner RG, Nelson ZW, Gottschling DE. Ubp10/Dot4p regulates the persistence of ubiquitinated histone H2B: distinct roles in telomeric silencing and general chromatin. Mol Cell Biol. 2005;25(14):6123–39.
Henry KW, Wyce A, Lo WS, Duggan LJ, Emre NC, Kao CF, Pillus L, Shilatifard A, Osley MA, Berger SL. Transcriptional activation via sequential histone H2B ubiquitylation and deubiquitylation, mediated by SAGA-associated Ubp8. Genes Dev. 2003;17(21):2648–63.
Chang B, Chen Y, Zhao Y, Bruick RK. JMJD6 is a histone arginine demethylase. Science. 2007;318(5849):444–7.
Shi Y, Lan F, Matson C, Mulligan P, Whetstine JR, Cole PA, Casero RA, Shi Y. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell. 2004;119(7):941–53.
Tsukada Y, Fang J, Erdjument-Bromage H, Warren ME, Borchers CH, Tempst P, Zhang Y. Histone demethylation by a family of JmjC domain-containing proteins. Nature. 2006;439(7078):811–6.
Marmorstein R, Trievel RC. Histone modifying enzymes: structures, mechanisms, and specificities. Biochim Biophys Acta. 2009;1789(1):58–68.
Berger SL, Kouzarides T, Shiekhattar R, Shilatifard A. An operational definition of epigenetics. Genes Dev. 2009;23(7):781–3.
Bao X, Tang J, Lopez-Pajares V, Tao S, Qu K, Crabtree GR, Khavari PA. ACTL6a enforces the epidermal progenitor state by suppressing SWI/SNF-dependent induction of KLF4. Cell Stem Cell. 2013;12(2):193–203.
Frye M, Fisher AG, Watt FM. Epidermal stem cells are defined by global histone modifications that are altered by Myc-induced differentiation. PLoS One. 2007;2(8):e763.
Kashiwagi M, Morgan BA, Georgopoulos K. The chromatin remodeler Mi-2beta is required for establishment of the basal epidermis and normal differentiation of its progeny. Development. 2007;134(8):1571–82.
LeBoeuf M, Terrell A, Trivedi S, Sinha S, Epstein JA, Olson EN, Morrisey EE, Millar SE. Hdac1 and Hdac2 act redundantly to control p63 and p53 functions in epidermal progenitor cells. Dev Cell. 2010;19(6):807–18.
Bannister AJ, Kouzarides T. Regulation of chromatin by histone modifications. Cell Res. 2011;21(3):381–95.
Ezhkova E, Pasolli HA, Parker JS, Stokes N, Su IH, Hannon G, Tarakhovsky A, Fuchs E. Ezh2 orchestrates gene expression for the stepwise differentiation of tissue-specific stem cells. Cell. 2009;136(6):1122–35.
Gdula MR, Poterlowicz K, Mardaryev AN, Sharov AA, Peng Y, Fessing MY, Botchkarev VA. Remodeling of three-dimensional organization of the nucleus during terminal keratinocyte differentiation in the epidermis. J Invest Dermatol. 2013;133(9):2191–201.
Vigano MA, Lamartine J, Testoni B, Merico D, Alotto D, Castagnoli C, Robert A, Candi E, Melino G, Gidrol X, Mantovani R. New p63 targets in keratinocytes identified by a genome-wide approach. EMBO J. 2006;25(21):5105–16.
Fessing MY, Mardaryev AN, Gdula MR, Sharov AA, Sharova TY, Rapisarda V, Gordon KB, Smorodchenko AD, Poterlowicz K, Ferone G, Kohwi Y, Missero C, Kohwi-Shigematsu T, Botchkarev VA. p63 regulates Satb1 to control tissue-specific chromatin remodeling during development of the epidermis. J Cell Biol. 2011;194(6):825–39.
Mardaryev AN, Gdula MR, Yarker JL, Emelianov VU, Poterlowicz K, Sharov AA, Sharova TY, Scarpa JA, Joffe B, Solovei I, Chambon P, Botchkarev VA, Fessing MY. p63 and Brg1 control developmentally regulated higher-order chromatin remodelling at the epidermal differentiation complex locus in epidermal progenitor cells. Development. 2014;141(1):101–11.
Sethi I, Sinha S, Buck MJ. Role of chromatin and transcriptional co-regulators in mediating p63-genome interactions in keratinocytes. BMC Genomics. 2014;15:1042.
Ong CT, Corces VG. Enhancer function: new insights into the regulation of tissue-specific gene expression. Nat Rev Genet. 2011;12(4):283–93.
Landt SG, Marinov GK, Kundaje A, Kheradpour P, Pauli F, Batzoglou S, Bernstein BE, Bickel P, Brown JB, Cayting P, Chen Y, DeSalvo G, Epstein C, Fisher-Aylor KI, Euskirchen G, Gerstein M, Gertz J, Hartemink AJ, Hoffman MM, Iyer VR, Jung YL, Karmakar S, Kellis M, Kharchenko PV, Li Q, Liu T, Liu XS, Ma L, Milosavljevic A, Myers RM, Park PJ, Pazin MJ, Perry MD, Raha D, Reddy TE, Rozowsky J, Shoresh N, Sidow A, Slattery M, Stamatoyannopoulos JA, Tolstorukov MY, White KP, Xi S, Farnham PJ, Lieb JD, Wold BJ, Snyder M. ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia. Genome Res. 2012;22(9):1813–31.
Giresi PG, Kim J, McDaniell RM, Iyer VR, Lieb JD. FAIRE (Formaldehyde-Assisted Isolation of Regulatory Elements) isolates active regulatory elements from human chromatin. Genome Res. 2007;17(6):877–85.
Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods. 2013;10(12):1213–8.
Song L, Crawford GE. DNase-seq: a high-resolution technique for mapping active gene regulatory elements across the genome from mammalian cells. Cold Spring Harb Protoc. 2010;2010(2):pdb prot5384.
Hansen RS, Thomas S, Sandstrom R, Canfield TK, Thurman RE, Weaver M, Dorschner MO, Gartler SM, Stamatoyannopoulos JA. Sequencing newly replicated DNA reveals widespread plasticity in human replication timing. Proc Natl Acad Sci U S A. 2010;107(1):139–44.
Zaret K. Micrococcal nuclease analysis of chromatin structure. Curr Protoc Mol Biol., Chapter 21:Unit. 2005;21:21.
Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods. 2008;5(7):621–8.
Carninci P, Kvam C, Kitamura A, Ohsumi T, Okazaki Y, Itoh M, Kamiya M, Shibata K, Sasaki N, Izawa M, Muramatsu M, Hayashizaki Y, Schneider C. High-efficiency full-length cDNA cloning by biotinylated CAP trapper. Genomics. 1996;37(3):327–36.
Fullwood MJ, Liu MH, Pan YF, Liu J, Xu H, Mohamed YB, Orlov YL, Velkov S, Ho A, Mei PH, Chew EG, Huang PY, Welboren WJ, Han Y, Ooi HS, Ariyaratne PN, Vega VB, Luo Y, Tan PY, Choy PY, Wansa KD, Zhao B, Lim KS, Leow SC, Yow JS, Joseph R, Li H, Desai KV, Thomsen JS, Lee YK, Karuturi RK, Herve T, Bourque G, Stunnenberg HG, Ruan X, Cacheux-Rataboul V, Sung WK, Liu ET, Wei CL, Cheung E, Ruan Y. An oestrogen-receptor-alpha-bound human chromatin interactome. Nature. 2009;462(7269):58–64.
Batut P, Gingeras TR. RAMPAGE: promoter activity profiling by paired-end sequencing of 5’-complete cDNAs. Curr Protoc Mol Biol, 104:Unit. 2013;25B:11.
Ruan X, Ruan Y. Genome wide full-length transcript analysis using 5’ and 3’ paired-end-tag next generation sequencing (RNA-PET). Methods Mol Biol. 2012;809:535–62.
Aebersold R, Mann M. Mass spectrometry-based proteomics. Nature. 2003;422(6928):198–207.
Jolma A, Kivioja T, Toivonen J, Cheng L, Wei G, Enge M, Taipale M, Vaquerizas JM, Yan J, Sillanpaa MJ, Bonke M, Palin K, Talukder S, Hughes TR, Luscombe NM, Ukkonen E, Taipale J. Multiplexed massively parallel SELEX for characterization of human transcription factor binding specificities. Genome Res. 2010;20(6):861–73.
Zhao Y, Granas D, Stormo GD. Inferring binding energies from selected binding sites. PLoS Comput Biol. 2009;5(12):e1000590.
Zykovich A, Korf I, Segal DJ. Bind-n-Seq: high-throughput analysis of in vitro protein-DNA interactions using massively parallel sequencing. Nucleic Acids Res. 2009;37(22):e151.
Dekker J, Rippe K, Dekker M, Kleckner N. Capturing chromosome conformation. Science. 2002;295(5558):1306–11.
Zhao Z, Tavoosidana G, Sjolinder M, Gondor A, Mariano P, Wang S, Kanduri C, Lezcano M, Sandhu KS, Singh U, Pant V, Tiwari V, Kurukuti S, Ohlsson R. Circular chromosome conformation capture (4C) uncovers extensive networks of epigenetically regulated intra- and interchromosomal interactions. Nat Genet. 2006;38(11):1341–7.
Dostie J, Richmond TA, Arnaout RA, Selzer RR, Lee WL, Honan TA, Rubio ED, Krumm A, Lamb J, Nusbaum C, Green RD, Dekker J. Chromosome Conformation Capture Carbon Copy (5C): a massively parallel solution for mapping interactions between genomic elements. Genome Res. 2006;16(10):1299–309.
Lieberman-Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, Amit I, Lajoie BR, Sabo PJ, Dorschner MO, Sandstrom R, Bernstein B, Bender MA, Groudine M, Gnirke A, Stamatoyannopoulos J, Mirny LA, Lander ES, Dekker J. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 2009;326(5950):289–93.
Cokus SJ, Feng S, Zhang X, Chen Z, Merriman B, Haudenschild CD, Pradhan S, Nelson SF, Pellegrini M, Jacobsen SE. Shotgun bisulphite sequencing of the Arabidopsis genome reveals DNA methylation patterning. Nature. 2008;452(7184):215–9.
Meissner A, Gnirke A, Bell GW, Ramsahoye B, Lander ES, Jaenisch R. Reduced representation bisulfite sequencing for comparative high-resolution DNA methylation analysis. Nucleic Acids Res. 2005;33(18):5868–77.
Jacinto FV, Ballestar E, Esteller M. Methyl-DNA immunoprecipitation (MeDIP): hunting down the DNA methylome. Biotechniques. 2008;44:1. 35, 37, 39 passim
Maunakea AK, Nagarajan RP, Bilenky M, Ballinger TJ, D’Souza C, Fouse SD, Johnson BE, Hong C, Nielsen C, Zhao Y, Turecki G, Delaney A, Varhol R, Thiessen N, Shchors K, Heine VM, Rowitch DH, Xing X, Fiore C, Schillebeeckx M, Jones SJ, Haussler D, Marra MA, Hirst M, Wang T, Costello JF. Conserved role of intragenic DNA methylation in regulating alternative promoters. Nature. 2010;466(7303):253–7.
Kothary R, Clapoff S, Darling S, Perry MD, Moran LA, Rossant J. Inducible expression of an hsp68-lacZ hybrid gene in transgenic mice. Development. 1989;105(4):707–14.
le T, A Fraser SE. Enhancer and gene traps for molecular imaging and genetic analysis in zebrafish. Dev Growth Differ. 2013;55(4):434–45.
Smale ST. Luciferase assay. Cold Spring Harb Protoc. 2010;2010(5):pdb prot5421.
Smale ST. Beta-galactosidase assay. Cold Spring Harb Protoc. 2010;2010(5):pdb prot5423.
Dickel DE, Zhu Y, Nord AS, Wylie JN, Akiyama JA, Afzal V, Plajzer-Frick I, Kirkpatrick A, Gottgens B, Bruneau BG, Visel A, Pennacchio LA. Function-based identification of mammalian enhancers using site-specific integration. Nat Methods. 2014;11(5):566–71.
Murtha M, Tokcaer-Keskin Z, Tang Z, Strino F, Chen X, Wang Y, Xi X, Basilico C, Brown S, Bonneau R, Kluger Y, Dailey L. FIREWACh: high-throughput functional detection of transcriptional regulatory modules in mammalian cells. Nat Methods. 2014;11(5):559–65.
Vockley CM, Guo C, Majoros WH, Nodzenski M, Scholtens DM, Hayes MG, Lowe WL, Reddy TE Jr. Massively parallel quantification of the regulatory effects of noncoding genetic variation in a human cohort. Genome Res. 2015;25(8):1206–14.
Inoue F, Ahituv N. Decoding enhancers using massively parallel reporter assays. Genomics. 2015;106(3):159–64.
Kwasnieski JC, Mogno I, Myers CA, Corbo JC, Cohen BA. Complex effects of nucleotide variants in a mammalian cis-regulatory element. Proc Natl Acad Sci U S A. 2012;109(47):19498–503.
Akhtar W, de Jong J, Pindyurin AV, Pagie L, Meuleman W, de Ridder J, Berns A, Wessels LF, Lohuizen v, M van Steensel B. Chromatin position effects assayed by thousands of reporters integrated in parallel. Cell. 2013;154(4):914–27.
Krivega I, Dean A. Enhancer and promoter interactions-long distance calls. Curr Opin Genet Dev. 2012;22(2):79–85.
Dean A. On a chromosome far, far away: LCRs and gene expression. Trends Genet. 2006;22(1):38–45.
Carter D, Chakalova L, Osborne CS, Dai YF, Fraser P. Long-range chromatin regulatory interactions in vivo. Nat Genet. 2002;32(4):623–6.
Tolhuis B, Palstra RJ, Splinter E, Grosveld F, de Laat W. Looping and interaction between hypersensitive sites in the active beta-globin locus. Mol Cell. 2002;10(6):1453–65.
Palstra RJ, Tolhuis B, Splinter E, Nijmeijer R, Grosveld F, de Laat W. The beta-globin nuclear compartment in development and erythroid differentiation. Nat Genet. 2003;35(2):190–4.
Kadauke S, Blobel GA. Chromatin loops in gene regulation. Biochim Biophys Acta. 2009;1789(1):17–25.
Hong JW, Hendrix DA, Levine MS. Shadow enhancers as a source of evolutionary novelty. Science. 2008;321(5894):1314.
Li G, Fullwood MJ, Xu H, Mulawadi FH, Velkov S, Vega V, Ariyaratne PN, Mohamed YB, Ooi HS, Tennakoon C, Wei CL, Ruan Y, Sung WK. ChIA-PET tool for comprehensive chromatin interaction analysis with paired-end tag sequencing. Genome Biol. 2010;11(2):R22.
Kiefer CM, Hou C, Little JA, Dean A. Epigenetics of beta-globin gene regulation. Mutat Res. 2008;647(1–2):68–76.
Song SH, Hou C, Dean A. A positive role for NLI/Ldb1 in long-range beta-globin locus control region function. Mol Cell. 2007;28(5):810–22.
Tripic T, Deng W, Cheng Y, Zhang Y, Vakoc CR, Gregory GD, Hardison RC, Blobel GA. SCL and associated proteins distinguish active from repressive GATA transcription factor complexes. Blood. 2009;113(10):2191–201.
Deng W, Lee J, Wang H, Miller J, Reik A, Gregory PD, Dean A, Blobel GA. Controlling long-range genomic interactions at a native locus by targeted tethering of a looping factor. Cell. 2012;149(6):1233–44.
Dixon JR, Selvaraj S, Yue F, Kim A, Li Y, Shen Y, Hu M, Liu JS, Ren B. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature. 2012;485(7398):376–80.
Sexton T, Yaffe E, Kenigsberg E, Bantignies F, Leblanc B, Hoichman M, Parrinello H, Tanay A, Cavalli G. Three-dimensional folding and functional organization principles of the Drosophila genome. Cell. 2012;148(3):458–72.
Symmons O, Uslu VV, Tsujimura T, Ruf S, Nassari S, Schwarzer W, Ettwiller L, Spitz F. Functional and topological characteristics of mammalian regulatory domains. Genome Res. 2014;24(3):390–400.
Lupianez DG, Kraft K, Heinrich V, Krawitz P, Brancati F, Klopocki E, Horn D, Kayserili H, Opitz JM, Laxova R, Santos-Simarro F, Gilbert-Dussardier B, Wittler L, Borschiwer M, Haas SA, Osterwalder M, Franke M, Timmermann B, Hecht J, Spielmann M, Visel A, Mundlos S. Disruptions of topological chromatin domains cause pathogenic rewiring of gene-enhancer interactions. Cell. 2015;161(5):1012–25.
Fraser J, Williamson I, Bickmore WA, Dostie J. An overview of genome organization and how we got there: from FISH to Hi-C. Microbiol Mol Biol Rev. 2015;79(3):347–72.
Amano T, Sagai T, Tanabe H, Mizushina Y, Nakazawa H, Shiroishi T. Chromosomal dynamics at the Shh locus: limb bud-specific differential regulation of competence and active transcription. Dev Cell. 2009;16(1):47–57.
Buffry AD, Mendes CC, McGregor AP. The functionality and evolution of eukaryotic transcriptional enhancers. Adv Genet. 2016;96:143–206.
Hagstrom KA, Meyer BJ. Condensin and cohesin: more than chromosome compactor and glue. Nat Rev Genet. 2003;4(7):520–34.
Parelho V, Hadjur S, Spivakov M, Leleu M, Sauer S, Gregson HC, Jarmuz A, Canzonetta C, Webster Z, Nesterova T, Cobb BS, Yokomori K, Dillon N, Aragon L, Fisher AG, Merkenschlager M. Cohesins functionally associate with CTCF on mammalian chromosome arms. Cell. 2008;132(3):422–33.
Wendt KS, Yoshida K, Itoh T, Bando M, Koch B, Schirghuber E, Tsutsumi S, Nagae G, Ishihara K, Mishiro T, Yahata K, Imamoto F, Aburatani H, Nakao M, Imamoto N, Maeshima K, Shirahige K, Peters JM. Cohesin mediates transcriptional insulation by CCCTC-binding factor. Nature. 2008;451(7180):796–801.
Rubio ED, Reiss DJ, Welcsh PL, Disteche CM, Filippova GN, Baliga NS, Aebersold R, Ranish JA, Krumm A. CTCF physically links cohesin to chromatin. Proc Natl Acad Sci U S A. 2008;105(24):8309–14.
Wallace JA, Felsenfeld G. We gather together: insulators and genome organization. Curr Opin Genet Dev. 2007;17(5):400–7.
Mishiro T, Ishihara K, Hino S, Tsutsumi S, Aburatani H, Shirahige K, Kinoshita Y, Nakao M. Architectural roles of multiple chromatin insulators at the human apolipoprotein gene cluster. EMBO J. 2009;28(9):1234–45.
Hou C, Dale R, Dean A. Cell type specificity of chromatin organization mediated by CTCF and cohesin. Proc Natl Acad Sci U S A. 2010;107(8):3651–6.
Seitan VC, Hao B, Tachibana-Konwalski K, Lavagnolli T, Mira-Bontenbal H, Brown KE, Teng G, Carroll T, Terry A, Horan K, Marks H, Adams DJ, Schatz DG, Aragon L, Fisher AG, Krangel MS, Nasmyth K, Merkenschlager M. A role for cohesin in T-cell-receptor rearrangement and thymocyte differentiation. Nature. 2011;476(7361):467–71.
Rao SS, Huntley MH, Durand NC, Stamenova EK, Bochkov ID, Robinson JT, Sanborn AL, Machol I, Omer AD, Lander ES, Aiden EL. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell. 2014;159(7):1665–80.
Jabbari S, Fitzmaurice T, Munoz F, Lafuente C, Zentner P, Bustamante JG. Cross-border collaboration in oncology: a model for United States-Mexico border health. Int J Radiat Oncol Biol Phys. 2015;92(3):509–11.
Vietri Rudan M, Barrington C, Henderson S, Ernst C, Odom DT, Tanay A, Hadjur S. Comparative Hi-C reveals that CTCF underlies evolution of chromosomal domain architecture. Cell Rep. 2015;10(8):1297–309.
Guo Y, Xu Q, Canzio D, Shou J, Li J, Gorkin DU, Jung I, Wu H, Zhai Y, Tang Y, Lu Y, Wu Y, Jia Z, Li W, Zhang MQ, Ren B, Krainer AR, Maniatis T, Wu Q. CRISPR inversion of CTCF sites alters genome topology and enhancer/promoter function. Cell. 2015;162(4):900–10.
de Wit E, Vos ES, Holwerda SJ, Valdes-Quezada C, Verstegen MJ, Teunissen H, Splinter E, Wijchers PJ, Krijger PH, de Laat W. CTCF binding polarity determines chromatin looping. Mol Cell. 2015;60(4):676–84.
Lee HK, Willi M, Wang C, Yang CM, Smith HE, Liu C, Hennighausen L. Functional assessment of CTCF sites at cytokine-sensing mammary enhancers using CRISPR/Cas9 gene editing in mice. Nucleic Acids Res. 2017;45(8):4606–18.
Whyte WA, Orlando DA, Hnisz D, Abraham BJ, Lin CY, Kagey MH, Rahl PB, Lee TI, Young RA. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell. 2013;153(2):307–19.
Poss ZC, Ebmeier CC, Taatjes DJ. The mediator complex and transcription regulation. Crit Rev Biochem Mol Biol. 2013;48(6):575–608.
Park SW, Li G, Lin YP, Barrero MJ, Ge K, Roeder RG, Wei LN. Thyroid hormone-induced juxtaposition of regulatory elements/factors and chromatin remodeling of Crabp1 dependent on MED1/TRAP220. Mol Cell. 2005;19(5):643–53.
Wang Q, Carroll JS, Brown M. Spatial and temporal recruitment of androgen receptor and its coactivators involves chromosomal looping and polymerase tracking. Mol Cell. 2005;19(5):631–42.
Krivega I, Dean A. LDB1-mediated enhancer looping can be established independent of mediator and cohesin. Nucleic Acids Res. 2017;45:8255.
Suzuki HI, Young RA, Sharp PA. Super-enhancer-mediated RNA processing revealed by integrative microRNA network analysis. Cell. 2017;168(6):1000–14.
Adam RC, Yang H, Rockowitz S, Larsen SB, Nikolova M, Oristian DS, Polak L, Kadaja M, Asare A, Zheng D, Fuchs E. Pioneer factors govern super-enhancer dynamics in stem cell plasticity and lineage choice. Nature. 2015;521(7552):366–70.
Folgueras AR, Guo X, Pasolli HA, Stokes N, Polak L, Zheng D, Fuchs E. Architectural niche organization by LHX2 is linked to hair follicle stem cell function. Cell Stem Cell. 2013;13(3):314–27.
Kadaja M, Keyes BE, Lin M, Pasolli HA, Genander M, Polak L, Stokes N, Zheng D, Fuchs E. SOX9: a stem cell transcriptional regulator of secreted niche signaling factors. Genes Dev. 2014;28(4):328–41.
Keyes BE, Segal JP, Heller E, Lien WH, Chang CY, Guo X, Oristian DS, Zheng D, Fuchs E. Nfatc1 orchestrates aging in hair follicle stem cells. Proc Natl Acad Sci U S A. 2013;110(51):E4950–9.
Chang CY, Pasolli HA, Giannopoulou EG, Guasch G, Gronostajski RM, Elemento O, Fuchs E. NFIB is a governor of epithelial-melanocyte stem cell behaviour in a shared niche. Nature. 2013;495(7439):98–102.
Klein RH, Lin Z, Hopkin AS, Gordon W, Tsoi LC, Liang Y, Gudjonsson JE, Andersen B. GRHL3 binding and enhancers rearrange as epidermal keratinocytes transition between functional states. PLoS Genet. 2017;13(4):e1006745.
Sethi I, Gluck C, Zhou H, Buck MJ, Sinha S. Evolutionary re-wiring of p63 and the epigenomic regulatory landscape in keratinocytes and its potential implications on species-specific gene expression and phenotypes. Nucleic Acids Res. 2017;45:8208.
Tsai MC, Manor O, Wan Y, Mosammaparast N, Wang JK, Lan F, Shi Y, Segal E, Chang HY. Long noncoding RNA as modular scaffold of histone modification complexes. Science. 2010;329(5992):689–93.
Rinn JL, Kertesz M, Wang JK, Squazzo SL, Xu X, Brugmann SA, Goodnough LH, Helms JA, Farnham PJ, Segal E, Chang HY. Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell. 2007;129(7):1311–23.
Mulder KW, Wang X, Escriu C, Ito Y, Schwarz RF, Gillis J, Sirokmany G, Donati G, Uribe-Lewis S, Pavlidis P, Murrell A, Markowetz F, Watt FM. Diverse epigenetic strategies interact to control epidermal differentiation. Nat Cell Biol. 2012;14(7):753–63.
Kim TK, Hemberg M, Gray JM. Enhancer RNAs: a class of long noncoding RNAs synthesized at enhancers. Cold Spring Harb Perspect Biol. 2015;7(1):a018622.
Lam MT, Cho H, Lesch HP, Gosselin D, Heinz S, Tanaka-Oishi Y, Benner C, Kaikkonen MU, Kim AS, Kosaka M, Lee CY, Watt A, Grossman TR, Rosenfeld MG, Evans RM, Glass CK. Rev-Erbs repress macrophage gene expression by inhibiting enhancer-directed transcription. Nature. 2013;498(7455):511–5.
Li W, Notani D, Ma Q, Tanasa B, Nunez E, Chen AY, Merkurjev D, Zhang J, Ohgi K, Song X, Oh S, Kim HS, Glass CK, Rosenfeld MG. Functional roles of enhancer RNAs for oestrogen-dependent transcriptional activation. Nature. 2013;498(7455):516–20.
Melo CA, Drost J, Wijchers PJ, van de Werken H, de Wit E, Oude Vrielink JA, Elkon R, Melo SA, Leveille N, Kalluri R, de Laat W, Agami R. eRNAs are required for p53-dependent enhancer activity and gene transcription. Mol Cell. 2013;49(3):524–35.
Lindblad-Toh K, Garber M, Zuk O, Lin MF, Parker BJ, Washietl S, Kheradpour P, Ernst J, Jordan G, Mauceli E, Ward LD, Lowe CB, Holloway AK, Clamp M, Gnerre S, Alföldi J, Beal K, Chang J, Clawson H, Cuff J, Di Palma F, Fitzgerald S, Flicek P, Guttman M, Hubisz MJ, Jaffe DB, Jungreis I, Kent WJ, Kostka D, Lara M, Martins AL, Massingham T, Moltke I, Raney BJ, Rasmussen MD, Robinson J, Stark A, Vilella AJ, Wen J, Xie X, Zody MC; Broad Institute Sequencing Platform and Whole Genome Assembly Team, Baldwin J, Bloom T, Chin CW, Heiman D, Nicol R, Nusbaum C, Young S, Wilkinson J, Worley KC, Kovar CL, Muzny DM, Gibbs RA; Baylor College of Medicine Human Genome Sequencing Center Sequencing Team, Cree A, Dihn HH, Fowler G, Jhangiani S, Joshi V, Lee S, Lewis LR, Nazareth LV, Okwuonu G, Santibanez J, Warren WC, Mardis ER, Weinstock GM, Wilson RK; Genome Institute at Washington University, Delehaunty K, Dooling D, Fronik C, Fulton L, Fulton B, Graves T, Minx P, Sodergren E, Birney E, Margulies EH, Herrero J, Green ED, Haussler D, Siepel A, Goldman N, Pollard KS, Pedersen JS, Lander ES, Kellis M. A high-resolution map of human evolutionary constraint using 29 mammals. Nature. 2011;478(7370):476–82.
Hoffman MM, Ernst J, Wilder SP, Kundaje A, Harris RS, Libbrecht M, Giardine B, Ellenbogen PM, Bilmes JA, Birney E, Hardison RC, Dunham I, Kellis M, Noble WS. Integrative annotation of chromatin elements from ENCODE data. Nucleic Acids Res. 2013;41(2):827–41.
Rinaldi L, Datta D, Serrat J, Morey L, Solanas G, Avgustinova A, Blanco E, Pons JI, Matallanas D, Von Kriegsheim A, Di C, L Benitah SA. Dnmt3a and Dnmt3b associate with enhancers to regulate human epidermal stem cell homeostasis. Cell Stem Cell. 2016;19(4):491–501.
Hadjur S, Williams LM, Ryan NK, Cobb BS, Sexton T, Fraser P, Fisher AG, Merkenschlager M. Cohesins form chromosomal cis-interactions at the developmentally regulated IFNG locus. Nature. 2009;460(7253):410–3.
Hemberg M, Gray JM, Cloonan N, Kuersten S, Grimmond S, Greenberg ME, Kreiman G. Integrated genome analysis suggests that most conserved non-coding sequences are regulatory factor binding sites. Nucleic Acids Res. 2012;40(16):7858–69.
Fisher S, Grice EA, Vinton RM, Bessling SL, McCallion AS. Conservation of RET regulatory function from human to zebrafish without sequence similarity. Science. 2006;312(5771):276–9.
Fanucchi S, Shibayama Y, Burd S, Weinberg MS, Mhlanga MM. Chromosomal contact permits transcription between coregulated genes. Cell. 2013;155(3):606–20.
Sander JD, Joung JK. CRISPR-Cas systems for editing, regulating and targeting genomes. Nat Biotechnol. 2014;32(4):347–55.
Kvon EZ, Kamneva OK, Melo US, Barozzi I, Osterwalder M, Mannion BJ, Tissieres V, Pickle CS, Plajzer-Frick I, Lee EA, Kato M, Garvin TH, Akiyama JA, Afzal V, Lopez-Rios J, Rubin EM, Dickel DE, Pennacchio LA, Visel A. Progressive loss of function in a limb enhancer during snake evolution. Cell. 2016;167(3):633–642.e611.
Diao YR, Li B, Meng ZP, Jung I, Lee AY, Dixon J, Maliskova L, Guan KL, Shen Y, Ren B. A new class of temporarily phenotypic enhancers identified by CRISPR/Cas9-mediated genetic screening. Genome Res. 2016;26(3):397–405.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer International Publishing AG, part of Springer Nature
About this chapter

Cite this chapter
Oh, I.Y., de Guzman Strong, C. (2018). Enhancer-Promoter Interactions and Their Role in the Control of Epidermal Differentiation. In: Botchkarev, V., Millar, S. (eds) Epigenetic Regulation of Skin Development and Regeneration. Stem Cell Biology and Regenerative Medicine. Humana Press, Cham. https://doi.org/10.1007/978-3-319-16769-5_10
Download citation
DOI: https://doi.org/10.1007/978-3-319-16769-5_10
Published:
Publisher Name: Humana Press, Cham
Print ISBN: 978-3-319-16768-8
Online ISBN: 978-3-319-16769-5
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)