
Abstract
αB-Crystallin, or HspB5, is a small molecular-weight heat shock protein expressed highly in cardiac and skeletal muscle with multifaceted cellular roles including, chaperone function towards essential myofibrillar components. Insights into protective roles played by αB-crystallin, as well as mutations in the gene encoding αB-crystallin, CRYAB, which resulted in human pathologies, have highlighted the critical functions of αB-crystallin in both skeletal and cardiac muscle, inter alia. Various human mutations in CRYAB appear to have tissue-specific effects, with loss of αB-crystallin only impacting skeletal muscle under basal conditions. This review aims to highlight the roles of αB-crystallin in skeletal and cardiac muscle homeostasis as well as under conditions of stress and disease, drawing insights from human pathologies resulting from CRYAB mutations, and to discuss the potential of using induced pluripotent stem cells to model αB-crystallin-opathies in vitro.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Cardiac muscle
- Skeletal muscle
- Myopathy
- CRYAB, HspB5
- Induced pluripotent stem cells (iPSCs)
- Protein aggregation
1 Introduction
αB-Crystallin, also known as HspB5, is a member of the small molecular-weight heat shock family (sHSPs) of molecular chaperones, a diverse family of proteins that are characterized by the presence of a conserved α-crystallin domain (Ignolia and Craig 1982; Kappé et al. 2002). The α-crystallin domain in αB-crystallin interacts with an adjacent monomer to form a dimeric building block, and further assembles into higher order oligomers (Bagneris et al. 2009; Jehle et al. 2010; Laganowsky et al. 2010). The CRYAB gene encodes αB-crystallin expression and primarily functions as a molecular chaperone (Horwitz 1992), among other things. Though its expression was originally thought to be confined to the lens, it was later realized that αB-crystallin is relatively ubiquitously and abundantly expressed in cardiac and skeletal muscle (Dubin et al. 1989). Enhancer elements modulating the CRYAB promoter account for its tissue specificity (Gopal-Srivastava et al. 1995; Gopal-Srivastava and Piatigorsky 1993). As described in this review, αB-crystallin plays essential functions in tissue maintenance during homeostasis and in stressed or pathological states of cardiac and skeletal muscle. Additionally, mutations in human CRYAB result in cardiac and/or skeletal myopathies, with causal mechanisms of the apparent tissue-specific effects of the mutations remaining unresolved. This review discusses the implications and requirements for αB-crystallin in both cardiac and skeletal muscle during homeostasis as well as under stressed or pathological conditions. We also invoke studies involving mutant forms of CRYAB present in patients to make inferences into tissue-specific requirements.
2 Structure and Function of αB-Crystallin
2.1 Constitutive and Inducible Expression
αB-Crystallin is classified as a Class I sHSP due to its ubiquitous expression (Taylor and Benjamin 2005). First discovered in 1894, it was originally thought to be a lens-specific protein (Morner 1894), until murine CRYAB was cloned and found to be expressed at high levels in the heart, skeletal muscle, kidney, and lung and low levels in the brain and spleen (Dubin et al. 1989). Muscle-specific expression of CRYAB is conferred by upstream enhancer elements that regulate promoter activity. Murine CRYAB cis-acting enhancer elements, identified by reporter expression driven by the CRYAB promoter, show regions required for expression in skeletal muscle, termed αBE-1, αBE-2, αBE-3, and MRF (Gopal-Srivastava and Piatigorsky 1993), which are also required for cardiac muscle expression along with an additional, unique element, αBE-4 (Gopal-Srivastava et al. 1995). The MRF site contains an E-box that in skeletal muscle is bound and activated by the bHLH myogenic regulatory factors (MRFs), including MyoD and myogenin (Gopal-Srivastava and Piatigorsky 1993). In cardiac muscle, the E-box of the MRF site may be bound by upstream stimulatory factor (USF) or an antigenically similar factor (Gopal-Srivastava et al. 1995). The cardiac-specific element, αBE-4, contains a reverse CArG box, which may be bound by serum response factor (SRF) or an alternate, antigenically similar protein (Gopal-Srivastava et al. 1995).
In addition to its constitutive expression, CRYAB is also inducible in response to multiple forms of stress including heat, oxidative stress, and inflammation, which are sensed by heat shock factor 1 (HSF1), a transcriptional activator that homo-trimerizes to interact with heat shock elements in the promoters of stress-response genes, including CRYAB (reviewed in (Morimoto 1998; Christians et al. 2002)). HSF1 deficient mice exhibit decreased basal levels of αB-crystallin in the normal heart, indicating HSF1 may be involved in regulation of CRYAB expression under non-stress conditions as well (Yan et al. 2002).
Continuous motor nerve stimulation of rabbit tibialis anterior muscle increases αB-crystallin levels, and this upregulation may involve interaction of the MRFs with the E-box in the CRYAB enhancer region (Neufer and Benjamin 1996). Eccentric contraction (lengthening contraction) in skeletal muscle also increases the levels of CRYAB along with other HSPs (Kostek et al. 2007; Thompson et al. 2001) resulting from mechanical and/or oxidative stress (Koh 2002). Mechanical load and nerve innervation of skeletal muscle also regulate the level of CRYAB expression as shown in experiments where suspension of the rat hindlimb decreases CRYAB mRNA levels, and denervation has differential effects on CRYAB mRNA levels in different types of skeletal muscle (Atomi et al. 1991). Passive stretch increases CRYAB mRNA levels in skeletal muscle (Atomi et al. 1991).
αB-Crystallin is the most abundant sHSP in the heart, making up 3 % of cardiac homogenates and its expression in the heart is limited to cardiomyocytes (Lutsch et al. 1997). Stress including ischemia/reperfusion (I/R) injury upregulates αB-crystallin in the heart (Martin et al. 1997; Ray et al. 2001).
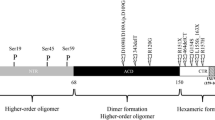
2.2 Structure and Oligomerization
sHSPs such as αB-crystallin contain an 80–100 residue, conserved α-crystallin domain (de Jong et al. 1998) comprised of an immunoglobulin-like β-sandwich, including strands β2−β9, which is critical for dimer formation through interaction of the extended β6 + 7 strand on adjacent monomers (Bagneris et al. 2009). Homo- or hetero-dimers assemble into large, oligomeric structures in the inactive state (Mymrikov et al. 2011) with an average of 40 subunits and a molecular mass of 800 kDa (Bloemendal 1981). Heat stimulates the rapid exchange of subunits, as determined by fluorescence resonance energy transfer experiments, and binding to large, denatured substrates greatly reduced the rate of subunit exchange (Bova et al. 1997). The variable c-terminal region of αB-crystallin contains a conserved I/V/L-X-I/V/L motif that binds the β4/8 groove of the adjacent monomer (Delbecq et al. 2012), contributing to dimer formation, and may also modulate chaperone activity (Bagneris et al. 2009; Laganowsky et al. 2010; Ghosh et al. 2006). The hydrophobic groove at the dimer interface is essential for substrate binding (Clark et al. 2011). The function of the variable N-terminus is less well-defined, though it is thought to impact oligomerization, containing three phosphorylation sites that can modulate the polydispersity of oligomers (Ecroyd et al. 2007) and reduce their size (Peschek et al. 2013).
2.3 Subcellular Localization
2.3.1 Cytoplasm
αB-Crystallin has disperse cytoplasmic localization and can bind to the intermediate filament protein desmin (Bennardini et al. 1992), as well as contractile proteins, including actin (Bennardini et al. 1992) and titin (Golenhofen et al. 2002; Bullard et al. 2004; Kotter et al. 2014). These interactions are stimulated in response to stress including I/R stress (Golenhofen et al. 1999, 2002) and stretch caused by eccentric contraction in skeletal muscle (Koh and Escobedo 2004; Kotter et al. 2014). Transgenic mice expressing the R120G mutant form of αB-crystallin in cardiomyocytes exhibit defects in mitochondrial organization, architecture, and respiration (Maloyan et al. 2005), which is attributable to breakdown of the desmin network (Wang et al. 2001). Specific mitochondrial alignment along adjacent sarcomeres is critical for maximal respiratory function in striated muscle, dependent upon cytoskeletal components, with cardiac and different types of skeletal muscle having differential arrangements and amounts of mitochondria (Milner et al. 2000; Reipert et al. 1999; Rambourg and Segretain 1980; Rappaport et al. 1998; Ogata and Yamaksak 1997). These data indicate that αB-crystallin, through interactions with cytoskeletal elements including desmin, among other things, may be crucial for maintaining organization and proper function of mitochondria.
2.3.2 Mitochondrial Interaction
Mitochondria are vital for both cardiac and skeletal muscle function, and as mentioned previously, the organization of mitochondria along myofibrils, which varies based on muscle type and is maintained through cytoskeletal interactions, is essential for maintaining maximal functional capacity (Milner et al. 2000; Reipert et al. 1999; Rambourg and Segretain 1980; Rappaport et al. 1998; Ogata and Yamaksak 1997). Maintaining mitochondrial integrity is crucial for inhibiting apoptosis and αB-crystallin has been shown to play a key role in this process during stress. αB-Crystallin associates with mitochondria, and this association increases with I/R stress, shown in rats (Mitra et al. 2013), and after exposure to hydrogen peroxide, shown in culture of mouse neonatal cardiomyocytes (Chis et al. 2012). Both phosphorylated and unphosphorylated αB-crystallin interact with the voltage-dependent anion channel (VDAC) and translocase of outer mitochondrial membrane 20 kDa (TOM 20) at the mitochondrial membrane as well as with caspase 3 and caspase 12 (Chis et al. 2012). It is thought that the interaction with VDAC is critical for preventing apoptosis through the mitochondrial pathway during I/R injury (Mitra et al. 2013). Temporal studies with I/R performed ex vivo in mouse hearts demonstrated that αB-crystallin translocates to the mitochondria during ischemia, becomes phosphorylated at Ser-59, and aids in maintaining mitochondrial membrane potential, thereby preventing apoptosis (Whittaker et al. 2009).
2.3.3 Nuclear Speckles
Nuclear speckles, located within the interchromatin space of the nucleoplasm, are proposed to be storage sites for splicing components and are associated with transcriptional activity (Spector and Lamond 2011). αB-Crystallin and other sHSPs, including Hsp27 (HspB1) localize to nuclear speckles in a variety of unstressed, transcriptionally active cell lines and primary cells (Vandenijssel et al. 2003). Overexpression of the R120G mutant form of αB-crystallin inhibited localization to nuclear speckles (Vandenijssel et al. 2003). These data suggest a nuclear role for αB-crystallin in modulating transcript processing, which may be important in R120G αB-crystallin-opathy (Vandenijssel et al. 2003).
A study utilizing mouse C2C12 myoblasts and differentiated myotubes showed differential nuclear speckle localization of αB-crystallin in response to heat shock (Adhikari et al. 2004). In the basal state, myoblasts showed expression of αB-crystallin in the cytoplasm and nucleus, with co-localization with lamin A/C and splicing factor SC-35, indicating nuclear speckle localization, and upon heat stress, myoblasts exhibited almost complete re-localization of αB-crystallin to the nucleus in nuclear speckles, returning to basal state following 3 h recovery. Interestingly, myotubes have αB-crystallin expression exclusively in the cytoplasm under basal and heat stressed conditions. This data may speak for a differential role of αB-crystallin in transcriptional modulation or cytoskeletal protection during differentiation and myogenesis.
2.3.4 Posttranslational Modifications
αB-Crystallin contains three phosphorylation sites (Ser-19, Ser-45, Ser-59), identified through experiments exposing cultured cells to various forms of stress (Kato et al. 1998; Ito et al. 1997). These sites are located within the n-terminal region, which can impact both structural and functional aspects of the sHSP, though the impact of phosphorylation on protection of the cell seems to be context dependent. Through studies employing phosphomimetic forms of αB-crystallin, it has been shown that phosphorylation at all three residues decreases oligomer size, with the dominant species being 12-mers and 6-mers, resulting in a more flexible n-terminal domain and increased chaperone activity toward malate dehydrogenase and p53 (Peschek et al. 2013). Additional studies have shown both increases and decreases in chaperone activity of αB-crystallin with phosphorylation depending on the substrate and conditions (Ahmad et al. 2008; Ecroyd et al. 2007), and the ratio of phosphorylated to non-phosphorylated forms of αB-crystallin present in oligomers may be key to regulating its wide range of chaperone activity (Ahmad et al. 2008).
Phosphorylation at Ser-45 may disrupt dimer formation, resulting in an increase in oligomers with an odd number of subunits (Aquilina et al. 2004). One study showed that phosphorylation at Ser-59 is required for nuclear import, and localization to nuclear speckles required phosphorylation at Ser-45 in HeLa cells (den Engelsman et al. 2005). Though a different study employing transient transfection of a phospho-defective form of αB-crystallin, demonstrated that phosphorylation was not required for nuclear localization (Vandenijssel et al. 2003). Phosphorylation at Ser-59 occurs in response to I/R stress in cardiomyocytes (Ito et al. 1997). A phosphomimetic at Ser-59 was shown to be necessary and sufficient for attenuating hyperosmotic or hypoxic stress-induced apoptosis in rat neonatal cardiomyocytes through inhibition of caspase 3 activation (Morrison et al. 2003). In differentiating C2C12 myoblasts, overexpression of wildtype αB-crystallin inhibits caspase 3 activation thereby blocking differentiation-induced apoptosis, and pseudophosphorylation of αB-crystallin at all three serine residues prevents αB-crystallin from performing this function (Kamradt et al. 2002). In light of these data showing varying effects of phosphorylation of αB-crystallin on its structure, localization, and function, it is likely that phosphorylation may be a mechanism for fine-tuning the activity of αB-crystallin, which is context dependent. In addition to phosphorylation, αB-crystallin undergoes O-GlcNAcylation at threonine 170, which has been linked to effects on αB-crystallin subcellular localization, protein-protein interactions, chaperone function, and degradation (Krishnamoorthy et al. 2013).
2.4 Roles
2.4.1 Chaperone
The major role of αB-crystallin is as a molecular chaperone. Chaperones bind hydrophobic regions of unfolded proteins, stabilizing the protein and preventing it from precipitating out of solution (Ellis and van der Vies 1991). αB-Crystallin, functions in an ATP-independent manner to bind severely compromised, aggregation-prone, late unfolding protein intermediates that are about to precipitate out of solution, and forms a stable, soluble complex that is resistant to aggregation (Carver et al. 1995; Rajaraman et al. 2001). Additionally, αB-crystallin transiently interacts with early unfolding intermediates and promotes their refolding (Rajaraman et al. 2001). Substrate recognition by αB-crystallin is not fully defined, but occurs through interaction of its hydrophobic regions, possibly various regions throughout the protein with exposed hydrophobic regions of unfolded proteins. This provides αB-crystallin with the ability to recognize a wide range of substrates (Basha et al. 2012).
The importance of αB-crystallin in maintaining cytoskeletal integrity is exemplified by its ability to bind and stabilize actin microfilaments, intermediate filaments, including desmin, vimentin, and GFAP, as well as microtubules reviewed in (Liang and MacRae 1997). In addition to binding cytoskeletal elements, αB-crystallin also has the ability to bind and prevent the aggregation of some growth factors, including FGF-2 and VEGF (Ghosh et al. 2007a, b). An in vitro study demonstrated the transient appearance of dimeric αB-crystallin interacting with the model destabilized substrate, α-lactalbumin, indicating that dimer release from the larger oligomers may be important for the chaperone function of αB-crystallin (Smirnova et al. 2013).
Desmin, a major intermediate filament protein in striated muscle, is responsible for connecting myofibrils together through Z-discs (Lockard and Bloom 1993). αB-Crystallin interacts with and functions as a chaperone for desmin (Perng et al. 1999a), and a mutant form of αB-crystallin, R120G, can increase its affinity for desmin resulting in aberrant desmin aggregation (Perng et al. 2004; Vicart et al. 1998). A study identifying that the interaction between αB-crystallin with desmin involved the β3 and β8 strands as well as c-terminal residues 155–165, also suggests that binding or cosedimentation of αB-crystallin with desmin does not necessarily increase αB-crystallin chaperone function towards desmin (Houck et al. 2011).
The muscle contractile protein titin, responsible for the elastic properties of muscle, contains spring-like immunoglobulin (Ig) domains that can unfold and allow contracted muscles to stretch (Minajeva et al. 2001), which can result in aggregation of the unfolded titin domains (Minajeva et al. 2001; Rief 1997). Titin and αB-crystallin were co-immunoprecipitated in pig hearts following ischemia and αB-crystallin was shown to bind to the I-band region of titin, with extraction of actin, in ischemic rat cardiomyocytes (Golenhofen et al. 2002). A following study more specifically identified that αB-crystallin binds to the N2B region of the cardiac titin isoform in physiologically stretched cardiomyocytes, and that higher stretching forces are required to unfold the titin domains in the presence of αB-crystallin (Bullard et al. 2004). Whereas binding of αB-crystallin appears to be specific to the N2B element of titin in cardiac muscle, in skeletal muscle, αB-crystallin shows scattered binding along the length of the I-band in rats, indicating it may bind to other Ig domains (Golenhofen et al. 2004). In vitro work shows that unfolded Ig domains, including cardiac N2B and skeletal N2A, of titin aggregate with increased aggregation under acidic conditions, and the presence of αB-crystallin prevents this aggregation (Kotter et al. 2014). αB-Crystallin was also localized to the Ig domain regions of titin in diseased, human cardiomyocytes and skeletal muscle, suggesting that this localization is necessary to prevent aggregation of unfolded Ig domains in overstretched muscle cells, and this interaction may also be relevant in skeletal muscle during exercise (Kotter et al. 2014). Additionally, in I/R experiments using isolated papillary muscles from CRYAB/HSPB2 double knockout (DKO) mouse hearts, it was speculated that more pronounced contracture in mutant muscles may be due to loss of αB-crystallin chaperone function of titin, thereby causing increased stiffness in response to ischemia (Golenhofen et al. 2006).
αB-Crystallin interacts with an f-box protein, FBX4, an important component of the SCF complex, which carries out ubiquitination of proteins, marking them for degradation (den Engelsman et al. 2003). This interaction is increased with phosphomimetics at Ser-19 and Ser-45 and also by the R120G mutation. These data suggest a potential role for αB-crystallin in directing substrate ubiquitination in the ubiquitin proteasome pathway, which may assist in maintaining cellular homeostasis by degrading proteins that will not refold properly. Activation of autophagy or the inflammatory response may also be a role for αB-crystallin in protein quality control as has been suggested for other sHSPs (Carra et al. 2008, 2009; Bruinsma et al. 2011).
2.4.2 Anti-apoptotic
Exogenous αB-crystallin expression in human lens epithelial cells prevented UVA-induced apoptosis through inhibition of the RAF/MEK/ERK pathway (Liu et al. 2004) via inhibition of RAS activation (Li et al. 2005). αB-Crystallin binds and modulates caspase 3 and Bax in the lens, regulating differentiation (Hu et al. 2012). It was also shown in retinal pigment epithelial cells that αB-crystallin is able to inhibit the translocation of Bax and Bcl-2 to the mitochondria (Mao et al. 2004). In cancer cell lines, αB-crystallin was identified as a target gene directly trans-activated by p53. αB-Crystallin binds to the DNA binding domain of p53 and reduced p53-induced apoptosis (Watanabe et al. 2009). In response to hydrogen peroxide treatment of C2C12 myogenic cells, αB-crystallin interacts with p53 in the cytoplasm, possibly preventing its translocation to the mitochondria and abrogating oxidative stress-induced apoptosis (Liu et al. 2007).
When myoblasts are stimulated to differentiate by growth factor deprivation, they will undergo apoptosis if they do not develop resistance to apoptosis, allowing differentiation to ensue. In differentiating C2C12 myoblasts, αB-crystallin is induced in those myoblasts that develop resistance to apoptosis and overexpression of αB-crystallin prevents differentiation-induced apoptosis through inhibition of caspase 3 activation (Kamradt et al. 2001, 2002). An independent experiment confirmed that overexpression of αB-crystallin decreased the levels of cleaved, activated caspase 3 in differentiating C2C12 myoblasts (Singh et al. 2010). These results suggest a critical role for αB-crystallin in the prevention of apoptosis during normal differentiation of myoblasts to myotubes. Additionally, DKO mice exhibit increases in both apoptosis and necrosis in response to I/R injury suggesting a protective role for one or both of these chaperones towards cardiomyocytes (Morrison et al. 2004). In αB-crystallin transgenic mice, Das and Dillmann have demonstrated ischemic cardioprotection (Ray et al. 2001).
2.4.3 Redox Modulation
Muscle contains high levels of mitochondria to comply with high energy requirements. The heart is made up of 55 % cardiomyocytes, with up to 35 % of the cardiomyocyte volume occupied by mitochondria and since mitochondria are the main source of reactive oxygen species (ROS), cells must adapt to deal with these harmful byproducts (Christians et al. 2012). Implications of αB-crystallin on redox state have mainly been explored through studies of the R120G mutant form. Transgenic mice overexpressing R120G αB-crystallin specifically in cardiomyocytes were shown to have a redox balance skewed towards reductive stress (Rajasekaran et al. 2007). In this model, R120G αB-crystallin-induced cardiomyopathy and aggregate formation were rescued by the intercross with mice having decreased levels of an antioxidative enzyme glucose-6-phosphate dehydrogenase, implicating an effect of the mutant form of αB-crystallin on redox state. αB-Crystallin cannot be thiolated due to its lack of cysteine residues, indicating that αB-crystallin does not undergo modification in response to extreme shifts of redox conditions.
3 Protective Effects of αB-Crystallin in Stressed or Pathological States
Many instances of protective roles for αB-crystallin have been described in the literature and correspond with its role as a stress response protein. Roles for αB-crystallin in I/R injury of the heart and exercise-induced injury in skeletal muscle will be discussed here.
3.1 Cardiac Muscle: Ischemia/Reperfusion
Neonatal and adult rat cardiomyocytes are protected from I/R injury by overexpression of αB-crystallin (Martin et al. 1997). Transgenic overexpression of αB-crystallin also protects the mouse heart in ex vivo I/R, decreasing the extent of infarction, lowering levels of oxidative stress, and decreasing apoptosis and necrosis (Ray et al. 2001). Conversely, DKO mouse hearts were more prone to I/R damage, and showed reduced contractile recovery, increased apoptosis and necrosis, and lower levels of the reduced form of glutathione, a protective molecule for oxidative damage (Morrison et al. 2004). To better understand the individual roles of αB-crystallin and HspB2 during I/R, Pinz and colleagues crossed DKO mice with mice transgenically overexpressing CRYAB and through I/R experiments, determined that αB-crystallin seems to be important for maintaining structure and diastolic function (Pinz et al. 2008). Through its mitochondrial interactions, as discussed previously, αB-crystallin may aid in maintaining mitochondrial integrity in response to I/R stress (Boelens 2014). In response to I/R, αB-crystallin also translocates to the mitochondria (Whittaker et al. 2009) and interacts with VDAC; this interaction may be critical for preventing apoptosis induced through the mitochondrial pathway (Mitra et al. 2013; Chis et al. 2012).
Sixty-minute cardiac ischemia in pigs resulted in overstretching of cardiomyocytes as observed by increased Z-disc spacing and increased distance between α-actinin and titin antibody staining (Golenhofen et al. 1999). In this study, αB-crystallin translocated from the cytoplasm to the I-band portion of titin in response to ischemia and following reperfusion, it remained bound to titin in cardiomyocytes that were no longer capable of contraction (Golenhofen et al. 1999). This result suggests that αB-crystallin is essential for mitigating the refolding of elastic titin domains in response to cardiomyocyte stretch as a result of ischemia, and αB-crystallin remains bound to the unfolded domains of titin in cardiomyocytes that are too damaged to recover contractile function.
3.2 Skeletal Muscle: Exercise
Eccentric contraction is the repeated stretching of actively contracting muscles and this can damage muscle fibers especially in untrained muscles, whereby damage occurs with concomitant inflammation (Koh 2002). Using ex vivo rabbit skeletal muscles, it was shown that desmin is lost in damaged fibers immediately after eccentric contraction and continues up to 3 days following injury (Friden and Lieber 2001). Dystrophin seems to be affected in a similar manner as a result of eccentric contraction in rat tibialis anterior muscles (Komulainen et al. 1998). An increase in free radical production in rabbit muscle occurred 24 h post-eccentric contraction, attributable to infiltrating inflammatory cells (Best et al. 1999).
Increases in the level of CRYAB were seen at 6 and 24 h after eccentric contraction (Kostek et al. 2007), which may be induced by mechanical and/or oxidative stress (Koh 2002). In a study involving human subjects using biopsies of the quadriceps muscle, αB-crystallin was upregulated 30 min following eccentric exercise and gradually decreased for 1 week after (Paulsen et al. 2007). Additionally, αB-crystallin responded immediately to eccentric exercise by binding to and fractioning with the cytoskeletal/myofibrillar proteins. Mice exposed to eccentric contractions showed immediate translocation of αB-crystallin from the soluble to insoluble fraction by western blot and cytosol to the Z-disc by immunostaining (Koh and Escobedo 2004). Phosphorylation of αB-crystallin was also observed during this time and may be important for its protective activity.
The temperature of muscles rises during exercise and may result in thermal injury. Chicken skeletal muscle myosin was unfolded by heat shock in vitro and in the presence of αB-crystallin, myosin retained enzymatic activity and aggregation was prevented (Melkani et al. 2006). αB-Crystallin was shown in vitro to prevent the acidic-induced aggregation of the N2A region of skeletal muscle titin, which contains Ig domains that are prone to unfolding during exercise (Kotter et al. 2014). It also is bound to the Ig domain regions of titin in skeletal muscle biopsies from patients with limb girdle muscular dystrophy type 2a; while in healthy human biopsies, αB-crystallin had cytosolic localization with faint Z-disc staining (Kotter et al. 2014). Ischemic rat skeletal muscle showed αB-crystallin bound along the length of the I-band, likely to the Ig domains of titin (Golenhofen et al. 2004). It is likely that this ability of αB-crystallin to protect titin Ig domains from aggregation upon unfolding due to stretch is also active during exercise (Kotter et al. 2014), especially eccentric contraction, where muscle is damaged from overstretching (Koh 2002).
4 αB-Crystallin-Opathies: Mutations and Human Pathologies
To date, 14 naturally occurring mutations in human CRYAB have been identified and published in the literature (Christians et al. 2012). Of these, ten are dominant mutations and four exhibit recessive inheritance patterns with disease pathologies ranging from skeletal myopathy, cardiomyopathy, cataracts, or some combination of the three. Eight of these mutations result in skeletal and/or cardiac myopathy (see Table 11.1) and can be classified as αB-crystallin-opathies (Selcen 2011; Sanbe et al. 2011; Christians et al. 2012). Variable penetrance and expressivity of the myopathy-causing mutations in CRYAB suggest genetic modifier and/or environmental factor contribution to the disease state (Christians et al. 2014). Additionally, for the dominant mutations, relative levels of the wildtype compared with the mutant form of αB-crystallin may also contribute to disease and could be modified by degradation, which may change with environmental stressors (Christians et al. 2014).
4.1 Point Mutations
First described in 1978 (Fardeau et al. 1978), the prototype and most well studied of the human mutations in αB-crystallin, arginine 120 mutated to a glycine (R120G) (Vicart et al. 1998), resulting in multisystem disorders and pathologies, was not identified until 1998 (Vicart et al. 1998). This disease affects a large, French pedigree and muscle biopsies characterized by electron microscopy revealed the buildup of dense, granulo-filamentous material within the sarcoplasm (Fardeau et al. 1978), which contained the intermediate filament protein desmin (Rappaport et al. 1988; Fardeau et al. 1978) as well as αB-crystallin, and therefore is classified as a desmin-related myopathy (DRM) (Vicart et al. 1998). Patient biopsies revealed a “rubbed-out” appearance when stained for myosin adenosine triphosphatase (ATPase) activity, potentially indicating a loss of myosin (Brady et al. 2001; Vicart et al. 1998; Fardeau et al. 1978). The R120G mutation (358 A>G) occurs in the α-crystallin domain and disrupts the structure as well as reduces the chaperone activity of αB-crystallin (Bova et al. 1999; Perng et al. 1999b). The binding affinity of αB-crystallin for desmin in vitro is increased by R120G, and results in the aberrant aggregation of desmin, preferentially during de novo desmin network synthesis rather than the aggregation of previously formed networks (Perng et al. 2004). This mutation shows closure of the hydrophobic groove present in αB-crystallin dimers, which may alter substrate binding as well as oligomerization (Clark et al. 2011). In vitro work shows that the R120G mutant form of αB-crystallin has complete loss of chaperone activity towards the N2B region of cardiac titin (Zhu et al. 2009).
Transgenic overexpression of R120G in the mouse heart portrays pathology similar to patients, showing dense desmin and αB-crystallin positive aggregates, and disruption of the cytoskeletal structure and mitochondrial architecture, resulting in cardiomyopathy and death in early adulthood (Wang et al. 2001). Overexpression of wildtype CRYAB, on the other hand, was relatively unremarkable (Wang et al. 2001). A knockin mouse model of R120G exhibits early onset cataracts and skeletal myopathy, with increased severity depending on dose (heterozygous versus homozygous knockin), though mortality was not increased (Andley et al. 2011), as is the case in the cardiac-specific overexpression of R120G (Wang et al. 2001). Cardiac function was not reported on in the publication of the R120G knockin animals (Andley et al. 2011).
Aggregates formed as a result of the R120G mutation contain amyloid oligomers and are classified as aggresomes (Sanbe et al. 2004), characteristic of neurodegenerative diseases, which result from transport of the aggregates along microtubules to the perinuclear region (Garcia-Mata et al. 2002). The dysfunction of mitochondria is one of the first defects observed in R120G overexpressing mouse hearts (Maloyan et al. 2005). Cardiomyocytes exhibit reduced mitochondrial oxygen consumption, changes in the permeability transition pore, and poor inner membrane potential, thereby activating apoptotic pathways and adversely affecting the hearts of transgenic animals. Transgenic overexpression of R120G in the heart also yields reductive stress, due to altered activity of enzymes that generate redox intermediates, including glucose-6-phosphate dehydrogenase, glutathione reductase, and glutathione peroxidase (Rajasekaran et al. 2007). These animals have elevated levels of autophagic activity by the age of 2 months, and this response is thought to be cytoprotective, possibly minimizing aggregate presence (Tannous et al. 2008). A study involving treatment of R120G transgenic mouse hearts with oxypurinol to rescue mitochondrial defects, determined that contractility was not rescued indicating further mechanical defects due to disarray of sarcomeres and accumulation of aggregates (Maloyan et al. 2009). Additionally, it has been suggested that aggregate formation and disease onset may be repressed at a young age due to competition with wildtype αB-crystallin (Andley et al. 2011; Vicart et al. 1998; Perng et al. 2004), which may also be the case for other dominant mutations described below.
The less studied ability of αB-crystallin to localize to nuclear speckles is eliminated by the R120G mutation, indicating a possible impact of the mutation on transcript processing (Vandenijssel et al. 2003). Overexpression of certain interacting partners has been found to attenuate the detrimental effects of R120G.
Interaction of R120G with BAG3, a co-chaperone involved in chaperone-assisted selective autophagy, increased R120G solubility, prevented its aggregation, and inhibited cell death induced by R120G overexpression in cell culture (Hishiya et al. 2011). Overexpression of HspB8 can inhibit R120G-induced aggresome formation and block the progression of cardiomyopathy (Sanbe et al. 2007, 2009). Voluntary exercise (Maloyan et al. 2007) and BCL-2 overexpression (Maloyan et al. 2009) also delay the onset and reduce the unfavorable effects of the R120G mutation.
The R120G disease state is likely due to a combination of loss and gain of function effects of the mutation (Sanbe et al. 2011). The broad range of roles played by αB-crystallin invites the potential for many factors to contribute to the R120G disease state, including: aggregation with desmin, loss of/altered chaperone function, altered mitochondrial architecture and function, and reductive stress. Since the initial discovery of R120G, other mutations have been identified in patient populations and can also be described as αB-crystallin-opathies.
A family with members harboring the dominant point mutation, D109H, in αB-crystallin exhibits similar multisystem pathology to patients with the R120G mutation (Sacconi et al. 2012). Residues D109 and R120 interact with each other during dimerization of αB-crystallin, and therefore disruption of this interaction may be responsible for the similar disease characteristics among patients with these two mutations (Sacconi et al. 2012) including effects on the hydrophobic groove, oligomerization (Clark et al. 2011), and desmin interaction (Perng et al. 2004). Overexpression of YFP-tagged constructs in HeLa cells showed similar aggregation of D109H and R120G as well as a similar decrease in aggregation and levels of apoptosis with Hsp27 co-overexpression (Raju and Abraham 2013), supporting a similar mechanism of action of the two mutations; however, further analysis is necessary.
A dominant glycine 154 to serine (G154S) mutation in a conserved residue was identified in a 48 year old patient diagnosed with mild dilated cardiomyopathy and slightly elevated skeletal creatine phosphokinase levels, indicating potential involvement of the skeletal muscle as well (Pilotto et al. 2006). The turnover rate of the protein is predicted to increase with this mutation (Pilotto et al. 2006). A second patient with this same mutation, G154S, presented in his sixties with progressive distal leg weakness and atrophy affecting mostly limb muscles, as well as mild difficulty swallowing (Reilich et al. 2010). The patient’s biopsy showed disrupted myofibrillar structure, vacuolization, and desmin/αB-crystallin positive aggregates. No cataracts, cardiac or respiratory involvement were noted in this patient. The G154S mutation appears to have affects in both cardiac and skeletal muscle, and the specific presentation of the patient’s symptoms may rely upon other confounding genetic or environmental factors causing stress in the muscle that result in the apparent tissue-specific disease manifestation. A potential increased turnover rate of this mutant form of the protein (Pilotto et al. 2006) may help to suppress symptoms exhibited by patients into later adulthood.
A patient with familial dilated cardiomyopathy, presenting after age 40, was identified to have a dominant, mutation of arginine 157 to a histidine residue (R157H), which was shown to decrease binding to the cardiac-specific N2B region of titin (Inagaki et al. 2006). This mutation, unlike the others, did not result in aberrant localization of αB-crystallin or in aggregation, and therefore, there may be an alternate disease mechanism causing dilated cardiomyopathy in this patient compared with the desmin-related cardiomyopathy in other patients with αB-crystallin-opathies. Reduced ability of αB-crystallin to bind to titin leaves titin vulnerable to stress and may predispose patients to heart failure (Inagaki et al. 2006). An in vitro chaperone assay showed that R157H maintained some chaperone activity towards the N2B region of titin, albeit reduced compared to wildtype αB-crystallin (Zhu et al. 2009). Interestingly, this patient and family members did not exhibit skeletal muscle weakness or disease, though αB-crystallin does bind and chaperone the Ig domains in the N2A region of titin in skeletal muscle (Kotter et al. 2014; et al. 2004), in addition to the cardiac N2B region (Kotter et al. 2014), which may indicate that αB-crystallin binds the alternate regions of titin in a different way and the R157H mutation specifically disrupts the association of αB-crystallin with the N2B region. In vitro binding assays using the skeletal N2A region of titin with R157H αB-crystallin could verify this. Alternatively, absence of specific environmental stress to the skeletal muscle may be responsible for apparent lack of skeletal muscle involvement. The R157H mutation may also have other yet-unstudied implications that leave cardiac muscle more vulnerable than skeletal muscle.
4.2 Dominant C-Terminal Truncation Mutations
Other than the aforementioned missense mutations, the remaining mutations in αB-crystallin causing myopathy are due to frameshift or nonsense mutations resulting in varying degrees of c-terminal truncation, some with additional, novel peptide sequences present. In vitro work using site-directed mutagenesis to mutate the two terminal lysine residues in αB-crystallin to leucine or glycine greatly reduced its chaperone activity, suggesting that the lysines in the c-terminus may be critical for interacting with unfolded proteins through charge-charge interactions (Plater et al. 1996). Loss of this ability by c-terminal truncation of αB-crystallin may prevent stable substrate binding and therefore reduce chaperone activity. It has also been suggested that the flexible c-terminal region folds over, and through the I/V/L-X-I/V/L motif, binds to the β4/8 groove of the adjacent monomer, contributing to dimer formation, and potentially blocking the substrate binding face of αB-crystallin, thereby modulating substrate binding and chaperone activity (Ghosh et al. 2006; Delbecq et al. 2012). Expression of only the α-crystallin domain of αB-crystallin results in dimer formation with retained chaperone activity, but the lost ability to oligomerize (Feil et al. 2001). The high potential of c-terminal truncated mutants to aggregate, suggests a critical role for the c-terminus in preventing self-aggregation of αB-crystallin possibly through promotion of oligomerization, and expression of the mutant forms may induce co-aggregation with wildtype αB-crystallin inducing a dominant negative effect, thereby inhibiting the chaperone function of wildtype αB-crystallin (Hayes et al. 2008). The myopathy-causing, dominant, c-terminal truncation mutations will now be described.
A patient harboring the dominant 464delCT mutation in CRYAB presented at age 52 with respiratory trouble due to reduced diaphragmatic movement, as well as leg weakness and difficulty swallowing, and died at age 58 due to respiratory failure (Selcen and Engel 2003). This mutation generates a frameshift resulting in eight missense codons before a premature stop codon. A 53 year old patient with a dominant 451C→T transition generating a nonsense mutation (Q151X) suffered for 10 years with slowly progressive leg weakness and atrophy and exhibited elevated creatine kinase levels (Selcen and Engel 2003). Neither patient had cataracts or cardiomyopathy. Abnormal fiber regions in both patients show intense desmin and αB-crystallin staining, and in both patients, the wildtype allele was preferentially expressed compared with the mutant forms.
Q151X results in extreme loss of protein stability and prevents oligomerization, while increasing in vitro chaperone activity towards citrate synthase and desmin (Hayes et al. 2008). The increase in chaperone activity may be due to potential loss of capping of the chaperone site by interaction with the c-terminal region (Delbecq et al. 2012; Ghosh et al. 2006), suggesting the c-terminus may be responsible for reducing αB-crystallin substrate binding for some substrates under normal conditions (Hayes et al. 2008). The 464delCT mutation results in the introduction of a novel peptide into the c-terminus resulting in decreased in vitro chaperone function towards desmin, as well as aggregation and loss of solubility that can be partially recovered through mixture with wildtype αB-crystallin (Hayes et al. 2008). Overexpression of Hsp27 also rescues the solubility and prevents aggregation of 464delCT, mediated by the ubiquitin proteasome system in H9C2 cells (a rat embryonic cardiomyocyte cell line) (Zhang et al. 2010).
4.3 Recessive Mutations
With mutations in CRYAB, especially the recessive mutations, a major question is to what extent the pathology observed is due to loss of function effects of αB-crystallin. DKO mice show progressive skeletal muscle deterioration with age following a normal development, with no apparent impact on the heart (Brady et al. 2001), except under stressful conditions such as I/R or transverse aortic constriction (TAC) (Morrison et al. 2004; Kumarapeli et al. 2008). These mice grow normally until 40 weeks, after which they lose weight, due to the inability to eat properly and exhibit severe kyphosis, hunched posture, both due to loss of corresponding musculature, with the tongue, head, and axial muscles being most greatly affected. Fatty replacement, macrophage infiltration, fibrosis, and vacuolization were observed in deteriorating muscles. Amorphous, flocculent, electron-opaque material was also noted corresponding with loss of myofibrils. Increased staining for desmin was also observed in affected fibers. Attempted determination of fiber type based on fiber type-specific myosin failed due to lack of myosin detection in deteriorating fibers (Brady et al. 2001), as confirmed in additional studies showing reduced levels of myosin heavy chain in skeletal muscle of DKO mice by western blot (Neppl et al. 2014). This may be similar to muscles of patients with the R120G mutation who have loss of myosin in their muscle cells (Vicart et al. 1998; Fardeau et al. 1978). Potentially, oxidative, slow-twitch fibers, which have the highest levels of αB-crystallin and are enriched in large truncal muscles, are preferentially degraded in the DKO mice (Brady et al. 2001) and in patients with recessive CRYAB mutations (Forrest et al. 2011; Del Bigio et al. 2011). Neppl and colleagues showed that 1 year old DKO mice have a significantly reduced number of satellite cells in their skeletal muscle and a reduced ability to regenerate muscle following cardiotoxin-induced injury, shown by decreased cross-sectional myofiber area and increased fibrosis 2 weeks post-injury, indicating a potential role for αB-crystallin in skeletal muscle regeneration (Neppl et al. 2014).
This DKO mouse model is confounded due to the double knockout nature. HspB2, or myotonic dystrophy protein kinase binding protein (MKBP), activates and protects myotonic dystrophy protein kinase (DMPK) (Suzuki et al. 1998). DMPK -/- mice show minor decreases in size of head and neck muscles with age (Jansen et al. 1996) and late onset myopathy (Reddy et al. 1996). It is, therefore, possible that through its activation and protection of DMPK and other potential functions of HspB2, the loss of HspB2 also contributes to the pathogenesis seen in DKO animals. Subsequent studies have attempted to define the specific roles for loss of αB-crystallin and HspB2. A genetic study comparing the effects of I/R or inotropic stimulation in the hearts of wildtype, DKO, CRYAB transgenic, and DKO crossed with CRYAB transgenic (effectively expressing αB-crystallin without HspB2) concluded that αB-crystallin is responsible for structural remodeling and mechanical maintenance, while HspB2 is tasked with energetic balance maintenance in the stressed heart (Pinz et al. 2008).
CRYAB transgenic mouse hearts subjected to TAC showed reduced NFAT transactivation and attenuated hypertrophic response, while DKO hearts exhibit increased NFAT transactivation at baseline and develop cardiac insufficiencies in response to TAC, leading the authors to conclude that αB-crystallin prevents cardiac hypertrophic responses, possibly through inhibition of NFAT signaling (Kumarapeli et al. 2008). A single, cardiac specific knockout of HSPB2 was also generated and shows under basal conditions that cardiac function, hypertrophic responses, and mitochondrial metabolism were unchanged; however, when animals were subjected to TAC, mitochondrial energetics were reduced (Ishiwata et al. 2012), in agreement with the previously suggested role for HspB2 in maintaining energetic balance (Pinz et al. 2008). To our knowledge, a whole body knockout of either CRYAB or HSPB2 has yet to be published and would further distinguish the distinct roles for each in both skeletal and cardiac muscle. Additionally, identification of recessive mutants in CRYAB affecting muscle, discussed below, may assist in identifying loss of αB-crystallin function phenotypes, though these mutants may also have gain of function effects since the mutant proteins are expressed to some extent.
Fatal, infantile onset muscular dystrophy was identified in a cohort of Canadian aboriginals all harboring a homozygous c.60C deletion in the n-terminus of CRYAB, predicting a serine to alanine mutation at residue 21 and a premature stop codon after 23 missense residues, with unaffected parents both being heterozygous for this mutation (Del Bigio et al. 2011). Patients developed rigid muscles with elevated serum creatine kinase levels and died of respiratory insufficiency shortly after birth, except one child surviving to 4 years of age on mechanical ventilation. Axial muscles were more severely affected than appendicular muscles, Z-disc disarray, dense inclusions, vacuole presence, immune cell infiltration, and necrotic and regenerating fibers were noted. Deteriorating fibers stained strongly for myotilin, desmin, which concentrated at the periphery of inclusions, and αB-crystallin (using an N-terminal specific antibody), which was highly expressed in inclusions and dimly throughout other regions of the fibers. Hearts examined in these patients were normal. This mutant mRNA is likely a target for nonsense-mediated mRNA decay due to the premature stop codon occurring upstream of the final splice junction (Amrani et al. 2006), which may attenuate its expression, though mutant protein is still expressed (Del Bigio et al. 2011). The authors hypothesize that the lack of association of αB-crystallin with titin in this case may be responsible for the severe muscle stiffness observed in patients.
A second recessive mutation in CRYAB, c.343delT, results in a frameshift mutation generating a predicted 127 amino acid protein, instead of the wildtype 175 amino acids (Forrest et al. 2011). The patient identified with this mutation, born from unrelated, heterozygous parents, presented starting at age 4 months with progressive feeding difficulties and respiratory distress, requiring ventilation, severe muscle stiffness affecting mostly axial muscles with some limb involvement, and elevated creatine kinase levels. No cardiomyopathy or cataracts were noted. Muscle biopsy showed increased vacuolization, increased lipid content, globular inclusions, fibrosis, and disrupted myofibrillar structure. Abnormal fibers were intensely positive for myotilin, desmin, and αB-crystallin, which was also detected in a 15 kDa truncated form by western blot. Appearance of symptoms following normal development suggests a role for αB-crystallin in remodeling of myofibrillar structure following contraction, rather than a developmental role.
Similarities between patients with these two recessive mutations in CRYAB and DKO mice, including skeletal muscle deterioration with fatty deposits, fibrosis, inflammation, and vacuolization, indicate potentially that the effects of these recessive mutants result from loss of αB-crystallin function. Patients develop more severe symptoms as infants, whereas the DKO mice develop progressive symptoms into adulthood. Additionally, patients exhibit severe muscle stiffness, which is not reported in the DKO mice. The phenotypic differences could be due to multiple factors including: the confounding effects of loss of HspB2 in the DKO mice, species variability of requirement for αB-crystallin in skeletal muscle, or potential additional gain of toxic function effects of the mutant protein expression in patients with these recessive mutations. Further examination is necessary to resolve these issues.
5 Differential Functions of αB-Crystallin in Skeletal and Cardiac Muscle
Tissue specificity of pathology correlating with the various mutations brings about interesting questions with regards to the roles and requirements for αB-crystallin in cardiac and skeletal muscle. Though the muscles are both striated, the structure and function is very different. Potentially, various mutations in αB-crystallin could affect tissues differently. The late-onset of disease with the majority of these mutations also suggests age-related stress and/or the presence of genetic modifiers may be key factors in disease manifestation. With variable patient lifestyles and different stress encounters, it is difficult to determine in the relatively small number of patients whether, given time and/or the correct stress, patients may also develop clinical symptoms in the yet-unaffected tissue. Nevertheless, we discuss in this section some potential reasons for tissue-specific impacts of αB-crystallin.
Detailed analysis of the cellular localization of αB-crystallin using immunohistochemistry of rat organs indicates potential expression of αB-crystallin in highly oxidative cells, as suggested by a correlation between αB-crystallin expression and markers of oxidative activity (Iwaki et al. 1990). All type 1 aerobic fibers (slow-twitch) and about half of type 2 anaerobic fibers (fast-twitch), corresponding to mostly type 2A fibers, were positive for αB-crystallin, with more intense staining in type 1 fibers. Additional studies confirmed higher expression of αB-crystallin in slow- compared to fast-twitch fibers (Golenhofen et al. 2004; Atomi et al. 2000). Variable expression in different types of skeletal muscle fibers may account for specific fiber-type degradation, but this would not account for the lack of cardiac involvement, since αB-crystallin levels are comparably high in the heart (Golenhofen et al. 2004). Higher αB-crystallin expression in oxidative muscle (cardiac and type 1 skeletal fibers) may also be due to requirements of αB-crystallin for maintaining redox balance (Rajasekaran et al. 2007).
αB-Crystallin plays a role in maintaining the desmin network in muscle, which is crucial for proper sarcomere alignment as well as mitochondrial architecture (Perng et al. 1999a; Lockard and Bloom 1993; Wang et al. 2001; Maloyan et al. 2005). Differences between muscle types in mitochondrial architecture and requirements for appropriate coupling (Milner et al. 2000; Reipert et al. 1999; Rambourg and Segretain 1980; Rappaport et al. 1998) could underlie tissue specificity of disease as well as exemplify normal roles for αB-crystallin.
Sarcomeric localization of αB-crystallin in ischemic skeletal muscle is similar to cardiac muscle, with I-band and intermediate filament localization, though at the I-band, αB-crystallin binds to titin along the length of the I-band region in skeletal muscle, where in cardiac muscle it binds to a narrow region (Golenhofen et al. 2004), identified as the N2B element of titin (Bullard et al. 2004; Golenhofen et al. 2002). Skeletal muscle is susceptible to stretch injury on a day-to-day basis as a result of exercise, which can unfold titin Ig domains (Friden and Lieber 2001; Koh 2002), whereas in cardiac muscle, titin plays a role in the maintenance of passive stiffness during diastole, and, in patients with heart failure, isoforms of titin switch to more compliant forms, with stretching of titin isoforms occurring in cardiomyocytes due to I/R injury and hypertrophic response in heart failure (Linke 2008; Golenhofen et al. 1999). Stretching induces unfolding of the elastic domains of titin (Rief 1997; Minajeva et al. 2001), which may require αB-crystallin to prevent aggregation (Bullard et al. 2004; Golenhofen et al. 2002, 2004; Kotter et al. 2014). Potentially the different isoforms of titin expressed in cardiac and skeletal muscle (Labeit and Kolmerer 1995), as well the apparent variable binding of αB-crystallin to the titin domains may be a reason for tissue specificity of disease. Or more simply, tissue susceptibility to stretch may also explain this phenomenon.
Development of cardiac and skeletal muscle appear not to require αB-crystallin, since DKO mice (Brady et al. 2001), as well as all patients with mutations in CRYAB, develop symptoms following normal development (see Table 11.1). The major role of αB-crystallin seems to be in the maintenance of muscle tissue as well as protection during stress, as described above. Figure 11.1 discusses the potential impacts of alterations in levels of αB-crystallin or mutations in CRYAB on various stages of skeletal muscle myogenesis. Maintenance of skeletal muscle requires the activation of resident, quiescent progenitor cells, known as satellite cells, identified by expression of the transcription factor Pax7, to sequentially express the myogenic regulatory factors (MRFs), including MyoD and myogenin, which regulate muscle differentiation (Weintraub 1993; Le Grand and Rudnicki 2007). Proliferating myoblasts expressing MyoD must turn off MyoD expression, exit the cell cycle, turn on myogenin, and fuse to form multinucleated myotubes (Weintraub 1993; Le Grand and Rudnicki 2007). Overexpression of αB-crystallin significantly delays this process in C2C12 myoblasts, with cells showing a defect in cell cycle exit and lower levels of MyoD (Singh et al. 2010). DKO mice exhibit reduced levels of satellite cells under basal conditions at 1 year of age, and unexpectedly, a cardiotoxin injury model of DKO mouse tibialis anterior muscles revealed a threefold increase in the percentage of satellite cells in response to injury; though the mice also showed decreased cross-sectional myofiber area and increased fibrosis (Neppl et al. 2014). This study suggests that loss of αB-crystallin results in an increase in proliferation, with lower levels of p21 and notch signaling molecules, possibly through modulation of Argonaute 2 activity, as shown by enrichment of miRNAs on Argonaute 2 in injured DKO skeletal muscles and co-immunoprecipitation of Argonaute 2 with αB-crystallin (Neppl et al. 2014). Argonaute 2 is an essential component of the RISC complex for miRNA-mediated silencing; the possibility of αB-crystallin as an allosteric regulator of the RISC complex may be a reason for its broad cellular impact (Neppl et al. 2014). The study by Neppl and colleagues suggests that with the loss of αB-crystallin, the regenerative response of satellite cells is skewed towards proliferation with inefficient differentiation to myotubes (Neppl et al. 2014). This is contradictory to data suggesting that overexpression of αB-crystallin in C2C12 myoblasts leads to increased proliferation through delayed exit from the cell cycle with a decrease in p21 expression (Singh et al. 2010). Both studies indicate a role for αB-crystallin in cell cycle regulation during skeletal muscle regeneration (Neppl et al. 2014; Singh et al. 2010). Additional experiments addressing the model system used (i.e. in vivo or cell culture, loss of αB-crystallin or overexpression) are necessary to reconcile the exact role for αB-crystallin in cell cycle regulation under basal and stressed conditions.

The schematic represents normal myogenic differentiation of satellite cells to myotubes, indicating relative expression levels of defining transcription factors throughout the process (Modified from Le Grand and Rudnicki 2007). Satellite cells/myogenic progenitor cells, expressing Pax7, are activated and enter the cell cycle to become proliferating myoblasts, with an increase in MyoD expression and concomitant decrease in Pax7 expression. Proliferating myoblasts must exit the cell cycle to fulfill differentiation to myocytes, which downregulate MyoD expression and upregulate myogenin. Myocytes then fuse into multi-nucleated myotubes, which maintain expression of myogenin. During myogenesis, αB-crystallin is expressed at low levels in proliferating myoblasts and is greatly upregulated upon differentiation to myotubes (Adhikari et al. 2004; Singh et al. 2010). The table to the right of the schematic indicates variations in myogenesis that occur as a result of overexpression of αB-crystallin, loss of αB-crystallin, recessive mutations in αB-crystallin, and dominant mutations in αB-crystallin. Red text indicates a testable hypothesis for that condition
Due to withdrawal of growth factors, myoblasts must also resist apoptosis when differentiating to myotubes. αB-crystallin is induced in and contributes to apoptosis-resistance in C2C12 myoblasts through inhibition of caspase 3 activation, and overexpression of αB-crystallin prevents apoptosis occurring as a result of differentiation (Kamradt et al. 2001, 2002). These studies suggest that αB-crystallin may be important for modulating myoblast differentiation during tissue regeneration by multifaceted means including impact on proliferation and cell cycle exit, levels of regulatory MyoD, and inhibition of apoptosis. Cardiac muscle does not have the luxury to regenerate as skeletal muscle does (Mercola et al. 2011), implicating that loss of αB-crystallin function may manifest in skeletal muscle instead of cardiac muscle under healthy conditions due to requirements in regenerative myogenesis.
6 Stem Cells as a Model Systems for Studying αB-Crystallin in Cardiac and Skeletal Muscle
Many model systems have been employed in the study of αB-crystallin as discussed above, including rodents and larger mammalian models, some with whole body or tissue-specific overexpression/knockout, human tissue biopsies from healthy and diseased patients, cell culture with primary cells or cell lines, as well as in vitro biochemical analysis. To more fully understand the differential implications of αB-crystallin in cardiac and skeletal muscle, it is crucial to use models in which both cell types can be analyzed in parallel. Animal models with whole body genetic modifications, cell culture using both cardiac and skeletal muscle cell lines, and in vitro experiments analyzing interactions with cardiac- and skeletal-specific isoforms will be useful in accomplishing this feat. Stem cells offer additional potential for analyzing the impact of αB-crystallin on cardiac and skeletal muscle. Induced pluripotent stem cells (iPSCs), a technology first developed in the lab of 2012 Nobel Prize winner, Shinya Yamanaka (Takahashi and Yamanaka 2006), are an invaluable tool offering the ability to model cell-autonomous disease using a patient’s own cells and the potential to screen drug compounds or provide cell therapy (Robinton and Daley 2012). Somatic cells are isolated from patients, which can be done now through minimally invasive procedures including blood draws and urine sample collection, and reprogrammed into induced pluripotent stem cells through a variety of techniques involving the overexpression of key transcription factors (Chou et al. 2011; Zhou et al. 2012). Once reprogrammed, these cells are pluripotent and can be directed to differentiate to either cardiac or skeletal muscle cells, which can be studied in culture (Hosoyama et al. 2014; Zhang et al. 2012; Lian et al. 2013; Darabi et al. 2012).
Our lab has previously generated and characterized mouse iPSC-derived cardiomyocytes from transgenic R120G CRYAB mice, and shown that cardiomyocytes derived from these cells exhibit αB-crystallin positive aggregates as well as activation of the hypertrophic response (Limphong et al. 2013), indicating that iPSCs have the potential to recapitulate at least some phenotypes observed in αB-crystallin-opathies. Many published examples of the use of iPSCs for modeling cardiac diseases exist in the literature, including long QT (Itzhaki et al. 2011) and LEOPARD syndromes (Carvajal-Vergara et al. 2010), with the list rapidly growing. Genetic manipulation is also feasible in iPSCs (Yusa et al. 2011; An et al. 2012; Fong et al. 2013), which allows for the generation of gene-corrected control cells from patient cells harboring mutations, or for insertion of mutations of interest into wildtype iPSC lines for analysis. Caveats of the use of iPSCs as a model include purity of differentiated cultures, which has improved drastically over time and is likely to continue improving with advancement of differentiation methods, lack of support cell presence in the culture (i.e. fibroblasts, endothelial cells, etc.) that may play a role in disease pathogenesis, and variability between iPSC lines with differentiation, which can be minimized through the use of genetic manipulation for the generation of appropriate controls. The iPSC system could be very effective for looking at cell-type specific impacts of αB-crystallin in mutant and wildtype form, which is a focus of pursuit in our laboratory.
7 Conclusions and Future Directions
Overall, much work has been done to describe the function of αB-crystallin in cardiac and skeletal muscle both in healthy and diseased states. The use of model systems that allow for direct comparison of the two cell/tissue types will better define differential roles of αB-crystallin in each tissue and help to determine stressors or genetic modifiers that may contribute to disease susceptibility. The study of mutant forms of αB-crystallin existing in diseased patients will provide insights into not only how the mutation affects the structure and function of αB-crystallin, but also into the function and requirements of wildtype αB-crystallin during tissue maintenance and in disease compromised tissues.
The use of iPSCs to model protein misfolding disorders such as αB-crystallin-opathies, provides not only an in vitro disease model with the ability to analyze various impacted cell types and determine mechanistic details of the disease, but also the potential for small molecule screening and possibly regenerative cell therapies to treat patients. Small molecules that reduce the presence of toxic protein aggregates may be beneficial in protein misfolding diseases and can be screened for using the clinically relevant cell type generated from iPSCs (Ebert et al. 2012). The probable impact of loss of αB-crystallin on satellite cells suggests the potential for cell therapy in transplant of gene-corrected autologous or unaffected, nonautologous muscle progenitor cells to prevent skeletal myopathy in patients with recessive, early onset αB-crystallin-opathies. Engraftment of human iPSC-derived skeletal muscle progenitor cells has been shown to seed the satellite compartment in dystrophic mice suggesting the potential for long term benefits from this type of cell therapy (Darabi et al. 2012). The rapidly advancing field of iPSCs will likely lend itself to mechanistic disease modeling, small molecule screening, and regenerative therapies in protein misfolding disorders in the years to come.
References
Adhikari AS, Sridhar Rao K, Rangaraj N, Parnaik VK, Mohan Rao C (2004) Heat stress-induced localization of small heat shock proteins in mouse myoblasts: intranuclear lamin A/C speckles as target for alphaB-crystallin and Hsp25. Exp Cell Res 299(2):393–403. doi:10.1016/j.yexcr.2004.05.032
Ahmad MF, Raman B, Ramakrishna T, Rao Ch M (2008) Effect of phosphorylation on alpha B-crystallin: differences in stability, subunit exchange and chaperone activity of homo and mixed oligomers of alpha B-crystallin and its phosphorylation-mimicking mutant. J Mol Biol 375(4):1040–1051. doi:10.1016/j.jmb.2007.11.019
Amrani N, Sachs MS, Jacobson A (2006) Early nonsense: mRNA decay solves a translational problem. Nat Rev Mol Cell Biol 7(6):415–425. doi:10.1038/nrm1942
An MC, Zhang N, Scott G, Montoro D, Wittkop T, Mooney S, Melov S, Ellerby LM (2012) Genetic correction of Huntington’s disease phenotypes in induced pluripotent stem cells. Cell Stem Cell 11(2):253–263. doi:10.1016/j.stem.2012.04.026
Andley UP, Hamilton PD, Ravi N, Weihl CC (2011) A knock-in mouse model for the R120G mutation of alphaB-crystallin recapitulates human hereditary myopathy and cataracts. PLoS one 6(3):e17671. doi:10.1371/journal.pone.0017671
Aquilina JA, Benesch JL, Ding LL, Yaron O, Horwitz J, Robinson CV (2004) Phosphorylation of alphaB-crystallin alters chaperone function through loss of dimeric substructure. J Biol Chem 279(27):28675–28680. doi:10.1074/jbc.M403348200
Atomi Y, Yamada S, Nishida T (1991) Early changes of αB-crystallin mRNA in rat skeletal muscle to mechanical tension and denervation. Biochem Biophys Res Commun 181(3):1323–1330
Atomi Y, Toro K, Masuda T, Hatta H (2000) Fiber-type-specific aB-crystallin distribution and its shifts with T3 and PTU treatments in rat hindlimb muscles. J Appl Physiol 88:1355–1364
Bagneris C, Bateman OA, Naylor CE, Cronin N, Boelens WC, Keep NH, Slingsby C (2009) Crystal structures of alpha-crystallin domain dimers of alphaB-crystallin and Hsp20. J Mol Biol 392(5):1242–1252. doi:10.1016/j.jmb.2009.07.069
Basha E, O’Neill H, Vierling E (2012) Small heat shock proteins and alpha-crystallins: dynamic proteins with flexible functions. Trends Biochem Sci 37(3):106–117. doi:10.1016/j.tibs.2011.11.005
Bennardini F, Wrzosek A, Chiesi M (1992) Alpha B-crystallin in cardiac tissue. Association with actin and desmin filaments. Circ Res 71(2):288–294. doi:10.1161/01.res.71.2.288
Best TM, Fiebig R, Corr DT, Brickson S, Ji L (1999) Free radical activity antioxidant enzyme and glutathione changes with muscle stretch injury in rabbits. J Appl Physiol 87:74–82
Bloemendal H (1981) The lens proteins. Molecular and cellular biology of the eye lens. Wiley, New York, pp 1–47
Boelens WC (2014) Cell biological roles of alphaB-crystallin. Prog Biophys Mol Biol. doi:10.1016/j.pbiomolbio.2014.02.005
Bova MP, Ding LL, Horwitz J, Fung BKK (1997) Subunit exchange of aA-crystallin. J Biol Chem 272(47):29511–29517
Bova MP, Yaron O, Huang Q, Ding L, Haley DA, Stewart PL, Horwitz J (1999) Mutation R120G in aB-crystallin, which is linked to a desmin-related myopathy, results in an irregular structure and defective chaperone-like function. Proc Natl Acad Sci U S A 96:6137–6142
Brady JP, Garland DL, Green DE, Tamm ET, Giblin FJ, Wawrousek EF (2001) aB-crystallin in lens development and muscle integrity: a gene knockout approach. IOVS 42(12):2924–2934
Bruinsma IB, de Jager M, Carrano A, Versleijen AA, Veerhuis R, Boelens W, Rozemuller AJ, de Waal RM, Verbeek MM (2011) Small heat shock proteins induce a cerebral inflammatory reaction. J Neurosci 31(33):11992–12000. doi:10.1523/JNEUROSCI. 0945-11.2011
Bullard B, Ferguson C, Minajeva A, Leake MC, Gautel M, Labeit D, Ding L, Labeit S, Horwitz J, Leonard KR, Linke WA (2004) Association of the chaperone alphaB-crystallin with titin in heart muscle. J Biol Chem 279(9):7917–7924. doi:10.1074/jbc.M307473200
Carra S, Seguin SJ, Lambert H, Landry J (2008) HspB8 chaperone activity toward poly(Q)-containing proteins depends on its association with Bag3, a stimulator of macroautophagy. J Biol Chem 283(3):1437–1444. doi:10.1074/jbc.M706304200
Carra S, Brunsting JF, Lambert H, Landry J, Kampinga HH (2009) HspB8 participates in protein quality control by a non-chaperone-like mechanism that requires eIF2{alpha} phosphorylation. J Biol Chem 284(9):5523–5532. doi:10.1074/jbc.M807440200
Carvajal-Vergara X, Sevilla A, D’Souza SL, Ang YS, Schaniel C, Lee DF, Yang L, Kaplan AD, Adler ED, Rozov R, Ge Y, Cohen N, Edelmann LJ, Chang B, Waghray A, Su J, Pardo S, Lichtenbelt KD, Tartaglia M, Gelb BD, Lemischka IR (2010) Patient-specific induced pluripotent stem-cell-derived models of LEOPARD syndrome. Nature 465(7299):808–812. doi:10.1038/nature09005
Carver JA, Guerreiro N, Nicholls KA, Truscott RJW (1995) On the interaction of a-crystallin with unfolded proteins. Biochem Biophys Acta 1252:251–260
Chis R, Sharma P, Bousette N, Miyake T, Wilson A, Backx PH, Gramolini AO (2012) Alpha-crystallin B prevents apoptosis after H2O2 exposure in mouse neonatal cardiomyocytes. Am J Physiol Heart Circ Physiol 303:H967–H978. doi:10.1152/ajpheart.00040.2012.-/-Crystallin
Chou BK, Mali P, Huang X, Ye Z, Dowey SN, Resar LM, Zou C, Zhang YA, Tong J, Cheng L (2011) Efficient human iPS cell derivation by a non-integrating plasmid from blood cells with unique epigenetic and gene expression signatures. Cell Res 21(3):518–529. doi:10.1038/cr.2011.12
Christians ES, Yan LJ, Benjamin IJ (2002) Heat shock factor 1 and heat shock proteins: critical partners in protection against acute cell injury. Crit Care Med 30(1):S43–S50
Christians ES, Ishiwata T, Benjamin IJ (2012) Small heat shock proteins in redox metabolism: implications for cardiovascular diseases. Int J Biochem Cell Biol 44(10):1632–1645. doi:10.1016/j.biocel.2012.06.006
Christians ES, Banerjee Mustafi S, Benjamin IJ (2014) Chaperones and cardiac misfolding protein diseases. Curr Protein Pept Sci 15(3):189–204
Clark AR, Naylor CE, Bagneris C, Keep NH, Slingsby C (2011) Crystal structure of R120G disease mutant of human alphaB-crystallin domain dimer shows closure of a groove. J Mol Biol 408(1):118–134. doi:10.1016/j.jmb.2011.02.020
Darabi R, Arpke RW, Irion S, Dimos JT, Grskovic M, Kyba M, Perlingeiro RC (2012) Human ES- and iPS-derived myogenic progenitors restore DYSTROPHIN and improve contractility upon transplantation in dystrophic mice. Cell Stem Cell 10(5):610–619. doi:10.1016/j.stem.2012.02.015
de Jong WW, Caspers GJ, Leunissen JAM (1998) Genealogy of the alpha-crystallin—small heatshock protein superfamily. Int J Biol Macromol 22:151–162
Del Bigio MR, Chudley AE, Sarnat HB, Campbell C, Goobie S, Chodirker BN, Selcen D (2011) Infantile muscular dystrophy in Canadian aboriginals is an alphaB-crystallinopathy. Ann Neurol 69(5):866–871. doi:10.1002/ana.22331
Delbecq SP, Jehle S, Klevit R (2012) Binding determinants of the small heat shock protein, alphaB-crystallin: recognition of the ‘IxI’ motif. EMBO J 31(24):4587–4594. doi:10.1038/emboj.2012.318
den Engelsman J, Keijsers V, de Jong WW, Boelens WC (2003) The small heat-shock protein alpha B-crystallin promotes FBX4-dependent ubiquitination. J Biol Chem 278(7):4699–4704. doi:10.1074/jbc.M211403200
den Engelsman J, Gerrits D, de Jong WW, Robbins J, Kato K, Boelens WC (2005) Nuclear import of {alpha}B-crystallin is phosphorylation-dependent and hampered by hyperphosphorylation of the myopathy-related mutant R120G. J Biol Chem 280(44):37139–37148. doi:10.1074/jbc.M504106200
Dubin RA, Wawrousek EF, Piatigorsky J (1989) Expression of the murine alpha B-crystallin gene is not restricted to the lens. Mol Cell Biol 9(3):1083–1091. doi:10.1128/mcb.9.3.1083
Ebert AD, Liang P, Wu JC (2012) Induced pluripotent stem cells as a disease modeling and drug screening platform. J Cardiovasc Pharmacol 60(4):408–416. doi:10.1097/FJC.0b013e318247f642
Ecroyd H, Meehan S, Horwitz J, Aquilina JA, Benesch JL, Robinson CV, Macphee CE, Carver JA (2007) Mimicking phosphorylation of alphaB-crystallin affects its chaperone activity. Biochem J 401(1):129–141. doi:10.1042/BJ20060981
Ellis RJ, van der Vies SM (1991) Molecular chaperones. Annu Rev Biochem 60(1):321–347. doi:10.1146/annurev.bi.60.070191.001541
Fardeau M, Godet-Guillain J, Tome FM, Collin H, Gaudeau S, Boffety C, Vernant P (1978) Une nouvelle affection musculaire familiale définie par l’accumulation intra-sarcoplasmique d’un matériel granulofilamentaire dense en microscopie electronique. [A new familial muscular disorder demonstrated by the intra-sarcoplasmic accumulation of a granulo-filamentous material which is dense on electron microscopy (author’s transl)]. Rev Neurol (Paris) 134:411–425
Feil IK, Malfois M, Hendle J, van Der Zandt H, Svergun DI (2001) A novel quaternary structure of the dimeric alpha-crystallin domain with chaperone-like activity. J Biol Chem 276(15):12024–12029. doi:10.1074/jbc.M010856200
Fong H, Wang C, Knoferle J, Walker D, Balestra ME, Tong LM, Leung L, Ring KL, Seeley WW, Karydas A, Kshirsagar MA, Boxer AL, Kosik KS, Miller BL, Huang Y (2013) Genetic correction of tauopathy phenotypes in neurons derived from human induced pluripotent stem cells. Stem Cell Rep 1(3):226–234. doi:10.1016/j.stemcr.2013.08.001
Forrest KM, Al-Sarraj S, Sewry C, Buk S, Tan SV, Pitt M, Durward A, McDougall M, Irving M, Hanna MG, Matthews E, Sarkozy A, Hudson J, Barresi R, Bushby K, Jungbluth H, Wraige E (2011) Infantile onset myofibrillar myopathy due to recessive CRYAB mutations. Neuromuscul Disord 21(1):37–40. doi:10.1016/j.nmd.2010.11.003
Friden J, Lieber RL (2001) Eccentric exercise-induced injuries to contractile and cytoskeletal muscle fibre components. Acta Physiol Scand 171:321–326
Garcia-Mata R, Gao Y, Sztul E (2002) Hassles with taking out the garbage: aggravating aggresomes. Traffic 3:388–396
Ghosh JG, Estrada MR, Clark JI (2006) Structure-based analysis of the beta8 interactive sequence of human alphaB crystallin. Biochemistry 45:9878–9886
Ghosh JG, Houck SA, Clark JI (2007a) Interactive sequences in the stress protein and molecular chaperone human alphaB crystallin recognize and modulate the assembly of filaments. Int J Biochem Cell Biol 39(10):1804–1815. doi:10.1016/j.biocel.2007.04.027
Ghosh JG, Shenoy AK, Clark JI (2007b) Interactions between important regulatory proteins and human alphaB crystallin. Biochemistry 46:6308–6317
Golenhofen N, Htun P, Ness W, Koob R, Schaper W, Drenckhahn D (1999) Binding of stress protein cryab to cardiac myofibrils correlates with the degree of myocardial damage during I/R in vivo. J Mol Cell Cardiol 31:569–580
Golenhofen N, Arbeiter A, Koob R, Drenckhahn D (2002) Ischemia-induced association of the stress protein alpha B-crystallin with I-band portion of cardiac titin. J Mol Cell Cardiol 34(3):309–319. doi:10.1006/jmcc.2001.1513
Golenhofen N, Perng MD, Quinlan RA, Drenckhahn D (2004) Comparison of the small heat shock proteins alphaB-crystallin, MKBP, HSP25, HSP20, and cvHSP in heart and skeletal muscle. Histochem Cell Biol 122(5):415–425. doi:10.1007/s00418-004-0711-z
Golenhofen N, Redel A, Wawrousek EF, Drenckhahn D (2006) Ischemia-induced increase of stiffness of alphaB-crystallin/HSPB2-deficient myocardium. Pflugers Arch 451(4):518–525. doi:10.1007/s00424-005-1488-1
Gopal-Srivastava R, Piatigorsky J (1993) The murine alpha B-crystallin/small heat shock protein enhancer: identification of alpha BE-1, alpha BE-2, alpha BE-3 and MRF control elements. Mol Cell Biol 13(11):7144–7152
Gopal-Srivastava R, Haynes JI II, Piatigorsky J (1995) Regulation of the murine aB-crystallin/small heath shock protein gene in cardiac muscle. Mol Cell Biol 15(12):7081–7090
Hayes VH, Devlin G, Quinlan RA (2008) Truncation of alphaB-crystallin by the myopathy-causing Q151X mutation significantly destabilizes the protein leading to aggregate formation in transfected cells. J Biol Chem 283(16):10500–10512. doi:10.1074/jbc.M706453200
Hishiya A, Salman MN, Carra S, Kampinga HH, Takayama S (2011) BAG3 directly interacts with mutated alphaB-crystallin to suppress its aggregation and toxicity. PLoS ONE 6(3):e16828. doi:10.1371/journal.pone.0016828
Horwitz J (1992) a-Crystallin can function as a molecular chaperone. Proc Natl Acad Sci U S A 89:10449–10453
Hosoyama T, McGivern JV, Van Dyke JM, Ebert AD, Suzuki M (2014) Derivation of myogenic progenitors directly from human pluripotent stem cells using a sphere-based culture. Stem Cells Transl Med. doi:10.5966/sctm.2013-0143
Houck SA, Landsbury A, Clark JI, Quinlan RA (2011) Multiple sites in alphaB-crystallin modulate its interaction with desmin filaments assembled in vitro. PLoS ONE 6(11):e25859. doi:10.1371/journal.pone.0025859.g001
Hu WF, Gong L, Cao Z, Ma H, Ji W, Deng M, Liu M, Hu XH, Chen P, Yan Q, Chen HG, Liu J, Sun S, Zhang L, Liu JP, Wawrousek E, Li DW (2012) αA- and αB-crystallins interact with caspase-3 and Bax to guard mouse lens development. Curr Mol Med 12(2):177–187
Ignolia TD, Craig EA (1982) Four small Drosophila heat shock proteins are related to each other and to mammalian alpha crystallin. Proc Natl Acad Sci U S A 79:2360–2364
Inagaki N, Hayashi T, Arimura T, Koga Y, Takahashi M, Shibata H, Teraoka K, Chikamori T, Yamashina A, Kimura A (2006) Alpha B-crystallin mutation in dilated cardiomyopathy. Biochem Biophys Res Commun 342(2):379–386. doi:10.1016/j.bbrc.2006.01.154
Ishiwata T, Orosz A, Wang X, Mustafi SB, Pratt GW, Christians ES, Boudina S, Abel ED, Benjamin IJ (2012) HSPB2 is dispensable for the cardiac hypertrophic response but reduces mitochondrial energetics following pressure overload in mice. PLoS one 7(8):e42118. doi:10.1371/journal.pone.0042118
Ito H, Okamoto K, Nakayama H, Isobe T, Kato K (1997) Phosphorylation of alphaB-crystallin in response to various types of stress. J Biol Chem 272:29934–29941
Itzhaki I, Maizels L, Huber I, Zwi-Dantsis L, Caspi O, Winterstern A, Feldman O, Gepstein A, Arbel G, Hammerman H, Boulos M, Gepstein L (2011) Modelling the long QT syndrome with induced pluripotent stem cells. Nature 471(7337):225–229. doi:10.1038/nature09747
Iwaki T, Kume-Iwaki A, Goldman JE (1990) Cellular distribution of alpha B-crystallin in non-lenticular tissues. J Histochem Cytochem 38(1):31–39. doi:10.1177/38.1.2294148
Jansen G, Groenen PJTA, Bachner D, Jap PHK, Coerwinkel M, Oerlemans F, van den Broek W, Gohlsch B, Pette D, Plomp JJ, Molenaar PC, Nederhoff MGJ, van Echteld CJA, Dekker M, Berns A, Hameister H, Wieringa B (1996) Abnormal myotonic dystrophy protein kinase levels produce only mild myopathy in mice. Nat Genet 13:316–324
Jehle S, Rajagopal P, Bardiaux B, Markovic S, Kuhne R, Stout JR, Higman VA, Klevit RE, van Rossum BJ, Oschkinat H (2010) Solid-state NMR and SAXS studies provide a structural basis for the activation of alphaB-crystallin oligomers. Nat Struct Mol Biol 17(9):1037–1042. doi:10.1038/nsmb.1891
Kamradt MC, Chen F, Cryns VL (2001) The small heat shock protein alpha B-crystallin negatively regulates cytochrome c- and caspase-8-dependent activation of caspase-3 by inhibiting its autoproteolytic maturation. J Biol Chem 276(19):16059–16063. doi:10.1074/jbc.C100107200
Kamradt MC, Chen F, Sam S, Cryns VL (2002) The small heat shock protein alpha B-crystallin negatively regulates apoptosis during myogenic differentiation by inhibiting caspase-3 activation. J Biol Chem 277(41):38731–38736. doi:10.1074/jbc.M201770200
Kappé G, Leunissen JM, Jong W (2002) Evolution and diversity of prokaryotic small heat shock proteins. In: Arrigo A-P, Müller WEG (eds) Small stress proteins, vol 28, Progress in molecular and subcellular biology. Springer, Berlin/Heidelberg, pp 1–17. doi:10.1007/978-3-642-56348-5_1
Kato K, Ito H, Kamei K, Inaguma Y, Iwamoto I, Saga S (1998) Phosphorylation of αB-crystallin in mitotic cells and identification of enzymatic activities responsible for phosphorylation. J Biol Chem 273:28346–28354
Koh TJ (2002) Do small heat shock proteins protect skeletal muscle from injury. Exerc Sports Sci Rev 30(3):117–121
Koh TJ, Escobedo J (2004) Cytoskeletal disruption and small heat shock protein translocation immediately after lengthening contractions. Am J Physiol Cell Physiol 286(3):C713–C722. doi:10.1152/ajpcell.00341.2003
Komulainen J, Takala TES, Kuipers H, Hesselink MKC (1998) The disruption of myofibre structures in rat skeletal muscle after forced lengthening contractions. Eur J Physiol 436:735–741
Kostek MC, Chen YW, Cuthbertson DJ, Shi R, Fedele MJ, Esser KA, Rennie MJ (2007) Gene expression responses over 24 h to lengthening and shortening contractions in human muscle: major changes in CSRP3, MUSTN1, SIX1, and FBXO32. Physiol Genomics 31:42–52. doi:10.1152/physiolgenomics.00151.2006.-Resistance
Kotter S, Unger A, Hamdani N, Lang P, Vorgerd M, Nagel-Steger L, Linke WA (2014) Human myocytes are protected from titin aggregation-induced stiffening by small heat shock proteins. J Cell Biol 204(2):187–202. doi:10.1083/jcb.201306077
Krishnamoorthy V, Donofrio AJ, Martin JL (2013) O-GlcNAcylation of alphaB-crystallin regulates its stress-induced translocation and cytoprotection. Mol Cell Biochem 379(1–2):59–68. doi:10.1007/s11010-013-1627-5
Kumarapeli AR, Su H, Huang W, Tang M, Zheng H, Horak KM, Li M, Wang X (2008) Alpha B-crystallin suppresses pressure overload cardiac hypertrophy. Circ Res 103(12):1473–1482. doi:10.1161/CIRCRESAHA.108.180117
Labeit S, Kolmerer B (1995) Titins: giant proteins in charge of muscle ultrastructure and elasticity. Science 270:293–296
Laganowsky A, Benesch JL, Landau M, Ding L, Sawaya MR, Cascio D, Huang Q, Robinson CV, Horwitz J, Eisenberg D (2010) Crystal structures of truncated alphaA and alphaB crystallins reveal structural mechanisms of polydispersity important for eye lens function. Protein Sci 19(5):1031–1043. doi:10.1002/pro.380
Le Grand F, Rudnicki MA (2007) Skeletal muscle satellite cells and adult myogenesis. Curr Opin Cell Biol 19(6):628–633. doi:10.1016/j.ceb.2007.09.012
Li DW, Liu JP, Mao YW, Xiang H, Wang J, Ma WY, Dong Z, Pike HM, Brown RE, Reed JC (2005) Calcium-activated RAF/MEK/ERK signaling pathway mediates p53-dependent apoptosis and is abrogated by alpha B-crystallin through inhibition of RAS activation. Mol Biol Cell 16(9):4437–4453. doi:10.1091/mbc.E05-01-0010
Lian X, Zhang J, Azarin SM, Zhu K, Hazeltine LB, Bao X, Hsiao C, Kamp TJ, Palecek SP (2013) Directed cardiomyocyte differentiation from human pluripotent stem cells by modulating Wnt/beta-catenin signaling under fully defined conditions. Nat Protoc 8(1):162–175. doi:10.1038/nprot.2012.150
Liang P, MacRae TH (1997) Molecular chaperones and the cytoskeleton. J Cell Sci 110:1431–1440
Limphong P, Zhang H, Christians E, Liu Q, Riedel M, Ivey K, Cheng P, Mitzelfelt K, Taylor G, Winge D, Srivastava D, Benjamin I (2013) Modeling human protein aggregation cardiomyopathy using murine induced pluripotent stem cells. Stem Cells Transl Med 2(3):161–166. doi:10.5966/sctm. 2012-0073
Linke WA (2008) Sense and stretchability: the role of titin and titin-associated proteins in myocardial stress-sensing and mechanical dysfunction. Cardiovasc Res 77(4):637–648. doi:10.1016/j.cardiores.2007.03.029
Liu JP, Schlosser R, Ma WY, Dong Z, Feng H, Liu L, Huang XQ, Liu Y, Li DW (2004) Human alphaA- and alphaB-crystallins prevent UVA-induced apoptosis through regulation of PKCalpha, RAF/MEK/ERK and AKT signaling pathways. Exp Eye Res 79(3):393–403. doi:10.1016/j.exer.2004.06.015
Liu S, Li J, Tao Y, Xiao X (2007) Small heat shock protein alphaB-crystallin binds to p53 to sequester its translocation to mitochondria during hydrogen peroxide-induced apoptosis. Biochem Biophys Res Commun 354(1):109–114. doi:10.1016/j.bbrc.2006.12.152
Lockard VG, Bloom S (1993) Trans-cellular desmin-lamin B intermediate filament network in cardiac myocytes. J Mol Cell Cardiol 25:303–309
Lutsch G, Vetter R, Offhauss U, Wieske M, Gröne H-J, Klemenz R, Schimke I, Stahl J, Benndorf R (1997) Abundance and location of the small heat shock proteins HSP25 and αB-crystallin in rat and human heart. Circulation 96:3466–3476
Maloyan A, Sanbe A, Osinska H, Westfall M, Robinson D, Imahashi K, Murphy E, Robbins J (2005) Mitochondrial dysfunction and apoptosis underlie the pathogenic process in alpha-B-crystallin desmin-related cardiomyopathy. Circulation 112(22):3451–3461. doi:10.1161/CIRCULATIONAHA.105.572552
Maloyan A, Gulick J, Glabe CG, Kayed R, Robbins J (2007) Exercise reverses preamyloid oligomer and prolongs survival in alphaB-crystallin-based desmin-related cardiomyopathy. Proc Natl Acad Sci U S A 104(14):5995–6000. doi:10.1073/pnas.0609202104
Maloyan A, Osinska H, Lammerding J, Lee RT, Cingolani OH, Kass DA, Lorenz JN, Robbins J (2009) Biochemical and mechanical dysfunction in a mouse model of desmin-related myopathy. Circ Res 104(8):1021–1028. doi:10.1161/CIRCRESAHA.108.193516
Mao YW, Liu JP, Xiang H, Li DW (2004) Human alphaA- and alphaB-crystallins bind to Bax and Bcl-X(S) to sequester their translocation during staurosporine-induced apoptosis. Cell Death Differ 11(5):512–526. doi:10.1038/sj.cdd.4401384
Martin JL, Mestril R, Hilal-Dandan R, Brunton LL, Dillmann WH (1997) Small heat shock proteins and protection against ischemic injury in cardiac myocytes. Circulation 96:4343–4348
Melkani GC, Cammarato A, Bernstein SI (2006) AlphaB-crystallin maintains skeletal muscle myosin enzymatic activity and prevents its aggregation under heat-shock stress. J Mol Biol 358(3):635–645. doi:10.1016/j.jmb.2006.02.043
Mercola M, Ruiz-Lozano P, Schneider MD (2011) Cardiac muscle regeneration: lessons from development. Genes Dev 25(4):299–309. doi:10.1101/gad.2018411
Milner D, Mavroidis M, Weisleder N, Capetanaki Y (2000) Desmin cytoskeleton linked to muscle mitochondrial distribution and respiratory function. J Cell Biol 150(6):1283–1297
Minajeva A, Kulke M, Fernandez JM, Linke WA (2001) Unfolding of titin domains explains the viscoelastic behavior of skeletal myofibrils. Biophys J 80:1442–1451
Mitra A, Basak T, Datta K, Naskar S, Sengupta S, Sarkar S (2013) Role of alpha-crystallin B as a regulatory switch in modulating cardiomyocyte apoptosis by mitochondria or endoplasmic reticulum during cardiac hypertrophy and myocardial infarction. Cell Death Dis 4:e582. doi:10.1038/cddis.2013.114
Morimoto RI (1998) Regulation of the heat shock transcriptional response: cross talk between a family of heat shock factors, molecular chaperones, and negative regulators. Genes Dev 12(24):3788–3796. doi:10.1101/gad.12.24.3788
Morner CT (1894) Untersuchung der Proteinsubstanzen in den lichtbrechenden Medien des Auges (Examination of the protein-substances in the refractory media of the eye). Hoppe Seyler’s Z Physiol Chem 18(61):106
Morrison LE, Hoover HE, Thuerauf DJ, Glembotski CC (2003) Mimicking phosphorylation of alphaB-crystallin on serine-59 is necessary and sufficient to provide maximal protection of cardiac myocytes from apoptosis. Circ Res 92(2):203–211. doi:10.1161/01.res.0000052989.83995.a5
Morrison LE, Whittaker RJ, Klepper RE, Wawrousek EF, Glembotski CC (2004) Roles for αB-crystallin and HSPB2 in protecting the myocardium from ischemia-reperfusion-induced damage in a KO mouse model. Am J Physiol Heart Circ Physiol 286:H847–H855
Mymrikov EV, Seit-Nebi AS, Gusev NB (2011) Large potentials of small heat shock proteins. Physiol Rev 91:1123–1159. doi:10.1152/physrev.00023.2010.-Modern
Neppl RL, Kataoka M, Wang DZ (2014) Crystallin-alphaB regulates skeletal muscle homeostasis via modulation of argonaute2 activity. J Biol Chem 289(24):17240–17248. doi:10.1074/jbc.M114.549584
Neufer PD, Benjamin IJ (1996) Differential expression of alpha B-crystallin and Hsp27 in skeletal muscle during continuous contractile activity. Relationship to myogenic regulatory factors. J Biol Chem 271(39):24089–24095. doi:10.1074/jbc.271.39.24089
Ogata T, Yamaksak Y (1997) Ultra-high-resolution scanning electron microscopy of mitochondria and sarcoplasmic reticulum arrangement in human red, white, and intermediate muscle fibers. Anat Rec 248:214–223
Paulsen G, Vissing K, Kalhovde JM, Ugelstad I, Bayer ML, Kadi F, Schjerling P, Hallen J, Raastad T (2007) Maximal eccentric exercise induces a rapid accumulation of small heat shock proteins on myofibrils and a delayed HSP70 response in humans. Am J Physiol Regul Integr Comp Physiol 293:R844–R853. doi:10.1152/ajpregu.00677.2006.-In
Perng MD, Cairns L, Vandenijssel P, Prescott A, Hutcheson AM, Quinlan RA (1999a) Intermediate filament interactions can be altered by HSP27 and alphaB-crystallin. J Cell Sci 112:2099–2112
Perng MD, Muchowski PJ, Vandenijssel P, Wu JS, Hutcheson AM, Clark JI, Quinlan RA (1999b) The cardiomyopathy and lens cataract mutation in alphaB-crystalln alters its protein structure, chaperone activity, and interaction with intermediate filaments in vitro. J Biol Chem 274:33235–33243
Perng MD, Wen SF, van den IP, Prescott AR, Quinlan RA (2004) Desmin aggregate formation by R120G alphaB-crystallin is caused by altered filament interactions and is dependent upon network status in cells. Mol Biol Cell 15(5):2335–2346. doi:10.1091/mbc.E03-12-0893
Peschek J, Braun N, Rohrberg J, Back KC, Kriehuber T, Kastenmüller A, Weinkauf S, Buchner J (2013) Regulated structural transitions unleash the chaperone activity of alphaB-crystallin. Proc Natl Acad Sci U S A 110(40):E3780–E3789
Pilotto A, Marziliano N, Pasotti M, Grasso M, Costante AM, Arbustini E (2006) AlphaB-crystallin mutation in dilated cardiomyopathies: low prevalence in a consecutive series of 200 unrelated probands. Biochem Biophys Res Commun 346(4):1115–1117. doi:10.1016/j.bbrc.2006.05.203
Pinz I, Robbins J, Rajasekaran NS, Benjamin IJ, Ingwall JS (2008) Unmasking different mechanical and energetic roles for the small heat shock proteins CryAB and HSPB2 using genetically modified mouse hearts. FASEB J 22(1):84–92. doi:10.1096/fj.07-8130com
Plater ML, Goode D, Crabbe MJC (1996) Effects of site-directed mutations on the chaperone-like activity of alphaB-crystallin. J Biol Chem 271(45):28558–28566
Rajaraman K, Raman B, Ramakrishna T, Rao Ch M (2001) Interaction of human recombinant alphaA- and alphaB-crystallins with early and late unfolding intermediates of citrate synthase on its thermal denaturation. FEBS Lett 497:118–123
Rajasekaran NS, Connell P, Christians ES, Yan LJ, Taylor RP, Orosz A, Zhang XQ, Stevenson TJ, Peshock RM, Leopold JA, Barry WH, Loscalzo J, Odelberg SJ, Benjamin IJ (2007) Human alpha B-crystallin mutation causes oxido-reductive stress and protein aggregation cardiomyopathy in mice. Cell 130(3):427–439. doi:10.1016/j.cell.2007.06.044
Raju I, Abraham EC (2013) Mutants of human alphaB-crystallin cause enhanced protein aggregation and apoptosis in mammalian cells: influence of co-expression of HspB1. Biochem Biophys Res Commun 430(1):107–112. doi:10.1016/j.bbrc.2012.11.051
Rambourg A, Segretain D (1980) Three-dimensional electron microscopy of mitochondria and endoplasmic reticulum in the red muscle fiber of the rat diaphragm. Anat Rec 197(1):33–48
Rappaport L, Contard F, Samuel JL, Delcayre C, Marotte F, Tome F, Fardeau M (1988) Storage of phosphorylated desmin in a familial myopathy. FEBS Lett 231(2):421–425
Rappaport L, Oliviero P, Samuel JL (1998) Cytoskeleton and mitochondrial morphology and function. Mol Cell Biochem 184:101–105
Ray PS, Martin JL, Swanson EA, Otani H, Dillmann WH, Das DK (2001) Transgene overexpression of aB crystallin confers simultaneous protection against cardiomyocyte apoptosis and necrosis during myocardial ischemia and reperfusion. FASEB J 15:393–402
Reddy S, Smith DBJ, Rich MM, Leferovich JM, Reilly P, Davis BM, Tran K, Rayburn H, Bronson R, Cros D, Balice-Gordon RJ, Housman D (1996) Mice lacking the myotonic dystrophy protein kinase develop a late onset progressive myopathy. Nat Genet 13:325–335
Reilich P, Schoser B, Schramm N, Krause S, Schessl J, Kress W, Muller-Hocker J, Walter MC, Lochmuller H (2010) The p.G154S mutation of the alpha-B crystallin gene (CRYAB) causes late-onset distal myopathy. Neuromuscul Disord 20(4):255–259. doi:10.1016/j.nmd.2010.01.012
Reipert S, Steinbock F, Fischer I, Bittner RE, Zeold A, Wiche G (1999) Association of mitochondria with plectin and desmin intermediate filaments in striated muscle. Exp Cell Res 252:479–491
Rief M (1997) Reversible unfolding of individual titin immunoglobulin domains by AFM. Science 276(5315):1109–1112. doi:10.1126/science.276.5315.1109
Robinton DA, Daley GQ (2012) The promise of induced pluripotent stem cells in research and therapy. Nature 481(7381):295–305. doi:10.1038/nature10761
Sacconi S, Feasson L, Antoine JC, Pecheux C, Bernard R, Cobo AM, Casarin A, Salviati L, Desnuelle C, Urtizberea A (2012) A novel CRYAB mutation resulting in multisystemic disease. Neuromuscul Dis 22(1):66–72. doi:10.1016/j.nmd.2011.07.004
Sanbe A, Osinska H, Saffitz JE, Glabe CG, Kayed R, Maloyan A, Robbins J (2004) Desmin-related cardiomyopathy in transgenic mice: a cardiac amyloidosis. Proc Natl Acad Sci U S A 101(27):10132–10136. doi:10.1073/pnas.0401900101
Sanbe A, Yamauchi J, Miyamoto Y, Fujiwara Y, Murabe M, Tanoue A (2007) Interruption of CryAB-amyloid oligomer formation by HSP22. J Biol Chem 282(1):555–563. doi:10.1074/jbc.M605481200
Sanbe A, Daicho T, Mizutani R, Endo T, Miyauchi N, Yamauchi J, Tanonaka K, Glabe C, Tanoue A (2009) Protective effect of geranylgeranylacetone via enhancement of HSPB8 induction in desmin-related cardiomyopathy. PLoS one 4(4):e5351. doi:10.1371/journal.pone.0005351
Sanbe A, Marunouchi T, Yamauchi J, Tanonaka K, Nishigori H, Tanoue A (2011) Cardioprotective effect of nicorandil, a mitochondrial ATP-sensitive potassium channel opener, prolongs survival in HSPB5 R120G transgenic mice. PLoS one 6(4):e18922. doi:10.1371/journal.pone.0018922
Selcen D (2011) Myofibrillar myopathies. Neuromuscular Dis NMD 21(3):161–171. doi:10.1016/j.nmd.2010.12.007
Selcen D, Engel AG (2003) Myofibrillar myopathy caused by novel dominant negative cryAB mutations. Ann Neurol 54(6):804–810
Singh BN, Rao KS, Rao Ch M (2010) Ubiquitin-proteasome-mediated degradation and synthesis of MyoD is modulated by alphaB-crystallin, a small heat shock protein, during muscle differentiation. Biochim Biophys Acta 1803(2):288–299. doi:10.1016/j.bbamcr.2009.11.009
Smirnova E, Chebotareva N, Gurvits B (2013) Transient transformation of oligomeric structure of alpha-crystallin during its chaperone action. Int J Biol Macromol 55:62–68. doi:10.1016/j.ijbiomac.2012.12.013
Spector DL, Lamond AI (2011) Nuclear speckles. Cold Spring Harb Perspect Biol 3(2):1–12. a000646. doi:10.1101/cshperspect.a000646
Suzuki A, Sugiyama Y, Hayashi Y, Nyu-i N, Yoshida M, Nonaka I, Ishiura S, Arahata K, Ohno S (1998) MKBP, a novel member of the small heat shock protein family, binds and activates the myotonic dystrophy protein kinase. J Cell Biol 140:1113–1124
Takahashi, Yamanaka (2006) Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126(4):663–676
Tannous P, Zhu H, Johnstone JL, Shelton JM, Rajasekaran NS, Benjamin IJ, Nguyen L, Gerard RD, Levine B, Rothermel BA, Hill JA (2008) Autophagy is an adaptive response in desmin-related cardiomyopathy. Proc Natl Acad Sci U S A 105(28):9745–9750. doi:10.1073/pnas.0706802105
Taylor RP, Benjamin IJ (2005) Small heat shock proteins: a new classification scheme in mammals. J Mol Cell Cardiol 38(3):433–444. doi:10.1016/j.yjmcc.2004.12.014
Thompson HS, Scordilis SP, Clarkson PM, Lohrer WA (2001) A single bout of eccentric exercise increases Hsp27 and HSC/HSP70 in human skeletal muscle. Acta Physiol Scand 171:187–193
Vandenijssel P, Wheelock R, Prescott A, Russell P, Quinlan R (2003) Nuclear speckle localisation of the small heat shock protein alphaB-crystallin and its inhibition by the R120G cardiomyopathy-linked mutation. Exp Cell Res 287(2):249–261. doi:10.1016/s0014-4827(03)00092-2
Vicart CA, Guicheney P, Li Z, Prévost MC, Faure A, Chateau D, Chapon F, Tomé F, Dupret JM, Paulin D, Fardeau M (1998) A missense mutation in the alphaB-crystallin chaperone gene causes a desmin-related myopathy. Nat Genet 20(1):92–95
Wang X, Osinska H, Klevitsky R, Gerdes AM, Nieman M, Lorenz J, Hewett T, Robbins J (2001) Expression of R120G- B-crystallin causes aberrant desmin and B-crystallin aggregation and cardiomyopathy in mice. Circ Res 89(1):84–91. doi:10.1161/hh1301.092688
Watanabe G, Kato S, Nakata H, Ishida T, Ohuchi N, Ishioka C (2009) alphaB-crystallin: a novel p53-target gene required for p53-dependent apoptosis. Cancer Sci 100(12):2368–2375. doi:10.1111/j.1349-7006.2009.01316.x
Weintraub H (1993) The MyoD family and myogenesis- redundancy, networks, and thresholds. Cell 75:1241–1244
Whittaker R, Gude GN, Sussman MA, Gottlieb RA, Glembotski CC (2009) Kinetics of the translocation and phosphorylation of alphaB-crystallin in mouse heart mitochondria during ex vivo ischemia. Am J Physiol Heart Circ Physiol 296:H1633–H1642. doi:10.1152/ajpheart.01227.2008.-/Bcrystallin
Yan LJ, Christians EC, Liu L, Xiao X, Sohal RS, Benjamin IJ (2002) Mouse heat shock transcription factor 1 deficiency alters cardiac redox homeostasis and increases mitochondrial oxidative damage. EMBO J 21(19):5164–5172
Yusa K, Rashid ST, Strick-Marchand H, Varela I, Liu PQ, Paschon DE, Miranda E, Ordonez A, Hannan NR, Rouhani FJ, Darche S, Alexander G, Marciniak SJ, Fusaki N, Hasegawa M, Holmes MC, Di Santo JP, Lomas DA, Bradley A, Vallier L (2011) Targeted gene correction of alpha1-antitrypsin deficiency in induced pluripotent stem cells. Nature 478(7369):391–394. doi:10.1038/nature10424
Zhang H, Rajasekaran NS, Orosz A, Xiao X, Rechsteiner M, Benjamin IJ (2010) Selective degradation of aggregate-prone CryAB mutants by HSPB1 is mediated by ubiquitin-proteasome pathways. J Mol Cell Cardiol 49(6):918–930. doi:10.1016/j.yjmcc.2010.09.004
Zhang J, Klos M, Wilson GF, Herman AM, Lian X, Raval KK, Barron MR, Hou L, Soerens AG, Yu J, Palecek SP, Lyons GE, Thomson JA, Herron TJ, Jalife J, Kamp TJ (2012) Extracellular matrix promotes highly efficient cardiac differentiation of human pluripotent stem cells: the matrix sandwich method. Circ Res 111(9):1125–1136. doi:10.1161/CIRCRESAHA.112.273144
Zhou T, Benda C, Dunzinger S, Huang Y, Ho JC, Yang J, Wang Y, Zhang Y, Zhuang Q, Li Y, Bao X, Tse HF, Grillari J, Grillari-Voglauer R, Pei D, Esteban MA (2012) Generation of human induced pluripotent stem cells from urine samples. Nat Protoc 7(12):2080–2089. doi:10.1038/nprot.2012.115
Zhu Y, Bogomolovas J, Labeit S, Granzier H (2009) Single molecule force spectroscopy of the cardiac titin N2B element: effects of the molecular chaperone alphaB-crystallin with disease-causing mutations. J Biol Chem 284(20):13914–13923. doi:10.1074/jbc.M809743200
Acknowledgments
Lindsey Barber provided excellent editorial assistance during preparation of this manuscript.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Mitzelfelt, K.A., Benjamin, I.J. (2015). Multifunctional Roles of αB-Crystallin in Skeletal and Cardiac Muscle Homeostasis and Disease. In: Tanguay, R., Hightower, L. (eds) The Big Book on Small Heat Shock Proteins. Heat Shock Proteins, vol 8. Springer, Cham. https://doi.org/10.1007/978-3-319-16077-1_11
Download citation
DOI: https://doi.org/10.1007/978-3-319-16077-1_11
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-16076-4
Online ISBN: 978-3-319-16077-1
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)