
Abstract
Eleven years ago, a secreted heme-thiolate peroxidase with promiscuity for oxygen transfer reactions was discovered in the basidiomycetous fungus, Agrocybe aegerita. The enzyme turned out to be a functional mono-peroxygenase that transferred an oxygen atom from hydrogen peroxide to diverse organic substrates (aromatics, heterocycles, linear and cyclic alkanes/alkenes, fatty acids, etc.). Later similar enzymes were found in other mushroom genera such as Coprinellus and Marasmius. Approximately one thousand putative peroxygenase sequences that form two large clusters can be found in genetic databases and fungal genomes, indicating the widespread occurrence of such enzymes in the whole fungal kingdom including all phyla of true fungi (Eumycota) and certain fungus-like heterokonts (Oomycota). This new enzyme type was classified as unspecific peroxygenase (UPO, EC 1.11.2.1) and placed in a separate peroxidase subclass. Furthermore, UPOs and related heme-thiolate peroxidases such as well-studied chloroperoxidase (CPO) represent a separate superfamily of heme proteins on the phylogenetic level. The reactions catalyzed by UPOs include hydroxylation, epoxidation, O- and N-dealkylation, aromatization, sulfoxidation, N-oxygenation, dechlorination and halide oxidation. In many cases, the product patterns of UPOs resemble those of human cytochrome P450 (P450) monooxygenases and, in fact, combine the catalytic cycle of heme peroxidases with the “peroxide shunt” of P450s. Here, an overview on UPOs is provided with focus on their molecular and catalytic properties.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
13.1 Introduction
Peroxygenase activities refer to the transfer of a peroxide-borne oxygen atom to substrates. Biocatalysts that preferably catalyze such reactions are classified in a separate sub-subclass, EC 1.11.2,Footnote 1 in the enzyme nomenclature system [www.chem.qmul.ac.uk/iubmb/enzyme/EC1/11/2/] (Fig. 13.1). The sub-subclass was approved in February 2011 and currently comprises four members, among which the unspecific peroxygenase (UPO, EC 1.11.2.1) is the most prominent because of its frequency in fungal organisms and promiscuity for oxygen transfer reactions.

Classification of enzymes using peroxide as the electron acceptor (EC 1.11; peroxidases and peroxygenases) according to the enzyme nomenclature system. Abbreviations: NADH-POD NADH peroxidase, POD peroxidase (phenol oxidizing), CPO chloroperoxidase, VP versatile peroxidase, DyP dye decolorizing peroxidase, UPO unspecific peroxygenase, MP myeloperoxidase, PSP plant seed peroxygenase, FAP fatty acid peroxygenase
The trivial name “peroxygenase” first appeared in the literature in 1977 in an article of Ishimarua and Yamazaki describing a new type of heme enzyme that catalyzes the hydroperoxide-dependent hydroxylation of several aromatic substrates (A) including indole, phenol and aniline in microsomes of pea seeds (Pisum sativum) [1]. The peroxygenase reaction can be illustrated in simplified form as shown in equation (eqn) 13.1.
where AH is the substrate, ROOH represents the hydroperoxide, R signifies an organic substituent or hydrogen atom, AOH designates the hydroxylated product and ROH depicts the reduced hydroperoxide or H2O. Nowadays, this enzyme that contains histidine-ligated heme and a caleosin-type calcium binding motif is classified under EC 1.11.2.3 as plant seed peroxygenase [2] that, among others, is thought to be involved in the synthesis of cutin [3].
In the P450 context, the term peroxygenase has been in use since the end of the 1980s [4–7] and is usually related to peroxide-driven substrate oxidation, a side activity that is also known as the “peroxide shunt” pathway [8–10]. Peroxygenase side activities have also been reported for a few dioxygenases [11, 12] as well as for tyrosinase [13]. Interestingly, in deviation from the typical monooxygenase cycle that works with reduced dinucleotides (NAD(P)H), there is one P450 type that prefers H2O2 over NAD(P)H. This “true P450-peroxygenase” (CYP152A1, P450BS, P450SPα, EC 1.11.2.4) is an intracellular enzyme found in bacteria such as Sphingomonas paucimobilis and Bacillus subtilis [14–16]. It preferably hydroxylates fatty acids (e.g. myristic acid) in the 2- and/or 3-position, as shown in eqns. 13.2 and 13.3, and was therefore designated as fatty acid peroxygenase (EC 1.11.2.4).Footnote 2
The fatty acid substrate can act as a decoy molecule, which widens the substrate spectrum of these peroxygenases. Thus, P450BSβ and P450SPα were shown to peroxygenate 1-methoxynaphthalene and styrene, respectively, in a carboxylic acid-dependent reaction [17, 18]. Interestingly, the decoy-molecule concept was later also successfully applied to classic P450s such as P450BM3 [19].
Mammalian myeloperoxidase (EC 1.11.2.2, formerly 1.11.1.7) is an additional member of the peroxygenase subclass and preferably oxidizes halides into hypohalites (eqn. 13.4), which in turn act as bactericidal agents in phagosomes [20].
Myeloperoxidase differs from catalytically similar fungal CPO (EC 1.11.1.10) in its preference for the formation of hypochlorite (HClO) over the chlorination of organic substrates under physiological conditions (pH 5–8).Footnote 3 In addition to halide oxidation, both myeloperoxidase and CPO have strong peroxidase (phenol oxidation) and moderate peroxygenase activities and, as an example, were reported to epoxidize styrene [21]. Beyond that, CPO epoxidizes linear alkenes [22], hydroxylates benzylic carbons to some extent [23], catalyzes sulfoxidations [24, 25] and converts indole to oxindole [26]. However, CPO is not capable of peroxygenating aromatic substrates or stronger C-H bonds as found in alkanes [27]. Nevertheless, from the phylogenetic point of view, CPO can be regarded as an ascomycetous peroxygenase specialized in halide oxidation (compare Fig. 13.2). The following sections will deal exclusively with fungal UPOs, focusing on their catalytic and molecular properties.

Neighbor-joining phylogentic tree of UPO/HTP-sequences using Jukes-Cantor genetic distances. Green – Basidiomycota, red – Ascomycota, blue – Oomycota, purple – Zygomycota, dark blue – Chytridiomycota and rose – Glomeromycota. The dotted lines separate UPO sequences of groups I and II
13.2 History and Occurrence of Unspecific Peroxygenases
The first enzyme of this type was described in 2004 as Agrocybe aegerita haloperoxidase for the respective fungus (syn. Agrocybe cylindracea, Cyclocybe aegerita) that belongs to the Basidiomycota (family Strophariaceae) and is commonly known as the Black poplar mushroom [28, 29]. The fungus grows preferably on wood of poplars (Populus spp.) and other broad-leaved trees and causes a moderate white rot. It is found in Europe, North America and Asia and prefers warm and mild climates. A. aegerita is a popular edible mushroom in Mediterranean countries, especially in Italy (ital. Pioppino or Piopparello), where it is also commercially cultured [30]. The first article had still not used the term peroxygenase and focused on the ability of the enzyme to oxidize halides and aryl alcohols [31]. Its unique oxygen atom transfer potential was recognized one year later by the hydroxylation of naphthalene [32], a reaction that later turned out to proceed via an initially-formed epoxide intermediate [33, 34]. Over the next few years, additional aromatic, heterocyclic and aliphatic substrates were found to be subjects of peroxygenation [27, 35, 36] (see also Sect. 13.4.3 below) and the name of the enzyme changed from haloperoxidase [31] via haloperoxidase-peroxygenase [33] to Agrocybe aegerita aromatic peroxygenase [37] and eventually to unspecific peroxygenase (UPOFootnote 4) [38]. Furthermore, UPOs are also referred to as heme-thiolate peroxidases (HTP), taking into account their characteristic heme ligation by a cysteinate and their relation to CPO [39–41].
The second UPO known as CraUPO was described for the Ink-cap Coprinellus (Coprinus) radians, a wood- and mulch-dwelling fungus that belongs to the family Psathyrellaceae closely related to the Strophariaceae [42]. As AaeUPO, CraUPO also oxidized naphthalene, aryl alcohols and bromide. Some differences between the two enzymes were observed with respect to the oxidation of aromatic rings vs. alkyl side chains or heteroatoms, as well as in the respective specific activities and kinetic data [43–45]. The third well-studied UPO known as MroUPO was that of the boreo-subtropical Pinwheel mushroom, Marasmius rotula, preferably colonizing twigs and belonging to the diverse basidiomycete family of Marasmiaceae. However, MroUPO was unable to oxidize halides. MroUPO exhibits a less pronounced aromatic ring-oxygenating activity [46] but instead oxidizes bulkier substrates than do the other UPOs [47, 48]. Besides these three UPO producers, there are several other mushroom species secreting UPOs (e.g. A. parasitica, A. chaxingu, A. alnetorum, Agaricus bisporus, Coprinus sp. DSM 14545, Coprinopsis verticillata, Auricularia auricula-judae, Mycena galopus). However, the purification or characterization of these enzymes has not been performed yet and the results have not been published. A recombinant UPO known as rCciUPO from the genome-sequenced model fungus, Coprinopsis cinerea, has recently been expressed at laboratory scale in Aspergillus oryzae [49].
More information on the occurrence of UPO-like enzymes can be gained from genetic databases where approximately 2,000 sequences of putative UPO enzymes are found. Figure 13.2 illustrates this diversity using a phylogenetic tree of UPOs (HTPs, respectively) covering 30 representative fungal species of different taxonomic and ecophysiological groups. Most sequences were retrieved from databases and genome projects but the tree also comprises 11 full sequences of A. aegerita, A. parasitica, C. radians, C. verticillata, M. rotula and Xylaria polymorpha generated in our laboratory, as well as the sequence of CPO from Leptoxyphium (Caldariomyces) fumago [40, 50, 51]. The majority of these sequences belongs to the Dikarya, i.e. Basidiomycota (353 sq.) and Ascomycota (580 sq.). However, other fungal phyla are also represented, such as the Mucoromycotina (“Zygomycetes”) by such common genera as Rhizopus and Mucor, the Chytridiomycota by a genome-sequenced Spizellomyces, the Glomeromycota by a Rhizophagus (syn. Glomus), and the Oomycota (fungus-like heterokonts/stramenopiles, water molds) by several species of the genus Phytophthora. The latter finding supports the hypothesis of certain mycologists that an extensive horizontal gene transfer had taken place between phytopathogenic ascomycetes and oomycetes early in evolution [52]. Interestingly, true yeasts such as Saccharomyces or fission yeasts such as Schizosaccharomyces do not have UPO genes. Also, plants including green algae (Viridiplantae) and animals (Metazoa) are obviously lacking such genes [40, 50].
A more detailed recent analysis of UPO-sequence data has revealed that there are two large groups of these enzymes: the “short and the long peroxygenases” (Fig. 13.2) [53]. The short UPO sequences of group I with an average molecular mass of the putative protein of 29 kDa are found in all fungal phyla, while the long sequences of group II with an average mass of 44 kDa and one internal disulfide bridge are present only in basidiomycetes and ascomycetes. Characterized MroUPO and CPO belong to group I and AaeUPO and CraUPO belong to group II. This also means that well-studied CPO, which has been an “orphan” among heme peroxidases for decades [27], is now one out of hundreds of fungal UPO/HTP enzymes. Differences between UPOs of groups I and II exist also in the active sites. In group I, a conserved histidine acts as the charge stabilizer whereas in the long enzymes (group II) of the AaeUPO type, an arginine occupies this position. There are conserved amino acids present in UPOs of groups I and II: -PCP-EHD-E- and -PCP-EGD-R-E-, respectively. Deviations may occur from the latter sequence pattern in long UPOs that do not belong to the AaeUPO subgroup (Figs. 13.2 and 13.3).

Separation of different AaeUPO forms by chromatofocusing on a mixed anion exchanger (Mono P). Major AaeUPO forms are highlighted in gray. Red line, absorbance at 420 nm; blue circles, UPO activity assayed with veratryl alcohol at pH 7.0; dotted line, concentration of eluting buffer (%); dashed green line, pH gradient (Modified according to [55])
UPOs are seemingly organized in gene clusters (multigene families) in fungal organisms. Thus, transcriptome studies on A. aegerita DSM 22459 have indicated (13 long and 3 short UPOs) the presence of at least 16 UPO sequences probably including several gene variants (Pecyna et al. 2013, unpublished results). In the genome of the common White button mushroom (Agaricus bisporus), even as much as 24 putative UPO sequences were identified [54]. Notably, among the A. aegerita sequences are both group II and a few group I UPOs. Albeit, the major AaeUPO forms expressed in soybean medium all belong to the closely-related group II enzymes that hardly differ in their catalytic properties [55]. At the moment, it is still impossible to say how many of these UPOs, and under what conditions, are actually translated and secreted and how this process is regulated on the molecular level.
Despite all the progress in understanding the catalytic mechanisms of UPOs and collecting their molecular data, the natural function of these enzymes in fungal organisms is not clear yet. Of course, the surpassing catalytic versatility may suggest an involvement in all kinds of detoxification reactions (i.e. detoxification of plant phytoalexins, microbial toxins, xenobiotic compounds) but other functions cannot be ruled out (e.g. involvement in lignin and humus degradation/modification or in biosynthetic pathways). The O-demethylation and cleavage of non-phenolic lignin model compounds (e.g. adlerol) by AaeUPO is at least an indication for UPOs participation in the oxidation of smaller lignin fragments emerging after the action of different enzymes, such as manganese peroxidase (EC 1.11.1.15), on the lignin polymer [56, 57].
13.3 Production, Purification and Properties
UPOs are typically produced with fungal wild-type strains in complex plant-based media rich in carbon and nitrogen. The growth medium must be optimized for each particular species and will vary considerably with respect to the concentration of individual ingredients. However, always soybean (or other legume) components have to be present in order to obtain sufficient amounts of UPOs. Thus, A. aegerita prefers slurries containing soybean meal (1–6 % w/w) and bactopeptone (0–2 %) [31], C. radians prefers mixtures of glucose (1–4 %) and soybean meal (1–3 %) [43], and M. rotula prefers soluble soybean peptone (4–5 %), yeast extract (4–5 %) and glucose (4 %) [46]. Though the structure of the soybean components triggering UPO production is not known, there are indications that seed storage proteins of the β-conglycinin and glycinin type, or their peptide fragments, are involved in the induction process (Pecyna, unpublished results).
Fungal fermentation can be carried out either simply in agitated or static culture flasks or in stirred-tank bioreactors under constant aeration [31, 43, 46]. As in the case of medium composition, culture parameters must be optimized for each fungal species/strain. UPO production in liquid cultures usually begins 5–12 days after inoculation with gently homogenized mycelium and reaches its maximum in the second to fourth week of cultivation (i.e. during secondary metabolism) [31, 43, 46]. There can be considerable differences in the overall production of UPO by individual fungal strains of the same species. Thus, among five strains of A. aegerita, only one strain (TM A1 = DSM 22459) secreted more than 1,500 unitsFootnote 5 per Liter (corresponding to 17 mg L−1 UPO protein), while all other strains produced only between 5 and 300 units [31]. At present, the highest amounts of UPO can be obtained with M. rotula that produces up to 445 mg L−1 UPO protein (corresponding to 41,000 U L−1), which is so far one of the highest levels of a secreted hemeprotein reported for a wild-type basidiomycete [46]. UPO activities (up to 120 U kg−1) are also detectable in solid-state cultures (e.g. beech-wood microcosms) of A. aegerita and C. radians but they have been too low to establish a functioning purification protocol [58].
UPOs are extracellular enzymes and can be concentrated by ultrafiltration of the culture liquid using appropriate membrane filters (e.g. 10-kDa cut-off). Purification of concentrated crude preparations is achieved by multistep fast protein liquid chromatography (FPLC) using different anion, cation and mixed-ion exchangers (e.g. Mono Q, S, P) and size exclusion chromatography (SEC) columns, depending on the particular UPO [31, 43, 46]. Alternative purification approaches on the basis of preparative isoelectric focusing turned out to be hardly suitable to obtain homogenous UPO fractions [55].
Usually, several UPO forms can be separated from fungal culture liquids. Figure 12.3 exemplarily shows the separation of the three major UPO forms (AaeUPO I–III) from A. aegerita grown in soybean slurry. The three forms have different isoelectric points (6.1, 5.6 and 5.2, respectively) but showed almost no differences in their catalytic properties, which suggests that these UPOs were considerably different glycosylated forms of the same protein and/or closely related gene products (e.g. of allelic sequences) rather than true UPO isoenzymes with different properties and functions [55].
Molecular masses and isoelectric points of characterized UPOs vary between 32 and 46 kDa as well as 3.8–6.1, respectively, and 16–42 % of mature UPOs are sugars of the high mannose type bound to up to six possible glycosylation sites [50, 59]. Table 13.1 summarizes some physical characteristics of UPOs from different fungi based on six original articles and unpublished results [31, 41, 43, 46, 55, 60]. UV-vis spectra of resting state UPOs are very similar to respective P450 spectra with Soret bands between 415 and 420 nm, which gives the purified UPOs a copper red color and sets them apart from CPO and other heme peroxidases [27]. The dithionite-reduced complex (ferrous UPO) shows a characteristic shift of the Soret band towards 450 nm when it comes into contact with CO, which is typical for heme-thiolate proteins [31, 43, 46, 61] (Fig. 13.4). UV-vis-spectral data of several UPOs are listed in Table 13.2, together with reference data for CPO, a P450 peroxygenase (P450SPα) and lignin peroxidase.

The successful crystallization of AaeUPO II (corresponding to the gene apo1 [50]) and solving of the crystal structure at 2.2 Å has recently been reported (Fig. 13.5) [59, 62]. The UPO protein contains ten α-helices and five very short β-sheets, a cysteinate-ligated heme as the prosthetic group and one disulfide bridge between Cys278 and Cys319 that stabilizes the C-terminal region after the last α-helix. One magnesium ion (probably structure-stabilizing) is located near the heme propionate and additionally coordinated by a glutamate (Glu122) and a serine (Ser126). The latter amino acids (E-XXX-S), as well as the PCP motif exposing a cysteinate as the proximal ligand to the heme (Fig. 13.6), are highly conserved in most UPOs along with another glutamic acid residue (Glu196) involved in acid-base catalysis and peroxide cleavage [50].

Left: Ribbon diagram of the molecular structure of AaeUPO; 10 α-helices (rainbow colored), 5 short β-sheets (purple). The red arrow marks the position of a disulfide bond (yellow); heme iron (brown ball), magnesium (blue ball). Right: solvent access surface of AaeUPO (colors represent electrostatic potentials: blue – positive, red – negative, dark grey – hydrophobic). The picture shows in the middle the channel that provides access to the heme (diameter of the entrance, ~7 Å); the arrow points at the iron (green) at the end of the heme channel (Based on [50, 59])

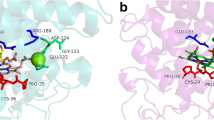
Figure 13.7 depicts the amino acid residues at the active sites of groups II and I UPOs by the example of AaeUPO and CPO. In both cases, a deprotonated glutamic acid residue (Glu196 in AaeUPO, Glu183 in CPO) near the heme abstracts a proton from the iron-bound peroxide to form compound 0 (Fig. 13.8), but the charge stabilizer of the glutamate is different. While an arginine (Arg189) functions as the charge stabilizer in AaeUPO, a histidine (His105) bears this role in CPO (compare also Sect. 13.4.3) [59]. Differences in the formation of compounds 0 and I, as well as in the behavior toward hydrogen peroxide, may be ascribed to these different residues. The heme channel of AaeUPO is ~7 Å in diameter and contains eight phenylalanines and one tyrosine residue, which make it rather hydrophobic and affined to aromatics. Current studies suggest that the molecular architecture of UPO heme channels is quite variable and that the hydrophobic character can also be brought about by aliphatic amino acids such as valine, leucine and isoleucine (Piontek, unpublished results).


13.4 Catalyzed Reactions and Reaction Mechanisms
13.4.1 Overview
The systematic name UPO according to the EC system is substrate: hydrogen peroxide oxidoreductase (RH-hydroxylating or -epoxidizing). Both the accepted name (unspecific peroxygenase) and the systematic name were chosen in analogy to P450 enzymes that are subsumed under EC 1.14.14.1Footnote 6 (unspecific monooxygenase, UMO) and act on a wide range of substrates including diverse xenobiotics, pharmaceuticals, alkanes and fatty acids [63, 64]. Many of these substrates are oxidized by UPOs in a similar manner, although the reaction requirements are different. While microsomal UMOs need molecular oxygen as a cosubstrate that has to be activated by two electrons delivered to the P450 monooxygenase from NAD(P)H via flavoproteins [65], extracellular UPOs need merely peroxide for efficient functioning [27], as shown in eqns. 13.5 and 13.6:
A summarizing overview of UPO-catalyzed reactions is shown in Fig. 13.9. The reaction portfolio, among others, includes alkane and alkyl hydroxylation, epoxidation of alkenes and aromatics, heteroatom oxygenation, O- and N-dealkylation and one-electron oxidation [36, 40]. Roughly calculated, over 300 compounds have been positively tested as UPO substrates and it is expected that even more substrates will be discovered. More details on particular reactions follow in Sect. 13.4.4.

Summarizing overview of UPO-catalyzed reactions
13.4.2 Reaction Cycle
The proposed reaction cycle of UPOs depicted in Fig. 13.10 is, in the first place, based on experimental data obtained with AaeUPO [66–68] and considers the comprehensive existing knowledge on P450s, CPO and heme-imidazole peroxidases such as horseradish peroxidase (HRP) [69–73]. It is assumed that this dual catalytic cycle applies for other UPOs/HTPs as well as CPO, with variation in the spectrum of oxidizable substrates (R-H vs. A-OH). It combines elements of the catalytic cycles of P450s and heme peroxidases [35, 36], in which compounds I and II are the key reactive intermediates that catalyze either the two-electron oxidation of a substrate molecule along with oxygen atom incorporation, or two one-electron oxidations resulting in the formation of two free diffusible substrate radicals (A-O•). In other words, UPOs oxygenate carbons in a similar manner as P450s (mono-peroxygenase pathway) and oxidize phenolics (peroxidase route) in a similar manner as prototypical heme peroxidases of the HRP type [74–77].

Proposed reaction cycle of UPOs with two routes: mono-peroxygenase pathway (left) and peroxidase route (right). Details are described in the text under Sect. 13.4.2
Now, we examine the catalytic properties of UPOs in more detail. Resting state UPO (i) contains a ferric heme that has a water molecule as the 6th (distal) ligand. There are indications that the substrate (R-H) can bind initially to the enzyme, because characteristic type I difference binding spectra [78] have been observed with different substrates such as veratryl alcohol, phenol and kaempferol [79], as well as with the pharmaceuticals dextromethorphan, diclofenac and propranolol [47]. However, this does not rule out that the substrate may also bind in later steps of the catalytic cycle, for example, after formation of compound I, as shown for (iii b ) and (iii c ) in Fig. 13.10 [66, 80]. UPO or the UPO-substrate complex then react with peroxide to form compound 0 (ii), a peroxo-complex that is heterolytically cleaved under electron re-arrangement to give the key compound I intermediate (oxo-ferryl cation radical complex) (iii) (see also Fig. 13.8). Compound I of AaeUPO and its kinetics of formation and decomposition have recently been studied using stopped-flow spectroscopy [66, 67]. In the mono-peroxygenation pathway, UPO compound I abstracts an electron and a proton (H-abstraction) from the substrate (R-H, e.g. an alkane) yielding protonated compound II (ferryl hydroxide complex) (iv) [69, 72]Footnote 7 and a substrate radical (R•) (located near the active site), which rapidly recombine to form a hydroxylated product (R-OH, e.g. an alcohol)–ferric enzyme complex. The product complex then dissociates with the release of the hydroxylated product, and a water molecule coordinates to the heme iron (i b ) to begin the catalytic cycle again. The cycle of the mono-peroxygenase pathway varies to some extent when epoxidation is the reaction under study. Based on P450 data and our own observations made during alkene oxidation by AaeUPO (see also Sect. 13.4.4.2), a modified compound II has to be proposed that binds the substrate (e.g. CH2=CH-R) as a radical via the ferryl oxygen [38, 81–83], i.e. [Heme]-FeIV-O-CH2-C•H-R (ferryl alkoxy radical complex). In this case, no H-abstraction would take place.
In the peroxidase route, both compounds I and II abstract one electron each from two substrate molecules (A-OH, e.g. a phenol), which are released as free radicals (A-O•, e.g. phenoxyl radicals) and can undergo coupling and/or disproportionation reactions [76]. It can be assumed that there are separate binding sites for one-electron oxidation substrates (A-OH) outside the heme channel, e.g. on the protein surface or at the heme channel entrance, as observed for ligninolytic peroxidases [59, 84, 85]. The route that is followed depends on the particular UPO enzyme and substrate, their redox potentials, the localization of the substrate binding site(s), the size of the heme channel and on the reaction pH [40, 67, 77]. In fact, there are differences between the characterized UPOs as well as UPOs and CPO regarding the substrates that can be oxygenated, as well as the extent to which the enzymes follow the mono-peroxygenase pathway or the peroxidase route. Some examples are presented in the following subsections.
13.4.3 UPO Assays
There are several spectrophotometric assays available to measure UPO activities (Fig. 13.11). They are based on the enzymes’ ability to oxidize alcohols to aldehydes, cleave ethers or to oxygenate aromatic rings. For routine measurements, the oxidation of veratryl alcohol to veratraldehyde is monitored at neutral pH. The reaction proceeds via initial hydroxylation of the benzylic carbon to give veratryl gem-diol (aldehyde hydrate) that is in equilibrium with veratraldehyde specifically absorbing at 310 nm [31]. Veratraldehyde is also formed in a second assay that uses the cleavage of methyl veratryl ether as a UPO-specific reaction (O-demethylation), leading to an unstable hemiacetal intermediate that spontaneously breaks down to veratraldehyde and methanol [86]. Demethylenation is a special case of O-dealkylation carried out with 5-nitro-1,3-benzodioxole as a substrate. Oxidation by UPO results in the formation of formic acid and 4-nitrocatechol. The latter product has the advantage that it specifically absorbs in the visible range at 425 nm (yellow color), which facilitates activity measurements in liquids with high background absorption in the UV range [87]. Aromatic ring oxygenation via initial epoxidation and subsequent spontaneous re-aromatization (phenol formation) can be monitored with naphthalene as a substrate at 303 nm [33, 34]. One-electron oxidations catalyzed by UPOs are assayed with classical peroxidase substrates such as ABTS or 2,6-dimethoxyphenol [31, 43, 46]. In addition to spectrophotometric measurements, it is also possible to determine UPO activities and kinetic data with HPLC or GC as was demonstrated, for example, for the oxidation of pyridine, ethylbenzene, benzene, cyclohexane and methylbutene [37, 38, 68, 88, 89].

13.4.4 Exemplary Reactions
13.4.4.1 Alkanes and Alkyl Groups
UPOs catalyze the hydroxylation of various linear, branched and cyclic alkanes as well as of alkyl groups (e.g. attached to aromatic rings) (Fig. 13.12). Most investigations were performed with AaeUPO [68], but two recent studies using peroxygenases from different fungi have shown that other UPOs can also efficiently hydroxylate alkanes, sometimes even with higher efficiency [49, 90]. Due to the low solubility of alkane substrates, reactions are usually performed in the presence of a co-solvent (e.g. acetone 4–60 % vol/vol).

UPO-catalyzed hydroxylation of alkanes and alkyls. Details are described in the text under Sect. 13.4.4.1
The size of linear alkane molecules that are oxidized by AaeUPO ranges from gaseous propane (C3) to viscous n-hexadecane (C16) [68]. The better-soluble fatty acids were even oxidized up to a chain length of C20 (arachidic acid) [41]. Alkanols hydroxylated in the 2- and 3-positions and hydroxy fatty acids with hydroxyl groups at (ω − 1) and (ω − 2) were the major products identified. The ratio between 2- and 3-alkanols depended on the chain length and amounted, for example, to 1:2 and 1.5:1 for the hydroxylation of n-pentane and n-heptane, respectively. In the latter case, an ee of 99.9 % was detected for the (R)-enantiomer [(R)-3-heptanol] [68]. In addition to monohydroxylated products, the corresponding alkanones were formed as minor over-oxidation products. With n-dodecane, n-tetradecane and n-hexadecane, hydroxylation was observed from both sides yielding small amounts of diols and their oxidation products (hydroxy-keto compounds and diketones) [41]. Traces of ω-hydroxylation products were only observed during the oxidation of fatty acids.
Branched alkanes up to a certain degree of branching are hydroxylated by AaeUPO as well, and often the tertiary carbons are preferably attacked. Thus, 2,3-dimethylbutane and isobutane were oxidized to the single products, 2,3-dimethylbutan-2-ol and 2-methylpropan-2-ol, respectively. The hydroxylation of 2,3,4-trimethylpentane yielded two products, 2,3,4-trimethylpentane-3-ol and 2,3,4-trimethylpentane-2-ol (Fig. 13.12) [68, 81]. Regarding the degree of branching, AaeUPO reaches its limit with 2,2,3,3-tetramethylbutene that is not subject to peroxygenation.
Cyclic alkanes from cyclopentane to cyclooctane are preferentially oxidized to form monohydroxylated products. Over-oxidation to the corresponding cycloalkanones is possible and depends on the reaction conditions and the UPO used. Recently, the optimization of cyclohexane oxidation via cyclohexanol to cyclohexanone has been reported using different UPOs, among which MroUPO was the most effective [90]. Over-oxidation of cyclohexane proceeds via a gem-diol intermediate (cyclohexane-1,1-diol) that spontaneously eliminates water (Fig. 13.13) [90]. In general, the oxidation of primary and secondary alcohols to carbonyls is a typical activity of all UPOs and leads to the formation of aldehydes and ketones (see also veratryl alcohol assay above) [36]. The aldehydes formed can be subjected to further oxidation, generating carboxylic acids (see also toluene oxidation below) [41].

Oxidation of cyclohexane via cyclohexanol and a hypothetical gem-diol to cyclohexanone (Modified according to [90])
The two-ring system of norcarane (bicyclo[4.1.0]heptane) represents a special case of cycloalkane oxidation, because it is a radical clock substrate that can be converted into a number of different products, whose ratios give information on the oxidation mechanism and the formation of an intermediate substrate radical (R•, compare (iv) Fig. 13.10) [66, 68]. With AaeUPO, the experiment yielded exo-2-norcarenol as a major product and five other products in smaller amounts including the rearrangement product 4-(hydroxymethyl)cyclohexane (Fig. 13.14). All these products have previously also been described for norcarane oxidation by P450s, which clearly points to an H-abstraction/oxygen rebound mechanism of oxygenation [91]. On the basis of these results, calculations revealed a lifetime of 9.4 ps for the substrate radical and an oxygen rebound rate of 2 × 1011 s−1 for the rebound reaction, which indicates a ~6-fold faster rebound reaction compared to similar functional P450s [68].

Norcarane oxidation by AaeUPO. (1) norcarane radical, (2) endo-2-norcaranol, (3) exo-2-norcaranol (major product), (4) methylcyclohexene radical, (5) cyclohexenyl methanol, (6) mesomeric forms of norcarane cation, (7) 3-cycloheptene-1-ol, (8) endo-3-norcaranol, (9) exo-3-norcaranol (Modified after [68])
Oxidation of the methyl group of toluene was one of the first UPO reactions studied in detail [32]. The molecule can be hydroxylated at both the methyl group and the aromatic ring, resulting in the formation of mixtures of benzyl alcohol, benzaldehyde and benzoic acid as well as p- and o-cresol, and methylhydroquinone. When 4-nitrotoluene was used as a substrate, the methyl group was oxidized in a similar way but ring hydroxylation was negligible [92]. In the case of toluene, the ratio of alkyl hydroxylation vs. aromatic oxygenation was 2:1 for AaeUPO and 26:1 for MroUPO [46]. Interestingly, the aromatic ring is no longer attacked by AaeUPO when alkyl benzenes with longer side chains are used as substrates [89, 93]. Thus, ethyl- and propylbenzene were hydroxylated exclusively at the benzylic carbon (Cα) to form (R)-1-phenylethanol and (R)-1-phenylpropanol, respectively. The reactions were highly enantioselective with an enantiomeric excess of >99 % for the (R)-isomers. With increasing alkyl-chain length (C4–C6), turnovers and ee values decrease along with an increase in the number and amount of by-products (e.g. ketones). The enzymatic preparation of (R)-1-phenylethanol was optimized using a fed-batch reaction design and resulted in a maximum TTN (total turnover number) of 43,000 and a space-time yield of ~60 g per Liter and day. Tetralin (cyclohexylbenzene) can be perceived as a benzene with a cyclic alkyl group and was in fact hydroxylated in a similar manner as ethyl-/propylbenzene with an ee of >99 % for tetralin-(R)-1-ol, which was much better compared to its aliphatic counterpart butylbenzene [89].
A set of ten model compounds, including alkylated benzoic acids, cycloaliphatic acids and a branched fatty acid, with ascending C-H bonding dissociation energies (BDEs; 83–100 kcal mol−1), was tested regarding oxidation by AaeUPO [66]. The study used a stopped-flow technique to generate AaeUPO compound I, and its activity was in turn studied kinetically. The plot of second-order rate constants for C-H hydroxylation by AaeUPO compound I vs. the BDE of the model compounds revealed a very distinct, non-linear correlation and a calculated upper limit of “hydroxylizability” of about 102 kcal mol−1, which corresponds to the C-H BDE of ethane. In fact, results of recent experiments have indicated that ethane is barely hydroxylated by AaeUPO whereas methane with a BDE of 107 kcal mol−1 is definitely not a substrate under normal conditions (Wang and Peter, unpublished results). Whether methane can be hydroxylated by UPO at elevated pressure is currently under investigation.
13.4.4.2 Alkenes and Aromatics
AaeUPO oxidizes various alkenes and alkenyls, in which both epoxidation and hydroxylation of the double bond’s adjacent carbons (allylic hydroxylation) can occur. In a recent study, 20 alkenes, among them propene and linear 1-alkenes up to C8, branched alkenes such as 2,3-dimethyl-2-butene, cyclohexene, butadiene and the two enantiomers of limonene, were oxidized by AaeUPO in that manner [38]. Considerable differences in conversion rates and product patterns were observed, depending on the size of the molecule and position of the double bond. Surprisingly, branched and cyclic alkenes were much better substrates than linear alkenes. Propene, branched butenes, buta-1,3-diene and cis- and trans-butene were epoxidized exclusively, while 1-alkenes (C4–C8) and cyclohexene were both hydroxylated and epoxidized, i.e. mixtures of 1-alken-3-ols and 1-alkenes epoxides (=1-alkyloxiranes = 1,2-epoxyalkanes) and 2-cyclohexen-1-ol and cyclohexene epoxide, respectively, were formed. However, no products were formed with both oxyfunctionalizations.
The oxidation of cis-2-butene and trans-2-butene yielded differing amounts of epoxidation products. When cis-2-butene was epoxidized to cis-2-butene epoxide, more than twice as much product was formed than during the oxidation of trans-2-butene to trans-2-butene epoxide under otherwise identical conditions. Better conversion of the cis-form than the trans-form of an alkenyl was also observed for the AaeUPO-catalyzed oxidation of styrene derivatives [89]. Thus, trans-β-Methylstyrene was oxidized to some extent but only at the terminal carbon. In contrast, cis-β-methylstyrene was almost completely oxidized to (1R,2S)-cis-β-methylstyrene epoxide (>99 % ee) as the sole product. Considering these results, it can be concluded that cis-trans isomerism strongly influences positioning of alkenes in the active site of UPOs and hence their oxidizibility.
Complex product patterns were observed as a result of the oxidation of the monoterpene limonene (1-isopropenyl-4-methyl-cyclohexane). Both enantiomers, (R)-(+)- and (S)-(−)-limonene, were rapidly oxidized by AaeUPO, which led to the formation of mixtures of alcohol (carveol) and epoxide products (1,2- and 8,9-limonene epoxides) with different ratios of enantiomers and diastereomers (Fig. 13.15) [38, 81].

Aromatic oxygenation was initially studied with naphthalene and toluene as substrates (see also Sect. 13.4.4.1) [32]. Naphthalene is regioselectively epoxidized by different UPOs to naphthalene 1,2-oxide that hydrolyzes in the presence of protons (pH <7.5) to 1- and 2-naphthol. The ratio of both naphthols varied, which depended on the pH, on the manner of H2O2 supply and surprisingly also, on the UPO used, indicating the possibility that the active sites somehow affect epoxide hydrolysis [33, 34, 43, 46, 55]. Other polycyclic aromatic hydrocarbons (PAHs) such as methylnaphthalenes, fluorene, anthracene, phenanthrene, pyrene and dibenzofuran were also subject to UPO-catalyzed oxygenation leading to mixtures of mono- and polyhydroxylated products [45]. Differences were observed in the efficiency of oxidation of aromatic vs. non-aromatic carbons. While AaeUPO clearly favors the attack on aromatic rings, CraUPO and MroUPO oxidize preferably alkyl side chains or methylene groups in non-aromatic rings (e.g. C9 in fluorene). In the case of AaeUPO, the upper limit of molecule size is reached with benzo[a]pyrene that is oxidized to a minor degree and only in the presence of high amounts of co-solvents (e.g. acetonitrile).
Eventually, benzene was also oxidized by AaeUPO, despite experimental difficulties due to its high volatility and low reactivity. The reaction proceeds via an initial epoxide intermediate that re-aromatizes in aqueous solution to form phenol. Identity of this intermediate as benzene epoxide (that is in equilibrium with oxepine) was proven by a freshly prepared authentic standard [88]. A second and third oxygenation was also observed and resulted in the formation of hydroquinone, catechol and 1,2,4-trihydroxybenzene.
Phenolic products formed during aromatic peroxygenations can be substrates of a subsequent peroxidative activity of UPOs (one-electron oxidations). The phenoxyl radicals formed tend to couple and polymerize. This can be prevented by adding radical scavengers such as ascorbic acid to the reaction mixture. Ascorbic acid reacts with phenoxyl radicals to yield ascorbyl radicals that in turn can disproportionate to dehydroascorbic acid and ascorbic acid. In other words, the phenoxyl radical abstracts one electron from ascorbic acid followed by rapid proton rebound that again produces the phenol (Fig. 13.16).

Re-reduction of phenoxyl radicals by ascorbic acid during UPO-catalyzed oxygenations yielding phenolic products. (1) phenolic substrate, (2) phenoxyl radical, (3) ascorbic acid (at pH 7), (4) ascorbyl radical, (5) dehydroascorbic acid
The re-reduction of phenoxyl radicals is of particular relevance when polyphenolic substrates such as flavonoids are oxygenated. Thus, in the presence of ascorbic acid, different flavones, flavonols, flavanones and isoflavones can be hydroxylated by AaeUPO, preferably at the C6 position. As an example, Fig. 13.17 shows the hydroxylation of quercetin, a pentahydroxyflavonol widely distributed in plants [94]. The reaction can proceed via a very unstable epoxide intermediate (7-oxybicyclo[4.1.0]hepta-2,4-diene-2,6,-diol) (Fig. 13.18) and yields quercetagetin (6-hydroxyquercetin) as the sole product. Initial epoxide formation could be demonstrated during the oxidation of unsubstituted flavone to 6-hydroxyflavone [94].

Regioselective oxidation of quercitin by AaeUPO in the presence of ascorbic acid via a hypothetical epoxide intermediate into 6-hydroxyquercitin (Based on [94])

Cleavage of methyl p-nitrobenzyl ether by AaeUPO in the presence of H2 18O2 resulting in the formation of 18O-labeled p-nitrobenzaldehyde (Based on [86])
In contrast to propylbenzene that is hydroxylated at the benzylic carbon (see above and [89]), 2-phenoxypropionic acid is not attacked in the side chain but exclusively in the para-position on the aromatic ring to yield 2-(4-hydroxyphenoxy)propionic acid. The latter compound is a herbicide precursor and is only formed in appreciable amounts in the presence of ascorbic acid. Chiral analyses after AaeUPO-catalyzed oxidation of racemic 2-phenoxypropionic acid revealed that both enantiomers were hydroxylated, but that the (R)-enantiomer was clearly the preferred substrate [95]. This interesting finding shows that the spatial orientation of polar side chains can influence the regioselectivity of UPOs and the extent of aromatic ring oxidation.
13.4.4.3 Dealkylation
UPOs catalyze O- and N-dealkylations of diverse ethers and secondary/tertiary amines, respectively. The mechanism involves, in both cases, initial hydroxylation of one of the heteroatoms’ adjacent carbons (e.g. methyl or methylene groups) giving rise to unstable intermediates (hemiacetals, hemiaminals: –HCOH–O–CH2– or –HCOH–NH-CH2–, respectively), which spontaneously cleave under release of water. Thus, hemiacetals yield alcohols/phenols and aldehydes, and hemiaminals generate primary or secondary amines and aldehydes. In both cases, the aldehydes indicative for this mechanism can be detected by their corresponding 2,4-dinitrohydrazone adducts [86, 96].
Ether cleavage occurred between aromatic and aliphatic molecules in alkyl aryl ethers (e.g. 1,4-dimethoxybenzene, 1,4-dipropoxybenzene) and in alicyclic and aliphatic ethers (e.g. tertrahydrofuran, dioxane, diisopropyl ether, methyl t-butyl ether) [86]. The incorporation of peroxide-borne oxygen into the carbonyl fission product was demonstrated using methyl p-nitrobenzyl ether [97, 98]Footnote 8 and H2 18O2 as substrate and cosubstrate, respectively (Fig. 13.18) [86].
As in the case of symmetrically deuterated n-hexane (hydroxylation of n-hexane-1,1,1,2,2,3,3-D7 to 3-hexanol-D7 and 3-hexanol-D6) [68], a strong intramolecular isotope effect [(kH/kD)obs >10] was observed during the O-demethylation of 1-methoxy-4-trideuteromethoxybenzene, indicating in both cases an H-abstraction/oxygen rebound mechanism for oxygen insertion (compare also Fig. 13.10) [86].
Substantial N-dealkylation (~60 %) was observed during N-methylaniline oxidation by AaeUPO along with ring hydroxylation, yielding phenolic products [79]. Other examples of N-dealkylated substrates are found among pharmaceuticals such as lidocaine, tamoxifen, methamphetamine and sildenafil [48]. With the two latter drugs (“Crystal meth” and Viagra), the N-demethylated metabolites, amphetamine and N-desmethyl sildenafil, respectively, formed in the human body by hepatic P450s (CYP2D6), are the actual effective ingredients [99, 100].
13.4.4.4 Additional Reactions and Scope of UPO Oxidations
UPOs are also capable of transferring oxygen to organic heteroatoms such as sulfur and nitrogen. For example, the heterocycle dibenzothiophene is oxidized at the sulfur atom to form the corresponding sulfoxide and sulfone [44]. Differences were observed in the product pattern between AaeUPO and CraUPO. While the former enzyme preferably hydroxylated the benzene rings of dibenzothiophene, the latter preferred the heterocyclic sulfur substrate [45]. In a similar reaction, UPO enantioselectively oxidized the side chain of thioanisole into the corresponding (R)-sulfoxide with high efficiency [101]. Pyridine and halo-, nitro- and cyanopyridines are oxidized by AaeUPO exclusively at the nitrogen atom to form the respective pyridine N-oxides. In contrast, methylated pyridines were oxygenated both at the methyl group and at the ring nitrogen [37].
In addition to epoxidation, the formation of naphthalene hydrates (i.e. 1- and 2-hydroxy-1,2-dihydronaphthalene) displays a side activity in the enzymatic transformation of 1,2-dihydronaphthalene by UPOs and accounts for up to 20 % of overall turnover. These arene hydrates decay into naphthalene via spontaneous aromatization. This reaction sequence represents a simple pathway for the selective synthesis of aromatic hydrocarbons via arene hydrates of conjugated cyclic dienes or cycloalkenyl benzenes [102].
AaeUPO shows strong bromide oxidation but, in contrast to CPO, only a very low chloride oxidation, even though (according to studies of compound I) its redox potential is higher than that of CPO [67]. The oxidation of halides (X−) is actually also an oxygen transfer reaction yielding reactive hypohalites (OX−) that in turn can halogenate organic substrates such as phenols [32]. In contrast to AaeUPO, MroUPO has almost no bromide oxidizing activity, indicating that not all peroxygenases have specific halide binding sites [46].
Halogens bound to a carbon atom undergoing hydroxylation are released as the corresponding halides, because the first intermediate (geminal halohydrin) is unstable. An example is the oxidation of chloromethylbenzene (benzylchloride) by AaeUPO that yields benzaldehyde and chloride [79]. The analogous reaction has been observed for benzylfluoride and studied with respect to cryptic stereoselectivity. It emerged from this study that AaeUPO displays a modest stereospecificity for benzylic pro-(R) C-H abstraction, although the fluorine atom displays a much-reduced steric influence relative to the methyl group when ethylbenzene is used as a substrate (Keddie and Kluge 2013, unpublished results).
The promiscuity of peroxygenases in oxygen atom transfer reactions becomes evident when the oxidation of pharmaceuticals and drugs is examined. All reactions mentioned previously can occur and, therefore, the enzymes catalyze, aromatic and aliphatic peroxygenations, O- and N-dealkylations and even cleavage of ester bonds, depending on the drug used. Altogether, more than 60 different pharmaceuticals and a number of illicit drugs have been shown to undergo oxidative modification by UPOs. Examples for the former agents are the painkillers diclofenac (phenyl hydroxylation) and ibuprofen (isopropyl hydroxylation), the antitussive dextromethorphan (O-demethylation), the β-blocker propranolol (naphthyl hydroxylation), the K+-channel blocker tolbutamide (benzylic hydroxylation), the anti-inflammatory aminophenazone (N,N-desmethylation) and the antiviral drug osaltamivir [47, 48]. Osaltamivir is a particularly interesting example, because this ethyl ester is exclusively cleaved by CraUPO [48]. The reaction is a special case of O-dealkylation and leads to the formation of acetaldehyde and osaltamivir carboxylate (Fig. 13.19). Among the drugs (of abuse) that are oxidized by UPOs are MDMA (“Ecstasy”, demethylenation), LSD (aromatic hydroxylation), THC (methylcyclohexenyl hydroxylation) and cocaine and codeine (N-desmethylation). UPOs have also successfully been used to prepare specifically labeled human drug metabolites and drug-drug interaction probes by using deuterated substrates as starting materials [96].

Cleavage of the antiviral drug osaltamivir to the respective carboxylate and acetaldehyde by CraUPO
A very recent study on the UPO-catalyzed transformation of 13 steroids revealed considerable differences between the three model UPOs. Whereas AaeUPO and CraUPO did not attack any steroid structure, MroUPO oxidized ten of the steroids by 50–100 %. In addition to hydroxylation products, there are mass-spectral indications that, in some cases (e.g. cortisone), the side chain can be removed by C-C bond cleavage ([47] and unpublished results). Whether similar complex reaction sequences are responsible for this cleavage, as in the case of P450s (CYP17A1) [103], is still under investigation.
As already indicated above, there are limitations in the performance of the currently known UPOs. In summary, the following structural characteristics prevent or impede an attack by UPOs: (1) molecular size (e.g. perylene, polyethylene glycol ≥ PEG7); (2) polarity of the substrate (e.g. rutin); (3) abstractability of hydrogen (e.g. biphenyl ether); and specific (still not understood) characteristics of the substrate, as in the case of coumarin.
13.4.4.5 Kinetic Data and Catalytic Performance
A summary of kinetic data of AaeUPO for a representative number of substrates and reaction types is given in Table 13.3. More information can be retrieved in the Handbook of Enzymes [104]. Most of the values are apparent, i.e. they were obtained by varying the concentration of one substrate while keeping the concentration of the second substrate (in most cases 1–2 mM H2O2) constant [37]. More precise bisubstrate kinetics, which facilitate steady-state conditions and circumvent interfering catalase activity by varying the concentration of both substrate and co-substrate (H2O2), have been reported for the cleavage of methyl 3,4-dimethoxybenzyl ether and the demethylenation of 5-nitro-1,3-benzodioxole (compare Fig. 13.11). The results obtained for these assay substrates are consistent with a ping-pong mechanism that is also characteristic for one-electron oxidations catalyzed by heme peroxidases [47, 69, 86, 105].
Catalytic efficiencies (kcat /Km), Michaelis-Menten constants (Km) and turnover numbers (kcat) of UPOs vary in a broad range (103–106 M−1 s−1, 101–104 μM, and 10−1–103 s−1, respectively). Most substrates, however, are oxidized with a catalytic efficiency of ~104–105 M−1 s−1, which again fits classical peroxidases rather than P450 monooxygenases. Compared to the latter, the turnover numbers of UPOs are generally higher (~10- to 1,000-times) whereas the substrate affinities are several times lower (Table 13.3).
The true peroxygenase nature of UPOs has experimentally been verified in various experiments by the incorporation of 18O from H2 18O2 into chemically diverse substrate molecules. The following reactions are just a few mentioned in this context: (1) toluene to benzyl alcohol, (2) benzaldehyde to benzoic acid [92], (3) naphthalene to naphthalene oxide and naphthols [34], (4) benzene to phenol [88], (5) pyridine to pyridine N-oxide [37] and (6) cyclohexane to cyclohexanol [90].
Ideally, the ratio between oxygenated product and peroxide consumed should be 1:1. In fact, there are examples where this ratio has been reached (e.g. tetrahydrofuran and methyl 3,4-dimethoxybenzyl ether cleavage) [86]. On the other hand, the ratio can be altered to the disadvantage of the substrate to be oxidized and much more peroxide is consumed than is actually necessary for peroxygenation. Then, the catalase activity of AaeUPO takes effect and consumes a substantial part of the peroxide without “productive” oxygen atom transfer (Fig. 13.20) [31, 106]. The reason for this activity may be attributed to improper binding of the substrate in the active site, thermodynamic/kinetic obstacles or by competing one-electron oxidations.

Catalase activity of AaeUPO. The reaction solution contained phosphate buffer (10 mM, pH 7), AaeUPO (0.2 μM) and H2O2 (2 mM). The graphs show the decrease in absorbance over time caused by H2O2 decay. Spectral time scans (every 6 s; left), decrease at 240 nm (right)
In summary, regarding their key oxygenating activities (oxygen transfer potential), the three characterized model UPOs and CPO can be grouped as follows [43, 46]: (1) aromatic oxygenation (AaeUPO > CraUPO > MroUPO; CPO does not oxygenate aromatics); (2) alkane/alkyl hydroxylation (MroUPO > AaeUPO ~ CraUPO >> CPO); (3) alkene/alkenyl epoxidation (AaeUPO > CraUPO ~ MroUPO > CPO); and (4) halide oxidation (CPO > AaeUPO > CraUPO ~ MroUPO).
13.5 Conclusions
M. J. Coon has called P450 enzymes “nature’s most versatile biological catalysts” in his excellent review from 2005 [107]. We do not wish to call this statement into question, as a number of P450 reactions such as the oxidation of coumarin [108], terminal alkane hydroxylation [109] or the aromatase reaction [110] have not yet been shown to be catalyzed by UPOs. However, fungal peroxygenases can at least approach the catalytic versatility of P450s and suitably supplement them in the field of biotechnology in the near future. Some applications of UPOs that are currently under development are biosensors for aromatic compounds [111, 112] as well as new procedures for the synthesis of pesticide precursors [113], drug metabolites [48, 95, 96], chiral alcohols [89] and even bulk chemicals [90].
Notes
- 1.
EC 1.11.2 With H2O2 as acceptor, one oxygen atom is incorporated into the product.
- 2.
- 3.
- 4.
Because of the discovery of many more unspecific peroxygenases, they should be systematically abbreviated by the capital letter of the genus plus the first and second letter of the epitheton and the acronym UPO: for example, AaeUPO = unspecific peroxygenase of Agrocybe aegerita.
- 5.
veratryl alcohol units (compare Sect. 13.4.3)
- 6.
- 7.
Note that in many, especially older publications on heme peroxidases, compound II is described as a (deprotonated) oxo-ferryl complex with a double bond between iron and oxygen (FeIV=O) corresponding to (v) in Fig. 13.10. In reality, both ferryl species (FeIV=O and FeIV-OH) of UPO compound II may be present as shown for CPO.
- 8.
Usually, oxygen in aldehyde functionalities rapidly exchanges in water via the corresponding aldehyde hydrates, which prevents the verification of oxygen insertion, but aromatic nitro groups as in p-nitrobenzaldehyde slow down the exchange.
References
Ishimaru A, Yamazaki I (1977) Hydroperoxide-dependent hydroxylation involving “H2O2-reducible hemoprotein” in microsomes of pea seeds. A new type enzyme acting on hydroperoxide and a physiological role of seed lipoxygenase. J Biol Chem 252:6118–6124
Hanano A, Burcklen M, Flenet M, Ivancich A, Louwagie M, Garin J, Blee E (2006) Plant seed peroxygenase is an original heme-oxygenase with an EF-hand calcium binding motif. J Biol Chem 281:33140–33151
Lequeu J, Fauconnier ML, Chammai A, Bronner R, Blee E (2003) Formation of plant cuticle: evidence for the occurrence of the peroxygenase pathway. Plant J 36:155–164
Salazar O, Cirino PC, Arnold FH (2003) Thermostabilization of a cytochrome P450 peroxygenase. ChemBioChem 4:891–893
Estabrook RW, Martin-Wixtrom C, Saeki Y, Renneberg R, Hildebrandt A, Werringloer J (1984) The peroxidatic function of liver microsomal cytochrome P-450: comparison of hydrogen peroxide and NADPH-catalysed N-demethylation reactions. Xenobiotica 14:87–104
McCallum GP, Weedon AC, Krug P, Bend JR (1996) Microsomal cytochrome P450 peroxygenase metabolism of arachidonic acid in guinea pig liver. J Pharm Exp Ther 278:1188–1194
Coon MJ, Vaz AD, Bestervelt LL (1996) Cytochrome P450 2: peroxidative reactions of diversozymes. FASEB J 10:428–434
Prasad S, Mitra S (2004) Substrate modulates compound I formation in peroxide shunt pathway of Pseudomonas putida cytochrome P450(cam). Biochem Biophys Res Commun 314:610–614
Nordblom GD, White RE, Coon MJ (1976) Studies on hydroperoxide-dependent substrate hydroxylation by purified liver microsomal cytochrome P-450. Arch Biochem Biophys 175:524–533
Sakaki T (2012) Practical application of cytochrome P450. Biol Pharm Bull 35:844–849
Zenser TV, Lakshmi VM, Hsu FF, Davis BB (1999) Peroxygenase metabolism of N-acetylbenzidine by prostaglandin H synthase. Formation of an N-hydroxylamine. J Biol Chem 274:14850–14856
Kuo HH, Mauk AG (2012) Indole peroxygenase activity of indoleamine 2,3-dioxygenase. Proc Natl Acad Sci U S A 109:13966–13971
Yamazaki S, Morioka C, Itoh S (2004) Kinetic evaluation of catalase and peroxygenase activities of tyrosinase. Biochemistry 43:11546–11553
Matsunaga I, Yamada M, Kusunose E, Miki T, Ichihara K (1998) Further characterization of hydrogen peroxide-dependent fatty acid α-hydroxylase from Sphingomonas paucimobilis. J Biochem 124:105–110
Lee DS, Yamada A, Sugimoto H, Matsunaga I, Ogura H, Ichihara K, Adachi S, Park SY, Shiro Y (2003) Substrate recognition and molecular mechanism of fatty acid hydroxylation by cytochrome P450 from Bacillus subtilis. Crystallographic, spectroscopic, and mutational studies. J Biol Chem 278:9761–9767
Matsunaga I, Sumimoto T, Ueda A, Kusunose E, Ichihara K (2000) Fatty acid-specific, regiospecific, and stereospecific hydroxylation by cytochrome P450 (CYP152B1) from Sphingomonas paucimobilis: substrate structure required for α-hydroxylation. Lipids 35:365–371
Shoji O, Wiese C, Fujishiro T, Shirataki C, Wunsch B, Watanabe Y (2010) Aromatic C-H bond hydroxylation by P450 peroxygenases: a facile colorimetric assay for monooxygenation activities of enzymes based on Russig’s blue formation. J Biol Inorg Chem 15:1109–1115
Fujishiro T, Shoji O, Kawakami N, Watanabe T, Sugimoto H, Shiro Y, Watanabe Y (2012) Chiral-substrate-assisted stereoselective epoxidation catalyzed by H2O2-dependent cytochrome P450SPα. Chem Asian J 7:2286–2293
Shoji O, Kunimatsu T, Kawakami N, Watanabe Y (2013) Highly selective hydroxylation of benzene to phenol by wild-type cytochrome P450BM3 assisted by decoy molecules. Angew Chem Int Ed 52:6606–6610
Gaut JP, Yeh GC, Tran HD, Byun J, Henderson JP, Richter GM, Brennan ML, Lusis AJ, Belaaouaj A, Hotchkiss RS, Heinecke JW (2001) Neutrophils employ the myeloperoxidase system to generate antimicrobial brominating and chlorinating oxidants during sepsis. Proc Natl Acad Sci U S A 98:11961–11966
Tuynman A, Spelberg JL, Kooter IM, Schoemaker HE, Wever R (2000) Enantioselective epoxidation and carbon-carbon bond cleavage catalyzed by Coprinus cinereus peroxidase and myeloperoxidase. J Biol Chem 275:3025–3030
Geigert J, Lee TD, Dalietos DJ, Hirano DS, Neidleman SL (1986) Epoxidation of alkenes by chloroperoxidase catalysis. Biochem Biophys Res Commun 136:778–782
Miller VP, Tschirretguth RA, Ortiz de Montellano P (1995) Chloroperoxidase-catalyzed benzylic hydroxylation. Arch Biochem Biophys 319:333–340
Colonna S, Gaggero N, Casella L, Carrea G, Pasta P (1992) Chloroperoxidase and hydrogen peroxide: an efficient system for enzymatic enantioselective sulfoxidations. Tetrahedron Asymmetry 3:95–106
Manoj KM, Hager LP (2001) Utilization of peroxide and its relevance in oxygen insertion reactions catalyzed by chloroperoxidase. Biochim Biophys Acta 1547:408–417
Zhang R, He Q, Chatfield D, Wang X (2013) Paramagnetic nuclear magnetic resonance relaxation and molecular mechanics studies of the chloroperoxidase-indole complex: insights into the mechanism of chloroperoxidase-catalyzed regioselective oxidation of indole. Biochemistry 52:3688–3701
Hofrichter M, Ullrich R (2006) Heme-thiolate haloperoxidases: versatile biocatalysts with biotechnological and environmental significance. Appl Microbiol Biotechnol 71:276–288
Stamets P, Chilton JS (1983) The mushroom cultivator: a practical guide to growing mushrooms at home. Agarikon Press, Olympia
IndexFungorum (2013) www.indexfungorum.org
Manzi P, Marconi S, Aguzzi A, Pizzoferrato L (2004) Commercial mushrooms: nutritional quality and effect of cooking. Food Chem 84:201–206
Ullrich R, Nüske J, Scheibner K, Spantzel J, Hofrichter M (2004) Novel haloperoxidase from the agaric basidiomycete Agrocybe aegerita oxidizes aryl alcohols and aldehydes. Appl Environ Microbiol 70:4575–4581
Ullrich R, Hofrichter M (2005) The haloperoxidase of the agaric fungus Agrocybe aegerita hydroxylates toluene and naphthalene. FEBS Lett 579:6247–6250
Kluge MG, Ullrich R, Scheibner K, Hofrichter M (2007) Spectrophotometric assay for detection of aromatic hydroxylation catalyzed by fungal haloperoxidase-peroxygenase. Appl Microbiol Biotechnol 75:1473–1478
Kluge M, Ullrich R, Dolge C, Scheibner K, Hofrichter M (2009) Hydroxylation of naphthalene by aromatic peroxygenase from Agrocybe aegerita proceeds via oxygen transfer from H2O2 and intermediary epoxidation. Appl Microbiol Biotechnol 81:1071–1076
Ullrich R, Hofrichter M (2007) Enzymatic hydroxylation of aromatic compounds. Cell Mol Life Sci 64:271–293
Hofrichter M, Ullrich R (2010) New trends in fungal biooxidation. In: Hofrichter M (ed) Industrial applications, 2nd edn. Springer-Verlag, Berlin, pp 425–449
Ullrich R, Dolge C, Kluge M, Hofrichter M (2008) Pyridine as novel substrate for regioselective oxygenation with aromatic peroxygenase from Agrocybe aegerita. FEBS Lett 582:4100–4106
Peter S, Kinne M, Ullrich R, Kayser G, Hofrichter M (2013) Epoxidation of linear, branched and cyclic alkenes catalyzed by unspecific peroxygenase. Enzyme Microb Technol 52:370–376
Ruiz-Dueñas FJ, Martínez AT (2010) Structural and functional features of peroxidases with a potential as industrial biocatalysts. In: Torres E, Ayala M (eds) Biocatalysis based on heme peroxidases as potential industrial biocatalysts, 1st edn. Springer-Verlag, Berlin, pp 37–59
Hofrichter M, Ullrich R, Pecyna MJ, Liers C, Lundell T (2010) New and classic families of secreted fungal heme peroxidases. Appl Microbiol Biotechnol 87:871–897
Gutiérrez A, Babot ED, Ullrich R, Hofrichter M, Martínez AT, del Río JC (2011) Regioselective oxygenation of fatty acids, fatty alcohols and other aliphatic compounds by a basidiomycete heme-thiolate peroxidase. Arch Biochem Biophys 514:33–43
Moncalvo J-M, Vilgalys R, Redhead SA, Johnson JE, James TY, Catherine Aime M, Hofstetter V, Verduin SJW, Larsson E, Baroni TJ, Greg Thorn R et al (2002) One hundred and seventeen clades of euagarics. Mol Phyl Evol 23:357–400
Anh DH, Ullrich R, Benndorf D, Svatos A, Muck A, Hofrichter M (2007) The coprophilous mushroom Coprinus radians secretes a haloperoxidase that catalyzes aromatic peroxygenation. Appl Environ Microbiol 73:5477–5485
Aranda E, Kinne M, Kluge M, Ullrich R, Hofrichter M (2009) Conversion of dibenzothiophene by the mushrooms Agrocybe aegerita and Coprinellus radians and their extracellular peroxygenases. Appl Microbiol Biotechnol 82:1057–1066
Aranda E, Ullrich R, Hofrichter M (2009) Conversion of polycyclic aromatic hydrocarbons, methyl naphthalenes and dibenzofuran by two fungal peroxygenases. Biodegradation 21:267–281
Gröbe G, Ullrich R, Pecyna MJ, Kapturska D, Friedrich S, Hofrichter M, Scheibner K (2011) High-yield production of aromatic peroxygenase by the agaric fungus Marasmius rotula. AMB Express 1:31
Poraj-Kobielska M (2013) Conversion of pharmaceuticals and other drugs by fungal peroxygenases. Ph.D. Thesis, TU Dresden; http://www.ihi-zittau.de/de/dnl/diss_poraj-kobielska_qucosa.4268.pdf
Poraj-Kobielska M, Kinne M, Ullrich R, Scheibner K, Kayser G, Hammel KE, Hofrichter M (2011) Preparation of human drug metabolites using fungal peroxygenases. Biochem Pharmacol 82:789–796
Babot ED, del Río JC, Kalum L, Martínez AT, Gutiérrez A (2013) Oxyfunctionalization of aliphatic compounds by a recombinant peroxygenase from Coprinopsis cinerea. Biotechnol Bioeng 110:2323–2332
Pecyna MJ, Ullrich R, Bittner B, Clemens A, Scheibner K, Schubert R, Hofrichter M (2009) Molecular characterization of aromatic peroxygenase from Agrocybe aegerita. Appl Microbiol Biotechnol 84:885–897
Nuell MJ, Fang GH, Axley MJ, Kenigsberg P, Hager LP (1988) Isolation and nucleotide sequence of the chloroperoxidase gene from Caldariomyces fumago. J Bacteriol 170:1007–1011
Richards TA, Soanes DM, Jones MD, Vasieva O, Leonard G, Paszkiewicz K, Foster PG, Hall N, Talbot NJ (2011) Horizontal gene transfer facilitated the evolution of plant parasitic mechanisms in the oomycetes. Proc Natl Acad Sci U S A 108:15258–15263
Kellner H, Luis P, Pecyna MJ, Barbi F, Kapturska D, Krüger D, Zak DR, Marmeisse R, Vandenbol M, Hofrichter M (2014) Widespread occurrence of expressed fungal secretory peroxidases in forest soils. PLoS One 9(4):e95557. doi:10.1371/journal.pone.0095557
Morin E, Kohler A, Baker AR, Foulongne-Oriol M, Lombard V, Nagy LG, Ohm RA, Patyshakuliyeva A, Brun A, Aerts AL, Bailey AM et al (2012) Genome sequence of the button mushroom Agaricus bisporus reveals mechanisms governing adaptation to a humic-rich ecological niche. Proc Natl Acad Sci U S A 109:17501–17506
Ullrich R, Liers C, Schimpke S, Hofrichter M (2009) Purification of homogeneous forms of fungal peroxygenase. Biotechnol J 4:1619–1626
Kinne M, Poraj-Kobielska M, Ullrich R, Nousiainen P, Sipilä J, Scheibner K, Hammel KE, Hofrichter M (2011) Oxidative cleavage of non-phenolic β-O-4 lignin model dimers by an extracellular aromatic peroxygenase. Holzforschung 65:673–679
Hatakka A, Lundell T, Hofrichter M, Maijala P (2003) Manganese peroxidase and its role in the degradation of wood lignin. In: Mansfield SD, Saddler JN (eds) Applications of enzymes to lignocellulosics, ACS Symposium series, 855th edn. American Chemical Society, Washington DC, pp 230–243
Liers C, Arnstadt T, Ullrich R, Hofrichter M (2011) Patterns of lignin degradation and oxidative enzyme secretion by different wood- and litter-colonizing basidiomycetes and ascomycetes grown on beech-wood. FEMS Microbiol Ecol 78:91–102
Piontek K, Strittmatter E, Ullrich R, Gröbe G, Pecyna MJ, Kluge M, Scheibner K, Hofrichter M, Plattner DA (2013) Structural basis of substrate conversion in a new aromatic peroxygenase: cytochrome P450 functionality with benefits. J Biol Chem 288:34767–34776
Anh DH (2008) Novel extracellular haloperoxidase-peroxygenases from the coprophilous fungi Coprinus radians and Coprinus verticillatus: production, purification and biochemical characterization. Ph.D. Thesis, International Graduate School of Zittau
Omura T (2005) Heme-thiolate proteins. Biochem Biophys Res Commun 338:404–409
Piontek K, Ullrich R, Liers C, Diederichs K, Plattner DA, Hofrichter M (2010) Crystallization of a 45 kDa peroxygenase/peroxidase from the mushroom Agrocybe aegerita and structure determination by SAD utilizing only the haem iron. Acta Crystallogr Sect F: Struct Biol Cryst Commun 66:693–698
Ullrich V, Kremers P (1977) Multiple forms of cytochrome P450. Arch Toxicol 39:41–50
Guengerich FP (1993) Metabolic reactions: types of reactions of cytochrome P450 enzymes. In: Schenkman J, Greim H (eds) Cytochrome P450, 1st edn. Springer, Berlin, pp 89–103
Meunier B, de Visser SP, Shaik S (2004) Mechanism of oxidation reactions catalyzed by cytochrome P450 enzymes. Chem Rev 104:3947–3980
Wang X, Peter S, Kinne M, Hofrichter M, Groves JT (2012) Detection and kinetic characterization of a highly reactive heme-thiolate peroxygenase compound I. J Am Chem Soc 134:12897–12900
Wang X, Peter S, Ullrich R, Hofrichter M, Groves JT (2013) Driving force for oxygen-atom transfer by heme-thiolate enzymes. Angew Chem Int Ed 52:9238–9241
Peter S, Kinne M, Wang X, Ullrich R, Kayser G, Groves JT, Hofrichter M (2011) Selective hydroxylation of alkanes by an extracellular fungal peroxygenase. FEBS J 278:3667–3675
Dunford HB (1999) Heme peroxidases. Wiley, New York
Ortiz de Montellano PR, De Voss JJ (2005) Substrate oxidation by cytochrome P450 enzymes. In: Ortiz De Montellano PR (ed) Cytochrome P450: structure, mechanism, and biochemistry, 3rd edn. Kluwer Academic/Plenum Publishers, New York, pp 183–245
Manoj KM, Hager LP (2008) Chloroperoxidase, a janus enzyme. Biochemistry 47:2997–3003
Stone KL, Hoffart LM, Behan RK, Krebs C, Green MT (2006) Evidence for two ferryl species in chloroperoxidase compound II. J Am Chem Soc 128:6147–6153
Kühnel K, Derat E, Terner J, Shaik S, Schlichting I (2007) Structure and quantum chemical characterization of chloroperoxidase compound 0, a common reaction intermediate of diverse heme enzymes. Proc Natl Acad Sci U S A 104:99–104
Hersleth HP, Ryde U, Rydberg P, Görbitz CH, Andersson KK (2006) Structures of the high-valent metal-ion haem-oxygen intermediates in peroxidases, oxygenases and catalases. J Inorg Biochem 100:460–476
Guengerich FP (2007) Mechanisms of cytochrome P450 substrate oxidation: MiniReview. J Biochem Mol Toxicol 21:163–168
Ortiz de Montellano PR (2010) Catalytic mechanisms of heme peroxidases. In: Torres E, Ayala M (eds) Biocatalysis based on heme peroxidases−peroxidases as potential industrial biocatalysts, 1st edn. Springer-Verlag, Berlin, pp 80–107
Krest CM, Onderko EL, Yosca TH, Calixto JC, Karp RF, Livada J, Rittle J, Green MT (2013) Reactive intermediates in cytochrome P450 catalysis. J Biol Chem 288:17074–17081
Lewis DFV (2001) Guide to cytochromes P450: structure and function, 2nd edn. Informa Healthcare, London
Kinne M (2010) The extracellular peroxygenase of the agaric fungus Agrocybe aegerita: catalytic properties and physiological background with particular emphasis on ether cleavage. Ph.D. Thesis, International Graduate School of Zittau; http://www.qucosa.de/fileadmin/data/qucosa/documents/6207/Diss_Kinne_final.pdf
Isin EM, Guengerich FP (2008) Substrate binding to cytochromes P450. Anal Bioanal Chem 392:1019–1030
Peter S (2013) Oxyfunctionalization of alkanes, alkenes and alkynes by unspecific peroxygenase (EC 1.11.2.1). Ph.D. Thesis, TU Dresden; http://www.ihi-zittau.de/de/dnl/dissertation_sebastian_peter.4267.pdf
de Visser SP, Ogliaro F, Harris N, Shaik S (2001) Multi-state epoxidation of ethene by cytochrome P450: a quantum chemical study. J Am Chem Soc 123:3037–3047
de Visser SP, Ogliaro F, Shaik S (2001) Stereospecific oxidation by compound I of cytochrome P450 does not proceed in a concerted synchronous manner. Chem Commun 22:2322–2323
Piontek K, Smith AT, Blodig W (2001) Lignin peroxidase structure and function. Biochem Soc Trans 29:111–116
Ruiz-Dueñas FJ, Morales M, Garcia E, Miki Y, Martínez MJ, Martínez AT (2009) Substrate oxidation sites in versatile peroxidase and other basidiomycete peroxidases. J Exp Bot 60:441–452
Kinne M, Poraj-Kobielska M, Ralph SA, Ullrich R, Hofrichter M, Hammel KE (2009) Oxidative cleavage of diverse ethers by an extracellular fungal peroxygenase. J Biol Chem 284:29343–29349
Poraj-Kobielska M, Kinne M, Ullrich R, Scheibner K, Hofrichter M (2012) A spectrophotometric assay for the detection of fungal peroxygenases. Anal Biochem 421:327–329
Karich A, Kluge M, Ullrich R, Hofrichter M (2013) Benzene oxygenation and oxidation by the peroxygenase of Agrocybe aegerita. AMB Express 3:5
Kluge M, Ullrich R, Scheibner K, Hofrichter M (2012) Stereoselective benzylic hydroxylation of alkylbenzenes and epoxidation of styrene derivatives catalyzed by the peroxygenase of Agrocybe aegerita. Green Chem 14:440–446
Peter S, Karich A, Ullrich R, Gröbe G, Scheibner K, Hofrichter M (2013) Enzymatic one-pot conversion of cyclohexane into cyclohexanone: comparison of four fungal peroxygenases. J Mol Catal B Enz. doi: 10.1016/j.molcatb.2013.1009.1016
Auclair K, Hu Z, Little DM, Ortiz De Montellano PR, Groves JT (2002) Revisiting the mechanism of P450 enzymes with the radical clocks norcarane and spiro[2,5]octane. J Am Chem Soc 124:6020–6027
Kinne M, Zeisig C, Ullrich R, Kayser G, Hammel KE, Hofrichter M (2010) Stepwise oxygenations of toluene and 4-nitrotoluene by a fungal peroxygenase. Biochem Biophys Res Commun 397:18–21
Churakova E, Kluge M, Ullrich R, Arends I, Hofrichter M, Hollmann F (2011) Specific photobiocatalytic oxyfunctionalization reactions. Angew Chem Int Ed 50:10716–10719
Barková K, Kinne M, Ullrich R, Hennig L, Fuchs A, Hofrichter M (2011) Regioselective hydroxylation of diverse flavonoids by an aromatic peroxygenase. Tetrahedron 67:4874–4878
Kinne M, Ullrich R, Hammel KE, Scheibner K, Hofrichter M (2008) Regioselective preparation of (R)-2-(4-hydroxyphenoxy)propionic acid with a fungal peroxygenase. Tetrahedron Lett 49:5950–5953
Poraj-Kobielska M, Atzrodt J, Holla W, Sandvoss M, Gröbe G, Scheibner K, Hofrichter M (2013) Preparation of labeled human drug metabolites and drug-drug interaction-probes with fungal peroxygenases. J Label Comp Radiopharm 56:513–519
Tien M, Kirk TK (1984) Lignin-degrading enzyme from Phanerochaete chrysosporium: purification, characterization, and catalytic properties of a unique H2O2-requiring oxygenase. Proc Natl Acad Sci U S A 81:2280–2284
Samuel D, Silver BL (1965) Oxygen isotope exchange reactions of organic compounds. In: Gold V (ed) Advances in physical organic chemistry. Academic Press, New York, pp 123–186
Maurer HH, Kraemer T, Springer D, Staack RF (2004) Chemistry, pharmacology, toxicology, and hepatic metabolism of designer drugs of the amphetamine (ecstasy), piperazine, and pyrrolidinophenone types: a synopsis. Ther Drug Monit 26:127–131
Langtry HD, Markham A (1999) Sildenafil. Drugs 57:967–989
Horn A (2009) The use of a novel peroxidase from the basidiomycete Agrocybe aegerita as an example of enatioselective sulfoxidation (Der Einsatz einer neuartigen Peroxidase des Basidiomyceten Agrocybe aegerita am Beispiel der enatioselektiven Sulfoxidation). Ph.D. Thesis, University of Rostock, Germany
Kluge M, Ullrich R, Scheibner K, Hofrichter M (2013) Formation of naphthalene hydrates in the enzymatic conversion of 1,2-dihydronaphthalene by two fungal peroxygenases and subsequent naphthalene formation. J Mol Catal B Enz. doi: 10.1016/j.molcatb.2013.1008.1017
Akhtar M, Wright JN, Lee-Robichaud P (2011) A review of mechanistic studies on aromatase (CYP19) and 17α-hydroxylase-17,20-lyase (CYP17). J Steroid Biochem Mol Biol 125:2–12
Schomburg D, Schomburg I (2013) Class 1, oxidoreductases, EC 1, vol S8, 2nd edn, Springer handbook of enzymes. Springer, Berlin/New York, pp 504–516
Segel IH (1993) Enzyme kinetics: behavior and analysis of rapid equilibrium and steady-state enzyme systems. Wiley, New York
Ullrich R (2008) Some special reactions of Agrocybe aegerita peroxygenase (AaP). Conference proceedings, Tampere (Finland), p 20
Coon MJ (2005) Cytochrome P450: nature’s most versatile biological catalyst. Annu Rev Pharmacol Toxicol 45:1–25
Pelkonen O, Sotaniemi EA, Ahokas JT (1985) Coumarin 7-hydroxylase activity in human liver microsomes. Properties of the enzyme and interspecies comparisons. Br J Clin Pharmacol 19:59–66
Johnston JB, Ouellet H, Podust LM, Ortiz de Montellano PR (2011) Structural control of cytochrome P450-catalyzed ω-hydroxylation. Arch Biochem Biophys 507:86–94
Yoshio O, Tadayoshi H, Fronckowiak M, Nobutaka Y, Yarborough C (1987) Aromatase. J Steroid Biochem 27:781–789
Yarman A, Peng L, Wu Y, Bandodkar A, Gajovic-Eichelmann N, Wollenberger U, Hofrichter M, Ullrich R, Scheibner K, Scheller F (2011) Can peroxygenase and microperoxidase substitute cytochrome P450 in biosensors. Bioanal Rev 3:67–94
Peng L, Wollenberger U, Kinne M, Hofrichter M, Ullrich R, Scheibner K, Fischer A, Scheller FW (2010) Peroxygenase based sensor for aromatic compounds. Biosens Bioelectron 26:1432–1436
Kinne M, Poraj-Kobielska M, Aranda E, Ullrich R, Hammel KE, Scheibner K, Hofrichter M (2009) Regioselective preparation of 5-hydroxypropranolol and 4′-hydroxydiclofenac with a fungal peroxygenase. Bioorg Med Chem Lett 19:3085–3087
Sundaramoorthy M, Terner J, Poulos TL (1995) The crystal structure of chloroperoxidase: a heme peroxidase-cytochrome P450 functional hybrid. Structure 3:1367–1377
Strittmatter E, Liers C, Ullrich R, Wachter S, Hofrichter M, Plattner DA, Piontek K (2013) First crystal structure of a fungal high-redox potential dye-decolorizing peroxidase: substrate interaction sites and long-range electron transfer. J Biol Chem 288:4095–4102
Dolge C, Sass A, Kayser G, Ullrich R, Hofrichter M (2011) Exploration of rCciAPO1 from Coprinopsis cinerea: first recombinant aromatic peroxygenase. Conference proceedings, Springer: Karlsruhe, p 132
Morris DR, Hager LP (1966) Chloroperoxidase. I. Isolation and properties of the crystalline glycoprotein. J Biol Chem 241:1763–1768
Farrell RL, Murtagh KE, Tien M, Mozuch MD, Kirk TK (1989) Physical and enzymatic properties of lignin peroxidase isoenzymes from Phanerochaete chrysosporium. Enzyme Microb Technol 11:322–328
Hollenberg PF, Hager LP (1973) The P-450 nature of the carbon monoxide complex of ferrous chloroperoxidase. J Biol Chem 248:2630–2633
Palcic MM, Rutter R, Araiso T, Hager LP, Dunford HB (1980) Spectrum of chloroperoxidase compound I. Biochem Biophys Res Commun 94:1123–1127
Imai Y, Matsunaga I, Kusunose E, Ichihara K (2000) Unique heme environment at the putative distal region of hydrogen peroxide-dependent fatty acid α-hydroxylase from Sphingomonas paucimobilis (Peroxygenase P450SPα). J Biochem 128:189–194
Koo LS, Tschirret-Guth RA, Straub WE, Moënne-Loccoz P, Loehr TM, Ortiz de Montellano PR (2000) The active site of the thermophilic CYP119 from Sulfolobus solfataricus. J Biol Chem 275:14112–14123
Sheng X, Horner JH, Newcomb M (2008) Spectra and kinetic studies of the compound I derivative of cytochrome P450 119. J Am Chem Soc 130:13310–13320
Baciocchi E, Manduchi L, Lanzalunga O (1999) Prochiral selectivity and deuterium kinetic isotope effect in the oxidation of benzyl alcohol catalyzed by chloroperoxidase. Chem Commun 17:1715–1716
Acknowledgments
We would like to thank K. Barkova, M. G. Kluge, S. Peter, C. Dolge and M. Poraj-Kobielska (TU Dresden-IHI Zittau, Germany) for still unpublished results on the catalytic properties of unspecific peroxygenases and our project partners L. Kalum and H. Lund (Novozymes A/S, Denmark) for enzyme samples as well as useful discussions. We acknowledge fruitful cooperations with the following colleagues: K. Piontek and D. Plattner (University of Freiburg, Germany) in the field of protein crystallography, X. Wang and J. T. Groves (Princeton University, USA) regarding stopped-flow techniques, and J. Atzrodt and W. Holla (Sanofi Frankfurt, Germany) in the field of drug metabolites. UPO work has been financially supported by the European Union (integrated projects Biorenew, Peroxicats and Indox), the Deutsche Bundestiftung Umwelt (DBU; projects AZ 1327 and AZ 13225) and the Bundesministerium für Forschung (BMBF, projects 0313433 and 0315877).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Hofrichter, M., Kellner, H., Pecyna, M.J., Ullrich, R. (2015). Fungal Unspecific Peroxygenases: Heme-Thiolate Proteins That Combine Peroxidase and Cytochrome P450 Properties. In: Hrycay, E., Bandiera, S. (eds) Monooxygenase, Peroxidase and Peroxygenase Properties and Mechanisms of Cytochrome P450. Advances in Experimental Medicine and Biology, vol 851. Springer, Cham. https://doi.org/10.1007/978-3-319-16009-2_13
Download citation
DOI: https://doi.org/10.1007/978-3-319-16009-2_13
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-16008-5
Online ISBN: 978-3-319-16009-2
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)