
Abstract
Several liver and biliary diseases are characterized by oxidative and endoplasmic reticulum stress, both of which can lead to inflammation, disruption of hormone signaling, and cell death. Endoplasmic reticulum stress results from the accumulation of unfolded proteins in the lumen and is linked to the unfolded protein response (UPR), a signaling pathway that reduces global protein synthesis and upregulates folding and degradation machinery. Protein folding in the endoplasmic reticulum lumen can also generate reactive oxygen species and oxidative stress can influence the UPR. In this chapter, the interactions between the endoplasmic reticulum, UPR, and oxidative stress will be discussed with special emphasis placed on these events in the liver.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Key words
- Non-alcoholic fatty liver disease
- Endoplasmic reticulum
- Protein folding
- Chaperones
- Inflammation
- Redox
- Cell stress
- Antioxidant
- Free fatty acids
- Ethanol
- Secretory pathway
- Hepatocytes
- Mitochondria
- Mitofusin
- Obesity
- Alzheimer disease
1 Introduction
Liver and biliary diseases can result from a wide variety of causes, including infectious agents, inherited defects, alcohol, toxins, and environmental insults. Common forms of liver and biliary diseases include viral hepatitis, alcoholic fatty liver disease, non-alcoholic fatty liver disease, and gallstones. Estimates suggest that hepatitis B and C virus are present in ~5 % and ~3 %, respectively, of the world’s population. Hepatocellular carcinoma is among the leading causes of cancer-related deaths and non-alcoholic fatty liver disease may affect 10–20 % of the population, largely due to the current worldwide obesity epidemic. Many liver and biliary diseases are characterized by both oxidative and endoplasmic reticulum stress, both of which can lead to inflammation, cell death, and global organ impairment. This chapter will discuss the endoplasmic reticulum (ER) and oxidative stress in the liver.
2 The Endoplasmic Reticulum
The ER is the largest continuous organelle in a eukaryotic cell and consists of an array of tubules (cisternae) that form a three-dimensional network (reticulum) stretching from the nuclear envelope to the cell surface. The smooth ER produces structural phospholipids and cholesterol, as well as significant amounts of triacylglycerol and cholesterol esters that have non-structural roles [1, 2]. The smooth ER is the main site of cholesterol synthesis, although much of this lipid is transported to other cellular organelles. Thus, the ER membrane is comprised of very low concentrations of cholesterol and complex sphingolipids [1]. It has been suggested that the loose packing of ER membrane lipids may provide an environment conducive to the insertion and transport of newly synthesized lipids and proteins [1]. This specialized lipid environment within the ER may have implications in diseases characterized by abnormal lipid accumulation, such as alcoholic and non-alcoholic fatty liver disease [3].
All eukaryotic cells contain a significant amount of rough ER, the site for protein folding and maturation. Proteins destined for secretion or insertion into membranes require modification, such as glycosylation and disulfide bond formation, which cannot be achieved in the cytosol [4]. The ER lumen provides a specialized environment for protein folding and maturation that is characterized by high concentrations of calcium, a low ratio (1:1–3:1) of reduced glutathione (GSH) to oxidized glutathione (GSSG), and a unique complement of molecular chaperones and folding enzymes [5]. The ER is also equipped with a quality control system that recognizes and degrades improperly folded proteins, termed ER-associated degradation (ERAD). ERAD can target and transport misfolded proteins from the ER lumen to the cytosolic proteasome machinery [6]. The unfolded protein response (UPR) monitors the ability of the ER lumen to match folding and degradation to the rate of entry of newly synthesized proteins and functions to restore ER homeostasis following periods of ER stress (i.e., accumulation of unfolded proteins within the ER lumen).
3 The Unfolded Protein Response
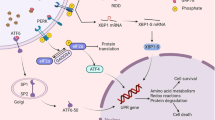
In mammalian cells, activation of the UPR (Fig. 5.1) generally involves three ER-localized proteins: inositol-requiring 1α (IRE1α), double-stranded RNA-dependent protein kinase-like ER kinase (PERK), and activating transcription factor-6 (ATF6) [7]. Each of these transmembrane proteins has an ER-luminal domain to sense unfolded proteins, a transmembrane domain for targeting to the ER membrane, and a cytosolic domain to transmit signals to the transcriptional and/or translational apparatus [8]. It is currently thought that in un-stressed cells all three proteins are maintained in an inactive state via their association with the ER protein chaperone glucose-regulated protein 78/immunoglobulin-heavy-chain-binding protein (GRP78). Subsequent to ER stress, GRP78 is released and sequestered on unfolded proteins, thereby allowing activation of PERK, IRE1α, and ATF6 [9]. PERK activation leads to phosphorylation of the α-subunit of the translation initiation factor eIF2 (p-eIF2α) and subsequent attenuation of translation initiation. Attenuation of translation also leads to selective translation of mRNAs containing open reading frames, such as activating transcription factor-4 (ATF4) [10, 11]. Increased expression of GADD34 (which also contains open reading frames), a member of the growth arrest and DNA damage family of proteins, is involved in dephosphorylation of eIF2α and therefore promotes reversal of translational attenuation [7]. IRE1α activation leads to splicing of X-box-binding protein-1 (XBP1s) mRNA and subsequent transcription of molecular chaperones (e.g., GRP78) and genes involved in ERAD (e.g., ER degradation-enhancing α—like protein (EDEM)) [11]. Activation of ATF6 leads to its release from the ER membrane, processing in the Golgi, and entry into the nucleus. Transcriptional targets of ATF6 include protein chaperones and XBP1 [8]. Thus, activation of the UPR initiates a spectrum of responses that include transient attenuation of global protein synthesis and an increased capacity for protein folding and degradation. This dual response may serve to not only minimize the increase in unfolded proteins in the ER lumen, but also the accumulation of chaperones and may be particularly relevant to cell types that produce a large amount of secreted proteins (e.g., β-cells) [12].

Overview of the mammalian unfolded response. The presence of unfolded proteins in the ER lumen leads to dimerization and autophosphorylation of PERK and IRE1α, and the release and proteolytic cleavage of ATF6 in the Golgi. PERK-mediated phosphorylation of eIF2α leads to transient attenuation of translation, but selective translation of mRNAs containing upstream open reading frames, such as ATF4. Increased transcription and translation of GADD34 subsequently leads to dephosphorylation of eIF2α and resumption of translation. Activation of IRE1α leads to the splicing of XBP1. XBP1s, ATF4, and the cleaved form of ATF6 lead to transcriptional activation of a number of gene targets related to protein folding and ER-associated degradation (see text). Reprinted with permission from ANTIOXIDANT AND REDOX SIGNALING (2011, volume 15, issue 2), published by Mary Ann Liebert, Inc., New Rochelle, NY
3.1 An Expanded View of the UPR
PERK is one of four protein kinases that can phosphorylate eIF2α; the other three are double-stranded RNA-activated protein kinase (PKR) which is activated in response to viral infection, general control non-derepressible 2 kinase (GCN2) which is activated in response to amino acid deprivation, and heme-regulated inhibitor kinase (HRI) which is primarily expressed in reticulocytes and appears to coordinate globin polypeptide synthesis with heme availability [13]. Protein kinase-mediated phosphorylation of eIF2α not only regulates translation, but also the activation of nuclear factor kappa-β (NFĸB), via reduction in the abundance of the NFĸβ inhibitor Iĸβ [11]. PERK can also phosphorylate nuclear erythroid 2 p45-related factor 2 (Nrf2), triggering the dissociation of Nrf2/Keap1 complexes and subsequent nuclear import of Nrf2 [14]. Thus, activation of this branch of the UPR links disruption of ER homeostasis to both inflammation, via NFκβ, and redox balance, via Nrf2 (see below).
IRE1α, in addition to catalyzing XBP1 splicing, has additional functions related to cellular signaling. Activated IRE1α can interact with the adaptor protein TNFR-associated factor 2 and lead to activation of c-Jun-NH2-terminal kinase and NFĸβ via apoptosis signaling-regulating kinase 1 [15]. IRE1α activation has also been linked to the activation of p38 mitogen-activated protein kinase and extracellular-regulated kinase [16–18]. These interactions suggest that the IRE1α branch of the UPR not only regulates adaptation to ER stress and cell survival via XBP1 splicing, but also activation of signaling pathways involved in inflammation, insulin action, and apoptosis. Regulated IRE1α-dependent decay of selected mRNAs can also reduce production of proteins destined for the ER lumen [19, 20].
4 The UPR and Antioxidant Defense
Oxidative stress is thought to be an important pathogenic event in many liver diseases. The ER provides a unique oxidizing environment for protein folding and disulfide bond formation. Each disulfide bond formed during oxidative protein folding produces a single reactive oxygen species. It has been estimated that secretory cells produce 3–6 million disulfide bonds per minute, thus protein folding in the ER is intimately linked to the generation of reaction oxygen species and potentially oxidative stress [21, 22]. Conversely, cellular oxidative stress can disrupt ER homeostasis and induce ER stress [23–25]. Therefore, it is not surprising that the UPR engages the antioxidant program via the transcription factor Nrf2 [26]. Nrf2 belongs to the Cap “n” Collar family of basic leucine zipper transcription factors and regulates the expression of antioxidant response element (ARE)-containing genes [26]. Nrf2 is highly expressed in the liver and kidney and is a substrate of the proximal UPR sensor PERK [27]. Importantly, Nrf2 deletion results in rapid onset and progression of steatohepatitis in mice provided a methionine-choline-deficient diet, often used to model components of non-alcoholic fatty liver disease [28]. In addition, Nrf2-deficient mice were characterized by increased mortality in response to endotoxin- and cecal ligation and puncture-induced septic shock [29]. As noted above, PERK-mediated phosphorylation of eIF2α also leads to the selective translation and upregulation of ATF4. Along with Nrf2, this transcription factor has been linked to the maintenance of cellular glutathione [14]. Thus, the PERK arm of the UPR appears to play a critical role in the defense against oxidative stress and the downstream substrate Nrf2 has been directly linked to steatohepatitis.
In addition to the PERK arm of the UPR, recent evidence has also linked the IRE1α-XBP1 branch of the UPR to the regulation of antioxidant defenses [30]. In this study, hydrogen peroxide-mediated cell death occurred more extensively in mouse embryonic fibroblast cells deficient in XBP1. XBP1 deficiency resulted in reduced catalase expression, and overexpression of XBP1 restored catalase expression in XBP1-deficient cells. Thus, XBP1 may provide protection from oxidative stress; however, whether this regulation occurs in hepatocytes is presently unknown.
5 The ER Lumen as a Source of Oxidative Stress
The ER lumen is an oxidizing environment characterized by a GSH:GSSG ratio of 1:1–3:1, much lower than the cytosolic ratio of 30:1–100:1 [31]. This environment is, in part, maintained by ER oxidase 1 (Ero1) and GSSG, and disulfide bond formation in the ER lumen appears to primarily result from electron transfer reactions involving Ero1, protein disulfide isomerase, and molecular oxygen. Hydrogen peroxide is a product of these transfer reactions and therefore disulfide bond formation and protein folding in the ER lumen are associated with the formation of reactive oxygen species [21, 32]. In addition, the luminal NADPH concentration may play an important antioxidant defense role in liver cells in a manner that appears to be independent of the thiol/disulfide redox system [33].
Very few studies have directly examined whether and how protein folding/misfolding influences oxidative stress. In one study, HIP-deficient cells that lack the ability to eliminate misfolded proteins from the ER were employed. Introduction of low levels of a mutant misfolded form of the vacuolar protein carboxypeptidase Y induced ER stress, accumulation of reactive oxygen species, and cell death [34]. Malhotra et al. utilized hydrodynamic delivery of FVIII (coagulation factor VIII, prone to misfolding in the ER lumen) DNA expression vectors into the tail vein of mice [35]. Accumulation of FVIII resulted in oxidative stress (monitored by dihydroethidine staining, malondialdehyde, GSH) and activation of the UPR in the liver. Treatment with butylated hydroxyanisole reduced accumulation of FVIII and attenuated oxidative stress and UPR activation. Taken together, these data are consistent with the notion that protein misfolding in the ER lumen can produce ROS and that ROS and accumulation of misfolded proteins induce ER stress and activate the UPR. In this context, ROS may be generated as a consequence of disulfide bond formation, depletion of cellular GSH, and/or mitochondrial oxidative phosphorylation.
6 Oxidative Stress as an Activator of the UPR
ROS can originate from exposure to irradiation and environmental pollutants and enzymatic reactions involving the mitochondrial respiratory chain, arachidonic acid pathway, cytochrome P450 family, glucose, amino acid, xanthine and NADP/NADPH oxidases, and nitric oxide synthase [36]. In cultured liver cells the combination of hydrogen peroxide generation using glucose oxidase and proteasome inhibition resulted in activation of the UPR and formation of inclusion bodies that was reduced by either pretreatment with N-acetyl-cysteine or the chemical chaperone, 4-phenylbutyrate [23].
Changes in nutrient flux, particularly fatty acid flux, may influence the functional capacity of the ER, in part, via effects on redox balance. Elevated free fatty acids, in particular saturated fatty acids, have been linked to activation of the UPR in a number of cell types, including hepatocytes [37–40]. Antioxidants, such as taurine, effectively reduce saturated fatty acid-mediated oxidative stress and UPR activation in both H4IIE liver cells and primary hepatocytes [41].
Ethanol impairs protein secretion in hepatocytes and the serum protein deficiency that can lead to clotting disorders, edema, and impaired iron delivery [42]. ER stress and activation of the UPR by ethanol is conserved across vertebrates and reductions in ER stress reduce alcohol-induced liver injury [42, 43]. Using a zebrafish larvae model, Tsedensodnom et al. demonstrated that ethanol exposure induced oxidative stress and that oxidative stress and low doses of ethanol synergize to induce the UPR in the liver [44]. In alcoholic liver disease, ROS is generated by ER-localized cytochrome P450’s and, perhaps, interactions between the ER and mitochondria (discussed below) [45]. Regardless of the source of ROS, these data are consistent with the notion that ROS can impair the secretory pathway in hepatocytes and lead to ER stress and UPR activation.
7 ER–Mitochondrial Interactions and Oxidative Stress
Protein folding in and clearance of aggregated proteins from the ER lumen requires energy, thus ER homeostasis is linked to mitochondrial bioenergetics and adenosine triphosphate (ATP) supply. Physical interactions between the ER and mitochondria have been documented and involve specific tethering proteins and elements of the cytoskeleton [46, 47]. One function of these physical interactions is to provide efficient calcium transfer from the ER to mitochondria, thereby regulating the activity of matrix dehydrogenases required for mitochondrial respiration and ATP production. In turn, ATP can be supplied to the ER lumen to support the energy requirements for protein folding and clearance [48, 49].
Several proteins have been identified as components of the ER–mitochondria tethering mechanism including mitofusin-1 and -2 [50]. In particular, mitofusin-2 appears to be enriched at ER–mitochondria contact sites, can influence ER morphology, and can directly tether ER and mitochondria via homo- and hetero-typic interactions [50]. Importantly, the absence of mitofusin-2 increased the distance between the ER and mitochondria and impaired mitochondrial calcium uptake [50]. Liver-specific ablation of mitofusin-2 in mice resulted in ER and oxidative stress, increased stress pathway signaling (c-Jun NH2 Terminal Kinase), and impaired insulin signaling [51]. Chemical chaperones or the antioxidant N-acetylcysteine improved insulin signaling and glucose homeostasis in liver-specific mitofusin-2 knockout mice [51]. It should be noted that hydrogen peroxide increased the expression of mitofusin-2 in cardiomyocytes and selective upregulation of mitofusin-2 was sufficient to induce myocyte apoptosis [52]. One interpretation of these data is that both reductions and increases in mitofusin-2 may interfere with mitochondrial fusion/fission, physical interactions between the ER and mitochondria, and potentially calcium and ATP transfer.
8 Summary
ER and oxidative stress are typically present in metabolic diseases, such as obesity and diabetes, neurodegenerative diseases, such as Alzheimer and Parkinson disease, and atherosclerosis. Protein folding in the ER lumen generates ROS and in turn, ROS can influence the fidelity of protein folding. The quality control system that responds to ER stress, the UPR, can regulate multiple pathways, including antioxidant defense. Thus, the UPR can be viewed as an adaptive mechanism that ultimately attempts to maintain cell survival and function. The ER is also structurally and functionally linked to mitochondria, and thus mitochondrial ATP and ROS may influence or interact with the ER lumen. Liver diseases are often characterized by both ER and oxidative stress and thus understanding how these two processes interact is critical to understanding the pathogenesis of these diseases.
References
van Meer G, Voelker DR, Feigenson GW (2008) Membrane lipids: where they are and how they behave. Nat Rev Mol Cell Biol 9:112–124. doi:10.1038/nrm2330
Gentile CL, Frye M, Pagliassotti MJ (2011) Endoplasmic reticulum stress and the unfolded protein response in nonalcoholic fatty liver disease. Antioxid Redox Signal 15:505–521. doi:10.1089/ars.2010.3790
Gentile CL, Pagliassotti MJ (2008) The role of fatty acids in the development and progression of nonalcoholic fatty liver disease. J Nutr Biochem 19:567–576. doi:10.1016/j.jnutbio.2007.10.001
Marciniak S, Ron D (2006) Endoplasmic reticulum stress signaling in disease. Physiol Rev 86:1133–1149
Schroder M, Kaufman RJ (2005) ER stress and the unfolded protein response. Mutat Res 569:29–63. doi:10.1016/j.mrfmmm.2004.06.056
Guerriero CJ, Brodsky JL (2012) The delicate balance between secreted protein folding and endoplasmic reticulum-associated degradation in human physiology. Physiol Rev 92:537–576. doi:10.1152/physrev.00027.2011
Rutkowski DT, Kaufman RJ (2004) A trip to the ER: coping with stress. Trends Cell Biol 14:20–28
Wu J, Kaufman RJ (2006) From acute ER stress to physiological roles of the Unfolded Protein Response. Cell Death Differ 13:374–384
Zhang K, Kaufman RJ (2008) From endoplasmic reticulum stress to the inflammatory response. Nature 454:455–462
Kaufman RJ (2002) Orchestrating the unfolded protein response in health and disease. J Clin Invest 110:1389–1398
Schroder M, Kaufman RJ (2005) The mammalian unfolded protein response. Annu Rev Biochem 74:739–789
Trusina A, Papa FR, Tang C (2008) Rationalizing translation attenuation in the network architecture of the unfolded protein response. Proc Natl Acad Sci U S A 105:20280–20285. doi:10.1073/pnas.0803476105
Kaufman RJ (1999) Stress signaling from the lumen of the endoplasmic reticulum: coordination of gene transcriptional and translational controls. Genes Dev 13:1211–1233
Cullinan SB, Zhang D, Hannink M, Arvisais E, Kaufman RJ, Diehl JA (2003) Nrf2 is a direct PERK substrate and effector of PERK-dependent cell survival. Mol Cell Biol 23:7198–7209
Urano F, Wang X, Bertolotti A, Zhang Y, Chung P, Harding HP, Ron D (2000) Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science 287:664–666
Hu P, Han Z, Couvillon AD, Kaufman RJ, Exton JH (2006) Autocrine tumor necrosis factor alpha links endoplasmic reticulum stress to the membrane death receptor pathway through IRE1alpha-mediated NF-kB activation and down-regulation of TRAF2 expression. Mol Cell Biol 26:3071–3084
Nguyen DT, Kebache S, Fazel A, Wong HN, Jenna S, Emadali A, Lee EH, Bergeron JJ, Kaufman RJ, Larose L, Chevet E (2004) Nck-dependent activation of extracellular signal-regulated kinase-1 and regulation of cell survival during endoplasmic reticulum stress. Mol Biol Cell 15:4248–4260
Hetz C, Glimcher LH (2009) Fine-tuning of the unfolded protein response: assembling the IRE1alpha interactome. Mol Cell 35:551–561
Hollien J, Weissman JS (2006) Decay of endoplasmic reticulum-localized mRNAs during the unfolded protein response. Science 313:104–107. doi:10.1126/science.1129631
Hollien J, Lin JH, Li H, Stevens N, Walter P, Weissman JS (2009) Regulated Ire1-dependent decay of messenger RNAs in mammalian cells. J Cell Biol 186:323–331. doi:10.1083/jcb.200903014
Santos CX, Tanaka LY, Wosniak J, Laurindo FR (2009) Mechanisms and implications of reactive oxygen species generation during the unfolded protein response: roles of endoplasmic reticulum oxidoreductases, mitochondrial electron transport, and NADPH oxidase. Antioxid Redox Signal 11:2409–2427. doi:10.1089/ARS.2009.2625
Shimizu Y, Hendershot LM (2009) Oxidative folding: cellular strategies for dealing with the resultant equimolar production of reactive oxygen species. Antioxid Redox Signal 11:2317–2331
Hanada S, Harada M, Kumemura H, Omary MB, Koga H, Kawaguchi T, Taniguchi E, Yoshida T, Hisamoto T, Yanagimoto C, Maeyama M, Ueno T, Sata M (2007) Oxidative stress induces the endoplasmic reticulum stress and facilitates inclusion formation in cultured cells. J Hepatol 47:93–102
Yokouchi M, Hiramatsu N, Hayakawa K, Okamura M, Du S, Kasai A, Takano Y, Shitamura A, Shimada T, Yao J, Kitamura M (2008) Involvement of selective reactive oxygen species upstream of proapoptotic branches of unfolded protein response. J Biol Chem 283:4252–4260
Xue X, Piao JH, Nakajima A, Sakon-Komazawa S, Kojima Y, Mori K, Yagita H, Okumura K, Harding H, Nakano H (2005) Tumor necrosis factor alpha (TNFalpha) induces the unfolded protein response (UPR) in a reactive oxygen species (ROS)-dependent fashion, and the UPR counteracts ROS accumulation by TNFalpha. J Biol Chem 280:33917–33925. doi:10.1074/jbc.M505818200
Cullinan SB, Diehl JA (2006) Coordination of ER and oxidative stress signaling: the PERK/Nrf2 signaling pathway. Int J Biochem Cell Biol 38:317–332
Moi P, Chan K, Asunis I, Cao A, Kan YW (1994) Isolation of NF-E2-related factor 2 (Nrf2), a NF-E2-like basic leucine zipper transcriptional activator that binds to the tandem NF-E2/AP1 repeat of the beta-globin locus control region. Proc Natl Acad Sci U S A 91:9926–9930
Sugimoto H, Okada K, Shoda J, Warabi E, Ishige K, Ueda T, Taguchi K, Yanagawa T, Nakahara A, Hyodo I, Ishii T, Yamamoto M (2010) Deletion of nuclear factor-E2-related factor-2 leads to rapid onset and progression of nutritional steatohepatitis in mice. Am J Physiol Gastrointest Liver Physiol 298:G283–G294
Thimmulappa RK, Lee H, Rangasamy T, Reddy SP, Yamamoto M, Kensler TW, Biswal S (2006) Nrf2 is a critical regulator of the innate immune response and survival during experimental sepsis. J Clin Invest 116:984–995
Liu Y, Adachi M, Zhao S, Hareyama M, Koong AC, Luo D, Rando TA, Imai K, Shinomura Y (2009) Preventing oxidative stress: a new role for XBP1. Cell Death Differ 16:847–857
Hwang C, Sinskey AJ, Lodish HF (1992) Oxidized redox state of glutathione in the endoplasmic reticulum. Science 257:1496–1502
Tu BP, Weissman JS (2004) Oxidative protein folding in eukaryotes: mechanisms and consequences. J Cell Biol 164:341–346. doi:10.1083/jcb.200311055
Szaraz P, Banhegyi G, Benedetti A (2010) Altered redox state of luminal pyridine nucleotides facilitates the sensitivity towards oxidative injury and leads to endoplasmic reticulum stress dependent autophagy in HepG2 cells. Int J Biochem Cell Biol 42:157–166. doi:10.1016/j.biocel.2009.10.004
Haynes CM, Titus EA, Cooper AA (2004) Degradation of misfolded proteins prevents ER-derived oxidative stress and cell death. Mol Cell 15:767–776. doi:10.1016/j.molcel.2004.08.025
Malhotra JD, Miao H, Zhang K, Wolfson A, Pennathur S, Pipe SW, Kaufman RJ (2008) Antioxidants reduce endoplasmic reticulum stress and improve protein secretion. Proc Natl Acad Sci U S A 105:18525–18530. doi:10.1073/pnas.0809677105
Malhotra JD, Kaufman RJ (2007) Endoplasmic reticulum stress and oxidative stress: a vicious cycle or a double-edged sword? Antioxid Redox Signal 9:2277–2293. doi:10.1089/ars.2007.1782
Listenberger LL, Han X, Lewis SE, Cases S, Farese RV Jr, Ory DS, Schaffer JE (2003) Triglyceride accumulation protects against fatty acid-induced lipotoxicity. Proc Natl Acad Sci U S A 100:3077–3082. doi:10.1073/pnas.0630588100
Unger RH, Zhou YT (2001) Lipotoxicity of beta-cells in obesity and in other causes of fatty acid spillover. Diabetes 50(Suppl 1):S118–S121
Feldstein AE, Werneburg NW, Canbay A, Guicciardi ME, Bronk SF, Rydzewski R, Burgart LJ, Gores GJ (2004) Free fatty acids promote hepatic lipotoxicity by stimulating TNF-alpha expression via a lysosomal pathway. Hepatology 40:185–194. doi:10.1002/hep.20283
Wei Y, Wang D, Topczewski F, Pagliassotti MJ (2006) Saturated fatty acids induce endoplasmic reticulum stress and apoptosis independently of ceramide in liver cells. Am J Physiol Endocrinol Metab 291:E275–E281. doi:10.1152/ajpendo.00644.2005
Gentile CL, Nivala AM, Gonzales JC, Pfaffenbach KT, Wang D, Wei Y, Jiang H, Orlicky DJ, Petersen DR, Pagliassotti MJ, Maclean KN (2011) Experimental evidence for therapeutic potential of taurine in the treatment of nonalcoholic fatty liver disease. Am J Physiol Regul Integr Comp Physiol 301:R1710–R1722. doi:10.1152/ajpregu.00677.2010
Howarth DL, Vacaru AM, Tsedensodnom O, Mormone E, Nieto N, Costantini LM, Snapp EL, Sadler KC (2012) Alcohol disrupts endoplasmic reticulum function and protein secretion in hepatocytes. Alcohol Clin Exp Res 36:14–23. doi:10.1111/j.1530-0277.2011.01602.x
Howarth DL, Passeri M, Sadler KC (2011) Drinks like a fish: using zebrafish to understand alcoholic liver disease. Alcohol Clin Exp Res 35:826–829. doi:10.1111/j.1530-0277.2010.01407.x
Tsedensodnom O, Vacaru AM, Howarth DL, Yin C, Sadler KC (2013) Ethanol metabolism and oxidative stress are required for unfolded protein response activation and steatosis in zebrafish with alcoholic liver disease. Dis Model Mech 6:1213–1226. doi:10.1242/dmm.012195
Marchi S, Patergnani S, Pinton P (2013) The endoplasmic reticulum-mitochondria connection: one touch, multiple functions. Biochim Biophys Acta. doi:10.1016/j.bbabio.2013.10.015
Hayashi T, Rizzuto R, Hajnoczky G, Su TP (2009) MAM: more than just a housekeeper. Trends Cell Biol 19:81–88. doi:10.1016/j.tcb.2008.12.002
Pizzo P, Pozzan T (2007) Mitochondria-endoplasmic reticulum choreography: structure and signaling dynamics. Trends Cell Biol 17:511–517. doi:10.1016/j.tcb.2007.07.011
Bravo R, Gutierrez T, Paredes F, Gatica D, Rodriguez AE, Pedrozo Z, Chiong M, Parra V, Quest AF, Rothermel BA, Lavandero S (2012) Endoplasmic reticulum: ER stress regulates mitochondrial bioenergetics. Int J Biochem Cell Biol 44:16–20. doi:10.1016/j.biocel.2011.10.012
Bravo R, Vicencio JM, Parra V, Troncoso R, Munoz JP, Bui M, Quiroga C, Rodriguez AE, Verdejo HE, Ferreira J, Iglewski M, Chiong M, Simmen T, Zorzano A, Hill JA, Rothermel BA, Szabadkai G, Lavandero S (2011) Increased ER-mitochondrial coupling promotes mitochondrial respiration and bioenergetics during early phases of ER stress. J Cell Sci 124:2143–2152. doi:10.1242/jcs.080762
de Brito OM, Scorrano L (2008) Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 456:605–610. doi:10.1038/nature07534
Sebastian D, Hernandez-Alvarez MI, Segales J, Sorianello E, Munoz JP, Sala D, Waget A, Liesa M, Paz JC, Gopalacharyulu P, Oresic M, Pich S, Burcelin R, Palacin M, Zorzano A (2012) Mitofusin 2 (Mfn2) links mitochondrial and endoplasmic reticulum function with insulin signaling and is essential for normal glucose homeostasis. Proc Natl Acad Sci U S A 109:5523–5528. doi:10.1073/pnas.1108220109
Shen T, Zheng M, Cao C, Chen C, Tang J, Zhang W, Cheng H, Chen KH, Xiao RP (2007) Mitofusin-2 is a major determinant of oxidative stress-mediated heart muscle cell apoptosis. J Biol Chem 282:23354–23361. doi:10.1074/jbc.M702657200
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Hudson, W.M., Pagliassotti, M.J. (2015). Oxidative Stress and the Unfolded Protein Response in the Liver. In: Albano, E., Parola, M. (eds) Studies on Hepatic Disorders. Oxidative Stress in Applied Basic Research and Clinical Practice. Humana Press, Cham. https://doi.org/10.1007/978-3-319-15539-5_5
Download citation
DOI: https://doi.org/10.1007/978-3-319-15539-5_5
Publisher Name: Humana Press, Cham
Print ISBN: 978-3-319-15538-8
Online ISBN: 978-3-319-15539-5
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)