
Abstract
Aging is a multifactorial process that depends on diverse molecular and cellular mechanisms, such as genome instability, epigenetic and transcriptional changes, loss of proteostasis, cell death and senescence, metabolic dysfunction, and inflammation. Enzymes of the family of poly(ADP-ribose) polymerases (PARPs) catalyze the synthesis of the biopolymer poly(ADP-ribose) (PAR), a drastic post-translational modification that plays significant roles in all of these processes. On the one hand, poly(ADP-ribosyl)ation (PARylation) contributes to genome and proteome homeostasis, as it participates in chromatin remodeling, genome maintenance, cell cycle control, and the regulation of the ubiquitin-proteasome system. On the other hand, PARPs and PARylation interfere with cellular and organismic energy metabolism, and act as mediators of inflammation, senescence and cell death. Therefore, PARylation is discussed both as a longevity assurance factor on the one hand and an aging-promoting factor on the other hand. Here we highlight the mechanisms underlying the various roles of PARylation in longevity and aging with a focus on molecular and cellular mechanisms.
This chapter represents an updated version of a recent review article that was published in Oxidative Medicine and Cellular Longevity under the Creative Commons Attribution License [1].
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
1 Introduction
Aging has been defined as a progressive post-maturational decline in physiological capacity, accompanied by an increased susceptibility to disease and an increased mortality risk [2]. It is important to keep in mind that aging is a pleiotropic and stochastic process and not restricted to a few distinct processes, but affects a plethora of molecular mechanisms that lead to accumulation of cellular damage and disturbed tissue homeostasis over time. Interestingly, many molecular mechanisms that are associated with aging are also involved in cancer biology, which supports a theory that aging and cancer are two sides of the same coin, with many mechanisms that protect from carcinogenesis contributing to aging in late life. Recently, a number of hallmarks of aging have been defined [3] mainly affecting the following processes: (i) genome maintenance (ii) epigenetics and transcription, (iii) proteostasis, (iv) cellular and organismic energy metabolism, (v) inflammation and immunity, and (vi) cell death, cellular senescence and stem cell regeneration. In general, mechanisms to maintain cellular homeostasis, such as genome maintenance and proteostasis are thought to counteract the aging process, whereas inflammation, senescence, and cell death are considered a driving force of human aging. As discussed below, the post-translational modification poly(ADP-ribosyl)ation (PARylation) is involved in all of these processes via a multitude of different, but often interconnected mechanisms. Due to the complexity and interrelation of these pro- and anti-aging mechanisms, there is no “simple”, unidirectional role for PARylation in aging and longevity. In contrast, there is ample evidence supporting a role for PARylation as a longevity assurance factor on the one hand, but also as an aging-promoting factor on the other hand. In this chapter, we will discuss the numerous cellular functions of PARylation in the context of mechanisms of longevity and aging and will put these into an organismic perspective by summarizing in vivo studies in mice and humans.
2 PARPs and PARylation
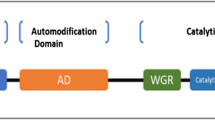
PARylation is a ubiquitous post-translational modification of proteins that occurs in most eukaryotic organisms. The reaction is carried out by enzymes of the family of poly(ADP-ribose) polymerases (PARPs) by using NAD+ as a substrate to synthesize the linear or branched biopolymer poly(ADP-ribose) (PAR), which consists of up to 200 ADP-ribose subunits (Fig. 6.1) [4]. PARP activation leads to covalent modification of various proteins with PAR, including PARPs themselves, as most of them catalyze their automodification. Individual proteins are either covalently modified or interact with PAR chains in a non-covalent fashion, or both. Covalent linkage is mediated through synthesis of PAR chains at glutamate, aspartate or lysine residues of the acceptor proteins [4]. Several hundreds of covalent PARylation target proteins have been identified that are involved in DNA repair and metabolism, transcription, chromatin organization, and mRNA processing [5, 6]. Apart from covalent modification, a wide range of proteins bind pre-existing PAR chains in a non-covalent fashion. The non-covalent PAR-protein interaction is mediated via at least five different PAR binding modules. Those include (i) a 20-amino-acid PAR binding motif (PBM), (ii) distinct macrodomains, (iii) a PAR-binding zinc finger, (iv) a WWE domain, and (v) a PAR-binding regulatory modif (pbR), all of which fulfill diverse cellular functions [7–13]. Whereas the PAR-binding macrodomains, zinc fingers, WWE domains, and the PbR are restricted to a limited number of human proteins (< 50), the 20-aa PBM has been identified in several hundred human protein sequences [8, 9]. This weakly conserved motif consists of (i) a cluster rich in basic amino acids and (ii) a pattern of hydrophobic amino acids interspersed with basic residues [8, 9].

The PARylation reaction. PARPs cleave the glycosidic bond of NAD+ between nicotinamide and ribose followed by the covalent modification of acceptor proteins with an ADP-ribosyl unit. PARPs also catalyze an adduct elongation, giving rise to linear polymers with chain lengths of up to 200 ADP-ribosyl units, characterized by their unique ribose (1’’ → 2’) ribose phosphate–phosphate backbone. At least some of the PARP family members also catalyze a branching reaction by creating ribose (1’’’ → 2’’) ribose linkages. (Reprinted from [1])
Similar to proteins that are targeted by covalent PARylation, most of the non-covalent PAR-binding proteins identified to date are involved in a wide spectrum of cellular mechanisms such as genome maintenance, chromatin remodeling, transcription, replication, RNA metabolism, inflammation, cell cycle control, and cell death [9, 14]. Both covalent PARylation as well as non-covalent protein-PAR interaction modulate protein function by modifying enzymatic activities or interactions with other macromolecules such as DNA, RNA, or proteins, thereby controlling and fine-tuning the spatio-temporal localization and activity of target proteins within the cell [15] (Fig. 6.2).

PARylation in the spatio-temporal control of protein function. It is important to note that PARylation can regulate protein function and localization in both directions. Depending on the specific target protein and the cellular condition, on the hand, PARylation can mediate protein complex assembly and recruitment of proteins to other macromolecules such as DNA and RNA; however, on the other hand PARylation can also induce disassembly of protein complexes and repulsion of proteins from DNA
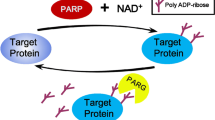
Importantly, the cellular existence of PAR is transient, since the polymer is rapidly hydrolyzed by PARPs’ catabolic counterpart, poly(ADP-ribose) glycohydrolase (PARG). PARG possess both exo- and endoglycosidic activities and is encoded by a single gene giving rise to at least five different splice variants with distinct subcellular localizations [16–20]. In addition, a second enzyme was identified with weak PARG activity, i.e., ADP-ribose-arginine protein hydrolase 3 (ARH3), with evidence that this enzyme is associated with PAR degradation in mitochondria [21, 22]. Both enzymes, PARG and ARH3 are not able to cleave off the last ADP-ribose moiety from target proteins. Recent studies demonstrated that macrodomain-containing proteins, such as MacroD1 and MacroD2, fulfill that task by acting as mono-ADP-ribosylhydrolases on the terminal, protein-proximal ADP-ribose ester linkage, releasing ADP-ribose and an unmodified amino acid that is readily available for the next round of ADP-ribosylation [23, 24].
The PARP gene family consists of 17 homologues in the human genome [4]. It is important to note that not all of these gene products are able to synthesize PAR, instead some PARPs act as mono-ADP-ribosyl transferases or are catalytically inactive. Thus far, PARylation capacity has been demonstrated experimentally for PARPs 1-3, vPARP (PARP4) and Tankyrases 1 and 2 (PARPs 5A and 5B, TNKS 1 and 2) [25]. In this chapter we will focus on the genuine “PARPs” with verified ability to catalyze the formation of PAR, which are briefly introduced in the following.
PARP1 is the founding member of the gene family. It exhibits key roles in the regulation of nuclear and cellular functions and can be activated either by DNA damage, post-translational protein modifications, or potentially by direct protein-protein interactions [26]. The strongest stimulation of PARP1 activity is mediated by its binding to DNA strand breaks, which induces its catalytic activation as a monomer or dimer by several hundred-fold [27–31]. Thus, measurements by quantitative isotope dilution mass spectrometry revealed that under physiological conditions ~ 3000 PAR molecules consisting of 10 ADP-ribose moieties exist; upon treatment of cells with genotoxins this number can rise to > 150,000 molecules [31]. Under these conditions, PARP1 accounts for > 75 % of the overall cellular PARylation capacity [32, 33].
The finding that PARP1-deficient cells still synthesized PAR led to the identification of an additional nuclear PARP, i.e. PARP-2, which can be also activated by binding to certain DNA structures [32, 33]. PARP-2 accounts for most of the residual nuclear PAR formation upon DNA damage and physically and functionally interacts with PARP1. PARP1 and PARP-2 exhibit, at least in part, redundant functions. This is supported by partially overlapping phenotypes of the corresponding single-gene knock-out mice and by the fact that double deficiency results in embryonic lethality in the mouse [34, 35]. Recently, functions of PARP-2 independent of PARP1 in genome maintenance, gene transcription, T cell development, and energy metabolism were reported [34, 36].
PARP-3 mainly resides in the nucleus [37], where it exhibits mono(ADP-ribosyl)ation and PARylation activity and is primarily involved in the control of cell division and DNA double strand break repair [38–40]. PARP3 -/- knockout mice are viable and fertile, and develop no obvious spontaneous phenotype until the age of 15 months. Interestingly, double deficiency in PARP1 -/- and PARP3 -/- significantly decreased survival rates after whole body irradiation compared to single knock-out mice [40].
PARP-4 also known as vault PARP (VPARP) is part of the cytoplasmic vault ribonucleoprotein complex, which has been implicated in multidrug resistance, and exhibits PARylation activity. vPARP has been localized to the nuclear pore and the mitotic spindle [41, 42]. VPARP-deficient mice show an increase in carcinogen-induced colon and lung tumor incidence as well as reduced tumor latency [43].
PARP5a and PARP5b, better known as tankyrases (TNKS) 1 and 2 are localized to multiple subcellular sites including cytoplasmic membrane compartments, telomeres and spindle poles. TNKS1 was reported to act as a positive regulator of telomere length and is required to resolve sister telomeres during mitosis (see below). Apart from this, TNKS1 was implicated in GLUT4 vesicle trafficking [42, 44]. Both tankyrases seem to exhibit at least in part redundant functions, since Tnks1 and Tnks2 single knock-out mice are viable, whereas double deficiency is embryonically lethal [45].
Apart from direct DNA damage-dependent activation, PARP activity is also regulated by posttranslational modifications such as phosphorylation, acetylation, and sumoylation [46–50]. Moreover, PARP activity is subject to regulation by direct protein-protein interactions [51–53]. DNA damage independent activation of PARPs holds in particular true for cytosolic PARPs, which play important roles in cell division and cellular stress response [54, 55].
In conclusion, three non-exclusive mechanisms of the cellular functions of PARPs can be distinguished: (i) Functions that rely on the enzymatic activity of PARPs and the subsequent covalent modification or non-covalent interaction of nuclear proteins with PAR. (ii) Direct interactions of proteins with PARPs via protein-protein interaction. And (iii) interference with the cellular NAD+ metabolism by excessive PARP stimulation and potential signaling functions of free PAR or its derivatives. Each of these three mechanisms contributes to the function of PARylation in various cellular processes as discussed below.
3 PARylation in Aging-Associated Molecular Mechanisms
There is a large body of evidence showing a positive correlation of PARylation capacity and mammalian longevity. Previously, we demonstrated that PARylation capacity in peripheral blood mononuclear cells (PBMCs) of 13 mammalian species strongly correlates with their maximum lifespan, e.g., maximum PARylation levels were five times higher in humans than in rodents [56]. Interestingly, these difference in PARylation are not associated with different enzyme levels, but are rather influenced by a higher PARylation capacity of the human PARP1 enzyme in comparison to its rodent orthologue [57]. Moreover, PARylation capacity in PBMCs declines with age in humans and rodents [56, 58]. Interestingly, humans exhibiting an exceptional long lifespan, i.e., centenarians, display a significantly higher PAR-ylation capacity than the average population [59], which is comparable to those of young subjects [60]. Moreover, in support of the view that PARP1 counteracts the aging process, is the finding that PARP1 -/- mice age moderately faster compared to wild-type animals [61].
On the other hand, the interaction of PARPs with key regulators of immune function, such as NF-κB, its drastic effects on NAD+ metabolism, and its potential to induce cell death may contribute to aging-promoting mechanisms. Consistently, PAR-ylation has been linked to many aging-associated inflammatory and degenerative diseases, which is supported by various studies demonstrating that pharmacological inhibition of PARylation as well as a genetic knock out of PARP1 in mice protects from such diseases.
It is tempting to speculate that the opposing effects of PARylation in cellular homeostasis on the one hand, and inflammation and cell death on the other hand, at least in part, explain the rather mild premature aging phenotype of PARP1 -/- mice. The ambivalent role of PARylation in aging and longevity is associated with its multifunctional role in many molecular and cellular mechanisms, such as (i) genome maintenance, (ii) epigenetics and transcriptions, (iii) proteostasis, (iv) cell death and senescence, (v) energy metabolism, and (vi) inflammation and immunity are discussed in the following sections (Fig. 6.3).

PARylation-related mechanisms of aging and longevity. For details see text. (Adapted from [62]). (Reprinted with permission of Elsevier)
3.1 Genome Maintenance
A large body of evidence supports the theory that genomic instability acts as a causative factor of aging, which is evident from the fact that most mouse models of premature aging as well as human progeria syndromes are related to dysfunctional genome maintenance [63]. This may be attributed to the fact that DNA serves as a blueprint for all cellular RNA and proteins. Consequently any acquired change in its sequence, which may arise from molecular damage, is permanent and thus may have irreversible consequences. For this reason, nature invested in a sophisticated network of various mechanisms to maintain genome integrity, such as DNA repair and cell cycle control. However, even if these mechanisms may be very efficient, they cannot cope with all the insults induced in the genome, leading to a gradual accumulation of DNA damage and mutations, thus contributing to organismic aging [63].
Multiple cellular studies support a role of PARylation as a cell survival factor upon genotoxic stimuli and a general caretaker of genomic stability. For instance, trans-dominant inhibition of PARP1 by overexpression of its DNA binding domain potentiates cytotoxicity upon treatment of cells with various genotoxins [64]. Consistent with this, overexpression studies demonstrated that PARP1 acts as a negative regulator of alkylation-induced sister chromatid exchange [65], and ex vivo supplementation of human PBMCs with the NAD+ precursor nicotinic acid enhances cellular PARylation and improves cell viability upon induction of genotoxic stress [66]. Furthermore, ample evidence for a role of PARylation in genome maintenance comes from a plethora of studies in three independently generated PARP1 knock-out mouse models. Thus, PARP1 -/- mice and cells derived from them are hypersensitive to DNA damaging agents and PARP1 -/- cells display increased spontaneous genomic instability as measured by the frequency of sister chromatid exchanges, chromosome aberrations and micronuclei formation [67–71]. Moreover, various studies supported the notion that PARP1 acts as a tumor suppressor gene, since PARP1 deficiency enhances carcinogenesis during aging and upon induction by DNA damaging agents [61, 72–75]. Consistently, data from human studies showed that a hypomorphic PARP1 polymorphism (V762A) serves as a risk factor for the development of several types of human cancers [76–83].
As discussed in the following sections, apart from its direct involvement in several DNA repair mechanisms, PARPs and PARylation participate in genome maintenance by regulating telomere length, chromatin structure, DNA replication, and cell cycle control (Fig. 6.4).

PARP1, some interaction partners, and their role in genomic maintenance. ATM indicates ataxia telangiectasia mutated; Bub3 Budding uninhibited by benzimidazoles 2; Cenpa/b centromeric protein a/b; CSB Cockayne syndrome type B; DEK DEK oncogene; DNA-Polβ DNA polymerase β; DNA-PK CS DNA-activated protein kinase catalytic subunit; HMGB1 high mobility group box 1; Ku70/80 Ku antigens 70/80 kDa subunit; MRE11 meiotic recombination 11; p21 cyclin-dependent kinase inhibitor 1A; p53 tumor suppressor protein p53; PCNA proliferating cell nuclear antigen; TRF2 telomeric repeat binding factor 2; WRN Werner syndrome protein; XRCC1 X-ray repair complementing defective in Chinese hamster 1; XPA xeroderma pigmentosum complementation group A. (Reprinted from [1])
3.1.1 DNA Repair
It is estimated that thousands of DNA damage lesions occur in a mammalian cell every day, all of which need to be repaired to ensure genomic stability and longevity. In mammals, at least six major DNA repair pathways exist, i.e. O6-methyl guanine methyltransferase (MGMT), base excision repair (BER), nucleotide excision repair (NER), mismatch repair (MMR), and DNA double strand break (DSB) repair including the sub-pathways homologous recombination (HR) and non-homologous end joining (NHEJ) [84]. Interestingly, defects in DNA repair lead to premature aging, but on the other hand, DNA repair mechanisms themselves can be subjected to age-related changes and deterioration [85]. PARylation is one of the first and certainly a very influential post-translational modification that is induced upon various forms of genotoxic stress, such as oxidative and alkylation damage, ionizing as well as UV irradiation, and affects several hundreds of target proteins with profound functions at all levels of cellular stress response [6, 86]. Of note, the recruitment of PARP1 to sites of DNA damage and induction of PARylation occurs within seconds and is one of the most immediate DNA damage responses [87, 88]. Except for the MGMT pathway, there is ample evidence that DNA damage dependent PARPs, i.e., PARPs 1-3 are involved in all known repair mechanisms, and therefore, these PARPs are considered as important caretakers of genomic stability with partially overlapping, but also distinct functions [26, 40].
Base excision and single strand break repair (BER/SSBR) is the major DNA repair pathway that acts on damage that occurs during cellular metabolism including damage from ROS, methylation, deamination, and hydroxylation. The levels of many of these lesions increase with age including the well-studied lesion 8-oxo-deoxyguanine (8-oxo-dG). Moreover, BER activity decreases with age in multiple tissues [89]. The core BER reaction is initiated by a DNA single strand break (SSB) upon excision of the damaged bases by DNA glycosylases [90].
PARP1 detects such SSB via its second zinc finger (ZFII), thus triggering its enzymatic activation [91, 92]. Moreover, PARP1 cooperates with certain DNA glycosylases that are important for the repair of oxidative DNA damage. For instance, PARP1 physically interacts with 8-oxo-dG-DNA glycosylase (OGG1), which further stimulates PARP1 activity, whereas activated PARP1 inhibits OGG1 activity indicating a reciprocal functional regulation of the two factors [93]. Similarly, PARP1 binds to the glycosylase NEIL1 which stimulates PARylation activity, whereas activated PARP1 inhibits incision activity of NEIL1. Interestingly, and consistent with the notion of compromised DNA repair during aging, PARP1 binds less efficiently to NEIL1 in old mice compared to young ones [94]. Another, important factor in BER/SSBR is the loading platform X-ray repair complementing factor 1 (XRCC1). Strikingly, its recruitment to SSBs is completely dependent on PARylation [95–97]. Thus, PARP1 and PAR are required for the assembly and stability of XRCC1 nuclear foci after DNA damage [96]. Furthermore, XRCC1 and PARP1 interact with DNA polymerase-β and DNA ligase III, forming a multiprotein complex consisting of the major BER factors [98–100]. As mentioned above, PARP1 and PARP-2 work at least partially in a redundant fashion which is evident from the fact that they homo- and heterodimerize and only double knock-out mice show embryonic lethality [35, 101]. Consistent with this idea, PARP-2 also participates in BER and interacts physically and functionally with XRCC1, DNA polymerase-β, and DNA ligase III. Recruitment studies indicate a role of PARP-2 in later steps of BER repair [102].
Nucleotide excision repair is responsible for the removal of bulky helix-distorting DNA adducts, which are caused by UV irradiation and endogenous metabolites [90]. Two distinct modes of NER are known: global genome repair (GGR) and transcription coupled repair (TCR). Whereas in TCR, DNA damage signaling is mediated via Cockayne syndrome group A and B proteins (CSA/CSB), GGR relies on the damage recognition by XPC and the UV-DDB complex (DDB1-DDB2-containing E3-ubiquitin ligase complex). Subsequent to DNA damage recognition, both sub-pathways merge into the same pathway, characterized by damage verification via XPA, DNA unwinding by the helicases XPB and XPD, excision of the damaged DNA fragment by the nucleases ERCC1/XPF and XPG, and DNA resynthesis and ligation via Pol δ/ε and ligase I/III, respectively. The functional role of the NER as a longevity assurance mechanism is impressively represented by the fact that patients with defects in a subset of NER proteins, i.e., CSA and CSB (Cockayne syndrome) and XPB, XPD, TTDA (trichothiodystrophy), as well as corresponding mouse models, show in some tissues a strong premature aging phenotype [84].
A role of PARP1 in NER is well established, and several NER factors, were identified as PAR binding factors, i.e., the DNA-dependent ATPase (CSB) protein, the DNA lesion recognition protein xeroderma pigmentosum group A (XPA), DDB2, and XPF [8, 103, 104]. Consistently, it has been reported that UVC light activates cellular PARylation [105], that PARP inhibition renders cells sensitive to UVC irradiation [106, 107], and that PARP inhibition sensitizes mice for the development of UVB-induced skin cancers [108]. With regards to potential underlying molecular mechanisms of these findings, it was reported that CSB physically interacts with PARP1 and its ATPase activity is inhibited by PARylation. Furthermore, the damage recognition protein DDB2 directly interacts with PARP1, which promotes PARP1 activation, subsequent chromatin relaxation, and recruitment of XPC and the chromatin-modifier ALC1 [106, 107, 109–111]. This may attract the central NER factor XPA, since this protein physically interact with PARP1 and PAR causing a reciprocal functional regulation of XPA and PARP1 at the site of the damage [103, 112, 113].
DNA double strand breaks (DSBs) arise from ionizing radiation, free radicals, chemicals, or during attempted replication of a SSB through collapsed replication forks. They represent the most cytotoxic form of DNA damage and, if unrepaired, they can trigger apoptosis, senescence, or genomic instability. Consistent with this, there is growing evidence that the number of DSB increases with age and that this profoundly affects cell and tissue homeostasis during the aging process [114]. Mammalian cells repair DSBs via two mechanisms: homologous recombination repair (HRR) utilizes the sister chromatid or chromosome for error-free repair of the DSB, whereas non-homologous end joining (NHEJ) reattaches free DNA ends without using a template. For this reason, NHEJ is prone to micro-deletions or insertions which can cause frameshift mutations [90]. If HRR or NHEJ is employed depends on the species, cell type, and cell cycle phase [115].
In both pathways, PARylation already participates at very early stages. Thus, PARP1 and the DSB sensing complexes MRN (MRE11/Rad50/NBS1) (involved in HR) and Ku70/80 (involved in NHEJ) were shown to interact with and compete for binding at free DNA ends, with PARP1 potentially guiding these proteins to the damaged site [87, 116]. PARP1 also physically and functionally interacts with two phosphatidyl inositol 3-like protein kinases, i.e., ATM (involved in HR) and DNA-PKcs (involved in NHEJ), which are crucial for DSB signaling [117–120]. With regards to NHEJ, two sub-types exist: the classical one, which is largely error-free and is initiated by the Ku complex; and a more error-prone alternative pathway. Several studies revealed that PARP1 is in particular responsible for the initiation of the alternative route and acts as a molecular switch between the two NHEJ sub-pathways [116, 121, 122]. It was suggested that PARP1 serves as a general DNA damage detecting molecule, which potentially also acts as a switch between NHEJ and the HRR [115, 123]. Consistent with this, several reports demonstrated an anti-recombinogenic activity of PARP1 [124–126]. However, the precise role of PARlyation in DSBR is very complex and so far it is not clear under which conditions PARylation supports HRR and under which conditions it induces a shift towards NHEJ and one of the two sub-pathways.
Another level of complexity is added by recent work demonstrating that SIRT6 is recruited to sites of DSBs. SIRT6 is one of seven mammalian sirtuins, which are homologues of the yeast Sir2 deacetylase that functions as a longevity regulator in yeast [127]. SIRT6 itself acts as an ADP-ribosylase and NAD+ -dependent deacetylase. A direct role of SIRT6 in mammalian lifespan regulation is suggested by the finding that SIRT6 deficiency in mice leads to shortened lifespan and an aging-like phenotype [128], whereas SIRT6 overexpression results in lifespan extension by 15 % in male mice [129]. On a molecular level, SIRT6 appears to be involved in BER and DSBR. Of note, SIRT6 interacts with PARP1 and stimulates its activity, thereby enhancing DSBR upon oxidative stress [130]. Furthermore, overexpression of SIRT6 in middle-aged and pre-senescent human fibroblasts with age-related decline in HRR capacity, led to restoration of HRR activity. Interestingly, this effect was dependent on functional PARP1, suggesting that PARP1 and SIRT6 cooperate to maintain efficient HRR in cells at young age [131].
Apart from PARP1 and PARP-2, recently PARP-3 joined the club of DNA damage dependent PARPs. In particular, PARP-3 appears to play an important role in DSBR. Thus, PARP-3 interacts with several NHEJ factors such as DNA-PKcs, Ku70/80, and DNA ligase IV [37]. Moreover, a series of recent studies demonstrated that PARP-3 acts in concert with PARP1 to control the relative contribution of HRR and NHEJ pathways [39, 132–134].
Taken together, these studies underscore that PARPs and PARylation act on multiple levels within the DNA repair network, but more work is necessary to define the exact molecular mechanisms by which PARylation participates in DNA repair and which role this may have during aging.
3.1.1.1 Telomere Maintenance
Telomeres are repetitive sequences at the end of the chromosomes and function as buffers to prevent loss of coding sequences during DNA replication. They are capped by a protein complex known as shelterin, which tightly regulates the telomeric structure by interaction with several DNA repair proteins and the telomere-elongating reverse transcriptase, i.e., telomerase. Deterioration of telomeres can be seen as a specific subform of genomic instability, as uncapped telomeres trigger a sustained DNA damage response. In accordance with this view, telomerase deficiency in humans is associated with several diseases, such pulmonary fibrosis, dyskeratosis congenita, and aplastic anemia, that are characterized by a loss of regenerative capacity of different tissues [135] and telomere shortening has been described as an important factor during normal human aging [136]. In line with this view, reactivation of telomerase can reverse tissue degeneration in aged telomerase-deficient mice [137].
The first PARP that was associated with telomere regulation was TNKS 1 [138]. TNKS1 regulates telomere length by modifying the shelterin component TRF1, thereby inhibiting its release from telomeres and blocking the access of telomerase to the end of the chromosomes [44]. In this respect, it has been shown in RNA interference experiments that TNKS1 and telomerase synergistically cooperate in telomere length regulation [139]. Proper TNKS1 activity in telomere maintenance is important also for overall genome maintenance, since TNKS1 knock-down or pharmacological inhibition sensitizes cells to ionizing irradiation-induced cell death, chromosome aberrations, and telomere fusions [140].
Apart from TNKS1, a role of PARP1 in the regulation of telomere length is well established. In vivo a substantial loss of telomeric DNA by 30 % was observed in the first generation of PARP1 -/- mice [141]. Gomez et al. reported that PARP1 is dispensable for the capping of normal telomeres, but is specifically recruited to eroded telomeres, where it might help to protect chromosomes against end to end fusions and genomic instability [142]. Our group demonstrated in a number of different cell culture systems that pharmacological inhibition of PARylation or knock-down of PARP1 via RNA interference leads to a rapid decrease in telomere length and stabilization at a lower level. Importantly, neither the length of the single-stranded telomeric overhang nor telomerase activity was affected by PARP1 inhibition. Interestingly, release from PARP inhibition led to a fast re-gain in telomere length in telomerase-positive cells indicating that PARP1 activity is an important determinant in telomere length regulation [143]. On a molecular level, the function of PARP1 in telomere length regulation presumably depends on its interaction with the telomeric repeat binding factor 2 (TRF2). TRF2 is another key component of the shelterin complex and is responsible for telomeric stability, length regulation, and suppression of unscheduled activity of the double-strand break repair machinery by maintaining the t-loop [144]. PARP1 interacts with and modifies TRF2, and the PARylation of TRF2 affects its binding to telomeric DNA [142, 145].
Another PARP1 interaction partner that is involved in telomere regulation is the RecQ helicase WRN [146]. Patients with the rare autosomal recessive disorder Werner syndrome (WS), in which the WRN gene is mutated, display genomic instability and telomere shortening on the cellular and premature aging on the organismic level with symptoms resembling normal human aging in many aspects including cataracts, graying of hair and alopecia, atherosclerosis, osteoporosis, and higher cancer incidence. The premature aging phenotype of these patients appears to be at least partially dependent on telomere length, since human symptoms were only recapitulated in mice with short telomeres, i.e., WRN/telomerase double knock-out mice [146, 147]. [NB. Mice usually exhibit considerably longer telomeres (~ 40 kb) than humans (5–15 kb)]. On a cellular level, fibroblasts derived from WS patients display genomic instability and a reduced replicative lifespan. This phenotype is in accordance with experimental data demonstrating that WRN is involved in multiple aspects of DNA metabolism, such as DNA replication, genomic maintenance, and telomere regulation [146]. WRN functions as a 3ʹ-5ʹ helicase and additionally as a 3ʹ-5ʹ exonuclease. Proper enzymatic activity of WRN seems to be crucial for maintaining genomic integrity, since pharmacological inhibition of WRN’s helicase activity causes DSBs and apoptosis [148]. WRN and PARP1 directly interact with each other physically and PARP1 modulates WRN’s exonuclease and helicase activities [149, 150]. In addition, we recently demonstrated that WRN interacts with PAR itself via at least one specific PAR binding motif and that this interaction inhibits WRN’s DNA binding affinity as well as all its enzymatic functions [151]. These results indicated that PARP1 and PARylation regulate WRN activity towards its substrates in time and space. Interestingly, as observed with other factors, the regulation of PARP1 and WRN appears to be reciprocal, because PARylation is impaired in WRN-deficient cells indicating that WRN is required for fully functional PARP1-dependent PARylation [152]. How exactly the interplay between PARP1 and WRN affects telomere maintenance mechanisms awaits further clarification. Moreover, obviously factors other than PARP1 and WRN are involved in these mechanisms, because WRN and PARP1 share many interaction partners, including DNA-PK, P53, and TRF2 (Fig. 6.5). For instance, PARP1, WRN, and DNA-PK (including Ku70/80 and DNA-PKcs) can form a complex, in which PAR-modified Ku70/80 inhibits WRN [153]. Furthermore, both PARP1 and WRN have a positive impact on telomere length, presumably by regulating the binding of TRF2 to the t-loop. Genetic cooperation between PARP1 and WRN was demonstrated in vivo, because mice with deficiencies in both proteins display higher rates of chromatid breaks, chromosomal rearrangements and cancer than each of the single mutant mice [154]. Moreover, double mutants appear to have reduced median and maximum lifespan, despite the fact that these mice were on a telomerase-positive genetic background and telomere lengths of single mutant MEFs did not differ significantly from the double mutant MEFs. This finding suggests that telomere-independent functions of WRN and PARP1 exist in the mouse to maintain organismic longevity. In conclusion, since PARP1 and WRN share many interaction partners and both proteins participate in other DNA repair pathways such as BER and NHEJ, they probably synergistically collaborate to maintain overall genomic stability and ensure longevity.

Interaction map between PARP1 and the Werner syndrome protein (WRN). The two proteins share many overlapping interaction pathways. There is a reciprocal interaction with DNA-PK (double-headed arrow) and p53, stimulation of base-excision repair (BER, one-headed arrow), and inhibition of TRF2-DNA binding (blocked arrow). PARP1 also inhibits WRN functions if in an unmodified state. (Modified from [144] with permission of Oxford University Press)
3.1.1.2 DNA Replication
The WRN helicase also participates in the response to replicative stress, a cellular stressor that was linked to mammalian aging, due to its ability to drive cells, including stem cells, into senescence and apoptosis [155, 156]. Replication forks contain several proteins such as helicases and polymerases, forming the so-called replisome. Usually progression of the replication fork continues until it encounters a replication fork barrier such as DNA-protein complexes or SSBs. In this case the replicative helicase progresses much more slowly, so that the fork is “stalled”. If this goes along with the disassembly of the replisome the fork “collapses” and a DSB is formed [157].
WRN and PARP1 are involved in the reactivation of stalled replication forks. Specifically, PARP1 binds to and is activated at stalled replication forks and mediates the recruitment of MRE11, a key component of the MRN complex. MRE11 may collaborate with WRN helicase to resect DNA ends for RAD51 loading and subsequent HR repair to promote replication fork restart after release from replication blocks [87, 157–159]. Interestingly, PARP1 not only mediates the recruitment of MRE11 to the stalled replication fork, but also controls its function, thereby protecting stalled replication forks from uncontrolled Mre11-dependent degradation [160]. In accordance with these data, PARylation is required for effective replication fork restart upon treatment of cells with sublethal doses of the replication-stress-inducing topoisomerase 1 inhibitor, camptothecin [161]. Specifically, PARP1 activity regulates the timing of replication fork restart by stabilizing forks in the regressed state and recruiting and controlling the RECQ helicase RECQ1 at the stalled fork, which in turn mediates repair and fork restart [162, 163].
3.1.2 Mitosis and Cell Cycle Control
After DNA replication is completed, proper mitotic regulation is crucial to ensure genomic integrity during cell proliferation [164]. During mitosis, the spindle pole formation requires the centrosome, whereas the centromere is the chromosomal region that organizes the kinetochore, thus enabling the attachment of the mitotic spindle microtubules. Strong evidence that mitotic spindle checkpoint proteins play an important role to ensure mammalian longevity is supported by studies demonstrating that mice with low levels of the mitotic checkpoint protein BubR1 and mice haploinsufficient for Bub3 and Rae1—another mitotic checkpoint gene—age prematurely [N.B.: A complete knockout of these genes results in embryonic lethality in the mouse] [165, 166]. On the other hand transgenic overexpression of BubR1 in mice protected cells from age-related aneuploidy and cancer and extended a healthy life-span in these mice [167]. Moreover, mutations in human BubR1 are often linked with the mosaic variegated aneuploidy (MVA) syndrome—a rare autosomal recessive disorder that is characterized by inaccurate chromosome segregation and aneuploidy. The disease is characterized by several segmental premature aging features, such as high cancer rates, facial dysmorphisms, short stature, cataracts, and death during childhood. Strikingly, a mouse model carrying a MVA mutation in one allele of BubR1 resulted in a premature aging phenotype and reduced lifespan, as well [168].
The first evidence for a role of PARylation in spindle regulation was obtained from a study with Xenopus laevis egg extracts showing that PAR itself is a component of the mitotic spindle and is required for its assembly and function, which was attributed to the enzymatic activity of TNKS1 [169, 170]. These authors suggested that PAR provides a dynamic cross-linking function at spindle poles by regulating the spindle pole protein NuMa, which promotes the assembly of exactly two poles [171]. Moreover, another study showed that TNKS1 modifies the mitotic kinetics regulator (Miki) at the Golgi apparatus in late G2 to prophase, which then translocates to mitotic centrosomes to induce downstream events that ensure appropriate prometaphase progression [172]. Furthermore, TNKS1 PARylates CPAP, which is required for procentriole formation, thereby regulating the CPAP levels during cell cycle to limit centriole elongation and ensure normal centrosome function [173].
In addition to TNKS1, PARP-3 is localized at the centrosomes during cell division and is involved in the regulation of G1/S cell cycle progression [174]. PARP-3 interacts with PARP1, which also resides in the centrosome during the cell cycle and it was shown that haploinsufficiency for PARP1 is related to centrosome duplication and chromosomal instability [174–177]. Moreover, PARP1 and PARP-2 are present at centromeres and interact with the constitutive centromere proteins CENPA, CENPB and the spindle check point protein Bub3 [178, 179]. The physical and functional relationship of PARP1 with the centrosome and the centromere links DNA damage surveillance to the mitotic spindle checkpoint. Importantly, apart from mitosis PARylation also plays a role in the regulation of other cell cycle phases. Thus, as discussed above, damaged replication forks during S-phase can activate PARylation. The PAR formed stimulates Chk1 kinase activity and PAR is required for efficient retention of Chk1 and phosphorylated Chk1 at the fork. To this end, Chk1-PAR interaction is important for proper S-phase checkpoint regulation with effects on Chk1 target proteins, such as p53 [7].
Because severe DNA damage or mitotic misregulation can cause genomic instability leading to tumor formation, a complex cellular security network has evolved to counteract carcinogenesis. This signaling network can stop the cell cycle at different stages, thereby either inducing DNA repair, or eradicating or neutralizing heavily damaged cells by apoptosis or senescence, respectively. To this end, apoptosis and senescence are powerful tumor-suppressive mechanisms, but on the other hand, both pathways can lead to depletion of the regenerative cell pool, thus promoting tissue degeneration and organ failure, which are hallmarks of aging [180]. One of the most important regulators of cell cycle progression and induction of senescence/apoptosis is the transcription factor p53. Consequently, mouse studies demonstrated that p53-deficiency leads to premature death due to tumor development, whereas constantly active p53 protects against cancer at the cost of a premature aging phenotype [180].
Consistent with the role of PARP1 and p53 as caretakers and guardians of the genome, PARP1 and p53 synergistically cooperate in vivo in telomere and chromosomal maintenance as well as in tumor suppression [75, 181–184]. Many functional interactions between PARP1 and p53 during DNA damage response and apoptosis exist, such as delayed p53 transactivation potential in PARP1-deficient cells [185–188]. In addition to its function as a positive regulator of gene expression, p53 also acts as a gene-specific transcriptional transrepressor. Interestingly, p53-mediated transrepression of the MTA1 gene (MTA1, metastasis associated protein 1), a component of a nucleosome remodeling complex which is associated with very aggressive tumor phenotypes, depends on functional PARylation of p53 [189]. On the other hand PARylation of p53 is also able to inhibit its binding to its transcriptional consensus sequence, indicating that multifaceted regulatory mechanisms exist between PARP1 and p53 [190, 191]. Kanai et al. suggested a mechanism of PARP1-dependent regulation of p53 activity: According to this study, PARylation induces structural changes in p53 that mask its nuclear export sequence, resulting in an accumulation of p53 in the nucleus, where it exerts its transactivational functions. Accordingly, a p53 mutant in which PAR acceptor sites were mutated, localized to the cytoplasm to a greater extent than wildtype P53 [192].
In conclusion, there is ample evidence that PARP1 modulates p53 stability, intra-cellular localization and transcriptional activity with likely implications in the induction of apoptosis and senescence on a cellular and therefore aging and longevity on an organismic level. However, studying the combined role of PARP1 and p53 in the aging process is complicated by the situation that mouse models with deficiencies in both tumor-suppressor genes show cancer-dependent premature death unrelated to other signs of premature aging. The development of sophisticated conditional mouse models with spatio-temporal controlled expression of PARP1 and p53 may represent an approach to overcome these hurdles.
Apart from the direct regulation of cell cycle proteins PARPs and PARylation are involved in cell cycle regulation through their role in chromatin remodeling and regulation of gene transcription. Thus, for instance, PARP-2 regulates cell cycle-related genes by controlling histone deacetylation and methylation independently of its PARylation activity [193] and PARP1 directly controls the action of the transcription factor SP1 during cell cycle progression [194]. The role of PARylation in chromatin regulation, epigenetics and transcription and how this may be connected to mechanisms of aging is discussed in the next section.
3.2 Chromatin Regulation, Epigenetics, and Transcription
In principle, all cells of the human body contain almost identical genomes, but show huge functional and phenotypical variability. For example, this becomes obvious when comparing hepatocytes, neurons and muscle cells. These phenotypic differences are largely regulated by epigenetic mechanisms. Epigenetics imply modifications of the cellular chromatin, i.e., DNA and associated proteins, such as DNA methylation and histone modifications. These marks are set during organismal development, but remain to a certain extent highly dynamic throughout a life-time. There is extensive evidence that aging is accompanied by a plethora of epigenetic changes [195] and that such epigenetic changes can affect the aging process at multiple levels, since they induce alterations in gene transcription networks and interfere with genome maintenance mechanisms [196].
DNA methylation occurs exclusively at cytosines in the mammalian genome and this predominantly happens in the context of the symmetrical CG dinucleotides, which are often localized as CpG islands (discrete 0.5–2 kb regions rich in CpG sites) in gene promoters, where CpG methylation is involved in transcriptional regulation. Promoter CpG island methylation is stable and self-perpetuated during cell division by enzymes of the family of DNA methyltransferases (DNMTs). Depending on the context, DNA methylation can be a dynamic process, since enzymes of the ten-eleven translocation (TET) family can sequentially oxidize 5-methylcytosine resulting in intermediates, such as 5-hydroxymethylcytosine that finally leads to demethylation [197]. Genome wide studies in aging cells and tissues have identified a stochastic DNA methylation drift. These drifts are thought to reflect the imperfect maintenance of epigenetic marks, generating epigenetic mosaicism in aging stem cells that could potentially disturb their regenerative potential, leading to stem cell exhaustion and focal proliferation defects that can contribute to cancer and aging [197].
PARylation affects DNA methylation at multiple levels and participates in the establishment and maintenance of genome methylation patterns. Thus, PAR non-covalently interacts with DNMT1, thereby inhibiting its enzymatic activity. According to these data, in the absence of PARylated PARP1, DNMT1 is free to methylate DNA, while if high levels of PARylated PARP1 are present, DNMT1 will be inhibited, preventing DNA methylation [198]. Interestingly, this seems to affect promoter activity of DNMT1 itself, since PARylated PARP1 occupies the Dnmt1 promoter, suggesting that PARylated PARP1 plays a role in protecting the promoter from methylation [199]. The chromatin insulator CTCF (CCCTC-binding factor) is another important regulator of gene transcription and chromatin structure. CTCF is able to activate PARP1 which then forms a complex with DNMT1 and inhibits its methylase activity at CTCF-bound CpGs [200]. Recent evidence suggests that not only methylation is regulated by PARylation, but also the demethylation process is under PARylation control. It was shown that active DNA demethylation is required for complete imprint erasure in primordial germ cells [201]. Interestingly, this study suggested that this mechanism is dependent on PARylation. Consistently, PARP activity enhances the expression of Tet1 hydroxylases, which is involved in DNA demethylation [202]. Furthermore, PARP1 deficiency led to a large increase in 5mC accumulation in MEFs during epigenetic reprogramming through iPS induction [203].
In conclusion, these findings suggest that PARylation actively contributes to the dynamics of DNA methylation establishing an epigenetic program that directs subsequent transcriptional induction at relevant loci during organismic development and aging.
Apart from DNA methylation, epigenetic changes of DNA-associated proteins and chromatin provide another level of control of replication, transcription and other fundamental cellular processes. Chromatin is a dynamic structure which is regulated by posttranslational modifications, such as acetylation, methylation, or PARylation. Such structural and functional alterations of chromatin are widely associated with aging from yeast to mammals [204]. The molecular mechanisms leading to chromatin disturbances in aging are largely unknown, but may be related to alterations in transcriptional programs thereby contributing to the aging process [204]. Moreover, DNA damage may lead to sustained alterations in chromatin structure. This potentially causes a positive feedback mechanism of DNA damage leading to chromatin rearrangements which, in turn, sensitizes DNA as a substrate for further damage. As discussed above, the histone deacetylase SIRT6, which is involved in BER and DSBR, gives an striking example that loss of function of an epigenetically relevant enzyme can lead to premature aging, whereas gain of function of such an enzyme can extend longevity in mice [3, 129, 205].
In terms of PARPs and PARylation, PARP1 acts as a structural and regulatory component of chromatin, both in undamaged cells and upon genotoxic stress. It may either regulate chromatin structure directly by PARylation of chromatin components, or indirectly by controlling the recruitment of chromatin remodeling factors [206]. Many PAR acceptor and binding proteins contribute to chromatin and nuclear architecture such as histones, lamins, high-mobility group (HMG) proteins, heterochromatin protein 1 (HP1), and the DEK protein [206–211]. It was proposed that PARP1 induces a histone shuttling mechanism, based on findings that PARylation of polynucleosomes causes relaxation of chromatin structure and that activity of PARG degrades PAR from modified histones [212–215]. According to this model, DNA-bound histones dissociate from DNA upon PARylation, causing an open chromatin structure and guiding repair factors to sites of DNA damage. Upon degradation of PAR by PARG, DNA reassociates with histones, thereby restoring the condensed chromatin structure. Moreover, upon DNA damage PARP1 activation leads to the recruitment of the histone variant macroH2A1.1 to the site of the damage, which transiently causes chromatin rearrangements and dynamically modulates the DNA damage response [13]. Kim et al. reported that PARP1 itself can function as a component of chromatin [216], i.e., histone H1 and PARP1 bind in a competitive and mutually exclusive manner to nucleosomes in vitro. Thereby, PARP1 promotes the local compaction of chromatin into higher-order structures, which are associated with transcriptional repression. The authors suggested that PARP1 modulates the chromatin architecture and gene transcription through its intrinsic enzymatic activity in a DNA-damage-independent manner; i.e., PARP1 activation and automodification triggers its release from chromatin, thereby facilitating chromatin decondensation and gene transcription by RNA polymerase II. Subsequent cellular studies demonstrated that PARP1 could replace histone H1 at RNA polymerase II-transcribed promoters, which was associated with actively transcribed genes [217].
In addition to a functional interplay between PARP1 with histones, an interesting physical and functional interaction exists between PARP1 and DEK. The DEK protein is a major non-histone chromatin component with functions in DNA metabolism and repair on a cellular, and carcinogenesis and autoimmunity on an organismic level. DEK is often found to be upregulated in tumor tissue, and high levels of DEK favor cell immortalization by inhibiting senescence and apoptosis. Consistently, DEK-deficient cells are prone to the induction of senescence in the response to genotoxic stress [218]. We and others have shown that PARP1 PARylates DEK. Moreover, DEK interacts with PAR in a non-covalent manner, which regulates its DNA binding affinity and multimerization with possible implications in response to genotoxic stress and gene transcription. In terms of gene transcription, DEK is released from chromatin upon PARylation to permit transcriptional initiation [208, 211, 219]. Whether DEK itself or its interplay with PARP1 have a direct role in aging mechanisms remains to be clarified.
Importantly, not only structural components of the chromatin are regulated by PARylation, PAR also serves as an important factor in the regulation of chromatin remodeling factors, such as ALC1 and NURD [12, 220–222]. For example, the recruitment of the NURD chromatin remodeling complex to sites of DNA lesions depends on the synthesis of PAR. Interestingly, this complex was identified as an important modulator of aging-associated chromatin defects, and loss of several NURD components and function was evident during human premature aging [223].
The role of PARP1 in gene transcription and chromatin remodeling was impressively demonstrated in a Drosophila study [224]. The authors revealed that PARP1 is crucial for puff formation in giant polytene chromosomes. Puff formation arises from local relaxation of the chromatin structure and is associated with actively transcribed regions [224]. Ju et al. provided interesting mechanistic evidence linking PARP1-dependent initiation of transcription and its function in DNA binding [225]. According to this work, PARP1 acts in concert with another binding partner, i.e., topoisomerase II. Topoisomerase II introduces a transient double strand break at the promoter, which leads to PARP1 binding and activation. The subsequent rapid but transient PARylation triggers chromatin relaxation and initiation of transcription. Furthermore, JIL-1 kinase mediated changes in nucleosome conformation trigger chromatin decondensation via PARylation. JIL-1 phosphorylates the C-terminus of the H2Av histone variant, which stimulates PARP1 enzymatic activity in the surrounding chromatin, leading to further modification of histones and chromatin loosening. The authors propose that chromatin loosening and associated initiation of gene expression is activated by phosphorylation of H2Av in a nucleosome positioned in promoter regions of PARP1 dependent genes [226].
Together, these findings suggest a functional interplay of PARylation and PARPs with chromatin components and associated remodeling factors, implying an active role of PARylation in chromatin function and transcriptional regulation during the aging process. Gene profiling data support such a hypothesis, since PARP1 deficiency alters expression of genes involved in cell cycle progression, DNA replication, oxidative stress, cancer initiation and aging [227, 228]. In addition to PARP1, PARP-2 appears to have overlapping as well as distinct functions from PARP1 in epigenetics and transcription [229]. The detailed spatial and temporal characteristics of these mechanisms in aging and longevity, however, remain to be determined.
3.3 Proteostasis
Besides maintaining genome integrity, homeostasis of the proteome plays an important role in aging and longevity, as accumulation of misfolded or damaged proteins are an important determinant of the aging process [230]. Many mechanisms exist that assure protein quality control in the cell, starting from supporting correct protein folding, such as heat shock family proteins, to several mechanisms of protein degradation such as the ubiquitin-proteasome and the autophagy-lysosomal system [231]. There is now accumulating evidence that some aspects of aging are related to a collapse of proteostasis [232]. Consistent with this view, experimental manipulations that improve proteostasis were able to delay aging in mammals [3, 233].
3.3.1 A Role for PARylation in Protein Folding and the Unfolded Protein Response
The endoplasmatic reticulum (ER) is one of the major cellular organelles involved in protein homeostasis. The major mechanism by which the ER ensures proper protein folding under stressed conditions is the unfolded protein response (UPR). The main function of this system is (i) to shut down further protein synthesis in order to prevent accumulation of misfolded proteins, (ii) to induce ER-associated chaperones to enhance proper protein folding, and (iii) to activate ER-associated degradation system to reduce the burden of misfolded proteins [231]. As a major proteostasis mechanism, many studies linked the UPR with aging and age-associated neurodegenerative diseases, such as Parkinson’s and Alzheimer’s diseases. So far the role of PARylation and PARPs in UPR is limited, but a recent interesting study suggested a role of PARP16, which acts as a mono-ADP-ribsyl-transferase in the UPR by activating the ER stress sensors PERK and IRE1α [234].
Another interesting role for PARylation in protein folding comes from a study that screened for compounds that rescue proper folding of the pathogenic F508 deletion of the cystic fibrosis transmembrane conductance regulator (CFTR). This study identified the natural compounds latonduines as F508del-CFTR correctors. Using chemical proteomics, several PARPs, i.e., PARPs 1-4, as well as TNKS1 and 2, were identified as latonduine binders. Functional analysis revealed that in particular PARP-3 activity is inhibited by latonduines [235]. In accordance with this, another study revealed that inhibiting PARPs, and in particular PARP1, activities restores F508del-CFTR trafficking in different cell lines and mouse embryonic fibroblasts [236]. Although the exact molecular mechanisms for these effects are not clear to date, these studies link PARylation to protein folding. Thus, in response to these studies the existence of a PARP-dependent proteostasis system has been suggested [237]. How exactly PARylation is involved in such mechanisms and how the PARylation-dependent regulation contributes to aging processes, needs to be clarified. Apart from a function in protein folding many other studies indicate that PARylation plays an important role in other proteostasis mechanisms, such as the ubiquitin-proteasome system.
3.3.2 PARylation in the Ubiquitin-Proteasome System
The ubiquitin proteasome network represents the major cellular pathway for the degradation of dynamically regulated or damaged proteins. Modification of proteins with poly-ubiquitin chains targets proteins for proteasomal degradation. The mammalian proteasome consists of a 20S core unit and 19S regulatory particles, located at the ends of the core unit, forming the 26S proteasome [231]. A role for the ubiquitin-proteasome system in mechanisms of aging is well established [238, 239]. In particular it has been shown that proteasomal activity declines with age and that this is associated with impaired capacity to remove damaged proteins [231]. For instance, transgenic mice with decreased proteasomal activity exhibited a shortened lifespan and developed age-related metabolic pathologies [240]. On the other hand, the longest-lived rodent, the naked mole-rat (lifespan of > 30 yrs) shows a three- to sixfold higher proteasome activity than laboratory mice, which promotes an efficient turnover and clearance of misfolded and damaged proteins [241].
A role of PARylation in proteasomal regulation is well established (Fig. 6.6) and this has been directly linked with an age-related loss of proteasome function [242]. Specifically, the 20S proteasome undergoes a rapid activation in response to oxidative stress, and this activation depends on the presence and activity of PARP1 [243–245]. Furthermore, inhibition of PARP1-dependent proteasome activation impaired the DNA repair capacity of cells suggesting an interesting link between the clearance of damaged proteins and the effectiveness of the DNA repair machinery [245]. Of note, replicative senescence of human fibroblasts is associated with dysfunctional stress-induced proteasomal activation in the nucleus [242], and this decline is due to a declined expression and activity of PARP1 both in cultured cells as well as in the skin of aged donors. These results indicate that PARP1 and PARylation play important roles in age-related dysfunction of the proteasome [242]. Apart from PARP1, TNKS activity has been associated with proteasome regulation (Fig. 6.6). Thus TNKS directly interacts with and modifies the proteasome regulator PI31, which reduces its affinity for a specific 20S proteasome subunit, thereby activating 20S proteasome activity. In addition the PI31 PARylation directly stimulates the assembly of the 26S proteasome assembly by promoting the binding of 19S regulatory particles [246].

PARylation in the control of the ubiquitin-proteasome system. The regulation of the ubiquitin-proteasome system is twofold: (i) direct effects of PARP1/TNKS1 on proteasome activity have been described. (ii) TNKS1/PARP1 can target certain proteins for proteasomal degradation through the PAR-dependent attraction of E3 ubiquitin ligases containing a WWE PAR binding sequence (e.g., Iduna/RNF146). For details see text. (Scheme based on [242–245, 247, 248, 257–261])
As it holds true for other cellular processes, the role of PARylation in the ubiquitin proteasome system is manifold, since PARylation not only regulates overall proteasome activity, but also promotes ubiquitination and targeting of specific proteins to proteasomal degradation [247]. Of central importance is the E3 ubiquitin ligase RNF146/Iduna. RNF146’s ligase activity requires non-covalent PAR binding via a WWE domain, thereby targeting proteins for ubiquitination and proteasomal degradation. RNF146 binds a number of proteins that are PARylated including PARP1/2, histones, XRCC1 and several other chromatin and DNA repair factors. Consistent with these findings, RNF146 facilitates DNA repair and promotes cell survival after induction of genotoxic stress [248]. However, no general conclusions can be drawn regarding the question if PARylation promotes or inhibits proteasomal degradation of proteins: Thus on the one hand, PAR-dependent ubiquitination of PARP1 targets it for proteasomal degradation [248], however, another study showed that PARylation of XRCC1 prevents its ubiquitination, despite the fact that it is an interaction partner of RNF146. These results indicate that the effect of PARylation on proteasomal degradation of target proteins highly depends on the target protein itself and the specific conditions and cell types studied. Apart from DNA repair, it has been shown that Wnt signaling is under the control PAR-dependent ubiquitination and proteasomal degradation (Fig. 6.6). The Wnt/β-catenin signaling pathway plays critical roles in embryonic development, stem cell biology, tissue homeostasis, and cancer development. Of note, Wnt/β-catenin signaling increases with aging and has a prominent role in many age-related conditions [249]. If Wnt/β-catenin signaling promotes or counteracts aging is a matter of debate [250]. On the one hand, downregulation of Wnt/β-catenin signaling causes cellular senescence in primary human cells [251]. On the other hand, Wnt/β-catenin signaling is increases in a mouse model of premature aging [252], and inhibition of Wnt/β-catenin signaling rescues the age-related impairment of muscle stem cell function and reduces tissue fibrosis [253, 254]. Moreover, Wnt/β-catenin signaling contributes to mesenchymal stem cell aging by induction of ROS and DNA damage response pathways, linking it to DNA damage and genomic instability [255, 256].
Axin is a scaffold protein of the β-catenin destruction complex and a negative regulator of the Wnt/β-catenin signaling pathway. Active Wnt signaling and translocation of β-catenin to the cell nucleus requires degradation of axin. A series of articles has shown that TNKS1 PARylates axin, which triggers the binding of RNF146, subsequent axin ubiquitination and its proteasomal degradation, thereby releasing β-catenin for nuclear translocation [257–261]. Overall, it appears as if the PARylation-directed ubiquitination and degradation mediated by RNF146/Iduna (and potentially also other E3 ubiquitin ligases comprising WWE domains) evolved as a general mechanism to control protein turnover that is analogous to phosphorylation-directed ubiquitination mediated by the SCF E3 ubiquitin complex [247]. It is very likely that such mechanisms contribute to proteostasis-dependent effects in organismic aging.
3.3.3 PARylation and Autophagy
In addition to proteasomal degradation, autophagy (‘self-digestion’) is another mechanism to turn-over proteins or whole organelles. It is based on the lysosomal degradation system and normally it is activated under stress conditions, such as starvation. Usually, it acts as a cytoprotective mechanism by removing damaged structures and mobilizing bioenergetic sources to ensure cellular survival and homeostasis [231]. However, in its most extreme form it can also lead to self-digestion of the whole cell and therefore to cell death (see below). The role of autophagy in aging and longevity has been extensively reviewed previously [262]. There is now convincing evidence that autophagy represents an aging-related mechanism, since, e.g., normal aging is often associated with a reduced autophagic capacity. Furthermore, genetic inhibition of autophagy induces age-related degenerative changes in mammals, while organismic model systems with increased life span often stimulated autophagic mechanisms [262].
Several studies analyzed the role of PARylation and in particular PARP1 in autophagy [263–268]. According to these studies, PARP1 exerts an active role in DNA-damage-induced autophagy and in the decision if a cell undergoes autophagy or cell death via necrosis. Specifically, PARP1 seems to promote autophagy through the AMPK-mTOR pathways. This pathway is of central importance in the regulation of autophagy, with AMPK generally considered a positive—and mTOR a negative regulator of autophagy. Both proteins exhibit key functions in aging-related mechanisms by controlling the intracellular response to insulin/IGF1 signaling, which plays an important role in molecular mechanisms of life-span extension by caloric restriction [269]. Interestingly, starvation-induced autophagy is linked to DNA damage formation in its early phase, potentially via ROS generation, and PARP1 deficiency strongly delays this autophagic response [267]. This study showed that PARP1 deficiency inhibited AMPK activation and prevented the complete loss of mTOR activity, thereby leading to a delay in autophagy and promoting apoptotic cell death. These results suggest a pro-sruvival role of autophagy and PARP1 activation after nutrient deprivation [267]. Interestingly, rapamycin, which targets the mTOR (mammalian target of rapamycin) pathway and leads to life-span extension in various organisms including mammals, inhibits stress-induced cellular PARylation, thereby linking PARylation with age-related mTOR signaling [270]. In the same line, another study showed that exposing cells to a DNA alkylating agent leads to PARP-dependent immediate drop in NAD+ and ATP levels, while AMP levels strongly increased. This led to activation of AMPK and inhibition of the mTOR pathway, thereby demonstrating that PARP1 and PARylation affect the energetic status of a cell by balancing the route of cell death in response to stress [271].
In conclusion, there is ample evidence that PARylation is involved in cellular proteostasis at multiple levels, including protein folding and maturation as well as protein turn-over via the ubiquitin proteasome and the autophagy pathways. In particular the latter one is closely connected to energy metabolism, another cellular process were PARylation plays an active role, as the PARylation substrate, NAD+, is of central importance in many cellular bioenergetic pathways.
3.4 Energy and NAD+ Metabolism
Dysregulated energy metabolism represents an important aspect in the aging process. This includes several molecular pathways such as insulin and IGF-1 signaling and other nutrient and energy response systems, such as mTOR and AMPK, as briefly discussed above. In general, there is strong support that anabolic signaling accelerates aging and decreased nutrient signaling extends longevity, which is evident from the fact that dietary restriction leads to prolonged life-span and health benefits in all species tested so far including in non-human primates. However, these benefits may come at the cost of reduced stress resistance and fertility [3, 272].
NAD+ is a central metabolic cofactor by functioning as an important redox factor and serving as a substrate for enzymes, such as PARPs as well as the class III deacetylases known as sirtuins [273]. There is ample evidence that NAD+ metabolism plays a crucial role in aging-dependent mechanisms. For example, NAD+ levels are reduced in aged animals, including mammals [274, 275]. Moreover, decreasing NAD+ levels causes a reduction in C. elegans lifespan. Conversely, genetic or pharmacological restoration of NAD+ prevents age-associated metabolic decline and extends longevity in C. elegans [275].
With regards to molecular causes and consequences of age-related changes in NAD+ levels, two classes of enzymes come into play, i.e., PARPs and the family of type III histone-deacetylases of sirtuins (i.e., in humans and mice SIRT1-7). Sirtuins regulate the energy homeostasis by controlling the acetylation status and activity of various enzymes and transcriptional regulators and have been identified to act as longevity factors in various species [276]. In vivo studies demonstrated that SIRT1-overexpressing mice are leaner, metabolically more active, show improved glucose tolerance, exhibit less inflammation, and are resistant to the development of certain types of cancers [277–281]. The action of PARPs and sirtuins is interrelated at three levels: (i) by competition for the common NAD+ substrate, (ii) by mutual posttranslational modifications, and (iii) by direct transcriptional effects [282]. Apart from the aging-relevant interaction of SIRT6 with PARylation (as discussed in Sec. 3.1.1), a crosstalk of PARPs with SIRT1 has been identified and characterized to date: PARP2 -/- mice exhibit increased SIRT1 activity and are protected against diet-induced obesity [283]. In this case, SIRT1 activity was not due to increased availability of NAD+. Instead, PARP-2 serves as a negative regulator of SIRT1 gene expression by controlling the SIRT1 promoter on a transcriptional level [283]. Furthermore, PARP1 and SIRT1 show an antagonistic interplay on a functional level [221, 284]. In contrast to the PARP-2/SIRT1 interaction, the interrelation of PARP1 with SIRT1 is based on the fact that both enzymes compete for NAD+ as a common substrate. This circumstance can be presumably attributed to the higher enzymatic activity of PARP1 compared to PARP-2. Apart from such indirect effects, PARP1 and SIRT1 directly interact with each other physically. In this regard, it has been shown that acetylation of PARP1 upon cellular stress induces its enzymatic activation, thereby potentially causing necrotic cell death via NAD+ /ATP depletion (see below). SIRT1, however, can reverse this acetylation, thereby deactivating PARP1 and promoting cell survival [221]. Consistently, PARP1 -/- mice exhibit increased NAD+ content and enhanced SIRT1 activity in various tissues. Consequently, PARP1 -/- mice phenocopy many aspects of SIRT1 activation, such as a higher mitochondrial content, increased energy expenditure, reduced body weight and protection against metabolic disease [285]. In line with these results, a study on human pelvic skin samples revealed that PARP activity and DNA damage significantly increased with age and inversely correlated with tissue NAD+ levels. On the other hand, sirtuin activity negatively correlated with age, but positively correlated with NAD+ levels [286]. Accordingly, this raises the hypothesis that age-related accumulation of DNA damage leads to chronic PARP activation, which reduces NAD+ levels and thereby compromises sirtuin activity. Strikingly, and in favor of such a scenario, PARP inhibition extended lifespan in C. elegans, and this lifespan extension was dependent on functional sirtuins [275].
3.5 Cell Death and Cellular Senescence
Cell death is a process that is important for the regulation of many physiological processes, such as organismic development, tissue homeostasis, and elimination of cells that encountered irreparable damage [287]. On the other hand, cell death pathways also play significant roles in pathophysiological processes, such as cancer, degenerative diseases and aging. Historically, two major mechanisms of mammalian cell death are distinguished, i.e., apoptosis and necrosis. [NB: In addition, in its most extreme form autophagy can also result in cell death (see above)]. Apoptosis is considered as the default pathway, where cell death occurs in a controlled manner resulting in the elimination of cells by macrophages without secondary damage of the surrounding cells. In contrast, necrosis is considered an uncontrolled process which leads to disruption of cells promoting tissue inflammation [288]. However, mounting evidence indicates that necrosis also occurs in a highly regulated manner [289]. Several transition states between the two pathways exist such as apoptosis inducing factor (AIF)-dependent cell death also known as parthanatos (named after PAR and Thanatos the Greek god of death) [290]. Cell death is an important factor contributing to organismic aging, because apoptosis can lead to depletion of the regenerative cell pool, while necrosis can cause chronic inflammatory conditions that promote age related-pathologies, such as cancer, atherosclerosis, and neurodegenerative diseases (see below).
It has been shown that H2O2 treatment of lymphocytes from young individuals mainly results in necrotic cell death, whereas lymphocytes from older donors undergo apoptosis [291]. Interestingly, in this study both kinds of cell death could almost completely be blocked by PARP inhibition. This result is consistent with the general view that PARP1 is involved in necrosis as well as in apoptosis at various levels, depending on the cell type and the intensity of DNA damage.
Excessive DNA damage, as it can be triggered by pathophysiological stimuli and during NF-κB-dependent inflammatory responses, can lead to PARP1 overactivation, which induces the depletion of cellular NAD+ pools and subsequently of ATP pools triggering bioenergetic failure [271, 292]. Interestingly, under specific conditions DNA repair mechanisms themselves in association with PARP1 activation can contribute to cell death induction, as it was shown that the DNA glycosylase MPG mediates excision of alkylation-induced DNA damage products resulting in strand breaks, subsequent PARP1 activation and necrotic cell death [293]. Moreover not only NAD+ depletion, but also NAD/ATP regeneration processes appear to play important roles in PARP-dependent necrosis, since cells that are depleted in ALKBH7, a mitochondrial ALKBH dioxygenase, exhibit rapid recovery from depleted intracellular NAD and ATP levels and are protected from PARP-dependent alkylation-induced cell death [294]. Future studies will provide further insight into the exact molecular mechanisms leading to PARP-dependent necrosis. Whatever these mechanisms are, current data suggests that PARP-dependent necrosis reinforces tissue inflammation leading to a vicious cycle of PARP1 activation, necrosis and inflammation contributing to age-related diseases.
The role of PARP1 in apoptosis is manifold depending on the cell cycle state. Two major types of apoptosis exist: caspase-dependent and caspase-independent apoptosis. On the one hand, in proliferating cells, PARP1 contributes to classical caspase-dependent apoptosis through its regulatory activity on p53 [295]. Here, after an initial synthesis of PAR, PARP1 is cleaved by caspases 3 and 7 in a 24 kD and an 89 kD fragment [296]. This occurs potentially to inactivate PARP1 and to preserve cellular ATP pools for the apoptosis program [297–299]. On the other hand, it was shown that PARP1 contributes to caspase-independent apoptosis by releasing AIF from the mitochondria in a cell death pathway named parthanatos (see above) [300, 301]. Here, PAR itself acts as a signaling molecule between the nucleus and mitochondria, where it binds to AIF in a non-covalent manner and then triggers its release. AIF then translocates to the nucleus, where it causes chromatin condensation, large scale DNA fragmentation, and finally cell death [302–304]. Importantly, it has been demonstrated recently that this mechanism is responsible for age-dependent dopaminergic neuron loss in a mouse model of Parkinson’s disease [305].
In conclusion, many interconnected cellular mechanisms have been proposed to be responsible for the involvement of PARP1 in cell death and associated age-related pathologies. First, PARP1 overactivation by severe DNA damage upon an initial pathological insult can lead to NAD+ and subsequent ATP depletion causing necrotic cell death due to a bioenergetic crisis [292]. Second, such an initial pathological insult or secondary necrotic disruption of cells can trigger an inflammatory response leading to further damage of the surrounding tissue, thereby supporting the aforementioned vicious cycle of DNA damage, subsequent PARP1 activation, and cell death potentiating inflammation and tissue damage. Third, the PAR-dependent release of apoptosis inducing factor (AIF) from the mitochondria resulting in caspase-independent apoptosis may contribute to some extent to PARP-dependent pathologies in particular neurodegenerative disorders [300, 303–305]. Over time these mechanisms can contribute to aging and the development of age-related pathological conditions.
Besides these detrimental effects, in particular apoptosis represents a classical tumor suppressive mechanism in the adult, since heavily damaged cells that are at risk to undergo malignant transformation, are eliminated from the body. In addition to cell death, cellular senescence exists as a second, alternative mechanism to functionally withdraw heavily damaged cells from the body. Senescence represents a state in which the cell is halted in a permanent cell cycle arrest, unable to divide, but still metabolically active. If a damaged cell dies or enters the state of cellular senescence largely depends on the cell type and the kind of stimulus the cell encountered [3]. Similar to cell death, on the organismic level cellular senescence is thought to be beneficial in the short run, since it withdraws heavily damaged cells from the body and therefore counteracts tumorigenesis. However, in the long run, this could lead to the depletion of the regenerative cell pool and thereby contribute to tissue aging at all levels. Accordingly, there is ample evidence that cellular senescence contributes to organismic aging [3].
A role for PARylation in cellular senescence is obvious based on the fact that many of the mechanism as discussed above, such as DNA damage signaling and cell cycle control, represent upstream mechanisms causing the induction of cellular senescence. Consistent with this notion, PARylation contributes to p53-dependent senescence in aging human fibroblasts and PARP inhibition extents the cellular life span [306].
Apart from depleting the regenerative cell pool, senescence contributes to aging via a second mechanism, as it is associate with a change in cellular physiology leading to the secretion of inflammatory cytokines, a paracrine effect known as senescence-associated secretory phenotype (SASP) [307, 308]. This can contribute to a pro-inflammatory state that may play a causative role in tissue aging [3]. Interestingly, PARylation itself contributes to this SAS phenotype of senescence cells [309], which points to a general role of PARylation in inflammatory processes as discussed in the next section.
3.6 Inflammation and Immunity
A direct relationship exists between physiological aging and increasing incidence of chronic inflammatory diseases. In its acute form inflammation acts as a protective mechanism in response to pathogen invasion or tissue damage and helps to restore physiological integrity and function. However, in its chronic form, inflammation can exert detrimental effects on the cellular as well as the organismic level [310]. The innate immune system, especially the mononuclear phagocyte system, is the most important mediator of chronic inflammation. Macrophages participate in the killing of invading microorganisms and emerging tumor cells through the production of reactive oxygen or nitrogen species (ROS and RNS). In addition, macrophages secrete cytokines, which play a key role in the regulation of multiple immune functions, especially inflammatory responses [310]. During aging, the continuous pressure on the immune system caused by repeated antigen stimulation, such as infections, food antigens, allergens, and self-antigens leads to an increase in activated cells and secretion of proinflammatory cytokines, such as TNFα [311]. These circulating proinflammatory factors may keep the immune system in a state of chronic low-level activation, a phenomenon described as ‘inflammaging’ [308, 312]. Eventually this causes ‘immunosenescence’, i.e., an age-related decline in the capacity of adaptive immunity, consisting of more specific responses carried out by B and T-cells [313]. Thus, with advanced age, the immune system undergoes a gradual remodeling in the attempt to re-establish a new balance that assures survival, however favoring the development of chronic inflammatory conditions [308, 312, 314, 315].
DNA damage and inflammation are inevitably linked by the production of reactive chemical species, such as ROS and RNS. Cellular ROS and RNS production occurs constantly under physiological as well as pathophysiological conditions as a consequence of electron leakage of the mitochondrial electron transport chain and via enzymes such as NADPH oxidase, nitric oxide synthases, and xanthine oxidase. The ‘free radical theory of aging’ posits that aging and its related diseases are the net consequence of free radical-induced damage and the inability to counterbalance these changes by antioxidative defenses and sufficient DNA repair [316]. Chronic inflammation results in the generation of a broad spectrum of ROS and RNS by activated macrophages and neutrophils, which damage cellular macromolecules including DNA [317, 318]. Conversely, the generation of ROS and RNS activates redox sensitive transcription factors, such as NF-κB, resulting in the generation of proinflammatory molecules. Altogether, this can trigger a positive feedback loop that amplifies the processes of inflammation, damage and destruction in target cells and organs, leading to an organismic decline and death over time. For example, chronic inflammation has been associated with an age-related decline in the function of hematopoietic and mesenchymal stem cells [319, 320] and has been implicated as a mediator of almost all of the aging-associated diseases, such as vascular diseases, diabetes, neurodegenerative diseases, and cancer [308, 310, 312, 315].
Various studies demonstrated that PARylation and in particular PARP1 participates in immunological processes, both in adaptive as well as innate immunity [321, 322]. Regarding its function in adaptive immunity, it is evident that, in general, lymphocytes exhibit high PARP1 expression levels, and specifically, it was shown that PARP1 modulates the differentiation and function of various subsets of T and B cells. Furthermore, both PARP1 and PARP14 deficient mice display reduced TH2 cell differentiation and impaired allergic responses [322].
Although our knowledge on the role of PARylation in adaptive immunity is growing, a huge body of evidence demonstrates its involvement in innate immunity and inflammation. The first evidence that PARylation is involved in the regulation of inflammation and the development of related pathologies was revealed by genetic studies in PARP1 -/- mice, because these animals (and to a lesser extent also PARP2 -/- mice) are protected from a series of inflammation and cell death-associated pathologies such as ischemic infarction, collagen-induced arthritis, and LPS-induced septic shock [71, 323]. Moreover, PARP1 -/- animals are resistant to MPTP-induced Parkinson’s disease and streptozotocin-induced diabetes mellitus [324–327]. Maybe the best studied process in this regard is the interaction of PARP1 with NF-κB. The transcription factor NF-κB is considered a master regulator in controlling gene expression upon proinflammatory stimuli. NF-κB is composed of dimeric combinations of Rel family members with the major subunits p65 and p50. In non-stimulated cells, NF-κB is located in the cytoplasm via the binding to the inhibitory IκB proteins. Upon proinflammatory stimuli, IκB proteins are phosphorylated by IκB kinases (IKK), which causes their degradation by the ubiquitin/proteasome system. Subsequently, NF-κB is translocated to the nucleus, where it can activate the transcription of a number of genes, especially inflammatory genes [328]. Apart from the regulation of NF-κB by its sub-cellular localization, its action is tightly regulated within the nucleus by posttranslational modifications and interaction with transcriptional co-factors. Importantly, NF-κB-dependent gene expression is associated with aging in the mouse as well as in humans [329]. Recently, it was shown that hyperactive NF-κB signaling contributes to premature aging in the mouse [128], and blocking of NF-κB in aged mice was sufficient to reverse some features of skin aging [329, 330]. In accordance with these studies, pharmacological inhibition of NF-κB prolongs lifespan of Drosophila melanogaster by ~ 15 % [331]. Cellular studies showed that NF-κB-dependent gene transcription can be induced by genotoxic stress and gene transcription studies in conditionally immortalized human fibroblast suggested that NF-κB signaling plays a causal role in the development of senescence [332]. In addition, NF-κB signaling was implicated in maintaining cellular senescence, because NF-κB-deficient fibroblasts escape senescence earlier and immortalize at a faster rate [333]. On the other hand, NF-κB-dependent gene transcription can be induced by genotoxic stress and is required for the transcription of many SASP factors [308]. In summary, there is substantial evidence that NF-κB plays a crucial role in aging and age-related diseases [334].
The expression and activation patterns of PARP1 and NF-κB are remarkably similar in various tissues. A direct role of PARP1 in NF-κB-mediated transcription was emphasized by the finding that expression of NF-κB-dependent pro-inflammatory mediators, such as TNFα, IL6, or iNOS is impaired in PARP1 -/- mice [323, 335]. PARP1 physically interacts with both major subunits of NF-κB, i.e., p65 and p50, and is required for NF-κB-dependent gene transcription (Fig. 6.7) [336]. Moreover, PARP1 is acetylated by the histone acetylase p300/CBP upon inflammatory stimuli, leading to a stronger association with NF-κB [49]. Subsequent expression of pro-inflammatory mediators such as iNOS lead to the production of highly reactive chemical species that, in turn, cause extensive DNA damage in the target cell, potentially supporting a positive-feedback mechanism. Importantly, in this study neither the DNA binding nor the enzymatic activity of PARP1 were necessary for direct transcriptional activation of NF-κB [337]. On the other hand, inhibition of PARP’s enzymatic activity is sufficient to decrease the expression of iNOS, IL6 and TNFα in cultured cells and to reduce the expression of inflammatory mediators in mice [338]. This is consistent with a recent study demonstrating that the PARP1-dependent activation of NF-κB occurs at two levels (Fig. 6.7): Thus, in addition to the nuclear co-activator function of PARP1 on NF-κB activity, this study identified PARP1 as a trigger for the translocation of NF-κB from the cytoplasm into the nucleus upon genotoxic stress (Fig. 6.7) [339]. According to this model, PARP1 is recruited to DNA strand breaks and is automodified with PAR. Upon dissociation into the nucleoplasm, PARP1 then rapidly forms a signalosome composed of the SUMO1 ligase PIASy, IKKγ (NEMO), and ATM. The signalosome is stabilized by a network of direct protein-protein interactions as well as by PAR binding of PIASy and ATM through PAR binding motifs. PAR degradation by PARG causes subsequent destabilization of the signalosome, resulting in IKKγ SUMOylation, translocation to the cytoplasm, phosphorylation of IκB proteins and NF-kB activation. This mechanism directly links the DNA damage signaling functions of PARP1 to its role in inflammation-related mechanisms. Interestingly, PARP1-NF-κB signaling seems also to contribute to the activation and maintenance of the secretory phenotype of senescent cells [309]. In consequence, the associated secretion of proinflammatory factors possibly changes the tissue microenvironment and forms a site of low level chronic inflammation with tumor and aging-promoting properties.

Evidence supporting a role of PARP1 and PARylation as a driving force of inflammation on an organismic level is given by the fact that PARP1 -/- mice are protected from several inflammation and cell death associated diseases and that PARP1 -/- mice and cells display lower expression levels of a whole spectrum of proinflammatory cytokines, adhesion molecules, and enzymes [323]. Consequently, given the role of inflammation during mammalian aging, PARP1 and PARylation were postulated to act as aging-promoting factors [340]. In line with this concept, PARP inhibition or ablation of gene transcription has beneficial effects on several age-related diseases, including aging-associated cardiac and vascular dysfunctions [338, 341, 342].
Conversely, the phenotype PARP1 -/- mice is mirrored by the phenotype of mice with ectopic expression of hPARP1 [343]. These mice develop sporadic obesity and show impaired glucose tolerance. Furthermore, hPARP1-expressing mice exhibit impaired survival rates, which is accompanied by premature development of several inflammation and age-associated pathologies, such as nephropathy, dermatitis, pneumonitis, myocardiopathy, and hepatitis. In support of this hypothesis, hPARP1 mice develop normocytic, normochromic anemia and show an increase in the fraction of circulating monocytes, which is suggestive of anemia of chronic inflammatory disease [344, 345]. Moreover, hPARP1 mice show typical signs of premature aging, such as early development of kyphosis and impaired hair regrowth. In addition to a potentially altered interplay between PARP1 and sirtuins in these mice, the pathological phenotype of hPARP1 mice might be related to an altered PARP1-NF-κB interaction leading to a continuous low-level increase in pro-inflammatory stimuli. Consistently, expression of NF-κB-dependent target genes, such as TNFα, IL1, and IL6, is dysregulated in hPARP1 animals. This may contribute to the premature development of typical age-related chronic diseases in these mice [343]. Interestingly, hPARP1 mice also display an impaired DNA repair capacity. These results from hPARP1 mice resemble the situation observed in the fungal aging model Podospora anserine. This organism exhibits one PARP homolog and when overexpressing this homolog this leads to increased sensitivity to DNA damaging agents, impaired growth and fertility and a shortened lifespan [346]. Together, these studies indicate that each organism has adapted optimal PARylation levels and responses for its respective molecular environment. Thus, reduced as well as increased PARP expression levels result in impaired genomic integrity.
4 Conclusions
Aging is a complex process which cannot be explained by a single pathway or even a set of closely related pathways. More likely, many diverse cellular functions will contribute to aging and they will do so in a highly inter-dependent manner [204]. As summarized here, this complexity is already represented at the level of a single post-translational modification. PARylation is a factor that connects DNA damage response, epigenetics, transcription, proteostasis, cell death and inflammatory mechanisms, all of which are closely associated with mammalian aging and longevity. Thus, under physiological conditions and mild stresses PARylation is thought to play an important role in genome and protein maintenance (Fig. 6.6). On the other hand, under pathophysiological conditions, PARylation can drive energy dysbalance, inflammation, and cell death, which contribute to the depletion of the regenerative cell pool and tissue dysfunction accumulating in the aging process.
There is ample evidence supporting a role of PARP1 as a longevity assurance factor on the one hand, but also as an aging-promoting factor on the other hand (Fig. 6.8). The dual role of PARP1 in longevity and aging might be reflected in the moderate premature aging phenotype observed in cohorts of PARP1 -/- mice [61]. Thus, it is reasonable to assume that overall aging in these mice is kept nearly in balance, due to compromised genomic integrity on the one hand, but reduced inflammatory status on the other hand. The generation of PARP1 -/- mice with tissue specific reconstitution of PARP1 expression may be a suitable model to test such a hypothesis: Tissue-specific re-expression of PARP1 in cells of the innate immune system on an otherwise PARP1 -/- background may lead to a more drastic accelerated aging phenotype, since PARP1-overexpressing cells of the innate immune system are expected to exhibit an enhanced inflammatory status, while cells of the remaining PARP1 -/- tissues are genomically unstable. Another possibility explaining the moderate premature aging phenotype of PARP1 -/- mice may be that alternative mechanisms are able to compensate for the PARP1 deficiency. Such potential backup mechanisms rely most likely on PARP-2 which shares some redundancy to PARP1, as it is evident by the finding that PARP1/ PARP2 double deficient mice are not viable. The generation of conditional and inducible double knockout mice may help to test this hypothesis.

Pleiotropic role of PARPs and PARylation in health and disease. For details see text. (Modified from [1])
Many theories of aging exist. Most of these are not mutually exclusive and although none of these is probably able to explain all characteristics of human aging, in all probability there is some truth in many of them. The ‘antagonistic pleitropy’ theory of aging postulates the existence of pleiotropic genes and mechanisms having opposite effects on fitness at different stages of age. Thus a gene or mechanism may be beneficial for survival in early life, when natural selection is strong, but harmful at later ages, when selection is weak or absent [347]. Mechanisms of DNA damage response as well as inflammation may support such a theory. DNA damage response, with its final end points, DNA repair, senescence and apoptosis, is clearly beneficial at young age, as these mechanisms prevent cancer development. However at older age, this may become detrimental, as depletion of the regenerative cell pool by senescence or apoptosis may contribute to tissue degeneration and aging. The same holds true for inflammation. At young age, inflammatory responses most likely fulfill beneficial functions, e.g., acting as a first line defense against infections. (N.B.: This is supported by the finding that some mouse models with deficiencies in NF-κB signaling are hypersensitive to infectious diseases [348]) However, at older age continuous pressure on the immune system caused by repeated antigen stimulation leads to remodeling of the immune system with pro-inflammatory properties reinforcing the aging process and the development of age-related disease. Because PARPs and PARylation fulfill key roles in opposing mechanisms such as DNA repair and inflammation, it is conceivable that functions of this post-translational modification act in some aspects in an antagonistic pleiotropic way, with beneficial functions in the youth and detrimental functions at old age (Fig. 6.8).
In conclusion, PARPs and the synthesis of poly(ADP-ribose) are emerging as central factors in general cellular stress response with functions in a plethora of molecular mechanisms, such as chromatin remodeling, transcription, DNA damage signaling, DNA repair, cell cycle regulation, proteostasis, cell death, and inflammation. As reviewed here, there is ample evidence that PARylation fulfills numerous direct as well as indirect roles in mechanisms of aging and longevity, which renders it an interesting factor to study in order to better define mechanisms of the aging process.
References
Mangerich A, Bürkle A (2012) Pleiotropic cellular functions of PARP1 in longevity and aging: genome maintenance meets inflammation. Oxid Med Cell Longev 2012:321653. doi:10.1155/2012/321653
Troen BR (2003) The biology of aging. Mt Sinai J Med 70(1):3–22
López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G (2013) The hallmarks of aging. Cell 153(6):1194–1217
Hottiger MO, Hassa PO, Luscher B, Schuler H, Koch-Nolte F (2010) Toward a unified nomenclature for mammalian ADP-ribosyltransferases. Trends Biochem Sci 35(4):208–219. doi:10.1016/j.tibs.2009.12.003
Zhang Y, Wang J, Ding M, Yu Y (2013) Site-specific characterization of the Asp- and Glu-ADP-ribosylated proteome. Nat Methods 10(10):981–984. doi:10.1038/nmeth.2603
Jungmichel S, Rosenthal F, Altmeyer M, Lukas J, Hottiger MO, Nielsen ML (2013) Proteome-wide Identification of Poly(ADP-Ribosyl)ation Targets in Different Genotoxic Stress Responses. Mol Cell. doi:10.1016/j.molcel.2013.08.026
Min W, Bruhn C, Grigaravicius P, Zhou ZW, Li F, Kruger A, Siddeek B, Greulich KO, Popp O, Meisezahl C, Calkhoven CF, Bürkle A, Xu X, Wang ZQ (2013) Poly(ADP-ribose) binding to Chk1 at stalled replication forks is required for S-phase checkpoint activation. Nat Commun 4:2993. doi:10.1038/ncomms3993
Pleschke JM, Kleczkowska HE, Strohm M, Althaus FR (2000) Poly(ADP-ribose) binds to specific domains in DNA damage checkpoint proteins. J Biol Chem 275(52):40974–40980
Gagne JP, Isabelle M, Lo KS, Bourassa S, Hendzel MJ, Dawson VL, Dawson TM, Poirier GG (2008) Proteome-wide identification of poly(ADP-ribose) binding proteins and poly(ADP-ribose)-associated protein complexes. Nucleic Acids Res 36(22):6959–6976. doi:10.1093/nar/gkn771
Ahel I, Ahel D, Matsusaka T, Clark AJ, Pines J, Boulton SJ, West SC (2008) Poly(ADP-ribose)-binding zinc finger motifs in DNA repair/checkpoint proteins. Nature 451(7174):81–85
Altmeyer M, Messner S, Hassa PO, Fey M, Hottiger MO (2009) Molecular mechanism of poly(ADP-ribosyl)ation by PARP1 and identification of lysine residues as ADP-ribose acceptor sites. Nucleic Acids Res. doi:gkp229 [pii] 10.1093/nar/gkp229
Gottschalk AJ, Timinszky G, Kong SE, Jin J, Cai Y, Swanson SK, Washburn MP, Florens L, Ladurner AG, Conaway JW, Conaway RC (2009) Poly(ADP-ribosyl)ation directs recruitment and activation of an ATP-dependent chromatin remodeler. Proc Natl Acad Sci U S A 106(33):13770–13774. doi:0906920106 [pii] 10.1073/pnas.0906920106
Timinszky G, Till S, Hassa PO, Hothorn M, Kustatscher G, Nijmeijer B, Colombelli J, Altmeyer M, Stelzer EH, Scheffzek K, Hottiger MO, Ladurner AG (2009) A macrodomain-containing histone rearranges chromatin upon sensing PARP1 activation. Nat Struct Mol Biol 16(9):923–929. doi:nsmb.1664 [pii] 10.1038/nsmb.1664
Krietsch J, Rouleau M, Pic E, Ethier C, Dawson TM, Dawson VL, Masson JY, Poirier GG, Gagne JP (2012) Reprogramming cellular events by poly(ADP-ribose)-binding proteins. Mol Aspects Med. doi:10.1016/j.mam.2012.12.005
Hakme A, Wong HK, Dantzer F, Schreiber V (2008) The expanding field of poly(ADP-ribosyl)ation reactions. ‘Protein Modifications: Beyond the Usual Suspects’ review series. EMBO Rep 9(11):1094–1100. doi:10.1038/emb\or.2008.191
Meyer-Ficca ML, Meyer RG, Coyle DL, Jacobson EL, Jacobson MK (2004) Human poly(ADP-ribose) glycohydrolase is expressed in alternative splice variants yielding isoforms that localize to different cell compartments. Exp Cell Res 297(2):521–532. doi:10.1016/j.yexcr.2004.03.050
Niere M, Kernstock S, Koch-Nolte F, Ziegler M (2008) Functional localization of two poly(ADP-ribose)-degrading enzymes to the mitochondrial matrix. Mol Cell Biol 28(2):814–824
Min W, Cortes U, Herceg Z, Tong WM, Wang ZQ (2010) Deletion of the nuclear isoform of poly(ADP-ribose) glycohydrolase (PARG) reveals its function in DNA repair, genomic stability and tumorigenesis. Carcinogenesis 31(12):2058–2065. doi:bgq205 [pii] 10.1093/carcin/bgq205
Barkauskaite E, Brassington A, Tan ES, Warwicker J, Dunstan MS, Banos B, Lafite P, Ahel M, Mitchison TJ, Ahel I, Leys D (2013) Visualization of poly(ADP-ribose) bound to PARG reveals inherent balance between exo- and endo-glycohydrolase activities. Nat Commun 4:2164. doi:10.1038/ncomms3164
Tucker JA, Bennett N, Brassington C, Durant ST, Hassall G, Holdgate G, McAlister M, Nissink JW, Truman C, Watson M (2012) Structures of the human poly (ADP-ribose) glycohydrolase catalytic domain confirm catalytic mechanism and explain inhibition by ADP-HPD derivatives. PLoS ONE 7(12):e50889. doi:10.1371/journal.pone.0050889
Oka S, Kato J, Moss J (2006) Identification and characterization of a mammalian 39-kDa poly(ADP-ribose) glycohydrolase. J Biol Chem 281(2):705–713
Niere M, Mashimo M, Agledal L, Doelle C, Kasamatsu A, Kato J, Moss J, Ziegler M (2012) ADP-ribosylhydrolase 3 (ARH3), not poly-ADP-Ribose glycohydrolase (PARG) isoforms, are responsible for degradation of mitochondrial matrix-associated poly-ADP-ribose. J Biol Chem. doi:M112.349183 [pii] 10.1074/jbc.M112.349183
Rosenthal F, Feijs KL, Frugier E, Bonalli M, Forst AH, Imhof R, Winkler HC, Fischer D, Caflisch A, Hassa PO, Luscher B, Hottiger MO (2013) Macrodomain-containing proteins are new mono-ADP-ribosylhydrolases. Nat Struct Mol Biol. doi:10.1038/nsmb.2521
Jankevicius G, Hassler M, Golia B, Rybin V, Zacharias M, Timinszky G, Ladurner AG (2013) A family of macrodomain proteins reverses cellular mono-ADP-ribosylation. Nat Struct Mol Biol. doi:10.1038/nsmb.2523
Gibson BA, Kraus WL (2012) New insights into the molecular and cellular functions of poly(ADP-ribose) and PARPs. Nat Rev Mol Cell Biol 13(7):411–424. doi:10.1038/nrm3376
Rouleau M, Patel A, Hendzel MJ, Kaufmann SH, Poirier GG (2010) PARP inhibition: PARP1 and beyond. Nat Rev Cancer 10(4):293–301. doi:10.1038/nrc2812
Pion E, Ullmann GM, Ame JC, Gerard D, de Murcia G, Bombarda E (2005) DNA-induced dimerization of poly(ADP-ribose) polymerase-1 triggers its activation. Biochemistry 44(44):14670–14681
Mendoza-Alvarez H, Alvarez-Gonzalez R (1993) Poly(ADP-ribose) polymerase is a catalytic dimer and the automodification reaction is intermolecular. J Biol Chem 268(30):22575–22580
Langelier MF, Planck JL, Roy S, Pascal JM (2012) Structural basis for DNA damage-dependent poly(ADP-ribosyl)ation by human PARP-1. Science 336(6082):728–732. doi:336/6082/728 [pii] 10.1126/science.1216338
Ali AA, Timinszky G, Arribas-Bosacoma R, Kozlowski M, Hassa PO, Hassler M, Ladurner AG, Pearl LH, Oliver AW (2012) The zinc-finger domains of PARP1 cooperate to recognize DNA strand breaks. Nat Struct Mol Biol 19(7):685–692. doi:10.1038/nsmb.2335nsmb.2335 [pii]
Martello R, Mangerich A, Sass S, Dedon PC, Bürkle A (2013) Quantification of cellular poly(ADP-ribosyl)ation by Stable isotope dilution mass spectrometry reveals tissue- and drug-dependent stress response dynamics. ACS Chem Biol 8(7):1567–1575. doi:10.1021/cb400170b
Shieh WM, Ame JC, Wilson MV, Wang ZQ, Koh DW, Jacobson MK, Jacobson EL (1998) Poly(ADP-ribose) polymerase null mouse cells synthesize ADP-ribose polymers. J Biol Chem 273(46):30069–30072
Ame JC, Rolli V, Schreiber V, Niedergang C, Apiou F, Decker P, Muller S, Hoger T, Menissier-de Murcia J, de Murcia G (1999) PARP-2, A novel mammalian DNA damage-dependent poly(ADP-ribose) polymerase. J Biol Chem 274(25):17860–17868
Yelamos J, Schreiber V, Dantzer F (2008) Toward specific functions of poly(ADP-ribose) polymerase-2. Trends Mol Med 14(4):169–178. doi:S1471-4914(08)00062-2 [pii] 10.1016/j.molmed.2008.02.003
Menissier de Murcia J, Ricoul M, Tartier L, Niedergang C, Huber A, Dantzer F, Schreiber V, Ame JC, Dierich A, LeMeur M, Sabatier L, Chambon P, de Murcia G (2003) Functional interaction between PARP-1 and PARP-2 in chromosome stability and embryonic development in mouse. Embo J 22(9):2255–2263
Bai P, Canto C (2012) The role of PARP-1 and PARP-2 enzymes in metabolic regulation and disease. Cell Metab. doi:S1550-4131(12)00320-8 [pii] 10.1016/j.cmet.2012.06.016
Rouleau M, McDonald D, Gagne P, Ouellet ME, Droit A, Hunter JM, Dutertre S, Prigent C, Hendzel MJ, Poirier GG (2007) PARP-3 associates with polycomb group bodies and with components of the DNA damage repair machinery. J Cell Biochem 100(2):385–401. doi:10.1002/jcb.21051
Loseva O, Jemth AS, Bryant HE, Schuler H, Lehtio L, Karlberg T, Helleday T (2010) Poly(ADP-ribose) polymerase-3 (PARP-3) is a mono-ADP ribosylase that activates PARP-1 in absence of DNA. J Biol Chem 285(11):8054–8060. doi:M109.077834 [pii] 10.1074/jbc.M109.077834
Boehler C, Gauthier LR, Mortusewicz O, Biard DS, Saliou JM, Bresson A, Sanglier-Cianferani S, Smith S, Schreiber V, Boussin F, Dantzer F (2011) Poly(ADP-ribose) polymerase 3 (PARP3), a newcomer in cellular response to DNA damage and mitotic progression. Proc Natl Acad Sci U S A. doi:1016574108 [pii] 10.1073/pnas.1016574108
De Vos M, Schreiber V, Dantzer F (2012) The diverse roles and clinical relevance of PARPs in DNA damage repair: current state of the art. Biochem Pharmacol 84(2):137–146. doi:10.1016/j.bcp.2012.03.018
Kickhoefer VA, Siva AC, Kedersha NL, Inman EM, Ruland C, Streuli M, Rome LH (1999) The 193-kD vault protein, VPARP, is a novel poly(ADP-ribose) polymerase. J Cell Biol 146(5):917–928
Mangerich A, Bürkle A (2011) How to kill tumor cells with inhibitors of poly(ADP-ribosyl)ation. Int J Cancer 128(2):251–265. doi:10.1002/ijc.25683
Raval-Fernandes S, Kickhoefer VA, Kitchen C, Rome LH (2005) Increased susceptibility of vault poly(ADP-ribose) polymerase-deficient mice to carcinogen-induced tumorigenesis. Cancer Res 65(19):8846–8852. doi:65/19/8846 [pii] 10.1158/0008-5472.CAN-05-0770
Riffell JL, Lord CJ, Ashworth A (2012) Tankyrase-targeted therapeutics: expanding opportunities in the PARP family. Nat Rev Drug Discov 11(12):923–936. doi:10.1038/nrd3868nrd3868 [pii]
Chiang YJ, Hsiao SJ, Yver D, Cushman SW, Tessarollo L, Smith S, Hodes RJ (2008) Tankyrase 1 and tankyrase 2 are essential but redundant for mouse embryonic development. PLoS ONE 3(7):e2639. doi:10.1371/journal.pone.0002639
Messner S, Schuermann D, Altmeyer M, Kassner I, Schmidt D, Schar P, Muller S, Hottiger MO (2009) Sumoylation of poly(ADP-ribose) polymerase 1 inhibits its acetylation and restrains transcriptional coactivator function. Faseb J 23(11):3978–3989. doi:fj.09-137695 [pii] 10.1096/fj.09-137695
Walker JW, Jijon HB, Madsen KL (2006) AMP-activated protein kinase is a positive regulator of poly(ADP-ribose) polymerase. Biochem Biophys Res Commun 342(1):336–341
Kauppinen TM, Chan WY, Suh SW, Wiggins AK, Huang EJ, Swanson RA (2006) Direct phosphorylation and regulation of poly(ADP-ribose) polymerase-1 by extracellular signal-regulated kinases 1/2. Proc Natl Acad Sci U S A 103(18):7136–7141. doi:10.1073/pnas.0508606103
Hassa PO, Haenni SS, Buerki C, Meier NI, Lane WS, Owen H, Gersbach M, Imhof R, Hottiger MO (2005) Acetylation of Poly(ADP-ribose) Polymerase-1 by p300/CREB-binding Protein Regulates Coactivation of NF-{kappa}B-dependent Transcription. J Biol Chem 280(49):40450–40464
Cohen-Armon M, Visochek L, Rozensal D, Kalal A, Geistrikh I, Klein R, Bendetz-Nezer S, Yao Z, Seger R (2007) DNA-independent PARP-1 activation by phosphorylated ERK2 increases Elk1 activity: a link to histone acetylation. Mol Cell 25(2):297–308
Midorikawa R, Takei Y, Hirokawa N (2006) KIF4 motor regulates activity-dependent neuronal survival by suppressing PARP-1 enzymatic activity. Cell 125(2):371–383
Berger F, Lau C, Ziegler M (2007) Regulation of poly(ADP-ribose) polymerase 1 activity by the phosphorylation state of the nuclear NAD biosynthetic enzyme NMN adenylyl transferase 1. Proc Natl Acad Sci U S A 104(10):3765–3770. doi:0609211104 [pii] 10.1073/pnas.0609211104
Guastafierro T, Cecchinelli B, Zampieri M, Reale A, Riggio G, Sthandier O, Zupi G, Calabrese L, Caiafa P (2008) CCCTC-binding factor activates PARP-1 affecting DNA methylation machinery. J Biol Chem 283(32):21873–21880. doi:M801170200 [pii] 10.1074/jbc.M801170200
Leung AK, Vyas S, Rood JE, Bhutkar A, Sharp PA, Chang P (2011) Poly(ADP-ribose) regulates stress responses and microRNA activity in the cytoplasm. Mol Cell 42(4):489–499. doi:S1097-2765(11)00319-4 [pii] 10.1016/j.molcel.2011.04.015
Vyas S, Chesarone-Cataldo M, Todorova T, Huang YH, Chang P (2013) A systematic analysis of the PARP protein family identifies new functions critical for cell physiology. Nat Commun 4:2240. doi:10.1038/ncomms3240
Grube K, Bürkle A (1992) Poly(ADP-ribose) polymerase activity in mononuclear leukocytes of 13 mammalian species correlates with species-specific life span. Proc Natl Acad Sci U S A 89(24):11759–11763
Beneke S, Scherr AL, Ponath V, Popp O, Bürkle A (2010) Enzyme characteristics of recombinant poly(ADP-ribose) polymerases-1 of rat and human origin mirror the correlation between cellular poly(ADP-ribosyl)ation capacity and species-specific life span. Mech Ageing Dev 131(5):366–369. doi:10.1016/j.mad.2010.04.003
Kunzmann A, Dedoussis G, Jajte J, Malavolta M, Mocchegiani E, Bürkle A (2008) Effect of zinc on cellular poly(ADP-ribosyl)ation capacity. Exp Gerontol 43(5):409–414
Muiras ML, Muller M, Schachter F, Bürkle A (1998) Increased poly(ADP-ribose) polymerase activity in lymphoblastoid cell lines from centenarians. J Mol Med 76(5):346–354
Chevanne M, Calia C, Zampieri M, Cecchinelli B, Caldini R, Monti D, Bucci L, Franceschi C, Caiafa P (2007) Oxidative DNA damage repair and parp 1 and parp 2 expression in Epstein-Barr virus-immortalized B lymphocyte cells from young subjects, old subjects, and centenarians. Rejuvenation Res 10(2):191–204. doi:10.1089/rej.2006.0514
Piskunova TS, Yurova MN, Ovsyannikov AI, Semenchenko AV, Zabezhinski MA, Popovich IG, Wang ZQ, Anisimov VN (2008) Deficiency in poly(ADP-ribose) polymerase-1 (PARP-1) accelerates aging and spontaneous carcinogenesis in mice. Curr Gerontol Geriatr Res
Veith S, Mangerich A (2014) RecQ helicases and PARP1 team up in maintaining genome integrity. Ageing Res Rev. Reprinted with permission of Elsevier
Vijg J, Suh Y (2013) Genome instability and aging. Annu Rev Physiol 75:645–668. doi:10.1146/annurev-physiol-030212-183715
Küpper JH, Müller M, Jacobson MK, Tatsumi-Miyajima J, Coyle DL, Jacobson EL, Bürkle A (1995) trans-dominant inhibition of poly(ADP-ribosyl)ation sensitizes cells against gamma-irradiation and N-methyl-Nʹ-nitro-N-nitrosoguanidine but does not limit DNA replication of a polyomavirus replicon. Mol Cell Biol 15(6):3154–3163
Meyer R, Müller M, Beneke S, Küpper JH, Bürkle A (2000) Negative regulation of alkylation-induced sister-chromatid exchange by poly(ADP-ribose) polymerase-1 activity. Int J Cancer 88(3):351–355
Weidele K, Kunzmann A, Schmitz M, Beneke S, Bürkle A (2010) Ex vivo supplementation with nicotinic acid enhances cellular poly(ADP-ribosyl)ation and improves cell viability in human peripheral blood mononuclear cells. Biochem Pharmacol. doi:S0006-2952(10)00444-2 [pii] 10.1016/j.bcp.2010.06.010
de Murcia JM, Niedergang C, Trucco C, Ricoul M, Dutrillaux B, Mark M, Oliver FJ, Masson M, Dierich A, LeMeur M, Walztinger C, Chambon P, de Murcia G (1997) Requirement of poly(ADP-ribose) polymerase in recovery from DNA damage in mice and in cells. Proc Natl Acad Sci U S A 94(14):7303–7307
Wang ZQ, Stingl L, Morrison C, Jantsch M, Los M, Schulze-Osthoff K, Wagner EF (1997) PARP is important for genomic stability but dispensable in apoptosis. Genes Dev 11(18):2347–2358
Masutani M, Nozaki T, Nishiyama E, Shimokawa T, Tachi Y, Suzuki H, Nakagama H, Wakabayashi K, Sugimura T (1999) Function of poly(ADP-ribose) polymerase in response to DNA damage: gene-disruption study in mice. Mol Cell Biochem 193(1–2):149–152
Trucco C, Oliver FJ, de Murcia G, Menissier-de Murcia J (1998) DNA repair defect in poly(ADP-ribose) polymerase-deficient cell lines. Nucleic Acids Res 26(11):2644–2649
Shall S, de Murcia G (2000) Poly(ADP-ribose) polymerase-1: what have we learned from the deficient mouse model? Mutat Res 460(1):1–15
Tong WM, Cortes U, Hande MP, Ohgaki H, Cavalli LR, Lansdorp PM, Haddad BR, Wang ZQ (2002) Synergistic role of Ku80 and poly(ADP-ribose) polymerase in suppressing chromosomal aberrations and liver cancer formation. Cancer Res 62(23):6990–6996
Nozaki T, Fujihara H, Watanabe M, Tsutsumi M, Nakamoto K, Kusuoka O, Kamada N, Suzuki H, Nakagama H, Sugimura T, Masutani M (2003) PARP-1 deficiency implicated in colon and liver tumorigenesis induced by azoxymethane. Cancer Sci 94(6):497–500
Tsutsumi M, Masutani M, Nozaki T, Kusuoka O, Tsujiuchi T, Nakagama H, Suzuki H, Konishi Y, Sugimura T (2001) Increased susceptibility of poly(ADP-ribose) polymerase-1 knockout mice to nitrosamine carcinogenicity. Carcinogenesis 22(1):1–3
Tong WM, Hande MP, Lansdorp PM, Wang ZQ (2001) DNA strand break-sensing molecule poly(ADP-Ribose) polymerase cooperates with p53 in telomere function, chromosome stability, and tumor suppression. Mol Cell Biol 21(12):4046–4054
Cottet F, Blanche H, Verasdonck P, Le Gall I, Schachter F, Bürkle A, Muiras ML (2000) New polymorphisms in the human poly(ADP-ribose) polymerase-1 coding sequence: lack of association with longevity or with increased cellular poly(ADP-ribosyl)ation capacity. J Mol Med 78(8):431–440
Lockett KL, Hall MC, Xu J, Zheng SL, Berwick M, Chuang SC, Clark PE, Cramer SD, Lohman K, Hu JJ (2004) The ADPRT V762A genetic variant contributes to prostate cancer susceptibility and deficient enzyme function. Cancer Res 64(17):6344–6348. doi:10.1158/0008-5472.CAN-04-033864/17/6344 [pii]
Zhang Q, Li Y, Li X, Zhou W, Shi B, Chen H, Yuan W (2008) PARP-1 Val762Ala polymorphism, CagA(+) H. pylori infection and risk for gastric cancer in Han Chinese population. Mol Biol Rep. doi:10.1007/s11033-008-9336-y
Zhang X, Miao X, Liang G, Hao B, Wang Y, Tan W, Li Y, Guo Y, He F, Wei Q, Lin D (2005) Polymorphisms in DNA base excision repair genes ADPRT and XRCC1 and risk of lung cancer. Cancer Res 65(3):722–726
Wang XG, Wang ZQ, Tong WM, Shen Y (2007) PARP1 Val762Ala polymorphism reduces enzymatic activity. Biochem Biophys Res Commun 354(1):122–126
Alanazi M, Pathan AA, Arifeen Z, Shaik JP, Alabdulkarim HA, Semlali A, Bazzi MD, Parine NR (2013) Association between PARP-1 V762A polymorphism and breast cancer susceptibility in saudi population. PLoS ONE 8(12):e85541. doi:10.1371/journal.pone.0085541
Hua R-X, Li H-P, Liang Y-B, Zhu J-H, Zhang B, Ye S, Dai Q-S, Xiong S-Q, Gu Y, Sun X-Z (2014) Association between the PARP1 Val762Ala Polymorphism and Cancer Risk: Evidence from 43 Studies. PLoS ONE 9(1):e87057. doi:10.1371/journal.pone.0087057
Roszak A, Lianeri M, Sowinska A, Jagodzinski PP (2013) Involvement of PARP-1 Val762Ala polymorphism in the onset of cervical cancer in caucasian women. Mol Diagn Ther. doi:10.1007/s40291-013-0036-5
Garinis GA, van der Horst GT, Vijg J, Hoeijmakers JH (2008) DNA damage and ageing: new-age ideas for an age-old problem. Nat Cell Biol 10(11):1241–1247. doi:ncb1108-1241 [pii] 10.1038/ncb1108–1241
Gorbunova V, Seluanov A, Mao Z, Hine C (2007) Changes in DNA repair during aging. Nucleic Acids Res
Gagne JP, Pic E, Isabelle M, Krietsch J, Ethier C, Paquet E, Kelly I, Boutin M, Moon KM, Foster LJ, Poirier GG (2012) Quantitative proteomics profiling of the poly(ADP-ribose)-related response to genotoxic stress. Nucleic Acids Res 40(16):7788–7805. doi:10.1093/nar/gks486
Haince JF, McDonald D, Rodrigue A, Dery U, Masson JY, Hendzel MJ, Poirier GG (2008) PARP1-dependent kinetics of recruitment of MRE11 and NBS1 proteins to multiple DNA damage sites. J Biol Chem 283(2):1197–1208
Mortusewicz O, Ame JC, Schreiber V, Leonhardt H (2007) Feedback-regulated poly(ADP-ribosyl)ation by PARP-1 is required for rapid response to DNA damage in living cells. Nucleic Acids Res 35(22):7665–7675
Xu G, Herzig M, Rotrekl V, Walter CA (2008) Base excision repair, aging and health span. Mech Ageing Dev 129(7–8):366–382
Hoeijmakers JH (2001) Genome maintenance mechanisms for preventing cancer. Nature 411(6835):366–374
Gradwohl G, Menissier de Murcia JM, Molinete M, Simonin F, Koken M, Hoeijmakers JH, de Murcia G (1990) The second zinc-finger domain of poly(ADP-ribose) polymerase determines specificity for single-stranded breaks in DNA. Proc Natl Acad Sci U S A 87(8):2990–2994
Molinete M, Vermeulen W, Bürkle A, Menissier-de Murcia J, Kupper JH, Hoeijmakers JH, de Murcia G (1993) Overproduction of the poly(ADP-ribose) polymerase DNA-binding domain blocks alkylation-induced DNA repair synthesis in mammalian cells. Embo J 12(5):2109–2117
Noren Hooten N, Kompaniez K, Barnes J, Lohani A, Evans MK (2011) Poly(ADP-ribose) polymerase 1 (PARP-1) binds to 8-oxoguanine-DNA glycosylase (OGG1). J Biol Chem 286(52):44679–44690. doi:10.1074/jbc.M111.255869
Noren Hooten N, Fitzpatrick M, Kompaniez K, Jacob KD, Moore BR, Nagle J, Barnes J, Lohani A, Evans MK (2012) Coordination of DNA repair by NEIL1 and PARP-1: a possible link to aging. Aging 4(10):674–685. doi:100492 [pii]
Masson M, Niedergang C, Schreiber V, Muller S, Menissier-de Murcia J, de Murcia G (1998) XRCC1 is specifically associated with poly(ADP-ribose) polymerase and negatively regulates its activity following DNA damage. Mol Cell Biol 18(6):3563–3571
El-Khamisy SF, Masutani M, Suzuki H, Caldecott KW (2003) A requirement for PARP-1 for the assembly or stability of XRCC1 nuclear foci at sites of oxidative DNA damage. Nucleic Acids Res 31(19):5526–5533
Campalans A, Kortulewski T, Amouroux R, Menoni H, Vermeulen W, Radicella JP (2013) Distinct spatiotemporal patterns and PARP dependence of XRCC1 recruitment to single-strand break and base excision repair. Nucleic Acids Res. doi:10.1093/nar/gkt025
Caldecott KW, Aoufouchi S, Johnson P, Shall S (1996) XRCC1 polypeptide interacts with DNA polymerase beta and possibly poly (ADP-ribose) polymerase, and DNA ligase III is a novel molecular ‘nick-sensor’ in vitro. Nucleic Acids Res 24(22):4387–4394
Leppard JB, Dong Z, Mackey ZB, Tomkinson AE (2003) Physical and functional interaction between DNA ligase IIIalpha and poly(ADP-Ribose) polymerase 1 in DNA single-strand break repair. Mol Cell Biol 23(16):5919–5927
Confer NF, Kumari SR, Alvarez-Gonzalez R (2004) Biochemical association of poly(ADP-ribose) polymerase-1 and its apoptotic peptide fragments with DNA polymerase beta. Chem Biodivers 1(10):1476–1486
Schreiber V, Ame J-C, Dolle P, Schultz I, Rinaldi B, Fraulob V, Menissier-de Murcia J, de Murcia G (2002) Poly(ADP-ribose) polymerase-2 (PARP-2) is required for efficient base excision DNA repair in association with PARP-1 and XRCC1. J Biol Chem 277(25):23028–23036
Mortusewicz O, Ame JC, Schreiber V, Leonhardt H (2007) Feedback-regulated poly(ADP-ribosyl)ation by PARP-1 is required for rapid response to DNA damage in living cells. Nucleic Acids Res 35(22):7665–7675
Fahrer J, Kranaster R, Altmeyer M, Marx A, Bürkle A (2007) Quantitative analysis of the binding affinity of poly(ADP-ribose) to specific binding proteins as a function of chain length. Nucleic Acids Res 35(21):e143. doi:10.1093/nar/gkm944
Thorslund T, von Kobbe C, Harrigan JA, Indig FE, Christiansen M, Stevnsner T, Bohr VA (2005) Cooperation of the Cockayne syndrome group B protein and poly(ADP-ribose) Polymerase 1 in the response to oxidative stress. Mol Cell Biol 25(17):7625–7636
Vodenicharov MD, Ghodgaonkar MM, Halappanavar SS, Shah RG, Shah GM (2005) Mechanism of early biphasic activation of poly(ADP-ribose) polymerase-1 in response to ultraviolet B radiation. J Cell Sci 118(Pt 3):589–599. doi:10.1242/jcs.01636
Pines A, Vrouwe MG, Marteijn JA, Typas D, Luijsterburg MS, Cansoy M, Hensbergen P, Deelder A, de Groot A, Matsumoto S, Sugasawa K, Thoma N, Vermeulen W, Vrieling H, Mullenders L (2012) PARP1 promotes nucleotide excision repair through DDB2 stabilization and recruitment of ALC1. J Cell Biol 199(2):235–249. doi:10.1083/jcb.201112132
Ghodgaonkar MM, Zacal N, Kassam S, Rainbow AJ, Shah GM (2008) Depletion of poly(ADP-ribose) polymerase-1 reduces host cell reactivation of a UV-damaged adenovirus-encoded reporter gene in human dermal fibroblasts. DNA Repair (Amst) 7(4):617–632
Epstein JH, Cleaver JE (1992) 3-Aminobenzamide can act as a cocarcinogen for ultraviolet light-induced carcinogenesis in mouse skin. Cancer Res 52(14):4053–4054
Robu M, Shah RG, Petitclerc N, Brind'amour J, Kandan-Kulangara F, Shah GM (2013) Role of poly(ADP-ribose) polymerase-1 in the removal of UV-induced DNA lesions by nucleotide excision repair. Proc Natl Acad Sci U S A. doi:10.1073/pnas.1209507110
Pines A, Mullenders LH, van Attikum H, Luijsterburg MS (2013) Touching base with PARPs: moonlighting in the repair of UV lesions and double-strand breaks. Trends Biochem Sci. doi:10.1016/j.tibs.2013.03.002
Luijsterburg MS, Lindh M, Acs K, Vrouwe MG, Pines A, van Attikum H, Mullenders LH, Dantuma NP (2012) DDB2 promotes chromatin decondensation at UV-induced DNA damage. J Cell Biol 197(2):267–281. doi:10.1083/jcb.201106074
Fischer JM, Popp O, Gebhard D, Veith S, Fischbach A, Beneke S, Leitenstorfer A, Bergemann J, Scheffner M, Ferrando-May E, Mangerich A, Bürkle A (2014) Poly(ADP-ribose)-mediated interplay of XPA and PARP1 leads to reciprocal regulation of protein function. FEBS J 281(16):3625–3641
King BS, Cooper KL, Liu KJ, Hudson LG (2012) Poly(ADP-ribose) contributes to an association between poly(ADP-ribose) polymerase-1 and xeroderma pigmentosum complementation group A in nucleotide excision repair. J Biol Chem 287(47):39824–39833. doi:10.1074/jbc.M112.393504
Li H, Mitchell JR, Hasty P (2008) DNA double-strand breaks: a potential causative factor for mammalian aging? Mech Ageing Dev 129(7–8):416–424. doi:10.1016/j.mad.2008.02.002
Shrivastav M, De Haro LP, Nickoloff JA (2008) Regulation of DNA double-strand break repair pathway choice. Cell Res 18(1):134–147
Wang M, Wu W, Rosidi B, Zhang L, Wang H, Iliakis G (2006) PARP-1 and Ku compete for repair of DNA double strand breaks by distinct NHEJ pathways. Nucleic Acids Res 34(21):6170–6182
Aguilar Quesada R, Munoz-Gamez JA, Martin-Oliva D, Peralta A, Valenzuela T, Martinez-Romero R, Quiles-Perez R, Menissier de Murcia J, De Murcia G, Ruiz de Almodovar M, Oliver FJ (2007) Interaction between ATM and PARP-1 in response to DNA damage and sensitization of ATM deficient cells through PARP inhibition. BMC Mol Biol 8(1):29
Haince JF, Kozlov S, Dawson VL, Dawson TM, Hendzel MJ, Lavin MF, Poirier GG (2007) Ataxia telangiectasia mutated (ATM) signaling network is modulated by a novel poly(ADP-ribose)-dependent pathway in the early response to DNA-damaging agents. J Biol Chem 282(22):16441–16453
Ruscetti T, Lehnert BE, Halbrook J, Le Trong H, Hoekstra MF, Chen DJ, Peterson SR (1998) Stimulation of the DNA-dependent protein kinase by poly(ADP-ribose) polymerase. J Biol Chem 273(23):14461–14467
Spagnolo L, Barbeau J, Curtin NJ, Morris EP, Pearl LH (2012) Visualization of a DNA-PK/PARP1 complex. Nucleic Acids Res 40(9):4168–4177. doi:gkr1231 [pii] 10.1093/nar/gkr1231
Audebert M, Salles B, Calsou P (2004) Involvement of poly(ADP-ribose) polymerase-1 and XRCC1/DNA ligase III in an alternative route for DNA double-strand breaks rejoining. J Biol Chem 279(53):55117–55126
Wray J, Williamson EA, Singh SB, Wu Y, Cogle CR, Weinstock DM, Zhang Y, Lee SH, Zhou D, Shao L, Hauer-Jensen M, Pathak R, Klimek V, Nickoloff JA, Hromas R (2013) PARP1 is required for chromosomal translocations. Blood 121(21):4359–4365. doi:10.1182/blood-2012-10-460527
Krietsch J, Caron MC, Gagne JP, Ethier C, Vignard J, Vincent M, Rouleau M, Hendzel MJ, Poirier GG, Masson JY (2012) PARP activation regulates the RNA-binding protein NONO in the DNA damage response to DNA double-strand breaks. Nucleic Acids Res. doi:gks798 [pii] 10.1093/nar/gks798
Dominguez-Bendala J, Masutani M, McWhir J (2006) Down-regulation of PARP-1, but not of Ku80 or DNA-PKcs’, results in higher gene targeting efficiency. Cell Biol Int 30(4):389–393. doi:10.1016/j.cellbi.2005.12.005
Waldman AS, Waldman BC (1991) Stimulation of intrachromosomal homologous recombination in mammalian cells by an inhibitor of poly(ADP-ribosylation). Nucleic Acids Res 19(21):5943–5947
Semionov A, Cournoyer D, Chow TY (2003) 1,5-isoquinolinediol increases the frequency of gene targeting by homologous recombination in mouse fibroblasts. Biochem Cell Biol 81(1):17–24
Kugel S, Mostoslavsky R (2014) Chromatin and beyond: the multitasking roles for SIRT6. Trends Biochem Sci 39(2):72–81. doi:10.1016/j.tibs.2013.12.002
Kawahara TL, Michishita E, Adler AS, Damian M, Berber E, Lin M, McCord RA, Ongaigui KC, Boxer LD, Chang HY, Chua KF (2009) SIRT6 links histone H3 lysine 9 deacetylation to NF-kappaB-dependent gene expression and organismal life span. Cell 136(1):62–74. doi:S0092-8674(08)01446-3 [pii] 10.1016/j.cell.2008.10.052
Kanfi Y, Naiman S, Amir G, Peshti V, Zinman G, Nahum L, Bar-Joseph Z, Cohen HY (2012) The sirtuin SIRT6 regulates lifespan in male mice. Nature 483(7388):218–221. doi:10.1038/nature10815
Mao Z, Hine C, Tian X, Van Meter M, Au M, Vaidya A, Seluanov A, Gorbunova V (2011) SIRT6 promotes DNA repair under stress by activating PARP1. Science 332(6036):1443–1446. doi:332/6036/1443 [pii] 10.1126/science.1202723
Mao Z, Tian X, Van Meter M, Ke Z, Gorbunova V, Seluanov A (2012) Sirtuin 6 (SIRT6) rescues the decline of homologous recombination repair during replicative senescence. Proc Natl Acad Sci U S A 109(29):11800–11805. doi:1200583109 [pii] 10.1073/pnas.1200583109
Beck C, Boehler C, Barbat JG, Bonnet ME, Illuzzi G, Ronde P, Gauthier LR, Magroun N, Rajendran A, Lopez BS, Scully R, Boussin FD, Schreiber V, Dantzer F (2014) PARP3 affects the relative contribution of homologous recombination and nonhomologous end-joining pathways. Nucleic Acids Res. doi:10.1093/nar/gku174
Rulten SL, Fisher AE, Robert I, Zuma MC, Rouleau M, Ju L, Poirier G, Reina-San-Martin B, Caldecott KW (2011) PARP-3 and APLF function together to accelerate nonhomologous end-joining. Mol Cell 41(1):33–45. doi:S1097-2765(10)00962-7 [pii] 10.1016/j.molcel.2010.12.006
Fenton AL, Shirodkar P, Macrae CJ, Meng L, Koch CA (2013) The PARP3- and ATM-dependent phosphorylation of APLF facilitates DNA double-strand break repair. Nucleic Acids Res 41(7):4080–4092. doi:10.1093/nar/gkt134
Armanios M, Blackburn EH (2012) The telomere syndromes. Nat Rev Genet 13(10):693–704. doi:10.1038/nrg3246
Blasco MA (2007) Telomere length, stem cells and aging. Nat Chem Biol 3(10):640–649. doi:10.1038/nchembio.2007.38
Jaskelioff M, Muller FL, Paik JH, Thomas E, Jiang S, Adams AC, Sahin E, Kost-Alimova M, Protopopov A, Cadinanos J, Horner JW, Maratos-Flier E, Depinho RA (2011) Telomerase reactivation reverses tissue degeneration in aged telomerase-deficient mice. Nature 469(7328):102–106. doi:10.1038/nature09603
Smith S, Giriat I, Schmitt A, de Lange T (1998) Tankyrase, a poly(ADP-ribose) polymerase at human telomeres. Science 282(5393):1484–1487
Zhang H, Yang MH, Zhao JJ, Chen L, Yu ST, Tang XD, Fang DC, Yang SM (2010) Inhibition of tankyrase 1 in human gastric cancer cells enhances telomere shortening by telomerase inhibitors. Oncol Rep 24(4):1059–1065
Dregalla RC, Zhou J, Idate RR, Battaglia CL, Liber HL, Bailey SM (2010) Regulatory roles of tankyrase 1 at telomeres and in DNA repair: suppression of T-SCE and stabilization of DNA-PKcs. Aging 2(10):691–708
d'Adda diFF, Hande MP, Tong WM, Lansdorp PM, Wang ZQ, Jackson SP (1999) Functions of poly(ADP-ribose) polymerase in controlling telomere length and chromosomal stability. Nat Genet 23(1):76–80
Gomez M, Wu J, Schreiber V, Dunlap J, Dantzer F, Wang Y, Liu Y (2006) PARP1 Is a TRF2-associated poly(ADP-ribose)polymerase and protects eroded telomeres. Mol Biol Cell 17(4):1686–1696
Beneke S, Cohausz O, Malanga M, Boukamp P, Althaus F, Bürkle A (2008) Rapid regulation of telomere length is mediated by poly(ADP-ribose) polymerase-1. Nucleic Acids Res 36(19):6309–6317
Beneke S, Bürkle A (2007) Poly(ADP-ribosyl)ation in mammalian ageing. Nucleic Acids Res 35(22):7456–7465
O’Connor MS, Safari A, Liu D, Qin J, Songyang Z (2004) The human Rap1 protein complex and modulation of telomere length. J Biol Chem 279(27):28585–28591
Rossi ML, Ghosh AK, Bohr VA (2010) Roles of Werner syndrome protein in protection of genome integrity. DNA Repair (Amst) 9(3):331–344. doi:10.1016/j.dnarep.2009.12.011
Chang S, Multani AS, Cabrera NG, Naylor ML, Laud P, Lombard D, Pathak S, Guarente L, DePinho RA (2004) Essential role of limiting telomeres in the pathogenesis of Werner syndrome. Nat Genet 36(8):877–882. doi:10.1038/ng1389ng1389 [pii]
Aggarwal M, Sommers JA, Shoemaker RH, Brosh RM Jr (2011) Inhibition of helicase activity by a small molecule impairs Werner syndrome helicase (WRN) function in the cellular response to DNA damage or replication stress. Proc Natl Acad Sci U S A 108(4):1525–1530. doi:1006423108 [pii] 10.1073/pnas.1006423108
von Kobbe C, Harrigan JA, Schreiber V, Stiegler P, Piotrowski J, Dawut L, Bohr VA (2004) Poly(ADP-ribose) polymerase 1 regulates both the exonuclease and helicase activities of the Werner syndrome protein. Nucleic Acids Res 32(13):4003–4014. doi:10.1093/nar/gkh72132/13/4003 [pii]
Adelfalk C, Kontou M, Hirsch-Kauffmann M, Schweiger M (2003) Physical and functional interaction of the Werner syndrome protein with poly-ADP ribosyl transferase. FEBS Lett 554(1–2):55–58. doi:S0014579303010883 [pii]
Popp O, Veith S, Fahrer J, Bohr VA, Bürkle A, Mangerich A (2013) Site-specific noncovalent interaction of the biopolymer poly(ADP-ribose) with the Werner syndrome protein regulates protein functions. ACS Chem Biol 8(1):179–188. doi:10.1021/cb300363 g
von Kobbe C, Harrigan JA, May A, Opresko PL, Dawut L, Cheng WH, Bohr VA (2003) Central role for the Werner syndrome protein/poly(ADP-ribose) polymerase 1 complex in the poly(ADP-ribosyl)ation pathway after DNA damage. Mol Cell Biol 23(23):8601–8613
Li B, Navarro S, Kasahara N, Comai L (2004) Identification and biochemical characterization of a Werner’s syndrome protein complex with Ku70/80 and poly(ADP-ribose) polymerase-1. J Biol Chem 279(14):13659–13667. doi:10.1074/jbc.M311606200M311606200 [pii]
Lebel M, Lavoie J, Gaudreault I, Bronsard M, Drouin R (2003) Genetic cooperation between the Werner syndrome protein and poly(ADP-ribose) polymerase-1 in preventing chromatid breaks, complex chromosomal rearrangements, and cancer in mice. Am J Pathol 162(5):1559–1569. doi:S0002-9440(10)64290-3 [pii] 10.1016/S0002-9440(10)64290–3
Ruzankina Y, Asare A, Brown EJ (2008) Replicative stress, stem cells and aging. Mech Ageing Dev 129(7–8):460–466. doi:S0047-6374(08)00075-4 [pii] 10.1016/j.mad.2008.03.009
Burhans WC, Weinberger M (2007) DNA replication stress, genome instability and aging. Nucleic Acids Res 35(22):7545–7556. doi:gkm1059 [pii] 10.1093/nar/gkm1059
Jones RM, Petermann E (2012) Replication fork dynamics and the DNA damage response. Biochem J 443(1):13–26. doi:BJ20112100 [pii] 10.1042/BJ20112100
Yang YG, Cortes U, Patnaik S, Jasin M, Wang ZQ (2004) Ablation of PARP-1 does not interfere with the repair of DNA double-strand breaks, but compromises the reactivation of stalled replication forks. Oncogene 23(21):3872–3882. doi:10.1038/sj.onc.12074911207491 [pii]
Bryant HE, Petermann E, Schultz N, Jemth AS, Loseva O, Issaeva N, Johansson F, Fernandez S, McGlynn P, Helleday T (2009) PARP is activated at stalled forks to mediate Mre11-dependent replication restart and recombination. Embo J 28(17):2601–2615. doi:emboj2009206 [pii] 10.1038/emboj.2009.206
Ying S, Hamdy FC, Helleday T (2012) Mre11-dependent degradation of stalled DNA replication forks is prevented by BRCA2 and PARP1. Cancer Res. doi:0008-5472.CAN-11-3417 [pii] 10.1158/0008-5472.CAN-11-3417
Ray Chaudhuri A, Hashimoto Y, Herrador R, Neelsen KJ, Fachinetti D, Bermejo R, Cocito A, Costanzo V, Lopes M (2012) Topoisomerase I poisoning results in PARP-mediated replication fork reversal. Nat Struct Mol Biol 19(4):417–423. doi:10.1038/nsmb.2258
Berti M, Chaudhuri AR, Thangavel S, Gomathinayagam S, Kenig S, Vujanovic M, Odreman F, Glatter T, Graziano S, Mendoza-Maldonado R, Marino F, Lucic B, Biasin V, Gstaiger M, Aebersold R, Sidorova JM, Monnat RJ Jr, Lopes M, Vindigni A (2013) Human RECQ1 promotes restart of replication forks reversed by DNA topoisomerase I inhibition. Nat Struct Mol Biol 20(3):347–354. doi:10.1038/nsmb.2501
Sharma S, Phatak P, Stortchevoi A, Jasin M, Larocque JR (2012) RECQ1 plays a distinct role in cellular response to oxidative DNA damage. DNA Repair (Amst) 11(6):537–549. doi:S1568-7864(12)00094-8 [pii] 10.1016/j.dnarep.2012.04.003
Scholey JM, Brust-Mascher I, Mogilner A (2003) Cell division. Nature 422(6933):746–752
Baker DJ, Jeganathan KB, Cameron JD, Thompson M, Juneja S, Kopecka A, Kumar R, Jenkins RB, de Groen PC, Roche P, van Deursen JM (2004) BubR1 insufficiency causes early onset of aging-associated phenotypes and infertility in mice. Nat Genet 36(7):744–749
Baker DJ, Jeganathan KB, Malureanu L, Perez-Terzic C, Terzic A, van Deursen JM (2006) Early aging-associated phenotypes in Bub3/Rae1 haploinsufficient mice. J Cell Biol 172(4):529–540
Baker DJ, Dawlaty MM, Wijshake T, Jeganathan KB, Malureanu L, van Ree JH, Crespo-Diaz R, Reyes S, Seaburg L, Shapiro V, Behfar A, Terzic A, van de Sluis B, van Deursen JM (2013) Increased expression of BubR1 protects against aneuploidy and cancer and extends healthy lifespan. Nat Cell Biol 15(1):96–102. doi:10.1038/ncb2643
Wijshake T, Malureanu LA, Baker DJ, Jeganathan KB, van de Sluis B, van Deursen JM (2012) Reduced life- and healthspan in mice carrying a mono-allelic BubR1 MVA mutation. PLoS Genet 8(12):e1003138. doi:10.1371/journal.pgen.1003138
Chang P, Jacobson MK, Mitchison TJ (2004) Poly(ADP-ribose) is required for spindle assembly and structure. Nature 432(7017):645–649
Chang P, Coughlin M, Mitchison TJ (2005) Tankyrase-1 polymerization of poly(ADP-ribose) is required for spindle structure and function. Nat Cell Biol 7(11):1133–1139. doi:ncb1322 [pii] 10.1038/ncb1322
Chang P, Coughlin M, Mitchison TJ (2009) Interaction between Poly(ADP-ribose) and NuMA contributes to mitotic spindle pole assembly. Mol Biol Cell 20(21):4575–4585. doi:E09-06-0477 [pii] 10.1091/mbc.E09-06-0477
Ozaki Y, Matsui H, Asou H, Nagamachi A, Aki D, Honda H, Yasunaga S, Takihara Y, Yamamoto T, Izumi S, Ohsugi M, Inaba T (2012) Poly-ADP ribosylation of Miki by tankyrase-1 promotes centrosome maturation. Mol Cell 47(5):694–706. doi:10.1016/j.molcel.2012.06.033
Kim MK, Dudognon C, Smith S (2012) Tankyrase 1 regulates centrosome function by controlling CPAP stability. EMBO Rep 13(8):724–732. doi:10.1038/embor.2012.86
Augustin A, Spenlehauer C, Dumond H, Menissier-De Murcia J, Piel M, Schmit AC, Apiou F, Vonesch JL, Kock M, Bornens M, De Murcia G (2003) PARP-3 localizes preferentially to the daughter centriole and interferes with the G1/S cell cycle progression. J Cell Sci 116(8):1551–1562
Kanai M, Tong WM, Wang ZQ, Miwa M (2007) Haploinsufficiency of poly(ADP-ribose) polymerase-1-mediated poly(ADP-ribosyl)ation for centrosome duplication. Biochem Biophys Res Commun 359(3):426–430
Kanai M, Uchida M, Hanai S, Uematsu N, Uchida K, Miwa M (2000) Poly(ADP-ribose) polymerase localizes to the centrosomes and chromosomes. Biochem Biophys Res Commun 278(2):385–389
Kanai M, Tong WM, Sugihara E, Wang ZQ, Fukasawa K, Miwa M (2003) Involvement of poly(ADP-Ribose) polymerase 1 and poly(ADP-Ribosyl)ation in regulation of centrosome function. Mol Cell Biol 23(7):2451–2462
Saxena A, Wong LH, Kalitsis P, Earle E, Shaffer LG, Choo KH (2002) Poly(ADP-ribose) polymerase 2 localizes to mammalian active centromeres and interacts with PARP-1, Cenpa, Cenpb and Bub3, but not Cenpc. Hum Mol Genet 11(19):2319–2329
Saxena A, Saffery R, Wong LH, Kalitsis P, Choo KH (2002) Centromere proteins Cenpa, Cenpb, and Bub3 interact with poly(ADP-ribose) polymerase-1 protein and are poly(ADP-ribosyl)ated. J Biol Chem 277(30):26921–26926
Reinhardt HC, Schumacher B (2012) The p53 network: cellular and systemic DNA damage responses in aging and cancer. Trends Genet 28(3):128–136. doi:S0168-9525(11)00199-5 [pii] 10.1016/j.tig.2011.12.002
Wesierska-Gadek J, Ranftler C, Schmid G (2005) Physiological ageing: role of p53 and PARP-1 tumor suppressors in the regulation of terminal senescence. J Physiol Pharmacol 56(Suppl 2):77–88
Tong WM, Ohgaki H, Huang H, Granier C, Kleihues P, Wang ZQ (2003) Null mutation of DNA strand break-binding molecule poly(ADP-ribose) polymerase causes medulloblastomas in p53(-/-) mice. Am J Pathol 162(1):343–352
Tong WM, Yang YG, Cao WH, Galendo D, Frappart L, Shen Y, Wang ZQ (2007) Poly(ADP-ribose) polymerase-1 plays a role in suppressing mammary tumourigenesis in mice. Oncogene 26: 3857-3867.
Beneke R, Moroy T (2001) Inhibition of poly(ADP-ribose) polymerase activity accelerates T-cell lymphomagenesis in p53 deficient mice. Oncogene 20(56):8136–8141
Kumari SR, Mendoza-Alvarez H, Alvarez-Gonzalez R (1998) Functional interactions of p53 with poly(ADP-ribose) polymerase (PARP) during apoptosis following DNA damage: covalent poly(ADP-ribosyl)ation of p53 by exogenous PARP and noncovalent binding of p53 to the M(r) 85,000 proteolytic fragment. Cancer Res 58(22):5075–5078
Valenzuela MT, Guerrero R, Nunez MI, Ruiz DAlmodovarJM, Sarker M, de Murcia G, Oliver FJ (2002) PARP-1 modifies the effectiveness of p53-mediated DNA damage response. Oncogene 21(7):1108–1116
Wang X, Ohnishi K, Takahashi A, Ohnishi T (1998) Poly(ADP-ribosyl)ation is required for p53-dependent signal transduction induced by radiation. Oncogene 17(22):2819–2825
Wieler S, Gagne JP, Vaziri H, Poirier GG, Benchimol S (2003) Poly(ADP-ribose) polymerase-1 is a positive regulator of the p53-mediated G1 arrest response following ionizing radiation. J Biol Chem 278(21):18914–18921
Lee MH, Na H, Kim EJ, Lee HW, Lee MO (2012) Poly(ADP-ribosyl)ation of p53 induces gene-specific transcriptional repression of MTA1. Oncogene. doi:10.1038/onc.2012.2onc20122 [pii]
Mendoza-Alvarez H, Alvarez-Gonzalez R (2001) Regulation of p53 sequence-specific DNA-binding by covalent poly(ADP-ribosyl)ation. J Biol Chem 276(39):36425–36430
Simbulan-Rosenthal CM, Rosenthal DS, Luo RB, Samara R, Jung M, Dritschilo A, Spoonde A, Smulson ME (2001) Poly(ADP-ribosyl)ation of p53 in vitro and in vivo modulates binding to its DNA consensus sequence. Neoplasia 3(3):179–188
Kanai M, Hanashiro K, Kim SH, Hanai S, Boulares AH, Miwa M, Fukasawa K (2007) Inhibition of Crm1-p53 interaction and nuclear export of p53 by poly(ADP-ribosyl)ation. Nat Cell Biol 9(10):1175–1183
Liang YC, Hsu CY, Yao YL, Yang WM (2013) PARP-2 regulates cell cycle-related genes through histone deacetylation and methylation independently of poly(ADP-ribosyl)ation. Biochem Biophys Res Commun. doi:10.1016/j.bbrc.2012.12.092
Yang L, Huang K, Li X, Du M, Kang X, Luo X, Gao L, Wang C, Zhang Y, Zhang C, Tong Q, Huang K, Zhang F, Huang D (2013) Identification of poly(ADP-ribose) polymerase-1 as a cell cycle regulator through modulating Sp1 mediated transcription in human hepatoma cells. PLoS ONE 8(12):e82872. doi:10.1371/journal.pone.0082872
Huidobro C, Fernandez AF, Fraga MF (2013) Aging epigenetics: causes and consequences. Mol Aspects Med 34(4):765–781. doi:10.1016/j.mam.2012.06.006
Burgess RC, Misteli T, Oberdoerffer P (2012) DNA damage, chromatin, and transcription: the trinity of aging. Curr Opin Cell Biol 24(6):724–730. doi:10.1016/j.ceb.2012.07.005
Issa JP (2014) Aging and epigenetic drift: a vicious cycle. J Clin Invest 124(1):24–29. doi:10.1172/jci69735
Caiafa P, Guastafierro T, Zampieri M (2008) Epigenetics: poly(ADP-ribosyl)ation of PARP-1 regulates genomic methylation patterns. Faseb J 23(3):672–678. doi:fj.08-123265 [pii] 10.1096/fj.08-123265
Zampieri M, Passananti C, Calabrese R, Perilli M, Corbi N, De Cave F, Guastafierro T, Bacalini MG, Reale A, Amicosante G, Calabrese L, Zlatanova J, Caiafa P (2009) PARP1 localizes within the Dnmt1 promoter and protects its unmethylated state by its enzymatic activity. PLoS ONE 4(3):e4717. doi:10.1371/journal.pone.0004717
Zampieri M, Guastafierro T, Calabrese R, Ciccarone F, Bacalini MG, Reale A, Perilli M, Passananti C, Caiafa P (2012) ADP-ribose polymers localized on Ctcf- PARP1-Dnmt1 complex prevent methylation of Ctcf target sites. Biochem J 441(2):645–652. doi:10.1042/BJ20111417
Kawasaki Y, Lee J, Matsuzawa A, Kohda T, Kaneko-Ishino T, Ishino F (2014) Active DNA demethylation is required for complete imprint erasure in primordial germ cells. Sci Rep 4:3658. doi:10.1038/srep03658
Ciccarone F, Klinger FG, Catizone A, Calabrese R, Zampieri M, Bacalini MG, De Felici M, Caiafa P (2012) Poly(ADP-ribosyl)ation acts in the DNA demethylation of mouse primordial germ cells also with DNA damage-independent roles. PLoS ONE 7(10):e46927. doi:10.1371/journal.pone.0046927PONE-D-12-10468 [pii]
Doege CA, Inoue K, Yamashita T, Rhee DB, Travis S, Fujita R, Guarnieri P, Bhagat G, Vanti WB, Shih A, Levine RL, Nik S, Chen EI, Abeliovich A (2012) Early-stage epigenetic modification during somatic cell reprogramming by PARP1 and Tet2. Nature 488(7413):652–655. doi:10.1038/nature11333
Pegoraro G, Misteli T (2009) The central role of chromatin maintenance in aging. Aging 1(12):1017–1022
Mostoslavsky R, Chua KF, Lombard DB, Pang WW, Fischer MR, Gellon L, Liu P, Mostoslavsky G, Franco S, Murphy MM, Mills KD, Patel P, Hsu JT, Hong AL, Ford E, Cheng HL, Kennedy C, Nunez N, Bronson R, Frendewey D, Auerbach W, Valenzuela D, Karow M, Hottiger MO, Hursting S, Barrett JC, Guarente L, Mulligan R, Demple B, Yancopoulos GD, Alt FW (2006) Genomic instability and aging-like phenotype in the absence of mammalian SIRT6. Cell 124(2):315–329
Rouleau M, Aubin RA, Poirier GG (2004) Poly(ADP-ribosyl)ated chromatin domains: access granted. J Cell Sci 117(6):815–825
Quenet D, Gasser V, Fouillen L, Cammas F, Sanglier-Cianferani S, Losson R, Dantzer F (2008) The histone subcode: poly(ADP-ribose) polymerase-1 ( PARP-1) and PARP-2 control cell differentiation by regulating the transcriptional intermediary factor TIF1beta and the heterochromatin protein HP1alpha. Faseb J 22(11):3853–3865. doi:fj.08-113464 [pii] 10.1096/fj.08-113464
Gamble MJ, Fisher RP (2007) SET and PARP1 remove DEK from chromatin to permit access by the transcription machinery. Nat Struct Mol Biol 14(6):548–555
Ditsworth D, Zong WX, Thompson CB (2007) Activation of poly(ADP)-ribose polymerase (PARP-1) induces release of the pro-inflammatory mediator HMGB1 from the nucleus. J Biol Chem 282(24):17845–17854
Messner S, Altmeyer M, Zhao H, Pozivil A, Roschitzki B, Gehrig P, Rutishauser D, Huang D, Caflisch A, Hottiger MO (2010) PARP1 ADP-ribosylates lysine residues of the core histone tails. Nucleic Acids Res 38(19):6350–6362. doi:gkq463 [pii] 10.1093/nar/gkq463
Kappes F, Fahrer J, Khodadoust MS, Tabbert A, Strasser C, Mor-Vaknin N, Moreno-Villanueva M, Bürkle A, Markovitz DM, Ferrando-May E (2008) DEK is a poly(ADP-ribose) acceptor in apoptosis and mediates resistance to genotoxic stress. Mol Cell Biol 28(10):3245–3257. doi:10.1128/MCB.01921-07
Poirier GG, de Murcia G, Jongstra-Bilen J, Niedergang C, Mandel P (1982) Poly(ADP-ribosyl)ation of polynucleosomes causes relaxation of chromatin structure. Proc Natl Acad Sci U S A 79(11):3423–3427
de Murcia G, Huletsky A, Lamarre D, Gaudreau A, Pouyet J, Daune M, Poirier GG (1986) Modulation of chromatin superstructure induced by poly(ADP-ribose) synthesis and degradation. J Biol Chem 261(15):7011–7017
Althaus FR (1992) Poly ADP-ribosylation: a histone shuttle mechanism in DNA excision repair. J Cell Sci 102(4):663–670
Realini CA, Althaus FR (1992) Histone shuttling by poly(ADP-ribosylation). J Biol Chem 267(26):18858–18865
Kim MY, Mauro S, Gevry N, Lis JT, Kraus WL (2004) NAD + -dependent modulation of chromatin structure and transcription by nucleosome binding properties of PARP-1. Cell 119(6):803–814
Krishnakumar R, Gamble MJ, Frizzell KM, Berrocal JG, Kininis M, Kraus WL (2008) Reciprocal binding of PARP-1 and histone H1 at promoters specifies transcriptional outcomes. Science 319(5864):819–821
Kavanaugh GM, Wise-Draper TM, Morreale RJ, Morrison MA, Gole B, Schwemberger S, Tichy ED, Lu L, Babcock GF, Wells JM, Drissi R, Bissler JJ, Stambrook PJ, Andreassen PR, Wiesmuller L, Wells SI (2011) The human DEK oncogene regulates DNA damage response signaling and repair. Nucleic Acids Res 39(17):7465–7476. doi:gkr454 [pii] 10.1093/nar/gkr454
Fahrer J, Popp O, Malanga M, Beneke S, Markovitz DM, Ferrando-May E, Bürkle A, Kappes F (2010) High-affinity interaction of poly(ADP-ribose) and the human DEK oncoprotein depends upon chain length. Biochemistry. doi:10.1021/bi1004365
Ahel D, Horejsi Z, Wiechens N, Polo SE, Garcia-Wilson E, Ahel I, Flynn H, Skehel M, West SC, Jackson SP, Owen-Hughes T, Boulton SJ (2009) Poly(ADP-ribose)-dependent regulation of DNA repair by the chromatin remodeling enzyme ALC1. Science 325(5945):1240–1243. doi:1177321 [pii] 10.1126/science.1177321
Rajamohan SB, Pillai VB, Gupta M, Sundaresan NR, Birukov KG, Samant S, Hottiger MO, Gupta MP (2009) SIRT1 promotes cell survival under stress by deacetylation-dependent deactivation of poly(ADP-ribose) polymerase 1. Mol Cell Biol 29(15):4116–4129. doi:10.1128/MCB.00121-09
Chou DM, Adamson B, Dephoure NE, Tan X, Nottke AC, Hurov KE, Gygi SP, Colaiacovo MP, Elledge SJ (2010) A chromatin localization screen reveals poly (ADP ribose)-regulated recruitment of the repressive polycomb and NuRD complexes to sites of DNA damage. Proc Natl Acad Sci U S A 107(43):18475–18480. doi:10.1073/pnas.1012946107
Pegoraro G, Kubben N, Wickert U, Gohler H, Hoffmann K, Misteli T (2009) Ageing-related chromatin defects through loss of the NURD complex. Nat Cell Biol 11(10):1261–1267. doi:ncb1971 [pii] 10.1038/ncb1971
Tulin A, Spradling A (2003) Chromatin loosening by poly(ADP)-ribose polymerase (PARP) at Drosophila puff loci. Science 299(5606):560–562. doi:10.1126/science.1078764299/5606/560 [pii]
Ju BG, Lunyak VV, Perissi V, Garcia-Bassets I, Rose DW, Glass CK, Rosenfeld MG (2006) A topoisomerase IIbeta-mediated dsDNA break required for regulated transcription. Science 312(5781):1798–1802
Thomas CJ, Kotova E, Andrake M, Adolf-Bryfogle J, Glaser R, Regnard C, Tulin AV (2014) Kinase-mediated changes in nucleosome conformation trigger chromatin decondensation via Poly(ADP-ribosyl)ation. Mol Cell. doi:10.1016/j.molcel.2014.01.005
Simbulan-Rosenthal CM, Ly DH, Rosenthal DS, Konopka G, Luo R, Wang Z-Q, Schultz PG, Smulson ME (2000) Misregulation of gene expression in primary fibroblasts lacking poly(ADP-ribose) polymerase. PNAS 97(21):11274–11279
Deschenes F, Massip L, Garand C, Lebel M (2005) In vivo misregulation of genes involved in apoptosis, development and oxidative stress in mice lacking both functional Werner syndrome protein and poly(ADP-ribose) polymerase-1. Hum Mol Genet 14(21):3293–3308
Szanto M, Brunyanszki A, Kiss B, Nagy L, Gergely P, Virag L, Bai P (2012) Poly(ADP-ribose) polymerase-2: emerging transcriptional roles of a DNA-repair protein. Cell Mol Life Sci. doi:10.1007/s00018-012-1003-8
Vilchez D, Simic MS, Dillin A (2014) Proteostasis and aging of stem cells. Trends Biol 24(3):161–170. doi:10.1016/j.tcb.2013.09.002
Cuanalo-Contreras K, Mukherjee A, Soto C (2013) Role of protein misfolding and proteostasis deficiency in protein misfolding diseases and aging. Int J Cell Biol 2013:638083. doi:10.1155/2013/638083
Taylor RC, Dillin A (2011) Aging as an event of proteostasis collapse. Cold Spring Harb Perspect Biol 3(5). doi:10.1101/cshperspect.a004440
Zhang C, Cuervo AM (2008) Restoration of chaperone-mediated autophagy in aging liver improves cellular maintenance and hepatic function. Nat Med 14(9):959–965. doi:10.1038/nm.1851
Jwa M, Chang P (2012) PARP16 is a tail-anchored endoplasmic reticulum protein required for the PERK- and IRE1alpha-mediated unfolded protein response. Nat Cell Biol. doi:10.1038/ncb2593ncb2593 [pii]
Carlile GW, Keyzers RA, Teske KA, Robert R, Williams DE, Linington RG, Gray CA, Centko RM, Yan L, Anjos SM, Sampson HM, Zhang D, Liao J, Hanrahan JW, Andersen RJ, Thomas DY (2012) Correction of F508del-CFTR trafficking by the sponge alkaloid latonduine is modulated by interaction with PARP. Chem Biol 19(10):1288–1299. doi:10.1016/j.chembiol.2012.08.014
Anjos SM, Robert R, Waller D, Zhang DL, Balghi H, Sampson HM, Ciciriello F, Lesimple P, Carlile GW, Goepp J, Liao J, Ferraro P, Phillipe R, Dantzer F, Hanrahan JW, Thomas DY (2012) Decreasing poly(ADP-ribose) polymerase activity restores DeltaF508 CFTR trafficking. Front Pharmacol 3:165. doi:10.3389/fphar.2012.00165
Gupta V, Balch WE (2013) Protein folding: salty sea regulators of cystic fibrosis. Nat Chem Biol 9(1):12–14. doi:10.1038/nchembio.1144
Hohn A, Jung T, Grune T (2014) Pathophysiological importance of aggregated damaged proteins. Free Radic Biol Med. doi:10.1016/j.freeradbiomed.2014.02.028
Breusing N, Grune T (2008) Regulation of proteasome-mediated protein degradation during oxidative stress and aging. Biol Chem 389(3):203–209. doi:10.1515/BC.2008.029
Tomaru U, Takahashi S, Ishizu A, Miyatake Y, Gohda A, Suzuki S, Ono A, Ohara J, Baba T, Murata S, Tanaka K, Kasahara M (2012) Decreased proteasomal activity causes age-related phenotypes and promotes the development of metabolic abnormalities. Am J Pathol 180(3):963–972. doi:10.1016/j.ajpath.2011.11.012
Rodriguez KA, Edrey YH, Osmulski P, Gaczynska M, Buffenstein R (2012) Altered composition of liver proteasome assemblies contributes to enhanced proteasome activity in the exceptionally long-lived naked mole-rat. PLoS ONE 7(5):e35890. doi:10.1371/journal.pone.0035890
Bakondi E, Catalgol B, Bak I, Jung T, Bozaykut P, Bayramicli M, Ozer NK, Grune T (2011) Age-related loss of stress-induced nuclear proteasome activation is due to low PARP-1 activity. Free Radic Biol Med 50(1):86–92. doi:10.1016/j.freeradbiomed.2010.10.700
Ullrich O, Ciftci O, Hass R (2000) Proteasome activation by poly-ADP-ribose-polymerase in human myelomonocytic cells after oxidative stress. Free Radic Biol Med 29(10):995–1004. doi:S0891-5849(00)00399-3 [pii]
Arnold J, Grune T (2002) PARP-mediated proteasome activation: a co-ordination of DNA repair and protein degradation? Bioessays 24(11):1060–1065
Catalgol B, Wendt B, Grimm S, Breusing N, Ozer NK, Grune T (2009) Chromatin repair after oxidative stress: Role of PARP-mediated proteasome activation. Free Radic Biol Med. doi:S0891-5849(09)00764-3 [pii] 10.1016/j.freeradbiomed.2009.12.010
Cho-Park PF, Steller H (2013) Proteasome regulation by ADP-ribosylation. Cell 153(3):614–627. doi:10.1016/j.cell.2013.03.040
Zhou ZD, Chan CH, Xiao ZC, Tan EK (2011) Ring finger protein 146/Iduna is a poly(ADP-ribose) polymer binding and PARsylation dependent E3 ubiquitin ligase. Cell Adh Migr 5(6):463–471. doi:10.4161/cam.5.6.18356
Kang HC, Lee YI, Shin JH, Andrabi SA, Chi Z, Gagne JP, Lee Y, Ko HS, Lee BD, Poirier GG, Dawson VL, Dawson TM (2011) Iduna is a poly(ADP-ribose) (PAR)-dependent E3 ubiquitin ligase that regulates DNA damage. Proc Natl Acad Sci U S A 108(34):14103–14108. doi:10.1073/pnas.1108799108
Naito AT, Shiojima I, Komuro I (2010) Wnt signaling and aging-related heart disorders. Circ Res 107(11):1295–1303. doi:10.1161/circresaha.110.223776
DeCarolis NA, Wharton KA Jr, Eisch AJ (2008) Which way does the Wnt blow? Exploring the duality of canonical Wnt signaling on cellular aging. Bioessays 30(2):102–106. doi:10.1002/bies.20709
Ye X, Zerlanko B, Kennedy A, Banumathy G, Zhang R, Adams PD (2007) Downregulation of Wnt signaling is a trigger for formation of facultative heterochromatin and onset of cell senescence in primary human cells. Mol Cell 27(2):183–196. doi:10.1016/j.molcel.2007.05.034
Liu H, Fergusson MM, Castilho RM, Liu J, Cao L, Chen J, Malide D, Rovira, II, Schimel D, Kuo CJ, Gutkind JS, Hwang PM, Finkel T (2007) Augmented Wnt signaling in a mammalian model of accelerated aging. Science 317(5839):803–806. doi:10.1126/science.1143578
Brack AS, Conboy MJ, Roy S, Lee M, Kuo CJ, Keller C, Rando TA (2007) Increased Wnt signaling during aging alters muscle stem cell fate and increases fibrosis. Science 317(5839):807–810. doi:10.1126/science.1144090
Naito AT, Sumida T, Nomura S, Liu ML, Higo T, Nakagawa A, Okada K, Sakai T, Hashimoto A, Hara Y, Shimizu I, Zhu W, Toko H, Katada A, Akazawa H, Oka T, Lee JK, Minamino T, Nagai T, Walsh K, Kikuchi A, Matsumoto M, Botto M, Shiojima I, Komuro I (2012) Complement C1q activates canonical Wnt signaling and promotes aging-related phenotypes. Cell 149(6):1298–1313. doi:10.1016/j.cell.2012.03.047
Zhang DY, Pan Y, Zhang C, Yan BX, Yu SS, Wu DL, Shi MM, Shi K, Cai XX, Zhou SS, Wang JB, Pan JP, Zhang LH (2013) Wnt/beta-catenin signaling induces the aging of mesenchymal stem cells through promoting the ROS production. Mol Cell Biochem 374(1–2):13–20. doi:10.1007/s11010-012-1498-1
Zhang DY, Wang HJ, Tan YZ (2011) Wnt/beta-catenin signaling induces the aging of mesenchymal stem cells through the DNA damage response and the p53/p21 pathway. PLoS ONE 6(6):e21397. doi:10.1371/journal.pone.0021397
Huang SM, Mishina YM, Liu S, Cheung A, Stegmeier F, Michaud GA, Charlat O, Wiellette E, Zhang Y, Wiessner S, Hild M, Shi X, Wilson CJ, Mickanin C, Myer V, Fazal A, Tomlinson R, Serluca F, Shao W, Cheng H, Shultz M, Rau C, Schirle M, Schlegl J, Ghidelli S, Fawell S, Lu C, Curtis D, Kirschner MW, Lengauer C, Finan PM, Tallarico JA, Bouwmeester T, Porter JA, Bauer A, Cong F (2009) Tankyrase inhibition stabilizes axin and antagonizes Wnt signalling. Nature 461(7264):614–620. doi:nature08356 [pii] 10.1038/nature08356
Zhang Y, Liu S, Mickanin C, Feng Y, Charlat O, Michaud GA, Schirle M, Shi X, Hild M, Bauer A, Myer VE, Finan PM, Porter JA, Huang SM, Cong F (2011) RNF146 is a poly(ADP-ribose)-directed E3 ligase that regulates axin degradation and Wnt signalling. Nat Cell Biol 13(5):623–629. doi:ncb2222 [pii] 10.1038/ncb2222
Bao R, Christova T, Song S, Angers S, Yan X, Attisano L (2012) Inhibition of tankyrases induces Axin stabilization and blocks Wnt signalling in breast cancer cells. PLoS ONE 7(11):e48670. doi:10.1371/journal.pone.0048670
Callow MG, Tran H, Phu L, Lau T, Lee J, Sandoval WN, Liu PS, Bheddah S, Tao J, Lill JR, Hongo JA, Davis D, Kirkpatrick DS, Polakis P, Costa M (2011) Ubiquitin ligase RNF146 regulates tankyrase and Axin to promote Wnt signaling. PLoS ONE 6(7):e22595. doi:10.1371/journal.pone.0022595
Morrone S, Cheng Z, Moon RT, Cong F, Xu W (2012) Crystal structure of a Tankyrase-Axin complex and its implications for Axin turnover and Tankyrase substrate recruitment. Proc Natl Acad Sci U S A 109(5):1500–1505. doi:10.1073/pnas.1116618109
Rubinsztein DC, Marino G, Kroemer G (2011) Autophagy and aging. Cell 146(5):682–695. doi:S0092-8674(11)00828-2 [pii] 10.1016/j.cell.2011.07.030
Wyrsch P, Blenn C, Bader J, Althaus FR (2012) Cell death and autophagy under oxidative stress: roles of poly(ADP-Ribose) polymerases and Ca(2+). Mol Cell Biol 32(17):3541–3553. doi:10.1128/MCB.00437-12
Huang Q, Shen HM (2009) To die or to live: the dual role of poly(ADP-ribose) polymerase-1 in autophagy and necrosis under oxidative stress and DNA damage. Autophagy 5(2):273–276. doi:7640 [pii]
Huang Q, Wu YT, Tan HL, Ong CN, Shen HM (2008) A novel function of poly(ADP-ribose) polymerase-1 in modulation of autophagy and necrosis under oxidative stress. Cell Death Differ. doi:cdd2008151 [pii] 10.1038/cdd.2008.151
Munoz-Gamez JA, Rodriguez-Vargas JM, Quiles-Perez R, Aguilar-Quesada R, Martin-Oliva D, de Murcia G, de Murcia JM, Almendros A, de Almodovar MR, Oliver FJ (2009) PARP-1 is involved in autophagy induced by DNA damage. Autophagy 5(1). doi:7272 [pii]
Rodriguez-Vargas JM, Ruiz-Magana MJ, Ruiz-Ruiz C, Majuelos-Melguizo J, Peralta-Leal A, Rodriguez MI, Munoz-Gamez JA, de Almodovar MR, Siles E, Rivas AL, Jaattela M, Oliver FJ (2012) ROS-induced DNA damage and PARP-1 are required for optimal induction of starvation-induced autophagy. Cell Res 22(7):1181–1198. doi:10.1038/cr.2012.70
Zhou ZR, Zhu XD, Zhao W, Qu S, Su F, Huang ST, Ma JL, Li XY (2013) Poly(ADP-ribose) polymerase-1 regulates the mechanism of irradiation-induced CNE-2 human nasopharyngeal carcinoma cell autophagy and inhibition of autophagy contributes to the radiation sensitization of CNE-2 cells. Oncol Rep 29(6):2498–2506. doi:10.3892/or.2013.2382
Speakman JR, Mitchell SE (2011) Caloric restriction. Mol Aspects Med 32(3):159–221. doi:10.1016/j.mam.2011.07.001
Fahrer J, Wagner S, Bürkle A, Konigsrainer A (2009) Rapamycin inhibits poly(ADP-ribosyl)ation in intact cells. Biochem Biophys Res Commun. doi:S0006-291X(09)01156-5 [pii] 10.1016/j.bbrc.2009.06.022
Ethier C, Tardif M, Arul L, Poirier GG (2012) PARP-1 Modulation of mTOR Signaling in Response to a DNA Alkylating Agent. PLoS ONE 7(10):e47978. doi:10.1371/journal.pone.0047978PONE-D-12-15496 [pii]
Fontana L, Partridge L, Longo VD (2010) Extending healthy life span-from yeast to humans. Science 328(5976):321–326. doi:328/5976/321 [pii] 10.1126/science.1172539
Mouchiroud L, Houtkooper RH, Auwerx J (2013) NAD metabolism: a therapeutic target for age-related metabolic disease. Crit Rev Biochem Mol Biol. doi:10.3109/10409238.2013.789479
Son N, Hur HJ, Sung MJ, Kim MS, Hwang JT, Park JH, Yang HJ, Kwon DY, Yoon SH, Chung HY, Kim HJ (2012) Liquid chromatography-mass spectrometry-based metabolomic analysis of livers from aged rats. J Proteome Res 11(4):2551–2558. doi:10.1021/pr201263q
Mouchiroud L, Houtkooper Riekelt H, Moullan N, Katsyuba E, Ryu D, Cantó C, Mottis A, Jo Y-S, Viswanathan M, Schoonjans K, Guarente L, Auwerx J (2013) The NAD + /Sirtuin Pathway Modulates Longevity through Activation of Mitochondrial UPR and FOXO Signaling. Cell 154(2):430–441
Houtkooper RH, Pirinen E, Auwerx J (2012) Sirtuins as regulators of metabolism and healthspan. Nat Rev Mol Cell Biol 13(4):225–238. doi:10.1038/nrm3293nrm3293 [pii]
Firestein R, Blander G, Michan S, Oberdoerffer P, Ogino S, Campbell J, Bhimavarapu A, Luikenhuis S, de Cabo R, Fuchs C, Hahn WC, Guarente LP, Sinclair DA (2008) The SIRT1 deacetylase suppresses intestinal tumorigenesis and colon cancer growth. PLoS ONE 3(4):e2020. doi:10.1371/journal.pone.0002020
Banks AS, Kon N, Knight C, Matsumoto M, Gutierrez-Juarez R, Rossetti L, Gu W, Accili D (2008) SirT1 gain of function increases energy efficiency and prevents diabetes in mice. Cell Metab 8(4):333–341. doi:S1550-4131(08)00255-6 [pii] 10.1016/j.cmet.2008.08.014
Bordone L, Cohen D, Robinson A, Motta MC, van Veen E, Czopik A, Steele AD, Crowe H, Marmor S, Luo J, Gu W, Guarente L (2007) SIRT1 transgenic mice show phenotypes resembling calorie restriction. Aging Cell 6(6):759–767. doi:ACE335 [pii] 10.1111/j.1474-9726.2007.00335.x
Pfluger PT, Herranz D, Velasco-Miguel S, Serrano M, Tschop MH (2008) Sirt1 protects against high-fat diet-induced metabolic damage. Proc Natl Acad Sci U S A 105(28):9793–9798. doi:0802917105 [pii] 10.1073/pnas.0802917105
Moynihan KA, Grimm AA, Plueger MM, Bernal-Mizrachi E, Ford E, Cras-Meneur C, Permutt MA, Imai S (2005) Increased dosage of mammalian Sir2 in pancreatic beta cells enhances glucose-stimulated insulin secretion in mice. Cell Metab 2(2):105–117. doi:S1550-4131(05)00176-2 [pii] 10.1016/j.cmet.2005.07.001
Canto C, Sauve AA, Bai P (2013) Crosstalk between poly(ADP-ribose) polymerase and sirtuin enzymes. Mol Aspects Med 34(6):1168–1201. doi:10.1016/j.mam.2013.01.004
Bai P, Canto C, Brunyanszki A, Huber A, Szanto M, Cen Y, Yamamoto H, Houten SM, Kiss B, Oudart H, Gergely P, Menissier-de Murcia J, Schreiber V, Sauve AA, Auwerx J (2011) PARP-2 regulates SIRT1 expression and whole-body energy expenditure. Cell Metab 13(4):450–460. doi:S1550-4131(11)00100-8 [pii] 10.1016/j.cmet.2011.03.013
El Ramy R, Magroun N, Messadecq N, Gauthier LR, Boussin FD, Kolthur-Seetharam U, Schreiber V, McBurney MW, Sassone-Corsi P, Dantzer F (2009) Functional interplay between PARP-1 and SirT1 in genome integrity and chromatin-based processes. Cell Mol Life Sci. doi:10.1007/s00018-009-0105-4
Bai P, Canto C, Oudart H, Brunyanszki A, Cen Y, Thomas C, Yamamoto H, Huber A, Kiss B, Houtkooper RH, Schoonjans K, Schreiber V, Sauve AA, Menissier-de Murcia J, Auwerx J (2011) PARP-1 inhibition increases mitochondrial metabolism through SIRT1 activation. Cell Metab 13(4):461–468. doi:10.1016/j.cmet.2011.03.004
Massudi H, Grant R, Braidy N, Guest J, Farnsworth B, Guillemin GJ (2012) Age-associated changes in oxidative stress and NAD+ metabolism in human tissue. PLoS ONE 7(7):e42357. doi:10.1371/journal.pone.0042357
Elkholi R, Chipuk JE (2014) How do I kill thee? Let me count the ways: p53 regulates PARP-1 dependent necrosis. Bioessays 36(1):46–51. doi:10.1002/bies.201300117
Hotchkiss RS, Strasser A, McDunn JE, Swanson PE (2009) Cell death. N Engl J Med 361(16):1570–1583. doi:361/16/1570 [pii] 10.1056/NEJMra0901217
Vanden Berghe T, Linkermann A, Jouan-Lanhouet S, Walczak H, Vandenabeele P (2014) Regulated necrosis: the expanding network of non-apoptotic cell death pathways. Nat Rev Mol Cell Biol 15(2):135–147. doi:10.1038/nrm3737
Broker LE, Kruyt FA, Giaccone G (2005) Cell death independent of caspases: a review. Clin Cancer Res 11(9):3155–3162
Behrens MI, Silva M, Schmied A, Salech F, Manzur H, Rebolledo R, Bull R, Torres V, Henriquez M, Quest AF (2011) Age-dependent increases in apoptosis/necrosis ratios in human lymphocytes exposed to oxidative stress. J Gerontol A Biol Sci Med Sci 66(7):732–740. doi:10.1093/gerona/glr039
Berger NA, Sims JL, Catino DM, Berger SJ (1983) Poly(ADP-ribose) polymerase mediates the suicide response to massive DNA damage: studies in normal and DNA-repair defective cells. Princess Takamatsu Symp 13:219–226
Calvo JA, Moroski-Erkul CA, Lake A, Eichinger LW, Shah D, Jhun I, Limsirichai P, Bronson RT, Christiani DC, Meira LB, Samson LD (2013) Aag DNA glycosylase promotes alkylation-induced tissue damage mediated by parp1. PLoS Genet 9(4):e1003413. doi:10.1371/journal.pgen.1003413
Fu D, Jordan JJ, Samson LD (2013) Human ALKBH7 is required for alkylation and oxidation-induced programmed necrosis. Genes Dev. doi:10.1101/gad.215533.113
Kanai M, Hanashiro K, Kim SH, Hanai S, Boulares AH, Miwa M, Fukasawa K (2007) Inhibition of Crm1-p53 interaction and nuclear export of p53 by poly(ADP-ribosyl)ation. Nat Cell Biol 9:1175–1183
Negri C, Donzelli M, Bernardi R, Rossi L, Bürkle A, Scovassi AI (1997) Multiparametric staining to identify apoptotic human cells. Exp Cell Res 234(1):174–177
Tewari M, Quan LT, O'Rourke K, Desnoyers S, Zeng Z, Beidler DR, Poirier GG, Salvesen GS, Dixit VM (1995) Yama/CPP32 beta, a mammalian homolog of CED-3, is a CrmA-inhibitable protease that cleaves the death substrate poly(ADP-ribose) polymerase. Cell 81(5):801–809
Lazebnik YA, Kaufmann SH, Desnoyers S, Poirier GG, Earnshaw WC (1994) Cleavage of poly(ADP-ribose) polymerase by a proteinase with properties like ICE. Nature 371(6495):346–347
Germain M, Affar EB, D'Amours D, Dixit VM, Salvesen GS, Poirier GG (1999) Cleavage of automodified poly(ADP-ribose) polymerase during apoptosis. Evidence for involvement of caspase-7. J Biol Chem 274(40):28379–28384
Yu SW, Wang H, Poitras MF, Coombs C, Bowers WJ, Federoff HJ, Poirier GG, Dawson TM, Dawson VL (2002) Mediation of poly(ADP-ribose) polymerase-1-dependent cell death by apoptosis-inducing factor. Science 297(5579):259–263
Cohausz O, Blenn C, Malanga M, Althaus FR (2008) The roles of poly(ADP-ribose)-metabolizing enzymes in alkylation-induced cell death. Cell Mol Life Sci 65(4):644–655
Wang Y, Kim NS, Haince JF, Kang HC, David KK, Andrabi SA, Poirier GG, Dawson VL, Dawson TM (2011) Poly(ADP-ribose) (PAR) binding to apoptosis-inducing factor is critical for PAR polymerase-1-dependent cell death (parthanatos). Sci Signal 4 (167):ra20. doi:10.1126/scisignal.2000902
Yu SW, Andrabi SA, Wang H, Kim NS, Poirier GG, Dawson TM, Dawson VL (2006) Apoptosis-inducing factor mediates poly(ADP-ribose) (PAR) polymer-induced cell death. Proc Natl Acad Sci U S A 103(48):18314–18319
Andrabi SA, Kim NS, Yu SW, Wang H, Koh DW, Sasaki M, Klaus JA, Otsuka T, Zhang Z, Koehler RC, Hurn PD, Poirier GG, Dawson VL, Dawson TM (2006) Poly(ADP-ribose) (PAR) polymer is a death signal. Proc Natl Acad Sci U S A 103(48):18308–18313
Lee Y, Karuppagounder SS, Shin JH, Lee YI, Ko HS, Swing D, Jiang H, Kang SU, Lee BD, Kang HC, Kim D, Tessarollo L, Dawson VL, Dawson TM (2013) Parthanatos mediates AIMP2-activated age-dependent dopaminergic neuronal loss. Nature Neurosci. doi:10.1038/nn.3500
Vaziri H, West MD, Allsopp RC, Davison TS, Wu YS, Arrowsmith CH, Poirier GG, Benchimol S (1997) ATM-dependent telomere loss in aging human diploid fibroblasts and DNA damage lead to the post-translational activation of p53 protein involving poly(ADP-ribose) polymerase. Embo J 16(19):6018–6033. doi:10.1093/emboj/16.19.6018
Rodier F, Coppe JP, Patil CK, Hoeijmakers WA, Munoz DP, Raza SR, Freund A, Campeau E, Davalos AR, Campisi J (2009) Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat Cell Biol 11(8):973–979. doi:ncb1909 [pii] 10.1038/ncb1909
Freund A, Orjalo AV, Desprez PY, Campisi J (2010) Inflammatory networks during cellular senescence: causes and consequences. Trends Mol Med 16(5):238–246. doi:S1471-4914(10)00046-8 [pii] 10.1016/j.molmed.2010.03.003
Ohanna M, Giuliano S, Bonet C, Imbert V, Hofman V, Zangari J, Bille K, Robert C, Bressac-de Paillerets B, Hofman P, Rocchi S, Peyron JF, Lacour JP, Ballotti R, Bertolotto C (2011) Senescent cells develop a PARP-1 and nuclear factor-{kappa}B-associated secretome (PNAS). Genes Dev 25(12):1245–1261. doi:10.1101/gad.625811gad.625811 [pii]
Medzhitov R (2010) Inflammation 2010: new adventures of an old flame. Cell 140(6):771–776. doi:S0092-8674(10)00242-4 [pii] 10.1016/j.cell.2010.03.006
de Gonzalo-Calvo D, Neitzert K, Fernandez M, Vega-Naredo I, Caballero B, Garcia-Macia M, Suarez FM, Rodriguez-Colunga MJ, Solano JJ, Coto-Montes A (2010) Differential inflammatory responses in aging and disease: TNF-alpha and IL-6 as possible biomarkers. Free Radic Biol Med 49(5):733–737. doi:S0891-5849(10)00336-9 [pii] 10.1016/j.freeradbiomed.2010.05.019
De Martinis M, Franceschi C, Monti D, Ginaldi L (2005) Inflamm-ageing and lifelong antigenic load as major determinants of ageing rate and longevity. FEBS Lett 579(10):2035–2039
Wang J, Geiger H, Rudolph KL (2011) Immunoaging induced by hematopoietic stem cell aging. Curr Opin Immunol 23(4):532–536. doi:S0952-7915(11)00058-6 [pii] 10.1016/j.coi.2011.05.004
Salminen A, Huuskonen J, Ojala J, Kauppinen A, Kaarniranta K, Suuronen T (2007) Activation of innate immunity system during aging: NF-kB signaling is the molecular culprit of inflamm-aging. Ageing Res Rev 7(2):83–105
Sarkar D, Fisher PB (2006) Molecular mechanisms of aging-associated inflammation. Cancer Lett 236(1):13–23. doi:S0304-3835(05)00365-4 [pii] 10.1016/j.canlet.2005.04.009
Harman D (2006) Free radical theory of aging: an update: increasing the functional life span. Ann N Y Acad Sci 1067:10–21. doi:1067/1/10 [pii] 10.1196/annals.1354.003
Mangerich A, Knutson CG, Parry NM, Muthupalani S, Ye W, Prestwich E, Cui L, McFaline JL, Mobley M, Ge Z, Taghizadeh K, Wishnok JS, Wogan GN, Fox JG, Tannenbaum SR, Dedon PC (2012) Infection-induced colitis in mice causes dynamic and tissue-specific changes in stress response and DNA damage leading to colon cancer. Proc Natl Acad Sci U S A 109(27):E1820–E1829. doi:10.1073/pnas.1207829109
Mangerich A, Dedon PC, Fox JG, Tannenbaum SR, Wogan GN (2013) Chemistry meets biology in colitis-associated carcinogenesis. Free Radic Res 47(11):958–986. doi:10.3109/10715762.2013.832239
Chambers SM, Shaw CA, Gatza C, Fisk CJ, Donehower LA, Goodell MA (2007) Aging hematopoietic stem cells decline in function and exhibit epigenetic dysregulation. PLoS Biol 5(8):e201. doi:06-PLBI-RA-2319 [pii] 10.1371/journal.pbio.0050201
Lepperdinger G (2011) Inflammation and mesenchymal stem cell aging. Curr Opin Immunol 23(4):518–524. doi:S0952-7915(11)00064-1 [pii] 10.1016/j.coi.2011.05.007
Bai P, Virag L (2012) Role of poly(ADP-ribose) polymerases in the regulation of inflammatory processes. FEBS Lett 586(21):3771–3777. doi:10.1016/j.febslet.2012.09.026
Rosado MM, Bennici E, Novelli F, Pioli C (2013) Beyond DNA repair, the immunological role of PARP-1 and its siblings. Immunology 139(4):428–437. doi:10.1111/imm.12099
Hassa PO, Hottiger MO (2002) The functional role of poly(ADP-ribose)polymerase 1 as novel coactivator of NF-kappaB in inflammatory disorders. Cell Mol Life Sci 59(9):1534–1553
Masutani M, Suzuki H, Kamada N, Watanabe M, Ueda O, Nozaki T, Jishage K, Watanabe T, Sugimoto T, Nakagama H, Ochiya T, Sugimura T (1999) Poly(ADP-ribose) polymerase gene disruption conferred mice resistant to streptozotocin-induced diabetes. Proc Natl Acad Sci U S A 96(5):2301–2304
Burkart V, Wang ZQ, Radons J, Heller B, Herceg Z, Stingl L, Wagner EF, Kolb H (1999) Mice lacking the poly(ADP-ribose) polymerase gene are resistant to pancreatic beta-cell destruction and diabetes development induced by streptozocin. Nat Med 5(3):314–319
Mandir AS, Przedborski S, Jackson-Lewis V, Wang ZQ, Simbulan-Rosenthal CM, Smulson ME, Hoffman BE, Guastella DB, Dawson VL, Dawson TM (1999) Poly(ADP-ribose) polymerase activation mediates 1-methyl-4-phenyl-1, 2,3,6-tetrahydropyridine (MPTP)-induced parkinsonism. Proc Natl Acad Sci U S A 96(10):5774–5779
Pieper AA, Brat DJ, Krug DK, Watkins CC, Gupta A, Blackshaw S, Verma A, Wang ZQ, Snyder SH (1999) Poly(ADP-ribose) polymerase-deficient mice are protected from streptozotocin-induced diabetes. Proc Natl Acad Sci U S A 96(6):3059–3064
Hayden MS, Ghosh S (2008) Shared principles in NF-kappaB signaling. Cell 132(3):344–362
Adler AS, Kawahara TL, Segal E, Chang HY (2008) Reversal of aging by NFkappaB blockade. Cell Cycle 7(5):556–559. doi:5490 [pii]
Adler AS, Sinha S, Kawahara TL, Zhang JY, Segal E, Chang HY (2007) Motif module map reveals enforcement of aging by continual NF-kappaB activity. Genes Dev 21(24):3244–3257. doi:gad.1588507 [pii] 10.1101/gad.1588507
Moskalev A, Shaposhnikov M (2011) Pharmacological inhibition of NF-kappaB prolongs lifespan of Drosophila melanogaster. Aging 3(4):391–394. doi:100314 [pii]
Rovillain E, Mansfield L, Caetano C, Alvarez-Fernandez M, Caballero OL, Medema RH, Hummerich H, Jat PS (2011) Activation of nuclear factor-kappa B signalling promotes cellular senescence. Oncogene 30(20):2356–2366. doi:onc2010611 [pii] 10.1038/onc.2010.611
Wang J, Jacob NK, Ladner KJ, Beg A, Perko JD, Tanner SM, Liyanarachchi S, Fishel R, Guttridge DC (2009) RelA/p65 functions to maintain cellular senescence by regulating genomic stability and DNA repair. EMBO Rep 10(11):1272–1278. doi:embor2009197 [pii] 10.1038/embor.2009.197
Tilstra JS, Clauson CL, Niedernhofer LJ, Robbins PD (2011) NF-kappaB in aging and disease. Aging Dis 2(6):449–465
Oliver FJ, Menissier-de Murcia J, Nacci C, Decker P, Andriantsitohaina R, Muller S, de la RG, Stoclet JC, de Murcia G (1999) Resistance to endotoxic shock as a consequence of defective NF-kappaB activation in poly (ADP-ribose) polymerase-1 deficient mice. Embo J 18(16):4446–4454. doi:10.1093/emboj/18.16.4446
Hassa PO, Hottiger MO (1999) A role of poly (ADP-ribose) polymerase in NF-kappaB transcriptional activation. Biol Chem 380(7–8):953–959. doi:10.1515/BC.1999.118
Hassa PO, Covic M, Hasan S, Imhof R, Hottiger MO (2001) The enzymatic and DNA binding activity of PARP-1 are not required for NF-kappa B coactivator function. J Biol Chem 276(49):45588–45597. doi:10.1074/jbc.M106528200M106528200 [pii]
Altmeyer M, Hottiger MO (2009) Poly(ADP-ribose) polymerase 1 at the crossroad of metabolic stress and inflammation in aging. Aging 1(5):458–469
Stilmann M, Hinz M, Arslan SC, Zimmer A, Schreiber V, Scheidereit C (2009) A nuclear poly(ADP-ribose)-dependent signalosome confers DNA damage-induced IkappaB kinase activation. Mol Cell 36(3):365–378. doi:S1097-2765(09)00692-3 [pii] 10.1016/j.molcel.2009.09.032
Aguilar-Quesada R, Munoz-Gamez JA, Martin-Oliva D, Peralta-Leal A, Quiles-Perez R, Rodriguez-Vargas JM, de Almodovar MR, Conde C, Ruiz-Extremera A, Oliver FJ (2007) Modulation of transcription by PARP-1: consequences in carcinogenesis and inflammation. Curr Med Chem 14(11):1179–1187
Radovits T, Seres L, Gero D, Berger I, Szabo C, Karck M, Szabo G (2007) Single dose treatment with PARP-inhibitor INO-1001 improves aging-associated cardiac and vascular dysfunction. Exp Gerontol 42(7):676–685
von Lukowicz T, Hassa PO, Lohmann C, Boren J, Braunersreuther V, Mach F, Odermatt B, Gersbach M, Camici GG, Stahli BE, Tanner FC, Hottiger MO, Luscher TF, Matter CM (2008) PARP1 is required for adhesion molecule expression in atherogenesis. Cardiovasc Res 78(1):158–166. doi:cvm110 [pii] 10.1093/cvr/cvm110
Mangerich A, Herbach N, Hanf B, Fischbach A, Popp O, Moreno-Villanueva M, Bruns OT, Bürkle A (2010) Inflammatory and age-related pathologies in mice with ectopic expression of human PARP-1. Mech Ageing Dev 131(6):389–404. doi:S0047-6374(10)00110-7 [pii] 10.1016/j.mad.2010.05.005
Zarychanski R, Houston DS (2008) Anemia of chronic disease: a harmful disorder or an adaptive, beneficial response? CMAJ 179(4):333–337. doi:179/4/333 [pii] 10.1503/cmaj.071131
Weiss G, Goodnough LT (2005) Anemia of chronic disease. N Engl J Med 352(10):1011–1023. doi:352/10/1011 [pii] 10.1056/NEJMra041809
Muller-Ohldach M, Brust D, Hamann A, Osiewacz HD (2011) Overexpression of Pa PARP encoding the poly(ADP-ribose) polymerase of Podospora anserina affects organismal aging. Mech Ageing Dev 132(1–2):33–42. doi:10.1016/j.mad.2010.11.003
Kirkwood TB (2005) Understanding the odd science of aging. Cell 120(4):437–447
Gerondakis S, Grumont R, Gugasyan R, Wong L, Isomura I, Ho W, Banerjee A (2006) Unravelling the complexities of the NF-kappaB signalling pathway using mouse knockout and transgenic models. Oncogene 25(51):6781–6799
Acknowledgements
Our experimental work was supported by the DFG-funded Collaborative Research Center (CRC) 969, the Konstanz Research School Chemical Biology (KoRS-CB), the International Research Training Group (IRTG) 1331 and the EU FP7 large-scale integrating project “European Study to Establish Biomarkers of Human Ageing” (MARK-AGE).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Mangerich, A., Bürkle, A. (2015). Multitasking Roles for Poly(ADP-ribosyl)ation in Aging and Longevity. In: Curtin, N., Sharma, R. (eds) PARP Inhibitors for Cancer Therapy. Cancer Drug Discovery and Development, vol 83. Humana Press, Cham. https://doi.org/10.1007/978-3-319-14151-0_6
Download citation
DOI: https://doi.org/10.1007/978-3-319-14151-0_6
Published:
Publisher Name: Humana Press, Cham
Print ISBN: 978-3-319-14150-3
Online ISBN: 978-3-319-14151-0
eBook Packages: MedicineMedicine (R0)