
Abstract
We have analyzed transient structures in deuterated acetylene isomerized by 10 fs 400 eV X-rays, and probed by coulomb explosion with a second delayed X-ray pulse. Structural changes are revealed through fragment kinetic energy release.
Access provided by Autonomous University of Puebla. Download conference paper PDF
Similar content being viewed by others
Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
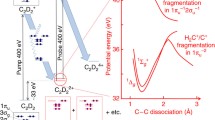
X-ray induced isomerization experiments in several molecules were carried out by the AMO75113 collaborationFootnote 1 using X-ray pulse pairs from the newly commissioned soft X-ray split and delay at the LCLS X-ray free electron laser [1]. We have analyzed X-ray-induced fragmentation energies in deuterated acetylene data in the linear DCCD isomer and the vinylidene CCD2 form to show how kinetic energy release correlates with isomerization.
The acetylene isomerization transition is a model for hydrogen migration in organic molecules. Relatively little is known about isomerization induced by photoionization of the 1 s shell. X rays above the carbon K-edge can induce core shell ionization followed by Auger relaxation, leading to the production of a molecular dication. Experiments at X-ray synchrotrons have shown strong evidence for X-ray dication formation accompanied by proton migration from the initial HCCH2+ configuration to the CCH22+ vinylidene metastable isomer [2]. These studies cannot resolve the motion of the protons within the molecule prior to dissociation. Photoelectron-photoion coincidence measurements at synchrotrons can clock proton migration times, and have concluded that they are faster than 60 fs [3]. This is at odds with the ~2 eV isomerization barrier between acetylene and vinylidene in the ground state of the dication, which suggests a longer isomerization time [4, 5].
Our studies used 10 fs, 400 eV X-ray pulse pairs. The first X-ray pulse ionizes a core electron and initiates the dynamics. The cation undergoes Auger relaxation in approximately 6 fs [5]. Deuteron migration dynamics is probed by a second X-ray pulse, which ionizes another core electron, leading to further Auger relaxation. The resulting tetracation is unstable, and dissociates promptly into several fragments. The three-layer delay-line anode detector can sort and resolve the position and arrival time of each ion fragment for up to 16 independent ions, therefore measuring the full momentum vectors. The pump and probe X-ray pulses were delayed by from 0 to 100 fs. The target gas density was maintained to a level where on the order of one molecule per shot was excited by two X-ray pulses (Fig. 1).

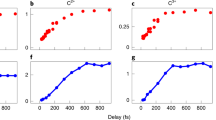
Momentum correlation may be used to find coincidences corresponding to the isomerization pathway after the X-ray photon absorption. Here, coincidences of C+ and CD2 + are found. For true coincidences the conservation of the momentum (along the time of flight axis in this case) results in the data lying on a line as shown above. In the study reported here, similar constraints were placed on four-particle coincidences that correspond to full dissociation of acetylene
Data in this study include events where four charged dissociation products were produced from the same molecule. We found that an effective filter to eliminate backgrounds with false coincidences was to require that the four particles be singly charged, two deuterons and two carbon ions, and that the total vector sum of the momenta of all fragments was approximately 0, to within 10 % of the peak momentum. Figure 2 shows how this works in the case of only two fragments, where the display is simpler. We applied this to the four-dimensional product space of four particle coincidences.

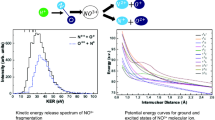
Dependence of the kinetic energy released (KER) on the ejection angle of the deuteron. Angles below π/2 correspond to V-type structures. These have significantly lower average energy and a sharper energy distribution than the A-type dissociations
The deuteron migration from acetylene to vinylidene can be detected by analyzing the individual ion momenta. For example, in this analysis we have used the following protocol: The four momentum vectors are \( \varvec{p}_{{\varvec{C}1}} \), \( \varvec{p}_{{\varvec{C}2}} \), \( \varvec{p}_{{\varvec{H}1}} \), and \( \varvec{p}_{{\varvec{H}2}} \).. Our event selection filter ensures that the vector sum of all momenta is zero: \( \varvec{p}_{{\varvec{C}1}} + \varvec{p}_{{\varvec{C}2}} + \varvec{p}_{{\varvec{H}1}} + \varvec{p}_{{\varvec{H}2}} = 0 \). We then define the molecular axis in the lab frame as the unit vector \( \hat{\varvec{A}} = \left( {\varvec{p}_{C1}} - {\varvec{p}_{\varvec{C}2} } \right)/\left| {\varvec{p}_{C1} }- {\varvec{p}_{\varvec{C}2} } \right| \). Finally, we look at the projection of the deuteron momenta on \( \hat{\varvec{A}} \). If the dot products \( \hat{\varvec{A}}\, \cdot \,\varvec{p}_{H1} \) and \( \hat{\varvec{A}}\, \cdot \,\varvec{p}_{H2} \) have different signs, then we identify that detected event as a molecule that has not undergone isomerization (the acetylene channel “A” or opposite-side deprotonation channel “P”). If, on the other hand, \( \hat{\varvec{A}}\, \cdot \,\varvec{p}_{H1} \) and \( \hat{\varvec{A}}\, \cdot \,\varvec{p}_{H2} \) have the same sign, then one deuteron has migrated to the other side of the molecule. This is the “V” channel that also includes the same-side deprotonation.
We find some striking physical features of the data sorted in this way. First the collection of A- and P-channel events have a total kinetic energy (KER) release distribution that is approximately 10 eV larger than V-channel events. This difference is so large that KER may be used as a method to separate non-isomerizing events from V-type events.
The ratio of V-type to non-isomerizing events is a strong function of the time delay between the pulses. The time-dependence is consistent with the LCLS X-ray autocorrelation time, and suggests that deuteron migration initiates on a time scale of approximately 20 fs or less following X-ray ionization.
Notes
- 1.
AMO75113 collaboration: V. Petrovic, C. Liekhus-Schmaltz, P. Bucksbaum, I. Tenney, T. Osipov, A. Belkacem, N. Berrah, R. Boll, C. Bomme, C. Bostedt, J. Bozek, S. Carron, R. Coffee, J. Devin, B. Erk, L. Fang, K. Ferguson, R. Field, L. Foucar, L. Frasinski, J.M. Glownia, M. Guehr, A. Kamalov, J. Krzywinski, H. Li, J. Marangos, T. Martinez, B. McFarland, S. Miyabe, B. Murphy, A. Natan, D. Rolles, A. Rudenko, A. Sanchez, M. Siano, E. Simpson, L. Spector, M. Swiggers, D. Walke, S. Wang, and T. Weber.
References
AMO75113 collaboration, in preparation (2014).
J. Laksman, D. Céolin, M. Gisselbrecht, S. E. Canton, and S. L. Sorensen, The Journal of Chemical Physics 131 (2009) 244305.
T. Osipov, C. L. Cocke, M. H. Prior, A. Landers, T. Weber, O. Jagutzki, L. Schmidt, H. Schmidt-Böcking, and R. Dörner, Physical Review Letters 90 (2003) 233002.
D. Duflot, J. M. Robbe, and J. P. Flament, The Journal of Chemical Physics 102 (1995) 355.
T. S. Zyubina, Y. A. Dyakov, S. H. Lin, A. D. Bandrauk, and A. M. Mebel, The Journal of Chemical Physics 123 (2005) 134320.
Acknowledgments
This research used the SLAC Linac Coherent Light Source, which is a DOE Office of Science User Facility. The members of the AMO75113 collaboration contributed advice and assistance. This research was supported by the National Science Foundation under Grant No. PHY-0649578.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer International Publishing Switzerland
About this paper
Cite this paper
Bucksbaum, P.H., Liekhus-Schmaltz, C.E., Tenney, I., Petrovic, V.S. (2015). Structure Dependence of Kinetic Energy Released in X-ray-Induced Fragmentation. In: Yamanouchi, K., Cundiff, S., de Vivie-Riedle, R., Kuwata-Gonokami, M., DiMauro, L. (eds) Ultrafast Phenomena XIX. Springer Proceedings in Physics, vol 162. Springer, Cham. https://doi.org/10.1007/978-3-319-13242-6_32
Download citation
DOI: https://doi.org/10.1007/978-3-319-13242-6_32
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-13241-9
Online ISBN: 978-3-319-13242-6
eBook Packages: Physics and AstronomyPhysics and Astronomy (R0)