
Abstract
New information concerning the effects of prolactin (PRL) on metabolic processes warrants reevaluation of its overall metabolic actions. PRL affects metabolic homeostasis by regulating key enzymes and transporters associated with glucose and lipid metabolism in several target organs. In the lactating mammary gland, PRL increases the production of milk proteins, lactose, and lipids. In adipose tissue, PRL generally suppresses lipid storage and adipokine release and affect adipogenesis. A specific case is made for PRL in the human breast and adipose tissues, where it acts as a circulating hormone and an autocrine/paracrine factor. Although its overall effects on body composition are both modest and species-specific, PRL may be involved in the manifestation of insulin resistance.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1.1 Introduction
Metabolic homeostasis of an individual is finely regulated by the nutritional status, energy expenditure, and hormonal signals. Peripheral organs such as the pancreas, liver, and adipose tissue, as well as many centers within the brain, respond to these changes and act coordinately to maintain metabolic stability. Prolactin (PRL) is a multifunctional pituitary hormone with more actions than all other pituitary hormones combined. These functions are broadly classified as reproductive, metabolic, osmoregulatory, and immunoregulatory. The actions of PRL are mediated by the PRL receptor (PRLR), which is expressed in all organs associated with metabolic regulation. Unique to humans, PRL is also produced at multiple extrapituitary sites, categorizing it as a classical circulating hormone as well as an autocrine/paracrine cytokine.
Whereas humans are the one species we wish to know most about, they are also the least accessible to experimental manipulations. Although some properties of PRL in humans are well documented, for example, the effects of drugs, prolactinoma formation, and variants of PRL and the PRLR, others remain obscure. By necessity, then, information derived from laboratory animals is essential for the understanding of PRL in human health and disease. Nonetheless, extrapolation from data obtained with rodents to humans should be done selectively and judiciously. Given that both PRL homeostasis and adipose tissue properties differ greatly among species, each section in this chapter contains an extensive comparison of these parameters in humans vs. rodents. To fully evaluate the role of PRL as a metabolic hormone with a focus on adipose tissue, the following topics are covered: (1) characteristics of PRL, (2) selected features of the PRLR, (3) adipose tissue properties, (4) expression and regulation of adipocyte PRL, and (5) metabolic actions of PRL.
1.2 Characteristics of PRL and Lactogens
1.2.1 General Features of Lactogens
Human PRL (hRPL), growth hormone (hGH), and placental lactogen (hPL), commonly referred to as lactogens, are members of the cytokine superfamily which includes over 20 proteins. Members of this family are defined by two criteria: (1) a tertiary structure of four antiparallel α helices in an up–up–down–down configuration (Fig. 1.1), and (2) binding to a nontyrosine kinase, single-pass transmembrane receptor [1]. Lactogens are made of a single polypeptide chain of 190–200 residues with 2–3 disulfide bridges. hPRL has three disulfide bridges, while hGH and hPL lack the N-terminal disulfide loop. hGH and hPL share 85 % sequence homology, but only 21–22 % homology with hPRL. In spite of the low homology at the primary amino acid sequence, the three-dimensional topology of the three human lactogens enables their binding to the hPRLR, as discussed below. Figure 1.1 also demonstrates that all three lactogens equally stimulate proliferation of Nb2 rat lymphocytes.

Left panel: Structural similarities of the three human lactogens: hPRL, hPL, and hGH. Right panel: The three human lactogens similarly increase proliferation of Nb2 rat lymphocytes, which have long served as the most sensitive bioassay for PRL
1.2.2 The PRL Protein
Humans express a single PRL gene, located on chromosome 6. In addition to the pituitary, hPRL is independently and differentially expressed at multiple sites that include the endometrium, myometrium, decidua, immune cells, brain, breast, prostate, skin, and adipose tissue [2]. Consequently, even when pituitary PRL release is severely impaired, humans are not deprived of their locally produced PRL. As judged by structural, biochemical, and functional criteria, extrapituitary and pituitary PRL proteins are identical, albeit their transcriptional regulation is dissimilar, as detailed in Sect. 1.5.
At extrapituitary sites PRL is produced at much smaller amounts than at the pituitary, and likely remains in the vicinity of the producing cells via its association with heparin-binding proteins. Two motifs in hPRL are implicated in heparin binding [3]. These are absent in hGH and hPL, rendering them incapable of binding to heparin. The heparin-binding properties of hPRL enhance its efficacy as a cytokine by enriching its local concentration in tissues with high content of glycosaminoglycans such as adipose tissue.
PRL in rodents is primarily expressed in the pituitary, although some PRL is detectable in the decidua [4] and the lactating mammary gland [5, 6]. Unlike humans, rodents express multiple PRL-related genes which are clustered on chromosome 13 in mice and chromosome 17 in rats. These have variable degrees of sequence homology and are expressed in the uterus and placenta [7]. Some of the PRL-like proteins play important roles during late pregnancy in rodents, when they compensate for the markedly reduced pituitary PRL release. Nonetheless, under nonpregnant, nonlactating conditions, the rodent pituitary is the sole source of PRL.
1.2.3 Growth Hormone and Placental Lactogens
Humans have five GH/PL-related genes clustered on chromosome 17 [8]. These include GH-N (normal GH), primarily expressed in the pituitary, and four GH/PL-related proteins: GH-V (variant GH), PL-A, PL-B, and variant PL, all of which are expressed in the placental syncytiotrophoblast [9]. Although hPRL and hGH show little sequence homology at the amino acid level, hGH binds not only to its cognate receptor (hGHR), but also to hPRLR, and can mimic some of PRL actions. In contrast, nonprimates’ GH binds only to GHR, while hPRL binds only to the PRLR. In spite of the higher sequence homology of hPLs to hGH than to hPRL, and their GH-like metabolic functions, hPLs bind to the hPRLR but not to hGHR, reviewed in [10]. Attempts to identify a unique receptor that binds hPL have not been successful. Unlike the multiplicity of GH/PL proteins in humans, mice and rats have only a single GH gene on chromosomes 11 and 10, respectively, which is primarily expressed in the pituitary.
1.2.4 Structural Diversity of PRL Proteins
The PRL protein can undergo a number of posttranslational modifications which include polymerization; proteolytic cleavage; glycosylation and phosphorylation; and impact on its stability, half-life, receptor binding, and bioactivity [11]. In addition to the 23 kDa PRL, human serum contains macroprolactin (> 100 kDa) and big PRL (40–60 kDa). Macroprolactin, a complex of monomeric PRL with IgG [12], is often elevated in hyperprolactinemic patients. Big PRL represents dimerized PRL, with an unclear relationship to macroprolactin. Proteolytic cleavage of PRL generates smaller fragments with different biological properties than the parent molecule. The N-terminal fragment, named 16 K PRL, has been best studied [13]. Recently, a family of N-terminal fragments of PRL, GH, and PL, named vasoinhibins, has been identified [14]. These peptides act on endothelial cells to suppress angiogenesis and promote vascular regression, and also play a role in tumorigenesis. Yet, the receptor(s) that mediates their action is unknown, and it is also unclear whether PRL produced at extrapituitary sites undergoes cleavage.
hPRL is N-glycosylated on Asn 34, comprising ~ 30 % of total pituitary PRL content. Glycosylated PRL is also abundant in serum, milk, and the amniotic fluid [11]. Glycosylation reduces the binding affinity of PRL to the receptor, and affects its proteolytic cleavage, tissue distribution, and clearance. Unlike nonmodified pituitary PRL, which is stored and released from secretory vesicles, glycosylated PRL is constitutively secreted [15]. Constitutive secretion is particularly applicable to adipose tissue which does not have secretory granules. Rat PRL lacks a consensus sequence for N-glycosylation and is O-glycosylated [16]. It constitutes > 50 % of serum PRL in rats, but only a minor component in the pituitary, indicating differential release rates or a longer half-life.
Phosphorylated PRL has been identified in many species, including rodents and humans [17]. PRL is phosphorylated on Ser 179 within the secretory granules of the rat pituitary, and its levels are altered under many physiological states. Studies with a molecular mimic of phosphorylated PRL (S179D) revealed that it acts as an agonist for cell differentiation and apoptosis, but as an antagonist for cell proliferation [18]. There is no information on whether extrapituitary PRL undergoes phosphorylation.
1.3 Selected Features of the PRLR
1.3.1 Cytokine-Type 1 Receptors
The cytokine-type receptors are single pass transmembrane proteins, devoid of intrinsic tyrosine kinase activity. Upon activation they are phosphorylated by a variety of cytoplasmic proteins. The receptors are subdivided into type I or type II, based on the number and spacing of cysteine and proline residues in their extracellular domain (ECD). The PRLR belongs to the type I subfamily which includes receptors for GH, leptin, a few interleukins, erythropoietin, leukemia inhibiting factor, and others [19]. Ligand binding to these receptors activates the Janus kinase-signal transducer and activator of transcription (Jak-Stat), as well as the mitogen-activated protein kinase (MAPK), and the phosphoinositide 3 kinase (PI3 K) signaling pathways.
1.3.2 Regulation of hPRLR Expression
The hPRLR gene is located on chromosome 5 in close proximity to the hGHR sequence. It is > 100 kb long and has 11 exons. Exons 1, 2, and part of exon 3 comprise the 5′-untranslated region (UTR), while the rest of the exons comprise the coding region [20]. The UTR has six alternative first exons that are expressed in a tissue-specific manner. Regardless of which first exon is utilized, all are spliced into a noncoding exon 2. Transcription of the hPRLR gene is regulated at different sites by alternative promoters, each driving a specific first exon [20]. Alternative splicing within the coding region yields several isoforms that differ in length of the cytoplasmic domain, as discussed below.
The PRLR is expressed in most tissues, with the highest expression in the liver, mammary gland, adrenal, and hypothalamus [21]. Receptor expression is altered in response to changes in circulating PRL and steroid hormones, which can increase or suppress receptor expression, depending on the cell context and the physiological conditions. PRL also induces proteolytic degradation of the PRLR through ubiquitination [22]. Impairment of this process in some malignant PRLR-expressing cells (e.g., breast cancer), results in increased stability of the PRLR, enhanced responsiveness to PRL, and increased tumorigenicity.
1.3.3 Structural Elements of hPRLR Protein
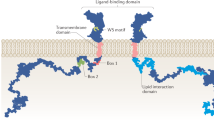
The PRLR protein consists of three distinct components: an ECD which binds the ligands, a short transmembrane domain™, and a variable intracellular domain (ICD) which links to second messengers [23, 24]. The ECD is ~ 200 amino acids long and contains two subdomains: an amino-terminal region called D1, and a membrane-proximal region called D2, both of which have type III fibronectin-like motifs. Two pairs of disulfide bonds in the D1 domain, and a “WS-motif” (Trp-Ser-X-Trp-Ser) in the D2 domain are critical for receptor folding and trafficking [25]. Within each species, the ECDs of all PRLR isoforms are identical. The two disulfide bonds in D1 are preserved in all species, but the WSxWS domain is seen in humans and rats but not in mice, which have a WSxWG. The ECD of the rat and mouse is 95 % homologous, differing only by 11 residues. The human ECD has 71 and 74 % homology to those in mice and rats, respectively [10]
1.3.4 PRLR Isoforms
Alternative splicing during transcription generates several PRLR isoforms, classified by the length of their ICD as “long,” “intermediate,” or “short.” The long PRLR is the major form through which PRL transmits its signals. It has an apparent mass of 90 kDa and is composed of 588 residues, 364 of which are in the ICD. The ICD contains ten tyrosine residues (only nine in rodents) whose location and adjacent residues determine whether they become phosphorylated following receptor activation [24].
Humans have more PRLR isoforms than rats and mice combined. Six isoforms of hPRLR have been identified [26]: long (85–90 kDa), intermediate (50 kDa), ΔS1 (70 kDa), short 1a (56 kDa), and short 1b (42 kDa). There is also a soluble PRL binding protein which contains only the ECD (32 kDa), and is generated by proteolysis and is present in the circulation. The isoforms are expressed at variable ratios in normal and malignant cells, and some have independent, site-specific biological activities [27]. When co-expressed with the long isoform, several short isoforms inhibit transcriptional responses to PRL, suggesting that they can act as dominant negatives and may provide protection against overstimulation by PRL ([28]; Fig. 1.2).

Comparison of PRLR isoforms in rodents vs. humans. The similar extracellular domain is designated by two subdomains, D1 and D2, which contain disulfide bonds and WS motif, respectively. Receptors are classified by the length of the intracellular domains as long (L), intermediate (I), and short (S). In addition, there is a receptor missing the D1 domain in the ECD (ΔS1), and another which contains only the extracellular domain (binding protein or BP). The intracellular domain contain two conserved regions, designated Box 1 and Box 2, which link the receptor to signaling molecules
A special case is an intermediate PRLR isoform that is expressed in rat Nb2 cells. Nb2 cells are a pre-T lymphocyte cell line which has served as the most sensitive bioassay for PRL for many decades. These cells encode a mutant receptor protein of 393 amino acids which lacks 198 residues in the cytoplasmic domain [29]. The mechanism by which PRL acts as a very potent mitogen for Nb2 cells, but as a rather weak mitogen in most other cells is unknown.
1.3.5 Ligand Binding
The secondary and tertiary configuration of both PRL and the PRLR, including specific residues in critical topological locations, determine receptor binding and activation. Yet, a structure-based explanation for the activation of the PRLR by different lactogens remains a major challenge. The rodent PRLR is activated by hPRL and hPL but not by hGH, while the hPRLR is activated by rPRL but not by mPRL [30]. The fact that human xenografts in nude mice do not respond to circulating mouse PRL is overlooked by many investigators. Such unresponsiveness hinders the ability to extrapolate data from mice to humans vis-à-vis the role of PRL in tumorigenesis and metabolic regulation. The species-dependent incompatibilities may be overcome with the recent generation of humanized rats [31] and mice [32] that express the hPRL gene, and the eventual crossing of hPRL-expressing mice to immune-deficient mice.
PRLR dimerization is obligatory for signal transmission, but whether PRL induces sequentialdimerization or binds to predimerized receptors is controversial [21, 25, 33]. While the former represents a long-held view, the latter has gained support, based on the identification of preformed dimers of other cytokine type 1 receptors. Two sites on PRL with different affinities are necessary for binding to the two receptors. The use of combinations of various constructs of the PRLR revealed that the TM domain is sufficient for dimerization in a ligand-independent fashion, but the interaction is strengthened by both the ECDand ICD [34]. A comprehensive cover of the different concepts of PRLR activation by its ligands can be found in a recent review [25].
1.3.6 Signal Transduction
The PRLR is devoid of intrinsic tyrosine kinase activity and utilizes the Jak2-Stat pathway as its main signaling cascade. The receptor-associated Jak2 is rapidly phosphorylated upon PRL binding and induces the phosphorylation of the receptor itself, associated kinases, and Stat proteins [23, 35]. Of the seven known Stat proteins, Stats 1, 3, and 5 can be activated by PRL, with Stats 5a and 5b especially important for mammary gland development and functions. Upon phosphorylation, Stat proteins hetero- or homodimerize via SH2–phosphotyrosine interactions. The activated dimers are translocated to the nucleus and bind to GAS (γ-interferon activation sequence) consensus elements on target genes. Milk proteins, that is, β-casein, lactalbumin, and whey acidic protein, are the best characterized PRLR-regulated genes downstream of the Jak-Stat pathway.
The Ras–Raf–MAPK pathway is also activated by PRL in many cells. Phosphorylation of Jak2 can recruit adaptor proteins such as Shc, Grb2, and SOS to the PRLR, resulting in the binding and activation of Ras and Raf. This leads to activation of the MAP kinase pathway, which is often associated with increased cell proliferation [36]. Activation of the PRLR also facilitates docking of Src family kinases, which in turn activates the PI3K/Akt pathway that mediates some of the antiapoptotic and metabolic actions of PRL [37] (Fig. 1.3).

The three major signaling transduction pathways of the long PRLR isoform: Jak2-Stat5a/b, PI3K/Akt, and MAPK which are activated by PRL in a cell-selective manner. See text for other explanations
Both the strength and duration of the PRL-induced signaling are regulated by members of the suppressors of cytokine signaling (SOCS) proteins, whose expression is rapidly induced following receptor activation [38]. The SOCS proteins interact either with the PRLR or with Jak2 and inhibit further signal activation. In sum, the signal transduction activated by the PRLR does not represent a linear progression but involves several interacting pathways that differ in predominance among the various PRLR-expressing cells.
1.4 Adipose Tissue Properties
1.4.1 Distinct Features of Adipose Tissue
Adipose tissue is an active organ that plays a pivotal role in metabolic, physiologic, and endocrine homeostasis [39, 40]. This highly specialized tissue of mesenchymal origin is comprised of multiple cell types held loosely together in a collagen matrix. The predominant cell is the terminally differentiated adipocyte, a very large cell whose size can be dramatically altered under various nutritional states. The stroma, often referred to as the stromal vascular fraction (SVF), contains pleuripotent stem cells, preadipocytes, endothelial cells, pericytes, mast cells, fibroblasts, and hematopoietic cells, primarily macrophages.
Obesity results from adipocyte enlargement through enhanced lipid accumulation (hypertrophy), as well as increased cell number (hyperplasia). The latter begins with recruitment of stem cells to the adipocyte lineage [41], followed by adipogenesis, which converts committed preadipocytes to mature adipocytes [42]. Approximately 10 % of fat cells are renewed annually irrespective of body mass index, without an increase in their overall number in adults. Death of adipocytes occurs by necrosis or apoptosis. Whether apoptosis is a critical factor which determines the number of adipocytes has been debated [43]. A therapeutic induction of apoptosis in adipocytes could become a valuable approach for treating obesity [44]. Obesity is also associated with recruitment of macrophages into adipose tissue, leading to increased production of proinflammatory cytokines such as TNFα and IL-6, and development of low-level inflammation [45].
1.4.2 White and Brown Adipocytes
Two types of adipose tissue are recognized: white adipose tissue (WAT) and brown adipose tissue (BAT). White adipocytes are characterized by a single, large lipid droplet occupying up to 90 % of the cell volume. The droplet consists of triacylglycerols (TAG) and cholesterol and is coated with specialized proteins, for example perilipins, which mediate interactions between the droplet and regulators of lipid metabolism. WAT secretes numerous important hormones/adipocytokines that include leptin, adiponectin, resistin, adipsin, TNFα and angiotensinogen [46, 47], as well as PRL [48], as detailed below. After menopause, WAT also becomes a significant source of estrogen, which is produced from circulating androgen precursors by the cytochrome P450 enzyme aromatase. In sum, the two main functions of WAT are storage and release of lipids, and production and secretion of adipokines. Together, they affect food intake, energy balance, insulin sensitivity, lipid and glucose metabolism, and cardiovascular functions [49].
Brown adipocytes contain multilocular lipid droplets and have a high density of mitochondria. They express uncoupling protein 1 (UCP-1) which generates heat by uncoupling electron transport from oxidative phosphorylation. BAT is specialized for nonshivering heat production [50]. It was recently discovered that functional BAT, once thought to exist mainly in infants, is also present in adults [51]. Brown adipocytes are detectable during cold exposure, and are associated with decreased adiposity. There is evidence for transdifferentiation between white and brown adipocytes. When stimulated, brown adipocytes enhance energy expenditure and increase glucose and fatty acid uptake. Thermogenesis in BAT is recognized as an important determinant in energy balance, and its therapeutic manipulation could be exploited in the treatment of obesity.
1.4.3 Fat Depots: Morphological and Functional Aspects
Human WAT is found in specific anatomical depots, which differ in morphology and functions and are classified as abdominal visceral (vis) and subcutaneous (sc) fat [52, 53]. The relative distribution of these depots determines body shape. There is sex dimorphism in the regional distribution of adipose tissue, with vis fat accounting for 6 % of body fat in women but 20 % in men, reflecting their greater propensity to accumulate excess abdominal fat. After menopause, fat distribution in women is closer to that of men, suggesting a role for sex steroids in anatomical deposition of fat [54]. The cellular composition of the depots varies, with vis fat having more macrophages and fewer preadipocytes than sc fat, whereas sc adipocytes in obese subjects are larger than their vis counterparts. The depots also differ in the secretion of adipokine, with vis fat secreting more IL-6 but less leptin and adiponectin than sc fat.
In humans, increased abdominal vis fat is a critical factor in the manifestation of the metabolic syndrome, which has become alarmingly pervasive in recent years. The metabolic syndrome is defined by glucose intolerance, hyperinsulinemia, hypertriglyceremia, altered serum lipoprotein levels, and hypertension. Vis fat is more sensitive to β-adrenergic agonists and less responsive to insulin than sc fat, making it more lipolytically active [52, 55]. As the output of vis fat drains into the hepatic portal blood, increased influx of free fatty acids (FFA) inhibits hepatic insulin clearance and contributes to hyperinsulinemia. Chronic elevation of FFA also impairs glucose metabolism and insulin sensitivity in liver and muscle, and reduces pancreatic β cell function [56]. Other factors, for example, increased production of some adipokines and inflammatory cytokines, and altered insulin receptor expression, link obesity to the metabolic syndrome [56].
Adipose depots in mice are present in several distinct locations: (1) gonadal fat surrounding the uterus and ovaries in females and the epididymis and testes in males; (2) abdominal fat, further subdivided into mesenteric, omental, and pericardial, (3) sc fat which also includes the mammary fat pads in females, and (4) brown adipose tissue, primarily seen in neonates in the neck and thoracic regions [57]. Of all fat depots in the mouse, the gonadal depots are the largest, comprising about 30 % of dissectible fat.
The mammary fat pad is especially relevant to any discussion on PRL and adipose tissue. The mammary gland contains myoepithelial and luminal epithelial cells that comprise the milk ducts and alveoli. These are embedded in a stroma composed of fibroblasts and adipocytes. Studies with rodents showed that the stroma provides signals to the epithelial cells that are critical for postnatal morphogenesis of the mammary gland [58]. Mammary adipocytes are especially active during the transitions from pregnancy to lactation, and into involution, and also contribute to tumorigenesis [59]. There are major differences in the distribution of mammary fat between humans and rodents. In rodents, stromal adipocytes are in close proximity to the epithelium, while human breast adipose tissue is interlaced with a network of fibroblasts and connective tissue, and a fibrous layer separates the epithelium from the adipocytes, reviewed in [10].
1.4.4 The Process of Adipogenesis
Adipocytes are derived from pluripotent mesenchymal stem cells (MSC), that can differentiate into adipocytes, myocytes, chondrocytes, and osteocytes [41]. Induction of MSC into the preadipocyte lineage occurs in response to stimulation/inhibition by bone morphogenic protein (BMP) family members, Wnt/β-catenin, and hedgehog signaling. White and brown adipocytes originate from different progenitors, with brown adipocytes characterized by the expression of Myf5, while white adipocytes are Myf5-negative [60].
Once committed, conversion of preadipocytes into mature adipocytes progresses through a well-coordinated, sequential activation/inactivation of a cascade of genes. This results in substantial structural, morphological, and functional changes (Fig. 1.4). Studies with murine preadipocytes, primarily 3T3-L1 cells, with supportive data from primary human preadipocytes, provided most of our knowledge of the molecular basis of adipogenesis. Under in vitro conditions, adipogenesis is initiated by exposure of preadipocytes to adipogenic-inducing hormones such as glucocorticoids, insulin/IGF-1, and cAMP activators [41, 61]. Although the time-course differs among the cellular models, most critical steps are common. These include an initial growth arrest, followed by clonal expansion, and a secondary arrest. Preadipocytes first withdraw from the cell cycle and become arrested at the G1/S stage. They then reenter the cell cycle and undergo 2–3 rounds of division, referred to as mitotic clonal expansion. The latter, however, may be a permissive, but not an absolute, requirement for differentiation of human preadipocytes.

Formation of mature adipocytes. Stem cells can differentiate into preadipocytes, chondrocytes, myoblasts, and osteoblasts. Preadipocytes undergo mitotic expansion, followed by cell cycle arrest and adipogenesis, which results in the formation of terminally-differentiated mature adipocytes. Brown adipocytes develop from another cell lineage, with trans-differentiation occurring between white and brown adipocytes. Obesity results primarily from hypertrophy by enhanced lipid accumulation, and is characterized by macrophage infiltration. Cell death occurs by necrosis or apoptosis
Adipogenesis progresses in well-orchestrated waves of genetic events [62]. Several transcription factors: peroxisome proliferator activated receptor γ (PPARγ) and CCAAT/enhancer binding proteins (C/EBP α, β, and γ), are central to the control of adipogenesis, while other transcription factors play supportive roles [41]. During early adipogenesis, the cells irreversibly exit the cell cycle and start losing their fibroblast-like morphology. As adipogenesis progresses, the cells accumulate TAGs in response to the increased expression of lipid synthesizing enzymes. The insulin receptor, the glucose transporter GLUT4, and key adipokines such as leptin and adiponectin, are also coordinately expressed. Adipogenesis is modulated by a plethora of hormones and local factors such as glucocorticoids, GH, IGF-1, TNFα, and IL-6 [62].
1.4.5 Lipid Metabolism: Lipogenesis vs. Lipolysis
Adipose tissue is the major site of lipid metabolism, which also occurs, to a lesser extent, in the liver [63, 64]. Based on weight, fat contains twice as many calories as proteins or carbohydrates, making energy storage in the form of TAGs highly efficient. Synthesis of TAGs requires assembly of three moieties of fatty acids with a molecule of glycerol. Fatty acids are available to the adipocytes from the circulation or from local synthesis [65], and glycerol is available as glycerol-3-phosphate from glycolysis and glyceroneogenesis [66]. The overall process of lipogenesis is affected by the frequency and composition of the diet and serum glucose and lipid levels, and is regulated by insulin, which increases lipogenesis, and GH and glucagon which decrease it.
Adipocytes store TAGs when energy supply is abundant, while during periods of caloric deficits, they hydrolyze them to FFA, which serve as fuel and metabolic precursors for other tissues [46]. Stored TAGs originate from two sources: (1) dietary fat in the form of chylomicrons from the intestines, very low-density lipoproteins (VLDLs) from the liver, and nonesterified fatty acids bound to albumin, and (2) de novo lipogenesis from carbohydrates [46, 64]. Under a typical western diet, stored lipids are primarily derived from dietary triglycerides, while de novo lipogenesis predominates under excess carbohydrate intake [63]. In human adipocytes, de novo lipogenesis is significantly less active than in rats because of differences in the expression of sterol regulatory element binding protein (SREBP-1), or the composition of diet in the two species [66].
Several key enzymes, some of which are affected by PRL (see below), are involved in TAG production. One is lipoprotein lipase (LPL), which is synthetized in adipocytes and secreted into adjacent endothelial cells. LPL hydrolyzes TAGs from circulating chylomicrons and lipoproteins into FFA, which are taken up by the adipocytes via membrane transport proteins [65]. Two other enzymes are pyruvate dehydrogenase (PDH), which generates acetyl-CoA, and acetyl-CoA carboxylase (ACC), which generates malonyl CoA, an essential intermediate in fatty acid biosynthesis. A critical lipogenic enzyme is fatty acid synthase (FAS), a multienzyme complex which catalyzes the production of palmitate from acetyl Co-A and malonyl Co-A into long chain fatty acids.
Lipolysis entails a stepwise breaking down of TAGs, first to into diacylglycerols (DAG) and then to monoacylglycerols (MAG), eventually yielding three molecules of FFAs and one molecule of glycerol. Hormone sensitive lipase (HSL), so named because of its high sensitivity to insulin and catecholamines, has long been considered the rate-limiting step in lipolysis. Following the discovery of adipocytes triglyceride lipase (ATGL) by three independent groups [67], it is now recognized that both enzymes act coordinately to regulate lipolysis.
Catecholamines and insulin increase and decrease lipolysis, respectively, by altering the cAMP/PKA signaling pathway in opposite directions [65, 68, 69]. Norepinephrine and epinephrine originate from the adrenal medulla and sympathetic nerve endings. They bind to α- and β-adrenergic receptors, which are classical seven-transmembrane receptors coupled to inhibitory (Gi) or stimulatory (Gs) G-proteins, respectively. Upon activation, Gs proteins stimulate adenylyl cyclase, increase cAMP and activate protein kinase A. Once activated, PKA phosphorylates both HSL, which translocates from the cytosol to the lipid droplet, and perillipin-1, which is displaced from the surface of the droplet into the cytosol. This coordinated event provides access of HSL to stored TAGs and enables their hydrolysis. In rodents, the β3-adrenergic receptor is the primary lipolytic mediator, while in humans, β1, β2, and α2 (an inhibitor of lipolysis) receptors, play more decisive regulatory roles [69].
Inhibition of lipolysis by insulin involves cAMP-dependent and cAMPindependent mechanisms [39, 65]. Insulin binds to its receptor and activates the insulin receptor substrate (IRS). This is followed by activation of PKB/Akt and phosphorylation of phosphodiesterase 3B (PDE3B), which degrades cAMP and deactivates PKA. Hence, the suppression of lipolysis by insulin results from reduced phosphorylation-mediated activation of HSL and perilipin. Insulin also inhibits lipolysis by stimulating phosphatase-1, which rapidly dephosphorylates and deactivates HSL. In humans, but not in rodents, natriuretic peptides stimulate lipolysis via a cGMP-dependent pathway that does not involve PDE-3B inhibition or cAMP production [70].
1.4.6 Selected Adipokines
Adipose tissue is an important endocrine organ whose hormones, the adipokines, regulate food intake, energy balance, insulin resistance, inflammatory responses, and blood pressure. In turn, the release of adipokines is influenced by the nutritional status, hormonal signals, and energy expenditure [39, 46, 71]. Since the discovery of leptin in 1994, a multitude of adipokines and adipocytokines has been identified. Some of these are exclusively produced by adipocytes, few are secreted by adipose stromal cells, and several are also produced to a variable degree by other organs. Here we focus only on leptin and adiponectin, which are affected by PRL.
Leptin is a 16-Kda protein produced primarily by mature adipocytes, and at low levels in the GI tract, muscle, mammary epithelium, placenta, and brain. Serum leptin levels increase in proportion to weight gain and decrease with weight loss, designating leptin as a “signal” of adiposity [72, 73]. The leptin receptor (Ob-R or LR) and the PRLR belong to the cytokine type 1 receptor family and share many features. Leptin utilizes Jak2/Stat 3 as its main signaling pathway, and also cross-talk with the insulin receptor through activation of IRS-1. Similar to the PRLR, the LR is alternatively spliced into secreted, short and long isoforms, with the long isoform serving as the primary mediator of leptin action. However, unlike the promiscuity of the PRLR which binds several lactogens, the LR responds only to leptin.
Leptin is a multifunctional hormone which acts on many peripheral organs and the brain. Much attention has been given to its ability to suppress appetite. Leptin can cross the blood–brain barrier through a saturable mechanism, and binds to receptors that are expressed in many sites within the brain [46]. Elevated leptin directly suppresses the orexigenic peptides neuropeptide Y and agouti-related peptide in the arcuate nucleus, while indirectly inhibiting melanin concentrating hormone and orexin in the lateral hypothalamus. Leptin also increases the proopiomelanocortin-derived anorectic peptide α-MSH. These coordinated actions ultimately lead to a reduction in food intake, increased energy expenditure, and increased thermogenesis.
The initial expectation that leptin could be used therapeutically to suppress appetite in obese patients has not materialized for several reasons. First, the short half-life of circulating leptin requires a very frequent delivery or a long-acting leptin formulation. More importantly, a prolonged rise in serum leptin levels in obesity induces leptin resistance, which results from lower leptin transport into the brain as well as reduced leptin signaling. Thus, leptin has diminished effects on food intake in obese patients [72]. Unlike rodents, in whom leptin is a major suppressor of appetite, the control of appetite in humans is more dominated by GI-derived hormones such as ghrelin, cholecystokinin, pancreatic polypeptide, peptide YY, and glucagon-like peptide [74].
Adiponectin is a major adipocytes-derived hormone which is not produced elsewhere. It circulates at very high levels, comprising as much as 0.01 % of total plasma proteins [75, 76]. Adiponectin is a 30-kDa protein with a complex structure, with some homology to collagen VIII. It circulates in many forms, from trimers to high-molecular weight decamers. In contrast to leptin, serum adiponectin is negatively correlated with fat mass, being low in obesity and high after weight loss. Adiponectin binds to two receptors (AdipoR1 and AdipoR2) which contain 7-transmembrane domains but are structurally and functionally distinct from the G-protein-coupled receptors [77]. AdipoR1 is primarily expressed in muscle and signals through AMP kinase, while AdipoR2 is expressed in liver and activates PPARα.
Adiponectin acts as an insulin sensitizer, and is classified as an antidiabetic, antiinflammatory, and antiatherogenic hormone [77]. In liver, adiponectin decreases FFA influx and reduces glucose output, while in muscle, it stimulates glucose utilization and fatty acid oxidation. Adiponectin has beneficial effects on the cardiovascular system, where it prevents atherosclerotic formation by inhibiting monocyte adhesion to the endothelium, and by suppressing transformation of macrophages into foam cells. The actions of adiponectin on the brain are not well defined. In rodents, intracerebral administration of adiponectin results in decreased body weight by increasing energy expenditure without altering food intake. In sum, low serum adiponectin is associated with insulin resistance and type 2 diabetes, while its elevation has the opposite effects. The ability of adiponectin to enhance insulin sensitivity and promote vascular health raises the prospect of its therapeutic use in the treatment of diabetes and cardiovascular diseases. However, biologically active recombinant adiponectin proteins are unstable and difficult to make, presenting a challenge for both research and clinical applications.
1.5 Expression and Regulation of Adipose PRL
1.5.1 The Discovery of Adipose PRL
PRL production in human adipose tissue was serendipitously discovered upon studying the potential role of local PRL in breast carcinogenesis [78]. Surgical specimens of normal and malignant breast tissue were separated into adipose and glandular explants and incubated for 10 days in serum-free media. Explants were analyzed for PRL gene expression by RT-PCR, and media were analyzed for secreted PRL by the Nb2 bioassay. Unexpectedly, breast adipose explants, intended to serve as negative controls, expressed and released 10–15 times more PRL than their glandular counterparts. To verify local synthesis rather than release from reuptake, explants were incubated with 35S-methionine, followed by immunoprecipitation, electrophoresis, and autoradiography [78]. The presence of metabolically-labeled PRL in both tissue extracts and conditioned media strongly supported de novo synthesis of PRL.
Another unexpected observation was a progressive rise in PRL release from adipose explants up to 7 days in culture, suggesting removal from inhibitory controls. PRL release from glandular explants was suppressed by progesterone, but neither estrogen nor progesterone altered its release from adipose explants [78], indicating dissimilar regulation of PRL in the two adjacent tissues. These findings raised several intriguing questions: (1) Is PRL synthesized in other adipose depots and, if so, is it affected by obesity? (2) Which cells synthesize PRL? (3) What is the nature of the inhibitor? and (4) What are the functions of local PRL?
To address these questions, vis and sc adipose tissue explants from morbidly obese and nonobese patients were placed in culture. Similar to the profile of PRL release from breast adipose tissue, PRL release from both types of explants showed time-dependent increases [79]. PRL release from sc explants from obese patients was significantly lower than that from lean patients, with no apparent difference between men and women. Isolated mature adipocytes had an identical pattern of PRL release to that from explants. Collectively, these data showed depot-specific control of PRL production which is markedly affected by obesity. The mechanism by which obesity causes a reduction in adipocyte PRL release and its functional consequences, remain to be determined. Adipocytes are the primary source of PRL in adipose tissue, although infiltrating macrophages which express PRL [80], could add to the overall adipose PRL output in obesity. PRL expression was undetectable in adipose tissue from rats, mice or 3T3-L1 and 3T3–442A murine preadipocyte cell lines, confirming the notion that adipocyte-derived PRL is unique to humans. Yet, infiltrating macrophages may carry out some PRL production in adipose tissue in obese rodents [80].
When compared on per cell basis, PRL release from a single adipocyte is many orders of magnitude lower than that from a pituitary lactotroph. However, the human pituitary weighs 1 g, while the weight of adipose tissue in obese individuals can exceed 100 kg. Consequently, the overall PRL production by adipose tissue could approach that of the pituitary. A relevant question is whether adipose PRL affects serum PRL levels. Given that hPRL binds heparin [3], most of the PRL secreted by adipocytes is presumably retained locally by proteoglycans, which are abundant in adipose tissue but low in the pituitary, making adipose PRL a true autocrine/paracrine factor. A recent study compared serum PRL levels in obese and lean patients and found higher basal serum PRL in women than men, but no effect of obesity [81]. Serum PRL levels did not correlate with BMI, and were unchanged after massive weight loss. Another study found lower serum PRL in obese than lean children [82]. Collectively, these data demonstrate that adipose-derived PRL has little, if any, effects, on circulating PRL levels.
1.5.2 Regulation of PRL Gene Expression
The human PRL gene consists of five coding exons. It is transcribed in the pituitary from a proximal promoter which depends on Pit-1 transcription factor for activation, and is regulated by dopamine, estrogen, neuropeptides, and some growth factors [83]. In contrast, expression of extrapituitary PRL is driven by a superdistal promoter, located 5.8 kb upstream of the pituitary start site. This promoter is silenced in the pituitary and does not depend on Pit-1. Exon 1a, serving as an alternative transcriptional start site (named decidual start site), is spliced into exon 1b, yielding an identical transcript to that of the pituitary except for a longer 5′ UTR (Fig. 1.5). The superdistal promoter extends − 3000 bp upstream of the decidual start site and is composed of a proximal region between − 350 and − 60 and a distal enhancer between − 2000 and − 1500 [84, 85]. The dissimilar control of the PRL gene in various tissues is exemplified by progesterone, which increases PRL expression in the endometrium, decreases it in the myometrium and breast, and has no effect on pituitary PRL.

The regulation of pituitary and extrapituitary PRL gene expression by the proximal and superdistal promoters, respectively. The PRL transcripts are identical except for a longer 5′ untranslated region (UTR) in the extrapituitary transcript. See text for other explanations
To map the active elements within the superdistal promoter, primary preadipocytes were transiently transfected with a luciferase reporter driven by a full-length decidual PRL promoter (− 3000/+ 66), or with progressively deleted mutants [86]. Transfection with either the full length promoter or the − 317 construct resulted in a 25-fold increase in luciferase activity above vector control. On the other hand, the − 1556 and − 675 constructs caused only five- to eightfold increases, suggesting presence of inhibitory elements between the proximal promoter and the distal enhancer. The two positive regulatory domains correspond to those in decidual cells [87], while the inhibitory region appears to be unique to adipocytes.
1.5.3 Factors Which Affect PRL Release
Knowledge of the control of PRL release in extrapituitary sites lags behind that of pituitary PRL for several reasons. First, unlike those of rodents, human tissues are not as readily available and show high variability among tissue donors. Second, PRL release from these sites is several orders of magnitude lower than pituitary PRL, requiring the use of more sensitive, but often less specific, bioassays. Third, there is no uniform mechanism for the control of PRL release, as each cell type utilizes different regulators. Fourth, there are no storage granules in most extrapituitary sites, implying constitutive PRL release rather than calcium-dependent exocytosis as in pituitary lactotrophs. Without vesicular storage, the main control of nonpituitary PRL is transcriptional, as is the case for most cytokines. In spite of the dissimilar regulation of pituitary and nonpituitary PRL, both are under inhibitory controls, reviewed in [10]. As detailed in Sect. 1.5.5., similar to the pituitary, dopamine serves as a physiological inhibitor of adipocyte PRL [88].
Both preadipocytes and mature adipocytes express and release PRL [86]. PRL release from freshly isolated preadipocytes was low and transiently increased during early adipogenesis. PRL expression was stimulated by many agents that elevate cAMP, including epinephrine, IBMX (3-isobutyl-1-methylxanthine), a phosphodiesterase inhibitor, isoproterenol, a β-adrenergic receptor agonist, PACAP (pituitary adenylate cyclase activating peptide), and vasoactive intestinal peptide (VIP). To identify the signaling pathways involved, preadipocytes were co-incubated with ligands and inhibitors of PKA, PI3K or MEK. All inhibitors blocked isoproterenol-stimulated PRL release, while the PKA inhibitor did not affect stimulation by PACAP [86]. These data indicate that PRL production in preadipocytes is stimulated by catecholamines and other cAMP activators via interacting signaling pathways.
1.5.4 LS14 Human Adipocyte Cell Line
Primary rodent adipocytes as well as 3T3-L1 and 3T3-F442A murine adipocyte cell lines express the PRLR and can be used to study PRL actions, but they do not produce PRL. To elucidate the control of PRL production, human adipocytes must be employed. Given the scarcity of human adipose tissue, the large variability among specimens, and the short life span of primary adipocytes, we sought a source of human adipocytes that meets the following criteria: immortality, inducible terminal differentiation, PRL release, and PRL response. After obtaining a surgically removed metastatic liposarcoma, we cloned a spontaneously immortalized adipocyte cell line which was named LS14 [89]. This cell line has been in extensive use since 2005.
To characterize the adipogenic nature of LS14 cells, expression of multiple genes was compared in LS14 and primary vis adipocytes before and after differentiation. Expression of aP2, GLUT4, HSL, LPL, and angiotensinogen was similarly induced during differentiation in both cell types. PPARγ was robustly expressed, Pref-1 was low, and UCP-1 was seen only in differentiated primary cells. Expression of adiponectin and leptin was seen in both LS14 and primary cells after differentiation, while IL-6 and TNFα were barely detected in differentiated LS14 cells. LS14 cells also express visfatin, resistin, and FIAF. Of the β-adrenergic receptors, only β2 was detected in LS14 cells, as well as insulin, estrogen (both ERα and ERβ), and dopamine receptors (Fig. 1.6).

Photograph of a lipid filled, fully differentiated LS14 human adipocyte. The various adipose markers and receptors, determined by RT-PCR are shown on the left, while secreted adipokines, enzyme activities and expression of receptor proteins are shown on the right side
The ability of LS14 cells to release of leptin, adiponectin, and IL-6 was confirmed by respective ELISAs. The release of FIAF was detected by Western blotting, while MMP-2 activity was verified by zymography. The use of fluorimetric enzyme assays confirmed that LS14 cells have functional lipid metabolizing enzymes. These complementary approaches validated the adipogenic nature of LS14 cells, established their resemblance, with few exceptions, to primary vis adipocytes, and verified their capacity not only to express, but also to release, key adipokines.
Like primary adipocytes, LS14 cells produce PRL and respond to PRL via the PRLR. PRL expression and release in both cell types increased markedly during early adipogenesis and peaked on days 5–7, follows by a decline (Fig. 1.7). Incubation of LS14 cells with exogenous PRL caused a dose-dependent inhibition of IL-6 [89]. Unlike many cells immortalized by genetic manipulation which often resist induced differentiation, LS14 cells undergo considerable morphological and functional differentiation under the appropriate culture conditions. The availability of LS14 cells opens up new avenues for research on human adipocyte biology, and adds to the small repertoire of non-pituitary PRL-producing human cell lines.

Left panel: induced differentiation of primary visceral preadipocytes into mature adipocytes over a 10-day period. Middle panel: PRL release from LS14 cells during adipogenesis, as determined by the Nb2 bioassay. Right panel: comparison of the expression of selected genes before (day 0) and after (day 10) differentiation in primary visceral adipocytes and LS14 cells
1.5.5 Dopamine: A Physiological Inhibitor of Adipocyte PRL
The time-dependent increase in adipocyte PRL release [79] resembled the progressive rise in PRL release from freshly incubated pituitary cells, which results from the removal of tonic inhibition by hypothalamic dopamine [90]. Yet, dopamine was initially ruled out as a putative inhibitor of adipocyte PRL because a ready source of dopamine to the adipocytes was not apparent, and there was no information whether dopamine receptors (DAR) are expressed in human adipose tissue. In addition, previous studies showed no effects of dopamine on PRL release from human decidual explants [91]. This was interpreted as insensitivity of the superdistal PRL promoter to dopamine rather than as a possible absence of DAR in this tissue. Dopamine binds to five 7-transmembrane, G-protein-coupled receptors, named D1R-D5R. D1R and D5R are classified by their ability to increase cAMP, while D2R, D3R, and D4R inhibit cAMP. The inhibition of pituitary PRL by dopamine occurs through activation of D2R [90].
Unlike the well-studied peripheral norepinephrine and epinephrine, the presence of dopamine in the general circulation has been overlooked by most investigators. The blood–brain barrier prevents transport of dopamine from the brain to the periphery, but small amounts of dopamine are produced by, and released from, the GI tract, adrenal medulla, and sympathetic nerve endings [92]. Little known is the fact that the major form of circulating dopamine in humans is the biologically inactive dopamine sulfate (DA-S). Sulfoconjugation is done in the GI tract by SULT1A3 sulfotransferase which is not expressed in rodents [93]. Basal serum DA-S levels at ≈ 10 nM exceeds by fivefold the combined levels of free dopamine, norepinephrine, or epinephrine. DA-S has a half-life of 3–4 h, compared with few minutes for unmodified dopamine [94]. Most importantly, unlike dopamine inactivation by deamination, O-methylation or glucuronidation, sulfoconjugation is reversible, and DA-S can be converted back to bioactive dopamine by arylsulfatase A (ARSA), a releasable lysosomal enzyme [95].
While pursuing the inhibitor of adipocyte PRL, our major assumption was that if human adipocytes express DAR and possess an active ARSA, circulating DA-S could serve as a readily available reservoir of dopamine for the adipocytes. Therefore, the objectives were to: (1) examine whether human adipocytes express DAR, (2) determine whether they have an active ARSA, and (3) examine if dopamine and DA-S affect adipose PRL expression and release [88]. A comprehensive approach was undertaken which included multiple analytical approaches as well as complementary cellular models: adipose tissue explants, primary adipocytes and two human adipocyte cell lines: LS14 and SW872, another liposarcoma-derived cell line from the ATCC.

Sources of dopamine (DA) and activation of D1R and D2R in human adipocytes. DA reaches the adipocytes from macrophages and nerve endings within adipose tissue. It is also available as dopamine sulfate (DA-S) which can be converted to bioactive DA by arylsulfatase A (ARSA). Acting via D2R and suppressing cAMP, DA inhibits PRL gene expression and release. Acting via D1R and activating both cGMP and/or MAPK, dopamine inhibits leptin and stimulates adiponectin and IL-6 release
Except for D3R, all other DAR are variably expressed at both the mRNA and protein levels in adipose tissue and adipocytes [88]. Expression of D1R decreases, while that of D2R increases during the first 3 days of adipogenesis. ARSA is expressed in adipocytes, and its enzymatic activity increases following adipogenesis. Dopamine at low nM concentrations suppresses cAMP, stimulates cGMP, and activates MAPK in adipocytes. Acting via D2R, both dopamine and DA-S inhibit PRL gene expression and release (Fig. 1.8). Dopamine shows a nonmonotonic dose-dependent inhibition of PRL, suggesting that the effects of inhibitory D2R at low dopamine doses is counteracted by stimulatory DAR at higher doses. The cAMP and/or MAPK signaling appear to be involved in mediating dopamine actions in adipocytes. Indeed, the superdistal PRL promoter has several cAMP responsive elements such as CREB and C/EBP, and two AP-1 sites which can respond to MAPK activation [96]. These data established dopamine as a suppressor adipocyte PRL via D2R through inhibition of cAMP and PKA. In addition to the suppression of adipocyte PRL, dopamine inhibits leptin and stimulates adiponectin and IL-6 release by binding to D1R and activating the cGMP/MAPK signaling (Fig. 1.8).
1.6 Metabolic Functions of PRL
1.6.1 Global Actions of PRL on Body Weight and Adiposity
Chronic elevation of PRL in rats is associated with increases in food intake but inconsistent changes in body weight. Suppression of PRL results in the opposite outcome, being most effective in lactating rats and least effective in males, reviewed in [48]. Injections of PRL into the paraventricular nucleus increased food intake, suggesting interaction with hypothalamic neurons that regulate appetite [48]. A more recent study reported that chronic intracerebral infusion of PRL increased food intake without altering body weight or estrous cyclicity [97]. The complex outcome was explained by an induction of leptin resistance via activated central PRLR. Many studies on PRL-leptin interactions took advantage of pregnant and lactating rats which are hyperphagic in adaptation for increased metabolic demands by fetuses and suckling young [98, 99]. High levels of PRL (early pregnancy and lactation) or placental lactogens (mid to late pregnancy) induced central leptin resistance by blocking its transport into the brain, and by reducing expression and signaling of its receptor, thus facilitating increased food intake.
Early studies with mice generated conflicting data. For example, elevated serum PRL, achieved by surgical or pharmacological manipulations, caused small increases in body weight and food intake with a slight decline in fat mass in males, but not females. A small decrease in retroperitoneal fat mass, but no change in body weight, was seen in PRL-overexpressing female, reviewed in [48]. Only minor changes in the overall metabolic phenotype were observed in our study with PRL-knockout mice [100]. PRL-deficiency did not affect the rate of weight gain, body composition, serum lipids, or adiponectin levels in either sex on low fat (LF) or high fat (HF) diets. Glucose tolerance was slightly impaired in very young PRL-knockout males, but not in females. Leptin was elevated only in males on LF diet. A different metabolic profile was seen in PRLR-knockout mice. The first report on a substantial decrease in weight gain and abdominal fat mass in old mice [101], was not confirmed in later studies with younger animals, attributing the weight loss in aging PRLR-deficient mice to the development of pituitary tumors.
More recent studies utilizing transgenic mice with altered PRLR, have clarified some of the above discrepancies. In one study, total PRLR deficiency was associated with resistance to HF-induced obesity due to enhanced energy expenditure and increased metabolic rate; these were attributed to the induction of brown adipocytes in several fat depots [102]. PRLR inactivation was associated with increased expression of genes that regulate brown adipocytes, suggesting that PRL suppresses transdifferentiation of white adipocytes into metabolically active brown adipocytes. Another study used mice that express only the long form of the PRLR [103]. These mice showed increased accumulation of visceral fat in older males without a significant change in body weight. The increased epididymal fat was attributed to suppression of leptin and diminished lipolysis. However, explanations for the sex-dependent changes, as well as evidence for lack of production of short PRLR isoforms were not provided.
In humans, sustained PRL elevation, caused by antipsychotic drugs [104] or prolactinomas [105], leads to increased weight, which can be ameliorated by normalization of serum PRL. Unexpectedly, the reduction in body weight in response to bromocriptine was more effective in men than women [106]. However, weight loss was not seen in all patients, was modest and delayed, and did not correlate well with the rapid and marked suppression of serum PRL levels. At present, there is no strong evidence that PRL at normal circulating levels is a major factor in human obesity. This still leaves open the possibility that certain individuals are more responsive to PRL due to variations in PRLR expression, presence of PRLR isoforms, and/or altered PRL signaling. Polymorphism in a site adjacent to the PRL gene was associated with increased risk of obesity in men but not women [107], but the relevance of this finding to the role of PRL in obesity is unclear.
1.6.2 Mammary Gland Metabolism
PRL is expressed and released by both adipose and glandular compartments of the normal human breast [78]. Recent evidence shows that activation of the PI3K/Akt pathway in the mouse mammary epithelium induced expression of autocrine PRL which is required for the initiation of lactation [6, 108]. In rats, PRL expression, determined by RT-PCR and in situ hybridization, was seen in alveolar and ductal epithelial cells in late pregnancy and throughout lactation [109]. PRLR expression in the rat mammary gland is low during most of pregnancy, increases on day 21, just before parturition, and continues to rise during lactation [110]. Both long and short PRLR isoforms are detectable in ducts and alveoli of the lactating mouse mammary gland, with some PRLR immunostaining seen near lipid droplets, suggesting expression by adipocytes [111]. In the same study, a strong PRLR immunostaining was seen in ductal epithelium of breast tissue from normal, nonpregnant, nonlactating women, and a lower staining in myoepithelial cells. Unfortunately, virtually nothing is known about expression of PRL or PRLR in the human breast during pregnancy or lactation.
Studies with rodents and ruminants were instrumental in developing the concept that during lactation, PRL acts as a physiological sensor which responds to high metabolic demands for milk production by partitioning nutrients away from adipose tissue into the mammary gland [112]. In the lactating mammary gland, PRL affects the synthesis of all milk constituents: proteins, lactose, and lipids. Here, we focus on lipids only. Compared to adipose tissue, lipid metabolism in the nonlactating mammary gland is negligible. However, at the onset of lactation, lipid production is blunted in adipose tissue and increases manyfold in the mammary gland, which produces TAGs from dietary fatty acids and de novo synthesis. The epithelial cells sequester fatty acids from adjacent adipocytes for de-novo lipogenesis [58]. As detailed in a previous review [48], PRL strongly enhances mammary lipid production by affecting the activities of many lipid biosynthetic enzymes: lipoprotein lipase (LPL), pyruvate dehydrogenase (PDH), acetyl-CoA carboxylase (ACC), and fatty acid synthase (FAS).
Several issues that are relevant to PRL and the mammary gland should be considered. One is local PRL production in the lactating mammary gland in rodents [5, 6] and human breast [78]. It is unknown whether local PRL emulates circulating PRL or fulfills distinct roles, because of selective phosphorylation, glycosylation, or cleavage. Another issue is the presence of large amounts of heavily glycosylated PRL in human milk [113]. Future studies should examine in more detail the synthesis, bioactivity, and transport of PRL into milk. Notably, the developing human fetus is exposed to very high levels of PRL from the amniotic fluid and the fetal pituitary, reviewed in [114], but the biological significance of high PRL availability to the fetus remains elusive. Milk PRL may represent a continuum of PRL availability to the newborn, who can absorb intact proteins through the GI tract for several days after birth [115].
1.6.3 Adipogenesis
Based on the belief that the PRLR is not expressed in adipose tissue, it was initially proposed that PRL does not directly regulate adipocyte functions [112]. As reviewed previously [10, 48], this concept has been revised following the reports that PRLR is expressed in both brown and white adipose tissue in all species examined. Expression of long and short PRLR isoforms increases manyfolds during differentiation of rat epididymal preadipocytes [116]. In human breast preadipocyte, PRLR shows an initial decrease, followed by an increase during adipogenesis [86]. PRLR, but not GHR, was markedly induced following differentiation of 3T3-L1 cells [117], which temporally coincided with a robust activation of Stat5a and 5b [118]. PRL upregulates the expression of its receptor in epididymal adipocytes [116], and increases Stat5a and 5b activity in differentiated 3T3-L1 cells [117].
Fetal bovine serum, which contains large quantities of lactogenic hormones and is required for efficient differentiation of 3T3-L1 cells, can be replaced by either GH or PRL [119]. PRL enhances the expression of C/EBPβ and PPARγ, two key transcription factors in adipogenesis. Furthermore, ectopic expression of the PRLR in NIH-3T3 cells increases the efficacy of adipocyte conversion when stimulated with PRL and a PPARγ ligand [120]. Studies with PRLR-deficient mice are also supportive. Receptor deficiency results in reduced size of fat depots, which was due to a lower adipocyte number rather than to a change in their volume [121]. PRL also plays a role in the differentiation, or transdifferentiation, of brown adipocytes [122].
Stat5 appears to be particularly critical for adipogenesis. Stat5 activation increases early in adipogenesis, and induces both the expression and activation of PPARγ, while targeting multiple genes that are associated with lipid and glucose metabolism as well as insulin signaling in mature adipocytes [123]. It is difficult, however, to assign a commanding role for PRL in Stat5 activation because it is equally induced by GH.
1.6.4 Lipid Metabolism and Adipokine Release
There is only sparse and inconsistent information on the involvement of PRL in lipid metabolism in adipose tissue under nonlactating conditions. Various rodent models with altered PRL/PRLR provide indirect, and often weak, support to this effect [60, 100, 103]. Studies with human subjects with hyperprolactinemia have not produced compelling evidence either. As illustrated in Fig. 1.9, PRL suppressed lipogenesis by inhibiting LPL activity [124], reducing GLUT4 expression, and lowering malonyl-CoA concentrations [125]. In fully differentiated 3T3-L1 adipocytes, PRL downregulated FAS expression [126].

Overall actions of PRL on lipid metabolism and adipokine release from adipose tissue. PRL inhibits lipid synthesis by suppressing Glut4, lipoprotein lipase (LPL), and fatty acid synthase (FAS), but it also inhibits lipolysis and the release of free fatty acids (FFA). Both adiponectin and IL-6 are inhibited by PRL while both stimulatory and inhibitory effects of PRL on leptin have been reported
A confounding problem in many in vitro studies is the use of supraphysiological doses of PRL. For example, PRL inhibited lipolysis in rat epididymal adipose explants in a dose-dependent manner within a narrow physiological range, while a higher dose of PRL resulted in a nonmonotonic curve [100]. Loss of linear dose–response relationships at high doses has been observed in some PRL target tissues and can lead to erroneous interpretation if only a single high dose is used. At high concentrations, PRL can downregulate the receptor, hinders receptor dimerization, activates dominant negative short receptors, or induces SOCS.
Direct effects of PRL on lipolysis vary among species, showing inhibition of isoproterenol-stimulated lipolysis in rat and human adipose tissues, but having no effects on lipolysis in mouse adipose explants [100, 116]. The anti-lipolytic effect of PRL in rat epididymal adipose explants takes several hours, suggesting transcriptional regulation rather than altered cAMP levels or phosphorylation of HSL and/or perilipin, as is the case with catecholamines and insulin.
Data on the effects of PRL on adipokines vary with the species and the experimental model, i.e., whether conducted in vivo or in vitro, representing indirect vs. direct effects, respectively. Such considerations are well illustrated by the variable data on the effects of PRL on leptin (Fig. 1.9). For example, serum leptin levels are lower in PRLR-deficient mice [101, 103], and are elevated in PRL-overexpressing mice [127]. However, an inhibitory effect of PRL is suggested by higher serum leptin levels in male PRL-knockout mice [100]. In rats, elevated serum PRL levels, achieved by pituitary grafts or PRL injections, increased serum leptin levels [128], while hyperprolactinemic patients had higher [129] or unchanged [130] serum leptin.
Data on leptin, based on in vitro studies are either inconsistent or have inherent limitations. PRL inhibits insulin-stimulated leptin release in mouse white adipocytes [127], but potentiates the effect of insulin in brown adipocytes [131]. Incubation of rat adipose tissue explants with PRL caused dose-dependent inhibition of leptin release [116]. Unfortunately, leptin expression in 3T3-L1 cells is severely downregulated, while the presence of autocrine PRL in human adipocytes confounds studies on its effect on leptin release. Presently, it is difficult to reach a clear conclusion to what extent PRL contributes to the control of leptin release.
Adiponectin is also affected by PRL. An inhibitory effect of PRL on adiponectin release is supported by the reduced serum adiponectin levels in both PRL transgenic and PRL-treated mice [132, 133]. PRL, however, is unlikely a major regulator of adiponectin in mice since deficiencies in either PRLR [133] or PRL [100] have no effect on serum adiponectin levels. Studies with human adipose tissue explants and isolated mature adipocytes show direct inhibitory effect of PRL on adiponectin release [133, 134]. However, a similar inhibitory relationship has not been observed in hyperprolactinemia patients.
1.7 Conclusions and Future Directions
PRL should be recognized as a metabolic hormone whose actions are not confined to the lactating mammary gland. Globally, excess PRL correlates with changes in food intake and body weight in some species, with marginal effects on fat deposition. Emerging data suggest that PRL plays a role in whole body insulin sensitivity through its stimulatory effect on insulin release and regulation of adipokine release. The recent finding of lower PRL release from human sc adipose tissue in obese vs. lean individuals suggests that adipose PRL may be involved in obesity-related complications, and should be further explored (Fig. 1.10).

The overall metabolic actions of PRL. PRL can reach target organs through the blood from the pituitary or through local production in adipose tissue and breast adipocytes
After being overlooked for a long time, the metabolic aspects of PRL have recently come into focus, in tune with the growing interest in obesity and diabetes. The rat may be a better model than the mouse for analyzing some metabolic aspects of PRL in live animals. On the other hand, the large repertoire of murine and human primary adipocytes and cell lines that express the PRLR provide an excellent opportunity to study interactions between PRL and metabolic hormones such as insulin, glucocorticoids, and catecholamines which affect adipogenesis, glucose, and lipid metabolism. Comparisons should also be made between the actions of PRL and GH, its sister molecule.
Being an emerging field with little fundamental knowledge, there are multiple challenges for future research. These include examination of PRL action on insulin release and β-cell functions in males and nonpregnant females, and explorations of PRL effects on the liver, a key organ in metabolic homeostasis which expresses high levels of the PRLR. Another issue of great interest is whether PRL is involved in human obesity and insulin resistance via its capacity to alter the production and release of adipokines, such as leptin and adiponectin.
To better establish whether PRL plays a role in lipid metabolism more comprehensive and methodological studies are urgently needed. Notably, available data reveal that PRL affects adipocyte functions in males, indicating that its impact on metabolic homeostasis is broader than previously appreciated. Although the PRLR is highly expressed in the liver, surprisingly little work has focused on potential actions of PRL in this tissue, which is so central to metabolic homeostasis. Given that PRL regulates enzymes and transporters associated with glucose and lipid metabolism in other target organs, future studies should examine its direct effects on hepatic tissue.
References
Trott JF, Vonderhaar BK, Hovey RC (2008) Historical perspectives of prolactin and growth hormone as mammogens, lactogens and galactagogues for the future! J Mammary Gland Biol Neoplasia 13:3–11
Ben-Jonathan N, Mershon JL, Allen DL, Steinmetz RW (1996) Extrapituitary prolactin: distribution, regulation, functions, and clinical aspects. Endocr Rev 17:639–669
Khurana S, Kuns R, Ben-Jonathan N (1999) Heparin-binding property of human prolactin: a novel aspect of prolactin biology. Endocrinology 140:1026–1029
Prigent-Tessier A, Tessier C, Hirosawa-Takamori M, Boyer C, Ferguson-Gottschall S, Gibori G (1999) Rat decidual prolactin. Identification, molecular cloning, and characterization. J Biol Chem 274:37982–37989
Steinmetz RW, Grant AL, Malven PV (1993) Transcription of prolactin gene in milk secretory cells of the rat mammary gland. J Endocrinol 136:271–276
Chen CC, Stairs DB, Boxer RB, Belka GK, Horseman ND, Alvarez JV et al (2012) Autocrine prolactin induced by the Pten-Akt pathway is required for lactation initiation and provides a direct link between the Akt and Stat5 pathways. Genes Dev 26:2154–2168
Alam SM, Konno T, Dai G, Lu L, Wang D, Dunmore JH et al (2007) A uterine decidual cell cytokine ensures pregnancy-dependent adaptations to a physiological stressor. Development 134:407–415
Forsyth IA, Wallis M (2002) Growth hormone and prolactin–molecular and functional evolution. J Mammary Gland Biol Neoplasia 7:291–312
Newbern D, Freemark M (2011) Placental hormones and the control of maternal metabolism and fetal growth. Curr Opin Endocrinol Diabetes Obes 18:409–416
Ben-Jonathan N, LaPensee CR, LaPensee EW (2008) What can we learn from rodents about prolactin in humans? Endocr Rev 29:1–41
Sinha YN (1995) Structural variants of prolactin: occurrence and physiological significance. Endocr Rev 16:354–369
Fahie-Wilson MN, John R, Ellis AR (2005) Macroprolactin; high molecular mass forms of circulating prolactin. Ann Clin Biochem 42:175–192
Clapp C, Martial JA, Guzman RC, Rentier-Delure F, Weiner RI (1993) The 16-kilodalton N-terminal fragment of human prolactin is a potent inhibitor of angiogenesis. Endocrinology 133:1292–1299
Clapp C, Gonzalez C, Macotela Y, Aranda J, Rivera JC, Garcia C et al (2006) Vasoinhibins: a family of N-terminal prolactin fragments that inhibit angiogenesis and vascular function. Front Horm Res 35:64–73
Pellegrini I, Gunz G, Grisoli F, Jaquet P (1990) Different pathways of secretion for glycosylated and nonglycosylated human prolactin. Endocrinology 126:1087–1095
Bollengier F, Mahler A, Braet C, Claeyssens M, Vanhaelst L (2001) Glycosylated rat prolactin: isolation and structural characterization. Arch Physiol Biochem 109:180–190
Walker AM (2007) S179D prolactin: antagonistic agony! Mol Cell Endocrinol 276:1–9
Ueda E, Ozerdem U, Chen YH, Yao M, Huang KT, Sun H et al (2006) A molecular mimic demonstrates that phosphorylated human prolactin is a potent anti-angiogenic hormone. Endocr Relat Cancer 13:95–111
Kossiakoff AA (2004) The structural basis for biological signaling, regulation, and specificity in the growth hormone-prolactin system of hormones and receptors. Adv Protein Chem 68:147–169
Hu ZZ, Zhuang L, Meng J, Tsai-Morris CH, Dufau ML (2002) Complex 5′ genomic structure of the human prolactin receptor: multiple alternative exons 1 and promoter utilization. Endocrinology 143:2139–2142
Bernichtein S, Touraine P, Goffin V (2010) New concepts in prolactin biology. J Endocrinol 206:1–11
Plotnikov A, Varghese B, Tran TH, Liu C, Rui H, Fuchs SY (2009) Impaired turnover of prolactin receptor contributes to transformation of human breast cells. Cancer Res 69:3165–3172
Clevenger CV, Furth PA, Hankinson SE, Schuler LA (2003) The role of prolactin in mammary carcinoma. Endocr Rev 24:1–27
Swaminathan G, Varghese B, Fuchs SY (2008) Regulation of prolactin receptor levels and activity in breast cancer. J Mammary Gland Biol Neoplasia 13:81–91
Brooks CL (2012) Molecular mechanisms of prolactin and its receptor. Endocr Rev 33:504–525
Meng J, Tsai-Morris CH, Dufau ML (2004) Human prolactin receptor variants in breast cancer: low ratio of short forms to the long-form human prolactin receptor associated with mammary carcinoma. Cancer Res 64:5677–5682
Binart N, Bachelot A, Bouilly J (2010) Impact of prolactin receptor isoforms on reproduction. Trends Endocrinol Metab 21:362–368
Qazi AM, Tsai-Morris CH, Dufau ML (2006) Ligand-independent homo- and hetero-dimerization of human prolactin receptor variants: inhibitory action of the short forms by heterodimerization. Mol Endocrinol 20:1912–1923
Ali S, Pellegrini I, Kelly PA (1991) A prolactin-dependent immune cell line (Nb2) expresses a mutant form of prolactin receptor. J Biol Chem 266:20110–20117
Utama FE, LeBaron MJ, Neilson LM, Sultan AS, Parlow AF, Wagner KU et al (2006) Human prolactin receptors are insensitive to mouse prolactin: implications for xenotransplant modeling of human breast cancer in mice. J Endocrinol 188:589–601
Semprini S, McNamara AV, Awais R, Featherstone K, Harper CV, McNeilly JR et al (2012) Peritonitis activates transcription of the human prolactin locus in myeloid cells in a humanized transgenic rat model. Endocrinology 153:2724–2734
Christensen HR, Murawsky MK, Horseman ND, Willson TA, Gregerson KA (2013) Completely humanizing prolactin rescues infertility in prolactin knockout mice and leads to human prolactin expression in extrapituitary mouse tissues. Endocrinology 154:4777–4789
Frank SJ (2002) Receptor dimerization in GH and erythropoietin action–it takes two to tango, but how? Endocrinology 143:2–10
Gadd SL, Clevenger CV (2006) Ligand-independent dimerization of the human prolactin receptor isoforms: functional implications. Mol Endocrinol 20:2734–2746
Goffin V, Bernichtein S, Touraine P, Kelly PA (2005) Development and potential clinical uses of human prolactin receptor antagonists. Endocr Rev 26:400–422
Gutzman JH, Rugowski DE, Schroeder MD, Watters JJ, Schuler LA (2004) Multiple kinase cascades mediate prolactin signals to activating protein-1 in breast cancer cells. Mol Endocrinol 18:3064–3075
Dominguez-Caceres MA, Garcia-Martinez JM, Calcabrini A, Gonzalez L, Porque PG, Leon J et al (2004) Prolactin induces c-Myc expression and cell survival through activation of Src/Akt pathway in lymphoid cells. Oncogene 23:7378–7390
Hennighausen L, Robinson GW (2008) Interpretation of cytokine signaling through the transcription factors STAT5A and STAT5B. Genes Dev 22:711–721
Ahima RS (2006) Adipose tissue as an endocrine organ. Obesity (Silver Spring) 14(Suppl 5):242S–249S
Ailhaud G (2006) Adipose tissue as a secretory organ: from adipogenesis to the metabolic syndrome. C R Biol 329:570–577
Tang QQ, Lane MD (2012) Adipogenesis: from stem cell to adipocyte. Annu Rev Biochem 81:715–736
Rosen ED, Walkey CJ, Puigserver P, Spiegelman BM (2000) Transcriptional regulation of adipogenesis. Genes Dev 14:1293–1307
Herold C, Rennekampff HO, Engeli S (2013) Apoptotic pathways in adipose tissue. Apoptosis 18:911–916
Arner P, Spalding KL (2010) Fat cell turnover in humans. Biochem Biophys Res Commun 396:101–104
Ortega FJ, Fernandez-Real JM (2013) Inflammation in adipose tissue and fatty acid anabolism: when enough is enough! Horm Metab Res 45:1009–1019
Harwood HJ Jr (2012) The adipocyte as an endocrine organ in the regulation of metabolic homeostasis. Neuropharmacology 63:57–75
Poulos SP, Hausman DB, Hausman GJ (2010) The development and endocrine functions of adipose tissue. Mol Cell Endocrinol 323:20–34
Ben-Jonathan N, Hugo ER, Brandebourg TD (2006) LaPensee CR. Focus on prolactin as a metabolic hormone. Trends Endocrinol Metab 17:110–116
Trayhurn P, Beattie JH (2001) Physiological role of adipose tissue: white adipose tissue as an endocrine and secretory organ. Proc Nutr Soc 60:329–339
Zafrir B (2013) Brown adipose tissue: research milestones of a potential player in human energy balance and obesity. Horm Metab Res 45:774–785
Peschechera A, Eckel J (2013) “Browning” of adipose tissue–regulation and therapeutic perspectives. Arch Physiol Biochem 119:151–160
Wajchenberg BL, Giannella-Neto D, da Silva ME, Santos RF (2002) Depot-specific hormonal characteristics of subcutaneous and visceral adipose tissue and their relation to the metabolic syndrome. Horm Metab Res 34:616–621
Lafontan M (2012) Historical perspectives in fat cell biology: the fat cell as a model for the investigation of hormonal and metabolic pathways. Am J Physiol Cell Physiol 302:C327–C359
Bloor ID, Symonds ME (2014) Sexual dimorphism in white and brown adipose tissue with obesity and inflammation. Horm Behav
Arner P (2001) Regional differences in protein production by human adipose tissue. Biochem Soc Trans 29:72–75
Potenza MV, Mechanick JI (2009) The metabolic syndrome: definition, global impact, and pathophysiology. Nutr Clin Pract 24:560–577
Cinti S (2005) The adipose organ. Prostaglandins Leukot Essent Fatty Acids 73:9–15
Hovey RC, Aimo L (2010) Diverse and active roles for adipocytes during mammary gland growth and function. J Mammary Gland Biol Neoplasia 15:279–290
Su Y, Shankar K, Rahal O, Simmen RC (2011) Bidirectional signaling of mammary epithelium and stroma: implications for breast cancer–preventive actions of dietary factors. J Nutr Biochem 22:605–611
Carre N, Binart N (2014) Prolactin and adipose tissue. Biochimie 97:16–21
Gregoire FM (2001) Adipocyte differentiation: from fibroblast to endocrine cell. Exp Biol Med (Maywood) 226:997–1002
MacDougald OA, Mandrup S (2002) Adipogenesis: forces that tip the scales. Trends Endocrinol Metab 13:5–11
Letexier D, Pinteur C, Large V, Frering V, Beylot M (2003) Comparison of the expression and activity of the lipogenic pathway in human and rat adipose tissue. J Lipid Res 44:2127–2134
Jeffcoat R (2007) Obesity—a perspective based on the biochemical interrelationship of lipids and carbohydrates. Med Hypotheses 68:1159–1171
Large V, Peroni O, Letexier D, Ray H, Beylot M (2004) Metabolism of lipids in human white adipocyte. Diabetes Metab 30:294–309
Proenca AR, Sertie RA, Oliveira AC, Campaaa AB, Caminhotto RO, Chimin P et al (2014) New concepts in white adipose tissue physiology. Braz J Med Biol Res 47:192–205
Zechner R, Kienesberger PC, Haemmerle G, Zimmermann R, Lass A (2009) Adipose triglyceride lipase and the lipolytic catabolism of cellular fat stores. J Lipid Res 50:3–21
Ahmadian M, Wang Y, Sul HS (2010) Lipolysis in adipocytes. Int J Biochem Cell Biol 42:555–559
Wang S, Soni KG, Semache M, Casavant S, Fortier M, Pan L et al (2008) Lipolysis and the integrated physiology of lipid energy metabolism. Mol Genet Metab 95:117–126
Lafontan M, Moro C, Berlan M, Crampes F, Sengenes C, Galitzky J (2008) Control of lipolysis by natriuretic peptides and cyclic GMP. Trends Endocrinol Metab 19:130–137
Trujillo ME, Scherer PE (2006) Adipose tissue-derived factors: impact on health and disease. Endocr Rev 27:762–778
Wauman J, Tavernier J (2011) Leptin receptor signaling: pathways to leptin resistance. Front Biosci (Landmark Ed) 16:2771–2793
Munzberg H, Bjornholm M, Bates SH, Myers MG Jr (2005) Leptin receptor action and mechanisms of leptin resistance. Cell Mol Life Sci 62:642–652
Perry B, Wang Y (2012) Appetite regulation and weight control: the role of gut hormones. Nutr Diabetes 2:e26
Li FY, Lam KS, Xu A (2012) Therapeutic perspectives for adiponectin: an update. Curr Med Chem 19:5513–5523
Turer AT, Scherer PE (2012) Adiponectin: mechanistic insights and clinical implications. Diabetologia 55:2319–2326
Kershaw EE, Flier JS (2004) Adipose tissue as an endocrine organ. J Clin Endocrinol Metab 89:2548–2556
Zinger M, McFarland M, Ben-Jonathan N (2003) Prolactin expression and secretion by human breast glandular and adipose tissue. J Clin Endocrinol Metab 88:689–696
Hugo ER, Borcherding DC, Gersin KS, Loftus J, Ben-Jonathan N (2008) Prolactin release by adipose explants, primary adipocytes, and LS14 adipocytes. J Clin Endocrinol Metab 93:4006–4012
Bouckenooghe T, Sisino G, Aurientis S, Chinetti-Gbaguidi G, Kerr-Conte J, Staels B et al (2014) Adipose tissue macrophages (ATM) of obese patients are releasing increased levels of prolactin during an inflammatory challenge: a role for prolactin in diabesity? Biochim Biophys Acta 1842:584–593
Ernst B, Thurnheer M, Schultes B (2009) Basal serum prolactin levels in obesity–unrelated to parameters of the metabolic syndrome and unchanged after massive weight loss. Obes Surg 19:1159–1162
Chirico V, Cannavo S, Lacquaniti A, Salpietro V, Mandolfino M, Romeo PD et al (2013) Prolactin in obese children: a bridge between inflammation and metabolic-endocrine dysfunction. Clin Endocrinol (Oxf) 79:537–544
Featherstone K, White MR, Davis JR (2012) The prolactin gene: a paradigm of tissue-specific gene regulation with complex temporal transcription dynamics. J Neuroendocrinol 24:977–990
Pohnke Y, Kempf R, Gellersen B (1999) CCAAT/enhancer-binding proteins are mediators in the protein kinase A- dependent activation of the decidual prolactin promoter. J Biol Chem 274:24808–24818
Watanabe K, Kessler CA, Bachurski CJ, Kanda Y, Richardson BD, Stanek J et al (2001) Identification of a decidua-specific enhancer on the human prolactin gene with two critical activator protein 1 (AP-1) binding sites. Mol Endocrinol 15:638–653
McFarland-Mancini M, Hugo E, Loftus J, Ben-Jonathan N (2006) Induction of prolactin expression and release in human preadipocytes by cAMP activating ligands. Biochem Biophys Res Commun 344:9–16
Brar AK, Kessler CA, Handwerger S (2002) An Ets motif in the proximal decidual prolactin promoter is essential for basal gene expression. J Mol Endocrinol 29:99–112
Borcherding DC, Hugo ER, Idelman G, De SA, Richtand NW, Loftus J et al (2011) Dopamine receptors in human adipocytes: expression and functions. PLoS ONE 6:e25537
Hugo ER, Brandebourg TD, Comstock CE, Gersin KS, Sussman JJ, Ben-Jonathan N (2006) LS14: a novel human adipocyte cell line that produces prolactin. Endocrinology 147:306–313
Ben-Jonathan N (1985) Dopamine: a prolactin-inhibiting hormone. Endocr Rev 6:564–589
Golander A, Barrett J, Hurley T, Barry S, Handwerger S (1979) Failure of bromocriptine, dopamine, and thyrotropin-releasing hormone to affect prolactin secretion by human decidual tissue in vitro. J Clin Endocrinol Metab 49:787–789
Goldstein DS, Swoboda KJ, Miles JM, Coppack SW, Aneman A, Holmes C et al (1999) Sources and physiological significance of plasma dopamine sulfate. J Clin Endocrinol Metab 84:2523–2531
Ghosh D (2007) Human sulfatases: a structural perspective to catalysis. Cell Mol Life Sci 64:2013–2022
Eldrup E (2004) Significance and origin of DOPA, DOPAC, and dopamine-sulphate in plasma, tissues and cerebrospinal fluid. Dan Med Bull 51:34–62
Strobel G, Werle E, Weicker H (1990) Isomer specific kinetics of dopamine beta-hydroxylase and arylsulfatase towards catecholamine sulfates. Biochem Int 20:343–351
Marano RJ, Ben-Jonathan N (2014) Minireview: extrapituitary prolactin: an update on the distribution, regulation, and functions. Mol Endocrinol 28:622–633
Naef L, Woodside B (2007) Prolactin/leptin interactions in the control of food intake in rats. Endocrinology 148:5977–5983
Lisboa PC, Passos MC, Dutra SC, Bonomo IT, Denolato AT, Reis AM et al (2006) Leptin and prolactin, but not corticosterone, modulate body weight and thyroid function in protein-malnourished lactating rats. Horm Metab Res 38:295–299
Augustine RA, Grattan DR (2008) Induction of central leptin resistance in hyperphagic pseudopregnant rats by chronic prolactin infusion. Endocrinology 149:1049–1055
LaPensee CR, Horseman ND, Tso P, Brandebourg TD, Hugo ER, Ben-Jonathan N (2006) The prolactin-deficient mouse has an unaltered metabolic phenotype. Endocrinology 147:4638–4645
Freemark M, Fleenor D, Driscoll P, Binart N, Kelly P (2001) Body weight and fat deposition in prolactin receptor-deficient mice. Endocrinology 142:532–537
Auffret J, Viengchareun S, Carre N, Denis RG, Magnan C, Marie PY et al (2012) Beige differentiation of adipose depots in mice lacking prolactin receptor protects against high-fat-diet-induced obesity. FASEB J 26:3728–3737
Le JA, Wilson HM, Shehu A, Devi YS, Aguilar T, Gibori G (2011) Prolactin activation of the long form of its cognate receptor causes increased visceral fat and obesity in males as shown in transgenic mice expressing only this receptor subtype. Horm Metab Res 43:931–937
Baptista T, Lacruz A, Meza T, Contreras Q, Delgado C, Mejias MA et al (2001) Antipsychotic drugs and obesity: is prolactin involved? Can J Psychiatry 46:829–834
Greenman Y, Tordjman K, Stern N (1998) Increased body weight associated with prolactin secreting pituitary adenomas: weight loss with normalization of prolactin levels. Clin Endocrinol (Oxf) 48:547–553
Berinder K, Nystrom T, Hoybye C, Hall K, Hulting AL (2011) Insulin sensitivity and lipid profile in prolactinoma patients before and after normalization of prolactin by dopamine agonist therapy. Pituitary 14:199–207
Nilsson L, Olsson AH, Isomaa B, Groop L, Billig H, Ling C (2011) A common variant near the PRL gene is associated with increased adiposity in males. Mol Genet Metab 102:78–81
Oliver CH, Watson CJ (2013) Making milk: a new link between STAT5 and Akt1. JAKSTAT 2:e23228
Iwasaka T, Umemura S, Kakimoto K, Koizumi H, Osamura YR (2000) Expression of prolactin mRNA in rat mammary gland during pregnancy and lactation. J Histochem Cytochem 48:389–396
Jahn GA, Edery M, Belair L, Kelly PA, Djiane J (1991) Prolactin receptor gene expression in rat mammary gland and liver during pregnancy and lactation. Endocrinology 128:2976–2984
Ueda EK, Huang K, Nguyen V, Ferreira M, Andre S, Walker AM (2011) Distribution of prolactin receptors suggests an intraductal role for prolactin in the mouse and human mammary gland, a finding supported by analysis of signaling in polarized monolayer cultures. Cell Tissue Res 346:175–189
Flint DJ, Binart N, Kopchick J, Kelly P (2003) Effects of growth hormone and prolactin on adipose tissue development and function. Pituitary 6:97–102
Ellis LA, Picciano MF (1995) Bioactive and immunoreactive prolactin variants in human milk. Endocrinology 136:2711–2720
Ben-Jonathan N, Munsick RA (1980) Dopamine and prolactin in human pregnancy. J Clin Endocrinol Metab 51:1019–1025
Grosvenor CE, Picciano MF, Baumrucker CR (1993) Hormones and growth factors in milk. Endocr Rev 14:710–728
Brandebourg TD, Bown JL, Ben-Jonathan N (2007) Prolactin upregulates its receptors and inhibits lipolysis and leptin release in male rat adipose tissue. Biochem Biophys Res Commun 357:408–413
Fleenor D, Arumugam R, Freemark M (2006) Growth hormone and prolactin receptors in adipogenesis: STAT-5 activation, suppressors of cytokine signaling, and regulation of insulin-like growth factor I. Horm Res 66:101–110
Stephens JM, Morrison RF, Pilch PF (1996) The expression and regulation of STATs during 3T3-L1 adipocyte differentiation. J Biol Chem 271:10441–10444
Stewart WC, Baugh JE Jr, Floyd ZE, Stephens JM (2004) STAT 5 activators can replace the requirement of FBS in the adipogenesis of 3T3-L1 cells. Biochem Biophys Res Commun 324:355–359
Nanbu-Wakao R, Fujitani Y, Masuho Y, Muramatu M, Wakao H (2000) Prolactin enhances CCAAT enhancer-binding protein-beta (C/EBP beta) and peroxisome proliferator-activated receptor gamma (PPAR gamma) messenger RNA expression and stimulates adipogenic conversion of NIH-3T3 cells. Mol Endocrinol 14:307–316
Flint DJ, Binart N, Boumard S, Kopchick JJ, Kelly P (2006) Developmental aspects of adipose tissue in GH receptor and prolactin receptor gene disrupted mice: site-specific effects upon proliferation, differentiation and hormone sensitivity. J Endocrinol 191:101–111
Carre N, Solomon G, Gertler A, Binart N (2014) Effects of high affinity leptin antagonist on prolactin receptor deficient male mouse. PLoS ONE 9:e91422
Zhao P, Stephens JM (2013) Identification of STAT target genes in adipocytes. JAKSTAT 2:e23092
Ling C, Svensson L, Oden B, Weijdegard B, Eden B, Eden S et al (2003) Identification of functional prolactin (PRL) receptor gene expression: PRL inhibits lipoprotein lipase activity in human white adipose tissue. J Clin Endocrinol Metab 88:1804–1808
Nilsson LA, Roepstorff C, Kiens B, Billig H, Ling C (2009) Prolactin suppresses malonyl-CoA concentration in human adipose tissue. Horm Metab Res 41:747–751
Hogan JC, Stephens JM (2005) The regulation of fatty acid synthase by STAT5A. Diabetes 54:1968–1975
Ling C, Billig H (2001) PRL receptor-mediated effects in female mouse adipocytes: PRL induces suppressors of cytokine signaling expression and suppresses insulin-induced leptin production in adipocytes in vitro. Endocrinology 142:4880–4890
Gualillo O, Lago F, Garcia M, Menendez C, Senaris R, Casanueva FF et al (1999) Prolactin stimulates leptin secretion by rat white adipose tissue. Endocrinology 140:5149–5153
Balci H, Akgun-Dar K, Gazioglu N, Kapucu A, Bolayirli M, Oz B (2009) The relationship between prolactin (PRL), leptin, nitric oxide (NO), and cytokines in patients with hyperprolactinemia. Pituitary 12:170–176
Atmaca A, Bilgici B, Ecemis GC, Tuncel OK (2013) Evaluation of body weight, insulin resistance, leptin and adiponectin levels in premenopausal women with hyperprolactinemia. Endocr 44:756–761
Viengchareun S, Bouzinba-Segard H, Laigneau JP, Zennaro MC, Kelly PA, Bado A et al (2004) Prolactin potentiates insulin-stimulated leptin expression and release from differentiated brown adipocytes. J Mol Endocrinol 33:679–691
Combs TP, Berg AH, Rajala MW, Klebanov S, Iyengar P, Jimenez-Chillaron JC et al (2003) Sexual differentiation, pregnancy, calorie restriction, and aging affect the adipocyte-specific secretory protein adiponectin. Diabetes 52:268–276
Nilsson L, Binart N, Bohlooly Y, Bramnert M, Egecioglu E, Kindblom J et al (2005) Prolactin and growth hormone regulate adiponectin secretion and receptor expression in adipose tissue. Biochem Biophys Res Commun 331:1120–1126
Asai-Sato M, Okamoto M, Endo M, Yoshida H, Murase M, Ikeda M et al (2006) Hypoadiponectinemia in lean lactating women: Prolactin inhibits adiponectin secretion from human adipocytes. Endocr J 53:555–562
Acknowledgments
This work was supported by NIH grants ES02909 and CA096613, DOD grants BC05725 and AR110050, CCTST, and Ride Cincinnati Pilot Grants
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Ben-Jonathan, N., Hugo, E. (2015). Prolactin (PRL) in Adipose Tissue: Regulation and Functions. In: Diakonova, PhD, M. (eds) Recent Advances in Prolactin Research. Advances in Experimental Medicine and Biology, vol 846. Springer, Cham. https://doi.org/10.1007/978-3-319-12114-7_1
Download citation
DOI: https://doi.org/10.1007/978-3-319-12114-7_1
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-12113-0
Online ISBN: 978-3-319-12114-7
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)