
Abstract
Indoleamine 2,3-dioxygenase (IDO) plays a key role in immune homeostasis via depletion of tryptophan and accumulation of kynurenines and is recognized as an important factor contributing to suppression of antitumor immune responses. However, the possibility of harnessing the IDO pathway for the therapy of autoimmune disease represents an intriguing possibility, and in this review, we highlight recent research on the involvement of IDO in immune-mediated inflammatory diseases, with a focus on rheumatoid arthritis. Inhibition of IDO was shown to exacerbate experimental arthritis and increase numbers of pathogenic Th1 and Th17 cells in the joints and draining lymph nodes. Analysis of the kinetics of expression of kynurenine pathway enzymes in animal models also pointed to a potential role for tryptophan metabolites in disease resolution and administration of l-kynurenine or [3,4-dimethoxycinnamonyl]-anthranilic acid (a synthetic derivative of 3-hydroxyanthranilic acid) reduced the severity of disease. A more recent study has identified an association between defective regulatory T cells in rheumatoid arthritis with reduced capacity to activate the kynurenine pathway. These findings suggest that strategies to activate IDO in a targeted manner may be effective in the therapy of autoimmune disease.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
Introduction
The degradation of the essential amino acid l-tryptophan by indoleamine 2,3-dioxygenase (IDO) is the first and rate-limiting step in the kynurenine pathway. IDO is encoded by an evolutionarily ancient gene that precedes the development of the adaptive immune system. In 1984, Pfefferkorn observed that the pro-inflammatory cytokine interferon-γ (IFN-γ) induced tryptophan degradation and prevented the growth of Toxoplasma gondii [1, 2]. In the intervening years, much research has focused on investigating the link between tryptophan degradation and innate immune responses. However, Munn and colleagues, who discovered that the activity of IDO could prevent the rejection of allogenic fetuses in pregnant mice, revealed that IDO-induced tryptophan depletion limited the supply of tryptophan for proliferating T cells, which in turn prevented a T cell response against the developing fetus [3]. This evidence suggested that the kynurenine pathway, as well as having a role in the innate immune system, also plays a major role in the adaptive immune system. In this chapter, we will focus on the role of the kynurenine pathway in inflammatory diseases, using rheumatoid arthritis (RA) as an example to highlight the potential of manipulating this pathway therapeutically.
The Regulation of IDO Expression During Inflammation
In mammals, three genes encode enzymes that catalyze oxidative degradation of tryptophan: IDO1, IDO2, and tryptophan 2,3-dioxygenase (TDO). Each enzyme catalyzes the same reaction, the cleavage of the 2,3 double bond in the indole ring [4]. TDO expression is mainly confined to the liver where it is involved in the homeostatic regulation of tryptophan and is not induced in response to immune stimulation. IDO1 and IDO2 are expressed in antigen-presenting cells (APCs) of the immune system and respond to a variety of inflammatory signals.
IDO1 and IDO2 share significant identity at the protein level but are not related structurally to TDO. The IDO genes are well conserved and contain several inflammatory response sequences [5], such as multiple response elements for interferon type I (IFN-α/β) and interferon type II (IFN-γ) signaling [6–9]. A number of inflammatory signals can induce IDO expression. For example, signal transducer and activator of transcription 1 (STAT1) and IFN-regulatory factor (IRF1) function in response to IFN-γ signaling to induce IDO, with a failure to induce IDO occurring in mice lacking either IFN-γ or IFR1 [10, 11]. Lipopolysaccharide (LPS), interleukin 1 (IL-1), and tumor necrosis factor (TNF) act synergistically with IFN-γ to enhance IDO expression in vitro [12]. However, other cytokines have been shown to act negatively. For example, TGF-β signaling can abrogate the effect of IFN-γ-induced IDO by reducing mRNA stability [13]. Similarly, administration of IL-10 both in vivo and in vitro leads to a decrease in the expression of IDO in DCs and splenic DCs [14].
Interestingly, an unexpected finding that IDO can be induced in DCs in response to ligation of CD80/86 by soluble recombinant cytotoxic T-lymphocyte-associated protein 4 fusion protein (CTLA-4-Ig) demonstrated that other pathways in addition to cytokines could regulate IDO expression [15]. IDO induction by ligation of CD80/86 has been demonstrated in numerous diseases in both humans and mice [16–18]. This observation also suggested the possibility that CTLA-4+ regulatory T cells (Tregs) may also regulate the induction of IDO in DCs. Indeed, this has been supported by observations that both human and mouse CD4+CD25+ Tregs induce IDO both in vivo and in vitro in a CTLA-4 dependent mechanism [16, 19, 20].
Tryptophan Depletion as a Mechanism to Regulate the Immune System
Tryptophan is an essential amino acid found in low abundance that is involved in protein synthesis. Local depletion of tryptophan by IDO is one proposed mechanism of immunomodulatory action for this pathway. T cell proliferation is critical for an effective immune response, and it has been shown that inhibition of T cell proliferation by tryptophan depletion represents a critical component of the immunosuppressive function of IDO, which is mediated through IDO expressing macrophages and DCs [21, 22]. Munn et al. have shown that a consequence of tryptophan depletion is the activation of the kinase GCN2, in T cells that act as a mediator for several tryptophan depletion effects, such as reduced proliferation and increased anergy [23]. GCN2 is a stress response kinase that is activated by elevations in uncharged tRNA in response to amino acid starvation [24]. Activation of GCN2 results in cell-cycle arrest, differentiation, and apoptosis [23, 25]. T cells that do not upregulate GCN2 proliferate normally in response to IDO positive DCs and are not rendered anergic [23]. However, whether tryptophan degradation represents the major mechanism for the immunosuppressive action of IDO activity remains questionable, since results that support this have been obtained using in vitro assays and may not be reproduced in vivo. Moreover, tryptophan levels in tissue are expected to be replaced faster than the local degradation rate [26]. Therefore the hypothesis that tryptophan degradation is the primary explanation for the immunosuppressive effect of IDO needs to be validated in vivo.
The Regulation of the Immune System Through Tryptophan Catabolites
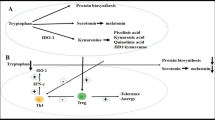
The breakdown of tryptophan by IDO leads to the formation of l-formylkynurenine, which is further degraded to l-kynurenine by formamidase. l-kynurenine acts as a substrate for a number of other enzymes that result in further breakdown into kynurenic acid, anthranilic acid, and 3-hydroxykynurenine. Further breakdown of 3-hydroxykynurenine by kynureninase produces 3-hydroxyanthranilic acid (Fig. 7.1). Each of these different tryptophan catabolites can differentially regulate the immune system, with most studies focusing on their anti-proliferative and apoptotic effects on T cells. l-kynurenine, 3-hydroxykynurenine, and 3-hydroxyanthranilic acid have all been shown to suppress CD4+ and CD8+ T cell proliferation and induce cell death [27, 28]. Moreover, tryptophan catabolites can preferentially induce apoptosis of Th1 cell clones but not Th2 cell clones, raising the possibility that tryptophan catabolites can alter the Th1/2 cell balance and potentially prevent a Th1-mediated response [28]. In addition to T cells, kynurenines also suppress natural killer (NK) cell function and can induce B cell death [27, 29], highlighting that the kynurenine pathway is involved in the suppression of a number of diverse inflammatory processes. A direct effect on APCs may also represent another mechanism for tryptophan catabolite-mediated immune suppression. DCs appear to be nonresponsive to kynurenines; however, macrophage-derived APCs are susceptible to apoptosis following culture with 3-hydroxyanthranilic acid [30].

The kynurenine pathway showing the catabolism of tryptophan
In addition to direct apoptotic effects of kynurenine and its metabolites, tryptophan catabolites may also be instrumental in skewing the immune system away towards anti-inflammatory responses. For example, IDO+ plasmacytoid DCs (pDCs) promote the de novo differentiation of Tregs from naïve T cells [19]. This differentiation also occurs when naïve T cells are cultured in low tryptophan/high kynurenine conditions resulting in a downregulation of the TCRζ chain and generation of a Treg phenotype [31]. In addition to the generation of Tregs, IDO+ DCs also promote the expansion of nTregs [32]. In this study, it was shown that IDO contributes to LPS and TNFα + poly(I:C) stimulated DC maturation, since inhibition of IDO resulted in a failure to induce mature DCs.
Kynurenine Targets of the Immune System
Kynurenine has been identified as the endogenous ligand for the aryl hydrocarbon receptor (AHR) [33]. Ligation of the AHR results in the promotion of Tregs and the suppression of pathogenic T cells. For example, co-cultures of AHR deficient DCs and naïve T cells led to reduced differentiation of Tregs [34]. In contrast, the addition of l-kynurenine promotes the differentiation of Tregs and suppresses Th17 differentiation [34]. Kynurenine metabolites may also mediate their inflammatory responses through other pathways. Kynurenic acid is the ligand of the orphan G-protein-coupled receptor 35 (GPR35), which is expressed on a number of inflammatory cells, and the interaction of kynurenic acid with GPR35 inhibits LPS-driven TNFα production in monocytes [35]. Another kynurenine metabolite, 3-hydroxyanthranillic acid, was found to reduce Th1 and Th17 cells in experimental autoimmune encephalomyelitis (EAE) [36], inhibit the generation of nitric oxide synthase in macrophages, and promote the production of TGF-β [37]; however, its primary molecular target is currently unknown.
Rheumatoid Arthritis
Rheumatoid arthritis (RA) is a chronic inflammatory disease of the joints that affects approximately 1% of the world’s population and remains a major cause of morbidity and mortality. The disease usually manifests itself as swelling of the joints, pain, and stiffness, with ankylosis developing in many cases [37]. A key observation made many years ago was that specific alleles of the class II major histocompatibility complex (MHC) confer susceptibility to RA [38]. In particular, susceptibility to RA was found to be associated with certain HLA-DRB1 alleles [39]. Subsequently, genome-wide association studies (GWAS) have revealed a number of other non-HLA susceptibility alleles that have also been implicated in disease, such as PTPN22, PADI4, CD40, and CTLA-4 [40], many of which are associated with immune responses. The involvement of inflammatory cells as drivers of RA pathogenesis is further emphasized by the presence of highly activated immune cells, particularly CD4+ T cells within the joints of RA patients.
The Role of the Kynurenine Pathway in Animal Models of RA
Animal models of RA have been extensively used to investigate the efficacy of preclinical therapeutics, understand the biology underpinning RA, and investigate the genetic susceptibility involved in the development of RA. Many models of arthritis exist; however, the collagen-induced arthritis (CIA) model has been widely investigated, since it shares many pathological features of RA, such as synovial hyperplasia, mononuclear cell infiltration, and cartilage degradation [41, 42]. Similar to RA, the genetic susceptibility in CIA is strongly linked with the MHC class II genes, in particular the susceptibility is highest in mouse strains that bear MHC types I-Aq and I-Ar [43]. Analysis of the levels of IDO mRNA expression by quantitative q-PCR in the spleen, lymph nodes, and paws of CIA mice has shown that there is significant increase in the expression of IDO following arthritis onset in the lymph nodes, but not in the spleen or paws [44]. Further analysis of the expression of IDO in these lymph nodes revealed that it was mainly confined to dendritic cells [45]. To understand the role played by the kynurenine pathway in arthritis, the levels of tryptophan and its metabolites were measured through the course of CIA [46]. The tryptophan concentration in lymph nodes decreased progressively during the development and resolution of arthritis, while the concentrations of kynurenine increased, indicating increased IDO activity [45]. Measurement of the downstream kynurenine metabolites revealed increased presence of anthranilic acid and 3-hydroxyanthranilic acid only during the resolution stage of arthritis [45]. This raises the possibility that the downstream kynurenine metabolites may play a role in disease resolution, since it is known that 3-hydroxyanthranilic acid plays a role in inhibiting T cell responses [47].
To investigate the role of IDO in CIA in more detail, the progression of CIA was monitored in mice treated with the IDO inhibitor, 1-methyl tryptophan (1-MT). Administration of 1-MT following disease onset increased the severity of CIA, in terms of increased paw thickness and enhanced humoral and cellular responses [44, 48]. A more comprehensive investigation into the role of IDO in CIA has been performed using IDO knockout (Indo −/−) mice [49]. One of the most important findings to emerge from this work was an earlier onset of arthritis in the Indo −/− mice compared to wild-type mice [44]. Indo −/− mice showed increased clinical severity, which was accompanied by a significant increase in bone erosion and cellular infiltration. Analysis of the cytokine production in Indo −/− mice revealed higher production of IL-17 and IFN-γ in the lymph nodes and FACS analysis showed increased frequency of Th1 and Th17 cells in the paws of the Indo −/− mice [44]. These findings highlight the importance of IDO expression in CIA and confirm the important role that IDO plays in regulating the pathogenic Th1/Th17 responses.
In comparison to CIA, studies in the K/BxN animal model, which is an antibody-mediated model of arthritis, have shown that administration of 1-MT results in the amelioration of arthritis due to reduced level of B cell activity [50]. Further analysis revealed that the effect of 1-MT was to inhibit differentiation of autoreactive B cells into autoantibody-secreting cells, without influencing the activation or survival of these cells [51]. These results suggest that the concept of IDO having a purely immunosuppressive function needs re-evaluation, since it may have differential effects depending upon which cells play a significant part in each model.
The Role of the Kynurenine Pathway in RA
Patients with RA display significantly decreased levels of circulating tryptophan and increased levels of kynurenine when compared to healthy individuals [52–54]. In addition, decreased levels of 3-hydroxykynurenine, 3-hydroxyanthranilic acid, and xanthurenic acid are detected in RA patients, while levels of kynurenic acid are normal when compared to healthy individuals [52]. Increased kynurenine may be associated with disease progression or increased as a result of treatment. For example, increased tryptophan correlates with increased disease progression [54]. However, another study found that RA patients had lower levels of kynurenine in both the synovial fluid and peripheral blood [55]. The discrepancy between these studies may be explained by differences in the patient demographics, in particular, exposure to disease-modifying anti-rheumatic drugs (DMARDs). To investigate differences in the kynurenine pathway early in disease, we have used a cohort of DMARD-naïve RA patients. Levels of tryptophan and kynurenine in serum were determined using HPLC-UV and we found no significant difference in the expression of tryptophan; however, a significant reduction in circulating levels of kynurenine was observed when compared to healthy controls (Fig. 7.2). These results suggest that low levels of kynurenine may contribute to the pathogenicity of RA in the early stages of disease.

Kynurenine and tryptophan levels in the human serum of DMARD-naïve RA patients. A reduction in the serum kynurenine levels was observed between DMARD-naïve RA patients and healthy individuals. n.s. = not significant, *P > 0.05 as measured using an Students t test
The Molecular Mechanisms Underpinning Differences in the Kynurenine Expression in RA
Further insight into the function of the kynurenine pathway in RA may give a better understanding of the pathogenesis of RA. Understanding the precise molecular mechanisms underpinning the differences in kynurenine expression described above may ultimately lead to new therapeutic targets. Evidence has begun to emerge to suggest that synovial joint but not peripheral blood isolated T cells from RA patients are less responsive to IDO-mediated suppression by IDO+ DCs [56]. Synovial T cells had increased expression of tryptophanyl-tRNA-synthetase (TTS), a cytoplasmic enzyme that mediates the association of tryptophan to tRNA that acts as a reservoir of tryptophan for protein synthesis. This led to enhanced intracellular storage of tryptophan and a resistance to IDO-mediated deprivation of tryptophan [56]. The resistance of T cells to IDO-mediated deprivation of tryptophan could be overcome by using blocking antibodies against IFN-γ or TNFα, which act to reduce the enhancement of TTS, suggesting that resistance to IDO activation represents a mechanism by which autoreactive T cells are maintained in the synovial joint.
Work from our own laboratory suggests that there is an intrinsic defect in the ability to induce expression of IDO in RA patients. We were the first to suggest this following investigation centered on examining dysfunctional Tregs from the peripheral blood of DMARD-naïve RA patients. Tregs act as potent inducers of the IDO pathway [19, 57], mediated in part through the ligation of CD80/86 by CTLA-4 on APCs [15]. We found that DMARD-naïve Tregs were unable to induce IDO expression in APCs following co-culture, which was associated with a reduction in both total and surface CTLA-4 expression [58]. The mechanism underpinning this reduction was through methylation of a single CpG within the Ctla4 promoter. Methylation of this site prevented the binding of the transcription factor and activator of CTLA-4, nuclear factor of activated T cells (NFAT), which in turn led to a reduction in the transcriptional activity of Ctla4 gene (Fig. 7.3). Overall, we identified that defective Treg function leads to an inability to induce IDO expression in APCs, leading to reduced local and systemic kynurenine levels and an inability to suppress effector T cells responses [58, 59].

Defective activation of the kynurenine pathway in DMARD-naïve patients with rheumatoid arthritis. The molecular mechanism underpinning reduced kynurenine expression in patients with RA is mediated through methylation of an NFAT binding site within the Ctla4 promoter of Tregs. This results in a reduction in CTLA-4 protein expression. In co-culture with APCs, the reduction of CTLA-4 on RA Tregs fails to induce IDO expression, leading to reduced activation of the kynurenine pathway. Reprinted with permission from Macmillan Publishers Ltd: Nature reviews rheumatology [59]
The Kynurenine Pathway and Other Inflammatory Diseases
As discussed previously, inhibition of IDO promotes disease progression in animal models of RA. This finding is consistent with studies in other murine models of inflammatory diseases. For example, treatment with 1-MT, the competitive inhibitor of IDO, accelerated disease in EAE and type 1 diabetes (T1D) [60, 61]. Grohmann and colleagues have shown that nonobese diabetic (NOD) mice exhibit a defect in IDO-mediated T cell suppressive function mediated though excessive production of peroxynitrite by DCs, which could be reversed with CTLA-4 fusion protein-mediated IDO induction [62, 63]. Similar to findings in RA, this suggests that defects in the ability to induce IDO in DCs of the NOD mice underlie the susceptibility to T1D and signaling though the CTLA-4/CD80/86 pathway can restore IDO-mediated suppressive function. However, it is unknown whether the inability to induce IDO is a primary cause of disease or mediated through secondary upstream effects. Moreover, these findings have not been explored in the context of human T1D so the relevance to human disease is currently unknown. In parallel with synovial T cells from RA patients that are resistant to IDO-mediated suppression, increased IDO resistance is also a feature of T1D. T cells from NOD mice display increased resistance to Treg-mediated suppression [64], which is also seen in the synovial T cells of patients with RA [65].
In EAE, amelioration of disease following stem cell transfer is mediated, in part, through the induction of IDO+ CD11c+ DCs, an effect that is abrogated following administration of the IDO inhibitor 1-MT [66]. Thus, inhibition of IDO increased disease progression and prevented disease resolution. Additionally, in both human disease and in animal models of cystic fibrosis, deficiencies in the ability to activate IDO contribute to a failure to eradicate pathogenic fungi from the lungs. Defective IDO expression was linked to an imbalance in Treg/Th17 responses, which could be corrected with the addition of tryptophan metabolites [67]. Therefore, although IDO does not contribute to the pathophysiology of cystic fibrosis, it plays a significant role in the ability to prevent the infection associated with the disease.
Targeting the Kynurenine Pathway Therapeutically for Inflammatory Disease
Evidence showing that the kynurenine pathway exerts immunosuppressive effects on the immune system has led to the idea that therapeutic manipulation of this pathway might be beneficial for treating inflammatory diseases. In order to determine whether kynurenine itself could be useful as a potential therapy, the progression of CIA was evaluated in CIA mice following treatment with l-kynurenine or vehicle alone following onset of arthritis. l-kynurenine significantly reduced the clinical and histological scores of disease [44], suggesting that kynurenine administration may have the potential to modify the progression of RA. However, it still needs to be established whether the therapeutic effects of l-kynurenine administration are mediated through kynurenine or one of its metabolites. Tranilast, a clinically approved 3-hydroxyanthranillic acid derivative used to treat allergy, has also been shown to reduce clinical and histological disease in CIA [68]. Therefore, suggesting that at least some of the therapeutic effects of l-kynurenine administration may be mediated through 3-hydroxyanthranillic acid. In addition to strategies designed to increase the systemic circulating levels of kynurenines, it has also been suggested that strategies designed to increase kynurenine levels at sites of inflammation may be beneficial for inflammatory diseases such as RA [69]. Xue et al. [70] have shown that in vitro administration of the DNA methyltransferase inhibitor zebularine given in combination with IFN-γ to human THP-1 cells results in demethylation of the IDO1 promoter and an increase in the activation of the IDO pathway.
Conclusion
Overall, it is clear that the kynurenine pathway plays a role in the pathogenesis of many inflammatory diseases. The contribution of the kynurenine pathway to the prevention of inflammation in murine models of disease is becoming clearer. Moreover, it has been demonstrated that administration of kynurenine and its downstream metabolites in murine models may have therapeutic potential for treating inflammatory conditions. However, more work is needed to ascertain whether therapeutic manipulation of this pathway in humans can provide real clinical benefits. However, the consequences of upregulating IDO activity systematically are likely to be detrimental; therefore, strategies for upregulating the pathway in a tissue specific manner require further elaboration. In this regard, the studies of Xue et al. [70] are potentially informative as they demonstrate a synergistic effect between zebularine and IFN-γ, a cytokine expressed specifically at sites of inflammation. Given the burden of autoimmune disease on society, understanding and exploiting the IDO pathway remains an important objective for the future.
Abbreviations
- 1-MT:
-
1-methyl tryptophan
- APCs:
-
Antigen-presenting cells
- AHR:
-
Aryl hydrocarbon receptor
- CIA:
-
Collagen-induced arthritis
- CTLA-4:
-
Cytotoxic T-lymphocyte-associated protein 4
- DC:
-
Dendritic cell
- DMARDs:
-
Disease-modifying anti-rheumatic drugs
- EAE:
-
Experimental autoimmune encephalomyelitis
- GPR35:
-
G-protein-coupled receptor 35
- GWAS:
-
Genome-wide association studies
- IDO:
-
Indoleamine 2,3-dioxygenase
- IFN:
-
Interferon
- IL-1:
-
Interleukin 1
- IRF1:
-
Interferon-regulatory factor
- LPS:
-
Lipopolysaccharide
- MHC:
-
Major histocompatibility complex
- NK:
-
Natural killer
- NOD:
-
Nonobese diabetic
- NFAT:
-
Nuclear factor of activated T cells
- pDCs:
-
Plasmacytoid DCs
- Tregs:
-
Regulatory T cells
- RA:
-
Rheumatoid arthritis
- STAT1:
-
Signal transducer and activator of transcription 1
- TCR:
-
T cell receptor
- Th:
-
T helper
- TGF:
-
Transforming growth factor
- TDO:
-
Tryptophan 2,3-dioxygenase
- TTS:
-
Tryptophanyl-tRNA-synthetase
- TNF:
-
Tumor necrosis factor
- T1D:
-
Type 1 diabetes
References
Pfefferkorn ER. Interferon gamma blocks the growth of Toxoplasma gondii in human fibroblasts by inducing the host cells to degrade tryptophan. Proc Natl Acad Sci U S A. 1984;81(3):908–12.
Pfefferkorn ER, Guyre PM. Inhibition of growth of Toxoplasma gondii in cultured fibroblasts by human recombinant gamma interferon. Infect Immun. 1984;44(2):211–16.
Munn DH, Zhou M, Attwood JT, Bondarev I, Conway SJ, Marshall B, et al. Prevention of allogeneic fetal rejection by tryptophan catabolism. Science. 1998;281(5380):1191–3.
Grohmann U, Fallarino F, Puccetti P. Tolerance, DCs and tryptophan: much ado about IDO. Trends Immunol. 2003;24(5):242–8.
Suzuki T, Yokouchi K, Kawamichi H, Yamamoto Y, Uda K, Yuasa HJ. Comparison of the sequences of Turbo and Sulculus indoleamine dioxygenase-like myoglobin genes. Gene. 2003;308:89–94.
Taylor MW, Feng GS. Relationship between interferon-gamma, indoleamine 2,3-dioxygenase, and tryptophan catabolism. FASEB J. 1991;5(11):2516–22.
Mellor AL, Munn DH. Tryptophan catabolism and T-cell tolerance: immunosuppression by starvation? Immunol Today. 1999;20(10):469–73.
Dai W, Gupta SL. Regulation of indoleamine 2,3-dioxygenase gene expression in human fibroblasts by interferon-gamma. Upstream control region discriminates between interferon-gamma and interferon-alpha. J Biol Chem. 1990;265(32):19871–7.
Hassanain HH, Chon SY, Gupta SL. Differential regulation of human indoleamine 2,3-dioxygenase gene expression by interferons-gamma and -alpha. Analysis of the regulatory region of the gene and identification of an interferon-gamma-inducible DNA-binding factor. J Biol Chem. 1993;268(7):5077–84.
Chon SY, Hassanain HH, Gupta SL. Cooperative role of interferon regulatory factor 1 and p91 (STAT1) response elements in interferon-gamma-inducible expression of human indoleamine 2,3-dioxygenase gene. J Biol Chem. 1996;271(29):17247–52.
Silva NM, Rodrigues CV, Santoro MM, Reis LF, Alvarez-Leite JI, Gazzinelli RT. Expression of indoleamine 2,3-dioxygenase, tryptophan degradation, and kynurenine formation during in vivo infection with Toxoplasma gondii: induction by endogenous gamma interferon and requirement of interferon regulatory factor 1. Infect Immun. 2002;70(2):859–68.
Fujigaki S, Saito K, Sekikawa K, Tone S, Takikawa O, Fujii H, et al. Lipopolysaccharide induction of indoleamine 2,3-dioxygenase is mediated dominantly by an IFN-gamma-independent mechanism. Eur J Immunol. 2001;31(8):2313–18. doi:10.1002/1521-4141(200108)31:8<2313::AID-IMMU2313>3.0.CO;2-S.
Yuan W, Collado-Hidalgo A, Yufit T, Taylor M, Varga J. Modulation of cellular tryptophan metabolism in human fibroblasts by transforming growth factor-beta: selective inhibition of indoleamine 2,3-dioxygenase and tryptophanyl-tRNA synthetase gene expression. J Cell Physiol. 1998;177(1):174–86. doi:10.1002/(SICI)1097-4652(199810)177:1<174::AID-JCP18>3.0.CO;2-D.
Jung ID, Lee MG, Chang JH, Lee JS, Jeong YI, Lee CM, et al. Blockade of indoleamine 2,3-dioxygenase protects mice against lipopolysaccharide-induced endotoxin shock. J Immunol. 2009;182(5):3146–54. doi:10.4049/jimmunol.0803104.
Grohmann U, Orabona C, Fallarino F, Vacca C, Calcinaro F, Falorni A, et al. CTLA-4-Ig regulates tryptophan catabolism in vivo. Nat Immunol. 2002;3(11):1097–101. doi:10.1038/ni846.
Mellor AL, Baban B, Chandler P, Marshall B, Jhaver K, Hansen A, et al. Cutting edge: induced indoleamine 2,3 dioxygenase expression in dendritic cell subsets suppresses T cell clonal expansion. J Immunol. 2003;171(4):1652–5.
Munn DH, Sharma MD, Mellor AL. Ligation of B7-1/B7-2 by human CD4+ T cells triggers indoleamine 2,3-dioxygenase activity in dendritic cells. J Immunol. 2004;172(7):4100–10.
Mellor AL, Chandler P, Baban B, Hansen AM, Marshall B, Pihkala J, et al. Specific subsets of murine dendritic cells acquire potent T cell regulatory functions following CTLA4-mediated induction of indoleamine 2,3 dioxygenase. Int Immunol. 2004;16(10):1391–401. doi:10.1093/intimm/dxh140.
Fallarino F, Grohmann U, Hwang KW, Orabona C, Vacca C, Bianchi R, et al. Modulation of tryptophan catabolism by regulatory T cells. Nat Immunol. 2003;4(12):1206–12. doi:10.1038/ni1003.
Baban B, Hansen AM, Chandler PR, Manlapat A, Bingaman A, Kahler DJ, et al. A minor population of splenic dendritic cells expressing CD19 mediates IDO-dependent T cell suppression via type I IFN signaling following B7 ligation. Int Immunol. 2005;17(7):909–19. doi:10.1093/intimm/dxh271.
Munn DH, Shafizadeh E, Attwood JT, Bondarev I, Pashine A, Mellor AL. Inhibition of T cell proliferation by macrophage tryptophan catabolism. J Exp Med. 1999;189(9):1363–72.
Grohmann U, Puccetti P. The immunosuppressive activity of proinflammatory cytokines in experimental models: potential for therapeutic intervention in autoimmunity. Curr Drug Targets Inflamm Allergy. 2002;1(1):77–87.
Munn DH, Sharma MD, Baban B, Harding HP, Zhang Y, Ron D, et al. GCN2 kinase in T cells mediates proliferative arrest and anergy induction in response to indoleamine 2,3-dioxygenase. Immunity. 2005;22(5):633–42. doi:10.1016/j.immuni.2005.03.013.
Wek SA, Zhu S, Wek RC. The histidyl-tRNA synthetase-related sequence in the eIF-2 alpha protein kinase GCN2 interacts with tRNA and is required for activation in response to starvation for different amino acids. Mol Cell Biol. 1995;15(8):4497–506.
Harding HP, Novoa I, Zhang Y, Zeng H, Wek R, Schapira M, et al. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol Cell. 2000;6(5):1099–108.
Terness P, Chuang JJ, Opelz G. The immunoregulatory role of IDO-producing human dendritic cells revisited. Trends Immunol. 2006;27(2):68–73. doi:10.1016/j.it.2005.12.006.
Terness P, Bauer TM, Rose L, Dufter C, Watzlik A, Simon H, et al. Inhibition of allogeneic T cell proliferation by indoleamine 2,3-dioxygenase-expressing dendritic cells: mediation of suppression by tryptophan metabolites. J Exp Med. 2002;196(4):447–57.
Fallarino F, Grohmann U, Vacca C, Bianchi R, Orabona C, Spreca A, et al. T cell apoptosis by tryptophan catabolism. Cell Death Differ. 2002;9(10):1069–77. doi:10.1038/sj.cdd.4401073.
Frumento G, Rotondo R, Tonetti M, Damonte G, Benatti U, Ferrara GB. Tryptophan-derived catabolites are responsible for inhibition of T and natural killer cell proliferation induced by indoleamine 2,3-dioxygenase. J Exp Med. 2002;196(4):459–68.
Morita T, Saito K, Takemura M, Maekawa N, Fujigaki S, Fujii H, et al. L-tryptophan-kynurenine pathway metabolite 3-hydroxyanthranilic acid induces apoptosis in macrophage-derived cells under pathophysiological conditions. Adv Exp Med Biol. 1999;467:559–63.
Fallarino F, Grohmann U, You S, McGrath BC, Cavener DR, Vacca C, et al. The combined effects of tryptophan starvation and tryptophan catabolites down-regulate T cell receptor zeta-chain and induce a regulatory phenotype in naive T cells. J Immunol. 2006;176(11):6752–61.
Hill M, Tanguy-Royer S, Royer P, Chauveau C, Asghar K, Tesson L, et al. IDO expands human CD4 + CD25high regulatory T cells by promoting maturation of LPS-treated dendritic cells. Eur J Immunol. 2007;37(11):3054–62. doi:10.1002/eji.200636704.
Mezrich JD, Fechner JH, Zhang X, Johnson BP, Burlingham WJ, Bradfield CA. An interaction between kynurenine and the aryl hydrocarbon receptor can generate regulatory T cells. J Immunol. 2010;185(6):3190–8. doi:10.4049/jimmunol.0903670.
Nguyen NT, Kimura A, Nakahama T, Chinen I, Masuda K, Nohara K, et al. Aryl hydrocarbon receptor negatively regulates dendritic cell immunogenicity via a kynurenine-dependent mechanism. Proc Natl Acad Sci U S A. 2010;107(46):19961–6. doi:10.1073/pnas.1014465107.
Tiszlavicz Z, Nemeth B, Fulop F, Vecsei L, Tapai K, Ocsovszky I, et al. Different inhibitory effects of kynurenic acid and a novel kynurenic acid analogue on tumour necrosis factor-alpha (TNF-alpha) production by mononuclear cells, HMGB1 production by monocytes and HNP1-3 secretion by neutrophils. Naunyn Schmiedebergs Arch Pharmacol. 2011;383(5):447–55. doi:10.1007/s00210-011-0605-2.
Yan Y, Zhang GX, Gran B, Fallarino F, Yu S, Li H, et al. IDO upregulates regulatory T cells via tryptophan catabolite and suppresses encephalitogenic T cell responses in experimental autoimmune encephalomyelitis. J Immunol. 2010;185(10):5953–61. doi:10.4049/jimmunol.1001628.
Feldmann M, Brennan FM, Maini RN. Rheumatoid arthritis. Cell. 1996;85(3):307–10.
Winchester R. The molecular basis of susceptibility to rheumatoid arthritis. Adv Immunol. 1994;56:389–466.
Orozco G, Rueda B, Martin J. Genetic basis of rheumatoid arthritis. Biomed Pharmacother. 2006;60(10):656–62. doi:10.1016/j.biopha.2006.09.003.
Suzuki A, Kochi Y, Okada Y, Yamamoto K. Insight from genome-wide association studies in rheumatoid arthritis and multiple sclerosis. FEBS Lett. 2011;585(23):3627–32. doi:10.1016/j.febslet.2011.05.025.
Trentham DE. Collagen arthritis as a relevant model for rheumatoid arthritis. Arthritis Rheum. 1982;25(8):911–16.
Brand DD, Latham KA, Rosloniec EF. Collagen-induced arthritis. Nat Protoc. 2007;2(5):1269–75. doi:10.1038/nprot.2007.173.
Brand DD, Whittington KB, Rosloniec EF. I-Aq and I-Ap bind and present similar antigenic peptides despite differing in their ability to mediate susceptibility to autoimmune arthritis. Autoimmunity. 2001;34(2):133–45.
Criado G, Simelyte E, Inglis JJ, Essex D, Williams RO. Indoleamine 2,3 dioxygenase-mediated tryptophan catabolism regulates accumulation of Th1/Th17 cells in the joint in collagen-induced arthritis. Arthritis Rheum. 2009;60(5):1342–51. doi:10.1002/art.24446.
Williams RO. Exploitation of the IDO pathway in the therapy of rheumatoid arthritis. Int J Tryptophan Res. 2013;6 Suppl 1:67–73. doi:10.4137/IJTR.S11737.
Kolodziej L. An exploratory study of the interplay between decreased concentration of tryptophan, accumulation of kynurenines, and inflammatory arthritis. IUBMB Life. 2012;64(12):983–7. doi:10.1002/iub.1092.
Bauer TM, Jiga LP, Chuang JJ, Randazzo M, Opelz G, Terness P. Studying the immunosuppressive role of indoleamine 2,3-dioxygenase: tryptophan metabolites suppress rat allogeneic T-cell responses in vitro and in vivo. Transpl Int. 2005;18(1):95–100. doi:10.1111/j.1432-2277.2004.00031.x.
Szanto S, Koreny T, Mikecz K, Glant TT, Szekanecz Z, Varga J. Inhibition of indoleamine 2,3-dioxygenase-mediated tryptophan catabolism accelerates collagen-induced arthritis in mice. Arthritis Res Ther. 2007;9(3):R50. doi:10.1186/ar2205.
Baban B, Chandler P, McCool D, Marshall B, Munn DH, Mellor AL. Indoleamine 2,3-dioxygenase expression is restricted to fetal trophoblast giant cells during murine gestation and is maternal genome specific. J Reprod Immunol. 2004;61(2):67–77. doi:10.1016/j.jri.2003.11.003.
Scott GN, DuHadaway J, Pigott E, Ridge N, Prendergast GC, Muller AJ, et al. The immunoregulatory enzyme IDO paradoxically drives B cell-mediated autoimmunity. J Immunol. 2009;182(12):7509–17. doi:10.4049/jimmunol.0804328.
Pigott E, Mandik-Nayak L. Addition of an indoleamine 2,3,-dioxygenase inhibitor to B cell-depletion therapy blocks autoreactive B cell activation and recurrence of arthritis in K/BxN mice. Arthritis Rheum. 2012;64(7):2169–78. doi:10.1002/art.34406.
Forrest CM, Kennedy A, Stone TW, Stoy N, Darlington LG. Kynurenine and neopterin levels in patients with rheumatoid arthritis and osteoporosis during drug treatment. Adv Exp Med Biol. 2003;527:287–95.
Schroecksnadel K, Kaser S, Ledochowski M, Neurauter G, Mur E, Herold M, et al. Increased degradation of tryptophan in blood of patients with rheumatoid arthritis. J Rheumatol. 2003;30(9):1935–9.
Schroecksnadel K, Winkler C, Duftner C, Wirleitner B, Schirmer M, Fuchs D. Tryptophan degradation increases with stage in patients with rheumatoid arthritis. Clin Rheumatol. 2006;25(3):334–7. doi:10.1007/s10067-005-0056-6.
Parada-Turska J, Zgrajka W, Majdan M. Kynurenic acid in synovial fluid and serum of patients with rheumatoid arthritis, spondyloarthropathy, and osteoarthritis. J Rheumatol. 2013;40(6):903–9. doi:10.3899/jrheum.121035.
Zhu L, Ji F, Wang Y, Zhang Y, Liu Q, Zhang JZ, et al. Synovial autoreactive T cells in rheumatoid arthritis resist IDO-mediated inhibition. J Immunol. 2006;177(11):8226–33.
Puccetti P, Grohmann U. IDO and regulatory T cells: a role for reverse signalling and non-canonical NF-kappaB activation. Nat Rev Immunol. 2007;7(10):817–23. doi:10.1038/nri2163.
Cribbs AP, Kennedy A, Penn H, Read JE, Amjadi P, Green P, et al. Regulatory T cell function in rheumatoid arthritis is compromised by CTLA-4 promoter methylation resulting in a failure to activate the IDO pathway. Arthritis Rheumatol. 2014. doi:10.1002/art.38715.
Bernard NJ. Rheumatoid arthritis: who knows why regulatory T cells are defective in RA … IDO. Nat Rev Rheumatol. 2014;10(7):381. doi:10.1038/nrrheum.2014.96.
Sakurai K, Zou JP, Tschetter JR, Ward JM, Shearer GM. Effect of indoleamine 2,3-dioxygenase on induction of experimental autoimmune encephalomyelitis. J Neuroimmunol. 2002;129(1–2):186–96.
Saxena V, Ondr JK, Magnusen AF, Munn DH, Katz JD. The countervailing actions of myeloid and plasmacytoid dendritic cells control autoimmune diabetes in the nonobese diabetic mouse. J Immunol. 2007;179(8):5041–53.
Grohmann U, Fallarino F, Bianchi R, Orabona C, Vacca C, Fioretti MC, et al. A defect in tryptophan catabolism impairs tolerance in nonobese diabetic mice. J Exp Med. 2003;198(1):153–60. doi:10.1084/jem.20030633.
Fallarino F, Bianchi R, Orabona C, Vacca C, Belladonna ML, Fioretti MC, et al. CTLA-4-Ig activates forkhead transcription factors and protects dendritic cells from oxidative stress in nonobese diabetic mice. J Exp Med. 2004;200(8):1051–62. doi:10.1084/jem.20040942.
You S, Belghith M, Cobbold S, Alyanakian MA, Gouarin C, Barriot S, et al. Autoimmune diabetes onset results from qualitative rather than quantitative age-dependent changes in pathogenic T-cells. Diabetes. 2005;54(5):1415–22.
van Amelsfort JM, Jacobs KM, Bijlsma JW, Lafeber FP, Taams LS. CD4(+)CD25(+) regulatory T cells in rheumatoid arthritis: differences in the presence, phenotype, and function between peripheral blood and synovial fluid. Arthritis Rheum. 2004;50(9):2775–85. doi:10.1002/art.20499.
Matysiak M, Stasiolek M, Orlowski W, Jurewicz A, Janczar S, Raine CS, et al. Stem cells ameliorate EAE via an indoleamine 2,3-dioxygenase (IDO) mechanism. J Neuroimmunol. 2008;193(1–2):12–23. doi:10.1016/j.jneuroim.2007.07.025.
Iannitti RG, Carvalho A, Cunha C, De Luca A, Giovannini G, Casagrande A, et al. Th17/Treg imbalance in murine cystic fibrosis is linked to indoleamine 2,3-dioxygenase deficiency but corrected by kynurenines. Am J Respir Crit Care Med. 2013;187(6):609–20. doi:10.1164/rccm.201207-1346OC.
Inglis JJ, Criado G, Andrews M, Feldmann M, Williams RO, Selley ML. The anti-allergic drug, N-(3’,4’-dimethoxycinnamonyl) anthranilic acid, exhibits potent anti-inflammatory and analgesic properties in arthritis. Rheumatology (Oxford). 2007;46(9):1428–32. doi:10.1093/rheumatology/kem160.
Opitz CA, Wick W, Steinman L, Platten M. Tryptophan degradation in autoimmune diseases. Cell Mol Life Sci. 2007;64(19–20):2542–63. doi:10.1007/s00018-007-7140-9.
Xue ZT, Sjogren HO, Salford LG, Widegren B. An epigenetic mechanism for high, synergistic expression of indoleamine 2,3-dioxygenase 1 (IDO1) by combined treatment with zebularine and IFN-gamma: potential therapeutic use in autoimmune diseases. Mol Immunol. 2012;51(2):101–11. doi:10.1016/j.molimm.2012.01.006.
Funding
This work was supported by the Kennedy Trust for Rheumatology Research.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Cribbs, A.P., Williams, R.O. (2015). Role of the Kynurenine Pathway in Immune-Mediated Inflammation. In: Mittal, S. (eds) Targeting the Broadly Pathogenic Kynurenine Pathway. Springer, Cham. https://doi.org/10.1007/978-3-319-11870-3_7
Download citation
DOI: https://doi.org/10.1007/978-3-319-11870-3_7
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-11869-7
Online ISBN: 978-3-319-11870-3
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)