
Abstract
Insomnia is a common clinical condition portrayed by difficulty in initiating or maintaining sleep, or non-restorative sleep with impairment of daytime functioning, such as irritability or fatigue during wakefulness. This ailment is one of the most rampant health concerns; however, it represents an everyday struggle to clinicians because of its many potential causes, unfamiliarity with behavioral treatments, and concerns about pharmacologic treatments. The etiology and pathophysiology of insomnia involve genetic, environmental, behavioral, and physiological factors culminating in hyperarousal.
Current pharmacological treatment for insomnia exists in the form of benzodiazepine receptor agonist drugs (GABA-A receptor). Nonetheless, the use of these hypnotic medications must be carefully monitored for adverse effects and concerns persist regarding their safety and limited efficacy.
The recent advances made in elucidating the processes of sleep/wake regulation have altered the way that insomnia is approached. Current studies have highlighted new targets for drug discovery. One of the most promising ones is the orexin (hypocretin) system. Orexin neuropeptides regulate transitions between wakefulness and sleep by promoting arousal through activation of cholinergic/monoaminergic neural pathways. This has led to a swift development of a novel class of drugs that antagonize the physiological effects of orexin. These pharmacological agents may lead to new therapies for insomnia without the side effect profile of benzodiazepines (e.g., impaired cognition, disturbed arousal, and motor balance difficulties); however, antagonizing the orexin system may produce an entirely new plethora of side effects.
Despite the impending side effect profile of orexin antagonists, these drugs will inevitably supplement or replace conventional BZD receptor agonists for treating insomnia.
In this chapter, we will appraise the role of orexin antagonists for the treatment of insomnia.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Introduction
Pharmacological treatment is often characterized by ambiguities regarding diagnosis, selection of adequate treatment, and assessment of outcomes for individual patients. Likewise, multiple efficacious treatments are often available for a particular condition. This is indeed true for insomnia, with multiple behavioral and pharmacologic treatments demonstrating short-term and long-term efficacy data. However, what is considerably more challenging is trying to determine which treatment is best suited for which particular patient, based on the characteristics of their disorder, and the availability of treatments.
During the past half century, the pharmaceutical industry has invested a great deal of effort in the approval and usage of the very popular central nervous system (CNS) agents that enhance signaling of γ-aminobutyric acid (GABA), an inhibitory neurotransmitter (Zisapel 2012). These endeavors have succeeded in ascertaining the popularity of the GABA compounds as sleep aids; nevertheless, there are several issues that still raise many questions as to their use, namely, the fact that all of these compounds are classified as controlled substances and the label change that was required by the Food and Drug Administration (FDA).
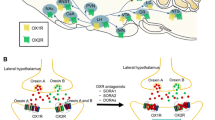
Over the past several years, a novel mechanism for modulating sleep that has gained significant popularity is the orexin/hypocretin system, due to its highly conserved nature and its ability to regulate arousal and wakefulness (Fig. 1).

Orexinergic control of sleep and wakefulness. (a) Awake state. Orexin neurons send excitatory projections to wake-active neurons, which send inhibitory feedback projections to orexin neurons. This system might maintain the activity of wake-active neurons. A small decrease in the activity of wake-active neurons results in decreased inhibitory influence to orexin neurons. Orexin neurons, therefore, are disinhibited and increase their excitatory influence on wake-active neurons to maintain their activity. These wake-active neurons send inhibitory projections to the preoptic area (POA) sleep center and excitatory projections to the thalamus and cerebral cortex. (b) Sleep state. GABAergic neurons in the sleep center are activated and send inhibitory projections to wake-active neurons and orexin neurons to maintain a sleep state. The figures represent the functional interaction between orexin neurons, wake-active centers, and sleep-active centers during various states of sleep and wakefulness. Arrows show excitatory and lines show inhibitory input. The thickness of arrows and lines represent relative strength of input. Circle sizes represent relative activities of each group of neurons. Modified from Lu and Zee, Clin Chest Med 31 (2010) 309–318
Orexins (also known as hypocretins) were first described in 1999 (de Lecea et al. 1998; Peyron et al. 1998; Sakurai et al. 1998) and, shortly, their absence was associated with the development of the sleep disorder narcolepsy (Chemelli et al. 1999; Lin et al. 1999; Thannickal et al. 2000). Since then, orexins have been intensely studied for their role in the sleep–wake cycle (SWC) primarily as wake-promoting neurotransmitters.
Orexin-producing neurons are found in the lateral hypothalamus (LH), where they synthesize two excitatory neuropeptides called orexin A and B (alternatively known as hypocretin 1 and 2) cleaved from a common protein precursor called prepro-orexin (prepro-hypocretin) (Sakurai et al. 1998). Orexinergic neurons extensively innervate the CNS, specifically areas known for their role in promoting arousal like the locus coeruleus (LC), the tuberomamillary nucleus (TMN), the basal forebrain (BF), the cerebral cortex, and the dorsal raphe (DR) (Peyron et al. 1998). Orexins exert their actions through their interaction with two G protein-coupled receptors called OX1R and OX2R (hcrtR1 and hcrtR2, respectively). These receptors have different affinities for the orexin peptides, while orexin A binds both receptors, orexin B selectively binds to OX2R (Sakurai et al. 1998).
Orexin-excited neurons become more excitable through an inhibition of potassium channels, including GIRK (G protein-regulated inward rectifier) channels; in addition, signaling through the orexin receptors can induce a rapid and sustained rise in intracellular calcium through voltage-gated calcium channels, through transient receptor potential channels, or from intracellular stores (Mieda et al. 2013). In addition to these postsynaptic effects, orexins can also act presynaptically on nerve terminals to induce release of GABA or glutamate, thus generating more complicated effects on downstream neurons (Mieda et al. 2013). Through these many mechanisms, orexins are thought to generally excite neurons that promote many aspects of arousal.
Furthermore, orexin receptors are differentially located throughout the CNS; the LC mainly expresses OX1R, the TMN exclusively expresses OX2R, while the DR, BF, and cerebral cortex express both receptors (Marcus et al. 2001; Mieda et al. 2013). The divergent distribution and affinities of orexin receptors suggest they play different functions in the conservation of wakefulness. This has been studied using different strains of transgenic mice, such as knockouts (KO) for either one of the orexin receptors, or both (double knockouts, DKO). These mice show varying degrees of sleep disturbances; while OX1R KO mice do not exhibit any obvious behavioral alterations (Sakurai 2007), OX2R KO mice manifest some features of narcolepsy, including an inability to sustain wakefulness (Willie et al. 2003). DKO mice display the most profoundly disturbed sleep phenotype of all three models: narcolepsy with cataplexy (transient episodes of behavioral arrest) (Kalogiannis et al. 2011). The robust narcoleptic phenotype in DKO mice indicates a synergistic role between OX1R and OX2R in the maintenance of wakefulness. Therefore, both the OX1R and OX2R are essential in the process of keeping a stable sleep/wakefulness cycle, with a larger contribution of OX2R.
To further characterize the role of orexin receptors, selective orexin receptor KO mice were stimulated with intracerebroventricular (ICV) infusions of orexin A. Specific stimulation of OXR1 in OXR2 KO mice produced a moderate improvement in wakefulness and suppression of nREM, whereas the stimulation of OX2R in OX1R KO mice resulted in a greatly enhanced wakefulness (Mieda et al. 2011). This suggests that OX1R plays an important role in suppressing the instigation of nREM sleep, while OX2R has a major role in promoting wakefulness.
In another direction, overexpression of components of the orexinergic system also disrupts the SWC. For example in the zebrafish, overexpression of orexinergic neurons has been shown to induce an insomnia-like phenotype (Prober et al. 2006). Mice that overexpress prepro-orexin display sleep abnormalities, which include fragmentation of nREM sleep, reduced REM sleep, and increased motor activity during REM sleep, suggesting an inability to maintain sleep states.
The excitatory effects of the orexin peptides on these arousal centers are hypothesized to stabilize and maintain wakefulness (Fig. 1) and if we take into consideration that the activation of the orexinergic system promotes wakefulness and that its disruption brings about sleep disturbances, orexin antagonists could offer a very effective therapeutic alternative for insomnia.
Given that orexins essentially promote arousal, orexin antagonists have the potential to selectively promote sleep and cause fewer side effects. Dependence and abuse should be less of a concern, as animal studies have shown that orexin antagonists actually reduce drug seeking. Imbalance and falls should not be a problem, as there is no evidence that the orexin system affects balance or gait directly. Consequences of an overdose should not be too concerning, as orexin antagonists should not significantly depress respiration or affect blood pressure.
Potential side effects of orexin antagonists, including disinhibition of REM sleep, are discussed below, but overall, many researchers anticipate that these drugs should promote sleep without many of the side effects encountered with current medications.
2 Orexin Receptor Antagonism: A Novel Approach for Treating Insomnia
The newest molecules in the pipeline for the treatment of insomnia are orexin antagonists. Drugs targeting insomnia ideally promote sleep throughout the night, maintain normal sleep architecture, and are devoid of residual effects associated with morning sedation. These features of an ideal compound are not only dependent upon pharmacokinetics, receptor binding kinetics, potency, and pharmacodynamic activity, but also upon a compound’s mechanism of action. There are many orexin antagonists currently being studied for the treatment of insomnia and they fall into one of two categories: single orexin receptor antagonists (SORAs) and dual orexin receptor antagonists (DORAs).
2.1 Single Orexin Receptor Antagonists (SORAs)
Evidence from experiments conducted in transgenic models of orexin receptor KO mice suggests that SORAs targeting OX1R will not promote sleep as effectively as those aimed at OX2R; nonetheless, OX1R antagonism could serve as a complementary treatment for insomnia or other sleep-related disorders.
2.1.1 OX1R
Of the available SORAs, SB-334867 was the first drug designed to selectively antagonize OX1R. This SORA is able to counteract the suppression of REM sleep after ICV infusion of OXA in rats (Smart et al. 2001; Smith et al. 2003); however, it does not decrease wakefulness, or increase the amount of time spent in sleep, nor does it reduce sleep latency by itself at any given dose. Also, SB-334867 at 3 and 30 mg/kg increased cumulative nREM during the first 4 and 6 h following administration (Morairty et al. 2012). SB-334867 is classified as a selective OX1R antagonist, but unspecific binding to adenosine and serotonin receptors has been reported; it also affects monoamine and norepinephrine transporters at high concentrations (Lebold et al. 2013).
Although the effect of SB-334867 on sleep induction was poor, this molecule has proven to be useful for the treatment of other conditions, such as substance abuse, withdrawal, obesity, and panic disorder (White et al. 2005; Johnson et al. 2010; Jupp et al. 2011; Smith and Aston-Jones 2012).
Other selective OX1R antagonists include SB-408124, SB-674042, and the newest AK-335827. So far, neither SB-408124 nor AK-335827 has been found to promote sleep (Dugovic et al. 2009; Steiner et al. 2013). In the case of SB-408124 however, insufficient brain penetration was observed and this could account in part for the absence of observable effects (Morairty et al. 2012).
There are few studies characterizing the effect of these antagonists; nonetheless, there is some evidence that they can be useful in the treatment of substance abuse and withdrawal, and have potential for treating obesity and panic disorder (Table 1). For example, it has been shown that subcutaneous administration of SB-408124 lowers the release of dopamine in the nucleus accumbens (Dugovic et al. 2009), and orally administered AK-335827 has anxiolytic effects (Steiner et al. 2013).
It is interesting that despite the lack of sleep-promoting effects of OX1R SORAs on their own, these compounds have the capacity to thwart the sleep-inhibiting effects of ICV orexin infusion (Smith et al. 2003). Strikingly, they can also reduce the sleep-promoting effects of other antagonists, as observed under the co-administration of OX1R and OX2R antagonists, which has a milder sleep-promoting effect than when the OX2R antagonist is administered by itself (Dugovic et al. 2009). This could be due to the high concentrations used in these experiments (30 mg/kg) and the unspecific binding that follows.
2.1.2 OX2R
Type 2 orexin receptors are selectively expressed in both the paraventricular nucleus (PVN) and the TMN. The PVN is part of the hypothalamic-pituitary-adrenal axis (HPA), and the overactivation of the HPA has been proposed to be involved in the etiology of primary insomnia. Withholding the orexinergic stimuli to the HPA could help prevent the development of a vicious cycle of overactivation that could lead to chronic insomnia. Additionally, the TMN, a histaminergic nucleus, has a major role in the arousal effect observed after orexinergic stimulation (Huang et al. 2001). Inhibition of the TMN with orexinergic antagonists could facilitate the induction of sleep by allowing the sleep-promoting nuclei to prevail.
OX2R antagonists are less common than the other classes. Among the few available molecules that have been studied in the context of sleep promotion are EMPA, TCS-OX2-29, and JNJ-10397049. These antagonists have been more successful at diminishing wakefulness than OX1R antagonists (Table 1).
EMPA is the least effective sleep-promoting OX2R SORA ever studied. While intraperitoneal administration of EMPA (100 mg/kg) has been shown to selectively increase cumulative nREM sleep during the first 4 and 6 h after administration, these increases are not accompanied by any significant increase in REM sleep or reduction in latencies for either sleep stage (Morairty et al. 2012). On the other hand, rats that received an ICV infusion of TCS-OX2-29 (40 nmol) increased their total sleep time by 7 % in comparison to controls that received saline infusions. Interestingly, this effect was secondary to a selective increase in REM sleep (Kummangal et al. 2013).
Intraperitoneal administration (5, 25, or 50 mg/kg) of JNJ-10397049 6 h into the dark phase produced a robust increase in total sleep time, traced to increases in both REM and nREM sleep (Gozzi et al. 2011). Similar results have been observed with subcutaneous injections (Dugovic et al. 2009). Starting at doses of 3 mg/kg, administration of JNJ-10397049 2 h into the light phase significantly decreased the latency to nREM sleep while increasing the length of each bout. At higher concentrations (30 mg/kg), this drug also induced a decrease in REM sleep latency without noticeable changes in its duration. Overall, 3 mg/kg of JNJ-10397049 increased total sleep time by 42 % while keeping the proportion of nREM/REM sleep observed in vehicle-treated animals.
Furthermore, microdialysis assays showed that this compound reduces histamine release in the LH. As mentioned earlier, release of histamine in the TMN is fundamental for the wake-promoting effects of OXA ICV infusions (Dugovic et al. 2009; Huang et al. 2001).
Animal studies support the notion that OX2R antagonists are helpful as sleep-inducing agents (Table 1). Further research is needed to determine the degree of sleep generation achieved by these compounds in different species, including humans. It is possible that the sleep-promoting effect of selectively antagonizing OX2R is less pronounced than the one observed with DORAs, but it may also be more specific, which would be worth investigating.
2.2 Dual Orexin Receptor Antagonists (DORAs)
It had been long suspected that antagonizing both orexin receptors would elicit the most powerful sleep-promoting effects; therefore, many of the studies around orexin antagonists have focused on DORAs. So far, evidence has proven this to be the case, to the point that DORAs are the only orexin antagonists currently undergoing clinical trials in the hope that the FDA will approve them for the treatment of insomnia.
2.2.1 Almorexant
ACT-078573 (almorexant) is the most widely studied DORA and one of the first to enter phase III clinical trials (NCT00608985). Almorexant is a reversible, selective dual OX1 and OX2 receptor antagonist.
In wild-type mice, the administration of almorexant 15 min before lights-out reduced the amount of time spent awake, while increasing the length of nREM and REM sleep bouts in a dose-dependent manner (Mang et al. 2012). Notably, the proportion of REM sleep observed after almorexant administration during the dark phase was in the range of that observed during the light phase with vehicle treatment.
Further studies in KO mice determined that the sleep-inducing effect of almorexant was related to the stimulation of OX2R and not OX1R. This conclusion was reached after the authors did not observe any changes in the amount of sleep in OX2R KO, but did for OX1R KO mice. Interaction with sites other than orexin receptors that could account for the changes in sleep times was discarded when no changes were observed in the SWC of DKO mice.
When administered in healthy humans, almorexant increased deep sleep and REM sleep (unlike GABA receptor modulators which decrease REM sleep). In a Phase II study of 161 patients with primary insomnia, almorexant 400 mg significantly reduced LPS (mean treatment effect—18 min; p = 0.02) and WASO (mean treatment effect—54 min; p < 0.001) compared to placebo. Dose-related increases in TST were found in all (400, 200, and 100 mg) almorexant groups compared with placebo. The most commonly reported adverse effects in the almorexant-treated patients were dizziness, nausea, fatigue, headache, and dry mouth. Almorexant had no effect on subjective WASO at any dose, but objective WASO decreased in a dose-dependent fashion. The higher almorexant doses (400 and 200 mg) were associated with some residual effects on next-day performance as evidenced by increased mean reaction times and lower scores on the digit span test. At doses up to 1,000 mg, there were no reports of cataplexy or narcolepsy in almorexant-treated patients (Hoever et al. 2012).
Actelion’s first Phase III study of almorexant (RESTORA 1) was a 2-week randomized, double-blind trial in 707 patients with primary insomnia. Patients were randomized to almorexant 100 mg, almorexant 200 mg, placebo, or zolpidem 10 mg (active comparator). The primary endpoint was change from baseline to Day 1 and 2 in WASO. Both almorexant groups showed statistically significant improvements in wake after sleep onset with median reductions of 29 and 40 min in the 100 and 200 mg groups, respectively, versus 11 min in the placebo group (Renzulli et al. 2011).
Doses of and above 200 mg elicited decreased alertness, with increased reports of fatigue, drowsiness, sleepiness, and sleep efficiency, measured as an increase in SWS and REM sleep (Brisbare-Roch et al. 2007). In patients with primary insomnia, it proved to be effective for boosting sleep, increasing total sleep time, and reducing both REM sleep latency and the frequency of awakening (Hoever et al. 2012). This effect was dose dependent, with the most notorious effect on sleep architecture achieved at doses of 400 mg; doses of 100 and 200 mg had modest effects on sleep, with fewer adverse effects (e.g., headache, dizziness, blurred vision).
Although almorexant was generally well tolerated in this study, safety signals led to further investigations of the clinical safety of almorexant. These expanded studies led to the discontinuation of almorexant development for undisclosed human tolerability issues (Tables 1 and 2). Currently, almorexant is in a new phase of clinical trials in order to evaluate its effect on cognitive performance (NCT01243060).
2.2.2 SB-649868
SB-649868 is a potent orally active DORA manufactured by the same pharmaceutical company as almorexant. There is also evidence for the effectiveness of SB-649868 in promoting sleep, in both animal studies and human trials.
When administered to rats, it elicited an increase in total sleep time (related to increases of both nREM and REM sleep) and reduced sleep latencies at doses of 10 and 30 mg. Moreover, the effect of SB-649868 on motor coordination was null, given that the rotarod model of coordination failed to reveal any motor impairment in rats treated with this compound, even when the orexin antagonist was administered concurrently with ethanol (Di Fabio et al. 2011). Compared to almorexant, the in vivo efficacy of this compound is excellent; thus it has been moved on to clinical trials.
The administration of SB-649868 to healthy volunteers who participated in a noise-disturbed sleep study showed that this compound is effective at inducing somnolence and fatigue at 10 and 30 mg doses (Bettica et al. 2012). Furthermore, patients diagnosed with PI reported that SB-649868 significantly improved the quality of sleep (10, 30, and 60 mg) while objectively increasing total sleep time, reducing sleep latency, and suppressing nighttime awakenings. During this study, the most common complaints were headaches, dry mouth, and nasopharyngitis; the number of complaints increased in a dose-dependent manner. Phase II clinical trials of SB-649868 have been completed (NCT00426816) (Table 1).
2.2.3 Suvorexant
Another promising DORA is the potent suvorexant (MK-4305, Merk), a compound variation from the diazepam series.
Animal studies have shown that suvorexant reduces active wake time by increasing nREM and REM sleep in rats, dogs, and monkeys. In all cases, these effects were achieved at much lower doses (10 mg) than with almorexant (Winrow et al. 2011).
It has been determined that its median peak plasma concentrations occur approximately 2 h after administration and are not affected by food. It has a volume of distribution of 105.9 L and is highly protein bound (99.5 %). It is primarily metabolized by the cytochrome P450 (CYP3A4) enzyme system, with some contribution from CYP2C19 into M9, an inactive metabolite. Finally, it is eliminated primarily via inactive metabolites in the feces; there is no renal elimination. The half-life is approximately 12.2 h on average (range 8–19 h) (Table 2). Steady-state plasma concentrations occur in about 3 days with daily administration (Sun et al. 2013).
This molecule is currently under evaluation for approval by the FDA. Merck’s application to the FDA for approval included 32 studies that enrolled more than 900 subjects (healthy and those with insomnia).
Healthy participants received one of three doses of suvorexant (10, 50, or 100 mg) or placebo and were evaluated in two 8-h (polysomnography) PSG recording sessions in a general laboratory setting. Different parameters were analyzed in order to assess the efficacy of suvorexant administration; these included: sleep onset, or latency to persistent sleep (LPS); sleep maintenance, or waking after sleep onset (WASO); sleep efficiency (SE); and total sleep time (TST).
The lowest dose (10 mg) reduced the number of awakenings after sleep onset, and at higher doses (50 mg) it reduced sleep latency, while increasing SE and TST. High doses (50 and 100 mg) elicit undesirable side effects such as an increase in reaction time, difficulty waking up, and reduced alertness following awakening; in addition, it led to mild complaints of headaches and somnolence (Sun et al. 2013).
With the purpose of assessing the effectiveness of orexin receptor antagonism as a novel approach for treating insomnia, men and women, 18–64 years of age with primary insomnia, based on Diagnostic and Statistical Manual of Mental Disorders, 4th edition, text revision (DSM-IV-TR) criteria, were enrolled in this clinical study. They were given 10- and 30-mg tablets, along with their corresponding placebos, in order to achieve doses of 10, 20, 40, and 80 mg per dose.
Suvorexant reduced sleep latency and increased the time patients spent asleep after a single administration without reducing the number of awakenings after sleep onset. The increase in total sleep time was mostly attributable to an increase in REM sleep (Herring et al. 2012). The most frequent adverse effects were somnolence, headaches, dizziness, and abnormal dreams, all of which occurred in a dose-dependent manner. In addition, there were no next-day residual effects, no rebound insomnia, complex sleep-related behaviors, or withdrawal effects after 4 weeks. Instead, during this study there were a few reports of sleep paralysis (n = 59, at 40 mg), and at high doses (80 mg), excessive daytime sleepiness (n = 61) and hypnagogic hallucinations (n = 61) (Herring et al. 2012). These are symptoms of narcolepsy, and should be carefully monitored due to the close association between narcolepsy and the orexinergic system.
In general, suvorexant was well tolerated and, because the most consistently effective dosages were 30 and 40 mg, the pharmaceutical company manufacturing suvorexant submitted a dose range of 15–40 mg for FDA approval. However, the FDA requested a lower starting dose of 10 mg for the general population and a 5 mg dose for those taking concomitant CYP3A4 inhibitors (Table 2). Patients should avoid taking other CYP3A medications while they are taking suvorexant. Potent CYP3A inhibitors increase plasma concentrations, placing patients well above the desired therapeutic threshold. Suvorexant is a mild inhibitor of CYP3A, but when administered with CYP3A substrates, including oral contraceptives and warfarin, it had minimal effects. As of August 2014, the FDA approved suvorexant (Belsomra, Merk) in four different strengths -5, 10, 15, and 20 mg.
An advantage of suvorexant over previous insomnia therapies is the low potential for addiction or dependence.
2.3 Advantages of DORAs over Other Pharmacological Alternatives
Based on these preclinical evaluations of structurally divergent DORAs, suvorexant exhibits a pharmacokinetic profile predicted to be advantageous for the treatment of insomnia. Analysis of the time course of suvorexant plasma concentrations determined in an early Phase I clinical trial indicates that concentrations sufficient for efficacy in humans are restricted to normal sleep periods while efficacy is maintained for a range of doses. Given their high OX2R occupancy threshold for efficacy, DORAs require a plasma concentration sufficient to maintain occupancy in order to promote sleep throughout the resting period in humans (Table 2). For suvorexant, this is achieved with a plasma concentration T1/2 exceeding 8 h.
Although insomnia is a common disorder, its underlying mechanisms are still not fully understood. DORAs have been rationally designed specifically to promote sleep by blocking wakefulness and thus alleviate the symptoms of insomnia while minimizing potential off-target activity that occurs with widespread signaling via GABA-A receptor modulators. By using antagonist compounds like suvorexant to block the arousal-promoting effects of orexin signaling in the brain, the oscillation in endogenous orexin pathway activation (highest signaling during active periods and diminished signaling during normal sleep times) is modified to mimic more closely the expected physiological state in normal sleep and wakefulness (Fig. 1).
One potential advantage of DORAs over classic insomnia treatments, such as benzodiazepines, is the possibility of inducing a more physiological sleep (Lanoir and Killam 1968; Borbély et al. 1985; Gaillard et al. 2009). For instance, while DORAs enhance REM sleep, benzodiazepines have proven to suppress this sleep stage. In addition, orexin antagonists appear to have a better side effect profile, with mild complaints of headaches and dizziness being the most common. The only exception appears to be almorexant, given the surprising suspension of clinical trials. Although the reasons for halting clinical trials have not been disclosed to the public, it is conceivable that the high doses required to achieve therapeutic effects could also cause more severe adverse effects, not observed in other drugs that require doses ten times smaller.
2.4 Considerations for Administering Orexin Antagonists
One of the most important questions when characterizing an orexin antagonist is whether or not it elicits narcoleptic symptoms. Thus far, orexin antagonists have not been observed to cause cataplexy in animal models or in human patients. Up until now, reports of human patients complaining of sleep paralysis or hypnagogic hallucinations have been scarce, only occurring with high doses of suvorexant. As clinical trials progress, medical practitioners should still be on the alert for symptoms of narcolepsy.
Also, because orexin antagonists have a novel mechanism of action, they have the potential to improve insomnia in patients who have found other agents ineffective. Clinical studies now under way should better define the benefits and limitations of orexin receptor antagonists.
Thus far, little information is publicly available on the adverse effects of orexin antagonists, but on the basis of the available evidence and the predicted effects of orexin blockade, one may anticipate that orexin antagonists will have a better adverse event profile than many currently available hypnotics. Orexin antagonists may have their own unique set of concerns, and additional clinical data are needed. However, unlike benzodiazepines, they should have little potential for abuse or unsteady gait, and unlike sedating antidepressants, they are unlikely to cause autonomic side effects such as orthostasis.
Morning or daytime sleepiness may be a concern for drugs that continue to block orexin signaling upon awakening. Possibly, this sleepiness would present differently from how it presents with benzodiazepines because people with narcolepsy often feel alert upon awakening and then sleepy later in the day. Thus, in clinical trials, it will be important to monitor sleepiness during the entire day, not just in the morning.
Unlike other hypnotics, orexin antagonists may cause some dysregulation of REM sleep as is encountered in narcolepsy. Hypnagogic hallucinations and sleep paralysis could occur around the onset of sleep or upon awakening, although they have not been reported. In general, these symptoms are disturbing but fleeting, and they should be manageable with patient education and dose reduction. The potential for cataplexy is a bigger concern because a sudden fall could produce injury, yet people with narcolepsy rarely have cataplexy during their sleep period, and no cataplexy was observed with almorexant in rats, dogs, and humans despite high levels of receptor blockade. However, these studies may have overlooked a tendency for cataplexy because it is usually triggered by strong, positive emotions (e.g., laughing at a great joke) and it is hard to elicit in the lab. In addition, animals and subjects were allowed to sleep in most studies, which could have masked any tendency toward cataplexy. Most likely, cataplexy will be a concern only in unusual circumstances inconsistent with intended clinical use, such as if a patient is wide awake and socializing after taking a high dose of an orexin antagonist.
Considering that orexins promote wakefulness and suppress REM sleep, one might find it odd and surprising that some people with narcolepsy can have moderately fragmented sleep, sleepwalking during nREM sleep, and movements during REM sleep known as REM sleep behavior disorder. Possibly, these symptoms are a consequence of chronic orexin deficiency or injury to neurons other than those producing orexins. As these symptoms are hard to explain with current models, predicting whether they may occur with orexin antagonists is difficult. However, in dogs, almorexant increased twitching of distal parts of the limbs during sleep, and clinical studies should monitor for movements or other disruptions of sleep.
Orexin signaling seems to enhance activity in the mesolimbic pathways that regulate reward and motivation, and reduced activity in this system could worsen mood or motivation. People with narcolepsy may have a higher prevalence of depression, although it is unknown if this is a direct consequence of reduced orexin signaling or a response to the challenge of having a chronic illness. Alternatively, better sleep in patients with depression could improve mood. As many patients with insomnia have depression, clinicians should watch for any changes in mood.
Benzodiazepines can depress respiration and worsen obstructive sleep apnea or severe lung disease, and overdose can be fatal, especially if used in combination with alcohol or other sedatives (Gaillard et al. 2009). As orexin knockout mice have relatively normal baseline ventilation (Kalogiannis et al. 2011), it seems unlikely that orexin antagonists would significantly reduce respiratory drive. However, they may reduce the response to hypercarbia. High levels of CO2 increase respiratory rate and tidal volume, and orexin antagonists can blunt this response, especially during wakefulness. Thus it may be wise to closely evaluate orexin antagonists in patients with hypercarbia, such as individuals with severe chronic obstructive pulmonary disease (COPD) or respiratory muscle weakness.
Whether orexin affects appetite or metabolism in humans remains unclear, but people and mice with narcolepsy tend to be slightly overweight despite apparently eating less than normal. Thus orexin deficiency may lower metabolic rate, and orexin antagonists could promote mild weight gain if administered chronically. In practice, orexin antagonists will be mainly given at night, and normal orexin signaling during the day should offset any reductions in metabolism or hunger at night.
Sedating antidepressants can produce unsteady gait, dizziness, and falls, but these are unlikely to be concerns with orexin antagonists as the cerebellum and vestibular nuclei essentially lack orexin fibers and receptors (Peyron et al. 1998). Orexin knockout mice run at a normal speed, and rats treated with almorexant balance well on a rotating rod. Studies of almorexant in humans have shown only small increases in body sway 1–3 h after dosing, so it seems unlikely that orexin antagonists will substantially increase the risk of falls.
Orexin antagonists should probably be avoided in patients with narcolepsy because they could worsen some of the patients’ symptoms. These compounds might also have a higher risk of producing narcolepsy-like effects during the day in other disorders in which the orexin-producing neurons are injured, such as Parkinson’s disease and severe traumatic brain injury. Thus in those patients, clinicians might consider initiating treatment with low doses.
Some researchers have questioned whether there is value in producing behavioral effects similar to narcolepsy, but overall, it appears that orexin antagonists should promote sleep with fewer and less harmful side effects than many current hypnotics. Ongoing clinical trials are watching closely for any symptoms related to dysregulation of REM sleep, and watching for fragmented sleep, movements during sleep, and worsening mood will also be important. Daytime sedation should not be much of a concern for compounds with favorable kinetics that permit normal orexin signaling during the day.
In addition, orexin enhances activity in mesolimbic pathways regulating reward and motivation; reduced activity in the orexin system could theoretically worsen mood or motivation. This would dictate cautious use in patients with underlying mood disorders especially since there were reports of suicidal ideation in high-dose suvorexant studies. Orexin receptor antagonists may offer yet another viable option for the pharmacological management of insomnia. Longer-term studies and head-to-head comparisons with existing hypnotics will be crucial to determine the benefit–risk ratio of these agents.
Conclusion
Although benzodiazepines and non-benzodiazepines are effective for insomnia, their adverse effect profiles and recommended limitations on long-term use may prompt patients and clinicians to seek other options. Patients who experience both sleep onset and sleep maintenance insomnia may be particularly challenging to treat. The recent discovery of orexins and their receptors has led to the development of new therapy targets. Evidence suggests that some of these medications offer a sustained benefit for patients with symptoms of chronic insomnia (Suvorexant in particular, which has a unique clinical profile).
We know of no evidence that people with insomnia have abnormally high orexin tone, but under most conditions, any reduction in orexin signaling should make it easier to fall asleep and to return to sleep.
Since orexin activity is highest during active wakefulness, not during sleep periods, these agents may be ineffective for certain types of insomnia. These drugs might be especially effective in shift workers or individuals with jet lag who are trying to sleep during their biological active period when orexin tone is high. They may also be helpful in the many insomnia patients who have high sympathetic tone because high sympathetic tone delays the onset of sleep and sympathetic activation may promote arousal by exciting the orexin neurons. These agents may also be chosen preferentially in the elderly to avoid gait disturbances and confusion and in those with substance abuse histories to avoid dependence and abuse. Benzodiazepines can cause imbalance and confusion, and orexin antagonists may be a good choice for some older patients because they may be less likely to cause these side effects. On the other hand, drugs that block orexin signaling may be less effective in people suffering from insomnia caused by anxiety or pain; there is no evidence that orexin antagonists reduce anxiety or raise sensory thresholds as benzodiazepines do.
The better understanding of the biology of the orexin/hypocretin system has promoted drug discovery programs in several pharmaceutical companies, resulting in a series of patents and compounds with different selectivity and in vitro characteristics. Targeting the orexin receptor is now entering a translational pipeline, and we believe that orexin antagonists as a treatment modality in insomnia management will be the gold standard in the near future, especially for DORAs.
References
Bettica P, Squassante L, Zamuner S, Nucci G, Danker-Hopfe H, Ratti E (2012) The orexin antagonist SB-649868 promotes and maintains sleep in men with primary insomnia. Sleep 35:1097–1104
Borbély AA, Mattmann P, Loepfe M, Strauch I, Lehmann D (1985) Effect of benzodiazepine hypnotics on all-night sleep EEG spectra. Hum Neurobiol 4:189–194
Brisbare-Roch C, Dingemanse J, Koberstein R, Hoever P, Aissaoui H, Flores S et al (2007) Promotion of sleep by targeting the orexin system in rats, dogs and humans. Nat Med 13:150–155
Chemelli R, Willie J, Sinton CM (1999) Narcolepsy in orexin knockout mice: molecular genetics of sleep regulation. Cell 98:437–451
de Lecea L, Kilduff T, Peyron C, Gao X, Foye P, Danielson P et al (1998) The hypocretins: hypothalamus-specific peptides with neuroexcitatory activity. Proc Natl Acad Sci USA 95:322–327
Di Fabio R, Pellacani A, Faedo S, Roth A, Piccoli L, Gerrard P et al (2011) Discovery process and pharmacological characterization of a novel dual orexin 1 and orexin 2 receptor antagonist useful for treatment of sleep disorders. Bioorg Med Chem Lett 21:5562–5567
Dugovic C, Shelton JE, Aluisio LE, Fraser IC, Jiang X, Sutton SW et al (2009) Blockade of orexin-1 receptors attenuates orexin-2 receptor antagonism-induced sleep promotion in the rat. J Pharmacol Exp Ther 330:142–151
Equihua AC, De la Herrán-Arita AK, Drucker-Colín R (2013) Orexin receptor antagonists as therapeutic agents for insomnia. Front Pharmacol 4:163
Gaillard JM, Schulz P, Tissot R (2009) Effects of three Benzodiazepines (Nitrazepam, Flunitrazepam and Bromazepam) on sleep of normal subjects, studied with an automatic sleep scoring system. Pharmacopsychiatry 6:207–217
Gozzi A, Turrini G, Piccoli L, Massagrande M, Amantini D, Antolini M et al (2011) Functional magnetic resonance imaging reveals different neural substrates for the effects of orexin-1 and orexin-2 receptor antagonists. PLoS ONE 6:e16406
Herring WJ, Snyder E, Budd K, Hutzelmann J, Snavely D, Liu K et al (2012) Orexin receptor antagonism for treatment of insomnia: a randomized clinical trial of suvorexant. Neurology 79:2265–2274
Hoever P, Dorffner G, Beneš H, Penzel T, Danker-Hopfe H, Barbanoj MJ et al (2012) Orexin receptor antagonism, a new sleep-enabling paradigm: a proof-of-concept clinical trial. Clin Pharmacol Ther 91:975–985
Huang ZL, Qu WM, Li WD, Mochizuki T, Eguchi N, Watanabe T et al (2001) Arousal effect of orexin A depends on activation of the histaminergic system. Proc Natl Acad Sci USA 98:9965–9970
Johnson PL, Truitt W, Fitz SD, Minick PE, Dietrich A, Sanghani S et al (2010) A key role for orexin in panic anxiety. Nat Med 16:111–115
Jupp B, Krivdic B, Krstew E, Lawrence AJ (2011) The orexin receptor antagonist SB-334867 dissociates the motivational properties of alcohol and sucrose in rats. Brain Res 1391:54–59
Kalogiannis M, Hsu E, Willie JT, Chemelli RM, Kisanuki YY, Yanagisawa M et al (2011) Cholinergic modulation of narcoleptic attacks in double orexin receptor knockout mice. PLoS ONE 6:e18697
Kummangal BA, Kumar D, Mallick HN (2013) Intracerebroventricular injection of orexin-2 receptor antagonist promotes REM sleep. Behav Brain Res 237:59–62
Lanoir J, Killam EK (1968) Alteration in the sleep-wakefulness patterns by benzodiazepines in the cat. Electroencephalogr Clin Neurophysiol 25:530–542
Lebold TP, Bonaventure P, Shireman BT (2013) Selective orexin receptor antagonists. Bioorg Med Chem Lett 23:4761–4769
Lin L, Faraco J, Li R, Kadotani H, Rogers W, Lin X et al (1999) The sleep disorder canine narcolepsy is caused by a mutation in the hypocretin (orexin) receptor 2 gene. Cell 98:365–376
Lu BS, Zee PC (2010) Neurobiology of sleep. Clin Chest Med 31:309–318
Mang GM, Dürst T, Bürki H, Imobersteg S, Abramowski D, Schuepbach E et al (2012) The dual orexin receptor antagonist almorexant induces sleep and decreases orexin-induced locomotion by blocking orexin 2 receptors. Sleep 35:1625–1635
Marcus JN, Aschkenasi CJ, Lee CE, Chemelli RM, Saper CB, Yanagisawa M et al (2001) Differential expression of orexin receptors 1 and 2 in the rat brain. J Comp Neurol 435:6–25
Mieda M, Hasegawa E, Kisanuki YY, Sinton CM, Yanagisawa M, Sakurai T (2011) Differential roles of orexin receptor-1 and -2 in the regulation of non-REM and REM sleep. J Neurosci 31:6518–6526
Mieda M, Tsujino N, Sakurai T (2013) Differential roles of orexin receptors in the regulation of sleep/wakefulness. Front Endocrinol (Lausanne) 4:57
Morairty SR, Revel FG, Malherbe P, Moreau J-L, Valladao D, Wettstein JG et al (2012) Dual hypocretin receptor antagonism is more effective for sleep promotion than antagonism of either receptor alone. PLoS ONE 7:e39131
Peyron C, Tighe DK, van den Pol A, de Lecea L, Heller HC, Sutcliffe JG et al (1998) Neurons containing hypocretin (orexin) project to multiple neuronal systems. J Neurosci 18:9996–10015
Prober DA, Rihel J, Onah AA, Sung R-J, Schier AF (2006) Hypocretin/orexin overexpression induces an insomnia-like phenotype in zebrafish. J Neurosci 26:13400–13410
Renzulli C, Nash M, Wright M et al (2011) Disposition and metabolism of [14C]SB-649868, an orexin receptor antagonist, in humans. Drug Metab Dispos 39:215–227
Sakurai T (2007) The neural circuit of orexin (hypocretin): maintaining sleep and wakefulness. Nat Rev Neurosci 8:171–181
Sakurai T, Amemiya A, Ishii M, Matsuzaki I, Chemelli RM, Tanaka H et al (1998) Orexins and orexin receptors: a family of hypothalamic neuropeptides and G protein-coupled receptors that regulate feeding behavior. Cell 92:573–585
Smart D, Sabido-David C, Brough SJ, Jewitt F, Johns A, Porter RA et al (2001) SB-334867-A: the first selective orexin-1 receptor antagonist. Br J Pharmacol 132:1179–1182
Smith RJ, Aston-Jones G (2012) Orexin/hypocretin 1 receptor antagonist reduces heroin self-administration and cue-induced heroin seeking. Eur J Neurosci 35:798–804
Smith MI, Piper DC, Duxon MS, Upton N (2003) Evidence implicating a role for orexin-1 receptor modulation of paradoxical sleep in the rat. Neurosci Lett 341:256–258
Steiner MA, Gatfield J, Brisbare-Roch C, Dietrich H, Treiber A, Jenck F et al (2013) Discovery and characterization of ACT-335827, an orally available, brain penetrant orexin receptor type 1 selective antagonist. ChemMedChem 8:898–903
Sun H, Kennedy WP, Wilbraham D, Lewis N, Calder N, Li X et al (2013) Effects of suvorexant, an orexin receptor antagonist, on sleep parameters as measured by polysomnography in healthy men. Sleep 36:259–267
Thannickal TC, Moore RY, Nienhuis R, Ramanathan L, Gulyani S, Aldrich MS et al (2000) Reduced number of hypocretin neurons in human narcolepsy. Neuron 27:469–474
White CL, Ishii Y, Mendoza T, Upton N, Stasi LP, Bray GA et al (2005) Effect of a selective OX1R antagonist on food intake and body weight in two strains of rats that differ in susceptibility to dietary-induced obesity. Peptides 26:2331–2338
Willie JT, Chemelli RM, Sinton CM, Tokita S, Williams SC, Kisanuki YY et al (2003) Distinct narcolepsy syndromes in orexin receptor-2 and orexin null mice. Neuron 38:715–730
Winrow CJ, Gotter AL, Cox CD, Doran SM, Tannenbaum PL, Breslin MJ et al (2011) Promotion of sleep by suvorexant-a novel dual orexin receptor antagonist. J Neurogenet 25:52–61
Zisapel N (2012) Drugs for insomnia. Expert Opin Emerg Drugs 17(3):299–317
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer International Publishing Switzerland
About this chapter
Cite this chapter
De la Herrán-Arita, A.K., Equihua-Benítez, A.C., Drucker-Colín, R. (2015). Orexin/Hypocretin Antagonists in Insomnia: From Bench to Clinic. In: Guglietta, A. (eds) Drug Treatment of Sleep Disorders. Milestones in Drug Therapy. Springer, Cham. https://doi.org/10.1007/978-3-319-11514-6_9
Download citation
DOI: https://doi.org/10.1007/978-3-319-11514-6_9
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-11513-9
Online ISBN: 978-3-319-11514-6
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)