
Abstract
Antiphospholipid syndrome (APS) is characterized by vascular thrombosis and/or pregnancy complications in association with the presence of antiphospholipid antibodies (aPL), i.e., anticardiolipin (aCL), anti-beta2-glycoprotein I (anti-β2GPI), and lupus anticoagulant (LA). In addition to these three aPL assays, a number of other laboratory tests have been proposed for APS diagnosis: antiprothrombin antibodies, anti-domain I and IV/V antibodies, anti-phosphatidylethanolamine antibodies, antibodies against other anionic phospholipids (such as phosphatidylserine, phosphatidylinositol, or phosphatidic acid), and antibodies against annexin A5 and annexin A2. aCL/anti-β2GPI and anti-annexin A5 antibodies of the IgA isotype have been also suggested. However, these additional assays are not included in the classification criteria, due to their poor sensitivity or specificity.
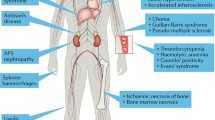
Moreover, besides arterial and venous thrombosis, a number of clinical manifestations that can be only in part explained by the thrombophylic state have been described: thrombocytopenia, heart valve disease, livedo reticularis, skin ulcers, nephropathy, and non-ischemic neurological features. These manifestations are associated with APS, but are not included in the clinical classification criteria.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Non-criteria APS manifestations
- Non-classification laboratory assays
- Anti-domain I 2GPI antibodies
- Anti-PS/PT antibodies
- Thrombocytopenia
6.1 Introduction
Antiphospholipid syndrome (APS) is an acquired autoimmune disease mainly characterized by vascular thrombosis and/or pregnancy complications in association with autoantibodies belonging to the antiphospholipid antibody family (aPL).
The laboratory classification criteria for APS require the presence of lupus anticoagulant (LA, detected by a clotting assay) and of medium/high positivity for anticardiolipin (aCL) antibodies (IgM or IgG isotype) or anti-β2-glycoprotein I antibodies (IgM or IgG isotype), which must be confirmed twice, at least 12 weeks apart [1, 2]. Several other autoantibodies have been described in APS patients that are not included in the formal criteria as their diagnostic and prognostic value is still unclear.
The clinical criteria include a history of arterial or venous thrombosis, early or late pregnancy loss, or severe prematurity due to (pre-)eclampsia or placental failure (which are the major features of obstetric APS) [3]. Deep venous thrombosis of the lower limbs is one of the most common manifestations of APS, and it can be complicated by pulmonary thromboembolism. Arterial involvement is less common and often involves the central nervous system circulation [2].
The clinical spectrum of APS has markedly broadened from the first description of APS and a variety of other clinical manifestations has been reported in aPL-positive patients in addition to thrombosis and obstetric morbidity. However, the formal classification criteria do not include these clinical findings because of their low prevalence or because they are not specific for APS, being common manifestations of other diseases [1].
6.2 Non-classification Laboratory Assays
Besides the three aPL assays currently included in the laboratory criteria for formal APS classification, several additional laboratory tests have been proposed for APS diagnosis [4, 5] (Table 6.1).
6.2.1 Anti-prothrombin Antibodies
Prothrombin (PT, also known as clotting factor II) is a vitamin K-dependent proenzyme that induces the conversion of fibrinogen to fibrin, via a prothrombinase complex. PT was first reported by Loeliger in 1959 as a possible cofactor for LA. Since then, many other authors have suggested that antibodies binding to PT could contribute to LA phenomenon and, at the present time, they are largely considered as one of the most important causes of the elongation of clotting time due to LA positivity, together with anti-β2GPI antibodies [6, 7].
Antibodies against prothrombin are usually detected by two different ELISAs (enzyme-linked immunosorbent assay) that employ human PT coated onto irradiated plates (aPT) or PT in complex with phosphatidylserine (aPS/PT) as antigen. These two assays seem to display different diagnostic and prognostic power, possibly attributable to their different ability to offer the antigen to antibody binding [6–8]. The real prevalence of aPT is still unknown, as it widely varies among studies, as a result of the variability of detection methods and the poor standardization among different laboratories. Also the clinical significance of aPT in both primary and secondary APS is still debated, as an apparent association with obstetric or thrombotic manifestations has not been definitely demonstrated [6]. On the other hand, aPS/PT antibodies strongly correlate with the presence of LA and are suggested to be highly specific for the diagnosis of APS [7, 8].
Most of the studies addressing the clinical significance of aPS/PT antibodies have demonstrated a significant association with thrombotic manifestations of APS, venous thrombosis above all, while the association with obstetric manifestations is still controversial. Actually, even if some authors reported that aPS/PT can be predictive of pregnancy morbidity in patients with systemic autoimmune diseases, most of the studies did not confirm this finding [9, 10].
Several studies have clearly demonstrated that multiple positive aPL tests are stronger predictor of thrombosis and pregnancy complications than single positivity. Accordingly, it has been suggested that the combination of routinely tested aPL (aCL, anti-β2GPI, and LA) with new (non-criteria) aPL assays would lead to a better risk stratification of patients. Among 23 possible combinations of six aPL assays (LA, aCL, anti-β2GPI, aPT, aPS/PT, and anti-phosphatidylethanolamine antibodies), the association of LA plus anti-β2GPI plus aPS/PT antibodies has recently been identified to display the best diagnostic accuracy for both vascular and obstetric APS [11].
A very recent and exhaustive review of the literature showed that both aPT and anti-PS/PT positivity increase the risk of thrombosis but that aPS/PT display the highest odds ratio (5.11 (95 % CI 4.2–6.3) vs 1.82 (95 % CI 1.44–2.75)). In line, the studies directly comparing aPT and aPS/PT and their odds ratio for thrombosis in 1196 patients demonstrate that aPS/PT antibodies are more strongly associated with both arterial and venous thrombosis than aPT antibodies [12].
Even if aPS/PT represent a very promising biomarker of APS, the lack of harmonization and standardization of the detection procedures and the low reproducibility of the results among laboratories are still unsolved problems. Promising data have been recently reported by Amengual et al. who have compared different assays for the detection of aPS/PT and found a good accuracy of both homemade and commercial ELISA kits and a high concordance of the results [13].
6.2.2 Antibodies to β2GPI Domains
β2GPI is a large anionic plasma glycoprotein, consisting of 326 amino acids, organized in five domains [14]. This protein is highly immunogenic, and it has been demonstrated that autoimmune patients can produce antibodies against several epitopes of the molecule, located in different domains. Antibodies directed to different β2GPI domains seem to display higher or lower clinical significance [15]. Anti-domain I (DI) antibodies were firstly described in 2002, but their importance has clearly emerged more recently [16]. Actually, even if there is growing evidence that domain I represents the immunodominant epitope of β2GPI, the clinical significance of anti-DI antibodies is still debated. De Laat et al. have shown that anti-β2GPI antibodies with DI specificity are associated with LA and that anti-DI positivity correlates with vascular thrombosis, with an OR for venous thrombosis ranging from 3.5 to 6.7 in different studies [17, 18]. In one of these studies, the group found a correlation between anti-DI antibodies and obstetric APS manifestations, even though to a lesser extent than with thrombosis [18]. However, the results of this study have to be carefully evaluated, as it showed no correlation between LA and miscarriages, conflicting with several previous publications and the known clinical LA predictive value for miscarriages [19, 20].
Moreover in a recent study by our group, a high prevalence of anti-DI antibodies was detected in both thrombotic and obstetric primary APS, albeit anti-DI IgG were not found to be predictive of these complications [21].
Antibodies directed to the other domains of β2GPI seem to have lesser predictive value for APS. For example, anti-domain IV (DIV) and domain V (DV) antibodies have been found in patients with chronic infections, such as leprosy, in children with atopic dermatitis and in aPL-positive asymptomatic carriers [22].
Thus, definite conclusions on the diagnostic and prognostic value of anti-DI antibodies cannot be drawn at present, as the data regarding the association with thrombosis are not univocal among different studies [17, 18, 21]. Moreover, a small but relevant proportion of anti-β2GPI-positive APS patients do not display anti-DI antibodies, suggesting that the assay for the whole molecule cannot be substituted up to now [21].
It has been proposed that the ratio between anti-β2GPI-DI and anti-β2GPI-D IV/V IgG antibody reactivities could add important information to discriminate between relevant anti-β2GPI positivity associated with an autoimmune disease (such as APS) and antibodies occurring in association with other pathologies, with less diagnostic and pathogenic value. If confirmed in larger studies, this finding would suggest that tests for antibodies against the different domains could help in the risk stratification of anti-β2GPI antibody-positive patients [23].
There are different methodologies that can be employed to detect these antibodies. Besides the two-step assay, three ELISAs and a CIA using different DI molecules or peptides have been reported [23, 24].
6.2.3 Other Antiphospholipid Antibodies
Phosphatidylethanolamine (PE) is a neutral phospholipid that constitutes the inner leaflets of biological membranes. PE has anticoagulant properties and the finding that PE can interfere with clotting time prolongation raised the hypothesis that aPE might be responsible for the LA phenomenon, even if a significant association between aPE and LA has not been demonstrated [25].
In in vitro experiments, PE has been found to be an essential cofactor for the protein C anticoagulant pathway. Moreover PE is an inhibitor of the factor Xa-prothrombin system [26].
Antibodies targeting PE have been reported in up to 43 % of APS patients [25]. In a population of women with a history of recurrent early pregnancy loss, aPE prevalence has been reported to range between 23 and 31.7 % [27]. In another study, the rate of aPE positivity in a population of patients with otherwise unexplained thrombotic events was 18 % when detected by ELISA and 30.5 % when tested using thin-layer immunostaining [25, 28]. Moreover, in a multicenter study on 270 thrombotic patients, 63 % of 40 aPE-positive subjects had no additional aPL laboratory tests [25]. On the basis of these data, some authors have proposed aPE as serological markers of seronegative APS. However, given the limited number of studies, the small sample size, and poor ELISA standardization, the clinical role of aPE is still not clear and aPE testing is still not recommended.
The diagnostic and prognostic role of several autoantibodies against negatively charged PLs (other than CL) have been also studied. Among them, most data regard phosphatidylserine (PS), phosphatidylinositol (PI), and phosphatidic acid (PA). In the past, aCL has been shown to cross-react with antibodies targeting both PS and PI. Further studies have demonstrated that aPS, aPI, and aPA antibodies mainly recognize a complex consisting of β2GPI, coupled to these negatively charged aPL [28]. Therefore, most of the cross-reactivity is due to autoantibodies actually reacting with β2GPI [5, 29].
There are no recent studies demonstrating that aPS, aPI, and aPA antibody testing significantly improves the diagnosis of APS [5]. Nevertheless, aPS antibody detection has been suggested to be useful in the context of pregnancy-related morbidity [29]. Recent available literature reports conflicting results. In one study aPS was not associated with recurrent pregnancy loss, while in another aPS IgG but not IgM were related to obstetric morbidity [5]. Moreover, in a cohort of women with a history of recurrent miscarriage, aPS was the only autoantibody detectable in 3.6 % of subjects [30]. The significant role of aPS in obstetric APS is also supported by in vivo studies on animal models. Two murine monoclonal antibodies targeting PS have been demonstrated to decrease human chorionic gonadotropin (hCG) secretion and to inhibit trophoblast invasion [30, 31]. Notably, one of these two aPS reacted with PS only, while the other was able to recognize both CL and PS, but no information about a possible cross-reactivity with β2GPI was available [30, 31]. More recently, active immunization with β2GPI-dependent aPS was able to induce fetal resorption in a murine model of APS [5]. However, data on humans are inconsistent and aPS assays are still not included in classification criteria.
Recently, a novel aPL assay (APhL IgG/IgM ELISA), using a mixture of negatively charged phospholipids as antigen (including PS, phosphatidic acid, and β2GPI), has been tested in APS patients [32]. Positivity for this commercial ELISA kit has been reported to be more predictive for APS than aCL. Particularly, APhL test showed higher positive and negative predictive values for APS in comparison to two commercially available aCL assays. Moreover, in the same study, antibodies against APhL have been associated with arterial events in a cohort of SLE patients. The authors suggest that the routine use of this assay could implement specificity, without losing sensitivity for APS [32].
Annexins are a family of proteins that bind Ca++ and phospholipids and display several different functions, including inhibition of coagulation processes in the vasculature and on trophoblasts [33]. Annexin V (AnnA5) is present on the intervillous surface of the placenta, forming a shield that prevents the activation of the coagulation cascade [33, 34]. Several studies have reported a significant reduction of annexin V binding on the placental tissue from patients with obstetric APS in comparison with normal controls. In addition, aPL have been shown to displace annexin V from both trophoblast and endothelial cell monolayers in in vitro studies [33, 34].
Recently, the determination of resistance to the anticoagulant effects of AnnA5 (AnnA5 resistance) has been proposed as a marker of APS. AnnA5 resistance has been found in a significantly higher proportion of APS patients in comparison to controls. Moreover, a significant reduction of AnnA5 anticoagulant activity was detected in a cohort of SLE children in comparison to pediatric controls [35]. Notably, the same cohort of patients displayed a significant increased prevalence of anti-DI antibodies (p = 0.014) compared to controls, and resistance to AnnA5 anticoagulant activity was found to inversely correlate with titers of anti-DI IgG antibodies [35]. Even if these promising data suggest that AnnV resistance could play a role in the identification of specific subsets of pathogenic aPL antibodies, further studies are needed to confirm this preliminary finding.
Annexin 2 (AnnA2) has been proposed as a target of aPL. Several studies have suggested that AnnA2 could represent a receptor mediating β2GPI binding to endothelial cells (ECs) [36]. Autoantibodies against AnnA2 have been described in patients with APS and severe thrombosis and/or pregnancy morbidity but also in some other autoimmune conditions (such as SLE and RA). Their clinical significance is unclear at the moment [36].
The clinical significance of aCL and anti-β2GPI antibodies of the IgA isotype in PAPS is still a controversial issue. IgA aCL and/or IgA anti-β2GPI antibodies have been reported in seronegative patients with a history of thrombosis and pregnancy morbidity. Particularly, IgA anti-β2GPI antibodies can potentially identify APS in patients who possess the clinical features of the disease but do not meet current laboratory criteria. In mouse models these antibodies were able to induce significantly larger thrombi and higher tissue factor levels compared to controls, demonstrating their pathogenic role. In a recent study of Mattia et al. on 84 PAPS patients, IgA aCL and IgA anti-β2GPI antibodies were found, respectively, in 19 and 50 % of patients. The mean titers of both IgA aCL and IgA anti-β2GPI antibodies were higher in the thrombotic patients, but only IgA anti-β2GPI were significantly associated with thrombosis. Isolated IgA anti-β2GPI antibody positivity was significantly prevalent in seven of the seronegative patients [37]. There are several reasons to explain why a number of studies failed to prove the usefulness of adding IgA aCL and IgA anti-β2GPI testing. In fact, these autoantibodies have a low prevalence and are mostly found in association with other aPL. Moreover, few accurate diagnostic tests are available for their detection [38]. Recently, Ruiz-García et al. found that mean levels of IgG, IgM, and IgA, both aCL and anti-β2GPI, antibodies were significantly higher in patients with clinical features of APS than in controls on a total of 156 patients fulfilling clinical criteria for APS. IgA anti-β2GPI was the most prevalent antibody in these patients [39].
6.3 Non-criteria Clinical Manifestations
In addition to thrombosis and pregnancy morbidity, a number of clinical manifestations have been described in aPL-positive patients. Non-thrombotic neurological features, thrombocytopenia, heart valve disease, microangiopathic nephropathy, livedo reticularis, and skin ulcers are some of the possible features that are not yet considered as classification criteria because of their low specificity (Table 6.2).
6.3.1 Skin
Skin involvement is common in APS patients, being skin ulcers and livedo reticularis the most frequent cutaneous manifestations [40]. Livedo reticularis (LR) is a blanching erythematous to violaceous netlike vascular pattern on the skin that can be secondary to numerous conditions, including APS. LR is present in up to 20–25 % of APS patients and has been originally described in association with arterial thrombosis. However, the relationship of LR with stroke or other types of arterial occlusion is still not clear as several studies did not confirm this original observation [41].
About 30–40 % of APS patients develop skin ulcers. Skin lesions are usually situated in the lateral face of the ankle or in pretibial area, display sharp margins, and are usually very painful. The pathogenesis of these dermatological manifestations is linked to fibrin deposition in the superficial dermal vessels lumen [41]. Skin ulcers are mainly associated with catastrophic APS and are usually concomitant with non-inflammatory purpura with microvascular occlusion [42].
Other skin manifestations anecdotally reported in the setting of APS include anetoderma, chronic venous ulcers, pseudovasculitis, superficial thrombophlebitis, superficial skin bullae, infarcts and distal gangrene, acrocyanosis, and relapsing polychondritis [43].
Anetoderma is an elastolytic disorder of unknown origin characterized by localized areas of flaccid skin, which can appear atrophic or protuberant. The occurrence of anetoderma in a skin region with no prior pathology (primary anetoderma) has been reported to be very suggestive for the presence of aPLs. In a study on 9 patients with primary anetoderma, aPLs were found in all 9 patients, with 4 patients also having APS [44].
6.3.2 Heart
Heart valve involvement has been reported in APS patients, with a prevalence ranging from 10 to 40 % of aPL-positive patients [45]. The prevalence can be even higher, up to 80 %, if highly sensitive techniques such as transesophageal echocardiography are used. Valve involvement can be characterized by several different alterations, including vegetations, valve thickening, and valvular dysfunction mainly affecting mitral and aortic valve [45]. Most patients are asymptomatic, but cerebrovascular accidents are more prevalent among patients with significant valve lesions [46].
Longitudinal studies have suggested that a small but significant proportion (7–25 %) of patients suffering from both primary and SLE-associated APS can develop valve vegetations or thickening during the course of disease but also that these alterations can disappear overtime [5]. Disease duration and a diagnosis of SLE-APS were independent factors associated with valvular disease progression in a cohort of 82 patients suffering from primary APS, SLE-APS, aPL-positive SLE, and SLE negative for aPL, followed up for 10 years. In this study, anticoagulation was not able to prevent the worsening of valvular involvement [47].
Less frequent cardiac manifestations are ventricular hypertrophy and dysfunction and pulmonary hypertension [48].
6.3.3 Kidney
Renal involvement is not very common in APS. However, thrombosis of the renal artery or its main branches has been reported [49]. The most typical nonischemic renal manifestation of APS is a small artery vasculopathy, involving both arterioles and glomerular capillaries, defined as aPL-associated nephropathy (aPLN). This histological entity has been described both in primary APS and SLE-APS. In the latter group it has been associated with pregnancy complications, extrarenal vascular thrombosis, and higher risk of renal failure [49]. aPLN can be clinically silent or manifest with systemic hypertension, proteinuria, and inconstant hematuria [5].
6.3.4 Thrombocytopenia
Thrombocytopenia is one of the most common laboratory abnormalities found in patients with APS [40, 48]. A variable degree of thrombocytopenia is observed in up to 40 % of patients with aPL [48]. As a low platelet count can manifest in a variety of autoimmune and non-autoimmune disease, this feature has not been included in the APS formal criteria [1]. Moderate thrombocytopenia is frequent in APS and generally does not modify the policy for treatment of thrombosis as several authors have clearly demonstrated that thrombocytopenia does not display a protective effect on the thrombotic risk of aPL [50]. Severe thrombocytopenia is relatively uncommon and it is seldom associated with bleeding events. A recent task force on non-criteria APS manifestations has concluded that thrombocytopenia should be incorporated in the clinical criteria of APS and that an international registry of aPL-positive patients with thrombocytopenia (“hematologic APS”) could be very useful. Moreover, the task force has proposed a multicentric, international, prospective long-term follow-up study on aPL-positive patients with thrombocytopenia, to assess the risk of thrombosis in this type of patients [51].
6.3.5 Neurological Manifestations
A wide variety of neurological manifestations has been described in association with aPL, in addition to cerebral ischemia [52]. Headache and migraine are common in APS, but the real correlation with aPL is still debated as these manifestations are very frequent also in the general population. In the past, several studies have reported a high incidence of seizures in APS patients, particularly in SLE-APS, and aPL positivity has been considered a risk factor for epilepsy in SLE patients [53]. However, more recent studies on a very large cohort of SLE patients did not confirm these previous findings [54].
Additional manifestations that have been associated to aPL positivity are chorea, transverse myelopathy, and Guillain-Barré syndrome. Moreover a clinical syndrome resembling multiple sclerosis (MS) has also been described in APS. Such patients can display magnetic resonance imaging (MRI) lesions similar to those observed in MS that can make the differential diagnosis very difficult [52].
The presence of cognitive impairment in SLE patients has been associated with aPL in the past. However, this association was not confirmed in several recent studies on very large SLE cohorts [55]. In primary APS the data on the real incidence of cognitive dysfunction are very limited. Only two studies have evaluated cognitive functions in primary APS. Both studies demonstrate a high incidence of cognitive defects, mainly involving attention, verbal fluency, memory, and visual learning [56, 57]. Recently our group has demonstrated a high prevalence of cognitive defects in a very well-characterized population of strongly positive APS patients, mostly involving frontal functions [58].
On the contrary, the presence of dementia as a consequence of chronic or recurrent ischemic events affecting small or large cerebral vessels has been clearly demonstrated in APS patients, with a prevalence ranging from 10 to 56 % in different studies [52].
6.3.6 Other Manifestations
Several other manifestations, such as vertigo or hearing loss due to middle ear involvement, myocardial dysfunction, and diffuse alveolar hemorrhage, have been anecdotally reported in APS patients [48].
In addition to the classical manifestations included in the classification criteria for APS, several other obstetric complications have been associated with aPL, such as intrauterine growth restriction and placental abruption [48]. However, these clinical features have not been included in the classification criteria because they can be present in several different conditions.
aPLs have also been addressed as a possible cause of infertility. Actually, patients suffering from infertility or with recurrent implantation failure after in vitro fertilization (IVF) display a significantly higher prevalence of aPL in comparison to the general female fertile population (20–30 % versus 1–3 %). However, data of the literature are not univocal and a clear demonstration of aPL as a cause of infertility has not been provided [59].
In conclusion, there are several laboratory and clinical features of APS that have not been included in the classification criteria. Nevertheless these clinical and serological characteristics have to be carefully addressed as they could help to assess the risk of complication (i.e., thrombosis and pregnancy morbidity) and to choose the appropriate therapy for a single patient.
References
Miyakis S, Lockshin MD, Atsumi M et al (2006) International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS). J Thromb Hemost 4:295–306
Ruiz-Irastorza G, Crowter M, Branch W et al (2010) Antiphospholipid syndrome. Lancet 376:1498–1509
D’Ippolito S, Meroni PL, Koike T et al (2014) Obstetric antiphospholipid syndrome: a recent classification for an old defined disorder. Autoimmun Rev. 13:901–908
Bertolaccini ML, Amengual O, Andreoli L et al (2014) 14th International Congress on Antiphospholipid Antibodies Task Force. Report on antiphospholipid syndrome laboratory diagnostics and trends. Autoimmun Rev. 13:917–930
Meroni PL, Chighizola CB, Rovelli F, Gerosa M (2014) Antiphospholipid syndrome in 2014: more clinical manifestations, novel pathogenic players and emerging biomarkers. Arthritis Res Ther 16:209
Sciascia S, Khamashta MA, Bertolaccini ML (2014) New tests to detect antiphospholipid antibodies: antiprothrombin (aPT) and anti-phosphatidylserine/prothrombin (aPS/PT) antibodies. Curr Rheumatol Rep 16:415
Pregnolato F, Chighizola C (2014) Phospholipid autoantibodies (non anticardiolipin)-anti-prothrombin antibodies. In: Shoenfeld Y, Gershwin E, Meroni PL (eds) The autoantibodies, 3rd edn. Elsevier, New York, pp 741–749
Pregnolato F, Chighizola CB, Encabo S et al (2013) Anti-phosphatidylserine/prothrombin antibodies: an additional diagnostic marker for APS? Immunol Res 56:432–438
Zigon P, Čučnik S, Ambrožič A et al (2013) Detection of antiphosphatidylserine/prothrombin antibodies and their potential diagnostic value. Clin Dev Immunol 724592
Vlagea A, Gil A, Cuesta MV et al (2013) Antiphosphatidylserine/prothrombin antibodies (aPS/PT) as potential markers of antiphospholipid syndrome. Clin Appl Thromb Hemost 19:289–296
Sciascia S, Murru V, Sanna G et al (2012) Clinical accuracy for diagnosis of antiphospholipid syndrome in systemic lupus erythematosus: evaluation of 23 possible combinations of antiphospholipid antibody specificities. J Thromb Haemost 10:2512–2518
Sciascia S, Sanna G, Murru V et al (2014) Anti-prothrombin (aPT) and anti-phosphatidylserine/prothrombin (aPS/PT) antibodies and the risk of thrombosis in the antiphospholipid syndrome: a systematic review. Thromb Haemost 111:354–364
Amengual O, Horita T, Binder W et al (2014) Comparative analysis of different enzyme immunoassays for assessment of phosphatidylserine-dependent antiprothrombin antibodies. Rheumatol Int. 34:1225–1230
de Groot PG, Meijers JCM (2011) β2-Glycoprotein I: evolution, structure and function. J Thromb Haemost 9:1275–1284
Shoenfeld Y, Krause I, Kvapil F et al (2003) Prevalence and clinical correlations of antibodies against six b2-glycoprotein I-related peptides in the antiphospholipid syndrome. J Clin Immunol 23:377–383
Iverson GM, Reddel S, Victoria EJ et al (2002) Use of single point mutations in domain I of B2-glycoprotein I to determine fine antigenic specificity of antiphospholipid autoantibodies. J Immunol 169:7097–7103
De Laat B, Pengo V, Pabinger I et al (2009) The association between circulating antibodies against domain I of beta2-glycoprotein I and thrombosis: an international multicenter study. J Thromb Haemost 7:1767–1773
De Laat B, Derksen RH, Urbanus RT, de Groot PG (2005) IgG antibodies that recognize epitope Gly40-Arg43 in domain I of beta 2-glycoprotein I cause LAC, and their presence correlates strongly with thrombosis. Blood 105:1540–1545
Ruffatti A, Tonello M, Visentin MS et al (2011) Risk factors for pregnancy failure in patients with anti-phospholipid syndrome treated with conventional therapies: a multicentre case–control study. Rheumatology 50:1684–1689
Erkan D, Patel S, Nuzzo M et al (2008) Management of the controversial aspects of the antiphospholipid syndrome pregnancies: a guide for clinicians and researchers. Rheumatology 47:iii23–iii27
Andreoli L, Nalli C, Borghi MO et al (2013) Domain I is the main specificity of anti-beta2 GPI in systemic autoimmune diseases. Arthritis Rheum 65:S4
Andreoli L, Nalli C, Motta M et al (2011) AntiB2glycoprotein I IgG antibodies from 1-year old healthy children born to mothers with systemic autoimmune diseases preferentially target domain 4/5: might it be the reason for their ‘innocent’ profile? Ann Rheum Dis 70:280–283
Chighizola CB, Gerosa M, Meroni PL (2014) New tests to detect antiphospholipid antibodies: anti-domain I beta-2-glycoprotein-I antibodies. Curr Rheumatol Rep 16:402
Willis R, Mahler M, Pregnolato F et al (2013) Clinical evaluation of two anti-Beta2glycoprotein I Domain 1 autoantibody assays to aid in the diagnosis and risk assessment of the antiphospholipid syndrome. Arthritis Rheum 65:S3
Staub HL, Bertolaccini ML, Khamashta MA (2012) Anti-phosphatidylethanolamine antibody, thromboembolic events and the antiphospholipid syndrome. Autoimmun Rev 12:230–234
Zhixin L, Wells CW, North PE (2011) Phosphatidylethanolamine at the luminal endothelial surface–implications for hemostasis and thrombotic autoimmunity. Clin Appl Thromb Hemost 17:158–163
Sugi T, Matsubayashi H, Inomo A et al (2004) Antiphosphatidylethanolamine antibodies in recurrent early pregnancy loss and mid-to-late pregnancy loss. J Obstet Gynaecol Res 30:326–332
Conti F, Alessandri C, Sorice M et al (2012) Thin-layer chromatography immunostaining in detecting anti-phospholipid antibodies in seronegative anti-phospholipid syndrome. Clin Exp Immunol 167:429–437
Bertolaccini ML, Amengual O, Atsumi T et al (2011) ‘Non-criteria’ aPL tests: report of a task force and preconference workshop at the 13th International Congress on Antiphospholipid Antibodies, Galveston, TX, USA. Lupus 20:191–205
Nayfe R, Uthman I, Aoun J et al (2013) Seronegative antiphospholipid syndrome. Rheumatology 52:1358–1367
Yodfat O, Blank M, Krause I, Shoenfeld Y (1996) The pathogenic role of anti-phosphatidylserine antibodies: active immunization with the antibodies leads to the induction of antiphospholipid syndrome. Clin Immunol Immunopathol 78:14–20
Suh-Lailam BB, Cromar A, Davis KW, Tebo AE (2012) APhL antibody ELISA as an alternative to anticardiolipin test for the diagnosis of antiphospholipid syndrome. Int J Clin Exp Pathol 5:210–215
Meroni PL, Gerosa M, Raschi E et al (2008) Updating on the pathogenic mechanisms of the antiphospholipid antibodies-associated pregnancy loss. Clin Rev Allergy Immunol 34:332–337
Meroni PL, Borghi MO, Raschi E et al (2011) Pathogenesis of antiphospholipid syndrome: understanding the antibodies. Nat Rev Rheumatol 7:330–339
Wahezi DM, Ilowite NT, Wu XX et al (2013) Annexin A5 anticoagulant activity in children with systemic lupus erythematosus and the association with antibodies to domain I of β2-glycoprotein I. Lupus 22:702–711
Luo M, Hajjar KA (2013) Annexin A2 system in human biology: cell surface and beyond. Semin Thromb Hemost 39:338–346
Mattia E, Ruffatti A, Tonello M et al (2014) IgA anticardiolipin and IgA anti- β2 glycoprotein I antibody positivity determined by fluorescence enzyme immunoassay in primary antiphospholipid syndrome. Clin Chem Lab Med. 52:1329–1333
Meijide H, Sciascia S, Sanna G et al (2013) The clinical relevance of IgA anticardiolipin and IgA anti-β2 glycoprotein I antiphospholipid antibodies: a systematic review. Autoimmun Rev 12:421–425
Ruiz-García R, Serrano M, Martínez-Flores JÁ et al (2014) Isolated IgA anti-β2 glycoprotein I antibodies in patients with clinical criteria for antiphospholipid syndrome. J Immunol Res 2014:704395
Erkan D, Lockshin MD (2010) Non-criteria manifestations of antiphospholipid syndrome. Lupus 19:424–427
Frances C (2010) Dermatological manifestations of Hughes’ antiphospholipid antibody syndrome. Lupus 19:1071–1077
Cervera R, Espinosa G (2012) Update on the catastrophic antiphospholipid syndrome and the CAPS registry. Semin Thromb Hemost 38:333–338
Thornsberry LA, LoSicco KI, English JC III (2013) The skin and hypercoagulable states. J Am Acad Dermatol 69:450–462
Hodak E, Feureman H, Molad Y et al (2003) Primary anetoderma: a cutaneous sign of antiphospholipid antibodies. Lupus 12:564–568
Zuily S, Huttin O, Mohamed S et al (2013) Valvular heart disease in antiphospholipid syndrome. Curr Rheumatol Rep 15:320–329
Cervera R (2005) Recent advances in antiphospholipid antibody-related valvulopathies. J Autoimmun 15:123–125
Kampolis C, Tektonidou M, Moyssakis I et al (2014) Evolution of cardiac dysfunction in patients with antiphospholipid antibodies and/or antiphospholipid syndrome: a 10-year follow-up study. Semin Arthritis Rheum 43(4):558–565
Cervera R, Piette JC, Font J et al (2002) Antiphospholipid syndrome: clinical and immunologic manifestations and patterns of disease expression in a cohort of 1,000 patients. Arthritis Rheum 46:1019–1027
Tektonidou MG (2009) Renal involvement in the antiphospholipid syndrome (APS)- nephropathy. Clin Rev Allergy Immunol 36:131–140
Atsumi T, Furukawa S, Amengual O et al (2005) Antiphospholipid antibody associated thrombocytopenia and the paradoxical risk of thrombosis. Lupus 14:499–504
Cervera R, Tektonidou MG, Espinosa G et al (2011) Task force on Catastrophic Antiphospholipid Syndrome (APS) and Non-criteria APS Manifestations (II): thrombocytopenia and skin manifestations. Lupus 20(2):174–181
Brey RL, Muscal E, Chapman J (2011) Antiphospholipid antibodies and the brain: a consensus report. Lupus 20:153–157
Sanna G, Bertolaccini ML, Cuadrado MJ et al (2003) Neuropsychiatric manifestations in systemic lupus erythematosus: prevalence and association with antiphospholipid antibodies. J Rheumatol 30:985–992
Hanly JG, Urowitz MB, Su L et al (2012) Seizure disorders in systemic lupus erythematosus results from an international, prospective, inception cohort study. Ann Rheum Dis 71:1502–1509
Hanly JG, Urowitz MB, Su L et al (2011) Autoantibodies as biomarkers for prediction of neuropsychiatric events in systemic lupus erythematosus. Ann Rheum Dis 70:1726–1732
Tektonidou MG, Varsou N, Kotoulas G et al (2006) Cognitive deficits in patients with antiphospholipid syndrome. Arch Intern Med 166:2278–2284
Kozora E, Erkan D, Zhang L et al (2014) Cognitive dysfunction in antiphospholipid antibody (aPL)-negative systemic lupus erythematosus (SLE) versus aPL-positive non-SLE patients. Clin Exp Rheumatol 32:34–40
Gerosa M, Poletti B, Pregnolato F et al (2009) Antiphospholipid syndrome and systemic lupus erythematosus: what’s new about cognitive impairment. J Neurol 256:S190
Buckingham KL, Chamley LW (2009) A critical assessment of the role of antiphospholipid antibodies in infertility. J Reprod Immunol 80:132–145
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Gerosa, M., Rovelli, F. (2015). Non-classification Criteria. In: Meroni, P. (eds) Antiphospholipid Antibody Syndrome. Rare Diseases of the Immune System. Springer, Cham. https://doi.org/10.1007/978-3-319-11044-8_6
Download citation
DOI: https://doi.org/10.1007/978-3-319-11044-8_6
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-11043-1
Online ISBN: 978-3-319-11044-8
eBook Packages: MedicineMedicine (R0)