
Abstract
In this chapter we aim to provide tools for clinical decision-making in treatment selection and implementation for pediatric epilepsy. Clinicians approach these treatment decisions considering a broad set of factors, including patient age, seizure semiology, EEG data, seizure etiology, side-effect profile, potential interaction with current medications, other patient risk factors, and feasibility of treatment monitoring. In this chapter, we discuss these considerations and the state of the science pertaining to the primary pharmacologic, dietary, and surgical treatment options for pediatric epilepsy. Specific attention will also be paid to a stepwise approach to the treatment of status epilepticus. The appendix of this chapter reviews dosing, side effects, and special considerations for each medication individually.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
Introduction
In this chapter, we outline the primary pharmacologic, dietary, and surgical treatments for pediatric epilepsy. Over the past two decades, there has been a significant increase in available treatment options. The medications currently available are diverse in structure and proposed mechanisms of action (Table 7.1) [1,2,3,4]. Treatment decisions are often based on the consideration of seizure semiology, epilepsy syndrome, and side effects. Efficacy data is relatively limited in pediatric epilepsy treatment. We aim to highlight available data, as well as review treatment practices from the MassGeneral for Children Pediatric Epilepsy Program. This chapter is intended to be an overview of seizure treatment options. For individual patient care needs, please review the medication dosing recommendations at your institution and collaborate with the pharmacy team to guide specific therapeutic choices and titration schedules.
Antiseizure Medication (ASM) Management
Initiation of Treatment
Following a first-time seizure, treatment is typically deferred. After a single unprovoked event, Stroink et al. found that 46% of children did not have a second event within a two-year follow-up [5, 6]. However, after two unprovoked seizures, Hauser et al. found that recurrence rates increased to about 75% in a population, across age groups [6, 7]. Treatment is therefore typically initiated after the second unprovoked seizure [6]. Patients with significant EEG abnormalities, or with a structural or metabolic etiology, are at higher risk for recurrence, and treatment may be initiated following a single event [8].
First ASM
The following parameters should be considered when choosing the first antiseizure medication: seizure type(s), epilepsy syndrome/etiology, age, comorbidities, and medication interactions [6]. To date, there are relatively few randomized control trials to support recommendations for first-line treatment in pediatric epilepsy. The following is a brief review of the data in select seizure types.
In a multicenter study evaluating initial treatment for children less than 3 years of age presenting with new onset non-syndromic epilepsy, levetiracetam was most often used first-line (62%) regardless of semiology [9]. It is important to note that this treatment practice is not based on efficacy data but rather consensus. The International League Against Epilepsy Treatment Guidelines review efficacy data for specific treatments as initial monotherapy for patients with newly diagnosed or untreated epilepsy [10]. Criteria for levels of evidence are presented in Table 7.2 [10].
For focal seizures, there is evidence to support treatment with oxcarbazepine (level A) [10]. For carbamazepine, phenobarbital, phenytoin, topiramate, valproate, and vigabatrin, evidence supports possible efficacy (level C) [10]. Finally, for clobazam, clonazepam, lamotrigine, and zonisamide, evidence supports potential efficacy (level D) for focal seizures [10].
For generalized tonic-clonic seizures, there is level C evidence to support treatment with carbamazepine, phenobarbital, phenytoin, topiramate, and valproate [10]. For absence seizures, there is level A evidence to support treatment with ethosuximide and valproate and level C evidence to support treatment with lamotrigine [10]. For juvenile myoclonic epilepsy, there is level D evidence for treatment with topiramate, levetiracetam, and valproate [10].
There is a growing body of literature to support treatment decisions based on epilepsy syndrome or genotype. A few examples will be briefly reviewed here. For patients with Dravet syndrome ~60% of whom have an SCN1A mutation, primary treatment recommendations include valproate, clobazam (and other benzodiazepines), stiripentol, and topiramate [11, 12]. For patients with myoclonic atonic epilepsy, primary pharmacologic treatment choices include valproate, ethosuximide, and topiramate [13,14,15,16]. For patients with Landau-Kleffner syndrome and epilepsy with continuous spike-wave during sleep (CSWS), high-dose oral diazepam, ACTH, or corticosteroids should be considered. Alternative treatments include lamotrigine, valproate, and topiramate [8]. See Chap. 4 for additional syndrome-specific treatment recommendations. General guidelines for treatment dosing are outlined in Table 7.3 [1,2,3, 17,18,19,20,21,22,23,24,25,26].
There are some medications that may increase seizure frequency in certain situations. This holds true primarily for treating generalized seizures with medications typically used for focal epilepsy. Glauser et al. review that carbamazepine, oxcarbazepine, phenobarbital, phenytoin, tiagabine, and vigabatrin are contraindicated in the treatment of absence seizures as they can exacerbate seizures in this context [10]. For juvenile myoclonic epilepsy, a similar set of treatments (with the addition of gabapentin and the exclusion of phenytoin) have been shown to exacerbate seizures [10]. In Dravet syndrome, sodium channel blockers, including carbamazepine, lamotrigine, and phenytoin, may worsen seizures and should be avoided [11, 27].
Second- and Third-Line Therapy
Kwan et al. found that 53% of patients across age groups failed initial monotherapy [28]. By comparison, in a pediatric cohort, Camfield et al. found improved outcomes with only 17% of patients showing failure of first-line therapy [29]. Poor response to first-line treatment portends poor long-term prognosis. With ineffective treatment response to initial monotherapy, only 11% eventually became seizure free [28]. Fig. 7.1 depicts seizure response rates across age groups following sequential medication trials [28]. In their review, Raspall-Chaure et al. describe that for patients who do not show a therapeutic response at an average ASM dose, only an additional 13–15% of patients will respond to an increase to maximal dosing [8]. Despite this fact, a given medication should be trialed near or just under maximal dosing before determining it a treatment failure. When the treatment choice has been determined to be ineffective, the clinician should reduce the dose of the first agent to the average therapeutic range and then add a second agent [8]. Once the second agent is therapeutic, the first-line agent should be slowly tapered and stopped, if possible, as monotherapy is typically the goal at this stage [8]. In many circumstances, however, a second agent is simply added on when the initial agent has shown incomplete efficacy. Again, poor early treatment response seems to portend poor prognosis. With failure of second-line treatment, less than 10% of individuals typically achieve long-term seizure freedom [8]. After two or three medications have been trialed as monotherapy, polytherapy is considered [8].

Response to medication in previously untreated epilepsy across age groups [28]
Drug-Resistant Epilepsy
In a prospective study of Dutch children, 12% of the cohort had a period of seizure intractability during the 15-year study interval [30]. Drug-resistant epilepsy is defined as failure to respond to two appropriately selected and trialed medications [6, 31]. At this point, dietary, surgical, and neuromodulating therapies should be considered. These non-pharmacologic treatments will be discussed later in the chapter.
There is no evidence to support increased efficacy with polytherapy compared to monotherapy [6]. Rational treatment theories recommend striving to choose two or more therapies that target different mechanisms of action. However, this hypothesis has limited data to support it as a superior treatment strategy. Human and animal data suggest three primary mechanistic combinations that may show improved clinical benefit: (1) combining a sodium channel blocker with a drug enhancing GABAergic inhibition; (2) combining two GABA mimetic agents; (3) combining an AMPA antagonist with an NMDA antagonist [32]. There are also a few specific ASM combinations that may have a synergistic effect in certain disease states. Examples of synergistic combinations include valproate with ethosuximide for absence seizures or myoclonic atonic epilepsy, valproate with lamotrigine for absence or myoclonic seizures, and stiripentol with clobazam for Dravet syndrome [11, 33,34,35,36].
With polytherapy, medication interactions are an important consideration. Many ASMs have interactions with each other as well as with many other classes of medications through hepatic enzyme-inducing or enzyme-inhibiting effects. A primary example is when valproate and lamotrigine are co-administered: valproate acts as a hepatic inhibitor of the metabolism of lamotrigine, and, as such, the lamotrigine dose typically needs to be significantly decreased to prevent toxicity (see appendix for detailed recommendations for valproate to lamotrigine cross titration) [1, 37, 38]. Table 7.4 highlights ASM interaction trends [1, 37]. Drug interactions should be evaluated by the treating provider whenever new medications are added and throughout the treatment course.
Treatment Side Effects
Side-effect profile is one of the primary considerations when selecting an ASM. There are two primary types of ASM side effects: neurotoxic and idiosyncratic. Neurotoxic side effects are a risk with nearly all ASMs and include symptoms such as somnolence, cognitive impairment, behavioral changes, vision changes, and ataxia [8]. These side effects are dose-dependent and are thought to be more common with older medications than the newer generations of ASMs [8]. Idiosyncratic reactions are unpredictable and may include rash, Stevens-Johnson syndrome (SJS), serum sickness, agranulocytosis, aplastic anemia, and hepatotoxicity [8, 39]. One of the most important idiosyncratic reactions is SJS, classically described with lamotrigine treatment but also seen with other ASMs. The risk of serious rash from lamotrigine can be significantly decreased with a slow titration schedule. Primary risk factors for developing a rash (serious or not) with lamotrigine treatment include: a history of another ASM-related rash, young age, and co-treatment with valproate [8, 40]. See Tables 7.5 [1,2,3, 11, 23, 41] and 7.6 [1,2,3, 11, 42
] for further details regarding medication side effects.
Laboratory Monitoring
Epilepsy treatment may include monitoring goal serum levels (Table 7.3) and/or following routine laboratory screening data, depending on the medication and patient risk factors. For many medications, routine laboratory screening is not indicated, felbamate being the notable exception. Among adults taking felbamate, there have been reports of fatal hepatic failure and aplastic anemia. Given the severity of a possible reaction, routine monitoring for agranulocytosis and hepatotoxicity is recommended [8, 43]. For other medications, laboratory evaluation is recommended in specific clinical circumstances depending on the medication, such as prior to surgery in patients taking valproate. Laboratory studies in this case should be sent to evaluate for hemostatic dysfunction (labs may include platelet count, PT, aPTT, TT, fibrinogen, vWF, factor XIII) [8, 44]. The clinician should have a very low threshold for checking laboratory tests if a patient presents with symptoms that could be explained by ASM toxicity. For example, a patient on valproate who presents with vomiting should have pancreatic and liver enzymes checked [8]. Routine ASM serum levels are not typically monitored for all medications. Some medications require drug level monitoring, including phenytoin, given its nonlinear kinetics, and carbamazepine, given the narrow therapeutic window [11, 45] Camfield et al. suggest that for patients on monotherapy with incomplete seizure control and no evidence of neurotoxicity, dosing may be increased without obtaining a medication level [45]. Similarly, for patients on monotherapy experiencing neurotoxicity, dosing should be decreased, again without obtaining a level. Clinical circumstances in which ASM levels may be helpful typically surround polytherapy [45]. If a patient presents with possible toxicity and/or uncontrolled seizures and it is unclear which medication may be causing the problem, ASM levels may provide guidance [8, 45].
Discontinuation of Treatment
When patients have been seizure free for 2 years, treatment discontinuation is typically considered. This clinical practice is supported by results of a Cochrane Review, which showed a 34% increased risk of seizure recurrence in patients for whom ASM treatment was discontinued prior to the two-year mark [46]. This rate of recurrence was further increased for patients with an abnormal EEG and/or focal seizures, so for this cohort, a minimum of 2 years prior to medication discontinuation was recommended [46]. In this same review, however, the authors noted that there remains insufficient data to guide the timing of medication discontinuation for patients with generalized epilepsies [46]. In a prospective pediatric study, among patients who had been seizure free for 2 years, 68% remained seizure free over the subsequent 2 years following discontinuation of medication [6, 47]. In this study, children with remote symptomatic epilepsy (defined as static encephalopathy prior to seizure history and/or having sustained a prior neurological insult) had a higher risk of recurrence compared to patients with idiopathic epilepsy [47]. Among this higher risk cohort, over 52% remained seizure free following discontinuation of treatment [47]. ASMs are typically tapered slowly, over a period of 6 weeks or more, and in some cases over 3–12 months [8, 11]. Slower tapers are implemented for benzodiazepines or phenobarbital, as these pose a higher risk of precipitating withdrawal seizures [11].
Deferring Treatment
Seizure treatment is not always indicated. Patients with childhood epilepsy with centrotemporal spikes and Panayiotopoulos syndrome often do not require treatment. The majority of patients with these epilepsy syndromes will have relatively few total lifetime events, most occur during sleep, and spontaneous remission is expected [48]. In these self-limited syndromes, indications for initiating therapy include: frequent seizures, young age (4 years or less), daytime seizures, generalized tonic-clonic seizures, a history of status epilepticus, or seizures causing significant distress/limiting quality of life for the patient or their family [48]. Treatment is also indicated if seizures are a likely component of an epileptic encephalopathy.
Febrile seizures are typically not treated with ASMs. Provoked seizures, such as those related to intoxication or trauma, are typically not treated [11]. Prophylactic treatment in the setting of concomitant intraparenchymal hemorrhage or subdural hemorrhage in high-risk populations remains controversial, given that the evidence to support this practice is mixed [49, 50].
Dietary Treatment
Dietary therapy is one of the most effective treatments available in the field of pediatric epilepsy. Consensus guidelines recommend offering dietary therapy to patients who have failed to respond to two appropriately selected and dosed anticonvulsants [51]. Dietary therapy is widely used in the treatment of both focal and generalized epilepsies. Therapeutic diets can also be effective in treating epileptic encephalopathies, such as infantile spasms [51,52,53,54,55]. The ketogenic diet is highly effective for patients across age groups and a range of seizure types and etiologies [56]. In a meta-analysis of three randomized controlled trials of the ketogenic diet, 52% of the participants in the intervention group showed a seizure frequency reduction of ≥50% [57].
The classic ketogenic diet is a high-fat, low-carbohydrate diet. With this treatment, patients are followed by a dietician who works with their neurologist. The ketogenic diet simulates a fasting state, and metabolism shifts from glycolysis toward fatty acid oxidation. All meals maintain a macronutrient ratio of 4:1 or 3:1 of fats to combined carbohydrates and protein. Meal preparation requires special attention to the above ratios, micronutrients, and fluid status. Maintaining the appropriate nutrient ratios often requires the use of a gram scale. Formula preparations are also commercially available.
Historically, the ketogenic diet has been difficult to tolerate for some patients. However, in recent years, the availability of compatible foods and recipe books created by patients and families has made this approach more palatable. Although data supporting the classic ketogenic diet is most robust, there are also other less restrictive diets that may be equally as effective. The low glycemic index treatment and the modified Atkins diet have been developed as liberalized versions of the classic ketogenic diet [58, 59]. These less restrictive diets allow more freedom in food choice, and less precision is required with meal preparation compared to the classic diet.
There are some seizure and metabolic disorders in which ketogenic diet is considered a primary therapy. Patients with pyruvate dehydrogenase deficiency may receive significant benefit in general neurologic outcomes and longevity with the ketogenic diet, as this therapy allows for a bypass of the metabolic defect and provides ketones as an alternative form of fuel to glucose [60]. Ketogenic diet is considered first-line therapy in patients with GLUT-1 glucose transporter deficiency, as it provides ketone bodies to the CNS, rather than glucose which cannot enter neurons [61]. The following populations may also be particularly responsive to the ketogenic diet: Angelman syndrome, complex 1 mitochondrial disorders, Dravet syndrome, myoclonic atonic epilepsy, febrile infection-related epilepsy syndrome, Ohtahara syndrome, and tuberous sclerosis complex [51].
Dietary therapy is contraindicated in patients with certain inborn errors of metabolism, including disorders of fatty acid mitochondrial transport, β-oxidation, and other mitochondrial cytopathies [56]. Gastrointestinal side effects are the most common in ketogenic diet therapy and typically occur during the initial first few weeks of treatment [51]. Additional side effects may include decreased linear growth, transient hypertriglyceridemia, metabolic acidosis, and nephrolithiasis [51]. Concomitant treatment with oral citrate salts may help prevent renal stones [62]. There have been rare cases of increased liver transaminases, hepatosteatosis, and cholelithiasis during the first year of treatment with the ketogenic diet [63]. Other rare but reported adverse effects with dietary therapy include cardiomyopathy, prolonged QTc interval, as well as pancreatitis [56].
The duration of epilepsy diet treatment is variable. Reasons to discontinue treatment include: lack of efficacy, poor compliance, and/or intolerable side effects [64]. If the patient has not demonstrated a positive treatment response, treatment is typically discontinued after 3–6 months. If a significant treatment response is seen (>50% decrease in seizure frequency), clinicians may consider discontinuing therapy after 2 years [51]. In one study, 80% of patients who became seizure free on the ketogenic diet remained so after treatment was discontinued [65]. Dietary therapy can be abruptly stopped in an emergency, but often the diet is weaned with gradual reintroduction of carbohydrates over a period of 1–3 months [51].
Epilepsy Surgery
For patients with drug-resistant epilepsy, epilepsy surgery should be considered. In recent years, there have been increased efforts to prevent the delay of surgical evaluation for pediatric patients with drug-resistant epilepsy [66]. Primary pathologies in pediatric epilepsy for which surgical intervention may be targeted include the following: cortical dysplasia, developmental brain tumors such as dysembryoplastic neuroepithelial tumor, ganglioglioma, perinatal injuries including stroke, hippocampal sclerosis, gliosis of any cause, tuberous sclerosis, hypothalamic hamartoma, Rasmussen’s encephalitis, and vascular malformations [66].
For lesional surgery, patients undergo an extensive workup to evaluate their candidacy for surgical resection. With this evaluation, teams aim to lateralize and localize an epileptic focus as well as determine the cortical function of the region at and surrounding it [67]. Phase 1 of the presurgical evaluation is largely noninvasive and may include some combination of the following: ictal and interictal EEG monitoring, magnetoencephalography (MEG), MRI/MRS, functional MR imaging, interictal positron emission tomography (PET), ictal/interictal single-photon emission computed tomography (SPECT), neuropsychological assessment, or intracarotid sodium amobarbital testing (Wada test). Phase 2 may include invasive monitoring with intracranial (subdural) or intracerebral electrodes, stereo-electroencephalography, electrocorticography, or cortical stimulation [67].
Lesional surgery is typically categorized as temporal and extra-temporal. Anterior temporal, selective mesial resection (amygdalohippocampectomy), and neocortical (extended) lesionectomy are the primary temporal resection strategies [68]. The most common primary temporal lesions are tumors, cortical dysplasia, and hippocampal sclerosis. Rates of seizure freedom following temporal resection range from 58% to 85% [68]. Surgical resection of extra-temporal lesions is often more challenging, given that the epileptic focus can be more difficult to localize and there is higher risk of damaging areas of eloquent cortex adjacent to the lesion [69]. Surgical planning may therefore involve more extensive invasive monitoring in extratemporal locations [69]. Typically, surgical outcomes are more guarded in extratemporal resection, but can still be quite successful. In a meta-analysis of 37 studies of pediatric extra-temporal epilepsy surgery, 56% of patients were seizure free post-operatively [69, 70].
Functional hemispherectomy is a surgical technique that has evolved from early iterations that involved complete resection of a hemisphere to modern disconnection approaches [71]. Surgical hemispheric disconnection is considered for patients with catastrophic hemispheric epilepsy most frequently in the setting of Rasmussen syndrome. It may also be employed for patients with hemiconvulsion-hemiplegia-epilepsy syndrome, Sturge-Weber syndrome, hemimegalencephaly, multilobar cortical dysplasia, perinatal infarction, or porencephalic cysts [71].
Corpus callosotomy is a surgical disconnection technique that aims to block the spread of epileptic discharges between the hemispheres [72]. This surgical procedure may be staged, with an initial disconnection of the anterior corpus callosum, preserving the connection at the splenium. If seizures persist, the patient may return to the operating room for complete disconnection. In some cases, it may be undertaken as a single-stage complete callosotomy [72]. This procedure is considered for patients with severe refractory epilepsy for whom lesion resection is not possible, due to bilateral foci or primary generalized seizures. Corpus callosotomy is most frequently used to target a reduction in atonic seizures, which can result in severe injury. Patients with atonic seizures related to Lennox-Gastaut syndrome are often considered for corpus callosotomy [72].
Patients with an epileptic focus in eloquent cortex may not be candidates for lesion resection but might be considered for surgical intervention with multiple subpial transection of the lesion. This surgical technique involves multiple vertical incisions made perpendicular to the surface of the cortex, resulting in the interruption of the short horizontal fiber intracortical connections [73]. The goal is to disrupt the synchronization and spread of epileptogenic discharges while preserving baseline cortical function [73]. Populations who may benefit from this technique include patients with Landau-Kleffner syndrome, cortical dysplasia, epilepsia partialis continua, and Rasmussen’s encephalitis [73].
A number of surgical techniques that have recently emerged may be less invasive than open resection. One example is stereotactic MRI-guided laser thermal ablation of the epileptogenic lesion [74, 75]. There are currently two FDA-approved systems, Visualase (Medtronic, Minneapolis, MN, USA), and NeuroBlate (Monteris Medical, Plymouth, NM, USA) [75]. In this technique, a laser probe is introduced through a burr hole in the skull and advanced with imaging guidance to the epileptic focus. Thermal ablation of the lesion is monitored in real time with MR-thermography [76]. One of the benefits is the ability to target deep lesions. There are few reports of the use of this technique in the pediatric epilepsy population, but the adult literature has shown some encouraging results [76]. Possible surgical targets for laser interstitial thermal therapy include hypothalamic hamartomas, cortical dysplasias (including insular lesions), corpora callosa, and periventricular heterotopias [76].
Finally, radiosurgical treatment for pediatric epilepsy is emerging as a viable treatment option. With the Gamma Knife technique, multiple stereotactic ionizing beams of radiation are targeted to the epileptic focus while limiting radiation exposure to the surrounding tissue [77]. This technique may be used for patients with mesial temporal lobe epilepsy with high risk for memory loss or who have previously undergone open surgery and had incomplete seizure control with prior resective surgery [77]. Radiosurgical intervention is also employed to treat hypothalamic hamartomas and cavernous malformations, and to perform callosotomies [77].
Neurostimulation
For patients with drug-resistant epilepsy who are not candidates for surgical resection, neuromodulation may be considered. The primary technology used in pediatric epilepsy is the vagus nerve stimulator (VNS). It is FDA-approved for patients 4 years and older for focal seizures, but it is commonly used for multiple seizure types and drug-resistant epilepsy syndromes [78, 79]. With VNS, a device is surgically placed subcutaneously in the left clavicular or lateral thoracic region similar to a cardiac pacemaker. An electrode is wrapped around the left vagus nerve and directs electrical impulses afferently into the brain. Efferent stimulation along the vagus nerve is blocked to prevent disruption of normal autonomic function. Stimulation is cycled at scheduled frequencies and can be programmed by clinicians using an external device. In the setting of aura or a seizure event, a magnet can be swiped over the device by the patient or caregiver to trigger a longer pulse stimulus to potentially abort or truncate the seizure [79]. Newer models (since July 2015) have an auto-detect feature that triggers a pulse of stimulation if there is an abrupt increase in heart rate, considered a proxy for seizure activity [78]. A meta-analysis of 74 clinical studies across age groups demonstrated 50% of patients had a greater than 50% reduction in seizures [79, 80]. Early side effects include hoarseness and cough, but these frequently improve with time [78]. There are low rates of surgical adverse events, but there is a risk of possible vocal cord paralysis related to the implantation procedure [78].
A second type of neuromodulation for refractory epilepsy is responsive neurostimulation. Though not currently approved for patients under 18 years, responsive neurostimulation is a technique that involves surgical implantation of a stimulator in the skull that is connected to up to two subdural or depth electrodes placed at the seizure focus [78]. The device collects continuous EEG data and provides cortical stimulation when abnormal electrical activity is detected prior to a seizure. NeuroPace, Inc. (Mountain View, CA, USA) was the first system to become FDA-approved [75, 79]. In a randomized control trial of adult patients, 59% of subjects responded to treatment at the 6-year mark, with a median reduction in seizures of 66% [79, 81]. Current use in pediatrics is off-label in specialized epilepsy centers [79].
Deep brain stimulation (DBS) is an additional neuromodulatory technique that shows some promising results but is not currently approved for use in pediatric patients [79]. Similar to the use of this technique in movement disorders, a generator is placed subcutaneously superficial to the pectoral muscles, and depth electrodes are placed in the brain. Options for electrode placement include the anterior or centromedian nucleus of the thalamus, hippocampus, globus pallidus, cerebellum, or the site of suspected seizure onset [78, 79, 82]. The exact mechanism by which DBS exerts its antiseizure effect is unknown. It has been proposed that DBS may disrupt the neural networks involved in seizure propagation [79, 83]. Pediatric efficacy data are limited, but one series of 13 pediatric subjects with electrodes in the centromedian nucleus of the thalamus demonstrated greater than 50% reduction in seizures in 92% of patients at 18 months [84]. In a systematic review of DBS treatment in 40 patients with drug-resistant epilepsy, 13% became seizure free and 85% showed reduction in seizure frequency [23, 85].
Status Epilepticus
Convulsive status epilepticus is the most common pediatric neurologic emergency [86]. Though the duration of seizure activity qualifying as status epilepticus has fluctuated over the years, the current consensus from the 2012 Neurocritical Care Society’s Guideline on the Evaluation and Management of Status Epilepticus and the 2016 American Epilepsy Society’s Guideline for Status Epilepticus Management defines status epilepticus as 5 minutes of continuous clinical or electrographic seizure activity [87,88,89]. Refractory status epilepticus is defined as persistent seizure activity following an initial abortive treatment (benzodiazepine) and an appropriate second antiseizure medication [88].
Treatment algorithms aim to stop both clinical and electrographic seizures within 60 minutes of presentation [88]. However, treatment should be initiated after 5 minutes of seizure activity, whether in the community or in the emergency department. Timing is critical. Chin et al. found for every minute that passed between the onset of status epilepticus and initiation of therapy, there was a 5% cumulative increase in the risk of developing refractory status epilepticus [86, 90]. Lewena et al. found that the first one or two medications administered were effective for 86% of cases when given within 20 minutes of seizure onset but were effective for only 15% of cases when seizure duration exceeded 30 minutes [86, 91]. The time from status onset to first, second, and third ASM dosing is positively correlated with the duration of refractory status epilepticus [92, 93]. One proposed mechanism for this phenomenon is that with prolonged status epilepticus, inhibitory GABA receptors on the neuronal cell membrane are internalized, making benzodiazepines less effective [86, 94, 95].
Etiology of Pediatric Status Epilepticus
Etiology is critical when approaching the question of treatment. Status epilepticus is classified into 5 categories: prolonged febrile convulsion, acute symptomatic (CNS infection, head injury, acute vascular accident), remote symptomatic (previous neurological abnormality), idiopathic epilepsy-related (known idiopathic epilepsy, or when the presentation allows a diagnosis of idiopathic epilepsy to be made), progressive encephalopathy (epilepsy secondary to a neurodegenerative process), and unclassified [96]. In a multicenter study of infants in the United Kingdom presenting in refractory convulsive status epilepticus, acute symptomatic causes were the most common (28.5%); in patients 1–5 years of age, prolonged febrile convulsions were the most common (33.8%); in patients over 5 years of age, remote symptomatic status was the most common (36–40%) [96]. A multicenter study in Australia and New Zealand reported that 67% of patients presenting in status had a history of prior seizure, and 35% carried a diagnosis of epilepsy. In this same study, 3% were found to have meningitis/encephalitis, and 1% had an electrolyte disturbance or hypoglycemia [97].
Status Epilepticus Treatment Pathway
Stabilization of the airway, breathing, and circulation, emergent diagnostic, and emergent treatment for status epilepticus should occur in tandem [89]. Patients should be assessed for airway protection and adequacy of oxygenation, ventilation, and perfusion. Peripheral intravenous access should be established. Finger-stick glucose should be checked, and the following STAT labs should be sent: complete metabolic panel, complete blood count with differential, PT/PTT, blood gas, toxicology screen, and anticonvulsant levels [88, 89]. Fig. 7.2 outlines a status epilepticus treatment reference [86,87,88,89, 98,99,100,101].

The American Academy of Neurology reported the following clinical data for pediatric patients in status epilepticus: low antiseizure medication levels (32%), neuroimaging abnormalities (8%), electrolyte disturbances (6%), inborn errors of metabolism (4%), ingestion (4%), central nervous system infections (13%), and positive blood cultures (3%) [89, 98].
First-Line Treatment
Status epilepticus treatment is classified as (1) emergent, (2) urgent, and (3) refractory [86]. Emergent antiseizure treatment should start with a benzodiazepine.
If the patient has IV/IO access
Lorazepam IV 0.1 mg/kg/dose (max 4 mg) or
Diazepam IV 0.15–0.2 mg/kg/dose (max 10 mg)
If the patient does not have IV access
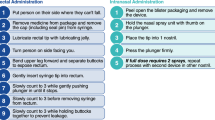
Midazolam IM 0.2 mg/kg/dose (MAX 10 mg) or
Midazolam intranasal 0.2 mg/kg/dose or
Midazolam buccal 0.5 mg/kg/doseor
Diazepam PR
2–5 years: 0.5 mg/kg
6–11 years: 0.3 mg/kg
≥ 12 years: 0.2 mg/kg (max 20 mg)
If the seizure persists for 5 minutes following the first treatment, a second benzodiazepine dose should be administered [89]. Lorazepam, diazepam, and midazolam have equivalent efficacy and risk of respiratory depression [87, 89]. Metabolic derangements, including hypoglycemia, hypocalcemia, hyponatremia, and hypomagnesemia should be treated expeditiously [89].
Second-Line Treatment
In a series of over 500 pediatric cases, 70% required additional treatment beyond benzodiazepines to control convulsive status epilepticus [89, 97]. If the seizure persists, urgent treatment with an intravenous loading dose of one of the following four agents is indicated: fosphenytoin, levetiracetam, phenobarbital, or valproate. Efficacy data are limited to guide rank order of these options. The Established Status Epilepticus Treatment Trial (ESETT), a randomized, blinded, adaptive trial, showed non-superiority between fosphenytoin, valproate, and levetiracetam for convulsive status epilepticus [102]. In this trial, these medications did not differ significantly with regard to safety [102]. Further analysis of the data showed no difference in the efficacy of the three options by age group, including a pediatric population (<18y) [103]. The American Epilepsy Society’s guidelines, when comparing valproate and phenobarbital, describe that valproate has equivalent efficacy but better tolerability [87, 88]. A meta-analysis of the four medications found phenytoin had lower efficacy (50%) when compared to levetiracetam (69%), phenobarbital (74%), and valproate (76%) [89, 104]. Lacosamide is an alternative agent that may be used for acute status management, although data in children are limited. For patients with epilepsy on standing ASM treatment, giving an IV loading dose of one of the patient’s medications may be prudent. Beyond this, clinicians should consider the medication side-effect profiles in the context of the patient’s current clinical circumstances and hemodynamics to help guide second-line agent selection. Valproate should be avoided in the setting of hepatic dysfunction, metabolic or mitochondrial disease, pancreatitis, or thrombocytopenia, or in patients less than 2 years of age with status epilepticus of unknown etiology [88, 89]. Given the risk for cardiac dysfunction, fosphenytoin should be avoided if a patient is in a low cardiac output state or at increased risk for arrhythmias. Similarly, particularly with rapid infusion, phenobarbital may precipitate hypotension, respiratory depression, or apnea and has the potential for prolonged sedative effect.
Urgent Workup
For patients with a history of trauma or malignancy, or in cases when the etiology of status is unknown, neuroimaging should be performed urgently [89]. Glucose and electrolytes should be evaluated immediately. Further laboratory workup to evaluate the cause of status may include lumbar puncture, urine toxicology screen, and a screen for inborn errors of metabolism. If infectious source is on the differential, CSF should be sent for cell counts, gram stain, culture, protein, glucose, and herpes simplex virus PCR, followed by the initiation of empiric coverage with antibiotics and acyclovir [89].
EEG Monitoring
EEG monitoring should be initiated within 15–60 minutes in cases where there is concern for ongoing seizure activity or the patient is not returning to baseline, raising concern for nonconvulsive status epilepticus [88, 89]. Long-term EEG monitoring may be indicated for 24–48 hours following presentation in status epilepticus depending on the clinical course.
Refractory Status Epilepticus Treatment
Refractory status epilepticus occurs in 10–40% of children with status epilepticus [89]. In the ESETT trial, approximately half of patients showed seizure cessation following initial loading dose of a second-line agent [102]. For persistent seizure activity, the Neurocritical Care Guidelines indicate that clinicians may consider giving a repeat loading dose of the selected second-line treatment or may move on to load with an alternative agent (phenobarbital, fosphenytoin, valproic acid, levetiracetam) [88, 89].
For persistent seizure activity following repeat loading doses, treatment transitions to the initiation of continuous infusion of anesthetic agents. At this point, patients should have a secured airway and be in a critical care unit with EEG in place. In a multicenter study of pediatric refractory status, midazolam (78%) was the first anesthetic agent used with an initial loading dose of 0.1–0.2 mg/kg and a continuous infusion rate of 0.1–0.2 mg/kg/hr [101]. EEG should be actively followed with possible repeat bolus doses of 0.1 mg/kg and titration of the midazolam infusion every 5–30 minutes by 0.05–0.1 mg/kg/hr, to a goal of seizure suppression. In the event that midazolam fails to achieve seizure control at a dose of 1 mg/kg/hour, a transition to pentobarbital should be considered. Pentobarbital treatment can be initiated with a loading dose of 2–5 mg/kg followed by a continuous infusion at a rate of 0.5–1 mg/kg/hour. The initial bolus may be repeated in the absence of seizure suppression. Titrate to seizure suppression on EEG, re-bolusing with 1–2 mg/kg and increasing the continuous infusion by 0.5 mg/kg/hr to a max rate of 5 mg/kg/hr. Alternative agents to consider include propofol and ketamine. Currently, there is no consensus standard of care for the rate of anesthetic titration and/or max dosing for pediatric refractory status epilepticus care. The above recommendations represent expert opinion.
The Neurocritical Care Society guidelines recommend 24–48 hours of electrographic seizure control prior to initiation of a wean of continuous infusion treatments for refractory status [88, 89]. Broader workup for refractory status epilepticus may include the following: antibodies or PCR for viral encephalitides, autoantibody testing, or testing for inborn errors of metabolism [89, 105]. EEG monitoring is indicated throughout anesthetic treatment and for at least 24 hours after treatment is weaned [89, 106]
Conclusions
Pediatric epilepsy treatment requires evaluation and consideration of multiple variables and longitudinal care. Our collective goal for each child should be seizure freedom. Becoming comfortable with treatment options and effectively optimizing the regimen in an ever-changing research and clinical landscape is a true life’s work. Unfortunately, head-to-head efficacy trials are limited, but there is a developing literature of syndrome-specific treatment and genotype-phenotype correlations to further guide treatment choice.
Dietary treatments have been shown to be as effective as any known pharmaceutical option and should be readily included among pediatric epilepsy treatment options. Our understanding of the therapeutic mechanisms and physiologic impact of dietary therapy continues to expand and will inform broader access and application. Evaluation for epilepsy surgery should be pursued in cases of refractory epilepsy without delay. Surgical techniques, specifically options for minimal and noninvasive intervention, continue to evolve offering the promise of improved seizure control and preserved function of surrounding eloquent cortex.
The further development of treatment pathways for status epilepticus, focusing on the emergent nature of the disease and the necessity for rapid event capture, aims to improve outcomes and prevent lasting harm.
This work focusing on eliminating seizures for children, at the bench and at the bedside, holds immeasurable promise for the future for patients and families, in the way of improved neurodevelopmental outcomes, systemic health and wellness, mitigating the risk of SUDEP, and improved quality of life.
Appendix: Antiseizure Medications
Acetazolamide
Section References: [1, 3, 107]
Acetazolamide is a broad-spectrum agent typically used for absence and focal seizures. It is also used in the treatment of GTCs, tonic/atonic, and myoclonic seizures. It may be used for catamenial epilepsy, Lennox-Gastaut syndrome (LGS), Landau-Kleffner/CSWS, and juvenile myoclonic epilepsy (JME).
Mechanism of Action
Inhibitor of brain carbonic anhydrase. This leads to increase in intracellular CO2 and decrease in intracellular pH, which results in depression of neuronal activity.
Dosing
< 12 years:
Start treatment with 3–6 mg/kg/day given in 1–2 doses
Increase by 3–6 mg/kg/day every 3–7 days
Goal of 10–20 mg/kg/day
≥ 12 years:
Start 250 mg/day given in 1–2 doses
Increase by 250 mg/day every 3–7 days
Goal of 250–1000 mg/day
Side Effects
Neurologic: somnolence/lethargy, blurred vision, paresthesia
Systemic: GI symptoms, metabolic acidosis, anorexia
Critical/Potentially Life-Threatening Side Effects
Systemic: Stevens-Johnson syndrome (SJS), toxic epidermal necrolysis (TEN), hepatotoxicity, renal failure, metabolic acidosis, nephrolithiasis, agranulocytosis, aplastic anemia
Lab Monitoring
May evaluate baseline and periodic sodium bicarbonate, but this is not routine practice.
Adrenocorticotropic Hormone (ACTH)
Section References: [1, 3, 8, 108, 109]
ACTH is typically used to treat infantile spasms (IS). It may also be used for severe epileptic encephalopathies including Landau-Kleffner syndrome, CSWS, LGS, Doose syndrome, Dravet syndrome, and Ohtahara syndrome. This medication must be administered intramuscularly.
Mechanism of Action
Stimulates cortisol release and inhibits cortisol-releasing hormone which has been reported as a proconvulsive neuropeptide.
Dosing
< 2 years:
Start 150 units/m2/day IM divided in 2 doses
Treatment course is typically a minimum of 2 weeks followed by a 2-week taper
Side Effects
Neurologic: irritability, insomnia
Systemic: hypertension, weight gain, hyperglycemia
Critical/Potentially Life-Threatening Side Effects
Systemic: myocardial dysfunction, immunosuppression, Pneumocystis pneumonia
Clinical/Lab Monitoring
Prior to treatment
Vital signs/blood pressure
UA, stool guaiac, CBC, Chem10 (electrolytes, BUN/Cr, Ca, Mg, phosphate)
Consider baseline echocardiogram
Throughout treatment
Blood pressure daily x1 week, then 3x weekly, urine glucose and stool guaiac 2x weekly
After 2–4 weeks: CBC with differential, Chem10
Consider follow-up echocardiogram
Avoid vaccines for 10 days prior to/during treatment. Treatment should include concomitant H2 antagonist for gut protection.
Brivaracetam
Section References: [1,2,3, 24, 110]
Brivaracetam is a broad-spectrum agent. The majority of data has been gathered in the treatment of focal seizures, but it has also shown promising results in the treatment of generalized seizures in adults [110].
Mechanism of Action
The mechanism is incompletely understood. It binds synaptic vesicle protein SV2A, involved in synaptic vesicle exocytosis. It is also a partial antagonist of sodium channels.
Dosing
<11 kg:
Start 1.5–3 mg/kg/day divided in 2 doses
Max 6 mg/kg/day
11–19 kg:
Start 1–2.5 mg/kg/day divided in 2 doses
Max 5 mg/kg/day
20–49 kg:
Start 1–2 mg/kg/day divided in 2 doses
Max 4 mg/kg/day
≥ 50 kg or ≥ 16 years:
Start 50–100 mg/day divided in 2 doses
Max 200 mg/day
Adding to levetiracetam does not likely provide additional benefit and may cause decreased efficacy.
Side Effects
-
Neurologic: somnolence/fatigue, irritability, ataxia/dizziness
-
General: GI symptoms
Critical/Potentially Life-Threatening Side Effects
-
Systemic: Hypersensitivity reaction
Lab Monitoring
-
Not required
Cannabidiol
Section References: [2,3,4, 20, 23, 111,112,113]
Cannabidiol is a broad-spectrum agent with suggested efficacy against both focal and generalized events. Treatment may be considered for LGS, Dravet syndrome, and tuberous sclerosis complex.
Mechanism of Action
The mechanism of action is incompletely understood. The antiseizure effect is likely not mediated by cannabinoid receptors [2, 111]. A potential mechanism has been proposed in which cannabidiol results in decreased intracellular calcium (via targets GPR55 and TRPV1), thereby reducing neuroexcitability. There may also be a therapeutic effect through modulation of adenosine-mediated signaling [4].
Dosing
-
Start 5 mg/kg/day divided in 2 doses
-
Increase by 5 mg/kg/day every 7 days
-
Goal 10–25 mg/kg/day
Side Effects
-
Neurologic: somnolence, irritability, insomnia
-
Systemic: GI symptoms, transaminitis, anorexia/weight loss
Critical/Potentially Life-Threatening Side Effects
-
Systemic: Hypersensitivity reaction
Lab Monitoring
Consider sending liver function tests (LFTs) at baseline, 1, 3, 6 months and then periodically; repeat labs within 1 month of dose change.
Carbamazepine
Section References: [1,2,3, 8, 11, 20, 114]
Carbamazepine is used to treat both focal seizures and primary GTCs. In some cases, it may cause seizure worsening for patients with idiopathic generalized epilepsies and/or epileptic encephalopathies. Carbamazepine should not be used to treat myoclonic, atonic, or absence seizures, as it may cause seizure exacerbation.
Mechanism of Action
Inhibits voltage-gated sodium channels, L-type calcium channels, and glutamate release.
Dosing
-
< 6 years:
-
Start 2–10 mg/kg/day divided in 2–3 (tab) or 4 (suspension) doses
-
Increase by 5–10 mg/kg/day every 7 days
-
Goal of 10–30 mg/kg/day
-
6–12 years:
-
Start 100 mg/day divided in 2 doses (tab) or 4 doses (suspension)
-
Increase by 100 mg/day every 7 days
-
Goal 600–1000 mg/day
-
> 12 years:
-
Start 200 mg/day divided in 2 doses (tab) or 4 doses (suspension)
-
Increase by 200 mg/day every 7 days
-
Goal of 800–1200 mg/day
Side Effects
-
Neurologic: somnolence, diplopia/blurred vision, ataxia/dizziness, headache
-
Systemic: GI symptoms, hyponatremia, leukopenia, rash, pruritis
Critical/Potentially Life-Threatening Side Effects
-
Neurologic: psychosis/mania
-
Systemic: SJS/TEN/hypersensitivity reaction, cardiac conduction disturbance, hyponatremia, bone marrow suppression, hypogammaglobulinemia
Lab Monitoring
Consider baseline CBC, Na, LFTs. Consider screening for HLA-B*1502 allele for patients with Asian ancestry due to potential increased risk for SJS/TEN. Repeat Na once treatment is therapeutic. Repeat Na and/or Chem10 periodically; LFTs and thyroid function tests as needed. Repeat CBC at 1–3 months after the start of treatment and every 6 months thereafter. Monitor for vitamin D deficiency with 25-hydroxyvitamin D levels.
Cenobamate
Cenobamate is typically used for the treatment of focal seizures.
Mechanism of Action
A tetrazole alkyl carbamate derivative, it acts as an inhibitor of voltage-gated sodium channels and potentially also enhances GABA inhibition.
Dosing
-
Adults:
-
Start 12.5 mg/daily
-
Week 3–4: 25 mg/daily
-
Week 5–6: 50 mg/daily
-
Week 7–8: 100 mg/daily
-
Week 9–10: 150 mg/daily
-
Week 11-ongoing: 200 mg/day
Side Effects
-
Neurologic: somnolence/fatigue, dizziness, headache, diplopia
Critical/Potentially Life-Threatening Side Effects
-
Systemic: decreased QT interval, hypersensitivity reaction
Lab Monitoring
Defer routine monitoring. Consider monitoring LFTs and potassium as clinically indicated.
Clobazam
Section References: [1,2,3, 11, 20, 41, 116]
Clobazam is a broad-spectrum agent. It has a therapeutic role as monotherapy as well as adjuvant therapy. It is notably effective in the treatment of Dravet syndrome and LGS. For Dravet, clobazam can be paired with stiripentol for synergistic effect. Tolerance, dependence, and/or excessive sedation are not typically clinical limitations.
Mechanism of Action
Clobazam is a 1–5 benzodiazepine, with a nitrogen at the 1 and 5 positions on the diazepine ring. This differs from other benzodiazepines used in epilepsy treatment which host nitrogen at the 1 and 4 positions. Clobazam is a GABAA agonist resulting in increased chloride ion permeability and neuronal inhibition. At high concentrations, benzodiazepines also influence sodium channel function.
Dosing
-
< 12 years:
-
Start 0.1–0.2 mg/kg/day daily at night
-
Increase by 0.1–0.25 mg/kg/day every 7 days
-
Goal of 0.8–2 mg/kg/day divided in 2 doses
-
≥12 years:
-
Start 5–10 mg/day at night
-
Increase by 5 mg/day every 7 days
-
Goal 40 mg/day given in 1–2 doses (max 80 mg/day)
Side Effects
-
Neurologic: somnolence, irritability/aggressiveness, ataxia, insomnia
-
Systemic: GI symptoms
Critical/Potentially Life-Threatening Side Effects
-
Systemic: SJS/TEN, respiratory depression
Lab Monitoring
Defer routine monitoring. May consider periodic monitoring of CBC and LFTs. Levels should be monitored when used with cannabidiol.
Clonazepam
Section References: [1,2,3, 11, 20, 117]
Clonazepam is a broad-spectrum agent used to treat both focal and generalized seizures. Efficacy against myoclonic and atonic seizures is most notable. It has a wide range of uses, particularly for epileptic encephalopathies, and is often employed as adjunctive therapy. It may also be used as an acute abortive treatment for seizure clusters.
Mechanism of Action
Clobazam is a type of benzodiazepine, a GABAA agonist resulting in increased chloride ion permeability and neuronal inhibition. At high concentrations, benzodiazepines also influence sodium channel function.
Dosing
-
≤ 12 years:
-
Start 0.01–0.02 mg/kg/day divided in 2–3 doses (max start 0.5 mg)
-
Increase by 0.05 mg/kg/day or ≤ 0.25 mg/day every 3–7 days
-
Goal of 0.1–0.2 mg/kg/day
-
> 12 years:
-
Start 0.25 mg at night
-
Increase by 0.25 mg/day every 7 days.
With titration, divide in 2–3 doses/day
-
Goal of 4–10 mg/day or 0.05–0.2 mg/kg/day
Side Effects
-
Neurologic: somnolence/fatigue, confusion, hyperexcitability, behavior changes, ataxia/impaired coordination
Critical/Potentially Life-Threatening Side Effects
-
Systemic: respiratory depression
Lab Monitoring
Defer routine monitoring. May consider periodic monitoring of CBC and LFTs.
Diazepam
Section References: [1, 3, 20, 118]
Diazepam is typically used as an acute abortive therapy for status epilepticus or seizure clusters.
Mechanism of Action
Diazepam is a type of benzodiazepine. GABAA agonist resulting in increased chloride ion permeability and neuronal inhibition. At high concentrations, benzodiazepines also influence sodium channel function.
Dosing
-
Abortive treatment options
-
PR dosing:
-
2–5 years: 0.5 mg/kg
-
6–11 years: 0.3 mg/kg
-
≥ 12 years: 0.2 mg/kg (max 20 mg)
-
-
Intranasal dosing:
-
6–11 years
-
10–18 kg: 5 mg in 1 nostril
-
19–37 kg: 10 mg in 1 nostril
-
38–55 kg: 7.5 mg x2, 1 spray in each nostril
-
56–74 kg: 10 mg x2, 1 spray in each nostril
-
-
≥ 12 years
-
14–27 kg: 5 mg, 1 nostril
-
28–50 kg: 10 mg, 1 nostril
-
51–75 kg: 7.5 mg x2, 1 spray in each nostril
-
≥ 76 kg: 10 mg x2, 1 spray in each nostril
-
-
-
IV dosing:
-
> 1 month
-
0.15–0.2 mg/kg/dose (max 10 mg/dose)
-
Side Effects
-
Neurologic: somnolence/fatigue, confusion, ataxia/dizziness
Critical/Potentially Life-Threatening Side Effects
-
Systemic: hypotension, respiratory depression
Lab Monitoring
Defer routine monitoring. May consider periodic monitoring of CBC and LFTs.
Eslicarbazepine Acetate
Section References: [1,2,3, 20, 119,120,121]
Eslicarbazepine acetate is used to treat focal seizures with or without secondary generalization. Like carbamazepine, eslicarbazepine acetate is contraindicated for primary generalized seizures due to the potential for exacerbation.
Eslicarbazepine acetate shares a chemical structure with carbamazepine and oxcarbazepine, a dibenzapine nucleus with a 5-carboxamide substitute, but has an acetate group at the 10,11 position. Eslicarbazepine acetate and oxcarbazepine are pro-drugs, both metabolizing to the active licarbazepine. This metabolism results in both S and R chiral forms [120]. Both compounds are metabolically active, but the S form may cross the blood–brain barrier more effectively and potentially be less toxic. Oxcarbazepine metabolizes to chiral ratio S:R of 4:1, whereas eslicarbazepine acetate metabolizes to a ratio of 20:1 [120]. There is evidence for improved side-effect profile and stable seizure control when transitioning from oxcarbazepine to eslicarbazepine acetate [121].
Mechanism of Action
Inhibits voltage-gated sodium channels. May also impact glutamate release.
Dosing
-
11–21 kg:
-
Start 200 mg/day
-
Increase by 200 mg/day every 7 days
-
Goal 400–600 mg/day dosed daily
-
22–31 kg:
-
Start 300 mg/day
-
Increase by 300 mg/day every 7 days
-
Goal 500–800 mg/day
-
32–38 kg:
-
Start 300 mg/day
-
Increase by 300 mg/day every 7 days
-
Goal 600–900 mg/day
-
> 38 kg:
-
Start 400 mg/day
-
Increase by 400 mg/day every 7 days
-
Goal 800–1200 mg/day
-
≥ 12 years:
-
Start 400 mg daily
-
Increase by 400 mg/day every 2 weeks
-
Max dose 1200 mg/day
Side Effects
-
Neurologic: somnolence, impaired attention, diplopia/blurred vision, tremor, ataxia/dizziness, headache
-
Systemic: GI symptoms, abnormal liver function, hyponatremia
Critical/Potentially Life-Threatening Side Effects
-
Systemic: SJS/TEN, cardiac conduction disturbance (AV block, prolonged PR interval), hyponatremia
Lab Monitoring
Consider baseline CBC, sodium, LFTs. Consider screening for HLA-B*1502 allele for patients with Asian ancestry due to potential increased risk for SJS/TEN. Repeat sodium once treatment is therapeutic. Repeat Chem10 and LFTs to monitor for renal and hepatic function every 12 months. Consider monitoring 25-hydroxyvitamin D levels.
Ethosuximide
Section References: [1,2,3, 122]
Ethosuximide is principally used to treat absence seizures. In rare cases, it may also be used to treat atonic or myoclonic seizures. Ethosuximide is ineffective and contraindicated in the treatment of focal seizures and generalized tonic-clonic-seizures.
Mechanism of Action
Reduces the number or the conductance of low-threshold (T-type) calcium channels in thalamic neurons.
Dosing
-
2–12 years:
-
Start 5–10 mg/kg/day
-
Increase by 5–10 mg/kg/day every 7 days
-
Goal 20–40 mg/kg/day in 1–3 doses (max 60 mg/kg/day or 2000 mg/day)
-
> 12 years:
-
Start 250 mg/day
-
Increase by 250 mg/day every 7 days
-
Goal 750–1500 mg/day divided in 2–3 doses
Side Effects
-
Neurologic: somnolence, headache
-
Systemic: hiccups, GI symptoms, anorexia/weight loss
Critical/Potentially Life-Threatening Side Effects
-
Systemic: SJS/TEN, systemic lupus erythematosus, agranulocytosis/aplastic anemia
Lab Monitoring
Baseline CBC with repeat at 2 months and then as needed thereafter.
Felbamate
Section References: [1,2,3, 11, 20, 123]
Felbamate is a broad-spectrum agent used to treat both focal and generalized epilepsy. It is effective in the treatment of tonic/atonic, myoclonic, and absence seizures as well as infantile spasms. It is reserved for patients with LGS and/or severe refractory seizures. It is not currently first-line therapy for any indication. This is due to the side-effect profile which includes a black box warning for aplastic anemia and hepatotoxicity. Dosing can be challenging due to narrow therapeutic range and significant medication interactions. Given this, close laboratory monitoring follow-up with the prescribing team is essential for treatment.
Mechanism of Action
Inhibits N-methyl-D-aspartate (NMDA)-induced intracellular calcium currents and excitatory activity. It also potentiates GABA activity.
Dosing
-
2–14 years:
-
Start 15 mg/kg/day divided in 3–4 doses
-
Increase by 15 mg/kg/day at every 7 days
-
Goal 15–45 mg/kg/day
-
> 14 years:
-
Start 1200 mg/day divided in 3–4 doses
-
Increase by 600–1200 mg/day every 7 days
-
Goal 3600 mg/day
May consider 20% reduction in concomitant ASMs (phenytoin/valproate/phenobarbital/carbamazepine) with the start of felbamate to avoid toxicity. Provide slower medication titration if the patient is not taking an enzyme-inducing ASM prior to the start of felbamate.
Side Effects
-
Neurologic: irritability, ataxia/dizziness, headache, insomnia
-
Systemic: GI symptoms, anorexia/weight loss
Critical/Potentially Life-Threatening Side Effects
-
Systemic: hepatotoxicity, aplastic anemia
Lab Monitoring
Recommend baseline CBC and LFTs with repeat CBC every 2 weeks and repeat LFTs every 4 weeks.
Fenfluramine
Section References: [23, 25, 41, 124]
Fenfluramine is an amphetamine derivative that has been shown to be effective for patients with LGS and Dravet syndrome.
It was initially FDA approved for weight loss but was removed from the market in 1997 with safety concerns for the development of pulmonary hypertension and valvular heart disease. Over the past two decades there have been reports of potential efficacy against seizures with a novel serotonergic mechanism [23]. Fenfluramine was reapproved by the FDA in 2020 for treatment for patients with Dravet syndrome. In the 2019 trial, there were no reported cases of pulmonary hypertension or valvular disease in children or young adults [41, 124]. In the United States, there is a REMS program to facilitate prescriber education, routine cardiac monitoring, and reporting of these potential adverse events.
Mechanism of Action
The mechanism is unknown. Modulation of NMDA receptor-mediated excitation and/or serotonergic effects are two mechanisms that have been proposed.
Dosing
-
≥ 2 years:
-
Start 0.2 mg/kg/day divided in 2 doses
-
Increase by 0.2 mg/kg/day every 4–7 days
-
Goal of 0.7 mg/kg/day (max 13 mg)
Side Effects
-
Neurologic: somnolence/fatigue
-
Systemic: GI symptoms, anorexia/weight loss
Critical/Potentially Life-Threatening Side Effects
-
Neurologic: serotonin syndrome
-
Systemic: cardiac valvular disease, pulmonary hypertension
Clinical Monitoring
Through the REMS program, cardiac echo is recommended at baseline prior to the start of treatment, every 6 months during treatment, and then once 3–6 months after the completion of treatment.
Fosphenytoin
Section References: [1,2,3, 125]
Fosphenytoin is a water-soluble prodrug of phenytoin. It is an IV formulation primarily used as an abortive therapy for status epilepticus. It may also be used as a substitute for phenytoin when enteral access is limited; this is typically short term.
Both phenytoin and fosphenytoin are used for focal seizures with or without secondary generalization. They may also be effective against some generalized seizures including primary GTC or for patients with mixed semiology, including tonic seizures. Fosphenytoin has the potential to worsen a subset of generalized seizures, typically myoclonic and absence. Fosphenytoin should be avoided for patients with LGS and progressive myoclonic epilepsies.
Mechanism of Action
Blocks voltage-gated sodium channels and modulates sustained repetitive firing. It also inhibits calcium channels and calcium sequestration and inhibits calcium-calmodulin protein phosphorylation.
Dosing
-
Status epilepticus:
-
Loading dose: 20 mg PE/kg IV
-
-
Seizure prophylaxis:
-
Loading dose: 10–15 mg PE/kg IV
-
Maintenance dosing: 4–5 mg/kg/day divided in 1–4 doses
-
With acute IV infusion, there is risk for myocardial dysfunction and hypotension.
With long-term use, the side-effect profile follows that of phenytoin (see below).
Gabapentin
Section References: [1,2,3, 11, 20, 126]
Gabapentin is primarily used to treat focal seizures with or without secondary generalization. In some cases, it may worsen generalized seizures.
Gabapentin is a GABA analog and structurally similar to pregabalin. Its effect as an antiseizure treatment, however, does not seem to be mediated by modulation of GABA.
Mechanism of Action
Binds to voltage-gated presynaptic calcium channels which inhibit inward calcium currents and decrease neurotransmitter release.
Dosing
-
3–5 years:
-
Start 10–15 mg/kg/day divided in 3 doses
-
Increase by 8–10 mg/kg/day daily for 3 days
-
Goal 40–50 mg/kg/day
-
6–12 years:
-
Start 10–15 mg/kg/day divided in 3 doses
-
Increase by 10 mg/kg/day daily for 3 days
-
Goal 25–35 mg/kg/day
-
> 12 years:
-
Start 300 mg/day daily
-
Increase by 300 mg/day daily divided in 3 doses
-
Goal 900–1800 mg/day (max 3600 mg/day)
Side Effects
-
Neurologic: somnolence/fatigue, diplopia/blurred vision, nystagmus, tremor, ataxia/dizziness
-
Systemic: GI symptoms, increased appetite/weight gain
Critical/Potentially Life-Threatening Side Effects
-
Systemic: pancreatitis, leukopenia
Lab Monitoring
Consider baseline LFTs and Chem10 to evaluate liver and renal function. Baseline CBC with repeat after 3 and 6 months of treatment.
Lacosamide
Section References: [1,2,3, 20, 127,128,129]
Lacosamide is primarily used to treat focal seizures. In some cases, it may be trialed for the treatment of refractory generalized seizures, but efficacy data are limited in pediatrics [128, 129]. With parenteral administration, it may be used for status epilepticus or if enteral access is limited.
Mechanism of Action
Enhanced slow inactivation of sodium channels, leading to stabilization of hyperexcitable neuronal membranes and inhibition of repetitive firing. A second proposed mechanism suggests that lacosamide may bind collapsin response mediator protein-2 (CRMP-2), a possible contributor to epileptogenesis.
Dosing
-
11–29 kg:
-
Start 2 mg/kg/day divided in 2 doses
-
Increase by 2 mg/kg/day every 7 days
-
Goal 6–12 mg/kg/day
-
30–49 kg:
-
Start 2 mg/kg/day divided in 2 doses
-
Increase by 2 mg/kg/day every 7 days
-
Goal 4–8 mg/kg/day
-
≥ 16 years or ≥ 50 kg:
-
Start 100 mg/day divided in 2 doses
-
Increase by 100 mg/day every 7 days
-
Goal 200–400 mg/day
Side Effects
-
Neurologic: diplopia/blurred vision, ataxia/dizziness/impaired coordination, headache
-
Systemic: GI symptoms
Critical/Potentially Life-Threatening Side Effects
-
Systemic: prolonged PR interval
Lab Monitoring
May consider baseline LFTs and Chem10 to evaluate liver and renal function with repeat labs every 12 months. May alternatively defer routine lab monitoring. Recommend baseline ECG to evaluate PR interval and identify potential preexisting arrhythmias.
Lamotrigine
Section References: [1,2,3, 20, 38, 130,131,132]
Lamotrigine is a broad-spectrum agent used to treat both focal and generalized seizures, including primary GTCs, tonic/atonic, and absence seizures. For generalized epilepsy syndromes with myoclonic seizures, specifically JME, lamotrigine can have mixed results with a subset showing worsening myoclonic seizures [3, 130, 132]. For patients with Dravet syndrome or progressive myoclonic epilepsy, lamotrigine is typically avoided [1].
Mechanism of Action
Inhibition of voltage-gated sodium channels limiting repetitive firing and stabilizing the neuronal membrane. The mechanism is incompletely understood, but there is a decrease in the release of excitatory neurotransmitters including glutamate and aspartate.
Dosing
-
2–12 years:
-
Week 1–2: 0.4 mg/kg/day
-
Week 3–4: 0.8 mg/kg/day
-
Increase by 0.4–0.8 mg/kg/day every 7–14 days thereafter
-
Goal 2–8 mg/kg/day divided in 2 doses
-
Concomitant treatment with valproate
-
Week 1–2: 0.15 mg/kg/day
-
Week 3–4: 0.3 mg/kg/day
-
Increase by 0.15–0.3 mg/kg/day every 7–14 days thereafter
-
Goal 1–5 mg/kg/day divided in 2 doses
-
Concomitant treatment with enzyme inducing ASMs (carbamazepine, phenytoin, phenobarbital, primidone)
-
Week 1–2: 0.6 mg/kg/day
-
Week 2–4: 1.2 mg/kg/day
-
Increase by 0.6–1.2 mg/kg/day every 7–14 days thereafter
-
Goal 5–15 mg/kg/day divided in 2 doses
-
> 12 years:
-
Week 1–2: 25 mg/day
-
Week 3–4: 50 mg/day
-
Increase by 50–100 mg/day every 7–14 days thereafter
-
Goal 100–400 mg/day divided in 2 doses
-
Concomitant treatment with valproate
-
Week 1–2: 25 mg every other day
-
Week 3–4: 25 mg/day
-
Increase by 25–50 mg/day every 7–14 days thereafter
-
Goal 100–200 mg/day divided in 2 doses
-
Concomitant treatment with enzyme-inducing ASMs
-
Week 1–2: 50 mg/day
-
Week 3–4: 100 mg/day
-
Increase by 50–100 mg/day every 7–14 days thereafter
-
Goal 200–500 mg/day divided in 2 doses
Steps for cross titration of valproate to lamotrigine [38]
-
1.
Hold valproate dose constant and titrate lamotrigine up to 200 mg/day as above (concomitant treatment with VPA).
-
2.
Maintain lamotrigine dose constant, and taper valproate by decrements of 500 mg/day every 7 days until a dose of 500 mg/day is reached.
-
3.
Increase lamotrigine to 300 mg and simultaneously decrease valproate to 250 mg for one week.
-
4.
Stop the valproate and increase lamotrigine by 100 mg/day each week until goal dose is reached.
Side Effects
-
Neurologic: somnolence, anxiety, irritability, ataxia/dizziness, headache, insomnia
Systemic: GI symptoms, rash
Critical/Potentially Life-Threatening Side Effects
-
Systemic: SJS/TEN, cardiac conduction abnormalities, hepatotoxicity, hematological abnormalities, hypogammaglobulinemia
Lab Monitoring
May consider baseline CBC with repeat at 3 and 6 months, and every 12 months thereafter; LFTs at baseline with repeat every 12 months.
Levetiracetam
Section References: [1,2,3, 20, 133]
Levetiracetam is a broad-spectrum agent used to treat both focal and generalized seizure types. The IV formulation may also be used as abortive therapy in status epilepticus.
Mechanism of Action
The mechanism is incompletely understood. It binds synaptic vesicle protein SV2A, involved in synaptic vesicle exocytosis. It inhibits high-threshold (N-type) calcium channels and may indirectly modulate GABA and glycine activity.
Dosing
-
Status epilepticus
-
60 mg/kg IV loading dose
-
-
Seizure treatment
-
< 12 years:
-
Start 20 mg/kg/day divided in 2 doses
-
Increase by 10–20 mg/kg/day every 7–14 days
-
Goal 30–100 mg/kg/day
-
-
≥ 12 years:
-
Start 500–1000 mg/day divided in 2 doses
-
Increase by 500 mg/day every 7–14 days
-
Goal 2000–3000 mg/day
Side Effects
-
Neurologic: somnolence, anxiety, irritability/emotional lability, asthenia, dizziness/ataxia
Critical/Potentially Life-Threatening Side Effects
-
Neurologic: suicide ideation/psychosis
-
Systemic: SJS/TEN, hepatotoxicity, pancytopenia, hypogammaglobulinemia
Lab Monitoring
-
Not required
Lorazepam
Section References: [1,2,3, 134]
Lorazepam is a broad-spectrum agent typically used as an acute abortive therapy for status epilepticus or intermittent use for seizure clusters.
Mechanism of Action
Lorazepam is a type of benzodiazepine, a GABAA agonist resulting in increased chloride ion permeability and neuronal inhibition. At high concentrations, benzodiazepines also influence sodium channel function.
Dosing
-
Status epilepticus
-
0.1 mg/kg IV
-
Side Effects
-
Neurologic: somnolence, cognitive dysfunction, dysarthria, ataxia/dizziness
Critical/Potentially Life-Threatening Side Effects
-
Systemic: respiratory depression, hypotension
Midazolam
Section References: [1,2,3, 26]
Midazolam is a broad-spectrum agent typically used as an acute abortive therapy for status epilepticus or intermittent use of seizure clusters.
Mechanism of Action
Midazolam is a type of benzodiazepine. GABAA agonist resulting in increased chloride ion permeability and neuronal inhibition. At high concentrations, benzodiazepines also influence sodium channel function.
Dosing
-
Acute abortive treatment/status epilepticus
-
Intranasal dosing:
-
0.2 mg/kg once (max 10 mg/dose)
-
-
≥ 12 years:
-
5 mg to one nostril, after 10 min, may repeat dose to the other nostril if needed
-
IM dosing:
-
0.2 mg/kg/dose (max 10 mg/dose)
-
-
IV dosing:
-
0.1–0.2 mg/kg/dose
-
Side Effects
-
Neurologic: somnolence, cognitive dysfunction, dysarthria, ataxia/dizziness
Critical/Potentially Life-Threatening Side Effects
-
Systemic: hypotension, respiratory depression
Oxcarbazepine
Section References: [1,2,3, 20, 135]
Oxcarbazepine is used to treat focal seizures with or without secondary generalization. Like carbamazepine, oxcarbazepine is contraindicated for primary generalized seizures due to the potential for exacerbation.
Oxcarbazepine shares a chemical structure with carbamazepine and eslicarbazepine acetate, a dibenzapine nucleus with a 5-carboxamide substitute, but differs with a keto group at the 10 position. Oxcarbazepine is a pro-drug, metabolizing to a 10-monohydroxy derivative.
Mechanism of Action
The 10-monohydroxy metabolite acts to block voltage-gated sodium channels. This stabilizes the membrane and prevents repetitive neuronal firing. It also increases potassium conductance, modulates high-threshold calcium channels, and reduces the release of glutamate.
Dosing
-
2–16 years:
-
Start 8–10 mg/kg/day divided in 2 (< 5y) or 3 (≥ 5y) doses
-
Increase by 5–10 mg/kg/day every 7 days
-
Goal 30–40 mg/kg/day (max 60 mg/kg/day)
-
or
-
< 20 kg: 600–900 mg/day
-
20–29 kg: 900–1200 mg/day
-
30–39 kg: 900–1500 mg/day
-
40–59 kg: 1500–1800 mg/day
-
> 16 years:
-
Start 300 mg/day divided in 2 doses
-
Increased by 150 mg/day every 2 days
-
Goal 1200–2400 mg/day
Side Effects
-
Neurologic: somnolence/lethargy, diplopia, ataxia/dizziness, headache
-
Systemic: GI symptoms, hyponatremia, rash
Critical/Potentially Life-Threatening Side Effects
-
Systemic: SJS/TEN/hypersensitivity reaction, cardiac arrhythmia, hyponatremia, agranulocytosis, aplastic anemia, pancytopenia, thrombocytopenia
Lab Monitoring
Consider baseline CBC, Na, LFTs. Consider screening for HLA-B*1502 allele for patients with Asian ancestry due to potential increased risk for SJS/TEN. Repeat Na at 1–2 months or once at treatment is therapeutic, and periodically thereafter. Repeat CBC at 3 and 6 months after the start of treatment. Consider monitoring 25–hydroxyvitamin D levels.
Perampanel
Section References: [1, 2, 17]
Perampanel is a broad-spectrum agent used to treat both focal and generalized seizures.
Mechanism of Action
Antagonist of the AMPA-type glutamate receptor, reducing intracellular calcium, resulting in reduced neuronal excitability.
Dosing
-
≥ 4 years:
-
Start 2 mg daily; or 4 mg daily if taking enzyme-inducing ASMs*
-
Increase by 2 mg/day every 7–14 days
-
Goal 4–8 mg/day
-
* carbamazepine, phenytoin, phenobarbital, primidone
Side Effects
-
Neurologic: somnolence/fatigue, irritability, ataxia/dizziness/gait disturbance, headache
-
Systemic: GI symptoms, weight gain
Critical/Potentially Life-Threatening Side Effects
-
Neurologic: psychosis, suicide ideation
-
Systemic: hypersensitivity reaction/DRESS
Lab Monitoring
Defer routine monitoring.
Phenobarbital
Section References: [1,2,3, 20, 42]
Phenobarbital is effective in the treatment of focal and primary generalized seizures. It is contraindicated for absence seizures. It is often used to treat neonatal seizures (see Chap. 5) and as an abortive therapy for status epilepticus across age groups.
Mechanism of Action
Binds the GABAA receptor leading to increased chloride ion permeability. This results in neuronal hyperpolarization and enhancing inhibition. May also result in a reduction in calcium-dependent action potentials.
Dosing
-
Status epilepticus:
-
15–20 mg/kg IV
-
-
Seizure treatment:
-
Infants:
-
5–6 mg/kg/day given in 1–2 doses
-
-
1–5 years:
-
3–8 mg/kg/day given in 1–2 doses
-
> 5 years to adolescents:
-
2–3 mg/kg/day given in 1–2 doses
-
> 12 years/adults:
-
1.5–4 mg/kg/day given in 1–2 doses
-
or
-
50–200 mg/day given in 1–2 doses
Side Effects
-
Neurologic: somnolence/lethargy, impaired attention/cognition, depression/mood changes, behavioral changes/hyperactivity, nystagmus, dysarthria, ataxia
-
Systemic: GI symptoms, decreased bone density, rash
-
Rare cases of connective tissue contractures/Dupuytren contracture
Critical/Potentially Life-Threatening Side Effects
-
Systemic: SJS/TEN/hypersensitivity reaction, respiratory depression/apnea (rapid IV infusion), hepatotoxicity, agranulocytosis/thrombocytopenia
Lab Monitoring
Drug levels, CBC, and LFTs should be monitored. Consider monitoring 25-hydroxyvitamin D levels.
Phenytoin
Section References: [1,2,3, 20, 136]
Phenytoin is used for focal seizures with or without secondary generalization. They may also be effective against some generalized seizures including primary GTC or for patients with mixed semiology. Phenytoin has the potential to worsen a subset of generalized seizures, typically myoclonic and absence. Phenytoin should be avoided for patients with LGS and progressive myoclonic epilepsies. For status epilepticus, fosphenytoin is preferred.
Mechanism of Action
Blocks voltage-gated sodium channels and modulates sustained repetitive firing. It also inhibits calcium channels and calcium sequestration and inhibits calcium-calmodulin protein phosphorylation.
Dosing
-
With the start of treatment, may consider enteral loading dose depending on the clinical circumstances.
-
Children:
-
Enteral loading dose:
-
15 mg/kg/day divided in 3 doses
-
24 hrs after loading dose: start 5 mg/kg/day given in 1–2 doses
-
-
or
-
Start 5 mg/kg/day given in 1–2 doses
-
Increase by 5 mg/kg/day every 3–4 weeks
-
Goal 5–10 mg/kg/day
-
-
Adults:
-
Enteral loading dose:
-
1 g divided in 3 doses
-
24 hrs after loading dose: start 300–400 mg/day given in 1–2 doses
-
-
or
-
Start 150–300 mg/day in 1–2 doses
-
Increase by 50 mg/day every 3–4 weeks
-
Goal 200–400 mg/day
Side Effects
-
Neurologic: somnolence, confusion, nystagmus, dysarthria, ataxia/dizziness
-
Systemic: GI symptoms, decreased bone density, hirsutism, rash, gingival hypertrophy
Critical/Potentially Life-Threatening Side Effects
-
Neurologic: suicide ideation, peripheral neuropathy
-
Systemic: SJS/TEN, systemic lupus erythematosus, hepatotoxicity, agranulocytosis/aplastic anemia, lymphadenopathy/lymphoma
Lab Monitoring
Recommend baseline LFTs and Chem10 to evaluate liver and kidney function; repeat screening labs every 12 months. Alternatively, may defer routine monitoring. Consider monitoring folate and 25-hydroxyvitamin D levels.
Pregabalin
Section References: [1,2,3, 18]
Pregabalin is used for focal seizures with or without secondary generalization. It has the potential to exacerbate primary generalized seizures, specifically myoclonus. Pregabalin is a broad-spectrum GABA analog and structurally similar to gabapentin. Its effect as an antiseizure treatment, however, does not seem to be mediated by modulation of GABA.
Mechanism of Action
Binds to voltage-gated presynaptic calcium channels, inhibiting inward calcium currents. This results in decreased release of neurotransmitters including glutamate, noradrenaline, and substance P.
Dosing
-
Infant to < 4 years:
-
Start 3.5–5 mg/kg/day divided in 2–3 doses
-
Increase by 3–5 mg/kg/day every 7 days
-
Max 14 mg/kg/day
-
4–16 years:
-
< 30 kg
-
Start 3.5 mg/kg/day divided in 2–3 doses
-
Increase by 3 mg/kg/day every 7 days
-
Max 14 mg/kg/day
-
-
≥ 30 kg
-
Start 2.5 mg/kg/day divided in 2–3 doses
-
Increase 3 mg/kg/day every 7 days
-
Max 10 mg/kg/day
-
-
≥ 17 years
-
Start 50–150 mg/day divided in 2–3 doses
-
Increase by 50–150 mg/day every 7 days
-
Goal 150–600 mg/day (max 600 mg/day)
Side Effects
-
Neurologic: somnolence, impaired attention, irritability/aggressiveness, tremor, ataxia/dizziness
-
Systemic: peripheral edema, GI symptoms, weight gain, xerostomia
Critical/Potentially Life-Threatening Side Effects
-
Systemic: hypersensitivity reaction, AV block, myocardial dysfunction, angioedema, rhabdomyolysis, hypoglycemia, neutropenia
Lab Monitoring
Consider baseline Chem10 to evaluate renal function in the setting of preexisting comorbidities and/or increased risk.
Primidone
Section References: [1,2,3, 20, 137]
Primidone is effective in the treatment of focal and primary generalized seizures. It is contraindicated for absence seizures.
Mechanism of Action
Primidone has two active metabolites, phenobarbital and phenylethylmalonamide. Primidone has been shown to provide an antiseizure effect independent from phenobarbital, but the mechanism of this effect is not understood.
Phenobarbital is described above; it binds the GABAA receptor leading to increased chloride ion permeability. This results in neuronal hyperpolarization and enhancing inhibition. It may also result in a reduction in calcium-dependent action potentials.
Dosing
-
Infants–8 years:
-
Start 1–2 mg/kg/day given once
-
Increase every 3 days
-
Goal 10–25 mg/kg/day (infants); 10–20 mg/kg/day (children) divided in 2–4 doses
-
or
-
Day 1–3: 50 mg/day at bedtime
-
Day 4–6: 100 mg/day divided in 2 doses
-
Day 7–9: 200 mg/day divided in 2 doses
-
Day 10: 375–750 mg/day divided in 3 doses
> 8 years – adults:
-
Day 1–3: 125 mg/day given once
-
Day 4–6: 200–250 mg/day divided in 2 doses
-
Day 7–9: 300–375 mg/day divided by 3 doses
-
Day 10: 750 mg/day divided by 3 doses
-
Goal 750–1500 mg/day or 10–20 mg/kg/day divided in 3–4 doses
Side Effects
-
Neurologic: somnolence/lethargy, impaired attention/cognition, depression/mood changes, behavioral changes/hyperactivity, nystagmus, dysarthria, ataxia
-
Systemic: GI symptoms, decreased bone density, rash
-
Rare cases of connective tissue contractures
Critical/Potentially Life-Threatening Side Effects
-
Neurologic: Acute toxic reaction (severe sedation, ataxia/dizziness, nausea/vomiting)
-
Systemic: SJS/TEN/hypersensitivity reaction, hepatotoxicity, agranulocytosis
Lab Monitoring
Defer routine monitoring. Consider monitoring 25-hydroxyvitamin D levels.
Rufinamide
Section References: [1,2,3, 20, 138]
Rufinamide is a broad-spectrum agent used to treat both focal and primary generalized seizures with a role in treatment of LGS and epileptic spams. As a triazole derivative, the chemical structural is unique among current ASMs.
Mechanism of Action
Blocks voltage-gated sodium channels after inactivation, prolonging recovery and preventing return to an active state. This persistent depolarization reduces bursts of high-frequency action potentials.
Dosing
-
≥ 4 years and < 30 kg:
-
Start 10 mg/day or 200 mg/kg/day divided in 2 doses
-
Increase by 10 mg/day or 200 mg/kg/day every 2–7 days
-
Goal 45 mg/day or 1000 mg/kg/day
-
Goal 400–600 mg/day *
-
* Concomitant treatment with valproate
-
Children ≥ 30 kg and adults:
-
Start 400 mg/day divided in 2 doses
-
Increase by 400 mg/day every 2 days
-
Goal
-
30–50 kg: 1800 mg/day
-
51–70 kg: 2400 mg/day
-
> 70 kg: 3200 mg/day
-
Side Effects
-
Neurologic: somnolence/fatigue, dizziness/ataxia, headache
-
Systemic: GI symptoms
Critical/Potentially Life-Threatening Side Effects
-
Neurologic: status epilepticus
-
Systemic: hypersensitivity reaction, decreased QTc interval
Lab Monitoring
Consider baseline LFTs and Chem10 for hepatic and renal function; repeat every 12 months. Alternatively, may defer routine monitoring. Recommend baseline ECG to evaluate QTc interval and identify potential preexisting arrhythmias.
Stiripentol
Section References: [1,2,3, 19, 23]
Stiripentol is used as an adjuvant therapy for synergistic effect with clobazam or valproate, typically for patients with Dravet syndrome and refractory GTCs. Co-treatment with carbamazepine, phenytoin, or phenobarbital is typically avoided due to risk of toxicity.
Mechanism of Action
Activates GABAA receptor with barbiturate-like mechanisms of increasing chloride ion permeability, neuronal hyperpolarization, and enhanced inhibition. It may also increase GABA levels by inhibiting synaptic uptake and/or inhibiting GABA transaminase. Stiripentol has additional indirect effects; as a CYP inhibitor, it may act to increase serum levels of concomitant ASMs such as clobazam.
Dosing
-
A broad range of dosing has been reported:
-
Start 25–50 mg/kg/day divided in 2–3 doses
-
Increase by 10 mg/kg/day every 7–14 days
-
Goal 75 mg/kg/day
-
-
May consider lower dosing to start:
-
Start 10–20 mg/kg/day divided in 2–3 doses
-
Increase over 2–4 weeks
-
Goal 50 mg/kg/day
Side Effects
-
Neurologic: somnolence, agitation/irritability, dysarthria, hypotonia, dystonia, tremor, ataxia, insomnia
-
Systemic: GI symptoms, anorexia/weight-loss
Critical/Potentially Life-Threatening Side Effects
-
Neurologic: suicide ideation
-
Systemic: aplastic anemia, leukopenia/neutropenia, thrombocytopenia
Lab Monitoring
Consider baseline LFTs and Chem10 for hepatic and renal function; repeat every 12 months.
Sulthiame
Sulthiame is used for focal seizures with or without secondary generalization and self-limited epilepsy with centrotemporal spikes. It is also used for epileptic encephalopathies, with specific efficacy for patients with CSWS and myoclonic epilepsies. It is not currently approved for use in the United States but is widely used in Europe.
Mechanism of Action
Inhibits voltage-gated sodium channels and glutamate release. Inhibits carbonic anhydrase which results in decreased inward calcium flow of NMDA receptors, thereby reducing neuronal excitability.
Dosing
-
Children:
-
Start 5 mg/kg/day divided in 2 doses
-
Increase by 5 mg/kg/day after 7 days
-
Goal 5–10 mg/kg/day
-
Adults:
-
Start 100–250 mg/day divided in 2 doses
-
Increase by 100 mg/day every 7 days
-
Goal 200–600 mg/day
Side Effects
-
Neurologic: paresthesia, ataxia/dizziness, headache
-
Systemic: hyperventilation, metabolic acidosis, anorexia/weight loss, rash
Critical/Potentially Life-Threatening Side Effects
-
SJS/TEN, renal dysfunction, metabolic acidosis, nephrolithiasis
Lab Monitoring
Consider baseline CBC and Chem10 to evaluate renal function.
Tiagabine
Section References: [1,2,3, 20, 139]
Tiagabine is used for focal seizures with or without secondary generalization. It may also be considered for epileptic spasms.
Mechanism of Action
Enhanced endogenous GABA effect by blocking GABA reuptake by inhibiting GABA transporter-1.
Dosing
-
Children > 12 years:
-
Start 0.1 mg/kg/day
-
Increase by 0.1 mg/kg/day every 7–14 days
-
Goal 0.5–2 mg/kg/day divided in 2–4 doses
-
Concomitant treatment with enzyme-inducing ASMs
-
> 12 years:
-
Start 0.25 mg/kg/day
-
Increase by 0.5–1 mg/kg/day every 7 days
-
Max 4–8 mg/kg/day divided in 3–4 doses
-
12–18 years:
-
Start 4 mg/day
-
Increase by 4 mg/day every 7 days
-
Max 32 mg/day divided in 2–4 doses
Side Effects
-
Neurologic: somnolence/fatigue, cognitive and attention impairment, anxiety, asthenia, tremor, dizziness, headache
-
Systemic: GI symptoms, ecchymosis
Critical/Potentially Life-Threatening Side Effects
-
Neurologic: nonconvulsive status or absence status epilepticus, suicide ideation
-
Concern for possible risk of visual field deficits but data is limited
-
Systemic: SJS/TEN
Lab Monitoring
Consider baseline CBC as well as LFTs and Chem10 for hepatic and renal function; repeat every 6–12 months. Alternatively, may defer routine monitoring.
Topiramate
Section References: [1,2,3, 20, 21]
Topiramate is a broad-spectrum agent used to treat both focal and primary generalized events, including primary GTCs, myoclonic and absence seizures. It is used for myoclonic syndromes including Dravet, progressive myoclonic epilepsies, myoclonic atonic epilepsy, and JME. It is also used for LGS and IS.
Mechanism of Action
Multiple described mechanisms of action including inhibition of voltage-gated sodium channels, L-type voltage-gated calcium channels, AMPA glutamate receptors, and carbonic anhydrase. It binds GABAA receptors mediating neuronal inhibition. Finally, topiramate may increase potassium channel conduction.
Dosing
-
Infants and children <12 years:
-
Start 0.5–1 mg/kg/day divided in 2–3 doses
-
Increase by 0.5–1 mg/kg/day every 7–14 days
-
Goal 3–9 mg/kg/day
-
2–16 years:
-
Start 1–3 mg/kg/day divided in 2–3 doses
-
Increase by 1–3 mg/kg/day every 7–14 days
-
Goal 5–9 mg/kg/day
-
≥ 17 years:
-
Start 25–50 mg/day divided in 2 doses
-
Increase by 25–50 mg/day every 7–14 days
-
Goal 100–400 mg/day
Side Effects
-
Neurologic: somnolence/fatigue, cognitive and attention impairment, anxiety, depression/mood changes, paresthesia, ataxia/dizziness
-
Systemic: metabolic acidosis, anorexia/weight loss, hypohidrosis
Critical/Potentially Life-Threatening Side Effects
-
Neurologic: acute myopia with secondary angle-closure glaucoma
-
Systemic: metabolic acidosis, nephrolithiasis, oligohydramnios/hyperthermia
Lab Monitoring
Consider CBC, LFTs, and Chem10 once maximum treatment dose is reached or at 3–6 months following the start of treatment. Alternatively, may defer routine monitoring.
Valproate
Section References: [1,2,3, 20, 140]
Valproate is a broad-spectrum agent used to treat both focal and primary generalized events including GTCs, tonic/atonic, myoclonic seizures, and absence seizures. It is used for JME and LGS. It may also be used in status epilepticus.
Mechanism of Action
Multiple described mechanisms of action including inhibition of voltage-gated sodium channels and T-type calcium channels, as well as increased potassium channel conduction. There is an increase in GABA concentrations by multiple proposed modalities including increased glutamic acid decarboxylase activity, inhibition of GABA transaminase or succinic semialdehyde dehydrogenase. Decreased expression of glutamate transporter-1 has also been described.
Dosing
-
Status epilepticus:
-
20–40 mg/kg IV
-
-
Seizure treatment:
-
≤ 12 years
-
Start 10–15 mg/kg/day divided in 2–3 doses
-
Increase by 5–15 mg/kg/day every 5–7 days
-
Goal 20–60 mg/kg/day
-
-
> 12 years
-
Start 200–500 mg/day divided in 2 doses
-
Increase by 200–500 mg/day every 3–7 days
-
Goal 1000–1500 mg/day
Side Effects
-
Neurologic: somnolence/lethargy, asthenia, tremor, ataxia/dizziness, headache
-
Systemic: GI symptoms, hyperammonemia, elevated liver enzymes, hypocarnitinemia, weight gain, decreased bone density, alopecia, ecchymosis
Critical/Potentially Life-Threatening Side Effects
-
Neurologic: hyperammonemic encephalopathy
-
Systemic: SJS/TEN, hepatotoxicity, pancreatitis, agranulocytosis, aplastic anemia, thrombocytopenia, hypogammaglobulinemia
Lab Monitoring
Consider baseline CBC and LFTs; repeat at 2 months after the start of treatment and every 6 months thereafter. Consider monitoring 25-hydroxyvitamin D levels.
Vigabatrin
Section References: [1,2,3, 11, 22, 141]
Vigabatrin is typically used to treat IS. For patients with tuberous sclerosis complex and IS, it is first line. It may also be used to treat refractory focal seizures with or without secondary generalization. It is contraindicated for primary generalized seizures including absence seizures.
Mechanism of Action
Increases synaptic GABA by irreversibly inhibiting GABA transaminase.
Dosing
-
Infants:
-
Start 50 mg/kg/day given in 2 doses
-
Increase by 25–50 mg/kg/day every 3 days
-
Max 150–200 mg/kg/day
-
2–16 years and ≤ 60 kg:
-
10–15 kg
-
Start 350 mg/day divided in 2 doses
-
Increase every 7 days
-
Goal 1050 mg/day
-
16–20 kg:
-
Start 450 mg/day divided in 2 doses
-
Increase every 7 days
-
Goal 1300 mg/day
-
21–25 kg:
-
Start 500 mg/day divided in 2 doses
-
Increase every 7 days
-
Goal 1500 mg/day
-
26–60 kg:
-
Start 500 mg/day divided in 2 doses
-
Increase every 7 days
-
Goal 2000 mg/day
-
≥ 17 years and/or > 60 kg:
-
Start 500–1000 mg/day given in 2 doses
-
Increase by 500 mg/day every 7 days
-
Goal 1000–3000 mg/day
Side Effects
-
Neurologic: Somnolence/fatigue, cognitive impairment, agitation/irritability, tremor, paresthesia, ataxia/dizziness, headache
-
MRI changes: restricted diffusion on T2 imaging of the basal ganglia, thalamus, brainstem, dentate
-
Systemic: GI symptoms, weight gain
A visual field deficit develops with vigabatrin treatment in about one third of cases [3, 141]. In the United States, there is a REMS program to support prescriber education and serial ophthalmologic monitoring.
Critical/Potentially Life-Threatening Side Effects
-
Neurologic: status epilepticus, psychosis
-
Systemic: hypersensitivity reaction, angioedema
Lab Monitoring
Consider baseline CBC as well as LFTs and Chem10 to evaluate hepatic and renal function; repeat labs every 6–12 months thereafter.
Zonisamide
Section References: [1,2,3, 20, 108, 142]
Zonisamide is a broad-spectrum agent used to treat both focal and generalized events including primary GTCs, tonic/atonic, myoclonic, and absence seizures. It may be used for epileptic encephalopathies including IS, Ohtahara syndrome, progressive myoclonic epilepsies, and Dravet syndrome.
Mechanism of Action
Blocks voltage-gated sodium channels, T-type calcium channels, and carbonic anhydrase. Decreases GABA transporter-1 expression and glutamate release. Increases extracellular levels of dopamine and serotonin. Increased expression of excitatory amino-acid carrier-1 (EAAC-1).
Dosing
-
> 6 years:
-
Start 1–2 mg/kg/day given in 1–2 doses
-
Increase by 1–2 mg/kg/day every 7–14 days
-
Goal 5–12 mg/kg/day
-
Adults:
-
Start 100 mg/day given in 1–2 doses
-
Increase by 100 mg/day every 7–14 days
-
Goal 100–600 mg/day
Side Effects
-
Neurologic: somnolence/fatigue, cognitive and attention impairment, anxiety, depression, agitation/irritability, paresthesia, ataxia/dizziness
-
Systemic: GI symptoms, anorexia/weight loss, metabolic acidosis, rash, hypohidrosis
Critical/Potentially Life-Threatening Side Effects
-
Neurologic: status epilepticus, psychosis, acute myopia with secondary angle-closure glaucoma
-
Systemic: SJS/TEN, hepatotoxicity, metabolic acidosis, nephrolithiasis, agranulocytosis, aplastic anemia, anhidrosis, hyperthermia
Lab Monitoring
Consider baseline chemistry panel with repeat 3–6 months after the start of treatment. For patients with cognitive impairment, for whom it may be difficult to recognize early signs or symptoms of metabolic acidosis, plan for ongoing periodic lab monitoring of bicarbonate levels.
References
Patsalos PN, St. Louis EK. The epilepsy prescriber’s guide to antiepileptic drugs. 3rd ed. Cambridge, Cambridge University Press; 2018.
Schachter SC. Antiseizure drugs: Mechanism of action, pharmacology, and adverse effects [Internet]. Garcia P, Dashe J, editors. UpToDate. Waltham, MA: UpToDate, Inc.; 2021 [cited 2021 Feb 8]. Available from: http://www.uptodate.com.
Panayiotopoulos C. Pharmacopedia. In: A clinical guide to epileptic syndromes and their treatment. 2nd ed. Springer; Berlin 2010. p. 565–620.
Gray RA, Whalley BJ. The proposed mechanism of action of CBD in epilepsy. Epileptic Disord. 2020;22(January):10–5.
Stroink H, Brouwer OF, Arts WF, Geerts AT, Peters ACB, Van Donselaar CA. The first unprovoked, untreated seizure in childhood: A hospital based study of the accuracy of the diagnosis, rate of recurrence, and long term outcome after recurrence. Dutch study of epilepsy in childhood. J Neurol Neurosurg Psychiatry. 1998;64(5):595–600.
Zuberi SM, Symonds JD. Update on diagnosis and management of childhood epilepsies. J Pediatr (Rio J). 2015;91(6):S67–77.
Hauser WA, Rich SS, Lee JR, Annegers JF, Anderson VE. Risk of recurrent seizures after two unprovoked seizures. N Engl J Med. 1998;338(7):429–34.
Raspall-Chaure M, Neville BG, Scott RC. The medical management of the epilepsies in children: conceptual and practical considerations. Lancet Neurol. 2008;7(1):57–69.
Shellhaas RA, Berg AT, Grinspan ZM, Wusthoff CJ, Millichap JJ, Loddenkemper T, et al. Initial treatment for nonsyndromic early-life epilepsy: an unexpected consensus. Pediatr Neurol. 2017;75:73–9.
Glauser T, Ben-Menachem E, Bourgeois B, Cnaan A, Guerreiro C, Kälviäinen R, et al. Updated ILAE evidence review of antiepileptic drug efficacy and effectiveness as initial monotherapy for epileptic seizures and syndromes. Epilepsia. 2013;54(3):551–63.
Guerrini R. Epilepsy in children. Lancet. 2006;367(9509):499–524.
Nieto-Barrera M, Candau R, Nieto-Jimenez M, Correa A. Ruiz del Portal L. Topiramate in the treatment of severe myoclonic epilepsy in infancy. Seizure. 2000;9(8):590–4.
Wallace SJ. Myoclonus and epilepsy in childhood: A review of treatment with valproate, ethosuximide, lamotrigine and zonisamide. Epilepsy Res. 1998;29(2):147–54.
Covanis A, Gupta AK, Jeavons PM. Sodium valproate: monotherapy and polytherapy. Epilepsia. 1982;23(6):693–720.
Oguni H, Tanaka T, Hayashi K, Funatsuka M, Sakauchi M, Shirakawa S, et al. Treatment and long-term prognosis of myoclonic-astatic epilepsy of early childhood. Neuropediatrics. 2002;33(3):122–32.
Mikaeloff Y, De Saint-Martin A, Mancini J, Peudenier S, Pedespan JM, Vallée L, et al. Topiramate: Efficacy and tolerability in children according to epilepsy syndromes. Epilepsy Res. 2003;53(3):225–32.
Perampanel [Internet]. Lexi-Drugs, Pediatric and Neonatal. Hudson, Ohio: UpToDate, Inc.; 2020 [cited 2021 Feb 8]. Available from: https://online.lexi.com.
Pregabalin [Internet]. Lexi-Drugs, Pediatric and Neonatal. Hudson, Ohio: UpToDate, Inc.; 2021 [cited 2021 Feb 8]. Available from: https://online.lexi.com.
Stiripentol [Internet]. Lexi-Drugs, Pediatric and Neonatal. Hudson, Ohio: UpToDate, Inc.; 2020 [cited 2021 Feb 8]. Available from: https://online.lexi.com.
Wilfong A. Seizures and epilepsy in children: Initial treatment and monitoring [Internet]. Nordli DR, Dashe JF, editors. UpToDate. Waltham, MA: UpToDate, Inc.; 2021 [cited 2021 Feb 8]. Available from: http://www.uptodate.com.
Topiramate [Internet]. Lexi-Drugs, Pediatric and Neonatal. Hudson, Ohio: UpToDate, Inc.; 2021 [cited 2021 Feb 8]. Available from: https://online.lexi.com.
Vigabatrin [Internet]. Lexi-Drugs, Pediatric and Neonatal. Hudson, Ohio: UpToDate, Inc.; 2021 [cited 2021 Feb 8]. Available from: https://online.lexi.com.
Gonzalez-Giraldo E, Sullivan JE. Advances in the treatment of drug-resistant pediatric epilepsy. Semin Neurol. 2020;40(2):257–62.
Brivaracetam [Internet]. Lexi-Drugs, Pediatric and Neonatal. Hudson, Ohio: UpToDate, Inc.; 2021 [cited 2021 Oct 5]. Available from: https://online.lexi.com.
Fenfluramine [Internet]. Lexi-Drugs, Pediatric and Neonatal. Hudson, Ohio: UpToDate, Inc.; 2021 [cited 2021 Feb 8]. Available from: https://online.lexi.com.
Midazolam [Internet]. Lexi-Drugs, Pediatric and Neonatal. Hudson, Ohio: UpToDate, Inc.; 2021 [cited 2021 Feb 8]. Available from: https://online.lexi.com.
Guerrini R, Dravet C, Genton P, Belmonte A, Kaminska A, Dulac O. Lamotrigine and seizure aggravation in severe myoclonic epilepsy. Epilepsia. 1998;39(5):508–12.
Kwan P, Brodie MJ. Early identification of refractory epilepsy. N Engl J Med. 2000;342(5):314–9.
Camfield PR, Camfield CS, Gordon K, Dooley JM. If a first antiepileptic drug fails to control a child’s epilepsy, what are the chances of success with the next drug? J Pediatr. 1997;131(6):821–4.
Geerts A, Brouwer O, Stroink H, Van Donselaar C, Peters B, Peeters E, et al. Onset of intractability and its course over time: The Dutch study of epilepsy in childhood. Epilepsia. 2012;53(4):741–51.
Kwan P, Arzimanoglou A, Berg AT, Brodie MJ, Hauser WA, Mathern G, et al. Definition of drug resistant epilepsy: Consensus proposal by the ad hoc Task Force of the ILAE Commission on Therapeutic Strategies. Epilepsia. 2010;51(6):1069–77.
Deckers CLP, Czuczwar SJ, Hekster YA, Kewser A, Kubova H, Meinardi H, et al. Selection of antiepileptic drug polytherapy based on mechanisms of action: The evidence reviewed. Epilepsia. 2000;41(11):1364–74.
Rowan AJ, Meijer JWA, Beer Pawlikowski N, Van Der Geest P, Meinardi H. Valproate-ethosuximide combination therapy for refractory absence seizures. Arch Neurol. 1983;40(13):797–802.
Perucca E. The management of refractory idiopathic epilepsies. Epilepsia. 2001;42(SUPPL. 3):31–5.
Ferrie CD, Panayiotopoulos CP. Therapeutic interaction of lamotrigine and sodium valproate in intractable myoclonic epilepsy. Seizure. 1994;3(2):157–9.
Chiron C, Marchand MC, Tran A, Rey E, D’Athis P, Vincent J, et al. Stiripentol in severe myoclonic epilepsy in infancy: A randomised placebo-controlled syndrome-dedicated trial. Lancet. 2000;356(9242):1638–42.
Interactions [Internet]. Lexicomp. Hudson, Ohio: UpToDate, Inc.; [cited 2021 Feb 8]. Available from: https://online.lexi.com.
Sale ME, Natarajan S, Biton V, Vuong A, Hammer AE, Messenheimer JA, et al. A dosing algorithm for converting from valproate monotherapy to lamotrigine monotherapy in patients with epilepsy. Epilepsy Behav. 2005;6(1):63–70.
Zaccara G, Franciotta D, Perucca E. Idiosyncratic adverse reactions to antiepileptic drugs. Epilepsia. 2007;48(7):1223–44.
Hirsch LJ, Weintraub DB, Buchsbaum R, Spencer HT, Straka T, Hager M, et al. Predictors of lamotrigine-associated rash. Epilepsia. 2006;47(2):318–22.
Andrade DM, Nascimento F. Dravet syndrome: management and prognosis [Internet]. Nordli DR, Dashe JF, editors. UpToDate. Waltham, MA: UpToDate, Inc.; 2021 [cited 2021 Feb 8]. Available from: http://www.uptodate.com.
Phenobarbital [Internet]. Lexi-Drugs, Pediatric and Neonatal. Hudson, Ohio: UpToDate, Inc.; 2021 [cited 2021 Feb 8]. Available from: https://online.lexi.com.
French J, Smith M, Faught E, Brown L. Practice advisory: The use of felbamate in the treatment of patients with intractable epilepsy. Epilepsia. 1999;40(6):803–8.
Gerstner T, Teich M, Bell N, Longin E, Dempfle CE, Brand J, et al. Valproate-associated coagulopathies are frequent and variable in children. Epilepsia. 2006;47(7):1136–43.
Camfield P, Camfield C. Monitoring for adverse effects of antiepileptic drugs. Epilepsia. 2006;47(SUPPL. 1):31–4.
Strozzi I, Nolan SJ, Sperling MR, Wingerchuk DM, Sirven J. Early versus late antiepileptic drug withdrawal for people with epilepsy in remission. Cochrane Database Syst Rev. 2015;2015(2):CD001902.
Shinnar S, Berg AT, Moshé SL, Kang H, O’Dell C, Alemany M, et al. Discontinuing antiepileptic drugs in children with epilepsy: A prospective study. Ann Neurol. 1994;35(5):534–45.
Sánchez Fernández I, Loddenkemper T. Pediatric focal epilepsy syndromes. J Clin Neurophysiol. 2012;29(5):425–40.
Mahmoud SH, Buxton J. Seizures and choice of antiepileptic drugs following subarachnoid hemorrhage: A review. Can J Neurol Sci. 2017;44(6):643–53.
Cusack TJ, Carhuapoma JR, Ziai WC. Update on the treatment of spontaneous intraparenchymal hemorrhage: medical and interventional management. Curr Treat Options Neurol. 2018;20(1):1.
Kossoff EH, Zupec-Kania BA, Auvin S, Ballaban-Gil KR, Christina Bergqvist AG, Blackford R, et al. Optimal clinical management of children receiving dietary therapies for epilepsy: Updated recommendations of the International Ketogenic Diet Study Group. Epilepsia Open. 2018;3(2):175–92.
Kossoff EH, Pyzik PL, McGrogan JR, Vining EPG, Freeman JM. Efficacy of the ketogenic diet for infantile spasms. Pediatrics. 2002;109(5):780–3.
Hong AM, Turner Z, Hamdy RF, Kossoff EH. Infantile spasms treated with the ketogenic diet: Prospective single-center experience in 104 consecutive infants. Epilepsia. 2010;51(8):1403–7.
Eun SH, Kang HC, Kim DW, Kim HD. Ketogenic diet for treatment of infantile spasms. Brain Dev. 2006;28(9):566–71.
Prezioso G, Carlone G, Zaccara G, Verrotti A. Efficacy of ketogenic diet for infantile spasms: A systematic review. Acta Neurol Scand. 2018;137(1):4–11.
Kossoff EH, Zupec-Kania BA, Amark PE, Ballaban-Gil KR, Christina Bergqvist AG, Blackford R, et al. Optimal clinical management of children receiving the ketogenic diet: Recommendations of the International Ketogenic Diet Study Group. Epilepsia. 2009;50(2):304–17.
Sourbron J, Klinkenberg S, van Kuijk SMJ, Lagae L, Lambrechts D, Braakman HMH, et al. Ketogenic diet for the treatment of pediatric epilepsy: review and meta-analysis. Childs Nerv Syst. 2020;36(6):1099–109.
Pfeifer HH, Thiele EA. Low-glycemic-Index treatment : A liberalized ketogenic diet for treatment of. Neurology. 2005;1–3.
Kossoff EH, McGrogan JR, Bluml RM, Pillas DJ, Rubenstein JE, Vining EP. A modified Atkins diet is effective for the treatment of intractable pediatric epilepsy. Epilepsia. 2006;47(2):421–4.
Sofou K, Dahlin M, Hallböök T, Lindefeldt M, Viggedal G, Darin N. Ketogenic diet in pyruvate dehydrogenase complex deficiency: short- and long-term outcomes. J Inherit Metab Dis. 2017;40(2):237–45.
Sandu C, Burloiu CM, Barca DG, Magureanu SA, Craiu DC. Ketogenic diet in patients with GLUT1 deficiency syndrome. Maedica (Buchar). 2019;14(2):93–7.
McNally MA, Pyzik PL, Rubenstein JE, Hamdy RF, Kossoff EH. Empiric use of potassium citrate reduces kidney-stone incidence with the ketogenic diet. Pediatrics. 2009;124(2):e300–4.
Arslan N, Guzel O, Kose E, Yılmaz U, Kuyum P, Aksoy B, et al. Is ketogenic diet treatment hepatotoxic for children with intractable epilepsy? Seizure. 2016;43(June 2013):32–8.
Henderson CB, Filloux FM, Alder SC, Lyon JL, Caplin DA. Efficacy of the ketogenic diet as a treatment option for epilepsy: Meta-analysis. J Child Neurol. 2006;21(3):193–8.
Martinez CC, Pyzik PL, Kossoff EH. Discontinuing the ketogenic diet in seizure-free children: Recurrence and risk factors. Epilepsia. 2007;48(1):187–90.
Çataltepe O, Jallo G. Basic considerations of pediatric epilepsy surgery. In: Çataltepe O, Jallo GI, editors. Pediatric epilepsy surgery: preoperative assessment and surgical treatment. 2nd ed. New York: Thieme; 2020. p. 3–9.
Akman ÇI, Riviello JJ. Intractable epilepsy in children and selection of surgical candidates. In: Çataltepe O, Jallo GI, editors. Pediatric epilepsy surgery: preoperative assessment and surgical treatment. 2nd ed. New York: Thieme; 2019. p. 81–91.
Schramm J, Zentner J. Surgery for temporal lobe epilepsy. In: Arzimanoglou A, Helen Cross J, Gaillard WD, Holthausen H, Kyakar P, Kahane P, et al., editors. Pediatric epilepsy surgery. 2016. p. 347–59.
Russo G, Bhatia S, Ojemann JG. Extratemporal localization and eloquent areas. In: Arzimanoglou A, Helen Cross J, Gaillard WD, Holthausen H, Kyakar P, Kahane P, et al., editors. Pediatric epilepsy surgery. 2016. p. 361–79.
Englot DJ, Breshears JD, Sun PP, Chang EF, Auguste KI. Seizure outcomes after resective surgery for extra-temporal lobe epilepsy in pediatric patients: A systematic review. J Neurosurg Pediatr. 2013;12(2):126–33.
Çataltepe O. Hemipherectomy and hemipherotomy techniques in pediatric epilepsy surgery. In: Çataltepe O, Jallo GI, editors. Pediatric epilepsy surgery: preoperative assessment and surgical treatment. 2nd ed. New York: Thieme; 2019. p. 497–505.
Wong T-T, Kwan S-Y, Chang K-P, Tsai M-L, Hsieh KL-C. Corpus callosotomy. In: Çataltepe O, Jallo GI, editors. Pediatric epilepsy surgery: preoperative assessment and surgical treatment. 2nd ed. New York: Thieme; 2019. p. 581–8.
Tovar-Spinoza ZS, Rutka JT. Multiple subpial transections in children with refractory epilepsy. In: Çataltepe O, Jallo GI, editors. Pediatric epilepsy surgery: preoperative assessment and surgical treatment. 2nd ed. New York: Thieme; 2019. p. 603–7.
Curry D, Gadgil N. Steriotactic laser ablation for hypothalamic hamartomas. In: Çataltepe O, Jallo GI, editors. Pediatric epilepsy surgery: preoperative assessment and surgical treatment. 2nd ed. New York: Thieme; 2019. p. 640–4.
Karsy M, Guan J, Ducis K, Bollo RJ. Emerging surgical therapies in the treatment of pediatric epilepsy. Transl Pediatr. 2016;5(2):67–78.
Tovar-Spinoza ZS, Rutka JT. MRI-guided laser thermal therapy in pediatric epilepsy surgery. In: Çataltepe O, Jallo GI, editors. Pediatric epilepsy surgery: preoperative assessment and surgical treatment. 2nd ed. New York: Thieme; 2019. p. 645–52.
Régis J, Hamdi H, Chauvel P. Radiosurgical treatment for epilepsy. In: Çataltepe O, Jallo GI, editors. Pediatric epilepsy surgery: preoperative assessment and surgical treatment. 2nd ed. New York: Thieme; 2019. p. 625–39.
Nune G, DeGiorgio C, Heck C. Neuromodulation in the treatment of epilepsy. Curr Treat Options Neurol. 2015;17(10):5.
Starnes K, Miller K, Wong-Kisiel L, Lundstrom BN. A review of neurostimulation for epilepsy in pediatrics. Brain Sci. 2019;9(10):PMC6826633.
Englot DJ, Chang EF, Auguste KI. Vagus nerve stimulation for epilepsy: A meta-analysis of efficacy and predictors of response - A review. J Neurosurg. 2011;115(6):1248–55.
Bergey GK, Morrell MJ, Mizrahi EM, Goldman A, King-Stephens D, Nair D, et al. Long-term treatment with responsive brain stimulation in adults with refractory partial seizures. Neurology. 2015;84(8):810–7.
Li MCH, Cook MJ. Deep brain stimulation for drug-resistant epilepsy. Epilepsia. 2018;59(2):273–90.
Chiken S, Nambu A. Mechanism of deep brain stimulation: inhibition, excitation, or disruption? Neuroscientist. 2016;22(3):313–22.
Velasco AL, Velasco F, Jiménez F, Velasco M, Castro G, Carrillo-Ruiz JD, et al. Neuromodulation of the centromedian thalamic nuclei in the treatment of generalized seizures and the improvement of the quality of life in patients with Lennox-Gastaut syndrome. Epilepsia. 2006;47(7):1203–12.
Yan H, Toyota E, Anderson M, Abel TJ, Donner E, Kalia SK, et al. A systematic review of deep brain stimulation for the treatment of drug-resistant epilepsy in childhood. J Neurosurg Pediatr. 2019;23(3):274–84.
Abend NS, Loddenkemper T. Pediatric status epilepticus management. Curr Opin Pediatr. 2014;26(6):668–74.
Glauser T, Shinnar S, Gloss D, Alldredge B, Arya R, Bainbridge J, et al. Evidence-based guideline: treatment of convulsive status epilepticus in children and adults: report of the guideline committee of the American epilepsy society. Epilepsy Curr. 2016;16(1):48–61.
Brophy GM, Bell R, Claassen J, Alldredge B, Bleck TP, Glauser T, et al. Guidelines for the evaluation and management of status epilepticus. Neurocrit Care. 2012;17(1):3–23.
Smith DM, McGinnis EL, Walleigh DJ, Abend NS. Management of status epilepticus in children. J Clin Med. 2016;5(4):PMC4850470.
Chin RF, Neville BG, Peckham C, Wade A, Bedford H, Scott RC. Treatment of community-onset, childhood convulsive status epilepticus: a prospective, population-based study. Lancet Neurol. 2008;7(8):696–703.
Lewena S, Young S. When benzodiazepines fail: How effective is second line therapy for status epilepticus in children? EMA - Emerg Med Australas. 2006;18(1):45–50.
Sánchez Fernández I, Abend NS, Agadi S, An S, Arya R, Brenton JN, et al. Time from convulsive status epilepticus onset to anticonvulsant administration in children. Neurology. 2015;84(23):2304–11.
Bennett KS. Treatment of refractory status epilepticus in children: current practice and opportunities to improve care. Pediatr Crit Care Med. 2016;17:1006–7.
Sánchez Fernández I, Goodkin HP, Scott RC. Pathophysiology of convulsive status epilepticus. Seizure. 2019;68(March 2018):16–21.
Goodkin HP, Joshi S, Mtchedlishvili Z, Brar J, Kapur J. Subunit-specific trafficking of GABAA receptors during status epilepticus. J Neurosci. 2008;28(10):2527–38.
Tully I, Draper ES, Lamming CR, Mattison D, Thomas C, Martland T, et al. Admissions to paediatric intensive care units (PICU) with refractory convulsive status epilepticus (RCSE): A two-year multi-centre study. Seizure. 2015;29:153–61.
Lewena S, Pennington V, Acworth J, Thornton S, Ngo P, McIntyre S, et al. Emergency management of pediatric convulsive status epilepticus: A multicenter study of 542 patients. Pediatr Emerg Care. 2009;25(2):83–7.
Riviello JJ, Ashwal S, Hirtz D, Glauser T, Ballaban-Gil K, Kelley K, et al. Practice parameter: Diagnostic assessment of the child with status epilepticus (an evidence-based review) - Report of the quality standards subcommittee of the American academy of neurology and the practice committee of the child neurology society. Neurology. 2006;67(9):1542–50.
Zhao ZY, Wang HY, Wen B, Yang ZB, Feng K, Fan JC. A comparison of midazolam, lorazepam, and diazepam for the treatment of status epilepticus in children: a network meta-analysis. J Child Neurol. 2016;31(9):1093–107.
Prasad M, Krishnan PR, Sequeira R, Al-Roomi K. Anticonvulsant therapy for status epilepticus. Cochrane Database Syst Rev. 2014;2014(9):CD003723.
Tasker RC, Goodkin HP, Sánchez Fernández I, Chapman KE, Abend NS, Arya R, et al. Refractory status epilepticus in children: intention to treat with continuous infusions of midazolam and pentobarbital∗. Pediatr Crit Care Med. 2016;17(10):968–75.
Kapur J, Elm J, Chamberlain JM, Barsan W, Cloyd J, Lowenstein D, et al. randomized trial of three anticonvulsant medications for status epilepticus. N Engl J Med. 2019;381(22):2103–13.
Chamberlain JM, Kapur J, Shinnar S, Elm J, Holsti M, Babcock L, et al. Efficacy of levetiracetam, fosphenytoin, and valproate for established status epilepticus by age group (ESETT): a double-blind, responsive-adaptive, randomised controlled trial. Lancet. 2020;395(10231):1217–24.
Yasiry Z, Shorvon SD. The relative effectiveness of five antiepileptic drugs in treatment of benzodiazepine-resistant convulsive status epilepticus: A meta-analysis of published studies. Seizure. 2014;23(3):167–74.
Watemberg N, Segal G. A suggested approach to the etiologic evaluation of status epilepticus in children: What to seek after the usual causes have been ruled out. J Child Neurol. 2010;25(2):203–11.
Herman ST, Abend NS, Bleck TP, Chapman KE, Drislane FW, Emerson RG, et al. Consensus statement on continuous EEG in critically Ill adults and children, part I: Indications. J Clin Neurophysiol. 2015;32(2):87–95.
Acetazolamide [Internet]. Lexi-Drugs, Pediatric and Neonatal. Hudson, Ohio: UpToDate, Inc.; 2021 [cited 2021 Feb 8]. Available from: https://online.lexi.com.
Shbarou R. Current treatment options for early-onset pediatric epileptic encephalopathies. Curr Treat Options Neurol. 2016;18(10):1–9.
Corticotropin [Internet]. Lexi-Drugs, Pediatric and Neonatal. Hudson, Ohio: UpToDate, Inc.; 2021 [cited 2021 Feb 8]. Available from: https://online.lexi.com.
Kwan P, Trinka E, Van Paesschen W, Rektor I, Johnson ME, Lu S. Adjunctive brivaracetam for uncontrolled focal and generalized epilepsies: Results of a phase III, double-blind, randomized, placebo-controlled, flexible-dose trial. Epilepsia. 2014;55(1):38–46.
Cannabidiol [Internet]. Lexi-Drugs, Pediatric and Neonatal. Hudson, Ohio: UpToDate, Inc.; 2021 [cited 2021 Feb 8]. Available from: https://online.lexi.com.
Thiele EA, Bebin EM, Bhathal H, Jansen FE, Kotulska K, Lawson JA, et al. Add-on cannabidiol treatment for drug-resistant seizures in tuberous sclerosis complex: a placebo-controlled randomized clinical trial. JAMA Neurol. 2020;02114:1–9.
Head AA, Dpt PE, Arzimanoglou A, Brandl U, Cross JH, Gil-Nagel A, et al. Seminar in epileptology epilepsy and cannabidiol: a guide to treatment. Epileptic Disord. 2020;22(1):1–14.
Carbamazepine. Lexi-Drugs, Pediatric and Neonatal. Hudson, Ohio: UpToDate, Inc.; 2021.
Cenobamate [Internet]. Lexi-Drugs. Hudson, Ohio: UpToDate, Inc.; 2021 [cited 2021 Feb 8]. Available from: https://online.lexi.com.
Clobazam [Internet]. Lexi-Drugs, Pediatric and Neonatal. Hudson, Ohio: UpToDate, Inc.; 2021 [cited 2021 Feb 8]. Available from: https://online.lexi.com.
Clonazepam [Internet]. Lexi-Drugs, Pediatric and Neonatal. Hudson, Ohio: UpToDate, Inc.; 2021 [cited 2021 Feb 8]. Available from: https://online.lexi.com.
Diazepam [Internet]. Lexi-Drugs, Pediatric and Neonatal. Hudson, Ohio: UpToDate, Inc.; 2021 [cited 2021 Feb 8]. Available from: https://online.lexi.com.
Eslicarbazepine acetate [Internet]. Lexi-Drugs, Pediatric and Neonatal. Hudson, Ohio: UpToDate, Inc.; 2021 [cited 2021 Feb 8]. Available from: https://online.lexi.com.
Galiana GL, Gauthier AC, Mattson RH. Eslicarbazepine acetate: a new improvement on a classic drug family for the treatment of partial-onset seizures. Drugs R D. 2017;17(3):329–39.
Mäkinen J, Rainesalo S, Peltola J. Transition from oxcarbazepine to eslicarbazepine acetate: A single center study. Brain Behav. 2017;7(3):1–6.
Ethosuximide [Internet]. Lexi-Drugs, Pediatric and Neonatal. Hudson, Ohio: UpToDate, Inc.; 2021 [cited 2021 Feb 8]. Available from: https://online.lexi.com.
Felbamate [Internet]. Lexi-Drugs, Pediatric and Neonatal. Hudson, Ohio: UpToDate, Inc.; 2021 [cited 2021 Feb 8]. Available from: https://online.lexi.com.
Lagae L, Sullivan J, Knupp K, Laux L, Polster T, Nikanorova M, et al. Fenfluramine hydrochloride for the treatment of seizures in Dravet syndrome: a randomised, double-blind, placebo-controlled trial. Lancet. 2019;394(10216):2243–54.
Fosphenytoin [Internet]. Lexi-Drugs, Pediatric and Neonatal. Hudson, Ohio: UpToDate, Inc.; 2021 [cited 2021 Feb 8]. Available from: https://online.lexi.com.
Gabapentin [Internet]. Lexi-Drugs, Pediatric and Neonatal. Hudson, Ohio: UpToDate, Inc.; 2021 [cited 2021 Feb 8]. Available from: https://online.lexi.com.
Lacosamide [Internet]. Lexi-Drugs, Pediatric and Neonatal. Hudson, Ohio: UpToDate, Inc.; 2021 [cited 2021 Feb 8]. Available from: https://online.lexi.com.
Wechsler RT, Yates SL, Messenheimer J, Leroy R, Beller C, Doty P. Lacosamide for uncontrolled primary generalized tonic-clonic seizures: An open-label pilot study with 59-week extension. Epilepsy Res. 2017;130:13–20.
Hwang ST, Stevens SJ, Fu AX, Proteasa SV. Intractable generalized epilepsy: therapeutic approaches. Curr Neurol Neurosci Rep. 2019;19(4):16.
Morris GL, Hammer AE, Kustra RP, Messenheimer JA. Lamotrigine for patients with juvenile myoclonic epilepsy following prior treatment with valproate: Results of an open-label study. Epilepsy Behav. 2004;5(4):509–12.
Lamotrigine [Internet]. Lexi-Drugs, Pediatric and Neonatal. Hudson, Ohio: UpToDate, Inc.; 2021 [cited 2021 Feb 8]. Available from: https://online.lexi.com.
Biraben A, Allain H, Scarabin JM, Schück S, Edan G. Exacerbation of juvenile myoclonic epilepsy with lamotrigine [5]. Neurology. 2001;56(10):1424–5.
Levetiracetam [Internet]. Lexi-Drugs, Pediatric and Neonatal. Hudson, Ohio: UpToDate, Inc.; 2021 [cited 2021 Feb 8]. Available from: https://online.lexi.com.
Lorazepam [Internet]. Lexi-Drugs, Pediatric and Neonatal. Hudson, Ohio: UpToDate, Inc.; 2021 [cited 2021 Feb 8]. Available from: https://online.lexi.com.
Oxcarbazepine [Internet]. Lexi-Drugs, Pediatric and Neonatal. Hudson, Ohio: UpToDate, Inc.; 2021 [cited 2021 Feb 8]. Available from: https://online.lexi.com.
Phenytoin [Internet]. Lexi-Drugs, Pediatric and Neonatal. Hudson, Ohio: UpToDate, Inc.; 2021 [cited 2021 Feb 8]. Available from: https://online.lexi.com.
Primidone [Internet]. Lexi-Drugs, Pediatric and Neonatal. Hudson, Ohio: UpToDate, Inc.; 2021 [cited 2021 Feb 8]. Available from: https://online.lexi.com.
Rufinamide [Internet]. Lexi-Drugs, Pediatric and Neonatal. Hudson, Ohio: UpToDate, Inc.; 2021 [cited 2021 Feb 8]. Available from: https://online.lexi.com.
Tiagabine [Internet]. Lexi-Drugs, Pediatric and Neonatal. Hudson, Ohio: UpToDate, Inc.; 2021 [cited 2021 Feb 8]. Available from: https://online.lexi.com.
Valproate [Internet]. Lexi-Drugs, Pediatric and Neonatal. Hudson, Ohio: UpToDate, Inc.; 2021. Available from: https://online.lexi.com.
Maguire MJ, Hemming K, Wild JM, Hutton JL, Marson AG. Prevalence of visual field loss following exposure to vigabatrin therapy: A systematic review. Epilepsia. 2010;51(12):2423–31.
Zonisamide [Internet]. Lexi-Drugs, Pediatric and Neonatal. Hudson, Ohio: UpToDate, Inc.; 2021 [cited 2021 Feb 8]. Available from: https://online.lexi.com.
Author information
Authors and Affiliations
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2022 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Larson, A.M., Thibert, R.L., Thiele, E.A. (2022). Pediatric Epilepsy Treatment. In: Dredge, D.C. (eds) Handbook of Pediatric Epilepsy. Springer, Cham. https://doi.org/10.1007/978-3-319-08290-5_7
Download citation
DOI: https://doi.org/10.1007/978-3-319-08290-5_7
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-08289-9
Online ISBN: 978-3-319-08290-5
eBook Packages: MedicineMedicine (R0)