
Abstract
Selective protein degradation by the ubiquitin–proteasome pathway (UPP) is critical to cellular homeostasis, and dysregulation of the UPP has been associated with human diseases including cancer. Proteasome inhibition as a strategy for cancer treatment was validated by the US Food and Drug Administration approval of the proteasome inhibitor bortezomib for the treatment of multiple myeloma in 2003. After 10 years of success, bortezomib and its combinational therapies have become a staple for treating relapsed/refractory multiple myeloma. Unfortunately, bortezomib has several limitations, including, most notably, the emergence of resistance. To overcome bortezomib resistance, several approaches have been taken, including the development of novel second-generation proteasome inhibitors, application of rationalized bortezomib-based combinational therapies, and targeting sites outside the proteasomal core as well as factors involved in resistance mechanisms. Further understanding the mechanisms of resistance to proteasome inhibitors in human cancers will significantly improve current proteasome inhibitor therapies and patient care.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
1.1 Introduction
The ubiquitin–proteasome pathway (UPP) has gained considerable attention as a potential target for cancer therapeutics, owing to its extreme importance to normal cellular function and dysregulation in malignant cells. In fact, critical proteasomal target proteins are involved in processes important for carcinogenesis, including cell cycle progression, proliferation, and differentiation. The past decade has witnessed the emergence of proteasome inhibition as an effective therapeutic strategy for treating multiple myeloma. Bortezomib was the first proteasome inhibitor approved by the US Food and Drug Administration (FDA) in 2003, and the use of bortezomib and bortezomib-based combinational therapies has become a staple for the treatment of relapsed/refractory multiple myeloma. Unfortunately, further success of bortezomib has been hampered by tumor resistance (both intrinsic and acquired), severe toxicities, and low efficacy in solid tumors. To overcome these limitations, especially resistance, scientists have investigated the molecular mechanisms involved and developed novel strategies to improve proteasome inhibitor-based therapies and patient care. By improving chemical and biochemical properties, binding affinity and reversibility, potency and selectivity, several second-generation proteasome inhibitors have been developed, among them carfilzomib, which is more specific and less toxic than bortezomib, and has become the second FDA-approved proteasome inhibitor for multiple myeloma treatment. Other cutting-edge strategies to overcome bortezomib resistance include selectively targeting immunoproteasomes or sites outside the catalytic core (such as 19S deubiquitinases or ubiquitin E3 ligases) and developing novel combinational therapies. Definitively elucidating the mechanisms responsible for proteasome inhibitor resistance is key in designing new compounds to fully overcome this resistance.
1.2 Ubiquitin–Proteasome Pathway
The UPP is the major pathway responsible for regulating protein turnover in cells. The UPP is so critical to normal cellular function that its discoverers, Aaron Ciechanover and Avram Hershko, were awarded the 2004 Nobel Prize in Chemistry [1, 2]. Proteins degraded by the UPP are involved in many biological processes, including development, differentiation, proliferation, signal transduction, and apoptosis [3]. In addition to its critical role in protein homeostasis, the proteasome also functions in several non-proteolytic processes, such as transcription initiation and elongation [4], regulation of gene expression [5], and transcription-coupled nucleotide excision repair [6].
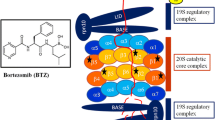
Protein degradation is carried out via two distinct steps: (1) conjugation of multiple ubiquitin molecules to the protein substrate and (2) degradation of the ubiquitin-tagged substrate by the 26S proteasome (Fig. 1.1) [7]. The 26S proteasome is a large (2.5 MDa), multi-subunit complex that is localized both in the cytosol and nucleus of cells [8–10]. The 26S proteasome is made up of the catalytic 20S core and one or two 19S regulatory caps (Fig. 1.2) [11, 12]. The 20S core is comprised of 28 subunits that form a barrel-like structure of four alternately stacked rings: two α-rings surrounding two β-rings, each with seven subunits [13–15]. The role of the α-subunits is to allow only unfolded proteins to enter the 20S core, while the β-subunits are responsible for the proteolytic activities of the proteasome, which are dependent on an amino-terminal nucleophilic Thr1 residue [15]: caspase or peptidyl-glutamyl peptide-hydrolyzing (PGPH)-like activity, carried out by β1, trypsin-like by β2 and chymotrypsin (CT)-like by β5 (Fig. 1.2) [15–17]. The 19S regulatory caps (700 kDa) can be divided into a base and a lid (Fig. 1.2); the base is responsible for the recognition and unfolding of ubiquitinated protein substrates, as well as opening the 20S pore and transport of protein substrates into the core, while the major responsibility of the lid component is deubiquitination of substrates before degradation. The base contains six ATPase subunits, Rpt1–6, which form a hexameric ring [18–20], as well as two non-ATPase subunits Rpn-1 and Rpn-2 [21, 22], and the lid consists of at least six non-ATPases, including Rpn-10/S5a and Rpn-13/Adrm1, which contain ubiquitin-interacting motifs (UIMs) [23]. Rpn-10/S5a has two UIMs that preferentially binds poly-ubiquitinated substrates [24], and Rpn-13/Adrm1 binds to the non-ATPase Rpn-2 to recruit deubiquitinating enzymes (DUBs) to the proteasome [25–27]. Deubiquitination is very highly regulated and is important for recycling ubiquitin molecules and controlling the rate of ubiquitin-dependent proteasomal degradation [27].

The ubiquitin–proteasome pathway. There are two steps in the UPP: ubiquitination and target degradation. The ubiquitination step is carried out by three distinct types of enzymes, E1 (ubiquitin activating), E2s (ubiquitin conjugating), and E3s (ubiquitin ligating). First, ubiquitin is activated by E1, the activated ubiquitin is transferred to an E2 for conjugation, and finally, an E3 ubiquitin-ligating enzyme aids in the transfer of active ubiquitin to lysine residues within the target protein. The target protein is then recognized, deubiquitinated, and translocated to the 26S proteasome by components of the 19S regulatory cap, followed by degradation into small peptide fragments, and the ubiquitin molecules are recycled

Proteasome structure. The 26S constitutive proteasome is comprised of a 20S catalytic core and one or two 19S regulatory caps. The 20S core contains four stacked rings—two α-rings surrounding two β-rings, each consisting of seven subunits. The catalytic activity is carried out by three β-subunits: β1, β2, and β5 responsible for caspase or peptidyl-glutamyl peptide-hydrolyzing (PGPH)-like, trypsin-like, and chymotrypsin (CT)-like, respectively
The ubiquitination step of the UPP is carried out by three distinct types of enzymes, E1 (ubiquitin activating), E2s (ubiquitin conjugating), and E3s (ubiquitin ligating). The first step in the pathway is ATP-dependent E1-mediated activation of ubiquitin, a small 76-amino-acid protein that is expressed ubiquitously and serves as a tag for protein substrates destined for various fates, including membrane trafficking, protein kinase activation, DNA repair and chromatin remodeling, as well as degradation by the UPP (Fig. 1.1) [28]. Activated ubiquitin is then transferred from E1 to an E2 enzyme, a group of enzymes responsible for ubiquitin conjugation, and then to an E3 ubiquitin-ligating enzyme, which aids in the transfer of active ubiquitin to lysine residues within the target protein (Fig. 1.1) [29, 30]. Following the conjugation of a sufficiently sized ubiquitin chain, which is four in most cases, except in rare cases such as mODC and HIF-1α, which require no ubiquitination for proteasomal degradation [31–33], the protein substrate is recognized, deubiquitinated, and translocated to the 26S proteasome by components of the 19S regulatory cap [34, 35]. Finally, the substrate is degraded into small peptide fragments and the ubiquitin molecules are recycled (Fig. 1.1) [36]. This process is tightly controlled and extremely crucial in the regulation of many cellular processes, including those involved in tumorigenesis [37], which makes it a promising target for anticancer therapeutic agents.
Because the UPP plays such a crucial role in normal cellular function, it is no surprise that it has also been implicated in the development, growth, and survival of various malignancies [38]. Thus, targeting factors involved in the synthesis and degradation of proteins, including the UPP, has been explored as a potential anticancer strategy [39]. Several studies have reported increased proteasome activity in various cancers, including colon, prostate, and leukemia [40–42], indicating that cancer cells may be more dependent on the UPP than normal cells and that targeting this pathway in the treatment of human cancer is a promising strategy. Specifically, inhibition of chymotrypsin (CT)-like activity has been associated with cell cycle arrest and apoptosis [43, 44], indicating that proteasome inhibition may effectively cause selective cell death in cancer cells, as well as sensitizing them to chemotherapeutics [45], with little toxicity in normal cells. Importantly, the use of proteasome inhibitors was validated by the US Food and Drug Administration (FDA) approval of bortezomib for the treatment of relapsed and refractory multiple myeloma and mantle cell lymphoma.
1.3 Proteasome Inhibitors
1.3.1 Early Inhibitors
Prior to the development and approval of bortezomib, numerous preclinical studies were carried out to validate the UPP as a valid druggable target. The most widely investigated early inhibitors include the peptide aldehydes, which are analogs of proteasome substrates that inhibit the CT-like activity of the proteasome and include MG-132 (Cbz-leu-leu-leucinal), MG-115 (Cbz-leu-leu-norvalinal), and ALLN (acetyl-leu-leu-norleucinal) [46, 47]. Importantly, in-depth studies using these complexes aided in elucidating the active site for compounds in this class, X-ray diffraction revealed that ALLN forms a hemiacetal complex with the N-terminal threonine hydroxyl groups of the catalytic β-subunits [14, 48]. Another peptide aldehyde inhibitor, PSI (Cbz-ile-glu(O-t-Bu)-alaleucinal), has been shown to suppress 26S proteasome-mediated proteolysis without affecting isopeptidase or ATPase activities [49]. These inhibitors are extremely potent (MG-132 K i = low nanomolar in purified proteasome; IC50 = low micromolar in cultured cells) and their inhibitory activities are reversible by their removal from the system [46, 47]. Interestingly, because peptide aldehydes are also able to inhibit calpains and some lysosomal cysteine proteases, certain degradative processes that were originally believed to be carried out by calpains were shown to actually be proteasomal processes.
Vinyl sulfone peptides have also been reported to be potent inhibitors of the proteasome in cell models [50]. These peptides exert their proteasome-inhibitory activity through covalent binding to the hydroxyl groups of the active site threonine within the β-subunits, and their use in human lymphoma cells resulted in proteasome inhibition followed by the appearance of distinct cell variants expressing a compensatory proteolytic system, which has not been clearly identified [51].
Other early inhibitors of the proteasome include lactacystin and its derivative clasto-lactacystin β-lactone, the active form to which it is converted in aqueous solution [52]. These are naturally occurring products that differ structurally from the peptide aldehydes and are much more specific. Lactacystin was first isolated from actinomycetes because of its ability to promote neurite outgrowth and block cell division in cultured neurons [53]. These compounds have a mode of action similar to that of the vinyl sulfones [54, 55].
Other naturally occurring metabolites that have been used in the preclinical setting as inhibitors of the proteasome include TMC-95A and argyrin A. TMC-95A is a cyclic tripeptide that was isolated from Apiospora montagnei. TMC-95A specifically binds via hydrogen bonds to all three catalytic β-subunits and causes inhibition in the low nanomolar range [56, 57]. The tumor growth suppression caused by argyrin A, a cyclic octapeptide derived from Archangium gephyra, has been attributed to the inhibition of proteasomal degradation of p27kip1 CDK inhibitor [58, 59]. Following the identification of these inhibitors, many other compounds were identified and designed to specifically target the tumor proteasome, ultimately resulting in the USFDA approval of bortezomib in 2003.
1.3.2 Bortezomib, the First Clinically Approved Proteasome Inhibitor in Preclinical Studies
Bortezomib (Velcade®) is a dipeptide boronic acid derivative that was first synthesized in 1995 by Myogenics Company and contains pyrazinoic acid, phenylalanine, and leucine in its structure. Bortezomib showed considerable apoptosis-inducing activity in a variety of tumor cell lines and animal models [60–62], and in 2003, seven years after its initial synthesis, bortezomib was approved by the USFDA for the treatment of relapsed multiple myeloma, and in 2006, it was approved for the treatment of mantle cell lymphoma. Bortezomib is able to enter nearly all tissues except brain and adipose, and is able to distribute to the plasma within 10 min of IV injection [63–66]. Furthermore, bortezomib is metabolized through intracellular cytochrome p450-mediated oxidative deboronation [67] and its half-life is more than 40 h [65].
Bortezomib is a reversible inhibitor of the 26S proteasome, with proteasome activity generally recovering within 72 h of administration [68]. Binding of the boronic acid group in bortezomib to the threonine hydroxyl group in the active site of the β5 subunit results in proteasome inhibition and, ultimately, cell death [69]. Bortezomib has been successful in hematological malignancies, but less than encouraging results have been observed in solid tumors [70, 71], limiting its use in the clinic.
Several preclinical studies demonstrated the potency of bortezomib against human tumor cells in vitro and in in vivo xenograft animal models. A standard NCI-60 screen revealed that bortezomib could potently inhibit cell proliferation [60] and induce apoptosis in many malignant cell lines, including multiple myeloma, prostate, pancreatic, renal and squamous cell carcinomas [72–77]. Importantly, the antitumor activity of bortezomib was observed in both chemoresistant and chemosensitive myeloma cells, and the sensitivity of resistant cells to chemotherapy was increased significantly when combined with a sublethal dose of bortezomib with no effect on normal hematopoietic cells [78, 79]. Additionally, in an in vitro study of four ovarian and three prostate cancer cell lines, bortezomib had comparable effects on cells derived from solid tumors and hematological malignancies [61]. Bortezomib was also able to potently inhibit the growth of multiple myeloma xenografts in mice [80].
Multiple targets of bortezomib have been identified in malignant cells, including the NF-κB signaling pathway. NF-κB is a p50/p65 heterodimer that usually exists in an inactive form in the cytoplasm bound to its inhibitory protein, IκB, and upon degradation of IκB, the NF-κB complex is activated and can translocate into the nucleus where it stimulates transcription of various genes including cytokines (IL-6, TNF-α), survival factors (IAPs, Bcl-XL), and insulin-like growth factor 1 (IGF-1), ultimately resulting in proliferation, resistance to apoptosis, and drug resistance in cancer cells [81]. Bortezomib is able to prevent degradation of IκB, blocking activation of NF-κB and suppressing expression of related cytokines and survival factors in drug-resistant multiple myeloma cells expressing increased NF-κB activity [78, 82]. In contrast, other studies have shown that the NF-κB pathway may not be important in bortezomib-mediated tumor cell death. Specifically, in a study of mice bearing human multiple myeloma cells, treatment with bortezomib was associated with NF-κB activation, rather than inhibition [83].
Another possible target of bortezomib is NOXA (Latin for damage) [84], a pro-apoptotic member of the Bcl-2 family [85] that is involved in p53-mediated apoptosis, gene expression of which is associated with direct activation of its promoter by p53 [85]. Thus, upregulation of p53 and subsequent Noxa gene expression may be one mechanism of chemo- or radiotherapy-induced apoptosis. Studies have shown that NOXA upregulation induces apoptosis through interaction with, and inhibition of anti-apoptotic Bcl-xL and Bcl-2 proteins, or through stimulation of other pro-apoptotic factors [86, 87]. Importantly, bortezomib treatment in myeloma and melanoma cell lines resulted in p53-independent induction of NOXA and blockade of NOXA with an antisense oligonucleotide caused only 30 % to 50 % reduction in bortezomib-induced apoptosis [84]. Bortezomib induces NOXA in various p53-defective tumor cell lines [88], and clinical studies indicate that bortezomib suppresses tumor growth in a p53-independent manner [11, 89]. Importantly, NOXA induction by bortezomib is selective to cancer cells over normal cells, with levels unaffected in normal melanocytes [84, 90, 91].
Still other mechanisms of bortezomib-mediated apoptosis include inhibition of angiogenesis in human myeloma, pancreatic and squamous cell cancer xenografts [77, 92]; induction of endoplasmic reticulum (ER) stress and generation of reactive oxygen species (ROS) [93, 94]; induction of extrinsic and intrinsic apoptotic pathways via activation of caspase-8 and caspase-9 [95, 96]; activation of the p38 mitogen-activated protein kinase (MAPK) pathway [97]; and disruption of the interaction between tumor cells and dendritic cells [98]. Multiple targets generally play important roles in bortezomib-mediated apoptosis in some cancer cells, while different targets may be critical in other cells.
1.3.3 Bortezomib in Clinical Trials
1.3.3.1 Phase I/II Trials
The promising preclinical data involving bortezomib resulted in a series of clinical trials that ultimately led to the USFDA approval of bortezomib as a treatment for multiple myeloma. One phase I trial of 27 patients with relapsed multiple myeloma investigated bortezomib as a single agent, and found that bortezomib induced a dose-dependent inhibition of 20S proteasome activity [99], confirming preclinical findings that bortezomib could inhibit proteasome activity in a dose- and time-dependent manner. Two other phase I studies evaluated bortezomib in combination with doxorubicin. In the first, 42 patients with advanced hematologic malignancies were enrolled to obtain preliminary response data and to determine the maximum tolerated dose (1.30 mg/m2) and dose-limiting toxicities (fatigue, thrombocytopenia, lymphopenia, nausea, constipation, peripheral neuropathy, and anemia) [100]. The other enrolled 22 patients with multiple myeloma, with eight patients achieving complete response (36 %) or near-complete response, and another eight partial responses (36 %) [100].
Additional phase I trials have investigated the effects of bortezomib either alone or in combination in solid tumors. Single-agent bortezomib showed antitumor activity in patients with advanced androgen-independent prostate cancer [101], but no significant responses were observed in patients with advanced metastatic breast cancer or neuroendocrine tumors [102, 103]. In combination with carboplatin, an overall response rate of 47 % was observed in recurrent ovarian or primary peritoneal cancer patients, but in combination with either docetaxel [104] or prednisone [105], hormone refractory and castrate-resistant metastatic prostate cancer patients achieved no significant responses. Therefore, while bortezomib has shown promise in hematological malignancies, it has proven quite ineffective against solid tumors.
The general success of phase I trials led to several phase II trials. In the SUMMIT (Study of Uncontrolled Multiple Myeloma Managed with Proteasome Inhibition Therapy) trial, 202 patients with relapsed or refractory myeloma with prior treatment were treated with 1.3 mg/m2 bortezomib on days 1, 4, 8, and 11 of a 3-week cycle for as many as eight cycles, and an overall response rate of 35 % was observed [106]. In the CREST (Clinical Response and Efficacy Study of Bortezomib in the Treatment of Relapsing Multiple Myeloma) trial, 67 patients with relapsed/refractory multiple myeloma were randomly divided to receive either 1.0 or 1.3 mg/m2 of bortezomib. The study ultimately showed that bortezomib was effective in relapsed multiple myeloma patients at a lower dose of 1.0 mg/m2 [107].
Two other phase II trials examined bortezomib in combination with other agents. One study reported a 95 % response rate in relapsed multiple myeloma patients treated with a combination of bortezomib, dexamethasone, and doxorubicin [108]. Another study in patients with symptomatic multiple myeloma with no prior treatment compared single-agent bortezomib to bortezomib in combination with dexamethasone. Of the 32 patients, 22 were treated with the combination, and an increased response was seen in 15 of 22 patients (68 %) [45].
Additionally, the effects of bortezomib against mantle cell lymphoma and non-Hodgkin’s lymphoma have also been investigated in clinical trials. A trial of patients with indolent non-Hodgkin’s lymphoma and mantle cell lymphoma showed a 58 % overall response rate as a result of bortezomib treatment [109]. Another study conducted in patients with pretreated and untreated mantle cell lymphoma revealed response rates of 46.2 % and 46.7 %, respectively, following treatment with 1.3 mg/m2 bortezomib, suggesting that bortezomib is an effective treatment for mantle cell lymphoma [110]. Finally, no significant response or survival advantage was observed in another phase II study evaluating the use of bortezomib and pemetrexed alone or in combination in advanced NSCLC with prior treatment, but bortezomib was better tolerated when given in combination with pemetrexed [111]. More clinical trials are being conducted to further explore the use of bortezomib in NSCLC. Unfortunately, phase II trials investigating the efficacy of bortezomib in solid tumors have yielded disappointing results.
1.3.3.2 Phase III Clinical Trials
Based on phase II trial results, a large international phase III trial in relapsed multiple myeloma patients with 1–3 prior therapies compared the effects of bortezomib to high-dose dexamethasone [75]. Patients (n = 669) received either 1.3 mg/m2 bortezomib (twice weekly for 2 weeks followed by a 1-week rest, intravenously) or high-dose dexamethasone (40 mg orally). Patients receiving bortezomib had a combined complete and partial response rate of 38 % compared to 18 % for the dexamethasone-treated patients, with median times to progression of 6.22 months in the bortezomib group versus 3.29 months in the dexamethasone group. Among patients taking bortezomib, the median time to progression was 6.22 months and 1-year survival rate was 80 %, while that for patients taking dexamethasone was 3.29 months and 66 % [75], demonstrating the advantage of bortezomib over dexamethasone in terms of response rate, time to progression, and survival.
Another study included 638 relapsed/refractory multiple myeloma patients who received 1.3 mg/m2 bortezomib and achieved an overall response rate of 67 % [112]. After completion of at least two cycles for progressive and four cycles for stable disease, 20 mg/day dexamethasone was added on the day of and after each bortezomib dose. Of the patients receiving dexamethasone, enhanced response was observed in 34 %, suggesting that bortezomib, alone or in combination with dexamethasone, is both safe and effective for the treatment of relapsed/refractory multiple myeloma in patients with prior treatment [112]. The APEX (Assessment of Proteasome Inhibition for Extending Remissions) trial assessed the impact of dose modification on the severity and reversibility of peripheral neuropathy associated with bortezomib treatment in patients with relapsed multiple myeloma [113]. Peripheral neuropathy could be improved by dose modification without adverse effects on the outcome in 37 % of patients (124/331) following several cycles of bortezomib treatment [113], indicating that bortezomib-induced peripheral neuropathy is not only manageable, but also reversible in most relapsed myeloma patients.
The efficacy of bortezomib in combination with conventional chemotherapeutics was conducted at 151 centers in 22 countries. Patients with untreated multiple myeloma (n = 682) were randomized to receive either a combination of bortezomib plus melphalan–prednisone or melphalan–prednisone alone [114]. Results revealed that bortezomib plus melphalan–prednisone may be a valuable frontline treatment option for myeloma patients [114]. Most recently, in the VISTA trial, bortezomib plus melphalan and prednisone was compared to melphalan and prednisone alone in multiple myeloma patients with no previous treatment. A prolonged follow-up (median = 36.7 months) indicated that bortezomib-based drugs as first-line treatments afford greater survival advantage than treatment with conventional drugs followed by salvage with bortezomib-based treatments [115]. Additionally, initial treatment with bortezomib, compared to initial treatment with traditional chemotherapeutics, resulted in less resistance to later therapies [115]. Overall, preclinical and clinical data evaluating the efficacy and safety of bortezomib have shown that the use of proteasome inhibitors as anticancer agents is a promising strategy that should be further investigated.
However, while bortezomib is successful in the clinic, toxicities and resistance have been reported, suggesting that further development of drugs like bortezomib is necessary. In fact, some second-generation proteasome inhibitors [116, 117] with different properties have been developed, with one, carfilzomib, being FDA approved. Additionally, inhibitors that specifically target the immunoproteasome (immunoproteasome-specific inhibitors, IPSIs) [117], as well as natural compounds that are able to inhibit the proteasome, may be sufficiently potent with significantly less adverse effects than currently approved drugs [118]. The use of these novel inhibitors may aid in overcoming bortezomib resistance or sensitizing resistant cells to bortezomib treatment, which could potentially result in increased clinical success.
1.3.4 The Second Clinically Approved Proteasome Inhibitor, Carfilzomib
Following the clinical success of bortezomib, the second-in-class proteasome inhibitor carfilzomib (Kyprolis®) was granted accelerated approval by the USFDA in July 2012 for the treatment of patients with MM progressing on or after treatment with bortezomib and an immunomodulatory agent. Carfilzomib is a peptide epoxyketone related to epoxomicin [119] that irreversibly inhibits the CT-like activity of the proteasome with high selectivity [120]. Preclinical studies revealed that carfilzomib inhibits CT-like activity in both the constitutive proteasome and the inducible immunoproteasome with IC50 values of 6 and 33 nM, respectively [121]. Carfilzomib was also extremely effective at suppressing tumor growth in cultured cell and tumor xenograft models, with prolonged proteasome inhibition for longer than one week in mice [121]. Importantly, carfilzomib was active against bortezomib-resistant cultured myeloma and patient plasma cells [119].
1.3.4.1 Phase I/II Trials
The data observed in cultured cell and xenograft models led to a series of clinical trials investigating the properties and efficacy of carfilzomib. In one phase I study, carfilzomib was administered on consecutive days twice weekly in patients with relapsed and refractory multiple myeloma or lymphoma. One hour following IV administration of 27 mg/m2 carfilzomib, CT-like activity in whole blood and peripheral blood mononuclear cells (PBMCs) was inhibited by approximately 85 % and 90 % on average, respectively, and this inhibition was sustained throughout the trial [122]. Another small phase I dose-escalation study evaluated the safety and efficacy of carfilzomib in relapsed or refractory myeloma and lymphoma, with patients (n = 29) receiving carfilzomib for five consecutive days within 14-day cycles [123]. One unconfirmed complete response, one partial response, and two minimal responses were observed with observable antitumor activity at or above 11 mg/m2 and a maximum tolerated dose of 15 mg/m2. Grade 1–2 toxicities included nausea, diarrhea, and fatigue in more than one-third of patients. At the highest dose administered (20 mg/m2), grade 3 febrile neutropenia and grade 4 thrombocytopenia were reported, and no grade 3 or 4 peripheral neuropathies were reported [123]. An additional phase I/II study investigated the tolerability, efficacy, and pharmacokinetic and pharmacodynamic profiles of carfilzomib in advanced solid tumors [124]. Carfilzomib was administered IV twice weekly on consecutive days within 28-day cycles. A small group of patients (n = 14) received carfilzomib during the phase I dose escalation, and the single dose-limiting toxicity was determined to be grade 3 fatigue at the highest administered dose (36 mg/m2). The maximum planned dose was determined based on the phase I cohort, and 65 additional patients then received carfilzomib at the maximum planned dose in a phase II study. The most common side effects were fatigue, nausea, anorexia, and dyspnea. No hepatotoxicity or ≥grade 2 peripheral neuropathy was reported. The half-life was determined to be <1 h, and one hour post treatment on day one of cycle two, proteasome CT-like activity in whole blood and PBMCs was inhibited by ≥80 %. Importantly, partial responses were reported in two patients (14 %) in the phase I study, with 21.5 % stable disease after four cycles in evaluable patients (n = 51) in the phase II cohort [124].
Another phase I/II trial included patients with newly diagnosed multiple myeloma (n = 53) and evaluated the efficacy and tolerability of the CRd combination treatment (carfilzomib + lenalidomide + dexamethasone) [125]. Carfilzomib was administered at 20, 27, or 36 mg/m2 twice weekly on consecutive days, lenalidomide was given at a dose of 25 mg/day daily for the first 21 days, and weekly dexamethasone was given at 40 mg during cycles 1–4 and 20 mg during any additional cycles. The maximum planned dose (carfilzomib 36 mg/m2) was expanded in the phase II study. Toxicities (grade 3–4) included anemia, thrombocytopenia, and neutropenia, hypophosphatemia, and hyperglycemia; no grade 3–4 peripheral neuropathy was observed and dose modification was not required in a majority of patients. A near-complete response was reported in 62 %, and complete response occurred in 42 % of patients (n = 53) after an average of 12 cycles. After a median follow-up of 13 months, the 24-month progression-free survival estimate was 92 % [125]. Thus, the combination of carfilzomib, lenalidomide, and dexamethasone is highly effective and well tolerated in treatment-naïve multiple myeloma patients.
Finally, a single-arm multicenter phase II was completed in relapsed/refractory multiple myeloma patients (n = 46) with at least two prior therapies [126]. Patients were given 20 mg/m2 carfilzomib IV on consecutive days twice weekly every 28 days for up to 12 cycles. The overall response and clinical benefit response rates were 16.7 % and 23.8 %, respectively, in the 42 evaluable patients, with seven partial responses. Median durations of response were 7.2 months and 13.8 months, respectively. Anemia, fatigue, and thrombocytopenia were the most common treatment-related adverse effects, and events of neuropathy were rare [126]. The promising results of this pilot study resulted in an amendment to test a higher dose in additional patients (PX-171-003-A1). During the PX-171-003-A1 study [127], patients (n = 266) received single-agent carfilzomib 20 mg/m2 IV twice weekly for 3 of 4 weeks in cycle one, followed by 27 mg/m2 for the remaining cycles (maximum = 12). The overall response rate (at least partial response) was 23.7 % with median duration of response of 7.8 months and median overall survival of 15.6 months. Manageable toxicities included anemia, nausea, fatigue, and thrombocytopenia, with grade 1–2 peripheral neuropathy in 12.4 % of evaluable patients [127]. Patients in this study had an average of five prior treatments, but the responses observed in this trial were quite durable and indicated that carfilzomib may be clinically beneficial in patients who fail on other chemotherapeutics, and based on this study, carfilzomib was approved by the FDA in July 2012 for relapsed and refractory multiple myeloma and mantle cell lymphoma.
1.3.4.2 Phase III Trials
Based on the promising phase I/II results, a randomized phase III study, FOCUS (Carfilzomib for Advanced Refractory Multiple Myeloma European Study), is being conducted to compare overall survival following single-agent carfilzomib treatment with best supportive care treatments in relapsed and refractory multiple myeloma patients who have received at least three previous treatments [128]. Enrolled patients (n ≈ 300) have responded to at least one prior therapy and are refractory to their most recent therapy. Patients were randomized to receive either IV carfilzomib, 20 mg/m2 on days 1–2 of cycle one, escalating to 27 mg/m2 on days 8, 9, 15, and 16 for the remaining cycles (up to 16), or an active BSC regimen consisting of corticosteroid treatment of prednisolone 30 mg, dexamethasone 6 mg, or equivalent every other day with optional oral cyclophosphamide 50 mg once daily. Treatment will continue until disease progression or unacceptable adverse events occur. The primary endpoint will be overall survival with secondary endpoints of progression-free survival, overall response rate, and safety [128]. Enrollment has been completed and the study has begun with anticipated completion in 2015 [NCT01302392; Onyx Therapeutics, Inc.].
Another randomized phase III, open-label, multicenter study is comparing CRd (carfilzomib + lenalidomide + dexamethasone) and Rd (lenalidomide + dexamethasone) regimens in patients with relapsed multiple myeloma [NCT01080391; Onyx Therapeutics, Inc.]. Approximately 750 patients have been randomized to receive either Rd (40 mg oral dexamethasone on days 1, 8, 15, and 22 + 25 mg oral lenalidomide on days 1–21 in 28-day cycles) or CRd (20 mg/m2 or 27 mg/m2 IV carfilzomib + 40 mg oral dexamethasone on days 1, 8, 15, and 22 + 25 mg oral lenalidomide on days 1–21 in 28-day cycles; carfilzomib will be discontinued after completion of 18 cycles). Progression-free survival is the primary endpoint of this study, and overall survival, overall response rate, response duration, disease control, safety, time to progression, and quality of life are secondary endpoints. Severity and incidence of adverse effects will also be compared between the two treatment regimens. This study is expected to be completed in early 2014 [NCT01080391; Onyx Therapeutics, Inc.].
1.4 Resistance to Proteasome Inhibitors
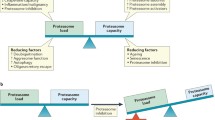
Unfortunately, although clinical success has been achieved with proteasome inhibitors, resistance has emerged as a limiting factor in their continued clinical use. Resistance to proteasome inhibitors, as well as other drugs, can be either inherent or acquired. Inherent resistance is resistance which exists within cells without any exposure to a drug. This type of resistance is fairly uncommon in cancer, but has been reported in regard to antibiotics. Acquired resistance occurs following exposure to a drug, generally by genetic mutations and overexpression of target proteins. The exact mechanisms by which cells become resistant to proteasome inhibitors have yet to be fully elucidated, but several studies have explored potential mechanisms (Fig. 1.3).

Potential mechanisms of proteasome inhibitor resistance. Several factors have been implicated in resistance to proteasome inhibitors. These include overexpression or mutation of β5 subunits, overexpression of heat shock proteins (HSPs), AKT pathway activation, altered expression of apoptosis- and growth-related proteins, altered autophagy pathways, and increased antioxidants
1.4.1 Inherent Resistance
In a study of relapsed/refractory acute leukemia patients who had progressed on prior treatments, bortezomib treatment resulted in minimal responses [129], suggesting the potential for inherent resistance to proteasome inhibitors due to prior treatments. The ECOG E2A02 trial conducted with newly diagnosed high-risk multiple myeloma patients showed no clinical response following single-agent bortezomib treatment [130]. Bortezomib also failed to show clinical benefit in several other hematological and solid tumors [129]. The lack of benefit from bortezomib as an initial treatment suggests that some tumors may simply be inherently resistant to treatment with proteasome inhibitors.
1.4.2 Acquired Resistance
While little is known about inherent resistance to proteasome inhibitors like bortezomib, a number of cell-based studies have elucidated putative mechanisms of acquired resistance either at the proteasome level or its downstream effectors. These include overexpression or mutation of the proteasomal β5 subunit, upregulation of heat shock proteins (HSPs), altered expression of apoptosis-related proteins, AKT pathway activation, overexpression of other growth-related proteins, altered autophagy pathways, and increased antioxidant levels (Fig. 1.3).
1.4.2.1 β5 Subunit Overexpression/Mutation
When human monocytic/macrophage THP1 cells were treated with increasing concentrations of bortezomib, up to 60-fold overexpression of proteasomal β5 subunit (PSMB5) protein was observed. Additionally, the overexpressed β5 contained an alanine–threonine mutation at position 49 in the highly conserved bortezomib-binding pocket [131]. Together the overexpression and mutation resulted in resistance to bortezomib as well as cross-resistance to β5-targeted cytotoxic peptides 4A6, MG132, MG262, and ALLN [131]. Interestingly, there were no marked changes in the baseline CT-like activity, and when the PSMB5 gene was silenced by siRNA, its sensitivity to bortezomib was restored [131]. A different set of mutations in the PSMB5 protein at positions 49 and 50, including Ala49Val, Ala49Thr, and Ala50Val, as well as the Ala49Thr mutation, were reported in T lymphoblastic lymphoma/leukemia cells developed from the Jurkat cell line when treated with increasing concentrations of bortezomib [132]. Similarly, human leukemia K562 cells have been shown to be more resistant to bortezomib compared to other leukemia and myeloma cell lines due to inherent overexpression of proteasomal β5. However, there is no direct evidence that these phenomena, i.e., mutations or β5 overexpression, are responsible for bortezomib resistance in vivo [133]. In fact, a multiple myeloma patient who rapidly developed resistance to bortezomib (evident by sudden and accelerated disease progression and death) had no mutations in the PSMB5 coding region, indicating that there may not be a correlation between bortezomib resistance and β5 mutation [134]. Further in-depth, large-scale studies are warranted to determine the role of β5 mutations and overexpression in in vivo resistance to proteasome inhibitors.
1.4.2.2 Upregulation of HSPs
The heat shock proteins are important in mediating resistance to apoptosis [135, 136], and many HSPs, especially HSP-72, are upregulated following proteasome inhibition. Several studies have reported dramatic HSP-72 upregulation resulting from treatment with a variety of proteasome inhibitors, including tripeptidyl aldehyde proteasome inhibitors, lactacystin and MG-132 [137–139]. Some studies demonstrated that proteasome inhibitor-mediated upregulation of HSP-72 was pro-apoptotic [138, 139], while others showed that MG-132 treatment caused an increase in HSP-72 expression and suppressed JNK activation, preventing JNK-mediated apoptosis by subsequent heat stress [139]. HSP-72 upregulation as a mechanism of proteasome inhibitor resistance was validated by a report showing that blocking HSP-72 by the introduction of an antisense oligonucleotide potentiated the apoptosis-inducing ability of MG-132 [140]. More recently, HSP-72 knockdown via siRNA was also shown to potentiate MG-132-induced cell death in prostate cancer cells [141].
Other HSP family members may also be involved in acquired resistance to proteasome inhibitors. Gene profiling of myeloma cells following bortezomib treatment revealed that several other HSPs are also induced by proteasome inhibition, including HSP-27, HSP-70, and HSP-90 [95, 142, 143]. One group demonstrated that bortezomib promotes increased phosphorylation of HSP-27 through activation of p38 and used p38 inhibitors and antisense-mediated downregulation of HSP-27 to reverse proteasome inhibitor resistance [142, 144]. HSP-70 has also been implicated in bortezomib resistance [143], and the flavonoid quercetin has been shown to inhibit HSP-70 mRNA and protein expression [145], suggesting that knockdown or inhibition of HSP-70 may also reverse acquired bortezomib resistance. Importantly, HSP-70 expression is high in pancreatic cancer cells, and inhibition of HSP-70 via quercetin treatment and siRNA knockdown both induced apoptosis in vitro [146].
Finally, HSP-90, which mediates the correct folding of various signal transduction intermediates, has also been implicated in proteasome inhibitor resistance [95]. In fact, synergistic cell death was observed in breast cancer cells treated with the combination of bortezomib and an HSP-90 inhibitor [147]. Additionally, in several preclinical multiple myeloma cell models, the combination resulted in increased apoptosis [147–149], but in pancreatic cancer the cell death resulting from combination treatment appears to be necrotic rather than apoptotic [150], indicating that the combination of HSP-90 antagonists with proteasome inhibitors must be further evaluated to more clearly understand their interactions with one another.
1.4.2.3 Altered Expression of Apoptosis-Related Proteins
As discussed previously, the Bcl-2 family members Bim [151] and Noxa [88] have been implicated in proteasome inhibitor-induced cell death in some cell types. While mutations causing inactivation of these proteins are rare in tumors [152, 153], cells may acquire resistance to proteasome inhibitors via epigenetic mechanisms. In fact, miR-17-92 and NFB2/p52 have recently been reported to repress Bim expression [154, 155], and Bmi-1-dependent methylation has been linked to Noxa expression [156]. The effects of Noxa and Bim could also be abrogated by overexpression of anti-apoptotic Bcl-2 proteins [157]. Small molecule inhibitors targeting Bcl-2, Bcl-xL (ABT-737), and MCL-1 (obatoclax) have been shown to significantly enhance bortezomib-induced cell death in various human cancer cell lines [158–160].
Other proteins that contribute to cell death, like p27, have also been reported to increase following proteasome inhibition [59, 161], and although inactivation of p27 through mutations is uncommon, its expression is often decreased due to increased Skp2 activity and proteasome-mediated degradation [162]. Methylation of p27 gene promoter occurs in almost 10 % of cancers [163], and proteasome inhibitor-resistant tumors may display increased methylation patterns that could contribute to the resistant phenotype. Additionally, p27 can be phosphorylated by AKT [164], which causes changes in its subcellular localization [164, 165], also potentially contributing to acquired proteasome inhibitor resistance.
1.4.2.4 Akt Pathway Activation
The pro-survival PI3K/Akt pathway is constitutively active in many cancers, and several pathways have been implicated in Akt activation, including amplification of PI3K [166] or Akt [167], growth factor receptor signaling [168], PTEN deletion [169], or mutation of Ras family members [170]. Akt activation, both constitutive and induced, can impair the activity of bortezomib [95, 171, 172]. Bortezomib can also directly activate Akt in some cell lines [173], and Akt inhibitors (both direct and indirect), including the PKC antagonist enzastaurin [174], PI3K inhibitors like perifosine, and the Raf inhibitor sorafenib [172] have been shown to enhance bortezomib-induced apoptosis. Additionally, Akt activation is regulated by receptor tyrosine kinase growth factor receptors like EGFR, and Akt activation can be reversed with selective RTK inhibitors in these cells [175], leading to increased bortezomib sensitivity [176, 177].
1.4.2.5 Overexpression of Other Growth-Related Proteins
Resistance to bortezomib has also been attributed to the overexpression of some proteins that are involved in cell growth, including interleukin-6 (IL-6) and insulin-like growth factor 1 (IGF-1), which are thought to confer resistance via activation of NF-κB through the PI3K/Akt and Raf/MEKKl pathways [178, 179]. IL-6 has been shown to play an important role in regulating drug sensitivity in multiple myeloma cells through inhibition of miRNA expression in bone marrow stromal cells (BMSCs) [180, 181]. In addition, IGF-1 receptor levels have also been shown to be high in multiple myeloma, and this overexpression, as well as increased IGF-1 levels, are associated with disease progression and poor patient prognosis [182, 183]. Increased IGF-1 signaling has been directly implicated in the resistance phenotype of bortezomib-resistant multiple myeloma cells with no β5 mutations. The role of IGF-1 signaling was further validated by gene expression profiling which showed that genes activated by IGF-1 were constitutively expressed in these bortezomib-resistant multiple myeloma cells. Importantly, blocking PI3K and mTOR downstream of IGF-1 partially overcame the bortezomib resistance. Direct inhibition of IGF-1R (insulin-like growth factor 1 receptor) was also able to sensitize cultured cells, in vivo models, and patient samples to bortezomib treatment [184], suggesting that combining bortezomib with IGF-1R inhibitors may be a promising strategy to prevent or overcome proteasome inhibitor resistance.
The receptor tyrosine kinase c-Met is also overexpressed in human myeloma cell lines and has been shown to promote drug resistance. One study showed that knockdown of c-Met in U266 human multiple myeloma cells enhanced their sensitivity to bortezomib via inhibition of the Akt/mTOR pathway [185]. Increased Akt/mTOR phosphorylation was also reported in bortezomib-resistant mantle cell lymphoma cells, and dual inhibition of PI3K and mTOR overcame acquired bortezomib resistance by suppressing the activated Akt/mTOR pathway [186]. Microarray analysis has also identified Rad (Ras associated with diabetes) as a potential factor in proteasome inhibitor resistance. Rad levels were increased in bortezomib-resistant Jurkat-R cells compared to parental controls, and knockdown resulted in induction of the mitochondrial apoptotic pathway via Noxa/Bcl-2, thus overcoming bortezomib resistance in these cells [187].
1.4.2.6 Altered Autophagy Pathways
Proteasome inhibitors are known to activate autophagy, but the exact role of autophagy in cancer cell death is a controversial one [188]. Studies have reported that inhibition of autophagy can both inhibit [189] and promote [190] proteasome inhibitor-mediated cell death depending on cell type. This may be due to the variable effects of these autophagy inhibitors, whereby they block macroautophagy but are unable to inhibit chaperone-mediated autophagy, which may play a critical role in clearing protein aggregates in some cells. These protein aggregates may be transferred to the lysosome via aggresomes during chaperone-mediated autophagy. HDAC6 is necessary for aggresome formation following proteasome inhibition, and HDAC inhibition has been shown to enhance proteasome inhibitor-induced cell death in proteasome inhibitor-sensitive cells and to reverse resistance in resistant cells [191, 192]. The combination of HDAC inhibition with proteasome inhibition has been extensively studied, and results suggest that this is the most promising combination. In fact, one phase I clinical trial investigating the combination of bortezomib and the pan-HDAC inhibitor SAHA was completed in patients with relapsed/refractory multiple myeloma [193], and another was completed in patients with solid tumors [NCT00310024; National Cancer Institute]. A phase II trial also investigated the combination in patients with progressive, recurrent glioblastoma [NCT00641706; National Cancer Institute], and results are forthcoming.
1.4.2.7 Increased Antioxidants
The production of ROS appears to play a role in cell death induced by some proteasome inhibitors, which suggests that antioxidant protection mechanisms may also contribute to proteasome inhibitor resistance. Sensitivity of multiple myeloma cells has been shown to increase following depletion of intracellular reduced glutathione by buthionine sulfoximine treatment [194]. Glutathione may promote resistance by acting as a cofactor for GSH-dependent enzymes; protein disulfide isomerase, glutathione peroxidase, and vitamin C, for example, inhibit toxicity induced by proteasome inhibitors, among other factors [195–199]. Thus, antioxidant levels may impact proteasome inhibitor sensitivity, so regulating these levels may be a strategy to overcome resistance.
1.5 Measures to Overcome Proteasome Inhibitor Resistance
1.5.1 Design of Novel Proteasome Inhibitors
Determination of exact molecular mechanisms of proteasome inhibitor resistance would help in the design of effective therapeutic strategies to overcome proteasome inhibitor resistance. Resistance at the proteasome level could be addressed by designing better, more potent inhibitors than bortezomib and carfilzomib. A new generation of irreversible proteasome inhibitors might be helpful in partially overcoming bortezomib resistance due to β5 overexpression. Targeting sites different than those targeted by bortezomib could also be explored for the design and development of next-generation proteasome inhibitors. Unfortunately, however, these next-generation inhibitors may not be effective in overcoming resistance due to downstream factors [133].
Bortezomib is administered intravenously and modification of its pharmacokinetic parameters affecting stability, metabolism, and tissue bioavailability may be a useful strategy for overcoming resistance [133]. Some orally bioavailable reversible second-generation inhibitors, like MLN9708 and CEP 18770, that can be hydrolyzed to an active form have been developed, and encouraging results in cultured cells and animal models have advanced these compounds to phase I clinical trials [200–202]. However, their similarities to bortezomib in terms of mode of action might hinder their ability to overcome bortezomib/carfilzomib resistance, but they may offer advantages in terms of pharmacokinetics and patient compliance due to their oral route of administration, dosing flexibility and convenience [133].
Structure–activity relationship (SAR) studies were conducted to develop orally bioavailable carfilzomib-like agents, and led to the discovery and development of ONX0912, a truncated carfilzomib analog with comparable potency, selectivity, and anticancer activities to its parent compound in vitro and in vivo in animal models [203, 204]. Carfilzomib and ONX0912 can be degraded by proteases and peptidases in the plasma due to their peptide-like structures, which decreases their half-life and efficacy [133]. Therefore, nonpeptidic, irreversible proteasome inhibitors like NPI0052 (salinosporamide/marizomib) with better bioavailability have been developed [205]. NPI0052 is a β-lactone-γ-lactam isolated from the marine bacterium Salinispora tropica that shows potent irreversible inhibition of all three proteolytic activities of the proteasome through the formation of very stable acyl-ester bonds. Due to its anticancer activities in cell culture and animal models, NPI0052 has advanced into clinical trials for hematological and solid tumor malignancies (Fig. 1.4) [116, 206]. Larger in vitro studies should be conducted to further enhance the understanding of mechanisms associated with resistance to proteasome inhibitors. These studies could lead to the development of personalized therapeutic approaches by identifying subgroups of patients who are more likely to respond well or fail to respond to particular proteasome inhibitors.

Potential strategies to overcome proteasome inhibitor resistance. Many strategies have been suggested for overcoming proteasome inhibitor resistance. These include designing novel proteasome inhibitors, targeting sites outside the catalytic center (such as the 19S regulatory cap, E1, E2s, or E3s), targeting the immunoproteasome, combination strategies (like proteasome inhibitors + HSP or HDAC inhibitors), and using metal-based or natural compounds as inhibitors
1.5.2 Combination Strategies
In addition to designing new compounds to target the proteasome, combining current proteasome inhibitors with distinct modes of action could be an effective strategy to overcome resistance to particular proteasome inhibitors. For instance, NPI0052 in combination with bortezomib has been shown to induce synergistic cytotoxicity in vitro in cultured multiple myeloma cells as well as in multiple myeloma cells isolated from patients, and in multiple myeloma mouse models (Fig. 1.4) [207]. Alternatively, proteasome inhibitors could be combined with other chemotherapeutic agents targeting factors downstream of the proteasome to enhance efficacy. In fact, lenalidomide and NPI0052 in combination display synergistic anti-multiple myeloma activities in cultured and patient multiple myeloma cells, as well as in tumor xenografts in mice [208].
Because HSP overexpression has been suggested as a potential mechanism by which cells become resistant to proteasome inhibitors, combining HSP inhibitors with proteasome inhibitors may sensitize resistant cells to proteasome inhibition (Fig. 1.4). In fact, the combination of the HSP-90 inhibitor tanespimycin (17-allylamino-17-demethoxy-geldanamycin [17-AAG]; geldanamycin analog) with bortezomib resulted in bortezomib-mediated cell death and tumor regression in multiple myeloma cell and xenograft models, respectively [148]. A phase I trial combining these drugs has also been completed in relapsed/refractory multiple myeloma patients, some of whom had progressed to bortezomib resistance [209]. An overall response rate of 27 % was achieved, suggesting that this combination is effective at reversing bortezomib resistance in multiple myeloma.
As discussed previously, some HDACs have also been implicated in proteasome inhibitor resistance, indicating that combining HDAC inhibitors with proteasome inhibitors may be a promising strategy to overcome proteasome inhibitor resistance (Fig. 1.4). Several preclinical studies have reported synergism between these types of drugs. For example, knockdown of HDAC1 enhanced bortezomib-mediated apoptosis, while overexpression of HDAC1 resulted in bortezomib resistance in multiple myeloma cells and treatment with the HDAC inhibitor romidepsin restored bortezomib sensitivity in HDAC1 overexpressing cells and tumor xenografts [210]. Additionally, the combination of bortezomib and tubacin (HDAC6-specific inhibitor) inhibited proliferation in cultured multiple myeloma cells [211]. Selective inhibition of HDAC6 by ACY-1215, both alone and in combination with bortezomib, has also been shown to suppress multiple myeloma cell growth in a xenograft mouse model, again suggesting a synergistic relationship [212]. Finally, the combination of bortezomib and the HDAC inhibitor SAHA (vorinostat) has also been evaluated in a phase I clinical trial in patients with advanced multiple myeloma, the results of which reported an overall response rate of 30 % in bortezomib-resistant patients [193]. Another phase I trial investigating the efficacy of SAHA in combination with bortezomib resulted in one partial remission and one minimal response among three bortezomib-resistant patients [213]. Another trial in multiple myeloma patients combined the natural HDAC inhibitor romidepsin with bortezomib, and 60 % partial and 8 % complete responses were achieved [214]. The Vantage 095 phase IIb trial reported an overall response rate of 18 % with a median duration of response of 6.3 months in patients with bortezomib-refractory relapsed multiple myeloma [215]. A randomized, double-blind phase III trial, Vantage 088, compared SAHA or placebo in combination with bortezomib in 637 myeloma patients and reported an overall response rate of 54 % in the group treated with the SAHA–bortezomib combination, compared with 41 % in the placebo group [216]. Thus, HDAC inhibition may also be a promising strategy for overcoming proteasome inhibitor resistance in refractory cancers.
1.5.3 Immunoproteasome-Specific Inhibitors
The immunoproteasome is an inducible proteasome variant primarily expressed in lymphocytes and monocytes, as well as in cells exposed to inflammatory cytokines such as interferon-γ (IFN-γ) and tumor necrosis factor-α (TNF-α). The immunoproteasome is involved in the production of peptides for major histocompatibility complex-1 (MHC-1). During assembly of the immunoproteasome, the constitutive β1, β2, and β5 subunits are replaced by β1i (PSMB9/LMP2), β2i (PSMB10/LMP-10/MECL1), and β5i (PSMB8/LMP7), respectively. These are associated with the 11S peptidase regulator and the remaining subunits of the constitutive proteasome, which leads to alterations in substrate specificity [217, 218]. Although there may be some functional redundancy between the two proteasome isoforms [219–221], CT-like (β5i) and trypsin-like (β2i) proteolytic activities are upregulated, and PGPH-like activity (β1i) is decreased in the immuno-20S compared to the constitutive 20S [222]. Many conventional proteasome inhibitors (e.g., bortezomib, carfilzomib, NPI0052) designed to target the constitutive proteasome are also able to inhibit the immunoproteasome and thus offer broader activity at the cost of specificity, which may contribute to adverse effects [223].
Therefore, specifically targeting the immunoproteasome in hematological malignancies might be a novel approach toward increasing effectiveness and reducing negative off-target effects (Fig. 1.4) [223, 224], which ultimately led to the development of IPSIs. One such agent, IPSI-001, has shown selectivity for the immunoproteasome over the constitutive proteasome in binding assays and has been shown to induce apoptosis in a dose-dependent manner in patient derived cells of hematologic malignancies. IPSI-001 was also able to overcome resistance to conventional chemotherapeutic agents like doxorubicin, melphalan and, most importantly, bortezomib in vitro [224]. Other agents like the β5i selective tripeptide epoxyketone-based immunoproteasome inhibitors PR-924 [225] and PR-957 [226] have also shown promising results in preclinical cell culture and animal studies. Synthetic analogs of the epoxyketone dihydroeponemycin were also developed as molecular probes to study the effects of β1i (LMP2) inhibition. The combination of β1i inhibitors with the β5i inhibitor lactacystin resulted in enhanced inhibition of total CT-like activity compared to each agent alone. These inhibitors have also shown growth inhibitory effects in PC-3 prostate cancer cells overexpressing β1i [227]. Taken together, these results indicate that targeting the immunoproteasome may be an effective strategy for overcoming resistance to conventional proteasome inhibitors.
1.5.4 Targeting Sites Other than the Catalytic Center
1.5.4.1 E1, E2s, and E3s
Targeting other factors in the UPP may also prove effective in overcoming resistance associated with inhibitors of the 20S catalytic core (Fig. 1.4). Although inhibiting the ubiquitin E1 enzyme was initially disregarded due to potential lethality, the identification of two natural E1 inhibitors, panepophenanthrin and himeic acid, has suggested that this may indeed be a viable strategy. Both inhibitors specifically inhibit the formation of E1 ubiquitin thioester intermediates [228, 229]. Additionally, PYR-41, a synthetic pyrazone derivative, with E1 inhibitory activity that prevents protein degradation and cytokine-mediated activation of NF-κB has also been developed [230]. Another compound, PYZD-4409 induced cell death in malignant cells as well as in a leukemia mouse model, potentially by a mechanism similar to ER stress induced by proteasome inhibitors [231]. Similarly, cell-based screening identified NSC624206 as an E1 inhibitor, though more studies are necessary to determine its molecular effects [232]. Following the observation that functional knockdown of the E2 Ubc13 results in increased p53 activity [233], inhibition of the E2 enzymes has also been explored. In fact, leucettamol A, a natural compound, has been reported to inhibit the interaction between the E2 Ubc13 and the inactive conjugating enzyme variant Uev1A, which is required for efficient poly-ubiquitin chain formation [234].
Perhaps one of the most widely researched strategies for targeting factors upstream of the proteasomal catalytic core is inhibition of ubiquitin E3 ligases (Fig. 1.4), likely due to their role in identifying target proteins for ubiquitination. E3 ligases are divided into one of three classes, RING, HECT, and U-box, based on domain structure and mechanisms of target recognition. The p53-specific RING-type E3 MDM2/HDM2 is a popular target for inhibition, due to its high frequency of overexpression in human cancers [235]. Indeed, nutlin-3, a MDM2 small molecule inhibitor, has been shown to suppress tumor progression in mouse xenograft models [236], suggesting that MDM2 is a promising target. Additionally, in bortezomib-sensitive multiple myeloma and epithelial carcinoma cells, nutlin-3 in combination with bortezomib resulted in additive and synergistic cytotoxic effects, respectively [237]. Some natural products, including chlorofusin and (-)-hexylitaconic acid, that inhibit the interaction between MDM2 and p53 have also been identified [238–241]. Interestingly, disulfiram and its derivatives have also been investigated for their ability to inhibit zinc finger- and RING-finger-containing ubiquitin E3 ligases [242]. Thus, inhibition of upstream UPP factors should be further investigated as this may be a viable strategy for overcoming resistance to 20S inhibitors.
1.5.4.2 19S Regulatory Subunit
Inhibition of proteasomal regulators may also be effective in overcoming resistance to conventional proteasome inhibitors, as this inhibition should only hinder some proteasomal functions (Fig. 1.4). Indeed, screening of a library of purine analog-capped peptoids identified RIP-1 (regulatory particle inhibitor peptoid-1) as an inhibitor of protein unfolding through targeting of the ATPase Rpt4 [243, 244]. Reports have indicated that ubistatin A is capable of blocking recruitment of ubiquitinated proteins to the 26S proteasome by binding ubiquitin chains, ultimately suppressing proteasome-mediated proteolysis [245], indicating that ubiquitin chain receptors may also be good drug targets. Inhibition of deubiquitinase activity of the regulatory particle could be another useful strategy, and b-AP15, a small molecular weight compound that inhibits deubiquitinating enzymes like USP14 and UCHL5, but not POH1, has shown anticancer activity in solid tumor models [246]. Thus, it is clear that factors regulating the 20S catalytic core are good drug targets, and further investigation into this strategy as a way to overcome inhibitors of the 20S core would be very worthwhile.
1.6 Nontraditional Options Targeting the 20S Core
1.6.1 Metals in Cancer Development and Therapy
Just as proteasome activity levels have been shown to be altered in cancer, so have levels of various metals like copper [247–252] and zinc [253–256], a discovery which has led to extensive research regarding the roles of these metals in the development of human cancers as well as their potential as anticancer therapeutics.
The discovery that some metal-based compounds, like cisplatin, possess potent anticancer properties, coupled with the importance of copper and zinc to essential biological processes like tumorigenesis, has led to the investigation into copper and zinc as metal centers in anticancer drugs. Since its discovery over four decades ago, cisplatin has cured over 90 % of testicular cancer cases, and it has also played a critical role in the treatment of various other cancers, including lymphoma, melanoma, bladder, cervical, and ovarian [257]. Unfortunately, although cisplatin use has proven effective, it has also been associated with toxicity and resistance, which has limited its use [258, 259] and prompted the search for less toxic metal-based drugs, including second-generation platinum drugs, as well as complexes containing metals like cobalt, copper, gallium, gold, tin, and zinc, among others.
1.6.2 Metal-Based Complexes as Proteasome Inhibitors
1.6.2.1 Gold-Containing Complexes
Because of the successful use of gold compounds in other diseases [260, 261], gold compounds have also been investigated for their potential anticancer activity (Fig. 1.4). Gold (I) complexes, including auranofin analogs, exhibited potent cytotoxic activity against B16 melanoma and P388 leukemia cells [262], but phosphine-gold(I) thiosugars were the most potent, and while active against leukemia in vivo, these analogs were completely inactive in solid tumor models [263]. Gold(III) complexes have also been investigated, in spite of initial trepidation due to their high redox activity and poor stability. Au(III) is expected to be reduced to Au(I) and metallic Au in the reducing tumor microenvironment, potentially making Au(III) complexes less effective [264]. Various Au(III) compounds with ligand platforms containing nitrogen atoms as donor groups [265], exhibiting a superior chemotherapeutic index, increased cytotoxicity, and fewer toxic side effects than cisplatin [264], were investigated for their antitumor abilities. One example is Au (DMDT) Br2, which significantly inhibited CT-like activity in purified 20S proteasome (IC50 = 7.4 μM) and 26S proteasome in intact MDA-MB-231 breast cancer cells (10–20 μM) and breast tumor xenografts [266]. Another gold(III) compound, AUL12, was also shown to exhibit potent proteasome-inhibitory and cell death-inducing activities in MDA-MB-231 breast cancer cells (IC50 = 4.5 μM, 70 % inhibition). Interestingly, treatment with this Au(III) compound was associated with redox processes, indicating that induction of oxidative stress may be partially responsible for the cytotoxicity of gold(III) compounds [267].
1.6.2.2 Metal Chelators as Proteasome Inhibitors
The success of metal-containing drugs, along with the functional importance of metals like copper and zinc to normal cellular function, has resulted in studies exploring chelation of these essential metals with chelators like dithiocarbamate and hydroxyquinolone compounds, several of which have been previously approved for the treatment of myriad diseases, such as AIDS, alcoholism, and bacterial and fungal infections [268–270].
1.6.2.2.1 Dithiocarbamates
Dithiocarbamate compounds, including disulfiram, are known to form metal complexes, a property that has been applied as a potential strategy to target the UPP in cancer. Disulfiram (tetraethylthiuram disulfide, DSF) is an irreversible aldehyde dehydrogenase inhibitor that is one of two drugs approved by the USFDA for the treatment of alcoholism [271–273]. When complexed with copper, disulfiram can potently inhibit both purified 20S (IC50 = 7.5 μM) and intact 26S proteasome in MDA-MB-231 breast cancer cell lysates (20 μM; >95 % inhibition), as well as inducing apoptosis in the cultured cells [274]. DSF alone, however, had no effect, which is unsurprising, since cultured cells do not express high levels of copper. Significant inhibition (74 %) of tumor growth was also observed in female athymic nude mice bearing MDA-MB-231 tumor xenografts, associated with an 87 % decrease in CT-like activity [274]. Together, the results indicate that the increased copper levels observed in human tumors may be exploited as an anticancer mechanism (Fig. 1.4).
The results of these and other preclinical studies of DSF have also led to a number of clinical trials investigating the use of DSF in humans. One phase I/II clinical trial evaluated the efficacy of DSF in stage IV metastatic melanoma patients [NCT00256230; UC-Irvine] and another examined the effects of DSF on PSA levels in recurrent prostate cancer patients [NCT01118741; Johns Hopkins University]. Two other trials investigated the effects of DSF in combination treatments. The first evaluated the toxicity profile and safety of coadministration of DSF and copper gluconate in refractory malignancies with liver metastases [NCT00742911; Huntsman Cancer Institute], and the other determined the effects of addition of DSF to current chemotherapeutic treatments in patients with non-small cell lung cancer [NCT00312819; Hadassah Medical Organization]. All of these trials have been completed, but results are as yet unavailable.
1.6.2.2.2 Hydroxyquinolones
Hydroxyquinolones are another class of metal-chelating compounds that have been investigated for their anticancer properties. One example is clioquinol (5-chloro-7-iodo-8-hydroxyuinoline, CQ), a lipophilic compound that can form stable complexes with copper (II) [275]. CQ has been shown to reduce and prevent the formation of amyloid plaques in Alzheimer’s disease transgenic mice [276], a discovery that led to two clinical trials that validated the efficacy of CQ in Alzheimer’s disease with no visible toxicity [277, 278]. Consequently, CQ is currently used for the treatment of Alzheimer’s and Huntington’s diseases [279, 280]. A CQ–Cu complex (1:1 molar ratio) inhibited both purified 20S (IC50 = 2.5 μM) and intact proteasome (20 μM) in LNCaP and C4-2B prostate cancer cells (82 % and 83 %, respectively). Additionally, mice bearing C4-2B xenografts treated with CQ exhibited significant tumor growth inhibition (66 %), as well as inhibition of angiogenesis and the proteasome and induction of apoptosis [281]. These data clearly indicate that compounds like DSF and CQ require copper to be transported into cancer cells in order to exert their proteasome-inhibitory and apoptosis-inducing abilities [282], but that when copper is present, they are quite potent proteasome inhibitors that are minimally toxic toward normal cells (Fig. 1.4) and therefore, they may be exploited as potential novel strategies for overcoming resistance to traditional proteasome inhibitors like bortezomib.
1.6.3 Natural Compounds as Proteasome Inhibitors
While much emphasis has been placed on the development of synthetic proteasome inhibitors to overcome resistance to proteasome inhibitors, the use of natural compounds and their analogs or derivatives might be a better strategy as many phytochemicals and marine products have shown proteasome-inhibitory and subsequent anticancer activities (Fig. 1.4) [283]. Some examples of natural products investigated for their potential as proteasome inhibitors include Withaferin A, celastrol, agosterols, green tea polyphenols, and apigenin. Withaferins are isolated from the medicinal plant “Indian winter cherry” or “Indian ginseng” (Withania somnifera), and have been widely used in traditional Indian “Ayurveda” medicine. Specifically, Withaferin A has been reported to possess anticancer abilities, which may be partly attributed to inhibition of CT-like activity [284]. Celastrol, a triterpene isolated from the Chinese “Thunder of God Vine” (Tripterygium wilfordii) has also shown proteasome-inhibitory activity leading to the accumulation of ubiquitinated proteins and proteasomal target proteins in both androgen receptor positive and negative prostate cancer cell lines [285]. Agosterols, isolated from the marine sponge Acanthodendrilla sp., have shown inhibition of CT-like activity in rat proteasome in the low micromolar range and also induced cytotoxicity in HeLa cervical cancer cells [286]. The proteasome-inhibitory, apoptosis-inducing activities of green tea polyphenols and apigenin have been more extensively studied, with both advancing to clinical trials.
1.6.3.1 Green Tea Polyphenols
Tea, derived from the Camellia sinensis plant, is the most popular beverage in the world after water. Tea comes in many varieties, including green, black, and oolong, all of which contain many beneficial compounds. The most potent of these are polyphenols, which are characterized by the presence of more than one phenol group per molecule and are believed to provide the coloring in many plants [287].
The most active polyphenol in tea is (-)-epigallocatechin-3-gallate, or (-)-EGCG, which has been shown to possess anticancer activity in several cancer types, including bladder, breast, and B-cell malignancies (Fig. 1.4) [288–290]. The proteasome-inhibitory activity of (-)-EGCG has been explored in vitro and in vivo. (-)-EGCG inhibited CT-like activity in both purified 20S (IC50 = 86–194 nM) and intact 26S (from Jurkat leukemia, LNCaP and PC-3 prostate cancer, and MCF-7 breast cancer cell extracts) proteasome, and increased ubiquitinated proteins, p27, and IκB-α were also observed [291]. Unfortunately, however, a recent study using experimental multiple myeloma models has revealed a direct interaction between (-)-EGCG and bortezomib that inhibits the efficacy of bortezomib [292], but whether green tea consumption affects the efficacy of bortezomib therapy in multiple myeloma patients needs to be confirmed.
The interesting preclinical data, coupled with the popularity of tea, have led to clinical trials using (-)-EGCG and other green tea polyphenols. One phase I trial evaluated the optimal dose and tolerability of (-)-EGCG in previously untreated, asymptomatic chronic lymphocytic leukemia patients, and results indicated that (-)-EGCG is tolerable and does result in some clinical benefit [293]. Another phase I study investigated the effects of (-)-EGCG supplementation on serum levels of prostate cancer biomarkers [294], with all prostate cancer-associated biomarkers, including hepatocyte growth factor (HGF), vascular endothelial growth factor (VEGF), insulin-like growth factor (IGF)-1, IGF-binding protein-3 (IGFBP-3), and prostate-specific antigen (PSA) decreasing significantly after treatment with no elevation of liver function enzymes. Therefore, even short-term (-)-EGCG treatment may be clinically beneficial to prostate cancer patients [294].
In addition, a number of phase I and II trials examining the effects of (-)-EGCG treatment in various types of cancer, including nonmetastatic bladder, breast, cervical, colorectal, prostate, non-small cell lung, and uterine carcinomas as well as multiple myeloma, are currently recruiting patients or are ongoing. Other studies are investigating the potential preventive effects of (-)-EGCG in patients at risk for cervical, esophageal, and lung cancers. Finally, still others are evaluating (-)-EGCG in combination with the EGFR inhibitor erlotinib in non-small cell lung cancer and premalignant lesions of the head and neck. The preclinical and clinical data, as well as the numerous recruiting/ongoing clinical trials, clearly support the use of (-)-EGCG as a chemopreventive or therapeutic agent, and its potential use following progression to resistance to other proteasome inhibitors should be explored.
1.6.3.2 Apigenin
Apigenin (5,7,4-trihydroxyflavone) has also been shown to possess antioxidant, antimutagenic, and chemopreventive properties (Fig. 1.4). Apigenin is a dietary flavonoid found in various natural products including celery seed, chamomile flowers, grapes, and parsley [295–298], and although the mechanism is not fully understood, chemoprevention by apigenin has been reported in several cancers including cervical [299], lung [300], prostate [301], and skin [302].
Studies have shown that the carbonyl carbon in the C4 position of apigenin binds to the β5 subunit in a suitable orientation for nucleophilic attack by the N-terminal Thr1 [303]. Apigenin potently inhibits CT-like activity of purified 20S (IC50 = 1.8–2.3 μM) and intact proteasome in Jurkat leukemia cell lysates (1–10 μM), with little to no toxicity in immortalized, non-transformed natural killer cells [303]. Proteasome inhibition-associated accumulation of ubiquitinated proteins and apoptosis-associated morphological changes, activation of caspase-3/-7, and cleavage of PARP were also observed, and similar proteasome inhibition and apoptosis induction were observed in breast cancer MBA-MD-231 cells and tumors, with no significant changes in body weight following apigenin treatment, indicating low toxicity in vivo [304]. The promising preclinical data have led to examination of the efficacy of apigenin in human patients. In one prospective study in patients with resected colon cancer or who had undergone polypectomy, one group received a flavonoid mixture and the other served as a matched control. The results suggested that dietary consumption of flavonoids like apigenin may reduce the risk of colorectal cancer [305]. This preventive effect has not been fully validated, however, and further studies are necessary to determine if apigenin could be used to overcome resistance to other proteasome inhibitors.
1.7 Conclusion
The clinical approval of the proteasome inhibitors bortezomib and carfilzomib was validation of the importance of the UPP as a critical anticancer therapeutic target. Unfortunately, intrinsic and acquired resistance in tumor cells is associated with the use of clinical proteasome inhibitors, so it is important to find novel strategies to overcome this resistance. Resistance may be due to a variety of factors including overexpression or mutation of the β5 subunit, overexpression of HSPs, altered expression of apoptosis- and growth-related proteins, Akt pathway activation, altered autophagy, and increased antioxidant levels. Toward the goal of overcoming this resistance, novel small molecules have been tested for their ability to selectively target and inhibit components of the UPP other than the catalytic 20S core. These novel targets include the 19S regulatory cap(s), deubiquitinating enzymes, and the enzymes involved in the ubiquitination cascade (E1, E2s, and E3s). Inhibitors containing metal centers and those derived from natural products may also be viable options for overcoming resistance associated with clinical proteasome inhibitors. Finally, targeting the specialized immunoproteasome also has potential as a valuable new strategy that may be more specific and could overcome resistance to constitutive 20S proteasome inhibitors. Therefore, the UPP is a promising target for cancer therapy, but further studies are necessary to develop inhibitors that can avoid the resistance associated with clinical proteasome inhibitors.
Abbreviations
- BMSC:
-
Bone marrow stromal cell
- CQ:
-
Clioquinol
- CT:
-
Chymotrypsin
- DSF:
-
Disulfiram
- DUB:
-
Deubiquitinating enzyme
- (-)-EGCG:
-
(-)-Epigallocatechin-3-gallate
- ER:
-
Endoplasmic reticulum
- FDA:
-
Food and Drug Administration
- HDAC:
-
Histone deacetylase
- HSP:
-
Heat shock protein
- IGF-1:
-
Insulin-like growth factor 1
- IL-6:
-
Interleukin-6
- IPSI:
-
Immunoproteasome-specific inhibitor
- MAPK:
-
Mitogen-activated protein kinase
- MHC:
-
Major histocompatibility complex
- NSCLC:
-
Non-small cell lung cancer
- PBMC:
-
Peripheral blood mononuclear cell
- PGPH:
-
Peptidyl-glutamyl peptide-hydrolyzing
- RIP-1:
-
Regulatory particle inhibitor peptoid-1
- ROS:
-
Reactive oxygen species
- SAR:
-
Structure–activity relationship
- TNF-α:
-
Tumor necrosis factor-α
- UIM:
-
Ubiquitin-interacting motif
- UPP:
-
Ubiquitin–proteasome pathway
References
Ciehanover A, Hod Y, Hershko A (1978) A heat-stable polypeptide component of an ATP-dependent proteolytic system from reticulocytes. Biochem Biophys Res Commun 81: 1100–1105
Hershko A, Ciechanover A, Heller H, Haas AL, Rose IA (1980) Proposed role of ATP in protein breakdown: conjugation of protein with multiple chains of the polypeptide of ATP-dependent proteolysis. Proc Natl Acad Sci U S A 77:1783–1786
Nalepa G, Rolfe M, Harper JW (2006) Drug discovery in the ubiquitin-proteasome system. Nat Rev Drug Discov 5:596–613
Baker SP, Grant PA (2005) The proteasome: not just degrading anymore. Cell 123:361–363
Collins GA, Tansey WP (2006) The proteasome: a utility tool for transcription? Curr Opin Genet Dev 16:197–202
Krogan NJ, Lam MH, Fillingham J, Keogh MC, Gebbia M, Li J, Datta N, Cagney G, Buratowski S, Emili A, Greenblatt JF (2004) Proteasome involvement in the repair of DNA double-strand breaks. Mol Cell 16:1027–1034
Ciechanover A (1998) The ubiquitin-proteasome pathway: on protein death and cell life. EMBO J 17:7151–7160
Rivett AJ (1998) Intracellular distribution of proteasomes. Curr Opin Immunol 10:110–114
Hirsch C, Ploegh HL (2000) Intracellular targeting of the proteasome. Trends Cell Biol 10:268–272
Wojcik C, DeMartino GN (2003) Intracellular localization of proteasomes. Int J Biochem Cell Biol 35:579–589
Adams J (2004) The proteasome: a suitable antineoplastic target. Nat Rev Cancer 4:349–360
Peters JM, Cejka Z, Harris JR, Kleinschmidt JA, Baumeister W (1993) Structural features of the 26 S proteasome complex. J Mol Biol 234:932–937
Baumeister W, Walz J, Zuhl F, Seemuller E (1998) The proteasome: paradigm of a self-compartmentalizing protease. Cell 92:367–380
Groll M, Ditzel L, Lowe J, Stock D, Bochtler M, Bartunik HD, Huber R (1997) Structure of 20S proteasome from yeast at 2.4 A resolution. Nature 386:463–471
Groll M, Heinemeyer W, Jager S, Ullrich T, Bochtler M, Wolf DH, Huber R (1999) The catalytic sites of 20S proteasomes and their role in subunit maturation: a mutational and crystallographic study. Proc Natl Acad Sci U S A 96:10976–10983
DeMartino GN, Slaughter CA (1999) The proteasome, a novel protease regulated by multiple mechanisms. J Biol Chem 274:22123–22126
Goldberg AL, Cascio P, Saric T, Rock KL (2002) The importance of the proteasome and subsequent proteolytic steps in the generation of antigenic peptides. Mol Immunol 39: 147–164
Nickell S, Beck F, Scheres SH, Korinek A, Forster F, Lasker K, Mihalache O, Sun N, Nagy I, Sali A, Plitzko JM, Carazo JM, Mann M, Baumeister W (2009) Insights into the molecular architecture of the 26S proteasome. Proc Natl Acad Sci U S A 106:11943–11947
da Fonseca PC, Morris EP (2008) Structure of the human 26S proteasome: subunit radial displacements open the gate into the proteolytic core. J Biol Chem 283:23305–23314
Hartmann-Petersen R, Tanaka K, Hendil KB (2001) Quaternary structure of the ATPase complex of human 26S proteasomes determined by chemical cross-linking. Arch Biochem Biophys 386:89–94
Fu H, Doelling JH, Rubin DM, Vierstra RD (1999) Structural and functional analysis of the six regulatory particle triple-A ATPase subunits from the Arabidopsis 26S proteasome. Plant J 18:529–539
Rubin DM, Glickman MH, Larsen CN, Dhruvakumar S, Finley D (1998) Active site mutants in the six regulatory particle ATPases reveal multiple roles for ATP in the proteasome. EMBO J 17:4909–4919
Finley D (2009) Recognition and processing of ubiquitin-protein conjugates by the proteasome. Annu Rev Biochem 78:477–513
Deveraux Q, Ustrell V, Pickart C, Rechsteiner M (1994) A 26S protease subunit that binds ubiquitin conjugates. J Biol Chem 269:7059–7061
Hamazaki J, Iemura S, Natsume T, Yashiroda H, Tanaka K, Murata S (2006) A novel proteasome interacting protein recruits the deubiquitinating enzyme UCH37 to 26S proteasomes. EMBO J 25:4524–4536
Qiu XB, Ouyang SY, Li CJ, Miao S, Wang L, Goldberg AL (2006) hRpn13/ADRM1/GP110 is a novel proteasome subunit that binds the deubiquitinating enzyme, UCH37. EMBO J 25:5742–5753
Yao T, Song L, Xu W, DeMartino GN, Florens L, Swanson SK, Washburn MP, Conaway RC, Conaway JW, Cohen RE (2006) Proteasome recruitment and activation of the Uch37 deubiquitinating enzyme by Adrm1. Nat Cell Biol 8:994–1002
Chen ZJ, Sun LJ (2009) Nonproteolytic functions of ubiquitin in cell signaling. Mol Cell 33:275–286
Adams J (2003) The proteasome: structure, function, and role in the cell. Cancer Treat Rev 29(suppl 1):3–9
Ciechanover A, Orian A, Schwartz AL (2000) Ubiquitin-mediated proteolysis: biological regulation via destruction. Bioessays 22:442–451
Jariel-Encontre I, Bossis G, Piechaczyk M (2008) Ubiquitin-independent degradation of proteins by the proteasome. Biochim Biophys Acta 1786:153–177
Hoyt MA, Zhang M, Coffino P (2003) Ubiquitin-independent mechanisms of mouse ornithine decarboxylase degradation are conserved between mammalian and fungal cells. J Biol Chem 278:12135–12143
Hoyt MA, Coffino P (2004) Ubiquitin-free routes into the proteasome. Cell Mol Life Sci 61:1596–1600
Coux O, Tanaka K, Goldberg AL (1996) Structure and functions of the 20S and 26S proteasomes. Annu Rev Biochem 65:801–847
Nandi D, Tahiliani P, Kumar A, Chandu D (2006) The ubiquitin-proteasome system. J Biosci 31:137–155
Ciechanover A (2006) The ubiquitin proteolytic system: from a vague idea, through basic mechanisms, and onto human diseases and drug targeting. Neurology 66:S7–S19
Adams J (2004) The development of proteasome inhibitors as anticancer drugs. Cancer Cell 5:417–421
Smith L, Lind MJ, Drew PJ, Cawkwell L (2007) The putative roles of the ubiquitin/proteasome pathway in resistance to anticancer therapy. Eur J Cancer 43:2330–2338
Dou QP, Li B (1999) Proteasome inhibitors as potential novel anticancer agents. Drug Resist Updat 2:215–223
Loda M, Cukor B, Tam SW, Lavin P, Fiorentino M, Draetta GF, Jessup JM, Pagano M (1997) Increased proteasome-dependent degradation of the cyclin-dependent kinase inhibitor p27 in aggressive colorectal carcinomas. Nat Med 3:231–234
Li B, Dou QP (2000) Bax degradation by the ubiquitin/proteasome-dependent pathway: involvement in tumor survival and progression. Proc Natl Acad Sci U S A 97:3850–3855
Kumatori A, Tanaka K, Inamura N, Sone S, Ogura T, Matsumoto T, Tachikawa T, Shin S, Ichihara A (1990) Abnormally high expression of proteasomes in human leukemic cells. Proc Natl Acad Sci U S A 87:7071–7075
An B, Goldfarb RH, Siman R, Dou QP (1998) Novel dipeptidyl proteasome inhibitors overcome Bcl-2 protective function and selectively accumulate the cyclin-dependent kinase inhibitor p27 and induce apoptosis in transformed, but not normal, human fibroblasts. Cell Death Differ 5:1062–1075
Lopes UG, Erhardt P, Yao R, Cooper GM (1997) p53-dependent induction of apoptosis by proteasome inhibitors. J Biol Chem 272:12893–12896
Orlowski RZ, Kuhn DJ (2008) Proteasome inhibitors in cancer therapy: lessons from the first decade. Clin Cancer Res 14:1649–1657
Rock KL, Gramm C, Rothstein L, Clark K, Stein R, Dick L, Hwang D, Goldberg AL (1994) Inhibitors of the proteasome block the degradation of most cell proteins and the generation of peptides presented on MHC class I molecules. Cell 78:761–771
Lee DH, Goldberg AL (1996) Selective inhibitors of the proteasome-dependent and vacuolar pathways of protein degradation in Saccharomyces cerevisiae. J Biol Chem 271: 27280–27284
Lowe J, Stock D, Jap B, Zwickl P, Baumeister W, Huber R (1995) Crystal structure of the 20S proteasome from the archaeon T. acidophilum at 3.4 A resolution. Science 268:533–539
Figueiredo-Pereira ME, Berg KA, Wilk S (1994) A new inhibitor of the chymotrypsin-like activity of the multicatalytic proteinase complex (20S proteasome) induces accumulation of ubiquitin-protein conjugates in a neuronal cell. J Neurochem 63:1578–1581
Bogyo M, McMaster JS, Gaczynska M, Tortorella D, Goldberg AL, Ploegh H (1997) Covalent modification of the active site threonine of proteasomal beta subunits and the Escherichia coli homolog HslV by a new class of inhibitors. Proc Natl Acad Sci U S A 94:6629–6634
Glas R, Bogyo M, McMaster JS, Gaczynska M, Ploegh HL (1998) A proteolytic system that compensates for loss of proteasome function. Nature 392:618–622
Dick LR, Cruikshank AA, Grenier L, Melandri FD, Nunes SL, Stein RL (1996) Mechanistic studies on the inactivation of the proteasome by lactacystin: a central role for clasto-lactacystin beta-lactone. J Biol Chem 271:7273–7276
Omura S, Matsuzaki K, Fujimoto T, Kosuge K, Furuya T, Fujita S, Nakagawa A (1991) Structure of lactacystin, a new microbial metabolite which induces differentiation of neuroblastoma cells. J Antibiot 44:117–118
Fenteany G, Standaert RF, Lane WS, Choi S, Corey EJ, Schreiber SL (1995) Inhibition of proteasome activities and subunit-specific amino-terminal threonine modification by lactacystin. Science 268:726–731
Craiu A, Gaczynska M, Akopian T, Gramm CF, Fenteany G, Goldberg AL, Rock KL (1997) Lactacystin and clasto-lactacystin beta-lactone modify multiple proteasome beta-subunits and inhibit intracellular protein degradation and major histocompatibility complex class I antigen presentation. J Biol Chem 272:13437–13445
Koguchi Y, Kohno J, Nishio M, Takahashi K, Okuda T, Ohnuki T, Komatsubara S (2000) TMC-95A, B, C, and D, novel proteasome inhibitors produced by Apiospora montagnei Sacc. TC 1093. Taxonomy, production, isolation, and biological activities. J Antibiot 53:105–109
Groll M, Koguchi Y, Huber R, Kohno J (2001) Crystal structure of the 20 S proteasome: TMC-95A complex: a non-covalent proteasome inhibitor. J Mol Biol 311:543–548
Sasse F, Steinmetz H, Schupp T, Petersen F, Memmert K, Hofmann H, Heusser C, Brinkmann V, von Matt P, Hofle G, Reichenbach H (2002) Argyrins, immunosuppressive cyclic peptides from myxobacteria. I. Production, isolation, physico-chemical and biological properties. J Antibiot 55:543–551
Nickeleit I, Zender S, Sasse F, Geffers R, Brandes G, Sorensen I, Steinmetz H, Kubicka S, Carlomagno T, Menche D, Gutgemann I, Buer J, Gossler A, Manns MP, Kalesse M, Frank R, Malek NP (2008) Argyrin a reveals a critical role for the tumor suppressor protein p27(kip1) in mediating antitumor activities in response to proteasome inhibition. Cancer Cell 14:23–35
Adams J, Palombella VJ, Sausville EA, Johnson J, Destree A, Lazarus DD, Maas J, Pien CS, Prakash S, Elliott PJ (1999) Proteasome inhibitors: a novel class of potent and effective antitumor agents. Cancer Res 59:2615–2622
Frankel A, Man S, Elliott P, Adams J, Kerbel RS (2000) Lack of multicellular drug resistance observed in human ovarian and prostate carcinoma treated with the proteasome inhibitor PS-341. Clin Cancer Res 6:3719–3728
Hideshima T, Richardson P, Chauhan D, Palombella VJ, Elliott PJ, Adams J, Anderson KC (2001) The proteasome inhibitor PS-341 inhibits growth, induces apoptosis, and overcomes drug resistance in human multiple myeloma cells. Cancer Res 61:3071–3076
Ryan DP, O’Neil BH, Supko JG, Rocha Lima CM, Dees EC, Appleman LJ, Clark J, Fidias P, Orlowski RZ, Kashala O, Eder JP, Cusack JC Jr (2006) A phase I study of bortezomib plus irinotecan in patients with advanced solid tumors. Cancer 107:2688–2697
Ryan DP, Appleman LJ, Lynch T, Supko JG, Fidias P, Clark JW, Fishman M, Zhu AX, Enzinger PC, Kashala O, Cusack J Jr, Eder JP (2006) Phase I clinical trial of bortezomib in combination with gemcitabine in patients with advanced solid tumors. Cancer 107:2482–2489
Nix D, Milton M, Pligavko C, et al (2003) Pharmacokinetics of the proteasome inhibitor bortezomib (Velcade) in male cynomolgus monkeys (abstract C245). AACR-NCI-EORTC molecular targets and cancer therapeutics discovery, biology, and clinical applications meeting
Nix D, Press R, Wehrman T, et al (2003) Tissue distribution and mass balance of bortezomib (Velcade) in non-human primates (abstract M1336). AAPS annual meeting and exposition
Kane RC, Bross PF, Farrell AT, Pazdur R (2003) Velcade: U.S. FDA approval for the treatment of multiple myeloma progressing on prior therapy. Oncologist 8:508–513
Schwartz R, Davidson T (2004) Pharmacology, pharmacokinetics, and practical applications of bortezomib. Oncology 18:14–21
Crawford LJ, Walker B, Ovaa H, Chauhan D, Anderson KC, Morris TC, Irvine AE (2006) Comparative selectivity and specificity of the proteasome inhibitors BzLLLCOCHO, PS-341, and MG-132. Cancer Res 66:6379–6386
Engel RH, Brown JA, Von Roenn JH, O’Regan RM, Bergan R, Badve S, Rademaker A, Gradishar WJ (2007) A phase II study of single agent bortezomib in patients with metastatic breast cancer: a single institution experience. Cancer Invest 25:733–737
Yang CH, Gonzalez-Angulo AM, Reuben JM, Booser DJ, Pusztai L, Krishnamurthy S, Esseltine D, Stec J, Broglio KR, Islam R, Hortobagyi GN, Cristofanilli M (2006) Bortezomib (VELCADE) in metastatic breast cancer: pharmacodynamics, biological effects, and prediction of clinical benefits. Ann Oncol 17:813–817
Adams J (2002) Preclinical and clinical evaluation of proteasome inhibitor PS-341 for the treatment of cancer. Curr Opin Chem Biol 6:493–500
Jagannath S, Durie BG, Wolf J, Camacho E, Irwin D, Lutzky J, McKinley M, Gabayan E, Mazumder A, Schenkein D, Crowley J (2005) Bortezomib therapy alone and in combination with dexamethasone for previously untreated symptomatic multiple myeloma. Br J Haematol 129:776–783
Kondagunta GV, Drucker B, Schwartz L, Bacik J, Marion S, Russo P, Mazumdar M, Motzer RJ (2004) Phase II trial of bortezomib for patients with advanced renal cell carcinoma. J Clin Oncol 22:3720–3725
Richardson PG, Sonneveld P, Schuster MW, Irwin D, Stadtmauer EA, Facon T, Harousseau JL, Ben-Yehuda D, Lonial S, Goldschmidt H, Reece D, San-Miguel JF, Blade J, Boccadoro M, Cavenagh J, Dalton WS, Boral AL, Esseltine DL, Porter JB, Schenkein D, Anderson KC, Assessment of Proteasome Inhibition for Extending Remissions I (2005) Bortezomib or high-dose dexamethasone for relapsed multiple myeloma. N Engl J Med 352:2487–2498
Shah SA, Potter MW, McDade TP, Ricciardi R, Perugini RA, Elliott PJ, Adams J, Callery MP (2001) 26S proteasome inhibition induces apoptosis and limits growth of human pancreatic cancer. J Cell Biochem 82:110–122
Sunwoo JB, Chen Z, Dong G, Yeh N, Crowl Bancroft C, Sausville E, Adams J, Elliott P, Van Waes C (2001) Novel proteasome inhibitor PS-341 inhibits activation of nuclear factor-kappa B, cell survival, tumor growth, and angiogenesis in squamous cell carcinoma. Clin Cancer Res 7:1419–1428
Ma MH, Yang HH, Parker K, Manyak S, Friedman JM, Altamirano C, Wu ZQ, Borad MJ, Frantzen M, Roussos E, Neeser J, Mikail A, Adams J, Sjak-Shie N, Vescio RA, Berenson JR (2003) The proteasome inhibitor PS-341 markedly enhances sensitivity of multiple myeloma tumor cells to chemotherapeutic agents. Clin Cancer Res 9:1136–1144
Berenson JR, Ma HM, Vescio R (2001) The role of nuclear factor-kappaB in the biology and treatment of multiple myeloma. Semin Oncol 28:626–633
LeBlanc R, Catley LP, Hideshima T, Lentzsch S, Mitsiades CS, Mitsiades N, Neuberg D, Goloubeva O, Pien CS, Adams J, Gupta D, Richardson PG, Munshi NC, Anderson KC (2002) Proteasome inhibitor PS-341 inhibits human myeloma cell growth in vivo and prolongs survival in a murine model. Cancer Res 62:4996–5000
Chauhan D, Anderson KC (2003) Mechanisms of cell death and survival in multiple myeloma (MM): therapeutic implications. Apoptosis 8:337–343
Feinman R, Koury J, Thames M, Barlogie B, Epstein J, Siegel DS (1999) Role of NF-kappaB in the rescue of multiple myeloma cells from glucocorticoid-induced apoptosis by bcl-2. Blood 93:3044–3052
Hideshima T, Ikeda H, Chauhan D, Okawa Y, Raje N, Podar K, Mitsiades C, Munshi NC, Richardson PG, Carrasco RD, Anderson KC (2009) Bortezomib induces canonical nuclear factor-kappaB activation in multiple myeloma cells. Blood 114:1046–1052
Qin JZ, Ziffra J, Stennett L, Bodner B, Bonish BK, Chaturvedi V, Bennett F, Pollock PM, Trent JM, Hendrix MJ, Rizzo P, Miele L, Nickoloff BJ (2005) Proteasome inhibitors trigger NOXA-mediated apoptosis in melanoma and myeloma cells. Cancer Res 65:6282–6293
Oda E, Ohki R, Murasawa H, Nemoto J, Shibue T, Yamashita T, Tokino T, Taniguchi T, Tanaka N (2000) Noxa, a BH3-only member of the Bcl-2 family and candidate mediator of p53-induced apoptosis. Science 288:1053–1058
Adams JM, Cory S (1998) The Bcl-2 protein family: arbiters of cell survival. Science 281:1322–1326
Gross A, McDonnell JM, Korsmeyer SJ (1999) BCL-2 family members and the mitochondria in apoptosis. Genes Dev 13:1899–1911
Nikiforov MA, Riblett M, Tang WH, Gratchouck V, Zhuang D, Fernandez Y, Verhaegen M, Varambally S, Chinnaiyan AM, Jakubowiak AJ, Soengas MS (2007) Tumor cell-selective regulation of NOXA by c-MYC in response to proteasome inhibition. Proc Natl Acad Sci U S A 104:19488–19493
Caravita T, de Fabritiis P, Palumbo A, Amadori S, Boccadoro M (2006) Bortezomib: efficacy comparisons in solid tumors and hematologic malignancies. Nat Clin Pract Oncol 3:374–387
Qin JZ, Xin H, Sitailo LA, Denning MF, Nickoloff BJ (2006) Enhanced killing of melanoma cells by simultaneously targeting Mcl-1 and NOXA. Cancer Res 66:9636–9645
Fernandez Y, Verhaegen M, Miller TP, Rush JL, Steiner P, Opipari AW Jr, Lowe SW, Soengas MS (2005) Differential regulation of noxa in normal melanocytes and melanoma cells by proteasome inhibition: therapeutic implications. Cancer Res 65:6294–6304
Nawrocki ST, Bruns CJ, Harbison MT, Bold RJ, Gotsch BS, Abbruzzese JL, Elliott P, Adams J, McConkey DJ (2002) Effects of the proteasome inhibitor PS-341 on apoptosis and angiogenesis in orthotopic human pancreatic tumor xenografts. Mol Cancer Ther 1:1243–1253
Fribley A, Wang CY (2006) Proteasome inhibitor induces apoptosis through induction of endoplasmic reticulum stress. Cancer Biol Ther 5:745–748
Fribley A, Zeng Q, Wang CY (2004) Proteasome inhibitor PS-341 induces apoptosis through induction of endoplasmic reticulum stress-reactive oxygen species in head and neck squamous cell carcinoma cells. Mol Cell Biol 24:9695–9704
Mitsiades N, Mitsiades CS, Poulaki V, Chauhan D, Fanourakis G, Gu X, Bailey C, Joseph M, Libermann TA, Treon SP, Munshi NC, Richardson PG, Hideshima T, Anderson KC (2002) Molecular sequelae of proteasome inhibition in human multiple myeloma cells. Proc Natl Acad Sci U S A 99:14374–14379
Strauss SJ, Higginbottom K, Juliger S, Maharaj L, Allen P, Schenkein D, Lister TA, Joel SP (2007) The proteasome inhibitor bortezomib acts independently of p53 and induces cell death via apoptosis and mitotic catastrophe in B-cell lymphoma cell lines. Cancer Res 67:2783–2790
Lioni M, Noma K, Snyder A, Klein-Szanto A, Diehl JA, Rustgi AK, Herlyn M, Smalley KS (2008) Bortezomib induces apoptosis in esophageal squamous cell carcinoma cells through activation of the p38 mitogen-activated protein kinase pathway. Mol Cancer Ther 7:2866–2875
Kukreja A, Hutchinson A, Mazumder A, Vesole D, Angitapalli R, Jagannath S, O’Connor OA, Dhodapkar MV (2007) Bortezomib disrupts tumour-dendritic cell interactions in myeloma and lymphoma: therapeutic implications. Br J Haematol 136:106–110
Orlowski RZ, Stinchcombe TE, Mitchell BS, Shea TC, Baldwin AS, Stahl S, Adams J, Esseltine DL, Elliott PJ, Pien CS, Guerciolini R, Anderson JK, Depcik-Smith ND, Bhagat R, Lehman MJ, Novick SC, O’Connor OA, Soignet SL (2002) Phase I trial of the proteasome inhibitor PS-341 in patients with refractory hematologic malignancies. J Clin Oncol 20:4420–4427
Orlowski RZ, Voorhees PM, Garcia RA, Hall MD, Kudrik FJ, Allred T, Johri AR, Jones PE, Ivanova A, Van Deventer HW, Gabriel DA, Shea TC, Mitchell BS, Adams J, Esseltine DL, Trehu EG, Green M, Lehman MJ, Natoli S, Collins JM, Lindley CM, Dees EC (2005) Phase 1 trial of the proteasome inhibitor bortezomib and pegylated liposomal doxorubicin in patients with advanced hematologic malignancies. Blood 105:3058–3065
Papandreou CN, Daliani DD, Nix D, Yang H, Madden T, Wang X, Pien CS, Millikan RE, Tu SM, Pagliaro L, Kim J, Adams J, Elliott P, Esseltine D, Petrusich A, Dieringer P, Perez C, Logothetis CJ (2004) Phase I trial of the proteasome inhibitor bortezomib in patients with advanced solid tumors with observations in androgen-independent prostate cancer. J Clin Oncol 22:2108–2121
Dou QP, Goldfarb RH (2002) Bortezomib (millennium pharmaceuticals). IDrugs 5:828–834
Kong X, Alvarez-Castelao B, Lin Z, Castano JG, Caro J (2007) Constitutive/hypoxic degradation of HIF-alpha proteins by the proteasome is independent of von Hippel Lindau protein ubiquitylation and the transactivation activity of the protein. J Biol Chem 282:15498–15505
Dreicer R, Petrylak D, Agus D, Webb I, Roth B (2007) Phase I/II study of bortezomib plus docetaxel in patients with advanced androgen-independent prostate cancer. Clin Cancer Res 13:1208–1215
Hainsworth JD, Meluch AA, Spigel DR, Barton J Jr, Simons L, Meng C, Gould B, Greco FA (2007) Weekly docetaxel and bortezomib as first-line treatment for patients with hormone-refractory prostate cancer: a Minnie Pearl Cancer Research Network phase II trial. Clin Genitourin Cancer 5:278–283
Richardson PG, Barlogie B, Berenson J, Singhal S, Jagannath S, Irwin D, Rajkumar SV, Srkalovic G, Alsina M, Alexanian R, Siegel D, Orlowski RZ, Kuter D, Limentani SA, Lee S, Hideshima T, Esseltine DL, Kauffman M, Adams J, Schenkein DP, Anderson KC (2003) A phase 2 study of bortezomib in relapsed, refractory myeloma. N Engl J Med 348:2609–2617
Jagannath S, Barlogie B, Berenson J, Siegel D, Irwin D, Richardson PG, Niesvizky R, Alexanian R, Limentani SA, Alsina M, Adams J, Kauffman M, Esseltine DL, Schenkein DP, Anderson KC (2004) A phase 2 study of two doses of bortezomib in relapsed or refractory myeloma. Br J Haematol 127:165–172
Oakervee HE, Popat R, Curry N, Smith P, Morris C, Drake M, Agrawal S, Stec J, Schenkein D, Esseltine DL, Cavenagh JD (2005) PAD combination therapy (PS-341/bortezomib, doxorubicin and dexamethasone) for previously untreated patients with multiple myeloma. Br J Haematol 129:755–762
O’Connor OA, Wright J, Moskowitz C, Muzzy J, MacGregor-Cortelli B, Stubblefield M, Straus D, Portlock C, Hamlin P, Choi E, Dumetrescu O, Esseltine D, Trehu E, Adams J, Schenkein D, Zelenetz AD (2005) Phase II clinical experience with the novel proteasome inhibitor bortezomib in patients with indolent non-Hodgkin’s lymphoma and mantle cell lymphoma. J Clin Oncol 23:676–684
Belch A, Kouroukis CT, Crump M, Sehn L, Gascoyne RD, Klasa R, Powers J, Wright J, Eisenhauer EA (2007) A phase II study of bortezomib in mantle cell lymphoma: the National Cancer Institute of Canada Clinical Trials Group trial IND.150. Ann Oncol 18:116–121
Scagliotti GV, Germonpre P, Bosquee L, Vansteenkiste J, Gervais R, Planchard D, Reck M, De Marinis F, Lee JS, Park K, Biesma B, Gans S, Ramlau R, Szczesna A, Makhson A, Manikhas G, Morgan B, Zhu Y, Chan KC, von Pawel J (2010) A randomized phase II study of bortezomib and pemetrexed, in combination or alone, in patients with previously treated advanced non-small-cell lung cancer. Lung Cancer 68:420–426
Mikhael JR, Belch AR, Prince HM, Lucio MN, Maiolino A, Corso A, Petrucci MT, Musto P, Komarnicki M, Stewart AK (2009) High response rate to bortezomib with or without dexamethasone in patients with relapsed or refractory multiple myeloma: results of a global phase 3b expanded access program. Br J Haematol 144:169–175
Richardson PG, Sonneveld P, Schuster MW, Stadtmauer EA, Facon T, Harousseau JL, Ben-Yehuda D, Lonial S, Goldschmidt H, Reece D, Blade J, Boccadoro M, Cavenagh JD, Boral AL, Esseltine DL, Wen PY, Amato AA, Anderson KC, San Miguel J (2009) Reversibility of symptomatic peripheral neuropathy with bortezomib in the phase III APEX trial in relapsed multiple myeloma: impact of a dose-modification guideline. Br J Haematol 144:895–903
San Miguel JF, Schlag R, Khuageva NK, Dimopoulos MA, Shpilberg O, Kropff M, Spicka I, Petrucci MT, Palumbo A, Samoilova OS, Dmoszynska A, Abdulkadyrov KM, Schots R, Jiang B, Mateos MV, Anderson KC, Esseltine DL, Liu K, Cakana A, van de Velde H, Richardson PG, Investigators VT (2008) Bortezomib plus melphalan and prednisone for initial treatment of multiple myeloma. N Engl J Med 359:906–917
Mateos MV, Richardson PG, Schlag R, Khuageva NK, Dimopoulos MA, Shpilberg O, Kropff M, Spicka I, Petrucci MT, Palumbo A, Samoilova OS, Dmoszynska A, Abdulkadyrov KM, Schots R, Jiang B, Esseltine DL, Liu K, Cakana A, van de Velde H, San Miguel JF (2010) Bortezomib plus melphalan and prednisone compared with melphalan and prednisone in previously untreated multiple myeloma: updated follow-up and impact of subsequent therapy in the phase III VISTA trial. J Clin Oncol 28:2259–2266
Potts BC, Albitar MX, Anderson KC, Baritaki S, Berkers C, Bonavida B, Chandra J, Chauhan D, Cusack JC Jr, Fenical W, Ghobrial IM, Groll M, Jensen PR, Lam KS, Lloyd GK, McBride W, McConkey DJ, Miller CP, Neuteboom ST, Oki Y, Ovaa H, Pajonk F, Richardson PG, Roccaro AM, Sloss CM, Spear MA, Valashi E, Younes A, Palladino MA (2011) Marizomib, a proteasome inhibitor for all seasons: preclinical profile and a framework for clinical trials. Curr Cancer Drug Targets 11:254–284
Kuhn DJ, Orlowski RZ, Bjorklund CC (2011) Second generation proteasome inhibitors: carfilzomib and immunoproteasome-specific inhibitors (IPSIs). Curr Cancer Drug Targets 11:285–295
Chen D, Frezza M, Schmitt S, Kanwar J, Dou QP (2011) Bortezomib as the first proteasome inhibitor anticancer drug: current status and future perspectives. Curr Cancer Drug Targets 11:239–253
Kuhn DJ, Chen Q, Voorhees PM, Strader JS, Shenk KD, Sun CM, Demo SD, Bennett MK, van Leeuwen FW, Chanan-Khan AA, Orlowski RZ (2007) Potent activity of carfilzomib, a novel, irreversible inhibitor of the ubiquitin-proteasome pathway, against preclinical models of multiple myeloma. Blood 110:3281–3290
Parlati F, Lee SJ, Aujay M, Suzuki E, Levitsky K, Lorens JB, Micklem DR, Ruurs P, Sylvain C, Lu Y, Shenk KD, Bennett MK (2009) Carfilzomib can induce tumor cell death through selective inhibition of the chymotrypsin-like activity of the proteasome. Blood 114:3439–3447
Demo SD, Kirk CJ, Aujay MA, Buchholz TJ, Dajee M, Ho MN, Jiang J, Laidig GJ, Lewis ER, Parlati F, Shenk KD, Smyth MS, Sun CM, Vallone MK, Woo TM, Molineaux CJ, Bennett MK (2007) Antitumor activity of PR-171, a novel irreversible inhibitor of the proteasome. Cancer Res 67:6383–6391
Alsina M, Trudel S, Furman RR, Rosen PJ, O’Connor OA, Comenzo RL, Wong A, Kunkel LA, Molineaux CJ, Goy A (2012) A phase I single-agent study of twice-weekly consecutive-day dosing of the proteasome inhibitor carfilzomib in patients with relapsed or refractory multiple myeloma or lymphoma. Clin Cancer Res 18:4830–4840
O’Connor OA, Stewart AK, Vallone M, Molineaux CJ, Kunkel LA, Gerecitano JF, Orlowski RZ (2009) A phase 1 dose escalation study of the safety and pharmacokinetics of the novel proteasome inhibitor carfilzomib (PR-171) in patients with hematologic malignancies. Clin Cancer Res 15:7085–7091
Papadopoulos KP, Burris HA III, Gordon M, Lee P, Sausville EA, Rosen PJ, Patnaik A, Cutler RE Jr, Wang Z, Lee S, Jones SF, Infante JR (2013) A phase I/II study of carfilzomib 2-10-min infusion in patients with advanced solid tumors. Cancer Chemother Pharmacol 72:861–868
Jakubowiak AJ, Dytfeld D, Griffith KA, Lebovic D, Vesole DH, Jagannath S, Al-Zoubi A, Anderson T, Nordgren B, Detweiler-Short K, Stockerl-Goldstein K, Ahmed A, Jobkar T, Durecki DE, McDonnell K, Mietzel M, Couriel D, Kaminski M, Vij R (2012) A phase 1/2 study of carfilzomib in combination with lenalidomide and low-dose dexamethasone as a frontline treatment for multiple myeloma. Blood 120:1801–1809
Jagannath S, Vij R, Stewart AK, Trudel S, Jakubowiak AJ, Reiman T, Somlo G, Bahlis N, Lonial S, Kunkel LA, Wong A, Orlowski RZ, Siegel DS (2012) An open-label single-arm pilot phase II study (PX-171-003-A0) of low-dose, single-agent carfilzomib in patients with relapsed and refractory multiple myeloma. Clin Lymphoma Myeloma Leuk 12:310–318
Siegel DS, Martin T, Wang M, Vij R, Jakubowiak AJ, Lonial S, Trudel S, Kukreti V, Bahlis N, Alsina M, Chanan-Khan A, Buadi F, Reu FJ, Somlo G, Zonder J, Song K, Stewart AK, Stadtmauer E, Kunkel L, Wear S, Wong AF, Orlowski RZ, Jagannath S (2012) A phase 2 study of single-agent carfilzomib (PX-171-003-A1) in patients with relapsed and refractory multiple myeloma. Blood 120:2817–2825
Hajek R, Bryce R, Ro S, Klencke B, Ludwig H (2012) Design and rationale of FOCUS (PX-171-011): a randomized, open-label, phase 3 study of carfilzomib versus best supportive care regimen in patients with relapsed and refractory multiple myeloma (R/R MM). BMC Cancer 12:415
Cortes J, Thomas D, Koller C, Giles F, Estey E, Faderl S, Garcia-Manero G, McConkey D, Ruiz SL, Guerciolini R, Wright J, Kantarjian H (2004) Phase I study of bortezomib in refractory or relapsed acute leukemias. Clin Cancer Res 10:3371–3376
Dispenzieri A, Jacobus S, Vesole DH, Callandar N, Fonseca R, Greipp PR (2010) Primary therapy with single agent bortezomib as induction, maintenance and re-induction in patients with high-risk myeloma: results of the ECOG E2A02 trial. Leukemia 24:1406–1411
Oerlemans R, Franke NE, Assaraf YG, Cloos J, van Zantwijk I, Berkers CR, Scheffer GL, Debipersad K, Vojtekova K, Lemos C, van der Heijden JW, Ylstra B, Peters GJ, Kaspers GL, Dijkmans BA, Scheper RJ, Jansen G (2008) Molecular basis of bortezomib resistance: proteasome subunit beta5 (PSMB5) gene mutation and overexpression of PSMB5 protein. Blood 112:2489–2499
Lu S, Yang J, Chen Z, Gong S, Zhou H, Xu X, Wang J (2009) Different mutants of PSMB5 confer varying bortezomib resistance in T lymphoblastic lymphoma/leukemia cells derived from the Jurkat cell line. Exp Hematol 37:831–837
Ruschak AM, Slassi M, Kay LE, Schimmer AD (2011) Novel proteasome inhibitors to overcome bortezomib resistance. J Natl Cancer Inst 103:1007–1017
Politou M, Karadimitris A, Terpos E, Kotsianidis I, Apperley JF, Rahemtulla A (2006) No evidence of mutations of the PSMB5 (beta-5 subunit of proteasome) in a case of myeloma with clinical resistance to Bortezomib. Leuk Res 30:240–241
Takayama S, Reed JC, Homma S (2003) Heat-shock proteins as regulators of apoptosis. Oncogene 22:9041–9047
Beere HM (2004) “The stress of dying”: the role of heat shock proteins in the regulation of apoptosis. J Cell Sci 117:2641–2651
Zhou M, Wu X, Ginsberg HN (1996) Evidence that a rapidly turning over protein, normally degraded by proteasomes, regulates hsp72 gene transcription in HepG2 cells. J Biol Chem 271:24769–24775
Bush KT, Goldberg AL, Nigam SK (1997) Proteasome inhibition leads to a heat-shock response, induction of endoplasmic reticulum chaperones, and thermotolerance. J Biol Chem 272:9086–9092
Meriin AB, Gabai VL, Yaglom J, Shifrin VI, Sherman MY (1998) Proteasome inhibitors activate stress kinases and induce Hsp72. Diverse effects on apoptosis. J Biol Chem 273:6373–6379
Robertson JD, Datta K, Biswal SS, Kehrer JP (1999) Heat-shock protein 70 antisense oligomers enhance proteasome inhibitor-induced apoptosis. Biochem J 344(Pt 2):477–485
Gabai VL, Budagova KR, Sherman MY (2005) Increased expression of the major heat shock protein Hsp72 in human prostate carcinoma cells is dispensable for their viability but confers resistance to a variety of anticancer agents. Oncogene 24:3328–3338
Chauhan D, Li G, Shringarpure R, Podar K, Ohtake Y, Hideshima T, Anderson KC (2003) Blockade of Hsp27 overcomes Bortezomib/proteasome inhibitor PS-341 resistance in lymphoma cells. Cancer Res 63:6174–6177
Shringarpure R, Catley L, Bhole D, Burger R, Podar K, Tai YT, Kessler B, Galardy P, Ploegh H, Tassone P, Hideshima T, Mitsiades C, Munshi NC, Chauhan D, Anderson KC (2006) Gene expression analysis of B-lymphoma cells resistant and sensitive to bortezomib. Br J Haematol 134:145–156
Hideshima T, Podar K, Chauhan D, Ishitsuka K, Mitsiades C, Tai YT, Hamasaki M, Raje N, Hideshima H, Schreiner G, Nguyen AN, Navas T, Munshi NC, Richardson PG, Higgins LS, Anderson KC (2004) p38 MAPK inhibition enhances PS-341 (bortezomib)-induced cytotoxicity against multiple myeloma cells. Oncogene 23:8766–8776
Hosokawa N, Hirayoshi K, Nakai A, Hosokawa Y, Marui N, Yoshida M, Sakai T, Nishino H, Aoike A, Kawai K et al (1990) Flavonoids inhibit the expression of heat shock proteins. Cell Struct Funct 15:393–401
Aghdassi A, Phillips P, Dudeja V, Dhaulakhandi D, Sharif R, Dawra R, Lerch MM, Saluja A (2007) Heat shock protein 70 increases tumorigenicity and inhibits apoptosis in pancreatic adenocarcinoma. Cancer Res 67:616–625
Mimnaugh EG, Xu W, Vos M, Yuan X, Isaacs JS, Bisht KS, Gius D, Neckers L (2004) Simultaneous inhibition of hsp 90 and the proteasome promotes protein ubiquitination, causes endoplasmic reticulum-derived cytosolic vacuolization, and enhances antitumor activity. Mol Cancer Ther 3:551–566
Mitsiades CS, Mitsiades NS, McMullan CJ, Poulaki V, Kung AL, Davies FE, Morgan G, Akiyama M, Shringarpure R, Munshi NC, Richardson PG, Hideshima T, Chauhan D, Gu X, Bailey C, Joseph M, Libermann TA, Rosen NS, Anderson KC (2006) Antimyeloma activity of heat shock protein-90 inhibition. Blood 107:1092–1100
Davenport EL, Moore HE, Dunlop AS, Sharp SY, Workman P, Morgan GJ, Davies FE (2007) Heat shock protein inhibition is associated with activation of the unfolded protein response pathway in myeloma plasma cells. Blood 110:2641–2649
Nawrocki ST, Carew JS, Dunner K Jr, Boise LH, Chiao PJ, Huang P, Abbruzzese JL, McConkey DJ (2005) Bortezomib inhibits PKR-like endoplasmic reticulum (ER) kinase and induces apoptosis via ER stress in human pancreatic cancer cells. Cancer Res 65:11510–11519
Tan TT, Degenhardt K, Nelson DA, Beaudoin B, Nieves-Neira W, Bouillet P, Villunger A, Adams JM, White E (2005) Key roles of BIM-driven apoptosis in epithelial tumors and rational chemotherapy. Cancer Cell 7:227–238
Mestre-Escorihuela C, Rubio-Moscardo F, Richter JA, Siebert R, Climent J, Fresquet V, Beltran E, Agirre X, Marugan I, Marin M, Rosenwald A, Sugimoto KJ, Wheat LM, Karran EL, Garcia JF, Sanchez L, Prosper F, Staudt LM, Pinkel D, Dyer MJ, Martinez-Climent JA (2007) Homozygous deletions localize novel tumor suppressor genes in B-cell lymphomas. Blood 109:271–280
Lee SH, Soung YH, Lee JW, Kim HS, Lee JH, Kim HS, Lee JH, Park JY, Cho YG, Kim CJ, Kim SY, Park WS, Kim SH, Lee JY, Yoo NJ (2003) Mutational analysis of Noxa gene in human cancers. Acta Pathol Microbiol Immunol Scand 111:599–604
Xiao C, Srinivasan L, Calado DP, Patterson HC, Zhang B, Wang J, Henderson JM, Kutok JL, Rajewsky K (2008) Lymphoproliferative disease and autoimmunity in mice with increased miR-17-92 expression in lymphocytes. Nat Immunol 9:405–414
Wang Z, Zhang B, Yang L, Ding J, Ding HF (2008) Constitutive production of NF-kappaB2 p52 is not tumorigenic but predisposes mice to inflammatory autoimmune disease by repressing Bim expression. J Biol Chem 283:10698–10706
Yamashita M, Kuwahara M, Suzuki A, Hirahara K, Shinnaksu R, Hosokawa H, Hasegawa A, Motohashi S, Iwama A, Nakayama T (2008) Bmi1 regulates memory CD4 T cell survival via repression of the Noxa gene. J Exp Med 205:1109–1120
Letai A, Bassik MC, Walensky LD, Sorcinelli MD, Weiler S, Korsmeyer SJ (2002) Distinct BH3 domains either sensitize or activate mitochondrial apoptosis, serving as prototype cancer therapeutics. Cancer Cell 2:183–192
Chauhan D, Velankar M, Brahmandam M, Hideshima T, Podar K, Richardson P, Schlossman R, Ghobrial I, Raje N, Munshi N, Anderson KC (2007) A novel Bcl-2/Bcl-X(L)/Bcl-w inhibitor ABT-737 as therapy in multiple myeloma. Oncogene 26:2374–2380
Nguyen M, Marcellus RC, Roulston A, Watson M, Serfass L, Murthy Madiraju SR, Goulet D, Viallet J, Belec L, Billot X, Acoca S, Purisima E, Wiegmans A, Cluse L, Johnstone RW, Beauparlant P, Shore GC (2007) Small molecule obatoclax (GX15-070) antagonizes MCL-1 and overcomes MCL-1-mediated resistance to apoptosis. Proc Natl Acad Sci U S A 104: 19512–19517
Paoluzzi L, Gonen M, Bhagat G, Furman RR, Gardner JR, Scotto L, Gueorguiev VD, Heaney ML, Manova K, O’Connor OA (2008) The BH3-only mimetic ABT-737 synergizes the antineoplastic activity of proteasome inhibitors in lymphoid malignancies. Blood 112:2906–2916
Williams SA, McConkey DJ (2003) The proteasome inhibitor bortezomib stabilizes a novel active form of p53 in human LNCaP-Pro5 prostate cancer cells. Cancer Res 63:7338–7344
Yang G, Ayala G, De Marzo A, Tian W, Frolov A, Wheeler TM, Thompson TC, Harper JW (2002) Elevated Skp2 protein expression in human prostate cancer: association with loss of the cyclin-dependent kinase inhibitor p27 and PTEN and with reduced recurrence-free survival. Clin Cancer Res 8:3419–3426
Worm J, Bartkova J, Kirkin AF, Straten P, Zeuthen J, Bartek J, Guldberg P (2000) Aberrant p27Kip1 promoter methylation in malignant melanoma. Oncogene 19:5111–5115
Shin I, Yakes FM, Rojo F, Shin NY, Bakin AV, Baselga J, Arteaga CL (2002) PKB/Akt mediates cell-cycle progression by phosphorylation of p27(Kip1) at threonine 157 and modulation of its cellular localization. Nat Med 8:1145–1152
Ogino S, Kawasaki T, Ogawa A, Kirkner GJ, Loda M, Fuchs CS (2007) Cytoplasmic localization of p27 (cyclin-dependent kinase inhibitor 1B/KIP1) in colorectal cancer: inverse correlations with nuclear p27 loss, microsatellite instability, and CpG island methylator phenotype. Hum Pathol 38:585–592
Ma YY, Wei SJ, Lin YC, Lung JC, Chang TC, Whang-Peng J, Liu JM, Yang DM, Yang WK, Shen CY (2000) PIK3CA as an oncogene in cervical cancer. Oncogene 19:2739–2744
Osaki M, Oshimura M, Ito H (2004) PI3K-Akt pathway: its functions and alterations in human cancer. Apoptosis 9:667–676
Thompson JE, Thompson CB (2004) Putting the rap on Akt. J Clin Oncol 22:4217–4226
Cantley LC, Neel BG (1999) New insights into tumor suppression: PTEN suppresses tumor formation by restraining the phosphoinositide 3-kinase/AKT pathway. Proc Natl Acad Sci U S A 96:4240–4245
Guerrero S, Casanova I, Farre L, Mazo A, Capella G, Mangues R (2000) K-ras codon 12 mutation induces higher level of resistance to apoptosis and predisposition to anchorage-independent growth than codon 13 mutation or proto-oncogene overexpression. Cancer Res 60:6750–6756
Shi Y, Yan H, Frost P, Gera J, Lichtenstein A (2005) Mammalian target of rapamycin inhibitors activate the AKT kinase in multiple myeloma cells by up-regulating the insulin-like growth factor receptor/insulin receptor substrate-1/phosphatidylinositol 3-kinase cascade. Mol Cancer Ther 4:1533–1540
Yu C, Friday BB, Lai JP, Yang L, Sarkaria J, Kay NE, Carter CA, Roberts LR, Kaufmann SH, Adjei AA (2006) Cytotoxic synergy between the multikinase inhibitor sorafenib and the proteasome inhibitor bortezomib in vitro: induction of apoptosis through Akt and c-Jun NH2-terminal kinase pathways. Mol Cancer Ther 5:2378–2387
Shi YY, Small GW, Orlowski RZ (2006) Proteasome inhibitors induce a p38 mitogen-activated protein kinase (MAPK)-dependent anti-apoptotic program involving MAPK phosphatase-1 and Akt in models of breast cancer. Breast Cancer Res Treat 100:33–47
Moreau AS, Jia X, Ngo HT, Leleu X, O’Sullivan G, Alsayed Y, Leontovich A, Podar K, Kutok J, Daley J, Lazo-Kallanian S, Hatjiharissi E, Raab MS, Xu L, Treon SP, Hideshima T, Anderson KC, Ghobrial IM (2007) Protein kinase C inhibitor enzastaurin induces in vitro and in vivo antitumor activity in Waldenstrom macroglobulinemia. Blood 109:4964–4972
Shrader M, Pino MS, Lashinger L, Bar-Eli M, Adam L, Dinney CP, McConkey DJ (2007) Gefitinib reverses TRAIL resistance in human bladder cancer cell lines via inhibition of AKT-mediated X-linked inhibitor of apoptosis protein expression. Cancer Res 67:1430–1435
Cascone T, Morelli MP, Morgillo F, Kim WY, Rodolico G, Pepe S, Tortora G, Berrino L, Lee HY, Heymach JV, Ciardiello F (2008) Synergistic anti-proliferative and pro-apoptotic activity of combined therapy with bortezomib, a proteasome inhibitor, with anti-epidermal growth factor receptor (EGFR) drugs in human cancer cells. J Cell Physiol 216:698–707
An J, Rettig MB (2007) Epidermal growth factor receptor inhibition sensitizes renal cell carcinoma cells to the cytotoxic effects of bortezomib. Mol Cancer Ther 6:61–69
Rajkumar SV, Richardson PG, Hideshima T, Anderson KC (2005) Proteasome inhibition as a novel therapeutic target in human cancer. J Clin Oncol 23:630–639
Siegel DS (2012) Relapsed/refractory multiple myeloma: defining refractory disease and identifying strategies to overcome resistance. Semin Hematol 49(Suppl 1):S3–S15
Wang X, Li C, Ju S, Wang Y, Wang H, Zhong R (2011) Myeloma cell adhesion to bone marrow stromal cells confers drug resistance by microRNA-21 up-regulation. Leuk Lymphoma 52:1991–1998
Zang MR, Li F, An G, Xie ZQ, Li CH, Yu Z, Xu Y, Qiu LG, Hao M (2012) Regulation of miRNA-15a/-16 expression on the drug resistance of myeloma cells. Zhonghua Yi Xue Za Zhi 92:1100–1103
Bataille R, Robillard N, Avet-Loiseau H, Harousseau JL, Moreau P (2005) CD221 (IGF-1R) is aberrantly expressed in multiple myeloma, in relation to disease severity. Haematologica 90:706–707
Chng WJ, Gualberto A, Fonseca R (2006) IGF-1R is overexpressed in poor-prognostic subtypes of multiple myeloma. Leukemia 20:174–176
Kuhn DJ, Berkova Z, Jones RJ, Woessner R, Bjorklund CC, Ma W, Davis RE, Lin P, Wang H, Madden TL, Wei C, Baladandayuthapani V, Wang M, Thomas SK, Shah JJ, Weber DM, Orlowski RZ (2012) Targeting the insulin-like growth factor-1 receptor to overcome bortezomib resistance in preclinical models of multiple myeloma. Blood 120:3260–3270
Que W, Chen J, Chuang M, Jiang D (2012) Knockdown of c-Met enhances sensitivity to bortezomib in human multiple myeloma U266 cells via inhibiting Akt/mTOR activity. Acta Pathol Microbiol Immunol Scand 120:195–203
Kim A, Park S, Lee JE, Jang WS, Lee SJ, Kang HJ, Lee SS (2012) The dual PI3K and mTOR inhibitor NVP-BEZ235 exhibits anti-proliferative activity and overcomes bortezomib resistance in mantle cell lymphoma cells. Leuk Res 36:912–920
Yeom SY, Lee SJ, Kim WS, Park C (2012) Rad knockdown induces mitochondrial apoptosis in bortezomib resistant leukemia and lymphoma cells. Leuk Res 36:1172–1178
Levine B, Yuan J (2005) Autophagy in cell death: an innocent convict? J Clin Invest 115: 2679–2688
Yang W, Monroe J, Zhang Y, George D, Bremer E, Li H (2006) Proteasome inhibition induces both pro- and anti-cell death pathways in prostate cancer cells. Cancer Lett 243:217–227
Ding WX, Ni HM, Gao W, Yoshimori T, Stolz DB, Ron D, Yin XM (2007) Linking of autophagy to ubiquitin-proteasome system is important for the regulation of endoplasmic reticulum stress and cell viability. Am J Pathol 171:513–524
Hideshima T, Bradner JE, Chauhan D, Anderson KC (2005) Intracellular protein degradation and its therapeutic implications. Clin Cancer Res 11:8530–8533
Nawrocki ST, Carew JS, Pino MS, Highshaw RA, Andtbacka RH, Dunner K Jr, Pal A, Bornmann WG, Chiao PJ, Huang P, Xiong H, Abbruzzese JL, McConkey DJ (2006) Aggresome disruption: a novel strategy to enhance bortezomib-induced apoptosis in pancreatic cancer cells. Cancer Res 66:3773–3781
Badros A, Burger AM, Philip S, Niesvizky R, Kolla SS, Goloubeva O, Harris C, Zwiebel J, Wright JJ, Espinoza-Delgado I, Baer MR, Holleran JL, Egorin MJ, Grant S (2009) Phase I study of vorinostat in combination with bortezomib for relapsed and refractory multiple myeloma. Clin Cancer Res 15:5250–5257
Nerini-Molteni S, Ferrarini M, Cozza S, Caligaris-Cappio F, Sitia R (2008) Redox homeostasis modulates the sensitivity of myeloma cells to bortezomib. Br J Haematol 141:494–503
Uehara T, Nakamura T, Yao D, Shi ZQ, Gu Z, Ma Y, Masliah E, Nomura Y, Lipton SA (2006) S-nitrosylated protein-disulphide isomerase links protein misfolding to neurodegeneration. Nature 441:513–517
Kalivendi SV, Cunningham S, Kotamraju S, Joseph J, Hillard CJ, Kalyanaraman B (2004) Alpha-synuclein up-regulation and aggregation during MPP+-induced apoptosis in neuroblastoma cells: intermediacy of transferrin receptor iron and hydrogen peroxide. J Biol Chem 279:15240–15247
Keller JN, Huang FF, Zhu H, Yu J, Ho YS, Kindy TS (2000) Oxidative stress-associated impairment of proteasome activity during ischemia-reperfusion injury. J Cereb Blood Flow Metab 20:1467–1473
Catley L, Anderson KC (2006) Velcade and vitamin C: too much of a good thing? Clin Cancer Res 12:3–4
Zou W, Yue P, Lin N, He M, Zhou Z, Lonial S, Khuri FR, Wang B, Sun SY (2006) Vitamin C inactivates the proteasome inhibitor PS-341 in human cancer cells. Clin Cancer Res 12:273–280
Kupperman E, Lee EC, Cao Y, Bannerman B, Fitzgerald M, Berger A, Yu J, Yang Y, Hales P, Bruzzese F, Liu J, Blank J, Garcia K, Tsu C, Dick L, Fleming P, Yu L, Manfredi M, Rolfe M, Bolen J (2010) Evaluation of the proteasome inhibitor MLN9708 in preclinical models of human cancer. Cancer Res 70:1970–1980
Lee EC, Fitzgerald M, Bannerman B, Donelan J, Bano K, Terkelsen J, Bradley DP, Subakan O, Silva MD, Liu R, Pickard M, Li Z, Tayber O, Li P, Hales P, Carsillo M, Neppalli VT, Berger AJ, Kupperman E, Manfredi M, Bolen JB, Van Ness B, Janz S (2011) Antitumor activity of the investigational proteasome inhibitor MLN9708 in mouse models of B-cell and plasma cell malignancies. Clin Cancer Res 17:7313–7323
Sanchez E, Li M, Steinberg JA, Wang C, Shen J, Bonavida B, Li ZW, Chen H, Berenson JR (2010) The proteasome inhibitor CEP-18770 enhances the anti-myeloma activity of bortezomib and melphalan. Br J Haematol 148:569–581
Zhou HJ, Aujay MA, Bennett MK, Dajee M, Demo SD, Fang Y, Ho MN, Jiang J, Kirk CJ, Laidig GJ, Lewis ER, Lu Y, Muchamuel T, Parlati F, Ring E, Shenk KD, Shields J, Shwonek PJ, Stanton T, Sun CM, Sylvain C, Woo TM, Yang J (2009) Design and synthesis of an orally bioavailable and selective peptide epoxyketone proteasome inhibitor (PR-047). J Med Chem 52:3028–3038
Lawasut P, Chauhan D, Laubach J, Hayes C, Fabre C, Maglio M, Mitsiades C, Hideshima T, Anderson KC, Richardson PG (2012) New proteasome inhibitors in myeloma. Curr Hematol Malig Rep 7:258–266
Chauhan D, Catley L, Li G, Podar K, Hideshima T, Velankar M, Mitsiades C, Mitsiades N, Yasui H, Letai A, Ovaa H, Berkers C, Nicholson B, Chao TH, Neuteboom ST, Richardson P, Palladino MA, Anderson KC (2005) A novel orally active proteasome inhibitor induces apoptosis in multiple myeloma cells with mechanisms distinct from Bortezomib. Cancer Cell 8:407–419
Fenical W, Jensen PR, Palladino MA, Lam KS, Lloyd GK, Potts BC (2009) Discovery and development of the anticancer agent salinosporamide A (NPI-0052). Bioorg Med Chem 17:2175–2180
Chauhan D, Singh A, Brahmandam M, Podar K, Hideshima T, Richardson P, Munshi N, Palladino MA, Anderson KC (2008) Combination of proteasome inhibitors bortezomib and NPI-0052 trigger in vivo synergistic cytotoxicity in multiple myeloma. Blood 111: 1654–1664
Chauhan D, Singh AV, Ciccarelli B, Richardson PG, Palladino MA, Anderson KC (2010) Combination of novel proteasome inhibitor NPI-0052 and lenalidomide trigger in vitro and in vivo synergistic cytotoxicity in multiple myeloma. Blood 115:834–845
Richardson PG, Chanan-Khan AA, Lonial S, Krishnan AY, Carroll MP, Alsina M, Albitar M, Berman D, Messina M, Anderson KC (2011) Tanespimycin and bortezomib combination treatment in patients with relapsed or relapsed and refractory multiple myeloma: results of a phase 1/2 study. Br J Haematol 153:729–740
Kikuchi J, Wada T, Shimizu R, Izumi T, Akutsu M, Mitsunaga K, Noborio-Hatano K, Nobuyoshi M, Ozawa K, Kano Y, Furukawa Y (2010) Histone deacetylases are critical targets of bortezomib-induced cytotoxicity in multiple myeloma. Blood 116:406–417
Hideshima T, Bradner JE, Wong J, Chauhan D, Richardson P, Schreiber SL, Anderson KC (2005) Small-molecule inhibition of proteasome and aggresome function induces synergistic antitumor activity in multiple myeloma. Proc Natl Acad Sci U S A 102:8567–8572
Santo L, Hideshima T, Kung AL, Tseng J-C, Tamang D, Yang M, Jarpe M, van Duzer JH, Mazitschek R, Cirstea D, Eda H, Scullen TA, Rodig S, Canavese M, Bradner J, Anderson KC, Jones S, Raje NS (2011) Selective HDAC6 inhibition via ACY-1215, either alone or in combination with bortezomib, restores osteoblast function and suppresses osteoclast differentiation in multiple myeloma. ASH Annual Meeting Abstracts 118:2908
Weber DM, Jagannath S, Mazumder A, Sobecks R, Schiller GJ, Gavino M, Sumbler C, McFadden C, Chen C, Ricker JL, Rizvi S, Oerth C, Brownell P, Hussein MA (2007) Phase I trial of oral vorinostat (suberoylanilide hydroxamic acid, SAHA) in combination with bortezomib in patients with advanced multiple myeloma. ASH Annual Meeting Abstracts 110:1172
Harrison SJ, Quach H, Link E, Seymour JF, Ritchie DS, Ruell S, Dean J, Januszewicz H, Johnstone R, Neeson P, Dickinson M, Nichols J, Prince HM (2011) A high rate of durable responses with romidepsin, bortezomib, and dexamethasone in relapsed or refractory multiple myeloma. Blood 118:6274–6283
Siegel DS, Dimopoulos MA, Yoon S-S, Laubach JP, Kaufman JL, Goldschmidt H, Reece DE, Leleu X, Durrant S, Offner FC, Cavo M, Nagler A, Jagannath S, Graef T, Houp J, Sun L, Howe J, Wear SM, Anderson KC (2011) Vantage 095: vorinostat in combination with bortezomib in salvage multiple myeloma patients: final study results of a global phase 2b trial. ASH Annual Meeting Abstracts 118:480
Dimopoulos MA, Jagannath S, Yoon S-S, Siegel DS, Lonial S, Hajek R, Facon T, Rosinol L, Blacklock HA, Goldschmidt H, Hungria V, Spencer A, Palumbo A, Reece DE, Graef T, Houp J, Sun L, Eid JE, Anderson KC (2011) Vantage 088: vorinostat in combination with bortezomib in patients with relapsed/refractory multiple myeloma: results of a global, randomized phase 3 trial. ASH Annual Meeting Abstracts 118:811
Heink S, Ludwig D, Kloetzel PM, Kruger E (2005) IFN-gamma-induced immune adaptation of the proteasome system is an accelerated and transient response. Proc Natl Acad Sci U S A 102:9241–9246
Kloetzel PM (2004) The proteasome and MHC class I antigen processing. Biochim Biophys Acta 1695:225–233
Noda C, Tanahashi N, Shimbara N, Hendil KB, Tanaka K (2000) Tissue distribution of constitutive proteasomes, immunoproteasomes, and PA28 in rats. Biochem Biophys Res Commun 277:348–354
Strehl B, Seifert U, Kruger E, Heink S, Kuckelkorn U, Kloetzel PM (2005) Interferon-gamma, the functional plasticity of the ubiquitin-proteasome system, and MHC class I antigen processing. Immunol Rev 207:19–30
Rivett AJ, Hearn AR (2004) Proteasome function in antigen presentation: immunoproteasome complexes, Peptide production, and interactions with viral proteins. Curr Protein Pept Sci 5:153–161
Gaczynska M, Rock KL, Goldberg AL (1993) Gamma-interferon and expression of MHC genes regulate peptide hydrolysis by proteasomes. Nature 365:264–267
Kuhn DJ, Orlowski RZ (2012) The immunoproteasome as a target in hematologic malignancies. Semin Hematol 49:258–262
Kuhn DJ, Hunsucker SA, Chen Q, Voorhees PM, Orlowski M, Orlowski RZ (2009) Targeted inhibition of the immunoproteasome is a potent strategy against models of multiple myeloma that overcomes resistance to conventional drugs and nonspecific proteasome inhibitors. Blood 113:4667–4676
Singh AV, Bandi M, Aujay MA, Kirk CJ, Hark DE, Raje N, Chauhan D, Anderson KC (2011) PR-924, a selective inhibitor of the immunoproteasome subunit LMP-7, blocks multiple myeloma cell growth both in vitro and in vivo. Br J Haematol 152:155–163
Muchamuel T, Basler M, Aujay MA, Suzuki E, Kalim KW, Lauer C, Sylvain C, Ring ER, Shields J, Jiang J, Shwonek P, Parlati F, Demo SD, Bennett MK, Kirk CJ, Groettrup M (2009) A selective inhibitor of the immunoproteasome subunit LMP7 blocks cytokine production and attenuates progression of experimental arthritis. Nat Med 15:781–787
Ho YK, Bargagna-Mohan P, Wehenkel M, Mohan R, Kim KB (2007) LMP2-specific inhibitors: chemical genetic tools for proteasome biology. Chem Biol 14:419–430
Sekizawa R, Ikeno S, Nakamura H, Naganawa H, Matsui S, Iinuma H, Takeuchi T (2002) Panepophenanthrin, from a mushroom strain, a novel inhibitor of the ubiquitin-activating enzyme. J Nat Prod 65:1491–1493
Tsukamoto S, Hirota H, Imachi M, Fujimuro M, Onuki H, Ohta T, Yokosawa H (2005) Himeic acid A: a new ubiquitin-activating enzyme inhibitor isolated from a marine-derived fungus, Aspergillus sp. Bioorg Med Chem Lett 15:191–194
Yang Y, Kitagaki J, Dai RM, Tsai YC, Lorick KL, Ludwig RL, Pierre SA, Jensen JP, Davydov IV, Oberoi P, Li CC, Kenten JH, Beutler JA, Vousden KH, Weissman AM (2007) Inhibitors of ubiquitin-activating enzyme (E1), a new class of potential cancer therapeutics. Cancer Res 67:9472–9481
Xu GW, Ali M, Wood TE, Wong D, Maclean N, Wang X, Gronda M, Skrtic M, Li X, Hurren R, Mao X, Venkatesan M, Beheshti Zavareh R, Ketela T, Reed JC, Rose D, Moffat J, Batey RA, Dhe-Paganon S, Schimmer AD (2010) The ubiquitin-activating enzyme E1 as a therapeutic target for the treatment of leukemia and multiple myeloma. Blood 115:2251–2259
Ungermannova D, Parker SJ, Nasveschuk CG, Chapnick DA, Phillips AJ, Kuchta RD, Liu X (2012) Identification and mechanistic studies of a novel ubiquitin E1 inhibitor. J Biomol Screen 17:421–434
Laine A, Topisirovic I, Zhai D, Reed JC, Borden KL, Ronai Z (2006) Regulation of p53 localization and activity by Ubc13. Mol Cell Biol 26:8901–8913
Tsukamoto S, Takeuchi T, Rotinsulu H, Mangindaan RE, van Soest RW, Ukai K, Kobayashi H, Namikoshi M, Ohta T, Yokosawa H (2008) Leucettamol A: a new inhibitor of Ubc13-Uev1A interaction isolated from a marine sponge, Leucetta aff. microrhaphis. Bioorg Med Chem Lett 18:6319–6320
Vassilev LT (2007) MDM2 inhibitors for cancer therapy. Trends Mol Med 13:23–31
Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, Kong N, Kammlott U, Lukacs C, Klein C, Fotouhi N, Liu EA (2004) In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 303:844–848
Ooi MG, Hayden PJ, Kotoula V, McMillin DW, Charalambous E, Daskalaki E, Raje NS, Munshi NC, Chauhan D, Hideshima T, Buon L, Clynes M, O’Gorman P, Richardson PG, Mitsiades CS, Anderson KC, Mitsiades N (2009) Interactions of the Hdm2/p53 and proteasome pathways may enhance the antitumor activity of bortezomib. Clin Cancer Res 15: 7153–7160
Duncan SJ, Gruschow S, Williams DH, McNicholas C, Purewal R, Hajek M, Gerlitz M, Martin S, Wrigley SK, Moore M (2001) Isolation and structure elucidation of Chlorofusin, a novel p53-MDM2 antagonist from a Fusarium sp. J Am Chem Soc 123:554–560
Clark RC, Lee SY, Boger DL (2008) Total synthesis of chlorofusin, its seven chromophore diastereomers, and key partial structures. J Am Chem Soc 130:12355–12369
Tsukamoto S, Yoshida T, Hosono H, Ohta T, Yokosawa H (2006) Hexylitaconic acid: a new inhibitor of p53-HDM2 interaction isolated from a marine-derived fungus, Arthrinium sp. Bioorg Med Chem Lett 16:69–71
Nakahashi A, Miura N, Monde K, Tsukamoto S (2009) Stereochemical studies of hexylitaconic acid, an inhibitor of p53-HDM2 interaction. Bioorg Med Chem Lett 19:3027–3030
Kona FR, Buac D, Burger A (2011) Disulfiram, and disulfiram derivatives as novel potential anticancer drugs targeting the ubiquitin-proteasome system in both preclinical and clinical studies. Curr Cancer Drug Targets 11:338–346
Lim HS, Archer CT, Kodadek T (2007) Identification of a peptoid inhibitor of the proteasome 19S regulatory particle. J Am Chem Soc 129:7750–7751
Lim HS, Cai D, Archer CT, Kodadek T (2007) Periodate-triggered cross-linking reveals Sug2/Rpt4 as the molecular target of a peptoid inhibitor of the 19S proteasome regulatory particle. J Am Chem Soc 129:12936–12937
Verma R, Peters NR, D’Onofrio M, Tochtrop GP, Sakamoto KM, Varadan R, Zhang M, Coffino P, Fushman D, Deshaies RJ, King RW (2004) Ubistatins inhibit proteasome-dependent degradation by binding the ubiquitin chain. Science 306:117–120
D’Arcy P, Linder S (2012) Proteasome deubiquitinases as novel targets for cancer therapy. Int J Biochem Cell Biol 44:1729–1738
Kuo SM, Huang CT, Blum P, Chang C (2001) Quercetin cumulatively enhances copper induction of metallothionein in intestinal cells. Biol Trace Elem Res 84:1–10
Rizk SL, Sky-Peck HH (1984) Comparison between concentrations of trace elements in normal and neoplastic human breast tissue. Cancer Res 44:5390–5394
Turecky L, Kalina P, Uhlikova E, Namerova S, Krizko J (1984) Serum ceruloplasmin and copper levels in patients with primary brain tumors. Klin Wochenschr 62:187–189
Nayak SB, Bhat VR, Upadhyay D, Udupa SL (2003) Copper and ceruloplasmin status in serum of prostate and colon cancer patients. Indian J Physiol Pharmacol 47:108–110
Diez M, Arroyo M, Cerdan FJ, Munoz M, Martin MA, Balibrea JL (1989) Serum and tissue trace metal levels in lung cancer. Oncology 46:230–234
Habib FK, Dembinski TC, Stitch SR (1980) The zinc and copper content of blood leucocytes and plasma from patients with benign and malignant prostates. Clin Chim Acta 104: 329–335
Gupta SK, Singh SP, Shukla VK (2005) Copper, zinc, and Cu/Zn ratio in carcinoma of the gallbladder. J Surg Oncol 91:204–208
Chakravarty PK, Ghosh A, Chowdhury JR (1986) Zinc in human malignancies. Neoplasma 33:85–90
Margalioth EJ, Schenker JG, Chevion M (1983) Copper and zinc levels in normal and malignant tissues. Cancer 52:868–872
Schwartz AE, Leddicotte GW, Fink RW, Friedman EW (1974) Trace elements in noraml and malignant human breast tissue. Surgery 76:325–329
Wong E, Giandomenico CM (1999) Current status of platinum-based antitumor drugs. Chem Rev 99:2451–2466
Galanski M, Arion VB, Jakupec MA, Keppler BK (2003) Recent developments in the field of tumor-inhibiting metal complexes. Curr Pharm Des 9:2078–2089
Galanski M, Jakupec MA, Keppler BK (2005) Update of the preclinical situation of anticancer platinum complexes: novel design strategies and innovative analytical approaches. Curr Med Chem 12:2075–2094
Forestier J (1935) Rheumatoid arthritis and its treatment with gold salts. The results of six years’ experience. J Lab Clin Med 20:827–840
Tiekink ER (2003) Phosphinegold(I) thiolates—pharmacological use and potential. Bioinorg Chem Appl 1(1):53–67
Mirabelli CK, Johnson RK, Hill DT, Faucette LF, Girard GR, Kuo GY, Sung CM, Crooke ST (1986) Correlation of the in vitro cytotoxic and in vivo antitumor activities of gold(I) coordination complexes. J Med Chem 29:218–223
Milacic V, Fregona D, Dou QP (2008) Gold complexes as prospective metal-based anticancer drugs. Histol Histopathol 23:101–108
Ronconi L, Giovagnini L, Marzano C, Bettio F, Graziani R, Pilloni G, Fregona D (2005) Gold dithiocarbamate derivatives as potential antineoplastic agents: design, spectroscopic properties, and in vitro antitumor activity. Inorg Chem 44:1867–1881
Messori L, Marcon G, Orioli P (2003) Gold(III) compounds as new family of anticancer drugs. Bioinorg Chem Appl 1:177–187
Milacic V, Chen D, Ronconi L, Landis-Piwowar KR, Fregona D, Dou QP (2006) A novel anticancer gold(III) dithiocarbamate compound inhibits the activity of a purified 20S proteasome and 26S proteasome in human breast cancer cell cultures and xenografts. Cancer Res 66:10478–10486
Zhang X, Frezza M, Milacic V, Ronconi L, Fan Y, Bi C, Fregona D, Dou QP (2010) Inhibition of tumor proteasome activity by gold-dithiocarbamato complexes via both redox-dependent and -independent processes. J Cell Biochem 109:162–172
Malaguarnera L, Pilastro MR, DiMarco R, Scifo C, Renis M, Mazzarino MC, Messina A (2003) Cell death in human acute myelogenous leukemic cells induced by pyrrolidinedithiocarbamate. Apoptosis 8:539–545
Reymond JC (1950) Fight against alcoholism; a new product: antabuse. J Prat Rev Gen Clin Ther 64:64–66
Schreck R, Meier B, Mannel DN, Droge W, Baeuerle PA (1992) Dithiocarbamates as potent inhibitors of nuclear factor kappa B activation in intact cells. J Exp Med 175:1181–1194
Johansson B (1992) A review of the pharmacokinetics and pharmacodynamics of disulfiram and its metabolites. Acta Psychiatr Scand Suppl 369:15–26
Vallari RC, Pietruszko R (1982) Human aldehyde dehydrogenase: mechanism of inhibition of disulfiram. Science 216:637–639
Meyer RE (1989) Prospects for a rational pharmacotherapy of alcoholism. J Clin Psychiatry 50:403–412
Chen D, Cui QC, Yang H, Dou QP (2006) Disulfiram, a clinically used anti-alcoholism drug and copper-binding agent, induces apoptotic cell death in breast cancer cultures and xenografts via inhibition of the proteasome activity. Cancer Res 66:10425–10433
Di Vaira M, Bazzicalupi C, Orioli P, Messori L, Bruni B, Zatta P (2004) Clioquinol, a drug for Alzheimer’s disease specifically interfering with brain metal metabolism: structural characterization of its zinc(II) and copper(II) complexes. Inorg Chem 43:3795–3797
Cherny RA, Atwood CS, Xilinas ME, Gray DN, Jones WD, McLean CA, Barnham KJ, Volitakis I, Fraser FW, Kim Y, Huang X, Goldstein LE, Moir RD, Lim JT, Beyreuther K, Zheng H, Tanzi RE, Masters CL, Bush AI (2001) Treatment with a copper-zinc chelator markedly and rapidly inhibits beta-amyloid accumulation in Alzheimer’s disease transgenic mice. Neuron 30:665–676
Regland B, Lehmann W, Abedini I, Blennow K, Jonsson M, Karlsson I, Sjogren M, Wallin A, Xilinas M, Gottfries CG (2001) Treatment of Alzheimer’s disease with clioquinol. Dement Geriatr Cogn Disord 12:408–414
Ritchie CW, Bush AI, Mackinnon A, Macfarlane S, Mastwyk M, MacGregor L, Kiers L, Cherny R, Li QX, Tammer A, Carrington D, Mavros C, Volitakis I, Xilinas M, Ames D, Davis S, Beyreuther K, Tanzi RE, Masters CL (2003) Metal-protein attenuation with iodochlorhydroxyquin (clioquinol) targeting Abeta amyloid deposition and toxicity in Alzheimer disease: a pilot phase 2 clinical trial. Arch Neurol 60:1685–1691
Ritchie CW, Bush AI, Masters CL (2004) Metal-protein attenuating compounds and Alzheimer’s disease. Expert Opin Investig Drugs 13:1585–1592
Nguyen T, Hamby A, Massa SM (2005) Clioquinol down-regulates mutant huntingtin expression in vitro and mitigates pathology in a Huntington’s disease mouse model. Proc Natl Acad Sci U S A 102:11840–11845
Chen D, Cui QC, Yang H, Barrea RA, Sarkar FH, Sheng S, Yan B, Reddy GP, Dou QP (2007) Clioquinol, a therapeutic agent for Alzheimer’s disease, has proteasome-inhibitory, androgen receptor-suppressing, apoptosis-inducing, and antitumor activities in human prostate cancer cells and xenografts. Cancer Res 67:1636–1644
Zhai S, Yang L, Cui QC, Sun Y, Dou QP, Yan B (2010) Tumor cellular proteasome inhibition and growth suppression by 8-hydroxyquinoline and clioquinol requires their capabilities to bind copper and transport copper into cells. J Biol Inorg Chem 15:259–269
Rentsch A, Landsberg D, Brodmann T, Bulow L, Girbig AK, Kalesse M (2013) Synthesis and pharmacology of proteasome inhibitors. Angew Chem Int Ed Engl 52:5450–5488
Yang H, Shi G, Dou QP (2007) The tumor proteasome is a primary target for the natural anticancer compound Withaferin A isolated from “Indian winter cherry”. Mol Pharmacol 71:426–437
Yang H, Chen D, Cui QC, Yuan X, Dou QP (2006) Celastrol, a triterpene extracted from the Chinese “Thunder of God Vine,” is a potent proteasome inhibitor and suppresses human prostate cancer growth in nude mice. Cancer Res 66:4758–4765
Tsukamoto S, Tatsuno M, van Soest RW, Yokosawa H, Ohta T (2003) New polyhydroxy sterols: proteasome inhibitors from a marine sponge Acanthodendrilla sp. J Nat Prod 66:1181–1185
Scalbert A, Williamson G (2000) Dietary intake and bioavailability of polyphenols. J Nutr 130:2073S–2085S
Shanafelt TD, Lee YK, Call TG, Nowakowski GS, Dingli D, Zent CS, Kay NE (2006) Clinical effects of oral green tea extracts in four patients with low grade B-cell malignancies. Leuk Res 30:707–712
Sartippour MR, Heber D, Ma J, Lu Q, Go VL, Nguyen M (2001) Green tea and its catechins inhibit breast cancer xenografts. Nutr Cancer 40:149–156
Kemberling JK, Hampton JA, Keck RW, Gomez MA, Selman SH (2003) Inhibition of bladder tumor growth by the green tea derivative epigallocatechin-3-gallate. J Urol 170:773–776
Nam S, Smith DM, Dou QP (2001) Ester bond-containing tea polyphenols potently inhibit proteasome activity in vitro and in vivo. J Biol Chem 276:13322–13330
Golden EB, Lam PY, Kardosh A, Gaffney KJ, Cadenas E, Louie SG, Petasis NA, Chen TC, Schonthal AH (2009) Green tea polyphenols block the anticancer effects of bortezomib and other boronic acid-based proteasome inhibitors. Blood 113:5927–5937
Shanafelt TD, Call TG, Zent CS, LaPlant B, Bowen DA, Roos M, Secreto CR, Ghosh AK, Kabat BF, Lee MJ, Yang CS, Jelinek DF, Erlichman C, Kay NE (2009) Phase I trial of daily oral Polyphenon E in patients with asymptomatic Rai stage 0 to II chronic lymphocytic leukemia. J Clin Oncol 27:3808–3814
McLarty J, Bigelow RL, Smith M, Elmajian D, Ankem M, Cardelli JA (2009) Tea polyphenols decrease serum levels of prostate-specific antigen, hepatocyte growth factor, and vascular endothelial growth factor in prostate cancer patients and inhibit production of hepatocyte growth factor and vascular endothelial growth factor in vitro. Cancer Prev Res (Phila) 2:673–682
Lepley DM, Li B, Birt DF, Pelling JC (1996) The chemopreventive flavonoid apigenin induces G2/M arrest in keratinocytes. Carcinogenesis 17:2367–2375
Janssen K, Mensink RP, Cox FJ, Harryvan JL, Hovenier R, Hollman PC, Katan MB (1998) Effects of the flavonoids quercetin and apigenin on hemostasis in healthy volunteers: results from an in vitro and a dietary supplement study. Am J Clin Nutr 67:255–262
Manach C, Scalbert A, Morand C, Remesy C, Jimenez L (2004) Polyphenols: food sources and bioavailability. Am J Clin Nutr 79:727–747
Kaiser CS, Rompp H, Schmidt PC (2004) Supercritical carbon dioxide extraction of chamomile flowers: extraction efficiency, stability, and in-line inclusion of chamomile-carbon dioxide extract in beta-cyclodextrin. Phytochem Anal 15:249–256
Zheng PW, Chiang LC, Lin CC (2005) Apigenin induced apoptosis through p53-dependent pathway in human cervical carcinoma cells. Life Sci 76:1367–1379
Liu LZ, Fang J, Zhou Q, Hu X, Shi X, Jiang BH (2005) Apigenin inhibits expression of vascular endothelial growth factor and angiogenesis in human lung cancer cells: implication of chemoprevention of lung cancer. Mol Pharmacol 68:635–643
Gupta S, Afaq F, Mukhtar H (2002) Involvement of nuclear factor-kappa B, Bax and Bcl-2 in induction of cell cycle arrest and apoptosis by apigenin in human prostate carcinoma cells. Oncogene 21:3727–3738
Birt DF, Mitchell D, Gold B, Pour P, Pinch HC (1997) Inhibition of ultraviolet light induced skin carcinogenesis in SKH-1 mice by apigenin, a plant flavonoid. Anticancer Res 17:85–91
Chen D, Daniel KG, Chen MS, Kuhn DJ, Landis-Piwowar KR, Dou QP (2005) Dietary flavonoids as proteasome inhibitors and apoptosis inducers in human leukemia cells. Biochem Pharmacol 69:1421–1432
Chen D, Landis-Piwowar KR, Chen MS, Dou QP (2007) Inhibition of proteasome activity by the dietary flavonoid apigenin is associated with growth inhibition in cultured breast cancer cells and xenografts. Breast Cancer Res 9:R80
Hoensch H, Groh B, Edler L, Kirch W (2008) Prospective cohort comparison of flavonoid treatment in patients with resected colorectal cancer to prevent recurrence. World J Gastroenterol 14:2187–2193
Acknowledgements
This work was partially supported by NIH 1R01CA20009, 3R01CA120009-04S1, 5R01CA127258-05 and NCI R21CA184788 (to Dr. Dou), and NIH Center grant P30 CA022453 (to Karmanos Cancer Institute).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Schmitt, S.M., Deshmukh, R.R., Dou, Q.P. (2014). Proteasome Inhibitors and Lessons Learned from Their Mechanisms of Action and Resistance in Human Cancer. In: Dou, Q. (eds) Resistance to Proteasome Inhibitors in Cancer. Resistance to Targeted Anti-Cancer Therapeutics. Springer, Cham. https://doi.org/10.1007/978-3-319-06752-0_1
Download citation
DOI: https://doi.org/10.1007/978-3-319-06752-0_1
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-06751-3
Online ISBN: 978-3-319-06752-0
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)