
Abstract
The sphingosine 1 phosphate receptor family has been studied widely since the initial discovery of its first member, endothelium differentiation gene 1. Since this initial discovery, the family has been renamed and the primary member of the family, the S1P1 receptor, has been targeted for a variety of disease indications and successfully drugged for the treatment of patients with relapsing multiple sclerosis. Recently, the three-dimensional structure of the S1P1 receptor has been determined by X-ray crystallography and the specifics of the sphingosine 1 phosphate ligand binding pocket mapped. Key structural features for the S1P1 receptor will be reviewed and the potential binding modes of additional pharmacologically active agents against the receptor will be analyzed in an effort to better understand the structural basis of important receptor–ligand interactions.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Introduction
Signal transduction is a fundamental process at the center of cellular activity and organismal function. The ability of cells to respond to signals in their environment allows adaptive responses central to survival. Cells are by nature isolated from their environments by means of a plasma membrane barrier which also facilitates the specific signals that impact the internal environment and behavior of the cell. This selectivity in signal transduction is achieved by means of a set of membrane proteins which control entry of reagents and information into the cytoplasm. One of the most important members of signal transduction set of membrane proteins are the G protein-coupled receptors (GPCRs).
GPCRs function through transmission of signals from the extracellular milieu to the cytoplasm of the cell where they are amplified by a variety of second messenger systems initiated by direct interaction with various G proteins or arrestins. As a family, the GPCRs recognize a wide spectrum of extracellular ligands including photons, ions, small organic molecules, peptides, proteins, and bioactive lipids. The GPCR family is one of the largest and most diverse membrane protein families consisting of more than 800 genes in the human genome. Each receptor is capable of recognizing specific ligands and transmitting the binding event to a wide variety of cytosolic signaling networks by means of conformational changes triggered by the specific ligand–receptor interactions (Kenakin and Onaran 2002). These receptor conformational changes are traditionally associated with three general pharmacological effects: inverse agonist, neutral antagonists, and agonist. Inverse agonists are ligands that alter the conformational landscape of the receptor so that it does not trigger any downstream signaling events. Inverse agonists are often classified as antagonists based on their ability to reduce agonist binding or signaling in a dose-dependent manner. Neutral antagonists are also often classified as antagonists based on their ability to reduce agonist binding or signaling, however, they do not alter the conformational landscape of the receptor. Agonists are compounds that alter the conformational landscape of the receptor to trigger a signaling event. It is often the case that the classification of a compound as an agonist depends on the assay used to measure signaling. Many compounds are capable of altering the conformational landscape of a receptor to signal along one pathway (i.e., arrestin) but not others (i.e., G protein) (Kenakin 2012). This phenomenon is known as ligand-biased signaling with the end result being that ultimate pharmacological classification of many compounds depends on the type of assay employed. Once triggered the downstream signaling networks result in a multitude of cellular responses that are dependent not only on the receptor and extracellular signal but also on the tissue type and cellular environment in which the signaling takes place.
Structural characterization of the GPCR superfamily has recently come of age with the solution of over 20 members (Cherezov et al. 2007; Warne et al. 2008; Jaakola et al. 2008; Chien et al. 2010; Wu et al. 2010; Granier et al. 2012; Hanson et al. 2012; Haga et al. 2012; Kruse et al. 2012; Manglik et al. 2012; Thompson et al. 2012; Wu et al. 2012; White et al. 2012; Zhang et al. 2012; Wacker et al. 2013; Wang et al. 2013a, b; Hollenstein et al. 2013; Siu et al. 2013; Tan et al. 2013), with the β2 adrenergic receptor (β2AR) having structural snapshots of multiple pharmacologically relevant signaling conformations (Cherezov et al. 2007; Rasmussen et al. 2011a, b). This wealth of structural information represents a relatively new addition to the corpus of knowledge developed around this family and will inevitably aid the multitude of ongoing efforts to develop pharmaceutics targeting the GPCR receptors.
From a global perspective, all of the structurally analyzed members of the GPCR family consist of a seven-helix bundle with the N-terminus oriented to the extracellular space of the receptor and the C-terminus the intracellular space. Adjacent helices are separated by loops termed extracellular loop (ECL) 1–3 and intracellular loops (ICL) 1–3. The extracellular loops are of variant lengths across the family and are highly divergent in terms of their primary, secondary, and tertiary structural characteristics. Intracellular loops one and two are similar in length and topology across the GPCR superfamily, while ICL3 is highly divergent and is probably unstructured in most GPCRs in the absence of G protein interactions.
From a more detailed perspective, GPCRs can be divided into four regions commonly referred to as domains (Fig. 1). The extracellular region controls access of the receptors’ ligand to the binding pocket. In some receptors the extracellular region also provides important contacts within the ligand binding pocket as well. The transmembrane region is responsible for the core functionality of the receptor family, namely signal transduction through ligand binding and conformational rearrangement. The details of the roles that these helices play for each receptor are now being determined with the benefit of the structural information being generated. The intracellular region forms the canonical allosteric interaction site for the GPCRs where the cytosolic signaling partners such as the G proteins or arrestin couple to the receptor. The fourth, ligand binding region is composed of specific residues within the transmembrane and extracellular domains and as its name implies is responsible for recognition of each receptor’s endogenous ligand. Each of these regions will be characterized in more detail in the context of the S1P1 receptor.

Structural overview of the S1P1 receptor and its four main regions. The position of the membrane is shown with white spheres
One can further stratify the function of the transmembrane region of GPCRs into ligand binding and signal transduction modules (Katritch et al. 2012). Comparing the structural deviation for the ligand binding module to the signal transduction module reveals greater structural diversity within the ligand binding region among the disparate GPCRfamily members. This of course is not surprising given that they each recognize distinct ligands but signal along similar pathways. Interestingly, when analyzing the β2AR structural representatives with an inverse agonist bound compared to an agonist and agonist plus G protein complex, the structural changes in the intracellular region associated with G protein binding are apparent, whereas conformational changes in the ligand binding module associated with agonist interactions are not significantly different (Cherezov et al. 2007; Rasmussen et al. 2011b).
The idea that the conformation of the ligand binding region of GPCRs need not undergo significant structural rearrangement during G protein signaling lends credence to the idea that structures with agonists bound to the conformationally inactive state of the receptor are valid starting points for analysis of agonist structure activity relationship (SAR) programs. Indeed, it should also be within range of modern molecular modeling techniques to predict agonist binding poses using an antagonist structure as an initial template. Thus, the utility of each new GPCR structure will extend well outside of the immediate pharmacological state it was solved in and support multiple efforts in pharmacology research and drug development.
The superfamily of GPCRs can be divided into six major classes based on sequence homology, termed A–F, with the class A family being the most prevalent and well represented among the structurally known GPCRs (Fredriksson et al. 2003). The class A family recognizes a diverse set of ligands from photons to peptides and proteins and is the grouping in which the sphingosine 1 phosphate (S1P) family of lipid binding receptors is classified.
Prior to deorphanization, the S1P and lysophosphatidic acid (LPA) family of receptors were collectively termed the endothelial differentiation gene (Edg) family of receptors. This naming was based on the first discovered member of the family, Edg1 which is involved in a set of immediate early response gene products cloned from human umbilical vein endothelial cells (Hla and Maciag 1990). Subsequent Edg family receptors were discovered and classified based on sequence similarity, however, they have no involvement in endothelial differentiation. The classification was formalized in 2002 to follow standard International Union of Pure and Applied Chemistry (IUPAC) naming conventions and Edg-1 became the S1P1 receptor based on its highest affinity endogenous ligand S1P and the chronological order of discovery within the family (Chun et al. 2002). At the time, there were five members of the S1P receptor family and three members of the closely related LPA receptor family. Currently, two additional LPA receptors have been characterized (Choi et al. 2010).
The S1P1 receptor plays a crucial role in lymphocyte trafficking and is expressed on both the lymphocytes themselves and the sinus-lining endothelium (Cahalan et al. 2011). S1P1 receptors are differentially regulated in different cell types with lymphocyte populations coupling to Gα2i and exhibiting rapid loss of cell surface-expressed receptor in response to agonist, in contrast to the endothelium-expressed receptor which has a ten-fold higher expression level and significant signaling reserve (Pham et al. 2008b; Cahalan et al. 2011; Arnon et al. 2011). Disruption of S1P1 receptor signaling can either result in an excursion of lymphocytes under low or transient agonist occupancy or a profound lymphopenia in the presence of high-affinity agonists which cause internalization of the receptor resulting in functional antagonism (Rosen et al. 2013). Similarly, it has been shown that high-affinity antagonists can induce a similar lymphopenia but with a significant increase in capillary leakage relative to agonists which may restrict the utility of antagonists for the S1P1 receptor in clinical settings (Sanna et al. 2006; Oo et al. 2011).
The ability of S1P1 receptor agonists to modulate immune responses by selectively arresting the trafficking of naïve and central memory T and B lymphocytes in peripheral lymphoid organs without affecting the trafficking of effector memory populations has prompted the development of agonists for a variety of autoimmune disorders with multiple sclerosis being the flagship indication for this class of compounds.
The high interest in S1P1 receptor pharmacological agents combined with a rapidly increasing confidence in the crystallizability of the GPCR superfamily in general (Hanson and Stevens 2009) ultimately led to the successful structure determination of the S1P1 receptor in its antagonist-bound state (Hanson et al. 2012). The details of this structure along with models of interactions for other ligands of various classes that bind to the S1P1 receptor will be discussed in this chapter.
2 Structural Analysis of the S1P1 Receptor
2.1 Structure Determination Process
The structural characterization of the S1P1 receptor was initiated after early success in the GPCR family on the β2AR (Cherezov et al. 2007) and adenosine A2a receptor (Jaakola et al. 2008). Both of these receptors employed the strategy of replacing one of the intracellular loops (ICL3) with a small soluble protein domain derived from T4-lysozyme (T4L) to facilitate crystal contact formation (Rosenbaum et al. 2007). A similar strategy was employed for the S1P1 receptor.
Full-length wild-type and T4L-fused receptor each with an N-terminal Flag epitope tag (to assess receptor expression levels) and a C-terminal 10x histidine tag (to facilitate receptor purification) proved intractable to further structural studies due to formation of higher-order oligomers. A serial deletion of the C-terminus of the receptor in four residue increments resolved this issue (Fig. 2a). Small-scale expression studies of this family of constructs revealed a dramatic improvement in reducing the oligomeric state of the receptor after extraction and purification as the C-terminus was shortened.

Select data from the protein chemistry and construct design effort for the S1P1 receptor. a A series of C-terminal truncations serially removing four amino acids at a time. The presence of the dimer in the SDS-PAGE gel was reduced and then disappeared for the optimal constructs 444, 445. b SEC stability analysis of junction adjustment mutations both with ligand (L) and without ligand (U). Each construct was compared to 445 and the quality was assessed based on peak profile without ligand after two days at 4 °C
After preliminary crystallization trials failed in lipidic cubic phase (LCP), further rounds of construct optimization were initiated. A series of combinatorial adjustments of the insertion point for the T4L fusion partner were tested for expression and stability in the presence and absence of ligand (Fig. 2b). Extraction and purification of each construct with and without the small molecule antagonist W146 (Sanna et al. 2006) (later renamed ML056 to avoid confusion with amino acid abbreviations) followed by analysis with size exclusion chromatography (SEC) (the position and width of an SEC peak gives a good indication of oligomeric state and sample homogeneity) showed a significant effect on stability as a function of fusion insertion point. The optimal construct was selected based on its SEC profile after heating (which should remain unchanged relative to unheated samples) in the absence of ligand and subsequently scaled up for crystallization trials.
The final crystallization construct was incorporated into the baculovirus genome which was then amplified to obtain a high-titer stock of recombinant baculovirus for infection of large-scale expression batches of Sf9 insect cells, to ultimately generate sufficient protein for crystallographic studies after purification. Extraction of the receptor from the insect plasma membrane was performed using high concentrations of dodecyl maltoside detergent. Purification of the recombinant S1P1 receptor was facilitated by the presence of the 10x histidine tag on the C-terminus of the receptor which was utilized for binding to immobilized metal affinity resin in a single-step chromatography protocol. After purification, the protein was concentrated to approximately 50 mg/mL. Receptor extraction, purification, and concentration were performed in the presence of saturating amounts of ML056.
Crystallization of the receptor was attempted using the LCP approach where protein samples of S1P1 receptor in complex with ML056 were reconstituted into monoolein which when hydrated forms a cubic mesophase capable of supporting crystallization of a wide variety of membrane proteins (Landau and Rosenbusch 1996; Caffrey 2000; Cherezov 2011). Resulting crystals were harvested directly from LCP matrix and flash frozen in liquid nitrogen for diffraction analysis.
X-ray diffraction data for approximately 400 crystal samples were collected and ultimately used in the final dataset. Due to the rapid onset of radiation damage, data collection was limited to a maximum of 6° oscillation per crystal of which only the first 1–2 frames diffracted to the maximum resolution. Data were processed using a novel method (Hanson et al. 2012) to extract individual reflections from decayed images and the structure was solved and refined by standard methods to 2.8Å. The final coordinates were deposited in the protein data bank under accession number 3V2Y. The supplemental material associated with the original publication of the S1P1 receptor structure contains complete details on the structure solution process (Hanson et al. 2012) which has since been used effectively in one other published GPCR structure determination effort (Hollenstein et al. 2013).
2.2 Structural Characterization
We will discuss in detail the extracellular, transmembrane, and ligand binding regions of the S1P1 receptor in this section. The lack of resolved intracellular loops with the exception of ICL1 combined with the presence of the T4L fusion protein inserted in ICL3 precludes any in-depth analysis of the intracellular region of the receptor.
2.2.1 Transmembrane Region
From a global perspective, the S1P1-T4L receptor structure (referred to as the S1P1 receptor for the remainder of the chapter) shares many common features with previous and subsequently determined receptors, including seven-transmembrane helices arranged in a structurally conserved bundle, and similar length and orientation of intracellular loop one. However, there are some important differences associated with the S1P1 receptor structure and presumably the family that lends itself to binding of its endogenous lipid ligand. We will examine these differences in more detail beginning with an analysis of the transmembrane region core.
In a recent review, Venkatakrishnan and colleagues examined conserved contact points among all of the GPCR structures determined to date (Venkatakrishnan et al. 2013). These contact points serve as a scaffold from which the diversity of the GPCR family is built and are used here to facilitate the comparison between the S1P1 receptor and other human class A GPCR structures. We use the core residues as a template for overlaying each of the human class A inverse agonist structures with the S1P1 receptor. The set was limited to restrict the interpretation to a single species and pharmacological state. The root mean square deviation (RMSD) for just the Cα atoms after aligning with the S1P1 receptor shows a significantly improved RMSD for the core residues compared to the entire transmembrane region (Fig. 3b). This type of overlay provides a superior template for visually assessing structural differences between the receptors’ transmembrane regions (Fig. 3a). Calculation of the root mean square fluctuation allows mapping of the structural deviations across the receptor set in order to understand the regions responsible for conveying the structural changes associated with recognition of ligand diversity across the family (Fig. 3c).

Analysis of the structural differences in the transmembrane (TM) region over the entire set of class A human GPCRs in the inverse agonist state. a Structural overlay focusing on TM VI, TM VII, and TM I. The shift in TM I for the S1P1 receptor could result in a larger gap between TM I and TM VII and is the largest helical divergence for the receptor. b Plot of the RMSD values of the receptor family in comparison with S1P1 receptor. The dark gray values are core RMSD, and the light gray values are transmembrane domain RMSD. Both are comparing only Cα atoms. c Graph of the root mean square fluctuation over the set of GPCRs
The position of the core contact residues are listed in Table 1 in their Ballesteros–Weinstein notation as well as residue number associated with the S1P1 receptor (Vroling et al. 2011). These contact positions are conserved throughout the determined class A structures and serve as an anchor point for our analysis of the bioactive lipid binding receptors as well as other class A GPCRs although the sequence identity at some of the positions can vary significantly (Table 1). These core contact residues can be further grouped based on spatial proximity to each other and the helices that are constrained by them. For instance, one cluster of core contacts links transmembrane (TM) I, II, and VII (Fig. 4a), cluster 1 consists of individual interaction groups 1, 2, 3, 4, 5, and 7. The second cluster of interactions links TM II, III, and IV through a series of four interaction groups 6, 8, 10 and a frequently observed interaction between position 2.45 and 4.50. These two positions interact through a hydrogen bond between an Asn (found in 60 % of class A GPCRs) or Ser (30 %) at 2.45 and the indole nitrogen of a Trp (found in 94 % of class A GPCRs) at position 4.50 (Fig. 4b).

Structural view of the clusters of core residues as defined in Table 1. Each cluster comprises a number of interacting groups that fit together to form a rigid scaffold. These clusters are distributed throughout the receptor helical bundle and help define the structural characteristics of the GPCR family
A third cluster of conserved contacts links together TM helices III, VI, and VII in the vicinity of the S1P1 receptor ligand binding pocket (Fig. 4c). These positions maintain important contacts between TM VI and TM III through two side chain-mediated interactions. The sequence conservation across the lipid binding receptors is shown in Table 1. Position 3.36 which is a Leu in the S1P1 receptor as well as 94 % of Edg family receptors interacts with the well-characterized Trp toggle switch at 6.48. In the Edg family, this location is 100 % conserved over all orthologs and homologs. Both these amino acids are adjacent to the canonical class A orthosteric binding pocket and as such contribute significant interactions to ligand binding. In contrast to the conservation of position 6.48 across class A GPCRs, position 3.36 is quite variable and has one of the highest sequence entropies of all of the core positions (Table 1). Importantly, the contrast between sequence entropy within the Edg family and overall entropy is the highest implicating this position as a major determinant of ligand specificity. It is reasonable based on the proximity of this cluster to the ligand binding pocket that ligand interactions also affect the strength of interaction between TM III and VI creating a potential linchpin for pharmacological activity at this position. Immediately below this important interaction is a second pair of residues whose conformation is often linked to receptor pharmacology (Fig. 4c). At the 3.40 and 6.44 positions are Val132 and Phe265; Val is conserved at 3.40 in the Edg family almost 70 % of the time, whereas overall class A receptors Val is found only 19 % of the time with Ile being the dominant amino acid. Phe is 100 % conserved at 6.44 in the Edg family and 77 % overall for class A receptors. It is likely based on this analysis that the interactions provided by this cluster serve an important role in maintaining the appropriate conformation of TM VI and that van der Waals interactions are the primary means by which this is achieved. This creates an environment sensitive to the shape and character of the binding pocket itself and by extension the nature of the bound ligand. During conformational switching, these contacts are maintained forcing a translation of small perturbations in the ligand binding pocket to the intracellular region by a series of compensatory movements. In S1P1 receptor SAR, it is known that the conformational switch between the antagonist state of the receptor and agonist state is driven by volume changes within the binding pocket (Davis et al. 2005). On the other side of TM VI, there are two groups of interactions that hold the top of TM VI and VII together (Fig. 4c). The first group consists of three interacting residues, Leu272:6.51, Phe296:7.38, and Leu297:7.39 which form a tight van der Waals cluster that is mediated mainly through mainchain and Cβ atoms and therefore relatively independent of the identity of the residue. The second group consists of Cys268:6.47 and Asn303:7.45. This interaction is dependent on side chain proximity and probably serves an important role in positioning the Asn residue for interacting through a hydrogen bond with the indole nitrogen of Trp269(6.48) (Fig. 4c). Asn is conserved in this position in 51 % of class A GPCRs and as an Asn, His, or Ser in 83 % of class A GPCRs, indicating that the ability of this position to serve as a hydrogen bond acceptor or donor is important for S1P1 receptor and GPCR function in general. This position also hydrogen bonds with a conserved network of tightly bound water molecules that play a role in the activation mechanism of GPCRs. However, the water network was not observed in the S1P1 receptor structure, probably due to resolution limitation. Finally, cluster number four consists of three interactions that constrain TM V relative to TM III and one interaction between TM V and TM VI (Fig. 4d). At the bottom of the TM III/TM V interface there are a series of interactions mediated by three residues. Arg223:5.60 forms a hydrogen bonding interaction with Tyr143:3.51 (Fig. 4d). An Arg residue at position 5.60 is highly conserved in the Edg family and somewhat conserved throughout the GPCR class A superfamily at 72 and 26 %, respectively. The lone interaction connecting TM V to TM VI occurs in this cluster and is mediated by a van der Waals contacts from the side chain of the amino acid at position 6.41 to the Cβ and mainchain atoms of position 5.54. Position 6.41 is a Leu in the case of S1P1 receptor and 90 % of Edg receptors, whereas it is a conserved as a Leu in 14 % of class A GPCRs. The most common residues in this position across the class A family are similar in hydrophobic side chain bulk with Val, Met, Leu, and Ile representing 82 % of class A GPCRs. The sequence conservation as function of Ballesteros–Weinstein position are determined over all species using the 7-TM explorer website http://gris.ulb.ac.be/cgi-bin2/xplor.py (Van Durme et al. 2006).
2.2.2 Extracellular Region
The extracellular region for all GPCRs consists of three loops: ECL1 between TM helices II and III, ECL2 between TM helices IV and V, and ECL3 between TM helices VI and VII. Optionally, there is a structured N-terminus that interacts with the ECLs such as with rhodopsin, CXCR4, CCR5, and S1P1 receptors (Fig. 5) (Palczewski et al. 2000; Wu et al. 2010; Hanson et al. 2012; Tan et al. 2013). In the case of the chemokine receptors CXCR4 and CCR5, this structured N-terminus participates in important interactions with the chemokine ligands. In the case of rhodopsin and the S1P1 receptor the structured N-terminus occludes the binding pocket, in the antagonist-bound state, cutting off access to the extracellular milieu (Fig. 5b). One possible role for this structured N-terminus is that it is a feature of the S1P1 receptor structure in general and its presence implies the ligand does not access the binding pocket from the extracellular space directly. Instead, it is possible that the ligand gains access to the binding pocket through the lipid membrane where there is an enlarged gap between TM I and TM VI. This gap is larger in the S1P1 receptor than other class A GPCRs largely due to a shift in the position of the extracellular end of TM I away from TM VII in the S1P1 receptor (Fig. 2a). This certainly makes sense in that the endogenous ligand is a lipid, more at home in a plasma membrane than the aqueous environment of the extracellular space. It is likely that access to the binding pocket is achieved by initial partitioning of the ligand into the plasma membrane where it then enters through an access channel formed by gaps in the apical section of the transmembrane bundle (Filipek et al. 2003; Schadel et al. 2003; Hurst et al. 2010, 2013). In addition, the limited access to the ligand binding site from the extracellular region is also supported from ligand binding pharmacology where S1P1 receptor ligands, including S1P itself, show slow saturation of receptor binding in the presence of excess ligand as well as slow off-rates (Rosen et al. 2009).

Overview of the extracellular region of the S1P1 receptor as viewed from the extracellular space looking down on the plane of the membrane. a Ribbon diagram representation of the extracellular region with the N-terminal capping helices and loops marked along with the disulfide bonds in ECL2 and ECL3. b Surface representation of the same view. The N-terminal capping helix packs tightly with the extracellular loops and prevents access of the ligand into the binding pocket directly from the extracellular space
2.2.3 Ligand Binding Region
The general region of the orthosteric binding pocket is roughly the same across the GPCR class. However, the details of binding within this region diverge considerably for different receptors and ligands (Katritch et al. 2012). Residue positions involved in binding pocket interactions are largely preserved but with each receptor class interacting specifically with a subset of approximately half of the potential contacts in the ligand binding cradle (Venkatakrishnan et al. 2013). The S1P1 receptor provides 18 residues from the transmembrane region for interactions with the ML056 antagonist along with three additional residues from ECL2 and two from the N-terminus, which are not factored into the referenced analysis (Table 1, Fig. 6).

Detailed S1P1 receptor ligand binding pocket interactions. ML056 is colored with green carbons. Interacting residues are rendered as sticks and colored according to region and binding pocket location. a Polar binding interactions are shown as orange carbons from the transmembrane region and blue or red carbons from ECL2 and N-terminus, respectively. b Hydrophobic interactions are shown as pink carbons. Residue labels designate the amino acid, S1P1 receptor number and Ballesteros–Weinstein index after the colon. c ML056 ligand interaction diagram showing all of the residues that are within 4Å of the ligand position in the structure
ML056 lies in an amphipathic pocket where the head group phosphonate interactions are largely polar in nature and the alkyl chain tail interactions are largely hydrophobic as would be expected (Fig. 6b and c). The polar interactions observed for ML056 largely confirm mutagenesis data establishing the importance of Arg120:3.28 and Glu121:3.29 which were identified as important residues for supplying interactions with the zwitterionic sphingosine head group (Parrill et al. 2000b; Jo et al. 2005). In addition, the phosphonate head group of ML056 is surrounded by a ring of positively charged and polar residues contributed by TM helices III and VII, ECL2, and the N-terminal capping helix. Together these residues form a pocket that provides charge complementarity and high-affinity interactions to the phosphate group of the sphingolipids (Fig. 6a). A feature of ML056 is a primary amine located in the beta position relative to the phosphonate group. This primary amine is likely protonated and charged at physiological pH, thus enhancing its interactions with Glu121:3.29 through salt bridge formation. In addition to Glu121, Asn101:2.60 and Tyr98:2.57 provide hydrogen bonding interactions with the primary amine and amide linkage of ML056, respectively (Fig. 6a). The phenyl aryl tail of ML056 inserts into a hydrophobic pocket consisting of residues from TM helices III, V, VI, and VII, as well as ECL2 (Fig. 6b). The pocket is lined with short aliphatic residues that define the shape and hydrophobicity of the pocket and four aromatic residues that provide the potential for specific interactions (Fig. 6c). The importance of some of these residues in the binding and signaling of the S1P1 receptor was determined previously through molecular modeling and mutagenesis; however, it is important to keep in mind that the antagonist interactions may differ from agonist interactions, and indeed, it has postulated that the antagonist aryl chain occupies a discrete pocket compared to the endogenous ligand (Parrill and Tigyi 2013).
3 Analyses of S1P1 Receptor Ligands
The S1P1 receptor represents the first example of a lipid binding GPCR being structurally determined. A good deal of biochemical and biophysical characterization of this receptor predated the actual structure solution. This type of information is critical for understanding and interpreting the experimental electron density maps and together they provide an important framework for analyzing additional compounds that bind to the S1P1 receptor. We review here an analysis of S1P1 receptor ligand space where we utilize the coordinates of the S1P1 receptor structure bound to ML056 to predict the binding poses of naturally occurring and synthetic antagonists and agonists.
3.1 S1P1 Receptor Antagonists
Perhaps the most straightforward extrapolation of the structural information is in using the receptor to analyze the binding mode of pharmacologically similar agents. In the case of the S1P1 receptor, we will analyze the antagonist space first as it requires little additional manipulation of the model to facilitate the discussion. The antagonist space has received little attention for the S1P1 receptor until quite recently due in part to the demonstrable success of agonists for immunomodulation as well as early reports of antagonists causing significant capillary leakage in vivo along with a dose competitive reversal of lymphopenia (Sanna et al. 2006). Antagonists were developed mainly as tool compounds, however, there were numerous reports on the application of certain antagonists for the S1P1 receptor typically for inhibition of angiogenesis and recent findings suggest that sufficiently potent antagonists have the same pharmacological effect on lymphocytes as agonists, a finding which may renew interest in developing pharmaceuticals that block the S1P1 receptor for immunomodulation in autoimmune indications such as rheumatoid arthritis, inflammatory bowel disease and multiple sclerosis.
3.1.1 Alkyl Phenyl Amide Phosphonates and Structural Analogs of FTY720-P
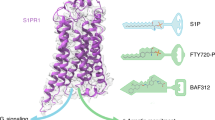
Initial efforts at developing antagonists for the S1P receptor family focused on generating structural analogs of the agonist FTY720-P, a phosphorylated and pharmacologically active metabolite of FTY720 (fingolimod) (see Sect. 3.2.2 for a description of FTY720-P) (Davis et al. 2005). These efforts resulted in antagonists with a very steep SAR profile and specificity for the S1P1 and S1P3 receptors. It was found during the course of this development effort that substitution around the central phenyl ring dictated the pharmacology observed for the compounds. For instance, para-substitution around the phenyl ring generated agonists for the S1P1 receptor, whereas meta-substitution coupled with progressive shortening of the aryl chain generated antagonists with varying degrees of receptor subtype specificity. Interestingly, meta-substituted 10 carbon aryl chain compound (VPC23069) possessed agonist pharmacology, whereas the 8 (VPC23019) and 6 (VPC23031) carbon aryl chains did not. Thus increasing hydrophobic volume on a meta-substituted compound or employing para-substitution could convert antagonist compounds into agonist (Davis et al. 2005). This family of compounds is similar to the ML056 antagonist and the SAR for the VPC series can be used to generate receptor models capable of docking agonist molecules for analysis. Indeed, this series of compounds was used to generate an agonist model of compound binding through induced fit docking protocols as reported in the initial structural analysis of the S1P1 receptor (Sherman et al. 2006; Hanson et al. 2012).
Analysis of the molecular interactions for VPC23019 after docking into the ML056-S1P1 receptor binding pocket shows a similar orientation compared to ML056 itself (Fig. 6). This is not unexpected as the compounds share a high degree of similarity, the main difference being a two-carbon extension of the meta-substituted aryl chain of VPC23019 compared to ML056, which is easily accommodated in the antagonist binding pocket.
More recently, the SAR around the hydrophobic region of the S1P1 receptor binding pocket, head group vinyl phosphonate analogs of FTY720-P showed pan antagonism for the S1P receptors on calcium mobilization assays, indicating that the modifications around the head group region can also have an effect on the exhibited pharmacology of the compounds. Interestingly, these compounds were still active on an extracellular signal-regulated kinase (ERK) phosphorylation assay raising the possibility of pathway bias (Valentine et al. 2010).
3.1.2 Non-lipid Antagonists
Recently, three lead-like non-lipidic compounds have been characterized as antagonists of the S1P1 receptor (Urbano et al. 2013). The first to be reported is a series of dipeptide, proline, triazole compounds that were optimized from a screening effort at Exelixis (Ibrahim et al. 2012). These compounds were shown to have in vivo efficacy against tumor growth with oral administration of mice implanted with MBA- MB-231T breast adenocarcinoma xenografts. Evaluation in higher order species demonstrated promising pharmacokinetic profiles in rat, dog, and cynomolgus monkey (Ibrahim et al. 2012).
The primary example of this compound series XL541 features a central tri-substituted proline ring with a 1,2,3-triazole substituent extending into the anionic binding region of the polar pocket (Fig. 7a). This triazole moiety provides the main ionic interactions driving compound binding while two additional substituents, a fluorophenyl and an oxydibenzene moiety, fill the putative access channel and hydrophobic tail region, respectively. An amide linkage to the oxydibenzene group presents the amide carbonyl functionality as a hydrogen bond acceptor for Tyr98:2.57 and the amide nitrogen as a hydrogen bond donor for Glu121:3.29 (Fig. 7a). The first ring of the oxydibenzene system is positioned similarly to the ML056 phenyl ring, whereas the terminal phenyl ring is positioned to interact with Phe125:3.33 (Fig. 7a).

Proposed molecular interactions for three antagonists docked into the ML056 binding pocket. Each antagonist is represented as a ligand interaction diagram showing all of the residues in the S1P1 receptor binding pocket within 4Å of the docked pose of the ligand (top) and structural interactions of each ligand (purple carbons) compared to ML056 (green carbons) (bottom). a Docking analysis of XL541. b Docking analysis of TASP0277308. c Docking analysis of NIBR-0213
The second series of antagonists under this category was discovered from a screening deck without optimization and reported to bind to the S1P1 receptor with a low nM IC50. This compound, TASP0277308, showed efficacy in a mouse collagen-induced arthritis model (Fujii et al. 2012a) and induces lymphopenia while suppressing swelling leukocyte infiltration and hyperplasia in a mouse contact hypersensitivity model (Fujii et al. 2012b). Finally, this antagonist has been tested and has shown efficacy for inhibition of VEGF-induced endothelial tube formation in vitro and suppressed tumor cell-induced angiogenesis in vivo (Fujii et al. 2012c).
The molecular interactions for TASP0277308 follow a similar pattern as other antagonists in the presentation of a polar anion to the phosphonate binding region of the ML056 pocket. In the case of TASP0277308, a sulfonamide moiety linked to a dichlorophenyl ring interacts at the top of the binding pocket. In addition, a 1,2,4-triazole acts as a protonated positively charged central ring which interacts through charge coupling with Glu121:3.29 (Fig. 7b). Finally, a para-substituted phenylpiperazine ring system fills the hydrophobic binding pocket with the phenyl ring lining up roughly with the ML056 phenyl and the piperazine ring interacting with Phe125:3.33 (Fig. 7b).
Finally, a series of N-biaryl(hetero) arylsulfonamide compounds originally reported in the patent literature and later optimized for pharmaceutics properties to produce NIBR-0213 (Berst et al. 2007; Quancard et al. 2012) have shown comparable efficacy to FTY720-P in mouse experimental autoimmune encephalomyelitus) models of human multiple sclerosis.
The molecular interactions for a docked NIBR-0213 which presents a carboxyl group as an anion for interacting with Arg120:3.28, Lys34 and Tyr29 (Fig. 7c). There are no predicted polar interactions with Glu121:3.29 or Asn101:2.60 which may be an important route for improving the potency of this compound series. This series of compounds features a meta-substituted phenyl ring that is predicted to bind in a similar position as the phenyl ring of ML056 (Fig. 7c) and a terminal 1-chloro-2-methylbenzene ring that interacts with Phe125:3.33 (Fig. 7c).
Together these data suggest that antagonists targeting the S1P1 receptor may have some benefit for indications involving immunomodulation, angiogenesis, and pain modulation (Welch et al. 2012), without the cardiac side effects observed with all of the S1P1 receptor agonists to varying degrees (Gergely et al. 2009; Schmouder et al. 2012; Fernandez et al. 2012; Zipp et al. 2012). However, it does appear that the antagonist class carries a greater burden of increased vascular permeability which may manifest as lung or macular edema (Sanna et al. 2006; Cahalan et al. 2013). The molecular interactions predicted for each of these antagonist compounds tracks closely with those of ML056 itself. Particularly important is the presence of a central aromatic ring with the correct orientation of substituents to mimic the meta-substitution pattern of ML056. This ensures adequate spacing between the ligand and Trp269:6.48 while maintaining hydrophobic or aromatic interactions with the rest of the lipid tail binding pocket. The final component is the correct presentation of polar or charged groups for interacting with the polar binding region of the binding pocket. Although occupation of all of these polar sites is not necessary, provided good van der Waals interactions are achieved in the lipid binding region.
3.2 S1P1 Receptor Agonists
The initial discovery of the Edg family of receptors and subsequent characterization of their endogenous ligands and role in the immune response, triggered development efforts for S1P1 receptor agonists in the treatment of immune-mediated pathologies beginning with solid organ transplant rejection through the approval of the non-selective S1P receptor prodrug agonist fingolimod for treatment of relapsing multiple sclerosis. Recently, a number of second-generation compounds with enhanced selectivity and pharmaceutics properties relative to fingolimod have entered the development and clinical trial pipeline (O’Sullivan and Dev 2013). These compounds can be roughly categorized into two types depending on their reliance on sphingosine-like head group interactions (class 1) or their independence on such interactions (class 2) (Hanson et al. 2012). We will examine the putative modeled binding mode for three agonist compounds, including the natural ligand S1P, using the antagonist-bound structural coordinates along with available mutagenesis data as a starting point.
3.2.1 Sphingosine 1-Phosphate
The endogenous signaling molecule for the S1P family of receptors, the zwitterionic lipid S1P is an important component of biological membranes and has evolved as a highly versatile signaling molecule regulating many cell responses such as proliferation (Zhang et al. 1991), apoptosis (Cuvillier et al. 1996), differentiation, migration (Hobson et al. 2001; Paik et al. 2001) and also immunological responses (Huwiler et al. 2000; Spiegel and Milstien 2003). Dysregulation of S1P itself has been implicated in a multitude of disease states including Alzheimers (Takasugi et al. 2011), pain (Coste et al. 2008; Welch et al. 2012), multiple sclerosis (Kułakowska et al. 2010), diabetes (Whetzel et al. 2006), and cancer (Xia et al. 2000; Ogretmen and Hannun 2004; LaMontagne et al. 2006; Visentin et al. 2006; Pyne and Pyne 2010) among others (O’Sullivan and Dev 2013). The lipid is generated from sphingomyelin as part of the sphingomyelin cycle which involves generation of ceramide, sphingosine, and finally sphingosine 1- phosphate (Fyrst and Saba 2010). S1P elicits its effect primarily through its actions on five S1P receptors (S1P1,2,3,4,5) (Huwiler and Pfeilschifter 2008). Analysis of the potential binding mode of S1P in the S1P1 receptor binding pocket will serve as a useful entry point for discussion of the potential differences and similarities between antagonist and agonist binding.
Modeling efforts to predict the binding mode of the endogenous ligand for S1P1 receptor have been ongoing prior to the solution of the receptor structure with varying degrees of success (Parrill et al. 2000a, b; Wang et al. 2001; Lim et al. 2004; Holdsworth et al. 2004; Inagaki et al. 2005; Deng et al. 2007; Pham et al. 2008a; Schürer et al. 2008; van Loenen et al. 2011). These modeling efforts have recently been reviewed in comparison with the antagonist-bound S1P1 receptor structure (Parrill and Tigyi 2013).
Using the structurally derived binding pocket as a starting point coupled with induced fit docking, there are essentially two possibilities for the orientation of the S1P molecule within the S1P1 receptor. The first possibility is that binding of long acyl chains is accommodated within the antagonist binding pocket or agonist induced expanded version. The hydrophobic volume of the long chain agonists trigger a conformational change associated with S1P1 receptor agonism. This is consistent with the SAR around the VPC antagonist compounds where sequential lengthening of the aryl chain resulted in a switch from agonist to antagonist pharmacology (Davis et al. 2005).
Docking of the S1P ligand into the antagonist binding pocket is straightforward with structurally derived coordinates able to accommodate S1P aryl chain while maintaining polar head group interactions (Fig. 8a). This is somewhat surprising given the finding that an agonist binding pocket requires an increased volume to accommodate agonist compounds. One explanation may be that the S1P aryl chain is by its nature very flexible and able to conform to many different binding pocket shapes albeit with varying degrees of associated conformational strain. Although the chain can be accommodated by the antagonist binding pocket of ML056, it is not optimal and is in a partially strained conformation based on the observed aryl chain dihedral angles measured after docking and minimization (Fig. 8a). The strain associated with nonoptimal torsional angles can be resolved by subtly changing the shape of the binding pocket through short-term molecular dynamics simulations. The entire system was first equilibrated in a phospholipid bilayer placed by alignment with the adenosine A2a receptor, water was added to the solvent accessible regions and ions were added to generate a charge neutral system. The simulation was carried out for 1.2 ns after equilibration to observe if the ligand would attain a more energetically favorable conformation and if there are any significant changes to the binding pocket in response (Shivakumar et al. 2010). Interestingly, much of the strain around the aryl chain torsion angles was resolved along with subtle changes in side chain positions of residues lining the binding cavity (Fig. 8a). Notably, the χ2 angle of Trp269:6.48 side chain changed from a value of 95° in the antagonist structure to an average angle of 124° in the molecular dynamics simulation (Fig. 8a).

Proposed binding poses for three agonist compounds. a S1P docks into the antagonist binding pocket while maintaining polar head group interactions, but with considerable strain in the aryl chain. The strain in the aryl chain of S1P (S1P antagonist) is mainly the result of the position of the W269 side chain which rotates out of the path of the extended aryl chain (S1P opt) during the course of molecular dynamics simulations to relieve the strain. b FTY720-P docks into the S1P-induced binding pocket with a similar pose compared to S1P itself. c RP-001 has increased rigidity compared to both S1P and FTY720-P but still docks within the S1P-induced binding pocket forming van der Waals interactions with the agonist position of W269 and polar interactions with E121 and R120
Based on this limited analysis the endogenous ligand could bind in roughly the same pocket as antagonist compounds. The conformational strain associated with this binding, however, will eventually be relieved by subtle conformational changes of the residues lining the pocket which will not only change the shape and characteristics of the cavity but also trigger a substantial receptor conformational change on the intracellular region associated with agonist signaling. The idea that subtle changes in the binding pocket can trigger the antagonist to agonist conformational switch has been validated with the recent structure of class A GPCRs with agonist bound and coupled to G protein (Rasmussen et al. 2011b).
3.2.2 Fty720-P
FTY720 (fingolimod) was synthesized in an effort to minimize the toxic effects of ISP-1, a fungal metabolite with immunosuppressive properties, which has been used in traditional Chinese herbal medicine as an eternal youth elixir (FUJITA et al. 1994; Napoli 2000). FTY720 was subsequently found to be effective in a variety of autoimmune and transplant models (Brinkmann et al. 2001). It is now known that FTY720 acts as a prodrug becoming phosphorylated in vivo through the action of sphingosine kinase 1 and 2. The phosphorylated active metabolite of FTY720 is termed FTY720-P(S) which is a non-selective agonist for S1P1,3,4, and 5 receptors (Mandala et al. 2002; Brinkmann 2002) and can function both as a receptor agonist and pharmacological functional antagonist in vivo (Gräler and Goetzl 2004; Oo et al. 2007; Ishii et al. 2009). The phenomenon of functional antagonism occurs when receptors are internalized and targeted to the polyubiquitination pathway destined for degradation as opposed to recycling back to the cell surface. It is thought that functional antagonism is a necessary property for S1P1 receptors to be effective in their role as immunomodulators. Since its discovery and characterization, FTY720 demonstrated efficacy in humans against multiple sclerosis, a neurodegenerative autoimmune disorder characterized by inflammation and demyelination in the central nervous system (Cohen et al. 2010; Kappos et al. 2010). FTY720 has completed a rigorous clinical trial program and is now indicated to treat patients with relapsing multiple sclerosis.
We examine here the potential binding mode of FTY720-P in the context of the S1P1 receptor in its modeled agonist-induced conformation. Because FTY720-P is a sphingolipid mimic the polar head group interactions are well characterized and should be similar to the ML056 crystal structure (Hanson et al. 2012). Both ML056 and FTY720-P have phenyl rings proximal to their polar head group, however, the substitution pattern around the phenyl ring of ML056 is meta relative to a 6-carbon aryl chain, whereas it is para for FTY720-P relative to its 8-carbon aryl chain. It has been shown with the VPC series of compounds that this substitution pattern can steer the pharmacology for the sphingolipid-like compounds where a meta-substituted 8-carbon aryl chain (VPC23019) is a potent antagonist on the S1P1 and S1P3 receptors, whereas the equivalent para-substituted compound (VPC24191) is an agonist (Davis et al. 2005; Welch et al. 2012). The molecular determinants of this pharmacology switch are speculative, however, it is interesting that the para-substitution pattern positions a significant increase in volume adjacent to W269:6.48 (Fig. 8b), similar to S1P. In the case of FTY720-P, resolution of this ligand strain through simple molecular dynamics simulations was not possible due to the large conformational shifts and a more thorough modeling effort is out of the scope of this chapter.
Interestingly, it appears that as rigidifying elements are added to the agonist compounds, fitting into the nonoptimally shaped antagonist binding pocket while maintaining polar head group interactions, necessitates placing more strain on the rotatable bonds. The conformational changes associated with releasing that strain through shifting to the agonist binding pocket become increasingly favorable energetically.
3.2.3 RP-001
A number of non-sphingolipid mimic compounds have been discovered as agonists for the S1P1 receptor (Sanna et al. 2004; Li et al. 2005; Vachal et al. 2006; Yan et al. 2006, 2007; Gonzalez-Cabrera et al. 2008; Saha et al. 2010) and a handful of these have been developed for testing in a clinical setting exemplified by RPC1063 (Hartung et al. 2013; Urbano et al. 2013). RPC1063 builds on the findings of CYM-5442 in that interactions mimicking the phosphate anion salt bridges are not necessary for generating picomolar agonist compounds at the S1P1 receptor. During the course of development of RPC1063, a number of compounds were created that added this salt bridge interaction for proof of concept purposes. One such compound, designated RP-001 was tested in vivo in a S1P1-eGFP knock-in mouse for its effects on the expression of the S1P1 receptor on both lymphocyte and endothelial populations of receptors (Cahalan et al. 2011).
The number of rotatable bonds for RP-001 is reduced compared to the FTY720-P and S1P which increases the rigidity and places an extra burden on the binding pocket geometry, such that the compound is not able to dock effectively in the ML056 binding pocket. This necessitates an agonist associated conformational change being modeled prior to docking and analysis of this compound. We use the S1P-induced binding pocket modeled above through molecular dynamics simulations, which allows placement of the compound with good polar and hydrophobic contacts that align well with mutagenesis for this series (Hanson et al. 2012). The carboxylic acid of RP-001 forms ionic and polar interactions with Y29 and R120:3.28, while the secondary amine which is likely protonated at physiological pH participates in hydrogen bonding interactions with N101:2.60 and charge pairs with E121:3.29. The central ring oxadiazole is positioned to interact indirectly (perhaps through water bridged hydrogen bonds) with E121:3.29 and Y98:2.57. The third cyano-isopropoxy benzene ring fits tightly between F125:3.33 and W269:6.48 with the cyano group appearing to pin W269 in the S1P-induced conformation (Fig. 8c).
4 Conclusions
The elucidation of the S1P1 receptor structure provided a framework for understanding the molecular interactions of not only the crystallized antagonist ligand ML056, but also a variety of antagonist and agonist sphingolipid and non-sphingolipid-like compounds. Compound development efforts and mutagenesis studies suggest the trigger for agonism on the S1P1 receptor is associated with an increase in binding pocket volume. This combined with the expectation that the polar interactions with the sphingosine zwitterion will be consistent across pharmacologies has allowed us to model an agonist binding pocket without the benefit of an agonist structure. This agonist binding pocket is capable of docking a variety of compounds including the Receptos 1063 series which, due to its lack of requirement for sphingosine-like interactions, was optimized to produce superior selectivity and pharmaceutics properties compared to compounds such as FTY720-P. We continue to use this structure and the models derived from it to advance our understanding of the molecular interactions employed by the S1P1 receptor agonists and potentially to design further improved compounds. Recently, it has been established that the S1P1 receptor agonists act, at least partially, through a functional antagonist mechanism. This finding introduces the possibility of designing agonist compounds that preferentially induce internalization of the receptor over other signaling pathways, a phenomenon known as biased ligand signaling (Xu et al. 2013; Healy et al. 2013). This concept could be an important mechanism for further improvements to the safety profile of immunomodulatory S1P1 receptor agonists, and indeed, many other classes of small molecule therapeutics designed to modulate G protein-coupled receptor pharmacology. Discovering the structural basis for biased ligand signaling could signify the next breakthrough in our understanding of GPCR pharmacology.
References
Arnon TI, Xu Y, Lo C et al (2011) GRK2-dependent S1PR1 desensitization is required for lymphocytes to overcome their attraction to blood. Science 333:1898–1903. doi:10.1126/science.1208248
Berst F, Grosche P, Janser P, Zecri F (2007) Biaryl sulfonamide derivatives. US Patent Office US 20100029609
Brinkmann V (2002) The immune modulator FTY720 targets sphingosine 1-phosphate receptors. J Biol Chem 277:21453–21457. doi:10.1074/jbc.C200176200
Brinkmann V, Pinschewer DD, Feng L, Chen S (2001) FTY720: altered lymphocyte traffic results in allograft protection. Transplantation 72:764–769
Caffrey M (2000) A lipid’s eye view of membrane protein crystallization in mesophases. Curr Opin Struct Biol 10:486–497
Cahalan SM, Gonzalez-Cabrera PJ, Nguyen N et al (2013) Sphingosine 1-phosphate receptor 1 (S1P1) upregulation and amelioration of experimental autoimmune encephalomyelitis by an S1P1 antagonist. Mol Pharmacol 83:316–321. doi:10.1124/mol.112.082958
Cahalan SM, Gonzalez-Cabrera PJ, Sarkisyan G et al (2011) Actions of a picomolar short-acting S1P1 agonist in S1P1-eGFP knock-in mice. Nat Chem Biol 7:254–256. doi:10.1038/nchembio.547
Cherezov V (2011) Lipidic cubic phase technologies for membrane protein structural studies. Curr Opin Struct Biol 21:559–566. doi:10.1016/j.sbi.2011.06.007
Cherezov V, Rosenbaum DM, Hanson MA et al (2007) High-resolution crystal structure of an engineered human beta2-adrenergic G protein-coupled receptor. Science 318:1258–1265. doi:10.1126/science.1150577
Chien EYT, Liu W, Zhao Q et al (2010) Structure of the human dopamine D3 receptor in complex with a D2/D3 selective antagonist. Science 330:1091–1095. doi:10.1126/science.1197410
Choi JW, Herr DR, Noguchi K et al (2010) LPA receptors: subtypes and biological actions. Annu Rev Pharmacol Toxicol 50:157–186. doi:10.1146/annurev.pharmtox.010909.105753
Chun J, Goetzl EJ, Hla T et al (2002) International union of pharmacology. XXXIV. Lysophospholipid receptor nomenclature. Pharmacol Rev 54:265–269
Cohen JA, Barkhof F, Comi G et al (2010) Oral fingolimod or intramuscular interferon for relapsing multiple sclerosis. N Engl J Med 362:402–415. doi:10.1056/NEJMoa0907839
Coste O, Brenneis C, Linke B et al (2008) Sphingosine 1-phosphate modulates spinal nociceptive processing. J Biol Chem 283:32442–32451. doi:10.1074/jbc.M806410200
Cuvillier O, Pirianov G, Kleuser B et al (1996) Suppression of ceramide-mediated programmed cell death by sphingosine-1-phosphate. Nature 381:800–803. doi:10.1038/381800a0
Davis MD, Clemens JJ, Macdonald TL, Lynch KR (2005) Sphingosine 1-phosphate analogs as receptor antagonists. J Biol Chem 280:9833–9841. doi:10.1074/jbc.M412356200
Deng Q, Clemas JA, Chrebet G et al (2007) Identification of Leu276 of the S1P1 receptor and Phe263 of the S1P3 receptor in interaction with receptor specific agonists by molecular modeling, site-directed mutagenesis, and affinity studies. Mol Pharmacol 71:724–735. doi:10.1124/mol.106.029223
Fernandez O, Pozzilli C, Freedman M, et al. (2012) Pharmacodynamic effect, safety and tolerability of ponesimod, a selective sphingosine 1-phosphate receptor-1 modulator, in patients with relapsing-remitting multiple sclerosis. ECTRIMS
Filipek S, Stenkamp RE, Teller DC, Palczewski K (2003) G protein-coupled receptor rhodopsin: a prospectus. Annu Rev Physiol 65:851–879. doi:10.1146/annurev.physiol.65.092101.142611
Fredriksson R, Lagerström MC, Lundin L-G, Schiöth HB (2003) The G-protein-coupled receptors in the human genome form five main families. Phylogenetic analysis, paralogon groups, and fingerprints. Mol Pharmacol 63:1256–1272. doi:10.1124/mol.63.6.1256
Fujii Y, Hirayama T, Ohtake H et al (2012a) Amelioration of collagen-induced arthritis by a novel S1P1 antagonist with immunomodulatory activities. J Immunol 188:206–215. doi:10.4049/jimmunol.1101537
Fujii Y, Ohtake H, Ono N et al (2012b) Lymphopenia induced by a novel selective S1P1 antagonist structurally unrelated to S1P. Biochim Biophys Acta Mol Cell Biol Lipids 1821:600–606. doi:10.1016/j.bbalip.2011.12.006
Fujii Y, Ueda Y, Ohtake H et al (2012c) Blocking S1P interaction with S1P1 receptor by a novel competitive S1P1-selective antagonist inhibits angiogenesis. Biochem Biophys Res Commun 419:754–760. doi:10.1016/j.bbrc.2012.02.096
Fujita T, Inoue K, Yamamoto S et al (1994) Fungal metabolites. Part 11. A potent immunosuppressive activity found in Isaria sinclairii metabolite. J Antibiot 47:208–215
Fyrst H, Saba JD (2010) An update on sphingosine-1-phosphate and other sphingolipid mediators. Nat Chem Biol 6:489–497. doi:10.1038/nchembio.392
Gergely P, Wallstrom E, Nuesslein-Hildesheim B, et al. (2009) Phase I study with the selective S1P1/S1P5 receptor modulator BAF312 indicates that S1P1 rather than S1P3 mediates transient heart rate reduction in humans. ECTRIMS
Gonzalez-Cabrera PJ, Jo E, Sanna MG et al (2008) Full pharmacological efficacy of a novel S1P1 agonist that does not require S1P-like headgroup interactions. Mol Pharmacol 74:1308–1318. doi:10.1124/mol.108.049783
Granier S, Manglik A, Kruse AC et al (2012) Structure of the δ-opioid receptor bound to naltrindole. Nature 485:400–404
Gräler MH, Goetzl EJ (2004) The immunosuppressant FTY720 down-regulates sphingosine 1-phosphate G-protein-coupled receptors. FASEB J 18:551–553. doi:10.1096/fj.03-0910fje
Haga K, Kruse AC, Asada H et al (2012) Structure of the human M2 muscarinic acetylcholine receptor bound to an antagonist. Nature 482:547–551. doi:10.1038/nature10753
Hanson MA, Roth CB, Jo E et al (2012) Crystal structure of a lipid G protein-coupled receptor. Science 335:851–855. doi:10.1126/science.1215904
Hanson MA, Stevens RC (2009) Discovery of new GPCR biology: one receptor structure at a time. Structure 17:8–14. doi:10.1016/j.str.2008.12.003
Hartung J, Olson A, Peach RJ, et al. (2013) Results of a thorough QT/QTc (TQT) study of orally administered RPC1063, a novel, selective S1P1 receptor agonist, in healthy adult volunteers. ECTRIMS
Healy LM, Sheridan GK, Pritchard AJ et al (2013) Pathway specific modulation of S1P1 receptor signalling in rat and human astrocytes. Br J Pharmacol 169:1114–1129. doi:10.1111/bph.12207
Hla T, Maciag T (1990) An abundant transcript induced in differentiating human endothelial cells encodes a polypeptide with structural similarities to G-protein-coupled receptors. J Biol Chem 265:9308–9313
Hobson JP, Rosenfeldt HM, Barak LS et al (2001) Role of the sphingosine-1-phosphate receptor EDG-1 in PDGF-induced cell motility. Science 291:1800–1803. doi:10.1126/science.1057559
Holdsworth G, Osborne DA, Pham TT et al (2004) A single amino acid determines preference between phospholipids and reveals length restriction for activation of the S1P4 receptor. BMC Biochem 5:12. doi:10.1186/1471-2091-5-12
Hollenstein K, Kean J, Bortolato A et al (2013) Structure of class B GPCR corticotropin-releasing factor receptor 1. Nature 499:438–443. doi:10.1038/nature12357
Hurst DP, Grossfield A, Lynch DL et al (2010) A lipid pathway for ligand binding is necessary for a cannabinoid G protein-coupled receptor. J Biol Chem 285:17954–17964. doi:10.1074/jbc.M109.041590
Hurst DP, Schmeisser M, Reggio PH (2013) Endogenous lipid activated G protein-coupled receptors: emerging structural features from crystallography and molecular dynamics simulations. Chem Phys Lipids 169:46–56. doi:10.1016/j.chemphyslip.2013.01.009
Huwiler A, Kolter T, Pfeilschifter J, Sandhoff K (2000) Physiology and pathophysiology of sphingolipid metabolism and signaling. Biochim Biophys Acta 1485:63–99
Huwiler A, Pfeilschifter J (2008) New players on the center stage: sphingosine 1-phosphate and its receptors as drug targets. Biochem Pharmacol 75:1893–1900. doi:10.1016/j.bcp.2007.12.018
Ibrahim MA, Johnson HWB, Jeong JW et al (2012) Discovery of a novel class of potent and orally bioavailable sphingosine 1-phosphate receptor 1 antagonists. J Med Chem 55:1368–1381. doi:10.1021/jm201533b
Inagaki Y, Pham TT, Fujiwara Y et al (2005) Sphingosine 1-phosphate analogue recognition and selectivity at S1P4 within the endothelial differentiation gene family of receptors. Biochem J 389:187–195. doi:10.1042/BJ20050046
Ishii M, Egen JG, Klauschen F et al (2009) Sphingosine-1-phosphate mobilizes osteoclast precursors and regulates bone homeostasis. Nature 458:524–528. doi:10.1038/nature07713
Jaakola V-P, Griffith MT, Hanson MA et al (2008) The 2.6 angstrom crystal structure of a human A2A adenosine receptor bound to an antagonist. Science 322:1211–1217. doi:10.1126/science.1164772
Jo E, Sanna MG, Gonzalez-Cabrera PJ et al (2005) S1P1-selective in vivo-active agonists from high-throughput screening: off-the-shelf chemical probes of receptor interactions, signaling, and fate. Chem Biol 12:703–715. doi:10.1016/j.chembiol.2005.04.019
Kappos L, Radue E-W, O’Connor P et al (2010) A placebo-controlled trial of oral fingolimod in relapsing multiple sclerosis. N Engl J Med 362:387–401. doi:10.1056/NEJMoa0909494
Katritch V, Cherezov V, Stevens RC (2012) Diversity and modularity of G protein-coupled receptor structures. Trends Pharmacol Sci 33:17–27. doi:10.1016/j.tips.2011.09.003
Kenakin T (2012) The potential for selective pharmacological therapies through biased receptor signaling. BMC Pharmacol Toxicol 13:3. doi:10.1186/2050-6511-13-3
Kenakin T, Onaran O (2002) The ligand paradox between affinity and efficacy: can you be there and not make a difference? Trends Pharmacol Sci 23:275–280
Kruse AC, Hu J, Pan AC et al (2012) Structure and dynamics of the M3 muscarinic acetylcholine receptor. Nature 482:552–556. doi:10.1038/nature10867
Kułakowska A, Żendzian-Piotrowska M, Baranowski M et al (2010) Intrathecal increase of sphingosine 1-phosphate at early stage multiple sclerosis. Neurosci Lett 477:149–152. doi:10.1016/j.neulet.2010.04.052
LaMontagne K, Littlewood-Evans A, Schnell C et al (2006) Antagonism of sphingosine-1-phosphate receptors by FTY720 inhibits angiogenesis and tumor vascularization. Cancer Res 66:221–231. doi:10.1158/0008-5472.CAN-05-2001
Landau EM, Rosenbusch JP (1996) Lipidic cubic phases: a novel concept for the crystallization of membrane proteins. Proc Natl Acad Sci 93:14532–14535
Li Z, Chen W, Hale JJ et al (2005) Discovery of potent 3,5-diphenyl-1,2,4-oxadiazole sphingosine-1-phosphate-1 (S1P1) receptor agonists with exceptional selectivity against S1P2 and S1P3. J Med Chem 48:6169–6173. doi:10.1021/jm0503244
Lim H-S, Park J-J, Ko K et al (2004) Syntheses of sphingosine-1-phosphate analogues and their interaction with EDG/S1P receptors. Bioorg Med Chem Lett 14:2499–2503. doi:10.1016/j.bmcl.2004.03.001
Mandala S, Hajdu R, Bergstrom J et al (2002) Alteration of lymphocyte trafficking by sphingosine-1-phosphate receptor agonists. Science 296:346–349. doi:10.1126/science.1070238
Manglik A, Kruse AC, Kobilka TS et al (2012) Crystal structure of the μ-opioid receptor bound to a morphinan antagonist. Nature 485:321–326. doi:10.1038/nature10954
Napoli KL (2000) The FTY720 story. Ther Drug Monit 22:47–51
O’Sullivan C, Dev KK (2013) The structure and function of the S1P1 receptor. Trends Pharmacol Sci 34:401–412. doi:10.1016/j.tips.2013.05.002
Ogretmen B, Hannun YA (2004) Biologically active sphingolipids in cancer pathogenesis and treatment. Nat Rev Cancer 4:604–616. doi:10.1038/nrc1411
Oo ML, Chang S-H, Thangada S et al (2011) Engagement of S1P1-degradative mechanisms leads to vascular leak in mice. J Clin Invest 121:2290–2300. doi:10.1172/JCI45403
Oo ML, Thangada S, Wu M-T et al (2007) Immunosuppressive and anti-angiogenic sphingosine 1-phosphate receptor-1 agonists induce ubiquitinylation and proteasomal degradation of the receptor. J Biol Chem 282:9082–9089. doi:10.1074/jbc.M610318200
Paik JH, Ss Chae, Lee MJ et al (2001) Sphingosine 1-phosphate-induced endothelial cell migration requires the expression of EDG-1 and EDG-3 receptors and Rho-dependent activation of alpha vbeta3- and beta1-containing integrins. J Biol Chem 276:11830–11837. doi:10.1074/jbc.M009422200
Palczewski K, Kumasaka T, Hori T et al (2000) Crystal structure of rhodopsin: a G protein-coupled receptor. Science 289:739–745
Parrill AL, Baker DL, Wang DA, et al. (2000a) Structural features of EDG1 receptor‐ligand complexes revealed by computational modeling and mutagenesis. Ann N Y Acad Sci 905:330–339. doi: 10.1111/j.1749-6632.2000.tb06573.x
Parrill AL, Tigyi G (2013) Integrating the puzzle pieces: the current atomistic picture of phospholipid-G protein coupled receptor interactions. Biochim Biophys Acta 1831:2–12. doi:10.1016/j.bbalip.2012.09.002
Parrill AL, Wang D, Bautista DL et al (2000b) Identification of Edg1 receptor residues that recognize sphingosine 1-phosphate. J Biol Chem 275:39379–39384. doi:10.1074/jbc.M007680200
Pham T-CT, Fells JI, Osborne DA et al (2008a) Molecular recognition in the sphingosine 1-phosphate receptor family. J Mol Graphics Modell 26:1189–1201. doi:10.1016/j.jmgm.2007.11.001
Pham THM, Okada T, Matloubian M et al (2008b) S1P1 receptor signaling overrides retention mediated by G alpha i-coupled receptors to promote T cell egress. Immunity 28:122–133. doi:10.1016/j.immuni.2007.11.017
Pyne NJ, Pyne S (2010) Sphingosine 1-phosphate and cancer. Nat Rev Cancer 10:489–503. doi:10.1038/nrc2875
Quancard J, Bollbuck B, Janser P et al (2012) A potent and selective S1P(1) antagonist with efficacy in experimental autoimmune encephalomyelitis. Chem Biol 19:1142–1151. doi:10.1016/j.chembiol.2012.07.016
Rasmussen SGF, Choi H-J, Fung JJ et al (2011a) Structure of a nanobody-stabilized active state of the β(2) adrenoceptor. Nature 469:175–180. doi:10.1038/nature09648
Rasmussen SGF, DeVree BT, Zou Y et al (2011b) Crystal structure of the β2 adrenergic receptor-Gs protein complex. Nature 477:549–555. doi:10.1038/nature10361
Rosen H, Gonzalez-Cabrera PJ, Sanna MG, Brown S (2009) Sphingosine 1-phosphate receptor signaling. Annu Rev Biochem 78:743–768. doi:10.1146/annurev.biochem.78.072407.103733
Rosen H, Stevens RC, Hanson M et al (2013) Sphingosine-1-phosphate and its receptors: structure, signaling, and influence. Annu Rev Biochem. doi:10.1146/annurev-biochem-062411-130916
Rosenbaum DM, Cherezov V, Hanson MA et al (2007) GPCR engineering yields high-resolution structural insights into beta2-adrenergic receptor function. Science 318:1266–1273. doi:10.1126/science.1150609
Saha AK, Yu X, Lin J et al (2010) Benzofuran derivatives as potent, orally active S1P1 receptor agonists: a preclinical lead molecule for MS. ACS Med Chem Lett 2:97–101. doi:10.1021/ml100227q
Sanna MG, Liao J, Jo E et al (2004) Sphingosine 1-phosphate (S1P) receptor subtypes S1P1 and S1P3, respectively, regulate lymphocyte recirculation and heart rate. J Biol Chem 279:13839–13848. doi:10.1074/jbc.M311743200
Sanna MG, Wang S-K, Gonzalez-Cabrera PJ et al (2006) Enhancement of capillary leakage and restoration of lymphocyte egress by a chiral S1P1 antagonist in vivo. Nat Chem Biol 2:434–441. doi:10.1038/nchembio804
Schadel SA, Heck M, Maretzki D et al (2003) Ligand channeling within a G-protein-coupled receptor. The entry and exit of retinals in native opsin. J Biol Chem 278:24896–24903. doi:10.1074/jbc.M302115200
Schmouder R, Hariry S, David OJ (2012) Placebo-controlled study of the effects of fingolimod on cardiac rate and rhythm and pulmonary function in healthy volunteers. Eur J Clin Pharmacol 68:355–362. doi:10.1007/s00228-011-1146-9
Schürer SC, Brown SJ, Gonzalez-Cabrera PJ et al (2008) Ligand-binding pocket shape differences between sphingosine 1-phosphate (S1P) receptors S1P1 and S1P3 determine efficiency of chemical probe identification by ultrahigh-throughput screening. ACS Chem Biol 3:486–498. doi:10.1021/cb800051m
Sherman W, Day T, Jacobson MP et al (2006) Novel procedure for modeling ligand/receptor induced fit effects. J Med Chem 49:534–553. doi:10.1021/jm050540c
Shivakumar D, Williams J, Wu Y et al (2010) Prediction of absolute solvation free energies using molecular dynamics free energy perturbation and the OPLS force field. J Chem Theory Comput 6:1509–1519. doi:10.1021/ct900587b
Siu FY, He M, de Graaf C et al (2013) Structure of the human glucagon class B G-protein-coupled receptor. Nature 499:444–449. doi:10.1038/nature12393
Spiegel S, Milstien S (2003) Sphingosine-1-phosphate: an enigmatic signalling lipid. Nat Rev Mol Cell Biol 4:397–407. doi:10.1038/nrm1103
Takasugi N, Sasaki T, Suzuki K et al (2011) BACE1 activity is modulated by cell-associated sphingosine-1-phosphate. J Neurosci 31:6850–6857. doi:10.1523/JNEUROSCI.6467-10.2011
Tan Q, Zhu Y, Li J et al (2013) Structure of the CCR5 chemokine receptor-HIV entry inhibitor maraviroc complex. Science 341:1387–1390. doi:10.1126/science.1241475
Thompson AA, Liu W, Chun E et al (2012) Structure of the nociceptin/orphanin FQ receptor in complex with a peptide mimetic. Nature 485:395–399. doi:10.1038/nature11085
Urbano M, Guerrero M, Rosen H, Roberts E (2013) Modulators of the sphingosine 1-phosphate receptor 1. Bioorg Med Chem Lett. doi:10.1016/j.bmcl.2013.09.058
Vachal P, Toth LM, Hale JJ et al (2006) Highly selective and potent agonists of sphingosine-1-phosphate 1 (S1P1) receptor. Bioorg Med Chem Lett 16:3684–3687. doi:10.1016/j.bmcl.2006.04.064
Valentine WJ, Kiss GN, Liu J et al (2010) (S)- FTY720-Vinylphosphonate, an analogue of the immunosuppressive agent FTY720, is a pan-antagonist of sphingosine 1-phosphate GPCR signaling and inhibits autotaxin activity. Cell Signal 22:1543–1553. doi:10.1016/j.cellsig.2010.05.023
Van Durme J, Horn F, Costagliola S et al (2006) GRIS: glycoprotein-hormone receptor information system. Mol Endocrinol 20:2247–2255. doi:10.1210/me.2006-0020
van Loenen PB, van Loenen PB, de Graaf C et al (2011) Agonist-dependent effects of mutations in the sphingosine-1-phosphate type 1 receptor. Eur J Pharmacol 667:105–112. doi:10.1016/j.ejphar.2011.05.071
Venkatakrishnan AJ, Deupi X, Lebon G et al (2013) Molecular signatures of G-protein-coupled receptors. Nature 494:185–194. doi:10.1038/nature11896
Visentin B, Vekich JA, Sibbald BJ et al (2006) Validation of an anti-sphingosine-1-phosphate antibody as a potential therapeutic in reducing growth, invasion, and angiogenesis in multiple tumor lineages. Cancer Cell 9:225–238. doi:10.1016/j.ccr.2006.02.023
Vroling B, Sanders M, Baakman C et al (2011) GPCRDB: information system for G protein-coupled receptors. Nucleic Acids Res 39:D309–D319. doi:10.1093/nar/gkq1009
Wacker D, Wang C, Katritch V et al (2013) Structural features for functional selectivity at serotonin receptors. Science 340:615–619. doi:10.1126/science.1232808
Wang C, Jiang Y, Ma J et al (2013a) Structural basis for molecular recognition at serotonin receptors. Science 340:610–614. doi:10.1126/science.1232807
Wang C, Wu H, Katritch V et al (2013b) Structure of the human smoothened receptor bound to an antitumour agent. Nature 497:338–343. doi:10.1038/nature12167
Wang DA, Lorincz Z, Bautista DL et al (2001) A single amino acid determines lysophospholipid specificity of the S1P1 (EDG1) and LPA1 (EDG2) phospholipid growth factor receptors. J Biol Chem 276:49213–49220. doi:10.1074/jbc.M107301200
Warne T, Serrano-Vega MJ, Baker JG et al (2008) Structure of a beta1-adrenergic G-protein-coupled receptor. Nature 454:486–491. doi:10.1038/nature07101
Welch SP, Sim-Selley LJ, Selley DE (2012) Sphingosine-1-phosphate receptors as emerging targets for treatment of pain. Biochem Pharmacol 84:1551–1562. doi:10.1016/j.bcp.2012.08.010
Whetzel AM, Bolick DT, Srinivasan S et al (2006) Sphingosine-1 phosphate prevents monocyte/endothelial interactions in type 1 diabetic NOD mice through activation of the S1P1 receptor. Circ Res 99:731–739. doi:10.1161/01.RES.0000244088.33375.52
White JF, Noinaj N, Shibata Y et al (2012) Structure of the agonist-bound neurotensin receptor. Nature 490:508–513. doi:10.1038/nature11558
Wu B, Chien EYT, Mol CD et al (2010) Structures of the CXCR4 chemokine GPCR with small-molecule and cyclic peptide antagonists. Science 330:1066–1071. doi:10.1126/science.1194396
Wu H, Wacker D, Mileni M et al (2012) Structure of the human κ-opioid receptor in complex with JDTic. Nature 485:327–332. doi:10.1038/nature10939
Xia P, Gamble JR, Wang L et al (2000) An oncogenic role of sphingosine kinase. Curr Biol 10:1527–1530. doi:10.1016/S0960-9822(00)00834-4
Xu H, McElvain M, Fiorino M et al (2013) Predictability of peripheral lymphocyte reduction of novel S1P1 agonists by in vitro GPCR signaling profile. J Biomol Screen. doi:10.1177/1087057113488629
Yan L, Huo P, Doherty G et al (2006) Discovery of 3-arylpropionic acids as potent agonists of sphingosine-1-phosphate receptor-1 (S1P1) with high selectivity against all other known S1P receptor subtypes. Bioorg Med Chem Lett 16:3679–3683. doi:10.1016/j.bmcl.2006.04.084
Yan L, Huo P, Hale JJ et al (2007) SAR studies of 3-arylpropionic acids as potent and selective agonists of sphingosine-1-phosphate receptor-1 (S1P1) with enhanced pharmacokinetic properties. Bioorg Med Chem Lett 17:828–831. doi:10.1016/j.bmcl.2006.10.057
Zhang C, Srinivasan Y, Arlow DH et al (2012) High-resolution crystal structure of human protease-activated receptor 1. Nature 492:387–392. doi:10.1038/nature11701
Zhang H, Desai NN, Olivera A et al (1991) Sphingosine-1-phosphate, a novel lipid, involved in cellular proliferation. J Cell Biol 114:155–167
Zipp F, Vollmer TL, Selmaj K, Bar-Or A (2012) Efficacy and safety of the S1P receptor agonist ONO-4641 in patients with relapsing-remitting multiple sclerosis: results of a 26-week, double-blind, placebo-controlled, Phase II trial (DreaMS). ECTRIMS
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Hanson, M.A., Peach, R. (2014). Structural Biology of the S1P1 Receptor. In: Oldstone, M., Rosen, H. (eds) Sphingosine-1-Phosphate Signaling in Immunology and Infectious Diseases. Current Topics in Microbiology and Immunology, vol 378. Springer, Cham. https://doi.org/10.1007/978-3-319-05879-5_2
Download citation
DOI: https://doi.org/10.1007/978-3-319-05879-5_2
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-05878-8
Online ISBN: 978-3-319-05879-5
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)