Indigo and Tyrian purple are two dyes that have been used historically worldwide, with their colors due mainly to the presence of indigoids. Indigoids are a fascinating class of natural products that are found in several plants and in marine organisms. This small family of natural products is composed of 13 members, with indigo being the most well-known and the main component of Indigo dyes. Their chemical structures rely on the connectivity of two indole moieties. Although there is a limited number of indigoids, they represent one of the oldest natural compound classes used by mankind and their chemical development and importance are intimately linked with the development of human society and the rise of the industrial revolution in the nineteenth century.
Their eye-opening hues, ranging from blue to deep purple, served as an early stimulus for the worldwide scientific community to identify the chemical basis of the color of the indigoids. Moreover, the rapid growth of the textile industry and the necessity of producing chemical dyes in large-scale quantities opened the way to several chemical access routes and promoted the high added-value of indigo. Interestingly, indirubin, the second major metabolite of Indigo dye, has later been discovered to be the active component of Danggui Longhui Wan, a formula used in Traditional Chinese Medicine for four millennia to treat the symptoms of leukemia, which acts by potently inhibiting cyclin-dependent kinases, a key family of proteins that regulate the cell cycle. This major discovery has stimulated the use of both indirubin and its marine relative, 6-bromoindirubin, as major scaffolds in medicinal chemistry for the study of biological processes and the development of active drug candidates. The aim of this contribution is to describe the fascinating story of indigoids and their evolution as important compounds in both textile science and medicinal chemistry.
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Introduction
Indigoids are a fascinating class of natural products in which their chemical structures rely on the connection of two indole moieties. The most well-known member is probably indigotin, the main component of indigo dye used all over the world. However indigoids have found over the centuries several applications outside of dye chemistry and are now lead compounds in medicinal chemistry. The development of the indigoids has therefore led to a particular chemistry, characteristic of the several chemical fields in which they were applied.
1.1 Natural Dyes
Early in his history, mankind began manufacturing pigments for practical, esthetic, cosmetic or spiritual applications. The discovery of the oldest ochre-producing workshop dating from 100,000 years ago (Blomos Cave, South Africa (1)) illustrates this old know-how. However, the number of such dyes was limited by the naturally occurring possibilities and the first pigments were principally red, yellow blue or black, with the other colors being created by the mixing of these “primary colors”. The chemical diversity of natural dyes varies from flavonoids (yellow dye), anthraquinones (red dye) through tannins (brown and black dyes) (2) to indigoids (blue and purple dyes). Their traditional sources could be divided in three categories: mineral, animal and vegetal. Mineral pigments are generally pure metals such as zinc (white), arsenic (green), or copper (green, blue, purple) at different oxidation states. They were used mainly for pictorial applications, and were of low interest for dyeing because of their instability or else because they were found to be toxic. Animal natural sources were also exploited such as insects as Kermes vermilio (cochineal). The Maya and Aztec civilizations from Central America produced the so-called carmine pigment, a red dye constituted of insoluble aluminum and calcium salts of carminic acid (1) (3). In the Mediterranean basin, the Murex shellfish family during antiquity provided the so-called “Tyrian purple”, as worn by emperors. The deep purple color was given by indigotin and indirubin derivatives belonging to the indigoid family, which are the subject of this contribution.
1.2 Dyes from Plants
Above all, the vegetable kingdom represents the main source of sustainable natural pigments and knowledge of their extraction has been available for millennia. Nowadays, new natural pigments have emerged, although few have had a traditional use throughout human history. The roots of Rubia tinctoria Salisb. (Rubiaceae) or madder were used by the Egyptians to produce the so-called “alizarin red” (4). This red pigment has been found in the Tutankhamun tomb and owes its color to the presence of the anthraquinone alizarin (2) (Fig. 1). Reseda luteola L. or weld (Brassicaceae) was used in France (thirteenth–seventeenth century) to dye yellow hats as a distinctive Jewish symbol. The yellow color was given by the flavonoid luteolin (3) (5) (Fig. 1) first isolated by the French chemist M. E. Chevreul. The so-called “black-catechu” black dye was obtained in India from the tree bark and heartwood of Acacia catechu Willd. (Leguminosae). This complex mixture contains especially tannins, and catechin (4) (Fig. 1) is the most abundant representative compound. The stigmata of the flowers of Crocus sativus L. have been used for a long time notably in Persia. By crushing them, an orange-red pigment was extracted, for which the main chromophore is the carotenoid crocin (5) (Fig. 1). The crushed flowers were then applied directly on the cloth to afford an orange colored product. Surprisingly and although it is widely represented in Nature, a green color has not been successfully extracted from natural sources. The abundant chlorophyll (6) (Fig. 1) is not sufficiently heat-stable to be correctly and permanently bound to textile fibers. Moreover, it loses its coordinating magnesium under basic conditions (washing) to form pheophytin (7) (Fig. 1, olive-green color). The clothes were thus dyed in green using the toxic cupric oxide (mineral), explaining the few green-dye substances found.
The most widely used color in human history undoubtedly has been blue, including indigo extracted from indigo-bearing plants such as Isatis (Europe, Asia), Indigofera (India), or Polygonum (Japan, Asia) species. Around 300 species of plants in these genera exist, but only a few were exploited for dyeing, as described later in this contribution. Traces of indigo production have been found from England to South America and through to India. Indigotin (8) (Fig. 1), the main indigo constituent, belongs to the family of indigoids mentioned above and will be discussed further below. It is the only natural dye for which an industrial synthesis has been successfully realized, leading to its great success, especially in the blue jeans industry.
Until recently, the use of natural dyes was very limited due to the development of methods worked out for their chemical synthesis. However, new interest in natural products combined with the overall environmental awareness that has developed in the last ten years has led to their “rediscovery”. Chlorophyll (7) is thus a useful tool known as an “eco-friendly” chemical to create more stable dyes (6). Indigo production took advantage of this “new” interest and local industries now produce it in an traditional way, as in the south of France (7). Nevertheless, its traditional production never stopped in India or in South America. Originally, artists such as Inge Boesken Kanold (8) found in Tyrian purple a new application as a high-class pigment in paintings. Indigo and Tyrian purple have symbolized the reputation of powerful people, made the fortunes of many merchants like the Phoenicians, and were a great source of benefits for European colonial empires. Their history has been deeply connected with the evolution of our society and they are still influential in our development. Moreover, the discovery of indirubin (9) (Fig. 1) as the main therapeutic compound of a traditional Chinese medicine (TCM) recipe has propelled this class of natural products from a dye to the field of medicinal chemistry.
2 Indigoid Family
2.1 Family Presentation
Indigoids are a natural class of organic compounds. They are bis-indole alkaloids connected by a double bond (of (Z) or (E) configuration) and can be substituted in their aromatic parts. “Indigo-blue” or indigotin (8) (Fig. 2) was the first representative of this family of natural products as the main chromophore of indigo dye. It is the symmetric dimer of two indole moieties connected by an (E)-double bond and exhibiting two internal hydrogen bonds. The (Z)-isomer (10) (Fig. 2) of indigotin (8) is also blue, but is found as a minor product during dye production (see Sect. 3.1.2, Chemistry). Isoindigo (11) (Fig. 2), the positional isomer of indigotin (8), is generally called “brown-indigo” due to its color in solution. Indirubin (9) (Fig. 2) is the second most important member of the indigoid group. It is an asymmetric compound and represents a combination of indigotin (8) and isoindigo (11) units. Isoindirubin (12) (Fig. 2), the (E)-isomer of indirubin, has also been identified in some indigo plants. The halogenated 6-bromo-, 6′-bromo- and 6,6′-bromoindirubins (13–15) (Fig. 2) were discovered in the Muricidae shellfish family (see Sect. 4.2.1) along with indigotin (8), indirubin (9), 6-bromoindigotin (16) and 6,6′-dibromoindigotin (17) (9). Recently, four new derivatives were identified in Hexaplex trunculus extracts using LC-MS/MS analysis: an imine group replaces the carbonyl at the 3′-position along with the presence of one or two bromine atoms (10). However, their unambiguous structures have not yet been elucidated. Two other brominated indigoids (18 and 19) (Fig. 2) have also been identified as constituents of the acorn worm, Ptychodera flava laysanica (11). Finally, the family of akashins A-C (20–22) (Fig. 2), the only example of naturally chlorinated indigoids, has been identified in a strain of the terrestrial Streptomyces sp. 48/1497 (12).
All members of the indigoid family are colored compounds. According to the type of isomer ((Z) or (E)), the symmetry and the degree of substitution, the resultant color ranges from blue (indigo) to red (indirubin) or purple for the marine brominated indigoids. Small structural modifications thus lead to a large range of colors all along the visible spectrum. Therefore, a simple question may be asked: what is the origin of the color?
2.2 Particular Electronic Effects: Origin of the Color
The origin of the color of indigotin (8) is a question that has stimulated scientific debate for more than a century, and a number of studies have been published on this subject. Summarized here are some of the most explicit ones.
The color of a molecule in solution or in solid phase is due to its ability to absorb light energy characterized by the maximum absorption wavelength (λmax) on raising the ground energy state to an excited state. The Plank relation (Eq. (1)) links the energy difference (ΔE) and the wavelength. As a consequence, ΔE decreases as the λmax increases.
$$ \varDelta E= hc/{\lambda}_{\max } $$
(1)
In the solid-phase state, indigotin (8) possesses a dark-blue color and values of λmax for amorphous and crystalline indigotin (8) have been measured at 640 and 680 nm, respectively. Weintsein and Wyman (13) have interpreted this large bathochromic effect with the formation of inter-molecular hydrogen bonds to form indigotin dimers (23) (or polymers), hence explaining the blue-dark aspect of solid indigotin (8). Primary rationalization studies led later by Lüttke and his collaborators (14) established the chromophoric unit of indigo as the so-called “H-shaped molecule” (24) (Fig. 3). This unit is localized on the hydrogen-bond area between the two indole nitrogens and the two oxygen atoms, confirming the hypothesis of dimerization through this region of the molecule.
Nonetheless, it has been shown that indigotin (8) maintains a strong “merocyanine” effect in solution. This is indicated by a strong bathochromic effect when the dielectric constant of the solvent is increased. In this case, the bathochromic effect is due to formation of hydrogen bonds with the solvent (15). Considering a polar solvent (MeOH/H2O/trifluoroacetic acid), the λmax values of indigotin (8), indirubin (9), and isoindigo (11) are 603, 552, and 500 nm (16). Referring to molecular orbital interactions, the excitation related to λmax corresponds to an HOMO → LUMO transition and moreover to a π → π* transition: the HOMO lies on the nitrogen atoms and the central double bond (ground state) whereas the LUMO lies mainly on the simple C-C bond and oxygen atoms (excited state) (17, 18). Indigoids are dyes of the donor/acceptor type: the two indole nitrogen atoms are electron donors while the two carbonyls groups are acceptors. Excitation due to light absorption delocalizes electrons through the bis-indole system. The representation of the resonance structures (17, 18) depicted in Schemes 1 and 2 illustrates the electron displacements and explains the differences of absorption. For indigotin (8) (Scheme 1), resonance forms lead to an extremely stable quadrupole structure: both the negative and positive charges are stabilized on both sides of the double bond. The energy difference between the ground state and the excited state is then small. Referring to the Planck equation (Eq. (1)), this implies a long wavelength (603 nm).
For indirubin (9), the resonance forms lead to another quadrupole structure (Scheme 2), which is less stabilized than the indigo form due to asymmetry. However, the resonance structure of indirubin cannot be considered as a proof and further explorations have been undertaken.
The charge distribution from the resonance structures of indirubin and indigo were confirmed by a computational calculation approach based on a molecular orbital interaction (17). While the charge transfer is equally distributed on both sides of the indigotin core (8), it is localized mainly on the left portion (“indigo part”) of indirubin (18) (9) (Fig. 4), destabilizing the charge equilibrium. This destabilization leads to a bigger ΔE and so to a lower absorption length of 552 nm for indirubin (9), in the red area of the visible spectrum.
Fig. 4
Charge transfer in indigotin (8) and indirubin (9). (Reproduction from reference (18) prepared by the authors.)
Indigoids possess a wide color range, resulting in high bathochromic effects due to their specific crystal network or solvent interactions. Those non-common electronic features still fascinate scientists and the use of these particular electronic effects is being applied for the development of new green organic electronics applications (19, 20).
3 Indigo and Its Relatives
Among all the natural dyes known and extracted by human beings, indigo is one of the oldest (21). Indigo stripes were found on the linen of the clothes of mummies from the 3rd millennium BCE (Before the Common Era) (22). Pre-Columbian buildings were decorated by Maya people with paintings produced around 800 BCE, with the so-called Maya-Blue being a pigment constituted of mineral oxides and indigo (23). A cuneiform tablet from the Ancient Babylonian times (~600 BCE), gives the first written instructions for wool dyeing and represents the earliest extraction procedure for indigo (22). The use of indigo spread worldwide, but no-one these days is able to explain where and when all this great history started.
The etymologic origin of the word indigo came from the ancient Greek ινδικόν (pronounced “indikon”), which was translated to indicum in Latin and means “something from India”. This supposes that the first contact with the pigment in South-East Europe should have happened through the large conquests carried out in the Ancient Greek and Persian empires. Besides the mythology and religious aspects, the use of indigo by the Ancient Egyptians indicates that the relevant extraction knowledge appeared early in human history. Even if the starting point is not clearly known, sources of indigo are, however, identified: indigo has been extracted from plants using diverse methods depending on the plant family and the region of the world where it was exploited. As mentioned above, more than 300 plant species are able to produce indigo. Depending on the plant source, the color varies from the palest sky blue to the deepest midnight blue. For reasons of clarity, only the three main species that contributed the most to the development and reputation of indigo around the world will be described.
3.1 Sources and Extraction of Indigo Around the World
3.1.1 Japan, Asia
3.1.1.1 History
In Japan, the leaves of Polygonum tinctorium Lour. (Polygonaceae) have been used as a traditional source for indigo production. This plant was also used in other areas of Asia, especially in mainland China, along with Isatis indigotica Fortune. The production of indigo was known in China since the Zhou period (1045–771 BCE) and indigo (sukumo in Japanese) was exported to Japan in the fifth or sixth century (24). In both countries, it was used to dye traditional costumes such as the kimono or simply everyday clothes. Its cultural importance was so encrusted in the Japanese society that the Japanese Ministry of Culture in 1978 protected indigo as a “National Treasure” and the dyers know-how was regarded as a “Living Treasure”. The Asian indigo-bearing plants were also utilized for another application in societies of the Far East, as the basis of a traditional medicine like Chinese Danggui Longhui Wan (see Sect. 5.1.1).
3.1.1.2 Extraction Process
Extraction of indigo from P. tinctorium reveals centuries of knowledge, with the tradition kept jealously by Japanese dyers. It consists of harvesting and fermenting leaves to increase the indigo precursor quantities. The whole procedure extending over the entire year has been described by R. Ricketts (24), and will not be discussed in detail in this contribution. The chemistry involved in the extraction process is included in the section on Indian plants (Sect. 3.1.3).
3.1.2 Europe
3.1.2.1 History
In Europe, indigo was produced predominantly from Isatis tinctoria L. (Brassicaceae), which is better known under the name of “woad” (Fig. 5). This biennial plant has been exploited in Western Europe since the Neolithic period (25), as attested to by the discovery of seeds in France. The Roman Emperor Caesar described the use of blue pigments by the Britons in England, but it has not been documented definitively that this blue color came from I. tinctoria.
The first textiles dyed with woad appeared during the Iron Age (~700 BCE) (25) and the beginning of the dye industry started during the Middle Ages. The main production areas of activity were concentrated in England (Lincolnshire and Somerset), Germany (Thuringia), Italy (Piedmont and Tuscany), and France (Gascony, Normandy, Britany, the Somme Basin). However, the main European area dedicated to indigo production was undoubtedly the Languedoc area in France, which led to some cities such as Toulouse, Albi, and Carcassone becoming extremely rich.
3.1.2.2 Extraction Procedure
Traditional extraction of indigo was performed from the plant leaves. After being harvested, the leaves were crushed and mixed with water to make a pulp and compressed into a pellet form (cocagne). After drying, the pellets were crushed into a powder and mixed with urine to induce oxidation. This oxidative stage leads to a paste, which once dried affords the dye or pastel, containing indigotin (8).
Currently, the extraction is conducted according to three major phases: maceration, oxidation, and precipitation. The leaves are immersed in a water bath (maceration phase) to extract indigo precursors; and the latter are then oxidized by stirring the bath (oxidation phase). The color of the water then switches from green to deep blue. The liquid is then allowed to stand and the dye is collected at the bottom of the bath by precipitation.
3.1.2.3 Chemistry
Indigo is not produced directly by woad. It is the result of a series of chemical reactions and several studies (16, 26–29) have identified these indigo precursors. The major precursor of indigo is isatan B (23) (indoxyl-β-ketogluconate). The minor precursors, indican (24) (indoxyl-β-d-glycoside) and isatan C (25) (16) (Fig. 6), have also been identified.
During the traditional preparation, the precursors are degraded by hydrolases released when the leaves are crushed. The liberated indoxyl (26) is then oxidized by the addition of urine to form the pastel.
Since the leaves are not crushed during the modern extraction procedure, isatan B (24), indican (25) and isatan C (26) are solubilized in the water bath. The maceration creates a medium rich in bacteria and sufficiently alkaline to break the glycosidic bond and liberate indoxyl (26). Under the influence of both aerial oxygen and basic conditions, indoxyl dimerizes to form indigotin (8) during the stirring step. However, indigotin (8) is not the only compound to be formed during the extraction process. Considering the nature of the precursors, side reactions occur and lead to the formation of indirubin (9), isoindigo (11), and isoindirubin (12) (16), as described in Scheme 3.
Scheme 3
Indigoid derivatives formed during the woad extraction process
Indigotin (8), the main coloring agent, is obtained by the dimerization of isatan B (23). Indirubin (9), the principal minor compound, is formed by the coupling of indoxyl (26) and isatin (27) with the latter resulting from the oxidation of indoxyl (26), due to the stirring conditions or on addition of urine. The decomposition of isatan C (25) leads to the formation of dioxindole (28), which can be oxidized to isatin (27). The dimerization of these two compounds affords isoindirubin (12). Finally, as a minor reaction, oxindole (29), an isomer of indoxyl (26), reacts with isatin (27) to yield isoindigo (11).
The biogenetic precursors of isatan B (23) and indican (24) have been studied by feeding young Isatis tincotria plants with [H3]-labeled-tryptophan and [C14]-labeled-acetate (30, 31). Indican (24) and isatan B (23) labeled on their indole (H3) and glucosidic parts (C14) have been isolated and identified, thereby proving their origin from indole. The shikimic acid pathway is probably involved in their biosynthesis (Scheme 4).
However, the pathway of the formation of indoxyl (26) from tryptophan is still obscure. Nonetheless, the biochemical synthesis of indigotin (8) from strains of E. coli (21) suggests that indigo precursors formation occurs through tryptophanase and dioxygenase.
The harvest time, the treatment, and the soil impurities (32, 33) affect the proportion of the constituents and so the resultant color. This could explain the pastel color differences observed all over Europe and moreover the assured quality of the French indigo. However, at the dawn of the sixteenth century, woad production in France was curtailed due to the arrival of a darker indigo along with the discovery of a sea route to India by Vasco de Gama.
3.1.3 India, South America, and French West Indies
3.1.3.1 History
From the beginning of the sixteenth century to the seventeenth century, the European pastel was supplanted by the more intense indigo from India. It was extracted mainly from Indigofera tinctoria L. (Fabaceae) or “true indigo”. This two-meter high tree is well acclimated to the tropical areas from South Asia and can be found in some parts of Africa. However, its original habitat is not known and according to the climate, it could be an annual, biennial, or perennial. More than 800 species of Indigofera exist, but most of them are not of interest for the production of dye. Indigofera tinctoria L. (India, Southeast Asia, Middle East Africa, Madagascar) and Indigofera suffruticosa Mill. (South America, French West Indies (Fig. 7)) remain the most widely used plants in this regard.
As one of the first suppliers of the Greco-Roman world, the dye industry in India had an important impact in society and economy. Except for exportations, blue-dyestuffs were manufactured for everyday life, or for ceremonial or religious purposes. The production of Indian indigo exploded when the Dutch East India Company and the British Empire took the control of India and its raw materials. The development of shipping routes to South America, as effected by the Spanish and the Portuguese kingdoms, marked the starting point of the introduction of indigo from I. suffruticosa on the European market. The commercialization of Indian and American indigo dislodged the French woad and initiated an economic war between the colonial empires, mainly between France and Great Britain. Due to the sugar crisis in nineteenth century, France relocated the production of indigo to the French West Indies (Fig. 8), on the islands of Marie-Galante (34) and Guadeloupe (35), where I. suffruticosa was present (or introduced).
Regardless of the place of production or the actual plant used, the extraction processes used for indigo samples have been quite similar. Thus, the leaves were placed in a large tank, maintained at the bottom by large wooden beams, soaked in water, and left to macerate. In India, slaves and later free workers entered the bath and stirred it with their legs or with wood sticks to create the correct oxidative conditions. In South America, workers manipulated wooden paddles outside the bath to stimulate the stirring process. In the French West Indies, a system of communicating baths was created: after maceration, the water went down to another tank and was stirred by wooden blades (named batterie in French) that were activated manually by slaves (34).
In all cases, after the stirring process, the dye was recovered at the surface of the water. Since the extraction step is the same as that for woad, the chemistry involved is then identical. What is the difference between European and Indian/American/Caribbean indigo? This will be answered in the next section of this chapter.
3.1.3.3 Chemistry
Studies (29, 36–38) of the plants used to produce indigo in India, America, Thailand, and Japan (Polygonum tinctorium) have shown that the main precursor is indican (24). The numbers of reactions possible is limited and thus so are the number of potential derivatives. While three different precursors are present in Isatis tinctoria and lead to the formation of four colored molecules, only one is present in the Indigofera species used, and this leads mainly to the formation of indigotin (8) and its isomer, indirubin (9) (Scheme 5). The concentration of indigotin (8) is then higher, thus resulting a more intensely blue and pure dye.
Scheme 5
Formation of the indigo and indirubin in Indigofera and Polygonum species
The struggle for the control of the indigo market lasted until the beginning of the nineteenth century. Independence movements in the different colonies, especially rebellious events occurring in India, diminished the external control by colonial empires. Moreover, the first chemical synthesis methods successfully adapted to an industrial scale ended trade in natural dyes and hence the prosperity of their local industries.
3.2 Chemical Synthesis of Indigo
Numerous syntheses of indigo are described in the literature. For reasons of clarity, this chapter will focus on the more relevant methods, starting from the first synthesis by Baeyer.
3.2.1 The First Syntheses: Baeyer, Drewsen, and BASF
The excessive cost in European markets of natural indigo dye, resulting from long transportation times, the emancipation of colonies, and the rise of the industrial revolution led to a reconsideration of the use of natural supplies. German chemists started developing chemical synthesis of indigo with the support of BASF and Hoechst, two important companies involved in the industrial development of indigo.
Adolf von Baeyer proposed the first synthesis of indigotin (8) from isatin (27) in 1870 (39) and its modification in 1879 (40) (Scheme 6). This method involved the chlorination of the 2-position of isatin by phosphorus chloride followed by dimerization under acidic-reductive conditions using hydrogen iodide (39) or Zn (40) in acetic acid (Scheme 6). However, isatin (27) was very expensive at this time, and Baeyer proposed in 1880 (41) a new pathway starting from the o-nitro-cinnamic acid (31) (Scheme 6). Although this method was patented, it was also too expensive to be exploited.
A third method was described later by Baeyer and Drewsen involving o-nitro-benzaldehyde (32) (42). This synthesis pathway is known as the Baeyer-Drewsen method (Scheme 7).
However, this method was impractical because of the difficulty in making the starting product. A major breakthrough was made in 1890 when Heumann discovered the route to indigotin (8) from aniline (43). Lucius and Brunning then improved his method. To date, this synthesis (Scheme 8) dating from 1925 and introduced by BASF, is the most widely used method and is still utilized today. This pathway involves the condensation of formaldehyde and hydrogen cyanide to form the corresponding cyanhydrin. The latter is then attacked by aniline (33) to form the corresponding 2-(phenylamino)acetonitrile (34). The hydrolysis of the cyano group leads to the β-aminoacid (35) being fused into indoxylate (36) by sodium amide. The oxidation step gives indigotin (8).
Diverse methods have been developed during the twentieth century (44). Most of these start from aniline (33), like the Lucius-Brunning method. One method of note involves the dimerization of indoxyl acetate, and was described in 1958 (45). It still has impact nowadays and an improvement has been applied in indirubin synthesis (Scheme 9).
Scheme 9
Method based on the dimerization of 3-acetoxyindole (41)
The Sandmeyer method starting from aniline (33) leads to isatin (27) via the formation of the intermediate isonitrosoacetanilide (37). Isatin (27) is then oxidized in anthranilic acid (38). The latter is engaged in the Lucius-Brunning reaction to afford diacetylindoxyl (40) through the formation of phenylglycine-o-carboxylic acid (39). Deacylation in alkaline conditions followed by the indoxyl acylation leads to 3-acetoxyindole (41). The formation of indigotin (8) is finally achieved by the dimerization of 41 in oxidative and basic conditions.
A study reported by Kaupp’s group (46) showed that the coupling reaction of indoxyl acetate in an alkaline medium generates an indoxyl radical (42) due to oxidative conditions. Two indoxyl radicals coupled to form leuco-indigo (43), a colorless compound. Leuco-indigo (43) is then quickly oxidized by air in indigotin (8) (Scheme 10).
The chemical synthesis of indigotin (8) led to the rapid development and internationalization of the textiles industry. The blue jeans production introduced earlier in America by Levi Strauss with denim indigo (produced in Nimes, France) has shown a great renewal. Nowadays, the global annual production is estimated at a billion pairs of jeans. The dyeing process involves the reduction of indigo to form the water-soluble leuco-indigo (43). Then, the cloth is steeped in the dye-containing water. On removal from the bath, subsequent oxidation in air colors the fabric blue.
Indigo has had great influence on human society. Its influence was so important that it led empires to fight to take over its control and to generate economic benefits. As the industrial revolution took place, indigo led to its own revolution through the development of the chemical synthesis methods and the rapid application by the textiles industry. Nowadays, new indigotin (8) derivatives have been synthesized and have found applications in medicinal chemistry.
4 Tyrian Purple
4.1 Legendary History
As I have no desire to tire you with endless lessons on a single subject, I will tell you instead the origins of purple […]. Hercules had a dog with him, as was the custom in the ancient world; as you know, dogs accompanied the heroes of old even when they entered an assembly. Hercules’ dog saw a purple mollusk crawling up a rock, poking out of its shell; he seized his flesh in his teeth, then ate it. Its blood dyed the dog’s lips the brightest red … The nymph, when Hercules was on her side, saw the dog with his lips this unusual color, and declared that she would refuse Hercules her love unless he presented her with garments even brighter than the lips of his dog. Hercules went back for the shell, extracted the blood, and gave the girl the gift she so desired, thereby acquiring the reputation in Tyre of inventor of the purple dye (Julius Pollux, 2nd century BCE) (47) (Fig. 9).
Fig. 9
Discovery of Tyrian purple by Hercules (Ref. (48))
Tyrian purple (Royal Purple, Imperial Purple, or Purple of the Ancients) is a natural purple dye for which the extraction and distribution was of economic and social importance in the Mediterranean Sea, starting from Antiquity to the fifteenth century of the Common Era. Although legend locates the first extraction in Tyre (Lebanon), documented evidence from the Minoan period (twenty-seventh–fifteenth century BCE) could place its first extraction in Crete (Greece). Undoubtedly, its first use took place during the seventeenth century BCE in Akrotiri of Thera (Santorini Island, Greece) (49, 50), as proved by HPLC-APCI analysis (51). Wall paintings discovered in this prehistoric city testified to its wealth during the Minoan Period. There is an evidence for the Royal Purple industry around the thirteenth century BCE in Serafand (Lebanon) (52). Its extensive production from the Phoenicians in the Tyre area dates back to 560 BCE and Tyrian purple has been widely used in Persia by the Achaeminian dynasty (559–330 BCE) (9, 53, 54). The meteoric rise of the dye justified its price: it was worth 10–20 times as much as its weight in gold (52). The blue Tekhelet, a variation of Tyrian purple mentioned in the Jewish bible, was used to dye the cord of the High Priest (52). After the conquests achieved by Alexander the Great (331 BCE) the eye-opening dye became well known in the north of Greece, especially in Macedonia (9). The beautiful color and its expensive value pushed the Roman emperor Nero to impose sumptuary laws and restrict its use for the ruling classes only, thereby becoming the symbol of power. Later, this law was extended to ceremonial purposes and the robes of popes and cardinals were also dyed with Tyrian purple. The fall of the Roman Empire slowed its production. Later, the fall of Constantinople in favor of the Ottoman Empire (1453 CE) and the decision of Pope Paul II to replace purple by carmine (1464 CE) (55) brought a fatal blow to the production of purple in Europe. William Cole from Bristol later rediscovered Purple of the Ancients in 1684 (56) on the shores of England. Nowadays, purple is still traditionally extracted in South America and its renewal has emerged in arts in Europe. 6-Bromoindirubin (13), one of the constituents of the dye, is actually a lead compound in medicinal chemistry research, thereby prolonging the legendary history of Tyrian purple.
4.2 Extraction
4.2.1 Sources
The purple dye is extracted from number of mollusks belonging to the family Muricidae. This worldwide family groups a wide variety of shaped and colored shellfish. Dye production is a characteristic of this conchylian family. In southeastern Europe, the highly prized Tyrian purple was obtained at great expense essentially from the two species Bolinus (Murex) brandaris and Hexaplex trunculus. It has been reported that 10,000 animals were needed to produce 1 g of the dye (54). In Britain and Ireland, the rediscovery of purple by W. Cole was possible owing to the occurrence of Nucella lapillus (dog whelk). Thais kiosquiformis (South America), Plicopurpura species (Mexico, Caribbean Sea, Atlantic Ocean), and Rapana venosa (Japan) have been subjected to dye production in their respective areas (55).
4.2.2 Extraction Process
The first surviving details of a purple dye existence were found on a Mycenaean Linear B tablet that dated from the thirteenth century BCE (50). Pliny the Elder was one of the first naturalists to describe the extraction in Naturalis Historia (57). The dye does not exist in the mollusk as indigo, as in an indigo-bearing plant. The purple color is generated from precursors, the so-called “chromogens”, contained in the hypobranchial gland. To access the vein, the shell should be broken carefully so as not to crush the organism inside. Once the vein is accessible, a colorless liquid can then be extracted, which once exposed to light becomes purple in color.
4.2.2.1 Central America-South America-Caribbean Sea
Thais kiosquiformis and Plicopurpura patula pansa were mainly used. Traditionally, the shellfish was impaled with a cactus needle and pressed to collect a milky liquid on a cotton piece. The shellfish was then allowed to stand on a rock while waiting to be extracted a second time. The organism was then withdrawn from the shell and compressed with a knife from head to tail and the liquid collected on a piece of cotton. This process is somewhat reminiscent of that used by farmers to take milk from cows: it has thus been baptized “shellfish milking”. Sometimes this operation was repeated three or four times before the death of the animal.
4.2.2.2 Japan
Few details have appeared in literature concerning purple dye extraction in Japan. It is known that the dye was extracted from Rapata venosa. The numbers of broken shells found in the area of Hitachi (55) attests to a similar process used as described in Sect. 4.2.2.1. Even though published information is scarce, traces of the dye have been identified on silk from Yoshinogari (Kyushu Island) (58).
4.2.2.3 Europe
H. trunculus and B. brandaris are the two most abundant species used in countries bordering the Mediterranean Sea in Europe and North-west Africa. N. lapillus is found preferentially in northern Europe (Atlantic Ocean). These three shellfish have been used widely used and two distinct methods have been described for dye production. The first one described by Pliny the Elder is an indirect extraction/dye process:
“The vein already mentioned is then extracted and about a sextarius of salt (3.17 kg) added to each hundred pounds (0.453 kg) of material. It should be soaked for three days, for the fresher extract, the more powerful the dye, then boiled in a leaden vessel. Next, five hundred pounds of dye-stuff, diluted with an amphora (30.28 L) of water, are subjected to an even more moderate heat by placing the vessels in a flue communicating with a distant furnace […] and a test is made about the tenth day by steeping a well-washed fleece […] The Tyrian color is obtained by first steeping the wool in a raw and unheated vat of pelagian extract, and then transferring to one of buccine…” (Pliny the Elder, Historia Naturalis, 1st century CE) (59).
The series of heating and dilution steps in differently coated containers implies that a series of chemical reactions was used to obtain the best dye (see Sect. 4.2.3.4). The processing of N. lapillus for dyeing was similar to that used in South America. The shell was broken and the vein penetrated. The viscous liquid was applied directly onto linen and left in the sun (56).
4.2.3 Chemistry
4.2.3.1 Composition
The main component of interest is 6,6′-dibromindigotin (17), and in some species such as R. venosa or those in the genus Plicopurpura, it can be found alone (58, 60, 61). It is often called “Tyrian purple” by reference to the dye and represents the real biomarker of true-purple dyes. The second main component is 6,6′-dibromoindirubin (15), which has been identified in H. trunculus (9, 62), N. lapillus (63), and B. brandaris (64). Two other brominated indirubins have been discovered: 6-bromoindirubin (13) and 6′-bromoindirubin (14). Traces of 6-bromoindigotin (16), indirubin (9), and indigotin (8) also have been found in H. trunculus (61, 62).
4.2.3.2 Precursors and Biosynthesis
Conchylian indigoids are formed next in a series of chemical reactions from precursors present in the hypobranchial glands. Identification of these precursors (Fig. 10) was a high scientific objective for dye chemists. It is suggested that four chromogens are always present in species in the family Muricidae, thanks to the contributions of Baker (65, 66) and Fouquet (67). These are indoxyl sulfates (44 and 45) and brominated indoxyl sulfates (46 and 47). Baker has found tyrindoxyl (48) in the glands of Dicathais orbita (65). However, paper chromatography (68) has shown that a wide variety of indoxyl sulfates exist in shellfish species. Baker and Duke have isolated a series of choline (50) and choline ester salts of tyrindoxyl sulfate (general structure (49)) from D. orbita and Mancinella keineri (69). Murexine salt (51) has been found to be the ultimate precursor of Tyrian purple in these species. Nonetheless, precursor differences have been observed according to the season (61) and the sex gender of the mollusk (70) making the identification more difficult.
The biosynthesis pathway leading to the formation of the precursors is not fully understood, but is assumed to be similar to that of indigo. The precursors may be formed from tryptophan through the action of a tryptophanase and/or other enzymes to indole (30). Indole (30) can then be oxidized by a diooxygenase to indoxyl (26), with the latter being converted to the corresponding sulfate. Alternatively, the pathway may convert tryptophan directly to indoxyl sulfate (44), following a new biosynthesis pathway. However, the origin of tryptophan or indoles in the mollusks could also be external to these organisms. Muricidae shellfish possess the ability to biosynthesize choline and its ester derivatives. Up to the present, no experimental proof has been obtained concerning their biosynthetic origin. The specific positions of bromine and sulfate groups suggest the need for specific enzymes. The absence of bromotryptophan in the hypobranchial glands (71) and evidence for post-translational bromination at the 6-position of tryptophan in a sea snail (Conus radiatus) (72) support this hypothesis. Moreover, bromoperoxidase and vanadium bromoperoxidase have been found in some algal species (73) and could be the enzymes involved (Scheme 11).
Scheme 11
Hypothetic biosynthesis of Tyrian purple precursors
Although the biosynthesis remains elusive, the precursor function of the mollusk has been clarified thanks to investigations by K. Benkendorff. She proposed a role in reproduction and defense for the indole derivatives and a de novo synthesis of indigoids inside the mollusk (71, 74).
4.2.3.3 Chemical Process
When exposed to light, these precursors undergo chemical and enzymatic transformation easily observed by applying the dye on fibers (Fig. 11). Several investigators have described color development from a white liquid becoming successively yellow, green, and then purple.
The nature of the intermediates has been the object of scientific debate (65, 75). However the formation of 6,6′-dibromoindigotin (17) and 6,6′-dibromoindirubin (15) is now accepted (Scheme 12).
Scheme 12
Formation of 6,6-dibromoindigotin (17) and 6,6′-dibromoindirubin (15)
Murexine-tyrindoxyl sulfate (51) (70) (white) undergoes a desulfonation to form tyrindoxyl (48), by action of a purpurase identified as arylsulfatase (67). Tyrindoxyl (48) under oxidative conditions leads to tyrindoleninone (52). Coupling between 48 and 52 affords the green intermediate tyriverdine (53) (76). Photolysis of the two methanethiol moieties yields 6,6′-dibromoindigo (17). 6,6′-Dibromoindirubin (15) is formed by the coupling reaction of tyroxindole (48) and 6-bromoisatin (54). The latter is generated by both oxidation of tyrindoleninone (52) and tyriverdine (53).
Formation of monobrominated indirubins and 6-bromoindigo (16) is, however, still obscure and there is no clear evidence of their formation to date. 6-Bromoindigo (16) probably arises from photodebromination of leuco-6,6′-dibromoindigo (55) formed by reduction of 6,6′-dibromoindigo (17) by tyrindoxyl (68). Leuco-6-bromoindigo (56) is then oxidized to give 6-bromoindigo (16) (Scheme 13).
The same mechanistic explanation is given for the formation of indirubin (9) (Scheme 14). 6,6′-Dibromoindirubin (15) is reduced by tyrindoxyl (48) to form leuco-6,6′-dibromoindirubin (57). The latter undergoes a photodebromination to afford two leuco- intermediates possessing a bromine atom at position 6 or 6′ (58 and 59). These intermediates are separately oxidized to give 6′-bromoindirubin (14) and 6-dibromoindirubin (13). Nonetheless, this hypothesis has not yet been proven experimentally.
Another hypothesis highlights the presence of non-brominated precursors in H. trunculus (Scheme 15). Sodium indoxyl sulfate (44) may be transformed in indoxyl (26) by action of purpurase. Indoxyl (26), by action of oxygen, could be transformed to isatin (27). Coupling between indoxyl (26) and 6-bromoisatin (54) affords 6-bromoindirubin (13) while the coupling of isatin (27) and tyrindoxyl (48) forms 6′-dibromoindirubin (14). As a minor reaction, indoxyl (26) and isatin (27) form indirubin (9) and the dimerization of indoxyl (26) leads to indigotin (8).
Scheme 15
Second hypothesis on formation of mono-brominated-indirubins
The Phoenician dye technique is reminiscent of vat dyeing, which has been described by Pliny the Elder (see Sect. 4.2.2). The dye was maintained in the leuco-form because of the water insolubility of 6,6′-dibromoindigo. The cloth was dipped inside and aerial oxidation led to the purple hue. A series of chemical reactions is involved and these are linked with dilution steps and differential heating in a specially coated vat. This implies the establishment of a reductive system, precursor stabilization, enzyme deactivation, and photochemical control. McGovern and Michel published a series of studies investigating the chemistry involved (54, 59, 77) that are summarized below.
“The best time to catch them is after the rising of the Dog star […] The vein already mentioned is then extracted and about a sextarius of salt (3.17 kg) added to each hundred pounds (0.453 kg) of material. It should be soaked for three days, for the fresher extract, the more powerful the dye, then boiled in a leaden vessel.”
Pliny described the mollusks being collected during a specified period resulting therefore in the presence of specific precursors. The boiling step must surely deactivate the purpurase (deactivated at 75 °C (59)). However, in Latin, Pliny used the word plumbo translated into ‘lead’, but this could be understood as “tin” as well. McGovern and Michel thus performed reduction in aqueous solution of 6,6′-dibromoindigo (17) (DBI) in the presence of different metals to form the corresponding leuco-DBI (56) (77). To control the efficiency of reduction, they sank a piece of filter paper into the liquid and observed the resultant color. They discovered that tin and lead can both reduce DBI but only tin in alkaline conditions (NaOH 1 N) at 90 °C could give the deep purple characteristic of Tyrian purple. However, Pliny never mentioned the use of alkalis despite the fact they were known at that time. Moreover, the additives described (salt, urine) are not alkaline enough to reduce DBI (17) under these conditions (59).
In addition, the reductive agent might be present in the mollusk (as 2-substituted indoxyls (59)) or formed during the process (e.g. methylmercaptan from tyriverdine photolysis (77)). Experience using 1-dodecanethiol at about 80 °C has partially confirmed this hypothesis (77). Interestingly, McGovern and Michel noted that a natural product present in B. brandaris collected in France was highly efficient in reducing indigoids without any alkaline conditions (77). Unfortunately, this product has not been identified yet. However, the concentrations of methylmercaptan and 2-substituted indoxyls are low and the addition of honey (attested to by ancient texts and notably by Plutarch) could preserve the reduced-form of the dye as proved by a reduction trial using dextrose.
“Five hundred pounds of dye-stuff […] are subjected to an even more moderate heat by placing the vessels in a flue communicating with a distant furnace […] a test is made around the tenth day”.
Stabilization of indoxyls must also be preserved and their oxidation to the insoluble indigoids blocked. At 40–50 °C and for long periods, the conditions described by Pliny, only the use of pure tin could prevent precipitation of the dye, confirming the use of tin-coated vat.
The authors showed that dark or subdued light could block the photolysis of diindoxyl. Several hypotheses could be proposed: the vat was covered, the dyers worked during the night or the insoluble by-products (other indigoids) formed a protective layer at the surface of water. Surprisingly, the text of Pliny does not mention the control of light and only one text refers to the effect of light without providing more precise details (59).
“The Tyrian color is obtained by first steeping the wool in a raw and unheated vat of pelagian extract, and then transferring to one of buccine […] Conchyliated garments are prepared by a similar method to the first but […] the dye is diluted simultaneously with urine and water”.
Here, “pelagian extract” corresponds to the Murex shellfish and “buccine” corresponds to an extract of Buccinum species (mollusk), which gives a red color. The famous Tyrian color was in fact a mix of two dyes, with the pelagian one serving to fix the buccine one.
All these studies were performed in the 1990s and do not answer all the questions. No recent studies have emerged since 1990. Additionally, difficulties are evident in chemically translate ancient texts. The decryption of the vat dyeing process of Tyrian purple used by the Phoenicians still remains to be chemically improved. However, the actual knowledge can be summarized as depicted in Scheme 16.
The chemical synthesis of 6,6′-dibromoindigo (17) has been as great a challenge as the synthesis of indigotin (8). While its structure was not totally elucidated, chemists first explored its synthesis to aim for the formation of new indigotin derivatives. Sachs and Kempf were the first chemists to propose a total synthesis (78) (Scheme 17).
Scheme 17
First proposed synthesis of Tyrian purple by Sachs and Kempf
The strategy involves the transformation of 2,4-dinitrotoluene (60) to the key intermediate 3-bromo-6-formyl-nitrobenzene (61) in five steps. The bromonitrobenzene is then submitted to an aldolization in the presence of acetone in alkaline medium. The β-hydroxyketone (62) encounters ring closure to form an indoxyl derivative. The latter dimerizes spontaneously to afford 6,6′-dibromoindigo (17). The major inconvenience of this synthesis is the number of steps to form the key intermediate. Moreover, the authors did not mention the yield for the last step.
Later, Sachs and Sichel (79) proposed a slight modification to improve the yield of the dimerization. In this procedure, the key intermediate is condensed with acetone in the presence of trisodium phosphate and the β-hydroxyketone cyclized in alkaline medium. Friedländer was the first investigator to discover the structure of 6,6′-dibromoindigo (17) in 1909 (80). He reported that 12,000 shellfish (M. brandaris) were needed to provide 1.4 g of the dye. He proposed more convenient synthesis methods (80, 81) starting from bromoanthranilic acid (63) (Scheme 18), with the latter being obtained from the oxidation of 2-amino-4-bromotoluene (64). Bromoanthranilic acid (63) reacts with chloroacetic acid to afford the corresponding phenylglycine (65). The following acetylation gives the N-acetyl-3-acetoxy-6-bromoindole (66). Alkaline hydrolysis under oxidative conditions leads to 6,6′-dibromoindigo (17). Although this method was more convenient than the Sachs strategy, the use of the expensive bromoanthranilic acid (63) as starting material represented an obstacle for industrial development.
As a consequence, Grandmongin and Seyder (82) proposed an alternative approach. 6,6′-Dibromoindigo (17) is formed by reduction of 6,6′-dinitroindigo (67), followed by bromination (Scheme 19).
Scheme 19
Synthesis of 6,6′-dibromoindigo (17) according to Grandmongin
The 6,6′-dinitroindigo (67) is prepared in the same conditions as described by Friedländer. The two nitro groups are then reduced in the presence of zinc in acidic conditions and the 6,6′-diaminoindigo (68) thus formed reacts with sodium nitrite in acidic conditions to form the corresponding bis-diazo compound (not shown in the Scheme). The latter is then brominated by cupric bromide. This method is the only example of bromine introduction at the end of the synthesis.
With the exception of the Grandmongin method, all the procedures described share the common starting product 2-amino-4-bromotoluene (64), which is then oxidized to the corresponding 3-bromo-6-formylnitrobenzene (61) or anthranilic acid. Therefore, synthesis of Tyrian purple has progressed through the perfection of the synthesis of intermediates. Torimoto cited in (74) proposed a new synthesis pathway (Scheme 20) to form 3-bromo-6-formyl-nitrobenzene (61) starting from 2-nitro-4-bromotoluene (69), thanks to the work of Barber (83).
Alternatively, Voss and Gerlach (84) proposed an approach starting from 1,4-dibromobenzene (70) based on selective lithiation at position-2 of 2,5-dibromonitrobenzene (71) to selectively introduce the formyl group in ortho-position of the nitro group to afford the key intermediate 61 (Scheme 21). The latter under basic and oxidative conditions affords the 6,6′-dibromoindigo (17). However, one of the major drawbacks of this method is to ensure a good control of temperature (−105 °C) for the lithiation.
Cooksey (57) developed a method to form the key intermediate by ameliorating the Sachs-Kempf method and applying it directly to the 2-nitro-4-bromotoluene (69) (Scheme 22). o-Nitro-toluene (72) is brominated at the meta-position of the nitro group by action of bromine in the presence of iron to form 69. The methyl group is then halogenated by action of N-bromosuccinimide and the bromine further displaced by pyridine to form the corresponding pyridinium salt (73). The latter is converted to the nitroso derivative 74, which by hydrolysis under strong acidic conditions affords the corresponding aldehyde (61). The key intermediate is then dimerized in 6,6′-dibromoindigo (17).
Surprisingly, few methods involving the transformation of indoles have been developed, probably because of the cost of the starting materials at the beginning of the twentieth century. Later, Majima (85) and his collaborators have proposed a method starting from plain indole (Scheme 23). The successive treatment of indole (30) with a Grignard reagent followed by ethylchloroformate affords the indole-3-carboxyethyl derivative (75). Position 6 is then brominated and the ester (76) is converted to the corresponding carboxylic acid (77). Dimerization occurs under alkaline and oxidative conditions to furnish 6,6′-dibromoindigo (17). Although this method is attractive it suffers from the disadvantage of a lack of regioselectivity for the carboxylation and bromination steps (86).
Finally, Tanoue (87) has recently developed a method based on a biosynthesis pathway (Scheme 24). The 6-bromoindole (78) is converted in the corresponding 3-acetoxy-6-bromoindole (79) in a two-step method developed by R. D. Arnold (88). Position 3 is first iodinated and then acetoxylated by action of silver acetate in acetic acid. The 3-acetoxy-indole (79) is then dimerized in an alcoholic alkaline medium. This procedure is the fastest synthesis pathway but it is also the most expensive.
Tyrian Purple, as for indigo, has been the subject of numerous studies due to its perfect color and its legendary history, which together have pushed chemists to discover the complex origin of the color and to understand the sophisticated vat dyeing technique of the Phoenicians. The challenge to develop a sustainable synthesis for industrial applications was a major objective after the synthesis of indigotin (8). However, with the industrial development of western countries, the use of natural dyes has declined. Blue and purple became common colors, having social significance or not (blue has been the color of working class), and the chemistry of indigotin (8) and 6,6′-dibromoindigo (17) has been largely abandoned (except by dye chemists).
5 Renewal of Interest of Indigoids in Medicinal Chemistry
Despite the fact that the use of natural dyes declined over a period of time in favor of synthetic dyes, the indigoids, and especially the indirubins have been subjected to a renewed interest over the last ten years. Indeed, at the end of the 1990s, indirubin (9) was found to be the main active constituent of a traditional Chinese medicine called Danggui Longhui Wan.
5.1 Discovery of the Biological Potency of Indirubin
5.1.1 Danggui Longhui Wan
Danggui Longhui Wan has been used for the treatment of chronic myelogenous leukemia (CML) in mainland China (62, 89). This medicinal formula consists of a mixture of eleven ingredients from traditional Chinese medicinal herbs, including Indigofera tinctoria, Angelica sinensis, Aloe vera, Moschus moschiferus, Phellodendron chinense, Saussurea lappa, Coptis chinensis, Gardenia jasminoides, Scutellaria baicalensis, Rheum palmatum, and Gentiana scabra (89). Qing Dai (Indigo naturalis) among these 11 ingredients is the active constituent of the dark blue powder that is extracted from indigo dye-producing plants (89, 90). Qing Dai is a mixture of the blue dye indigotin (8) and the red dye indirubin (9) (89–91). Indigotin (8) is inactive as the main component of Qing Dai, whereas indirubin (9) is a minor component with antitumor activity (92). Clinical studies for treatment of CML and chronic granulocytic leukemia have shown the efficacy of indirubin (9), which led to 26% complete remission and 33% partial remission out of 314 cases (89, 91). Low toxicity and minimal side effects have been detected in most of the patients evaluated (89, 91). However, indirubin (9) has low water solubility and bioavailability, and some patients have suffered from severe gastrointestinal side effects (91–93).
5.1.2 Mechanism of Action
Cyclin-dependent kinases (CDKs), a family of serine/threonine kinases, are key regulators of the cell cycle and form enzyme complexes with regulatory subunit cyclins for CDK activity (94–96). Selective modulations of CDKs have been validated extensively as attractive molecular targets for developing human cancer therapeutic agents (94–96). Originally, indirubin was identified as a potent modulator of CDK1/cyclin B, CDK2/cyclin A, CDK2/cyclin E and CDK5/p25, thereby mediating strong growth inhibition of human tumor cells (89, 91, 97). The analysis of the cell cycle suggests indirubin (9) arrests tumor cells at the G1/S or G2/M phases (Fig. 12), resulting in cell growth inhibition and eventually, induction of apoptosis (89, 91).
In an X-ray crystallography study of the CDK/indirubin complex, indirubin (9) was shown to bind competitively to the ATP-binding pocket as an ATP mimic in the catalytic domain of CDKs. Three hydrogen bonds are formed in the ATP-binding pocket by the NH-CO-N′H functionality of indirubin and are crucial for binding to the peptide backbone of CDKs (89, 98) (Figs. 13 and 14). Molecular modifications or substitutions of these groups of indirubin destroy the binding affinity to the ATP pocket of the catalytic domain and the ATP-competitive inhibition of CDKs.
Fig. 13
Indirubin-5-sulfonate bound in CDK2. (The picture was created from the PDB file 1E9H using Maestro software, Academic version 9.3.5., Schrödinger Inc.)
Indrubin-5-sulfonate in the ATP-binding pocket of CDK2. (The picture was created from the PDB file 1UV5 using Maestro software, Academic version 9.3.5., Schrödinger Inc.)
5.2.1 Discovery of the Potential of 6-Bromoindirubin
The biological promises of the constituents of Tyrian Purple secreted from the hypobranchial glands of Hexaplex trunculus were identified using a bioactivity- guided isolation method (62). The inhibitory potential of each extract and fraction towards a selected panel of protein kinases (GSK-3, CDK1/cyclin B and CDK5/p35) has been evaluated. 6-Bromoindirubin (13) (6-BI) was first isolated and recognized as a selective inhibitor of glycogen synthase kinase-3β (GSK-3β) (62, 99) (IC50 of 0.5 μM).
5.2.2 GSK-3β
Glycogen synthase kinase-3 (GSK-3) is a serine/threonine protein kinase that consists of two isoforms (GSK-3α and GSK-3β) with 97% homologous similarity (100). Originally, GSK-3 was discovered for its involvement in glycogen metabolism as the enzyme responsible for phosphorylation of glycogen synthase. It is the last link in the chain of phosphorylating agents triggered by the insulin-signaling pathway. The release of insulin inhibits GSK-3 signaling, leading to the storage of glucose (101, 102). Due to its preponderant presence in the brain, GSK-3 has further been pinpointed as a valuable target to treat neurodegenerative disorders, including Alzheimer’s disease by preventing over-phosphorylation of Tau protein (103–105), mood disorders (106) and schizophrenia (107). In addition, GSK-3 was identified as a key intermediate of several important biological processes such as the Wnt (100) and Hedgehog (Hh) (108) signaling pathways. Canonical Wnt signaling controls differentiation at the stages of embryonic and neuronal developments, the fate of stem cells, and neuroprotection (109) and is deregulated in cancer (110, 111). As for Wnt, Hh signaling is a complex pathway regulating embryonic development and tissue repair by controlling stem and progenitor cells (112). It is also deregulated in cancer (113). Therefore, GSK-3 appears to be an attractive target for cancer. Recently, GSK-3 activity has been proved to regulate also STAT signaling (114).
5.2.3 6BIO, a Biological Tool
Based on the observed results of 6-BI, a slight modification has been performed at the 3′-position and has led to the semi-synthetic analog, 6-bromoindirubin-3′-oxime (80) (6BIO, Fig. 15) (115).
(2Z,3′E)-6-Bromo-3′-(hydroxyimino)-(2′,3-biindolonylidene)-2-one, 6BIO (80) possesses an affinity 100-fold more potent than 13 towards GSK-3β (IC50 = 5 nM) (62) and a selectivity ratio CDK1/GSK-3 of 1/64 and CDK5/GSK-3 of 1/17 (62, 115). The crystal structure of 6BIO (62) (Fig. 16, PDB 1UV5) inside the ATP pocket of GSK-3 revealed the binding mode and lights up as a reason for such great affinity.
Fig. 16
6BIO bound in the ATP-binding pocket of GSK-3. (The picture was created from the PDB file 1UV5 using Maestro software, Academic version 9.3.5., Schrödinger Inc.)
The presence of the successive NH-CO-NH pattern of the H-shape part of 6BIO (80) is the key to ensure the strong anchoring of the molecule by creating three hydrogen bonds inside the cavity with the two critical residues of Val135 and Asp133 (Fig. 17). The oxime at the 3′-position assures the completion of the binding by interacting with amino side chains through a water molecule (115). Moreover, the presence of Leu132 above the molecule provides enough space and hydrophobicity for the bromine to interact through van der Waal’s forces. Indeed, the cavities of CDK2 and CDK5 contain a phenylalanine residue at the corresponding region of Leu132 thereby hindering the access of 6BIO (80) in their cavity (62). The perfect combination of key substituents on the indirubin skeleton and the shape of the protein residues confer the high selectivity of 6BIO (80) for the inhibition of GSK-3.
Fig. 17
Interactions of 6BIO inside the ATP-binding pocket of GSK-3. The H-bonds are shown as dotted lines. (The picture was created from the PDB file 1UV5 using Maestro software, Academic version 9.3.5., Schrödinger Inc.)
Due to the key role of GSK-3 in biological processes and its overregulation in diseases and the potent inhibitory activity of 6BIO (80), the latter exerts a broad range of biological activities (116, 117). However, discussed are only the most salient highlights.
5.2.3.1 Maintenance of Stem Cell Pluripotency
Embryonic stem cells are capable of self-renewal and differentiation in any type of cells (pluripotent cells) or into specialized cells. The Wnt pathway is notably the cellular signaling pathway controlling their fates. Indeed, numbers of its components were detected in human embryonic stem cells (118). The activation of Wnt needs GSK-3 to be switched off (109). Therefore, it has been proved that administration of 6BIO (80) to mouse and human embryonic stem cells was able to maintain their pluripotency (119). This important finding not only offers the possibility of creating sustainable in vivo models (120), but also opens the door to practical applications of regenerative medicine.
5.2.3.2 Antiparasitic Activity
As with all living organisms, parasites (Leishmania spp., Trypanosoma spp.) possess a kinome, although this is less developed than in the human body. The lower numbers of protein kinases present in these species play an important role in cell cycle progression and survival (121). For a few years, studies have been engaged to identify parasitic protein kinases as biological targets (122), for the development of potent therapeutic agents that are currently lacking to treat neglected tropical diseases (NTDs) (123). The structure of parasitic GSK-3 has been determined (124) and is marked as a valuable target (125) along with other kinases like CRK-3 (126), the parasitic parent of human CDKs. A screening of kinase inhibitors has revealed that indirubin derivatives possess interesting antiparasitic activity (127). Due to its demonstrated ability to interact with the ATP pocket of kinases, subsequently 6BIO (80) has later been evaluated in Leishmania donovani and identified as a potent CRK-3 inhibitor (128). As a result, 6BIO (80) was one of the most potent parasitic kinase inhibitors found and this has propelled indirubins as useful scaffold molecules for the study of NTDs.
5.2.3.3 Anticancer Activity
Among the 518 kinases composing the human kinome, more than 80% are serine/threonine kinases (129). Moreover, it is known than the ATP-site where indirubins bind is well conserved all over the kinome (129, 130). Investigations have shown that 6BIO (80) also inhibits several other kinases (131). Among its other targets, 6BIO (80) inhibited activities of JAKs in vitro and down-regulated constitutive activation of Stat3 signaling, associated with induction of apoptosis and suppression of tumor growth in vivo, in a human melanoma model study (132). In addition to these pharmacological effects on melanoma, 6BIO (80) induces apoptosis of aggressive breast tumor cells (133).
5.2.4 Novel Analogs of 6BIO
Novel 6BIO analogs containing extended amino-aliphatic chains on the 3′-oxime group have been designed in silico to accentuate the ATP mimicry. Their synthesis consists of two successive nucleophilic substitutions on the oxime (134). In a primary step, dibromoethane undergoes a first nucleophilic substitution by attack of the oxime in the presence of Et3N to form an intermediate (81). A second step involves the removal of the second bromine by the corresponding amines (Scheme 25). This versatile synthesis method then allowed the creation of a new assembly of 40 derivatives (134).
Enzyme inhibition assays showed substantial improvement of both potency and selectivity of the new analogs against GSK-3β. Furthermore, the secondary or tertiary amines could be converted to their corresponding salts (by addition of HCl/Et2O, Scheme 25) in order to enhance water solubility (134). These analogs exhibit strong antitumor activities against SH-SY5Y neuroblastoma cells (134) and a recent report highlights them as promising anti-Alzheimer’s agents by preventing phosphorylation of tau protein (135).
5.3 Indirubin Synthesis
As indirubin (9) was not chemically identified until the end of the nineteenth century and was not of equal importance for industrial development as indigotin (8), the development of methods for its synthesis was not a priority. However, as a natural by-product of indigo, indirubin (9) was first observed as a by-product formed during the synthesis of indigotin (8). Although Schunk had characterized the structure of this by-product and denominated it as “indirubin” (136), the synthesis of this bis-indole has required rather challenging chemistry to perform.
5.3.1 Baeyer Breakthrough
Baeyer first produced indirubin (9) by reduction of isatin (27) in 2-chloro-3H-indole-3-one (82), using phosphorus pentachloride (39, 40, 137) (Scheme 26). At that time, Baeyer noticed the formation of indigotin (8) in a low yield along with a red-colored product partially soluble in ethanol but not in water, which he called “indigo purpurin”.
A few years later, Baeyer carried out the coupling between indoxyl (26) and isatin (27) (138) and found the first access to the indirubin core, following therefore the biosynthesis pathway (Scheme 27). Nonetheless, indoxyl (26) was a difficult intermediate to synthesize as it exhibits considerable instability and dimerizes quickly to indigotin (8).
The significant advance that represented the two methodologies (activation of the 2-position of isatin and biomimetic pathways) developed by Baeyer laid the foundations for the development of more efficient pathways.
5.3.2 Activation of the 2-Position
5.3.2.1 The 2-Chloro-3H-indole-3-one Pathway
Although the first formation of indirubin (9) could be categorized as a “happy accident”, the reduction of isatin (27) by phosphorus pentachloride has nonetheless inspired chemists, who concentrated efforts towards the development of improved methodology. Indeed, Wahl (139) initially and Katritzky (140) later on successfully managed to control the formation of 2-chloro-3H-indol-3-one (82) from isatin (27). The intermediate was reacted directly with oxindole (29) in anhydrous pyridine (139) or toluene (140) to afford indirubin (9) in moderate to good yields (Scheme 28).
Scheme 28
Formation of indirubin through 2-chloro-3H-indol-3-one (82)
Although the activation of isatin (27) to 2-chloro-3H-indol-3-one (82) has been performed with relative success, the aforementioned intermediate is quite unstable and has to be used readily after its formation under drastic anhydrous conditions. Several attempts therefore have then been made in order to generate stabilized activated isatin (27). Wahl and collaborators have carried out the synthesis of a new type of intermediate through the formation of the corresponding silver salt (83). The latter is reacted with methyl iodide to furnish a stable 2-methoxy-3H-indol-3-one (84) (141). Finally, 84 is then condensed with oxindole (29) in acetic anhydride and dry benzene in the presence of a catalytic amount of sulfuric acid, to furnish indirubin (9) (Scheme 29). A second stabilization method later developed by Martinet consisted in the formation of a 2-phenyliminoisatin (85) (142) (Scheme 29). The intermediate is then reacted with isatin (27) to afford indirubin (9) under an atmosphere of H2S and in the presence of ammonia.
Later Albert defined a strategy involving the formation of a thioamide (86). The latter dimerizes to form indirubin (9) (65% yield) in the presence of sodium sulfite and sodium bicarbonate (143) (Scheme 30).
The use of isatin (27) to study the formation of indigoids has been of great value, and Ainley and Robinson in their efforts to synthesize epindoline derivatives proposed an elegant alternative to obtain indirubin (9) (144) (Scheme 31). The reaction between isatin (27) and o-nitrophenacyl bromide (87) in alkaline medium (EtONa) affords isatylidene-o-nitroacetophenone (88), probably by rearrangement of N-o- nitrophenacylisatin (89) under basic conditions. The isatylidene-o-nitroacetophenone (88) is then converted to indirubin (9) in an EtOH-ammonia solution or by reduction of the starting material using zinc dust. Despite the efforts of the authors, the mechanism of formation is not yet clearly understood.
Finally, a further method involves the reduction of isatin-3-oxime (90) with lithium aluminum hydride to form indirubin (9) (145) (44% yield), with the starting material being obtained from the condensation of isatin (27) and hydroxylamine (Scheme 32).
The biomimetic synthesis developed by Baeyer from indoxyl (26) indisputably has represented a major breakthrough and has been used by several researchers after its discovery (136, 146–148). However, the ability of indoxyl (26) to dimerize to yield indigotin (8) has led to the necessity of developing a more stable intermediate.
5.3.3.1 Stabilization at the 2-Position
Martinet in his studies on indole derivatives proposed an alternative to the iminoisatin previously developed. Indeed, he used the reactivity of the 2-position of indoxyl (26) to condense the latter with phenylhydrazine (91) to obtain a stable 2-phenylazoindoxyl (92) (149). The latter is then reduced under basic conditions and in the presence of sodium sulfite to afford indirubin (9) along with ammonia and aniline (Scheme 33).
5.3.3.2 Indoxyl Acetate: The Ultimate Intermediate
During the same period, Vorländer achieved the second major breakthrough by stabilizing the indoxyl with the synthesis of indoxyl diacetate on the one hand and indoxyl acetate (93) on the other (150). The latter is obtained from the esterification of indoxyl salt (94) in a degassed aqueous solution of acetic anhydride (Scheme 34). Indoxyl acetate (93) is then oxidized in the presence of hydrogen peroxide (30% solution) to form indirubin (9). This method inspired Spencer, who proposed an acidic hydrolysis using iron chloride (III) (151) (Scheme 34).
Finally, additional progress has been realized by Russell and Kaupp (46) by coupling indoxyl acetate (93) and isatin (27) in methanol in the presence of sodium carbonate. This pathway, as in Baeyer’s method, mimics the biosynthesis of indirubin (9) and has later been used widely for the generation of assemblies of derivatives (Scheme 35).
Scheme 35
Coupling between indoxyl acetate and isatin by Russell and Kaupp
Indigoids, including indirubins, have been obtained by feeding Polygonum tinctorium tissue cultures with indoles (152). Although this method was useful for elucidation of the biosynthesis mechanism of indigotin (8) and indirubin (9), its application for synthesis is, however, limited.
Considering the involvement of oxidative media in the natural generation of indigoids, the use of enzymes that oxidize indoles has come to the forefront. In this approach, mutant human cytochrome P450 (expressed as Escherichia coli-mediated oxidation has been performed using 5-methoxy- or 5-methylindole (153). This “eco-friendly” method has not only permitted the generation of indigotin (8) and indirubin (9), but it has provided also new analogs such as 5-methoxyindigo, 5′-methoxyindirubin, 5,5′-dimethoxyindigo, 5-methoxyindirubin, 5,5′-dimethoxyindirubin, 5-methylindirubin, and 5′-methylindirubin. This assembly was then evaluated for the inhibition of protein kinases (CDK1, CDK5, and GSK-3) and all the indirubin derivatives were more active than their parent compound, in terms of their preference for GSK-3. Despite its ecological benefits, this production procedure for indirubins suffers from a lack of selectivity.
Nevertheless, this enzymatic approach has inspired chemists, and a catalytic cobalt (II)-based oxidative degradation method was developed for indoles, leading to the selective formation of indigoids (Scheme 36) (154). Indeed, it is reported that the mechanism of oxidation by cytochrome P450 resembles chemical oxidation by hypervalent transition metal oxidants. The Co(II) complex was first prepared using glycine and 8-hydroxyquinoline as ligands (155) followed by the successive addition of indole and hydrogen peroxide. The decomposition of H2O2 generates the oxidative species (O2) expected to be involved in the study. The mixed ligand Co(II) complex and H2O2 led to the production of isatin (27), indigotin (8), and indirubin (9) in high yields and to the recovery of the catalyst.
Scheme 36
Catalytic decomposition of hydrogen peroxide and application to indigoid synthesis
5.4 Biological Applications of Synthetic Indirubins
5.4.1 Creation of Compound Assemblies
Due to the demonstrated importance of indirubins for biological studies and drug development, the necessity of developing large numbers of derivatives has become evident, in order to conduct structure-activity relationship studies (SAR). For quick and easy access, the methodology of Russell and Kaupp has been widely applied as it offers the maximum flexibility for the introduction of new substituents. Therefore, the dimerization of a correctly substituted isatin and the commercially available 3-acetoxyindole can lead to the synthesis of substituted indirubins. In this manner, a few indirubin analogs (89, 93, 98) and 30 new indirubins bearing chlorine, fluorine, iodine, or vinyl groups on the 6-position were obtained, but 5,6-disubstituted derivatives were synthesized initially (115). The selection of 5- or 7-substituted isatins opened the door to the creation of new libraries of synthetic analogs possessing diverse substituents at the 5- and 7-positions (156–160). Interestingly, the versatility of the method is conserved when starting from azaisatin or 3-acetoxy-5- or 6-substituted-indoles, leading, respectively, to azaindirubins (161, 162) and di-substituted indirubins (158, 163, 164). Overall, the general synthesis scheme can be represented as depicted in Scheme 37. Owing to this versatile synthesis method, the pharmacological benefits of indirubins have been explored rapidly as a result of this production method for promising indirubin analogs.
Scheme 37
General scheme of the synthesis of indirubin analogs
The 5- or 3′-positions of indirubin (9) can be modified for improving potency, selectivity, and solubility (93, 98). These chemically modified or substituted small molecules have been actively evaluated for their cytotoxic and antitumor activities using systems of purified enzymes in vitro, cell cultures, and in vivo models (89). Among synthetic indirubin analogs, indirubin-3′-monoxime (94) (IO, Fig. 23) and indirubin-5-sulfonate (95) (Fig. 18) show potent inhibitory activities against CDKs, and induce cell growth inhibition of human MCF-7 breast cancer cells (91, 92).
Several different analogs were synthesized based on the reactivity of the oxime. Indeed, under basic conditions, diverse side chains can be introduced at the 3′-position (e.g. glycerol or glucose) leading to the synthesis of derivative E804 (96) (Fig. 18) (93). Concerning the biological mechanism of action, a number of indirubin analogs potently block constitutively activated Stat3 signaling of human breast and prostate cancer cells. Particularly, in vitro biochemical kinase assay data suggest E804 containing a dihydroxypropyl 3′-oxime ether substituent as a potent inhibitor of Src kinase, down-regulating constitutively activated Stat3 or Stat5 in human breast cancer, CML, and T315I mutant Bcr-Abl expressing CML cells (93, 165). However, prolonged modulation against Src activity can cause a recovery of impaired Stat3 activation and thus tumor cell survival through altered JAK/Stat3 interaction (166, 167). This compensatory feedback implies that dual inhibition of JAKs and the Src family of kinases (SFKs) is probably a more favorable molecular target to inactivate downstream Stat3 signaling in cancer therapy (107, 168). On the basis of the previous findings, indirubin analog E738 (Fig. 18) was synthesized mainly to improve water solubility and bioavailability (91, 168). Unexpectedly, the small molecule E738 (97) appears to be a strong dual inhibitor of JAKs/Stat3 or SFKs/Stat3 signaling, associated with induction of apoptosis of human pancreatic cancer cells (107, 168).
5.4.3 7-Bromo-Indirubins
Isomers of 6BIO (80) have been developed with the introduction of bromine and other halogens in the 7-position (159). As for 6BIO, 7BIO (98) exerts a potent cytotoxic activity (131, 159) (Fig. 19). Surprisingly, the displacement of the bromine on the 7-position induces a shift in kinase selectivity. Indeed, 7-bromoindirubins have been proved to be potent Aurora B and C inhibitors (169). The aforementioned kinases are intimately involved in cell cycle regulation and are valuable biological targets for cancer cell killing. Furthermore, in vivo evidence suggests a water-soluble analog of 7-BIO (99), MLS-2438 (99) (Fig. 19), can suppress the survival of human melanoma cells associated with inhibition of Stat3 and Akt signaling.
The protein kinase DYRK1A is encoded by the DYRK1A gene localized on chromosome 21. It is involved in cell cycle progression and neurodegenerative diseases such as Down syndrome. This kinase has been poorly studied because of the lack of potent inhibitors. The promising scaffold provided by 7-BIO analogs resulted in the modification of the ring leading to the development of 7-bromo-5′- or 6′-substituted indirubins (163). Carboxylic acid, formyl, oxime, ethanol, and cyano groups have been introduced with success at the above-mentioned positions. Interestingly, the insertion of a tetrazole moiety (7-bromo-5′-tretazolindirubin (100)) as a bioisostere of the carboxylic acid has been carried out from the corresponding 7-bromo-5′-cyanoindirubin (101) using the stable trimethylsilyl azide (Scheme 38). To the present, this molecule is the first and only example of the introduction of an extra ring on the indirubin skeleton.
Scheme 38
Synthesis of the 7-bromo-5′-tetrazoloindirubin (100)
Moreover, a new series of 7-trifluoromethylindirubins has also been developed for biological investigation (Fig. 20). Interestingly, the presence of chemical entities on the positions 5′- and 6′- confers another type of kinase inhibition to the new analogs. In fact, di-substituted 7-BIO indirubins are potent and selective DYRK kinase inhibitors, and 7-bromo-3′-oximindirubin-5′-carboxylic acid (7-BIO-5′-COOH (102)) (Fig. 20) is the lead compound for this new series of inhibitors (163).
Interestingly, the binding mode of 7-BIO-5′-COOH was unexpected as the indirubin binds in an inverse fashion when compared to traditional anchoring (163) (Fig. 21). In this specific case, the bromine was exposed to both the solvent and the carboxylate group formed a salt bridge with the lysine 178 residue inside the cavity. Moreover, the traditional NH-CO-NH pattern was not entirely involved with binding as only the lactam ring components formed a hydrogen-bond with leucine 231. Hence, the new indirubins represent valuable biological tools to deepen knowledge on the role and the impact of DYRK kinases in the biological system.
Fig. 21
Zoom on the interactions of 102 in the DYRK cavity. (The pictures have been created from PDB file 3KVW using Maestro software, Academic version 9.3.5., Schrödinger Inc.)
Exploration of the 5-position was recently led to some success (170). Compounds such as 5-fluoro-indirubin-3′-oxime (103) (Fig. 22) have been recognized as potent FMS-like tyrosine kinase-3 (FLT3) kinase inhibitors (171) involved in cancer development and especially leukemia (172, 173).
As another novel family of synthetic indirubins, 5,5′-disubstituted indirubin-3′-oxime analogs, such as 5-nitro-5′-hydroxyindirubin-3′-oxime (104) (Fig. 20), also exhibit potent inhibition against CDK2/cyclin E in vitro, and in addition, an induction of apoptosis and in vivo efficacy (157, 164).
5.4.6 Conclusion
Although indirubins are minor compounds of ancient dyes, they provide interesting scaffolds with several applications in biology. These unexpected applications have allowed the discovery of powerful ATP-mimetic/ATP-competitive serine-threonine or tyrosine kinase inhibitors. Moreover, indirubins are modulators of GSK-3 signaling, which plays a key role of the maintenance of stem cell pluripotency. These crucial breakthroughs may contribute to the development of regenerative medicine therapies. Chemists involved with the design of indirubin derivatives might well observe that “a trip around the indirubin scaffold is a trip around the human kinome” (Fig. 23).
Fig. 23
Distribution of indirubin kinase selectivity around the human kinome
As mentioned in the first part of this contribution, the indigoid family includes natural halogenated-N-glycoside indigos. Akashins A, B, and C (20–22) have been isolated from Streptomyces sp GW48/1497 (12). Akashins are the first examples of natural 5,5′-disubstituted indigoids and N-glycoside indigos (even if synthetic compounds are included). As observed for their non-substituted parent molecule, akashins have low solubility in non-polar solvents even if the presence of the glycosidic residue enhances the solubility in methanol (but not in water). Nevertheless, akashins A, B, and C (20–22) possess cytotoxic activities against various human cancer cell lines (12) (colon carcinoma, melanoma, lung carcinoma, breast cancer, kidney tumor). Thus, their structures represent the first indigotin-based scaffolds to have been utilized in medicinal chemistry.
5.5.1.2 Synthesis of Akashins Derivatives
The challenging chemistry and the novelty of the structures of akashins led chemists to explore the total synthesis of their derivatives. Hein et al. first proposed an approach by O-glycosylation of N-benzylindigo (174). However, the key step consisting of a O → N rearrangement offers much less flexibility as it depends on the nature of the carbohydrate moiety (174) and its protective group. Therefore, the synthesis of protected pivaloyl-N-rhamnosylindigo (105) was achieved (Scheme 39), but the deprotection of the sugar moiety was unsuccessful.
Scheme 39
Synthesis of protected N-pivaloylrhamnosylindigo (105)
Later, the same group proposed a novel approach using the reactivity of dehydroindigo (106) (175). The latter was prepared from indigotin (8) by the oxidative addition of acetate groups on carbons C-2 and C-9 following by elimination of acetic acid in an alkaline medium. The reaction of dehydroindigo (106) with TMS protected l-rhamnoside and subsequent acetolysis provided N-(2,3,4-tri-O-acetyl-l-rhamnosyl)indigo (108). The deprotection of the sugar in the presence of a catalytic amount of t-BuONa in methanol finally afforded N-rhamnosylindigo (109) (Scheme 40a). This sequence of reactions has then been applied successfully to mannosyl and glycosyl derivatives (Scheme 40b). However, the total synthesis of the natural akashins still represents an interesting challenge for chemists.
In continuation of their efforts to obtain the akashins, Libnow et al. became involved in the synthesis of glycosidic indirubins.
5.5.2.1 Indirubin N-Glycosides
The investigators first proposed the synthesis of N-glycosylated indirubins (176), elegantly named as “red sugars”, adopting the Russell and Kaupp methodology. The key step relies on the synthesis of a N-glycosylation from aniline. The first step consists of the introduction of aniline at the anomeric position of rhamnose (110). After protecting the sugar moiety, the aniline is condensed in isatin (27) through an aluminum chloride-mediated cyclization in the presence of oxalyl chloride to form N-(2,3,4-tri-O-acetyl-α,β-l-rhamnopyranosyl)isatin (111). A pure sample of the β-isomer 112 was then reacted with indoxyl acetate, using an excess of sodium carbonate to ensure the deprotection of the rhamnose, leading to the desired N-(β-l-rhamnopyranosyl)indirubin (113) in a good yield (77%) (Scheme 41).
This efficient methodology has opened the way for the creation of a small assembly of derivatives (114–120, Fig. 24), including N-glucosyl- (e.g.117 and 118), N-galactosyl- (e.g.119 and 120), and N-mannosylindirubins (177). Moreover, some 5′-chloro-substituted indirubins (e.g.115), structurally closer to the akashins than previous analogs made, were also synthesized. The compounds were evaluated for their cytotoxic activities against four cell lines (5637, A-427, KYSE-70, MCF-7), giving the opportunity to perform a preliminary SAR analysis. Interestingly, the compounds exhibited a clear selectivity towards MCF-7 breast cancer cells. Furthermore, only the non-substituted N-rhamnosylindirubins exerted potent activity for MCT-5 cells, with IC50 values of between 0.67 and 0.76 μM.
Fig. 24
Assembly of N-glycosylindirubin derivatives developed by Libnow et al.
In order to extend their study of glycosyl indirubins, Libnow et al. succeeded in obtaining N′-glycosylindirubins by developing the first synthesis of N-glycosylated indoxyls (178). For this purpose, indoline (121) was first reacted with rhamnose. 1-N-rhamnosylindoline (122) was subjected to a DDQ-mediated dehydrogenation to provide the corresponding N-rhamnosylindole, which was directly protected (using BnBr (123) or MeI (124)). Next, the sugar-protected indole was used in the Arnold sequence consisting in the introduction of iodine in position-3 followed by its substitution using silver acetate to provide the protected N-rhamnosyl-3-acetoxyindole (125–126). The latter was condensed with isatin (27) using optimized conditions (piperidine, benzene) to form the protected N′-rhamnosylindirubin 127 and 128. Indeed, the application of the classical conditions (methanol, Na2CO3) resulted in the formation of complex mixtures due to the instability of the indoxyl under such conditions. Finally, the deprotection of the sugar in a sealed tube (the steric hindrance reduces the use of less drastic conditions) afforded for the first time the corresponding N′-rhamnosylindirubin (129) (Scheme 42).
Due to the positive biological attributes of indirubin (IO (94), especially), and as a result of its lack of solubility in biological media and slow metabolism in animals, the introduction of sugar moieties directly on the skeleton of this molecule is particularly attractive. However, the absence of substituents on nitrogen is critical for keeping the affinity of indirubin for the ATP-binding site of kinases. Eisenbrand and his group (179) were the first to explore the reactivity of the oxime at the 3′-position. Indirubin (9) was formed according to the method of Russell and Kaupp and IO (94) was obtained by action of hydroxylamine hydrochloride. Then, the coupling between acetylated O-chloroethylglucose (130) (prepared from an acetyl-protected glycosylfluoride (131)) and IO in basic conditions provided the corresponding monoxime ether containing an unprotected sugar moiety (132) (Scheme 43a). Another method (179) consists of a direct glycosylation of arabinose (133) using a haloalcohol and subsequent coupling reaction with IO (94) to form the corresponding indirubin-3′-(2-l-arabinopyranosyloxylethyl) oxime (134) (Scheme 43b). The major advantage of this procedure is the avoidance of a multi-step protection-deprotection sequence.
Finally, in order to increase the rate of metabolism of indirubin, it has been proved that the introduction of a methyl or a methoxy at the 5-position can be a valuable option. Moreover, after administration of 5-methylindirubin (135) (Fig. 25), the first metabolite identified was 5-methyl-6-hydroxyindirubin (136) (Fig. 25), showing increased solubility and consequent overall interest from a chemical point of view.
Considering the importance of glucuronide derivatives for cell penetration and metabolism (the glucuronide would be released in situ by action of glucuronidase), the synthesis of 6-glucuronated indirubins was conducted (Scheme 44). Thus, 6-hydroxy-5-methylisatin (137) was synthesized initially from 5-amino-2-methylphenol (138). After a first step of phenol and amine protection (with triisopropylsilyl and Boc groups), the protected compound was engaged in a metallocyclization (directed ortho-metallation) using diethyloxalate. The compound 6-hydroxy-5-methylisatin (137) was finally recovered after acidic hydrolysis. This derivative was then involved in a coupling reaction with indoxyl acetate to provide 136. The glycosylation step under Koenigs-Knorr conditions followed with the deprotection of the glycoside, and finally afforded 6-glucuronide-5-methylindirubin (139). This synthesis strategy has been patented (180) and the compound claimed as a potential anticancer candidate.
Scheme 44
Synthesis of 6-glucoronide-5-methylindirubin (139)
Isoindigo (11) has been considered for a long time as a by-product that alters the quality of indigo dye. Manufacturers (traditional approaches or the chemical industry) have used their skills to eliminate isoindigo (11) from their preparations. The extensive interest of indirubin (9) for its biological applications has stimulated medicinal chemists to pay more attention to this relatively forgotten member of the indigoid compound class.
5.6.1 Synthesis of Isoindigo
As described previously, major efforts have been directed towards the synthesis of indigotin (8) and indirubin (9). The first sustainable synthesis approach for isoindigo (11) was developed much later, when compared to indigotin (8) and indirubin (9). Wahl and his collaborator Bagard first proposed a quantitative synthesis starting from oxindole (29) in the presence of 2-methoxy-indol-3-one under acidic conditions (i.e. acetic acid and a catalytic amount of hydrochloric acid) (181, 182). This method was then improved with the replacement of 2-methoxy-indol-3-one by isatin (27) and now has been applied widely (16, 183–187). It now represents the most well-known approach to the synthesis of isoindigo (11) (Scheme 45).
Other investigators have proposed alternative pathways based on the activation of the 3-position of isatin with the introduction of diazomethane (188–190) or phosphine (191, 192) (for a subsequent Wittig reaction) or oxindole (29) with sulfine (193, 194) (using thionyl chloride), a disulfur bridge (3,3-oxindole dimers condensed under basic conditions) (195–198) and nitrososulfonate (199) (followed by acidic hydrolysis).
5.6.2 Isoindigo in Medicinal Chemistry
The side effects that were first observed after administration of natural indirubin (9), enticed medicinal chemists to develop new indigoid analogs, including isoindigo derivatives. However, as shown also for indirubin (9), isoindigo (11) has a low solubility in biological media, so the changes applied to its structure have been similar to those for indirubin (9).
5.6.2.1 Halogenated Isoindigo
The versatile chemical pathway developed some time ago by Wahl and Balard led to the synthesis of brominated isoindigo (182), but the investigators were not interested in evaluating biological activities at that time. Later on, several fluoro- (140), chloro- [(141) and (142)], and trifluoromethylisoindigo (143–145) (Fig. 26) analogs were synthesized and evaluated for their antiproliferative effects (186). The positive results obtained have highlighted the biological potential of this class of compounds, which were shown to be potent inducers of NAD(P)H-quinone oxidoreductase (NQO1).
In order to overcome the inherent solubility problems, glycosylisoindigo was developed. The lead compound in this series is Natura® (146). This N-glycosylisoindigo has been synthesized by Wang et al. in 2003 (200) from an acetyl-protected isatin N-glycoside. The latter is obtained by aluminum chloride-mediated ring closure from glycosyl aniline using oxalyl chloride (see Scheme 41). The reaction of N-glycosylisatin and with diverse oxindoles in acidic conditions converts the products into the desired isoindigo derivatives inclusive of N-methylated isoindigo (147) (Meisoindigo). Natura® and meisoindigo (Fig. 27) exerted good cytotoxicity towards various cancer cell lines. Interestingly, the acetyl-protected glycosylisoindigo was more active than its deprotected analog.
Based on those interesting results, Sassateli et al. have generated later an assembly of diversely substituted derivatives of glycosylisoindigo (Fig. 28) and have evaluated their antiproliferative activities (201–203).
Fig. 28
General structure of the chemical assembly generated by Sassateli et al.
A final group of compounds in this class was synthesized as previously described even though access to the N-glycosylation was redesigned. Starting from a substituted indoline (148), the investigators introduced the glucose moiety (149) and built the isatin (150) by successive oxidation of the 2,3-positions (DDQ then CrO3, Scheme 46). The coupling between the aforementioned isatin (150) and oxindole (29) followed by the deprotection of the sugar finally led to substituted isoindigo derivatives (151). These derivatives were then evaluated for their cytotoxic potency and exhibited moderate inhibitory activities.
Scheme 46
Novel access to N-glycosylation applied to N-glycosylisoindigo synthesis
The access to more soluble analogs has been a driving force in the medicinal chemistry of indigoids. Therefore, the same team proposed the synthesis of the new 7′-azaisoindigo (Scheme 47), which was diversely substituted on both aromatic parts of the molecule (Fig. 29) (204, 205).
To fulfill this objective, the corresponding 7-azaoxindoles have been prepared from 7-azaindole (152) (204) using pyridinium bromide perbromide (PBPB) following by catalytic hydrogenation. 7-Azaoxindole (153) has then served as a scaffold for the two-step introduction of bromine at the 5-position to form 5-bromo-7-azaoxindole (154). The investigators developed also various substituted oxindoles from simple oxindoles (e.g.155–156), the oxidation of indoles (e.g.157) or the selective reduction of isatins (e.g.158). The intermediates were then coupled with unsubstituted or substituted isatins to generate the resultant compound assembly.
Interestingly, the 5′-iodo-7′-azaisoindigo (159) prepared has been used lately as a key intermediate for a Sonogashira cross-coupling (205) in order to introduce various vinyl groups (Scheme 48).
Scheme 48
Sonogashira cross-coupling products from 5-iodo-7′-azaisoindigo (159)
As depicted previously, meisoindigo (147) (1-methylisoindigo) possesses an interesting cytotoxic profile, which has led later to the development of a new series of N-arylisoindigo derivatives (160) (185). The targeted compounds were synthesized using a microwave-assisted coupling reaction between a N-substituted isatin and oxindole (29) (Scheme 49). The introduction of diverse aryl substituents has been performed by simple nucleophilic substitution between isatin (27) and the corresponding halides under microwave irradiation. However, the target compounds exhibited low to moderate cytotoxic potency.
“Progress in the Chemistry of Organic Natural Compounds” is an excellent medium to document developments made in the investigation of the indigoid family of natural products. Dyes containing indigoids have been used in societies all around the world to symbolize power (Tyrian purple) and religious seniority (indigo, Tyrian Purple), but also as the color of both working class (indigo) and fashionable clothes (indigo). The commercial and industrial importance of these stimulated early chemists to develop skills to perform the sustainable synthesis of indigoids at low-cost that led to substantial benefits. Approaches to the synthesis of these compounds progressed alongside the concomitant availability of cheap starting materials, first from nitrobenzene, leading eventually biomimetic synthesis involving indoles and isatins. The complexity of the natural production of indigoids interested biochemists, who applied knowledge in their field to assist chemists in understanding their mechanism of formation. Chemists then acquired the ability to synthesize indigo and indirubin using enzymes (e.g. plant tissue cultures or cytochrome P450) or by developing chemical catalytic systems based on enzyme functions (e.g. cobalt-mediated oxidation).
Although indigoids were related closely to the dye chemistry, studies on traditional Chinese medicine led to the unexpected discovery that indigoids have the ability to alleviate the symptoms of certain types of leukemia, thus projecting interest in this class of natural products and their analogs in the field of medicinal chemistry. The indirubins and bromoindirubins became useful lead compounds in the development of selective protein kinases inhibitors. The availability of simple and versatile synthesis methods allowed the creation of a large assembly of indirubin derivatives (more than 400 compounds), providing an important group of bioactive substances to investigate in various biological activities, such as their effects on stem cells, in parasitology, and their potential use for cancer. Although indigotin is not an active compound, the discovery of akashins and the synthesis of their analogs has enhanced scientific interest in the indigoids. This work involved the development of various derivatives of isoindigo, a compound for a long time considered mainly as by-product that tended to alter the quality of the indigo dye. Moreover, the fascinating electronic structures of indigoids have been applied in material chemistry as new conductors (not covered in this review), and isoindigo-based polymers could serve as key components for solar panels in the future. In fact, indigoids have a place in many diverse branches of science, including organic chemistry, medicinal chemistry, material and physical chemistry, dye chemistry, biology, and textile science as well as the pictorial arts. There is no doubt that, in the future, members of the colorful family of indigoids will continue to have promising future applications for the benefit of mankind!
References
Henshilwood C, d’Errico F, Vanhaeren M, van Niekerk K, Jacobs Z (2004) Middle Stone Age Shell Beads from South Africa. Science 304:404
Jurasekova Z, del Puerto E, Bruno G, García-Ramos JV, Sanchez-Cortes S, Domingo C (2010) Extractionless Non-Hydrolysis Surface-Enhanced Raman Spectroscopic Detection of Historical Mordant Dyes on Textile Fibers. J Raman Spectrosc 41:1455
Cerrato A, De Santis D, Moresi M (2002) Production of Luteolin Extracts from Reseda luteola and Assessment of Their Dyeing Properties. J Sci Food Agric 82:1189
Karapanagiotis I, de Villemereuil V, Magiatis P, Polychronopoulos P, Vougogiannopoulou K, Skaltsounis AL (2006) Identification of the Coloring Constituents of Four Natural Indigoid Dyes. J Liq Chromatogr Relat Technol 29:1491
Surowiec I, Nowik W, Moritz T (2012) Mass Spectrometric Identification of New Minor Indigoids in Shellfish Purple Dye from Hexaplex trunculus. Dyes Pigm 94:363
Maskey RP, Grün-Wollny I, Fiebig HH, Laatsch H (2002) Akashins A, B, and C: Novel Chlorinated Indigoglycosides from Streptomyces sp. GW 48/1497. Angew Chem Int Ed 41:597
Klessinger M, Lüttke W (1963) Theoretische und Spektroskopische Untersuchungen an Indigo-Farbstoffen: Das Chromophore System der Indigo-Farbstoffe. Tetrahedron 19, Supplement 2:315
Jacquemin D, Preat J, Wathelet V, Perpete EA (2006) Substitution and Chemical Environment Effects on the Absorption Spectrum of Indigo. J Chem Phys 124:074104
Christie RM (2006) Indirubin: The Relationship Between Chemical Structure and Physical Properties, With Particular Reference to Color. In: Indirubin, The Red Shade of Indigo. Life in Progress Editions. Station Biologique, Roscoff, France, p 103
Yao M, Araki M, Senoh H, Yamazaki S-i, Sakai T, Yasuda K (2010) Indigo Dye as a Positive-Electrode Material for Rechargeable Lithium Batteries. Chem Lett 39:950
Irimia-Vladu M, Głowacki ED, Troshin PA, Schwabegger G, Leonat L, Susarova DK, Krystal O, Ullah M, Kanbur Y, Bodea MA, Razumov VF, Sitter H, Bauer S, Sariciftci NS (2012) Indigo - A Natural Pigment for High Performance Ambipolar Organic Field Effect Transistors and Circuits. Adv Mater 24:375
Ensley BD, Ratzkin BJ, Osslund TD, Simon MJ, Wackett LP, Gibson DT (1983) Expression of Naphthalene Oxidation Genes in Escherichia coli Results in the Biosynthesis of Indigo. Science 222:167
Ricketts R (2006) Polygonum tinctorium: Contemporary Indigo Farming and Processing in Japan. In: Indirubin, The Red Shade of Indigo. Life in Progress Editions. Station Biologique, Roscoff, France, p 7
Zech-Matterne V, Leconte L (2010) New Archaeobotanical Finds of Isatis tinctoria L. (Woad) from Iron Age Gaul and a Discussion of The Importance of Woad in Ancient Time. Veg Hist Archaeobot 19:137
Gilbert KG, Hill DJ, Crespo C, Mas A, Lewis M, Rudolph B, Cooke DT (2000) Qualitative Analysis of Indigo Precursors from Woad by HPLC and HPLC-MS. Phytochem Anal 11:18
Oberthür C, Schneider B, Graf H, Hamburger M (2004) The Elusive Indigo Precursors in Woad (Isatis tinctoria L.) – Identification of the Major Indigo Precursor, Isatan A, and a Structure Revision of Isatan B. Chem Biodiversity 1:174
Oberthür C, Graf H, Hamburger M (2004) The Content of Indigo Precursors in Isatis tinctoria Leaves‚ a Comparative Study of Selected Accessions and Post-Harvest Treatments. Phytochemistry 65:3261
Garcia-Macias P, John P (2004) Formation of Natural Indigo Derived from Woad (Isatis tinctoria L.) in Relation to Product Purity. J Agric Food Chem 52:7891
Laitonjam WS, Wangkheirakpam SD (2011) Comparative Study of the Major Components of the Indigo Dye Obtained from Strobilanthes flaccidifolius Nees. and Indigofera tinctoria Linn. Int J Plant Phys Biochem 3:108
Chanayath N, Lhieochaiphant S, Phutrakul S (2002) Pigment Extraction Techniques from the Leaves of Indigofera tinctoria Linn. and Baphicacanthus cusia Brem. and Chemical Structure Analysis of Their Major Components. CMJ Journal 1:149
Holt SJ, Sadler PW (1958) Studies in Enzyme Cytochemistry. III. Relationships between Solubility, Molecular Association and Structure in Indigoid Dyes. Proc R Soc Lond B Biol 148:495
Russell GA, Kaupp G (1969) Reactions of Resonance Stabilized Carbanions. XXXI. Oxidation of Carbanions. 4. Oxidation of Indoxyl to Indigo in Basic Solution. J Am Chem Soc 91:3851
Haubrichs R (2006) Natural History and Iconography of Purple Shells. In: Indirubin, The Red Shade of Indigo. Life in Progress Editions. Station Biologique, Roscoff, France, p 55
Sotiropoulou S (2004) La Pourpre dans l’Art Cycladique: Identification du Pigment dans les Peintures Murales d’Akrotiri (Théra, Grèce). Preist Alp 40:167
Sotiropoulou S, Karapanagiotis I (2006) Conchylian Purple Investigation in Prehistoric Wall Paintings of the Aegean Area. In: Indirubin, The Red Shade of Indigo. Life in Progress Editions. Station Biologique, Roscoff, France, p 71
Cole W (1685) A Letter from Mr William Cole of Bristol, to the Phil. Society of Oxford; Containing His Observations on the Purple Fish. Phil Trans 15:1278
Terada T (2008) Paper 138: Sea Snails in Contemporary Japanese Embroidery. In: American Textiles Society Symposium Proceedings, Textile Society of America 11th Biennial Symposium, Honolulu, Hawaii, USA, 4–7 September 2008, p 1
Michel RH, Lazar J, McGovern PE (1992) The Chemical Composition of the Indigoid Dyes Derived from The Hypobranchial Glandular Secretions of Murex Molluscs. J Soc Dyers Colour 108:145
Meijer L, Skaltsounis A-L, Magiatis P, Polychronopoulos P, Knockaert M, Leost M, Ryan XP, Vonica CA, Brivanlou A, Dajani R, Crovace C, Tarricone C, Musacchio A, Roe SM, Pearl L, Greengard P (2003) GSK-3 Selective Inhibitors Derived from Tyrian Purple Indirubins. Chem Biol 10:1255
Clark RJH, Cooksey CJ (1997) Bromoindirubins: The Synthesis and Properties of Minor Components of Tyrian Purple and The Composition of the Colorant from Nucella lapillus. J Soc Dyers Colour 113:316
Baker JT, Sutherland MD (1968) Pigments of Marine Animals: VIII. Precursors of 6,6′-Dibromoindigotin (Tyrian Purple) from the Mollusc Dicathais orbita Gmelin. Tetrahedron Lett 9:43
Baker JT, Duke CC (1973) Isolation from Marine Mollusks Hypobranchial Glands of 6-Bromo-2,2-bis(Methylthio)indolin-3-one and 6-Bromo-2-(methylthio)indoleninone as Alternative Precursors to Tyrian Purple. Tetrahedron Lett 27:2481
Baker JT, Duke CC (1976) Isolation of Choline and Choline Ester Salts of Tyrindoxyl Sulfate from the Marine Molluscs, Dicathais orbita and Mancinella keineri. Tetrahedron Lett 15:1233
Westley C, Benkendorff K (2008) Sex-Specific Tyrian Purple Genesis: Precursor and Pigment Distribution in the Reproductive System of the Marine Mollusc Dicathais orbita. J Chem Ecol 34:44
Westley CB, Vine KL, Benkendorff K (2006) A Proposed Functional Role for Indole Derivatives in Reproduction and Defence of the Muricidae (Neogastropoda: Mollusca) In: Indirubin, The Red Shade of Indigo. Life in Progress Editions. Station Biologique, Roscoff, France, p 31
Benkendorff K, Bremner J, Davis A (2000) Tyrian Purple Precursors in the Egg Masses of the Australian Muricid, Dicathais orbita: A Possible Defensive Role. J Chem Ecol 26:1037
Michel RH, McGovern PE (1990) The Chemical Processing of Royal Purple Dye: Ancient Descriptions as Elucidated by Modern Science, Part II. Archeomaterials 4:97
Voß G, Gerlach H (1989) Regioselektiver Brom/Lithium-Austausch bei 2,5-Dibrom-1-Nitrobenzol. – Eine Einfache Synthese von 4-Brom-2-Nitrobenzaldehyd und 6,6′-Dibromindigo. Chem Ber 122:1199
Majima R, Kotake M (1930) Synthetische Versuche in der Indol-Gruppe, VII: Über Nitrieren und Bromieren des β-Indol-Carbonsäure-Esters und Eine Neue Synthese des Farbstoffs des Antiken Purpurs. Ber Chem Ges 63:2237
Tanoue Y, Terada A, Sakata K, Hashimoto M, Morishita S-I, Hamada M, Kai N (2001) A Facile Synthesis of Tyrian Purple Based on a Biosynthetic Pathway. Fish Sci 67:726
Hoessel R, Leclerc S, Endicott JA, Nobel MEM, Lawrie A, Tunnah P, Leost M, Damiens E, Marie D, Marko D, Niederberger E, Tang W, Eisenbrand G, Meijer L (1999) Indirubin, the Active Constituent of a Chinese Antileukaemia Medicine, Inhibits Cyclin-Dependent Kinases. Nat Cell Biol 1:60
Damiens E, Baratte B, Marie D, Eisenbrand G, Meijer L (2001) Anti-Mitotic Properties of Indirubin-3′-monoxime, a CDK/GSK-3 Inhibitor: Induction of Endoreplication Following Prophase Arrest. Oncogene 20:3786
Marko D, Schatzle S, Friedel A, Genzlinger A, Zankl H, Meijer L, Eisenbrand G (2001) Inhibition of Cyclin-Dependent Kinase 1 (CDK1) by Indirubin Derivatives in Human Tumour Cells. Br J Cancer 84:283
Leclerc S, Garnier M, Hoessel R, Marko D, Bibb JA, Snyder GL, Greengard P, Biernat J, Wu Y-Z, Mandelkow E-M, Eisenbrand G, Meijer L (2001) Indirubins Inhibit Glycogen Synthase Kinase-3β and CDK5/P25, Two Protein Kinases Involved in Abnormal Tau Phosphorylation in Alzheimer’s Disease: A Property Common to Most Cyclin-Dependent Kinase Inhibitors? J Biol Chem 276:251
Davies TG, Tunnah P, Meijer L, Marko D, Eisenbrand G, Endicott JA, Noble MEM (2001) Inhibitor Binding to Active and Inactive CDK2: The Crystal Structure of CDK2-Cyclin A/Indirubin-5-sulfonate. Structure 9:389
Vougogiannopoulou K, Skaltsounis AL (2012) From Tyrian Purple to Kinase Modulators: Naturally Halogenated Indirubins and Synthetic Analogues. Planta Med 78:1515
Martinez A, Castro A, Dorronsoro I, Alonso M (2002) Glycogen Synthase Kinase-3 (GSK-3) Inhibitors as New Promising Drugs for Diabetes, Neurodegeneration, Cancer, and Inflammation. Med Res Rev 22:373
Mondragón-Rodríguez S, Perry G, Zhu X, Moreira PI, Williams S (2012) Glycogen Synthase Kinase 3: A Point of Integration in Alzheimer’s Disease and a Therapeutic Target? Int J Alzheimer’s Dis 2012:4
Picchini AM, Manji HK, Gould TD (2004) GSK-3 and Neurotrophic Signaling: Novel Targets Underlying The Pathophysiology and Treatment of Mood Disorders? Drug Discov Today Dis Mech 1:419
Freyberg Z, Ferrando SJ, Javitch JA (2010) Roles of the Akt/GSK-3 and Wnt Signaling Pathways in Schizophrenia and Antipsychotic Drug Action. Am J Psychiat 167:388
Takenaka K, Kise Y, Miki H (2007) GSK-3beta Positively Regulates Hedgehog Signaling Through Sufu in Mammalian Cells. Biochem Biophys Res Commun 353:501
Coni S, Infante P, Gulino A (2013) Control of Stem Cells and Cancer Stem Cells by Hedgehog Signaling: Pharmacologic Clues from Pathway Dissection. Biochem Pharmacol 85:623
Polychronopoulos P, Magiatis P, Skaltsounis A-L, Myrianthopoulos V, Mikros E, Tarricone A, Musacchio A, Roe SM, Pearl L, Leost M, Greengard P, Meijer L (2004) Structural Basis for the Synthesis of Indirubins as Potent and Selective Inhibitors of Glycogen Synthase Kinase-3 and Cyclin-Dependent Kinases. J Med Chem 47:935
Martin L, Magnaudeix A, Esclaire F, Yardin C, Terro F (2009) Inhibition of Glycogen Synthase Kinase-3β Downregulated Total Tau Proteins in Cultured Neurons and Its Reversal by the Blockade of Protein Phosphatase-2A. Brain Res 1252:66
Knockaert M, Blondel M, Bach S, Leost M, Elbi C, Hager GL, Nagy SR, Han D, Denison M, Ffrench M, Ryan XP, Magiatis P, Polychronopoulos P, Greengard P, Skaltsounis L, Meijer L (2004) Independent Actions on Cyclin-Dependent Kinases and Aryl Hydrocarbon Receptor Mediate the Antiproliferative Effects of Indirubins. Oncogene 23:4400
Sato N, Sanjuan IM, Heke M, Uchida M, Naef F, Brivanlou AH (2003) Molecular Signature of Human Embryonic Stem Cells and Its Comparison with the Mouse. Dev Biol 260:404
Sato N, Meijer L, Skaltsounis L, Greengard P, Brivanlou AH (2004) Maintenance of Pluripotency in Human and Mouse Embryonic Stem Cells Through Activation of Wnt Signaling by a Pharmacological GSK-3-Specific Inhibitor. Nat Med 10:55
Sato H, Amagai K, Shimizukawa R, Tamai Y (2009) Stable Generation of Serum- and Feeder-Free Embryonic Stem Cell-Derived Mice with Full Germline-Competency by Using a GSK-3 Specific Inhibitor. Genesis 47:414
Hassan P, Fergusson D, Grant KM, Mottram JC (2001) The CRK3 Protein Kinase is Essential for Cell Cycle Progression of Leishmania mexicana. Mol Biochem Parasitol 113:189
Xingi E, Smirlis D, Myrianthopoulos V, Magiatis P, Grant KM, Meijer L, Mikros E, Skaltsounis A-L, Soteriadou K (2009) 6-Br-5-Methylindirubin-3′-oxime (5-Me-6-BIO) Targeting the Leishmanial Glycogen Synthase Kinase-3 (GSK-3) Short Form Affects Cell-Cycle Progression and Induces Apoptosis-Like Death: Exploitation of GSK-3 for Treating Leishmaniasis. Int J Parasitol 39:1289
Liu L, Nam S, Tian Y, Yang F, Wu J, Wang Y, Scuto A, Polychronopoulos P, Magiatis P, Skaltsounis L, Jove R (2011) 6-Bromoindirubin-3′-oxime Inhibits JAK/STAT3 Signaling and Induces Apoptosis of Human Melanoma Cells. Cancer Res 71:3972
Nicolaou KA, Liapis V, Evdokiou A, Constantinou C, Magiatis P, Skaltsounis AL, Koumas L, Costeas PA, Constantinou AI (2012) Induction of Discrete Apoptotic Pathways by Bromo-Substituted Indirubin Derivatives in Invasive Breast Cancer Cells. Biochem Biophys Res Commun 425:76
Vougogiannopoulou K, Ferandin Y, Bettayeb K, Myrianthopoulos V, Lozach O, Fan Y, Johnson CH, Magiatis P, Skaltsounis A-L, Mikros E, Meijer L (2008) Soluble 3′,6-Substituted Indirubins with Enhanced Selectivity toward Glycogen Synthase Kinase-3 Alter Circadian Period. J Med Chem 51:6421
Martin L, Magnaudeix A, Wilson CM, Yardin C, Terro F (2011) The New Indirubin Derivative Inhibitors of Glycogen Synthase Kinase-3, 6-BIDECO and 6-BIMYEO, Prevent Tau Phosphorylation and Apoptosis Induced by the Inhibition of Protein Phosphatase-2A by Okadaic Acid in Cultured Neurons. J Neurosci Res 89:1802
Giovannini E, Lorenz T (1957) Reduktionen mit LiAlH4 in der Isatin-Reihe. 2. Mitteilung. Einwirkung von LiAlH4 auf Indol- und Isatin-Derivate. Helv Chim Acta 40:2287
Das PJ, Baruah A (2011) Oxidative Degradation of Indole Using Mixed Ligand Co(II) Complex and Hydrogen Peroxide. Selective Formation of Indole Dimers. Chem Biol Interface 1:355
Shivankar VS, Thakkar NV (2005) Decomposition of Hydrogen Peroxide in Presence of Mixed Ligand Cobalt(II) and Nickel(II) Complexes as Catalysts. J Sci Ind Res 64:496
Kim S-A, Kim Y-C, Kim S-W, Lee S-H, Min J-J, Ahn S-G, Yoon J-H (2007) Antitumor Activity of Novel Indirubin Derivatives in Rat Tumor Model. Clin Cancer Res 13:253
Moon MJ, Lee SK, Lee J-W, Song WK, Kim SW, Kim JI, Cho C, Choi SJ, Kim Y-C (2006) Synthesis and Structure-Activity Relationships of Novel Indirubin Derivatives as Potent Anti-Proliferative Agents with CDK2 Inhibitory Activities. Bioorg Med Chem 14:237
Beauchard A, Ferandin Y, Frère S, Lozach O, Blairvacq M, Meijer L, Thiéry V, Besson T (2006) Synthesis of Novel 5-Substituted Indirubins as Protein Kinases Inhibitors. Bioorg Med Chem 14:6434
Ferandin Y, Bettayeb K, Kritsanida M, Lozach O, Polychronopoulos P, Magiatis P, Skaltsounis A-L, Meijer L (2006) 3′-Substituted 7-Halogenoindirubins, a New Class of Cell Death Inducing Agents. J Med Chem 49:4638
Lee J-W, Moon MJ, Min H-Y, Chung H-J, Park E-J, Park HJ, Hong J-Y, Kim Y-C, Lee SK (2005) Induction of Apoptosis by a Novel Indirubin-5-nitro-3′-monoxime, a CDK Inhibitor, in Human Lung Cancer Cells. Bioorg Med Chem Lett 15:3948
Wang ZH, Dong Y, Wang T, Shang MH, Hua WY, Yao QZ (2010) Synthesis and CDK2 Kinase Inhibitory Activity of 7/7′-Azaindirubin Derivatives. Chin Chem Lett 21:297
Kritsanida M, Magiatis P, Skaltsounis A-L, Peng Y, Li P, Wennogle LP (2009) Synthesis and Antiproliferative Activity of 7-Azaindirubin-3′-oxime, a 7-Aza Isostere of the Natural Indirubin Pharmacophore. J Nat Prod 72:2199
Myrianthopoulos V, Kritsanida M, Gaboriaud-Kolar N, Magiatis P, Ferandin Y, Durieu E, Lozach O, Cappel D, Soundararajan M, Filippakopoulos P, Sherman W, Knapp S, Meijer L, Mikros E, Skaltsounis A-L (2013) Novel Inverse Binding Mode of Indirubin Derivatives Yields Improved Selectivity for DYRK Kinases. ACS Med Chem Lett 4:22
Choi S-J, Lee J-E, Jeong S-Y, Im I, Lee S-D, Lee E-J, Lee SK, Kwon S-M, Ahn S-G, Yoon J-H, Han S-Y, Kim J-I, Kim Y-C (2010) 5,5′-Substituted Indirubin-3′-oxime Derivatives as Potent Cyclin-Dependent Kinase Inhibitors with Anticancer Activity. J Med Chem 53:3696
Sen B, Saigal B, Parikh N, Gallick G, Johnson FM (2009) Sustained Src Inhibition Results in Signal Transducer and Activator of Transcription 3 (STAT3) Activation and Cancer Cell Survival via Altered Janus-Activated Kinase‚ STAT3 Binding. Cancer Res 69:1958
Johnson FM, Saigal B, Tran H, Donato NJ (2007) Abrogation of Signal Transducer and Activator of Transcription 3 Reactivation after Src Kinase Inhibition Results in Synergistic Antitumor Effects. Clin Cancer Res 13:4233
Nam S, Wen W, Schroeder A, Herrmann A, Yu H, Cheng X, Merz KH, Eisenbrand G, Li H, Yuan YC, Jove R (2013) Dual Inhibition of Janus and Src Family Kinases by Novel Indirubin Derivative Blocks Constitutively-Activated STAT3 Signaling Associated with Apoptosis of Human Pancreatic Cancer Cells. Mol Oncol 7:369
Myrianthopoulos V, Magiatis P, Ferandin Y, Skaltsounis A-L, Meijer L, Mikros E (2007) An Integrated Computational Approach to the Phenomenon of Potent and Selective Inhibition of Aurora Kinases B and C by a Series of 7-Substituted Indirubins. J Med Chem 50:4027
Choi SJ, Moon MJ, Lee SD, Choi S-U, Han S-Y, Kim Y-C (2010) Indirubin Derivatives as Potent FLT3 Inhibitors with Anti-Proliferative Activity of Acute Myeloid Leukemic Cells. Bioorg Med Chem Lett 20:2033
Han S-Y, Ahn JH, Shin CY, Choi S-U (2010) Effects of Indirubin Derivatives on the FLT3 Activity and Growth of Acute Myeloid Leukemia Cell Lines. Drug Dev Res 71:221
Hein M, Phuong NTB, Michalik D, Görls H, Lalk M, Langer P (2006) Synthesis of the First Deprotected Indigo N-Glycosides (Blue Sugars) by Reductive Glycosylation of Dehydroindigo. Tetrahedron Lett 47:5741
Libnow S, Methling K, Hein M, Michalik D, Harms M, Wende K, Flemming A, Köckerling M, Reinke H, Bednarski PJ, Lalk M, Langer P (2008) Synthesis of Indirubin-N′-Glycosides and Their Anti-Proliferative Activity Against Human Cancer Cell Lines. Bioorg Med Chem 16:5570
Libnow S, Hein M, Langer P (2009) The First N-Glycosylated Indoxyls and Their Application to the Synthesis of Indirubin-N-Glycosides (Purple Sugars). Synlett 2009:221
Papageorgiou C, Borer X (1988) Acid-Catalyzed Rearrangements for a Diastereoselective Entry into a New Fused Hexacyclic Heterocycle: (5RS,7aRS,12RS,14aRS)-4,5,7,7a,11,12,14,14a-Octahydro-5,12-dimethyldiindo[1,7-bc:1′,7′-gh][2,6]naphthyridine. Helv Chim Acta 71:1079
Menozzi C, Dalko PI, Cossy J (2007) Studies on Enol Carbonate Chemistry: Stereoselective Construction of Vicinal Quaternary Benzylic Centers in the Bis-Oxindole Series. Heterocycles 72:199
Wee XK, Yeo WK, Go M-L, Zhang B, Tan VBC, Lim KM, Tay TE (2009) Synthesis and Evaluation of Functionalized Isoindigos as Antiproliferative Agents. Bioorg Med Chem 17:7562
Zhang W, Go M-L (2009) Functionalized 3-Benzylidene-indolin-2-ones: Inducers of NAD(P)H-Quinone Oxidoreductase 1 (NQO1) with Antiproliferative Activity. Bioorg Med Chem 17:2077
Kloeck C, Jin X, Choi K, Khosla C, Madrid PB, Spencer A, Raimundo BC, Boardman P, Lanza G, Griffin JH (2011) Acylideneoxoindoles: A New Class of Reversible Inhibitors of Human Transglutaminase 2. Bioorg Med Chem Lett 21:2692
Sassatelli M, Bouchikhi F, Messaoudi S, Anizon F, Debiton E, Barthomeuf C, Prudhomme M, Moreau P (2006) Synthesis and Antiproliferative Activities of Diversely Substituted Glycosyl-Isoindigo Derivatives. Eur J Med Chem 41:88
Sassatelli M, Bouchikhi F, Aboab B, Anizon F, Fabbro D, Prudhomme M, Moreau P (2007) In Vitro Antiproliferative Activities and Kinase Inhibitory Potencies of Glycosyl-Isoindigo Derivatives. Anti-Cancer Drugs 18:1069
Bouchikhi F, Anizon F, Moreau P (2009) Synthesis, Kinase Inhibitory Potencies and in Vitro Antiproliferative Activity of Isoindigo and 7′-Azaisoindigo Derivatives Substituted by Sonogashira Cross-Coupling. Eur J Med Chem 44:2705
The authors would like to thank Mr. Tristan Yvon (Archeologist, Guadeloupe) for interesting discussions about the production of indigo in the French West Indies, Ms. Inge Boesken Kanold, an artist residing in France, for passionate discussions on the use of Tyrian Purple as a pigment for paintings, and, last but not least, Ms. Laure Bénard for her support and tolerance. This work was supported by the Commission of the European Community through the INsPiRE project (EU-FP7- REGPOT-2011-1, proposal 284460).
Author information
Authors and Affiliations
Department of Pharmacognosy and Natural Products Chemistry, School of Pharmacy, University of Athens, Panepistimiopoulis Zografou, GR-15771, Athens, Greece
Nicolas Gaboriaud-Kolar & Alexios-Leandros Skaltsounis
Molecular Medicine, Beckman Research Institute, City of Hope Comprehensive Cancer Center, 1500 East Duarte Road, Duarte, CA, 91010, USA
Gaboriaud-Kolar, N., Nam, S., Skaltsounis, AL. (2014). A Colorful History: The Evolution of Indigoids.
In: Kinghorn, A., Falk, H., Kobayashi, J. (eds) Progress in the Chemistry of Organic Natural Products 99. Progress in the Chemistry of Organic Natural Products, vol 99. Springer, Cham. https://doi.org/10.1007/978-3-319-04900-7_2


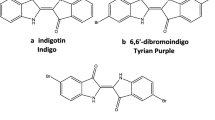



















{kind=link}
{kind=link}
{kind=link}



























































{kind=link}