
Abstract
The data on 6-fluorо-1,4-dihydroquinolin-4-oxo-3-carboxylic acids and their structural analogues accumulated in the literature for the last 10–15 years are reviewed. Synthetic approaches to the quinolone system, as well as all kind of structural modifications by incorporating substituents into 1–8 positions or by means of annelation have been discussed. The “structure-activity” relationships for antibacterial fluoroquinolones, as well as the data on other types of biological activity for the family of bi- and polycyclic fluoroquinolones are presented. The formation of complexes of fluoroquinolones with metals and their applications have been considered. The bibliography – 377 references.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others
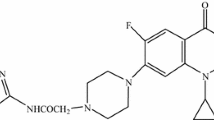

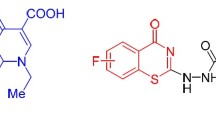
Keywords
1 Introduction
Nearly three decades passed since the time when the first representatives of the fluoroquinolone family of antibacterials, such as norfloxacin, pefloxacin, ciprofloxacin and ofloxacin had appeared in the world pharmaceutical market (Scheme 1).

Structure of some fluoroquinolone antibacterials
It is worth mentioning that the first drug in the series of quinolones, nalidixic acid (1-ethyl-1,4-dihydro-7-methyl-4-oxo-1,8-naphthyridin-3-carboxylic acid), bearing no fluorine atoms, was launched into medicinal practice in 1963.
Structural modification of the quinolone skeleton by incorporating of fluorine atoms at C-6 and other positions of the benzene ring resulted in a remarkable improvement of antimicrobial properties and opened new prospects in clinical treatment of infections. Indeed, compounds of the fluoroquinolone family proved to exhibit a high level of antibacterial activity and a wide spectrum which surpass many antibiotics, including the third generation of cephalosporin’s and other chemotherapeutic antibacterials [1–13]. Due to enhanced penetration ability through cell membranes and their effects on bacteria reproduction by inhibiting bacterial DNA-gyrase, fluoroquinolones possess a high antibacterial activity (Fig. 1) [6].

Inhibiting bacterial DNA-gyrase by fluoroquinolones (Reproduced with permission of publishing Folium [6])
It is extremely important that fluoroquinolones have a specific mechanism of action, different from antibiotics and other groups of antibacterials (cephalosporins, aminoglycosides, etc.), which allows one to apply fluoroquinolones for treatment of infectious diseases caused by strains resistant to many other classes of antibacterials drugs.
Depending on their behavior relative to bacteria enzymes of three types of fluoroquinolones can be distinguished:
-
the first type of fluoroquinolones inhibiting mainly the topoisomerase IV: norfloxacin, enoxacin, fleroxacin, ciprofloxacin, lomefloxacin, trovafloxacin, grepafloxacin, ofloxacin and levofloxacin;
-
the second type of fluoroquinolones which inhibit mainly the DNA-gyrase (nadifloxacin and sparfloxacin);
-
the third type of fluoroquinolones which have a double effect: they inhibit both topoisomerase IV and DNA-gyrase: gatifloxacin, pazufloxacin, moxyfloxacin, and clinafloxacin.
An important feature of fluoroquinolones is their selective biological action: suppressing bacterial DNA-gyrase, they don’t influence the mammalian DNA cell processes. In fact, quinolones don’t kill bacteria by inhibiting critical cellular processes, but rather break action of two essential enzymes, DNA-gyrase and topoisomerase IV, and use them by causing a rupture of two-spiral DNA.
During the last two decades the whole series of antibacterial fluoroquinolones have found their application in clinical practice, thus demonstrating a beginning of a new era in chemotherapy of bacterial infections. The vast majority of fluoroquinolones, launched into medical practice, are based on the bicyclic structure of 6-fluoro-4-oxo-1,4-dihydroquinolin-3-carboxylic acid. Annelation of the benzene ring, and carbo- or heterocyclic fragments to the quinolone skeleton usually allow one to enhance antibacterial activity of fused fluoroquinolones and their therapeutical properties; in some cases derivatives of this class become capable of exhibiting other types of activity, including antiviral and antineoplastic ones. The most known representatives of tricyclic fluoroquinolones appear to be ofloxacin and levofloxacin. For many years fluoroquinolones have been intensively studied worldwide as evidenced by numerous review articles and monographs [1–13].
2 Synthesis and Antibacterial Activity of Fluoroquinolones
2.1 Bicyclic Fluoroquinolones
There are two basic approaches which are commonly used for the synthesis of quinolin-4-one-3-carboxylic acids [4, 14]. The first one is based on use of fluorinated anilines (1, A = CH, CF) or 2-aminopyridines (1, A = N) as starting materials and involves their condensation with ethoxymethylene derivative of malonate, cyanoacetate or acetoacetate to form enamines 2. The intramolecular cyclization of compounds 2 with polyphosphoric acid (PPA) (the Gould-Jacobs reaction) affords the corresponding fluoroquinolones (3, A = CH, CF) or naphthyridones (3, A = N) (Scheme 2).

Synthesis of fluoroquinolones from fluorinated anilines
One of the key problems of the Gould-Jacobs reaction is a choice of high-boiling solvent. Diphenyl ether which has been applied for a long time is not appropriate due to environmental reasons. A good alternative of Ph2O seems to be a summer diesel fuel, which is cheaper than individual C12-C18 hydrocarbons, and allows one to carry out the process at 230–245 °С providing a good purity of the key intermediates in the synthesis of fluoroquinolones.
The second approach suggests use of fluorine-containing benzoyl derivatives (4, A = CF, CH) or their nicotinoyl analogs (4, A = N) as building-blocks (Scheme 3). The key intermediates in this case are benzoyl- or pyridinoyl acrylates 6 [6]. Cyclization of enaminones 7 can be carried out by heating in DMF in the presence of potassium carbonate, or in ethyl acetate with NaH. Other basic conditions can also be applied, including organic amines or amidines, 1,4-diazabicyclo[2.2.2]-octane (DABCO) or 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) [4, 15].

Synthesis of fluoroquinolones from fluorinated benzoyl derivatives
The method can be improved by use of the dimethylamino analogue of intermediate 7, which can be derived from the reaction of ethyl 3-dimethyl aminoacrylate with the corresponding fluorine-containing benzoyl chlorides followed by the displacement of the dimethylamino group with a suitable amine.
A great deal of research studies aimed at improvement of synthetic procedures leading to fluoroquinolones, enhancing their yields and quality of products, and reducing a number of steps and cost of the synthesis have been performed [16–31]. Improved synthetic procedures have been applied to obtain 1-ethyl-6-fluoro-7-(4-methylpiperazinyl)-4-oxo-1,4-dihydro-3-quinolinecarboxylic acid and 1-ethyl-6-fluoro-7-(piperazinyl-1)-4-oxo-1,4-dihydro-3-quinolinecarboxylic acid as well as their intermediates [18–21]. Further research studies on the synthesis of more active bicyclic fluoro-quinolones to expand a range of their biological activity, and to develop antibacterial drugs against resistant strains are in progress now.
2.1.1 Modification of the Position N(1)
The nitrogen atom N-1 and N-substituents are important features of the molecule of fluoroquinolones because of their considerable contribution into antibacterial activity. Replacement of the nitrogen atom with a carbon or oxygen in analogues of the oxolinic acid results in complete deactivation of these molecules. Modification of NH fluoroquinolones is usually based on N-alkylation reaction with the corresponding alkyl halide in the presence of a base. The first representatives of commercial fluoroquinolones bearing the ethyl group at N(1) are presented by norfloxacin, pefloxacin, and enoxacin; fleroxacin has N-fluoroethyl substituent, while amifloxacin contains the N-methylamino group. Research study on activity of the series of analogues of enoxacin, bearing C1-C5 aliphatic groups at N(1) have shown the preference of the N-ethyl group [32].
Modification of the N-ethyl group by means of incorporation of a fluorine atom (CH2CH2F, fleroxacin) appeared to be a reasonable approach [33]. Also conformationally restricted analogs of fleroxacin 9 and 10 have been synthesized (Scheme 4). The Z-isomers proved to be 2–32-fold more potent in vitro against gram-positive strains of bacteria then the corresponding E-isomers [34].

Structure of fleroxacin and analogs
Replacement of N-ethyl group with NHCH3 leads to a highly effective drug amifloxacin. Although it has not exhibited in vitro tests a considerable advantage in comparison with norfloxacin and pefloxacin, it shows a better pharmacokinetic profile, being equally active in both oral and parenteral administration.
It has been revealed that a high antibacterial activity of fluoroquinolones is associated with the presence of a small lipophilic group, such as, for instance, N-cyclopropyl substituent in position 1. Indeed, a number of commercially important fluoroquinolones bear the cyclopropyl fragment at N(1): ciprofloxacin, enrofloxacin, grepafloxacin, clinafloxacin, gatifloxacin, moxifloxacin (Scheme 5) [7].

Structure of amifloxacin and 1-cyclopropyl-fluoroquinolones
Incorporation of methyl or phenyl substituents in the cyclopropane ring, as well as the replacement of the cyclopropyl moiety with cyclobutyl or cyclopentyl ones diminishes the activity of these derivatives (Table 1) [7].
Further modification of the cyclopropyl fragment (for example, 2-fluorocyclopropyl derivatives 11) gives rise to optically active isomers, which differ considerably in their activities, as illustrated by the fact that cis-analogs are more active against gram-positive strains of bacteria, than the corresponding trans-isomers, for example, cis-isomer of fluoroquinolone 11 (R7 = 4-methyl-piperazin-1-yl) shows MIC 0,1 μg/ml against St. aur., while trans-isomer has only 1,56 μg/ml. New synthetic approaches enabling one to introduce at N-1 of fluoroquinolones a fluorine-containing cyclopropyl fragment with a certain stereo-configuration have been developed [35, 36].
Incorporation of benzyl or t-butyl groups at N-1 enhances antibacterial activity of fluoroquinolones [37, 38]. Monofluoro-t-butyl derivatives proved to possess a higher antibacterial activity than their non-fluorinated analogs. An opportunity to use 1-trifluoromethyl-1,2-ethylenediamines for modification of position 1 of fluoroquinolones (compounds 12) (Scheme 6) [39] has been shown.

Structure of fluoroquinolones 11 and 12
Derivatives of bicyclic pefloxacin 13 and 14 represent an interesting type of hybrid molecules, in which N-butylfluoroquinolone fragments are linked with the pyrimidine and purine heterocyclic bases (Scheme 7) [40].

Structure of fluoroquinolones 13 and 14
Fluoroquinolones 15 bearing the (hydroxyethoxy)methyl fragment, which is present in acyclovir, the known antiviral agent, can be regarded as acyclic analogs of nucleosides (Scheme 8) [41]. Also 5′-thioalkyl acyclic nucleosides of fluoroquinolones have been obtained by the reaction of mesylate 15 with methanethiolate- or thiophenolate anions [42].

Structure of fluoroquinolones 15 and 16
A series of new quinolones 16 bearing the fragments of natural amino acids have been synthesized. According to the data of preliminary biological studies these fluoroquinolones exhibit antibacterial activity against Bacillus subtilis and Staphylococus aureus [43].
Synthetic routes to new fluoroquinolones, containing in position 1 aryl substituent have also been described [44–46]. As a rule, a fluorophenyl substituent with one or two fluorine atoms has a favorable effect, increasing an activity of fluoroquinolones towards anaerobic bacteria. It has been found that 1-(5-amino-2,4-difluorophenyl)-8-R-substituted quinolones 17 possess a rather high antibacterial activity relative to Gram-positive and Gram-negative microorganisms (Scheme 9) [47]. 7-(Methylpiperazinyl)-6-fluoro-1-(4-fluorophenyl)-1,4-dihydro-4-oxo-3-qui-nolincarboxylic acids (difloxacin) has been established to be one of the most active fluoroquinolones in experiments in vitro against Chlamydia trachomatis and other intracellular parasites; also it demonstrates excellent pharmacokinetic properties. Also, the antibacterial drug linezolid 18 bearing at N-1 2-fluoro-(4-oxazolidon-1-yl)phenyl fragment has been developed [48] (Scheme 9). N-(5-Amino-2,4-difluorophenyl)-7-aminoazetidinyl-8-chloro-substituted fluoroquinolone has been found to possess a high antibacterial activity relative to Gram-positive and Gram-negative microorganisms; its activity against Strentococcus pneumoniae proved to be 30-fold higher than that of trovafloxacin.

Structure of fluoroquinolones 17 and 18
A number of researches were dedicated to incorporating of heterocyclic fragments in position 1 of fluoroquinolones in expectation of enhanced activity [49]. Indeed, 1-(6-amino-3,5-difluoropyridin-2-yl) substituted quinolone 19 (Scheme 10) proved to be rather promising for treatment of serious respiratory diseases and infections of the urinary tract. This fluoroquinolone has a wide range of antibacterial activity, including quinolone-sensitive and resistant staphylococcus and streptococcus, vancomicin-sensitive and resistant enterococcus, anaerobic bacteria and other infections [50], 20 was shown to be more active than ciprofloxacin [51] (Scheme 10).

Structure of fluoroquinolones 19 and 20
1-Trifluoromethylated fluoroquinolone shows antibacterial activity at the level of norfloxacin [52]. 1-Hydroxy-2-phenyl- and 1-hydroxy-2-methyl substituted quinolones have been obtained, however they have not shown a remarkable level of antibacterial activity [53, 54].
Analysis of the data of biological trials for N-substituted fluoroquinolones available in literature enables to conclude that compounds bearing in position 1 cyclopropyl, fluorophenyl or t-butyl fragments exhibit a higher level of antibacterial activity than their N-unsubstituted analogues.
2.1.2 Modification of the Position C(2)
Modifications of the C(2)-position are limited due to synthetic difficulties associated with direct introduction substituents at C-2. However, the synthesis of 2-phenylsubstituted fluoroquinolones has been developed [55], and 6-fluoro-quinolon-2-carboxylic acids have been obtained by cyclization of the corresponding 2-aminosubstituted 3-pentafluorobenzoyl acrylic acids [56]. 2-Thio substituted quinolones are widely used for the synthesis [a]- or [b]-annelated fluoroquinolones, such as thiazolo- and azethydinoquinolones [57–59]. Synthesis of 1-cyclopropyl-2-alkylthio-8-methoxyfluoroquinolones was described; however elucidation of their antibacterial activity revealed no regularities associated with incorporation of 2-alkylthio substituents [60]. All known 2-aza analogues of quinolones and naphthyridines, derivatives of cinnoline, have not exhibited any remarkable antibacterial activity.
2.1.3 Modification of the 3-Carboxyl Group
Modifications of the 3-carboxyl group appear to be worth only in those cases where these derivatives are considered as precursors of the corresponding carboxylic acids [61], however precursors not always exhibit activity in vivo. Replacement of the 3-carboxyl group with acyl, ethoxycarbonyl, methoxycarbonyl and other acidic fragments (hydroxamic, acetic, phosphonic, sulphinic or sulpho) results in complete loss or diminishes dramatically antibacterial activity of these compounds.
Functional properties of the carboxyl group have been used to modify it with osteofilic bisphosphonate fragments, as exemplified by structural modifications of moxi-, gati- and ciprofloxacin are developed [62]. Derivatives of these fluoroquinolones 21, containing bisphosphonate ester, thioester or amide groups have been obtained (Scheme 11). Their abilities to contact bones and to recycle thus active medicinal component have been studied. It has been shown that bisphosphonate derivatives of fluoroquinolones are osteotropic predecessors for prevention of osteomielit.

Structure of fluoroquinolones 21
Amides, hydrazides, and thiourea derivatives are important derivatives of fluoroquinolones [63–65]. It is worth noting that 7-chloroquinolones bearing the amide moiety at C-3 are rather active against B. subtilis and S. aureus. Also phenylthiourea derivatives proved to be more active against B. subtilis than the parent ciprofloxacin [64]. Synthesis of glycosylhydrazides and aminoacids on the basis of the corresponding hydrazido- and azido derivatives of 6-fluoroquinolin-4-one-3-carboxylic acids has been described [66].
Esters and hydrazides of 6-fluoroquinoline-4-oxo-3-carboxylic acids have been used for modification of the position 3 through the formation of heterocyclic fragments, such as oxadiazole, triazole, thiadiazole, benzofuropyrazoline, thiazolidine and others [67, 68]. Synthesis of fluoroquinolones containing in position 3 quinoxalinone, benzoxazinone and benzothiazinone fragments has recently been described [69, 70]. This synthesis was realized through interaction of fluoroquinolones bearing EtOC(O)C(O) residue with aromatic 1,2-binucleophiles. 3-Formyl- and acetyl derivatives of fluoroquinolones and also alcohols and amines have been obtained through transformation of amides [71].
It has been established that after oral administration of 3-formyl analogue of norfloxacin in mice the formyl group is metabolized rather fast into the carboxyl one, thus converting 3-formyl derivatives into norfloxacin. Due to a good solubility, a much higher level (at least two times) of the formyl derivative in blood serum can be reached, than on administration of norfloxacin, which at physiological рН values exists in the form of poor soluble zwitter-ionic form.
During the last two decades a lot of attention has been paid to development of “double mechanism” antibiotics. One of plausible approaches to such compounds is esterification of fluoroquinolone carboxylic acids with derivatives of cephalosporin and penicillin. Such combination allows one to expand a spectrum of antibacterial activity of beta-lactams conjugated with quinolones due to complementary mechanisms of their actions [7, 72].
Displacement of the carboxyl group in position 3 with hydrogen atom and the decarboxylation of fluoroquinolones have been discussed in the literature [73–76]. Since no decarboxylated fluoroquinolones have exhibited antibacterial activity, many authors have come to conclusion on the extremely importance of the 3-carboxy group.
2.1.4 Modification of the 4-Oxo Group
The oxo group can be modified through the formation of oximes, hydrazones, and semicarbazones, as exemplified by transformations of norfloxacin and other fluoroquinolones [73]. Specific methods are needed to convert fluoroquinolones into their 4-alkoxy analogues, due to a preferable N-alkylation of fluoroquinolones at position 1. Another modification is the synthesis of 4Н-1,4-benzothiazin-1-oxides and 1,1-dioxides [77] with various substituents in the benzene ring. However, these compounds proved to exhibit neither antibacterial activity, nor they inhibit DNA-gyrase. These results show that SO and SO2 groups in quinolones cannot be regarded as bioisosters of the carbonyl group.
It has to be concluded that the oxo group at C(4) is necessary for linkage of quinolones with DNA-gyrase, and elimination or replacement of the oxo fragment with other moieties lead to inactive compounds.
2.1.5 Modification of the Position С(5)
The most promising results have been received in those cases when the amino group was introduced at position 5 of fluoroquinolones. The detailed analysis of the “structure–activity” relationship for 5-substituted 1-cyclopropyl-6-fluoro-quinolones has shown that the positive effects of NH2 and CH3 groups are approximately identical, and these fluoroquinolones possess a wide range and high level of antibacterial activity [7]. Indeed, 7-(7-aminomethyl-5-azaspiro[2.4]heptan-5-yl)quinolone 22 proved to be 12 times more active against S. aureus HPC527 than ciprofloxacin [78, 79]. The methoxy derivative 23, and also its 8-methyl analogues show a high antibacterial activity towards a great deal of microorganisms [80] (Scheme 12). 5-Also acylaminoquinolones have been synthesized [81].

Structure of 5-aminofluoroquinolones 22, 23
In order to obtain multi-binding therapeutic agents that modulate enzymatic processes, two fluoroquinolone ligands were linked at positions 5 through 1,3-di-aminopropane bridge (compound 24) [82]. Fluoroquinolones bearing the hydrazino group in position 5 appear to be effective antimicrobials towards a number of pathogenic microorganisms; also they possess a good solubility in water relative to other fluoroquinolones [83]. 5-Methoxy- and 5-hydroxy-6-fluoro-1,8-naphthyridin-4-oxo-3-carboxylic acids (25a,b) are more active against S. pneumoniae 7257 than levofloxacin [84] (Scheme 13).

Structure of fluoroquinolones 24, 25
Incorporation of such substituents as Cl, Br, SH, SCH3, CHO into position 5 of 1-cyclopropyl-6,8-difluoro-7-(4-methylpiperazin-1-yl)-4-oxo-1,4-dihydro-3-quinolincarboxylic acids didn’t result in substantial increase of their activity. Some substituents at C(5) have a negative effect on antibacterial activity of fluoroquinolones which can possibly be explained by steric hindrance to interaction of the 4-oxo-3-carboxy-fragment of fluoroquinolone molecules with metal ions of the bacterial DNA-gyrase. However, a fluorine atom at C-5 with nearly the same space volume as a hydrogen one also diminishes the activity of fluoroquinolones, and it can’t be connected with its steric effect.
2.1.6 Modification of the Position С(6)
Replacement of a fluorine atom in position 6 with other substituents didn’t enhance their activity, at the same time it was shown that in order to obtain highly active antibacterial compounds the presence of fluorine atom at C(6) is not obligatory, it is more important to have in the quinolone skeleton the N(1)-cyclopropyl and C(7)-3-aminopyrrolidinyl pharmacophoric groups (Table 2) [85–88].
Studies of antibacterial activity of 6-fluoro-1-[(1R,2S)-2-fluorocyclopropan-1-yl]-8-methoxyquinolones and their C(6)-defluoro analogs showed that all of them are in 4–520 times more active against gram-positive bacteria, than trova-, moxi-, gati- or ciprofloxacin [89]. These quinolones have shown the indices of activity against Gram-negative bacteria E. coli and K. pneumoniae which are comparable with those of trova- and ciprofloxacin.
Incorporation of the nitrogen atom (derivatives of 1,6-naphthiridines) proved to diminish considerably the activity of quinolones.
2.1.7 Modification of the Position С(7)
A great deal on the chemistry of 6-fluoroquinolones concerns modification of the position 7. It is due to the fact that a halogen atom at C(7) undergoes easily nucleophilic displacement with N-, S-, O- and C-nucleophiles, thus allowing one to vary the structure of quinolones. Nearly all commercially important fluoroquinolones contain at C-7 the fragments of cycloalkylimines [90–94].
Quinolones bearing in position 7 small or linear substituents, such as H, OH, OEt, COOH, Cl, Me, NH2, NHR, NH-c-C3H5, NHNH2, SCH2CH2NH2 etc., have a relatively low activity against gram-positive microorganisms and are practically inactive towards the negative bacteria. Also 7-aza analogues of 6-fluoroquinolon-3-carboxylic acids, derivatives of 1,7-naphthyridines, didn’t show any remarkable antibacterial activity.
A lot of studies have been directed to the synthesis of fluoroquinolones, bearing a variety of piperazinyl substituents, since this part of quinolone molecule is of significant importance. Indeed, some representatives of 6-fluoroquinolones bearing at C(7) piperazine (norfloxacin, ciprofloxacin), 4-methylpiperazine (pefloxacin), 3-methylpiperazin (lomefloxacin, temafloxacin) proved to possess a much broader range of antibacterial activity, than those without the piperazine moiety, such as nalidixic and oxolinic acids.
In order to introduce the piperazine residue into position 7 of fluoroquinolones the reaction of 7-chloroquinolone with N-alkoxycarbonylpiperazine in high-boiling dipolar aprotic solvent followed by hydrolysis of alkoxycarbonyl group has been exploited. In some cases the borondiacetate complexes of fluoroquinolones have also been used for introduction of the piperazine fragment.
The difference in activity for R- and S-enantiomers of 7-(3-methylpiperazin-1-yl)quinolones, obtained from the corresponding (R)- and (S)-t-butyl-2-methylpiperazin-1-carboxylates, proved to be in the range from 2 to 64 folds in 52 % of cases [95]. In order to improve transport through biological membranes the piperazine moiety in norfloxacin was modified considerably and compound 26 was obtained [96]. To clarify the mechanism of antibacterial action of fluoroquinolones at the cellular level, two regioisomeric citrate-functionalized derivatives of ciprofloxacin 27a,b [97] (Scheme 14) have been obtained and studied.

Structure of fluoroquinolones 26, 27
Introduction of spiropiperazine or piperazinedione groups in position 7 of 1-cyclopropyl substituted fluoroquinolones has been shown to enhance their antimicrobial activity (compounds 28a,b) (Scheme 15) [98, 99].

Structure of fluoroquinolones 28
Also the piperazine fragment of fluoroquinolones was modified by introduction of a number of heterocyclic fragments, such as 2,6-diaminopyrimidinyl, 4,6-diamino-1,3,5-triazinyl, 2-aminothiazinyl, 1,3,4-thiadiazolyl, 2-furyl and other groups, thus allowing one to obtain more active antibacterial drugs [100–103].
Hybrid derivatives of fluoroquinolones bearing fragments of penicillin and cephalosporin antibiotics or uracils, for example compounds 29–31, proved to possess a wide spectrum and high level of antibacterial activity, including their potency against resistant to β-lactams strains [74, 104–107] (Scheme 16). High antibacterial activity has also been shown by 7-(N-aryl-2,2,2-trifluoroacetimidoyl)piperazinyl derivatives of fluoroquinolones [108].

Structure of fluoroquinolones 29–31
Influence of the second heteroatom in the piperazine ring is not so unequivocal. For instance, the replacement N(4) in the piperazine moiety of amifloxacin with O, S or CH2 fragments has been shown to diminish activity of these compounds in vitro and in vivo, however when the piperazine residue in norfloxacin was replaced with thiomorpholine a much more potent compound against Gram-positive bacteria has been obtained. 7-(3-Aminomorpholin-1-yl) and 7-[3-(or 4)-aminomethylpiperidin-1-yl]-derivatives proved also to possess a high activity against St. aur. (Table 3). 7-Azetidinyl substituted fluoroquinolones, in particular trans-3-amino-2-methyl-1-azetidinyl derivatives proved to be highly active antibacterial compounds [84, 109, 110].
A large group of highly active fluoroquinolones contains the pyrrolidine fragment in position 7, and, therefore, a considerable attention has been paid to the synthesis of 6-fluoro-7-pyrrololidinoquinolones with 3-amino-, 3-aminomethyl- or 3-(2-cyanomethylamino) substituents in the pyrrolidine ring [111–114]. As a rule, the compounds of this series possess a much higher activity towards Gram-positive microorganisms than the corresponding piperazine derivatives.
Fluoroquinolones 32a, containing alkyloximino substituent at C-4 and the aminomethyl fragment at position 3 of the pyrrolidine ring, exhibit a high antibacterial activity towards Gram-positive and Gram-negative microorganisms, including a methicillin-resistant strain of S. aureus (MRSA) [115–118]. Compounds 32b having an optically active center in the pyrrolidine ring and the methyloximino group proved to possess not only high antibacterial activity, but also a good pharmacokinetic profile [119, 120]. Also, the series of fluoroquinolones, containing spiropyrrolidine substituents at C-7, for example, compound 33a, have been obtained (Scheme 17) [121, 122].

Structure of fluoroquinolones 32, 33a
Effects of the chiral fragments, such as 1-(cis-2-fluorocyclopropyl) and 7-(7-amino-5-azaspiro[2.4]heptyl) substituents (compounds 32b, 33a) on antibacterial properties of the series of fluoroquinolones have been studied (Scheme 18) [123]. It has been shown that derivatives of 1-[(1R,2S)-2-fluorocyclopropyl]- and 7-[(7S)-amino-5-azaspiro[2.4]heptyl]-fluoroquinolones are more active towards a number of Gram-positive and Gram-negative bacteria, than other stereoisomers. The presence of spiropyrrolidine residue at C(7) of fluoroquinolones enhances their lipophilic properties, thus promoting a better assimilation on oral administration [98].

Structure of fluoroquinolones 33b, 34
Compounds 33b, 34 with the amino group attached to the spiropyrrolidine or cyclopropyl-substituted pyrrolidine fragment proved to exhibit broad spectrum of antibacterial activity (Scheme 18) [124–129]. Aminomethyl substituted pyrrolidines and their heterocyclic derivatives were incorporated into position 7 of fluoroquinolone [130–132]. Optically active derivatives of 7-(3-hydroxypyrrolidin-1-yl)-6-fluoroquinolones have been shown to be promising antibacterials [133–135].
One more residue which is frequently present in position 7 of active fluoroquinolones is piperidine [136–139]. Indeed, 1-cyclopropyl-6-fluoro-quinolones, containing (3S)-amino-(4R)-piperidinyl fragment in position 7, show a high activity towards resistant strains of Staphylococus aureus and Streptococus pneumoniae [140]. A number of substituents, such as 4-amino, 4-hydroxy, 3-aminomethyl, 4-aminomethyl and 3-methylamino were incorporated in the piperidinyl fragment [141, 142]. Novel 6-fluoroquinolones and naphthyridines with 4 (3)-alkoxyimino-3-aminomethyl-3-H(methyl)piperidinyl substituents, for instance 35, have been obtained (Scheme 19) [143–145]. They shown a high activity against all gram-positive organisms, including those resistant to fluoroquinolones. One of compounds of this series proved to be in 16–128, 2–32 and 4–8 times more active against fluoroquinolone-resistant MSSA, MRSA and MRSE than gemi-, cipro- and levofloxacin, respectively. Introduction of 4-(1Н-1,2,3-triazol-1-yl)piperidinyl residue in the structure of fluoroquinolone resulted in a good activity against S. aureus and S. epidermidis [146].

Structure of fluoroquinolones 35–38
A very promising modification of fluoroquinolones is introduction of bridged cyclic amines in position 7 [147–153]. A series new fluoroquinolones 36 was synthesized (Scheme 19), and one of compounds showed high activity against quinolone-sensitive and multi-resistant bacteria, especially towards Streptococcus pneumonia [154].
Trovafloxacin 37, the very active compound with a wide spectrum of action, contains 7-(1α, 5α, 6α)-3-azabicyclo[3.1.0]hexyl substituent (Scheme 19) [155, 156]. 6-Fluoro-1-[(1R, 2S)-2-fluorocyclopropan-1-yl]-4-oxoquinolin-3-carboxylic acids, containing in position 7 2-amino-8-azabicyclo[4.3.0]nonan-8-yl fragment have been shown to inhibit bacterial DNA topoisomerase IV very effectively [157]. A great deal of research are dedicated to the synthesis and biological tests of 7-di- and triazabicyclononyl substituted 6,8-difluoroquinolones, for instance 38 (Scheme 19) [158–163].
An effective way for introduction of a variety of heterocyclic fragments in the position 7 of the fluoroquinolone skeleton is the methodology of 1,3-dipolar cycloaddition reactions [164–167]. Indeed, the reaction of 7-azido derivative of 6-fluoroquinolone 39 with enamines of cyclic ketones and norbornene proceeds rather smoothly with the formation of the corresponding exo-1,2,3-triazolines 40 which undergo the cationic rearrangements into amidines 41 or aminonorbornane 42 [164, 165]. 7-Azido derivatives 39 are capable of reacting with heterocyclic amines to form new 7- fluoroquinolones (Scheme 20) [168].

1,3-Dipolar cycloaddition reactions of 7-azido derivative 39

The cycloaddition reactions of azomethine 43
The cycloaddition reaction of azomethine 43 with alkenes proceeds in regio- and stereoselective manner and represents a convenient way to obtain a variety of stereoisomeric 7-isoxazolidinyl quinolones 44–48 [166, 167] (Scheme 21).

Synthesis of fluoroquinolone 49
Synthesis of new hydroxybisphosphonate derivatives of ciprofloxacin 49 has been performed by using Cu-catalyzed 1,3-dipolar cycloaddition reaction between the corresponding azide and N-alkynyl substituted quinolone [169] (Scheme 22). Derivatives of gati- and moxifloxacin have been obtained similarly. All of these modified compounds maintained antibacterial activity of the starting quinolones and, in addition to that, exhibit osteotropic properties.
A number of 6-fluoroquinoline- and 6-fluoronaphthyridine-3-carboxylic acids, containing at C(7) rather complicated fragment of multilinе (compounds 50) have been synthesized (Scheme 23) [170]. Quinolones 50 exhibit a high activity against resistant bacteria, in particular, methicillin- and quinolone-resistant Staphylococcus, Streptococcus pneumoniae, etc.

Structure of fluoroquinolones 50, 51
Synthesis on the basis of organoelement compounds play an important role for modification of position 7 in fluoroquinolones [171]. As mentioned above, fluoroquinolones, containing hetaryl residues in position 7 are promising for medicinal chemistry [172]. In particular, a number of highly active fluoroquinolones have been obtained on the basis of 7-nitromethyl derivatives [173, 174]. The 7-(1,2,3,4-tetrahydropirrolo[1,2-a]pyrazin-7-yl) fragment has been incorporated in the structure of quinoline and naphthiridine carboxylic acids 51 through the carbon-carbon bond formation by reacting 7-halogeno or tosyl-substituted quinolones with the corresponding borates (Scheme 23) [175]. It should be noted that several compounds of this series have exhibited a high activity against ciprofloxacin-resistant bacteria of Streptococcus pneumoniae.
Thus, varying substituents in position 7 provides a good platform for development of novel antibacterial drugs. New opportunities for modification of position 7 are associated with design of hybrid molecules, as illustrated, for instance, by the development of the double action drugs containing both a fluoroquinolone and β-lactam antibiotic fragments.
2.1.8 Modification of Position С(8)
The nature of substituents in position 8 of fluoroquinolones also makes a certain impact on antibacterial activity. The key role of the 8-methoxy substituent is demonstrated by the fact that this fragment is a part of such effective drugs, as moxifloxacin and gatifloxacin [176–180]. Indeed, fluoroquinolone 52 shows a high activity against H. influenza and M. catarrhalis [181], while compound 53 is 4 times more active against S. pneumoniae than levofloxacin [182, 183]. 8-Methoxy-6-fluoroquinolone 54 has smaller side effects on the cardio-vascular system, than gatifloxacin (Scheme 24) [184].

Structure of fluoroquinolones 52–54
Fluoroquinolones, containing 8-methyl substituent usually demonstrate a high antibacterial activity, e.g. olamufloxacin is of great importance for treatment of urological diseases [185–188]. Also the cyano group in position 8 proved to be an appropriate substituent, as illustrated by the synthesis of 8-cyanoquinolones 55 and 56 [189] (Scheme 25). Indeed, compound 55 has been shown to possess a high antibacterial activity towards Gram-positive and Gram-negative bacteria [193], while 8-cyanoquinolone 56, containing the diazobicyclononane residue in position 7 is more active antibacterial compound than enrofloxacin (Scheme 25) [190]. Substituents NO2, NH2, SCH3, CF3 in position 8 have usually a negative impact on both in vitro and in vivo activities, especially towards Gram-negative microorganisms.

Structure of olamufloxacin and fluoroquinolones 55, 56
In order to obtain “structure-biological activity” relationships mathematic methods have been used [191–193]. Quantitative correlations between molecular structure and pharmacokinetic and pharmacodynamic characteristics of fluoroquinolones in combination with informative hemometric approach have been used to forecast anti-pneumococcus activity [194]. Elucidation of the structure – activity relationships in the series of fluoroquinolones is the subject of numerous publications [195–197]. Dependence of antibacterial activity on the nature of substituents has been established for several series of bicyclic fluoroquinolones [11, 198–200].
2.2 Polycyclic Fluoroquinolones
Modification of fluoroquinolones by annelation of carbo- or heterocyclic rings leads to fused polycyclic systems (Scheme 26).

Possible locations of additional rings in polycyclic fluoroquinolones
2.2.1 [a]-Annelated Fluoroquinolones
There are two principal approaches to the synthesis of [a]-annelated fluoro-quinolones. The first one suggests that an [a]-annelated ring is already involved in the structure of intermediates, such as aminoacrylates A or malonates B, followed by their cyclization into the corresponding fluoroquinolones. The second approach is based on use of 1- or 2-substituted quinolones C or D, which undergo intramolecular [a]-fusion (Scheme 27) [10].

Approaches to the synthesis of [a]-annelated fluoroquinolones
The first approach has been used to obtain [a]-annelated fluoroquinolones 57 and 58 from the correspondingly substituted ethyl acetates and 2-chlorobenzazoles or iminoesters (Scheme 28). 7-(1-Piperazinyl)- and 7-(4-methyl-1-piperazinyl)-benzothiazolo-[3,2-a]quinolones 57 have been established to exhibit rather good activity against a number of bacteria [201].

Synthesis of azolo[a]quinolones
Synthetic routes to [a]-fused quinolones of general formula 59 from the corresponding polyfluorobenzoyl chlorides and α-azahetaryl acetonitriles have been developed [202]. Heterocyclization of quinoxalones, containing polyfluoroaroyl fragment in position 3 in DMSO in the presence of triethylamine affords 60 (Scheme 29) [203].

Structure of fluoroquinolones 59, 60
The [a]-annelation in which the starting material is N-methylaminoquinolone has been described [204, 205]. Use of the 1,4-addition to the activated multiple bonds followed by the Michael intramolecular reaction leads to tetrahydropyrazolo[1,5-a]quinolones 61, which are oxidized into the corresponding pyrazolo[1,5-a]quinolones. Hexahydropyrrolo[1,2-a]quinolones 62 can be regarded as [3 + 2] adducts derived from the reactions of N-(ethoxycarbonyl)methyl substituted ethyl esters of di-, three- and tetrafluoro-4-oxo-1,4-dihydroquinolin-3-carboxylic acids with methylmetacrylate (Scheme 30) [206].

Structure of fluoroquinolones 61–64
Derivative of [1, 2, 4]triazino[1,6-a]quinoline 63 has been obtained from methyl 6-fluoro-4-oxo-1,4-dihydro-2-quinolincarboxylate through the N-amination followed by condensation of the corresponding aroyl isocyanate and cyclization of the obtained α-semicarbazidocarboxylate [207]. 8-Fluoro-4-hydroxy-1Н-[1,2,4]-triazino[4,5-a]-quinolin-1,6(2Н)-dione 64 has been obtained by condensation of 6-fluoro-4-oxo-1,4-dihydro-2-quinolinecarbohydrazide by action of phosgene [208]. 8-Fluoro-1,2-dihydro[1,4]oxazino[4,3-a]quinolin-4,6-dione was derived from intramolecular cyclization of 2-chloroethyl 6-fluoro-4-oxo-1,4-dihydro-2-quinolincarboxylate [209]. New tetracyclic system containing fluoroquinolone fragment 66 was obtained by intramolecular condensation of ethyl 3-acetyl-5-oxopyrazolo[1,5-a]quinolin-4-carboxylate 65 on heating [210] (Scheme 31).

Synthesis of tetracyclic fluoroquinolone 66
2-Mercapto-6-fluoroquinolin-3-carboxylic acids are considered as important intermediates in schemes leading to [a]-annelated fluoroquinolones, as shown by the synthesis of a number of thiazeto[a]quinolones 67 possessing a high level of antibacterial activity (Table 4) [211–213]. For instance, modification of position 7 of thiazeto[3,2-a]quinolones results in the formation of highly effective tricyclic antibacterials, such as prulifloxacin 68, which is metabolized in organisms into ulifloxacin 69 (Scheme 32) [214–217]. It is worth noting that decarboxylation of ulifloxacin drops down the antibacterial activity in 60–12,000 times. A similar phenomenon has been observed in case of cipro- and moxifloxacin [60], thus showing an extremely important role of the carboxyl group. The synthesis of thiazolo[3,2-a]-, [1,3]benzothiazino[3,2-a]- and [1,3]benzothiazino[1,2-a]quinolin-6-carboxylic acids has also been reported [218, 219].

Structure of thiazeto[a]quinolones 67–69
It should be noted that [a]-annelation of additional rings through the reactions of1- or 2-substituted fluoroquinolones has certain restrictions, while cyclocondensation of fluorinated benzoyl chlorides with C,N-bifunctional nucleophiles appears to be a more common method for the synthesis of a broad range of [a]-annelated fluoroquinolones. Incorporation of original bicyclic amines at position 7, as well as the synthesis of new derivatives through reactions of the carboxyl group are the main directions for modification of [a]-annelated fluoroquinolones.
2.2.2 [b]-Annelated Fluoroquinolones
The thesis concerning necessity of the carboxyl group in position 3 of fluoro-quinolones to provide their antibacterial properties is not in agreement with the data on activity of [b]-annelated isothiazolo-, pyrido-, pyrimido- and pyrazino-quinolones which stimulated research studies of this group of compounds [7]. Indeed, a whole number of oxoisothiazolo[5,4-b]quinolones possessing a high antibacterial activity (Table 5), for instance compound 70a and its analogues, have been obtained [220–224]. Also 9-cyclopropyl-6-fluoro-8-methoxy-7-(2-methylpyridin-4-yl)-9H-isothiazolo[5,4-b]-quinolin-3,4-dione has shown a high activity in vitro against methicillin-sensitive strains of Staphylococcus aureus (MRSA), high level of inhibiting of DNA-gyrase and topoisomerase IV of S. aureus, in combination with a neglect able effect on human topoisomerase II and low cytotoxicity [225, 226]. A series of 7-(3′-substituted) pyrrolidinyl-8-methoxyisothiazolo[b]quinolones 71 has been obtained and their antibacterial activity towards methicillin-sensitive Staphylococcus aureus (MSSA), methicillin-resistant Staphylococcus aureus (MRSA) and Escherichia coli, including stereochemical aspects and influence of substituents, has been elucidated [226].
The synthesis of 1-methyl-1,4-dihydro-9H-pyrazolo[4,3-b]quinoline-9-one 72, inhibitor of protein kinase C, has been performed by means of cyclization of 4-[(4-fluorophenyl)amino]-1-methyl-1Н-pyrazole-5-carboxylic acid (Scheme 33) [227]. The main trends in development of research studies in the field of [b]-annelated fluoroquinolones are dealt with use of these compounds for the synthesis of novel [i,j]-annelated systems, a varying of substituents at C-7, and also with obtaining of new 2-substituted fluoroquinolones.

Structure of [b]-annelated quinolones 70a-72
2.2.3 [с]- and [d,e]-Annelated Fluoroquinolones
The targeted synthesis of these types of fused fluoroquinolones has never been carried out, since the oxo-group in position 4 which is responsible for linkage of fluoroquinolones with DNA gyrase has to be eliminated [7].
2.2.4 [f]- and [g]-Annelated Fluoroquinolones
Both [f], and [g]-annelation results in loss of fluorine atom in position 6 the presence of which has long been associated with a high level of antibacterial activity of fluoroquinolones. However, a number of highly active compounds have been revealed in the series of oxazolo-, thiazolo- and imidazo[4,5-f] fused fluoroquinolones. For instance, derivative 73 (R3 = R4 = F) has shown a good activity against both Gram-positive, and Gram-negative bacteria [228]. According to in vitro biological tests 5-methoxyimidazo[4,5-f]quinolones 74 exceeds in activity the corresponding analogs of ofloxacin [229]. Furonaphthyridine 75 has found application as the basis to obtain antibacterials (Scheme 34) [230].

Structure of [f]-and [g]-annelated fluoroquinolones 73–75
2.2.5 [h]-Annelated Fluoroquinolones
6-Oxo-6,9-dihydro[1,2,5]oxadiazolo[3,2-h]quinolin-7-carboxylic acid 76 was synthesized from 7-azido-8-nitroquinolone [231]. A convenient method for the synthesis of 6-oxothiazolo[3,4-h]quinolin-7-carboxylic acids 77 has been suggested (Scheme 35) [232]. The structure of compounds 76 and 77 has been confirmed by X-ray crystallography. Biological tests of fluoroquinolone 77 have revealed that this compound possesses a high activity against Gram-positive bacilli and staphylococci, including methicillin-resistant strains, as well as Gram-negative bacteria (Table 6).

Structure of [h]-annelated fluoroquinolones 76, 77
A series of ethyl 2-R(Ar)-9-cyclopropyl-4-fluoro-6-oxo-1H-imidazo[4,5-h]quinoline-7-carboxylates 78 have been obtained through cyclocondensations of the corresponding 7,8-diamino quinolones [233]. Also a number of tetracyclic [h]-annelated fluoroquinolones, such as 1-cyclopropyl-6-fluoro-4-oxo-1,4-dihydro-pyrido[2,3-a]carbazole-3-carboxylic acids 79 and their thiene isosters have been obtained (Scheme 36) [198]. All derivatives proved to possess a high activity against Bacillus subtilus and Staphylococci.

Structure of [h]-annelated fluoroquinolones 78, 79
2.2.6 [i,j]-Annelated Fluoroquinolones
The most known representatives of tricyclic [i,j]-annelated fluoroquinolones are ofloxacin 80 and its analogues 81 (Scheme 37) [234]. Ofloxacin is well-known to clinical physicians, since more than 15 years it has been applied in medical practice. Ofloxacin has produced in two ready forms, peroral and injective ones, and both of them are characterized by a high clinical efficiency, wide range of indications for treatment, relative stability of the ofloxacin molecule in the process of bio-transformations in organism, and a low interference with drugs of other pharmacological groups. The oxygen atom in the oxazine ring is supposed to be an important element of the structure, thus providing an optimal antibacterial effect of this compound. Ofloxacin represents a racemic mixture of the right- and left-rotating optical isomers. The left-rotating enantiomer, levofloxacin, which proved to be much more active than its stereo analogue against nearly all bacteria, had been launched into medicinal practice in 1997. Inhibition of E. coli DNA gyrase by levofloxacin (I50 2,50 μg/ml) was shown to surpass inhibition of the same enzyme by ofloxacin (I50 6,20 μg/ml) [235].

Structure tricyclic [i,j]-annelated fluoroquinolones
The starting materials 82 for the synthesis of ofloxacin and its analogues have been obtained by interacting ethyl 2-(tetrafluorobenzoyl)-3-ethoxy acrylates with 2-aminopropanol [236]. It is clear that use of optically active S-(-)-2-aminopropanol enables one to obtain levofloxacin [237–241]. Another approach to fluoroquinolones 81 is cyclization of compounds 83, derived from condensation of the corresponding benzoxa(thia)zines with diethylethoxy methylenemalonate (Scheme 38). In this way the synthesis of levofloxacin has been realized from the (S)-isomer of 7,8-difluoro-2,3-dihydro-3-methyl-4H[1, 4]benzoxazine [242].

Synthesis of fluoroquinolones 81
During the last two decades the synthesis of levofloxacin and its S-(-)-pre-cursors has been improved considerably, and new approaches have been advanced [243–255]. In particular, kinetic resolution of 7,8-difluoro-2,3-dihydro-3-methyl-4H-[1,4]-benzoxazine racemate using naproxen, N-[sulphonylsubstituted]-(R)-proline and (2S)-(6-methoxynapht-2-yl)propionyl chloride, has been advanced [256–261]. The optically active (S)-isomer obtained by this method has been used for the synthesis of levofloxacin (S)-(-)-80 [256]. Also a new synthetic approach to (S)-isomer through catalytic reduction of 7,8-difluoro-3-methyl-2H-1,4-benzoxazine with use of chiral Bronsted acids as catalyst and substituted dihydropyridine as a source of hydrogen has been described [262].
A number of ofloxacin analogues modified in position 10, including the well-known antibacterial drug pazufloxacin 84, have been synthesized [263–265]. Some compounds of this series show a high activity towards a number of microorganisms, such as Shigella flexneri, Proteus vulgaris [263]. It is worth noting that (3S)-10-[Cis-(3S,4S)-3-amino-4-(fluoromethyl)pyrrolidin-1-yl]-9-fluoro-2,3-dihydro-3-methyl-7-oxo-7H-pyrido[1,2,3-d,e][1,4]benzoxazin-6-carboxylic acid 85 is more active than levofloxacin against Staphylococcus aureus 870307 [266]. An analogue of ofloxacin, containing a macrocyclic fragment in position 6 has been described [267]. All kinds of modifications of the structure of ofloxacin have been performed by varying substituents not only in positions 6 and 10, but also in the oxazine ring. In particular, compounds 86 show a comparable with ofloxacin activity against Gram-positive and negative microorganisms, and a high activity towards methicillin-resistant strain of S. aureus MR5867 [MIC 0,016–0,25 μg/ml for compound 86 (X = O, R = 3-cyclopropylaminomethyl-1-pyrrolidine)] (Scheme 39) [268].

Structure of fluoroquinolones 84–86
Marbofloxacin 87 is a representative of another promising group of tricyclic fluoroquinolones, pyridino[3,2,1-i,j]-1,3,4-benzoxadiazines, is widely used in veterinary practice (Scheme 40) [269].

Structure of fluoroquinolones 87–90
Synthetic methods to obtain other members of the family of [i,j]-annelated fluoroquinolones have been developed. For instance, derivatives of 1,3,4-thiadiazino[6,5,4-i,j]-, 1,3,4-oxadiazino[6,5,4-i,j]- and 1,2,4-triazino[5,6,1-i,j]-annelated quinolones 88a-c have been obtained by means of cyclization of 2-polyfluorobenzoyl acrylates bearing hydrazide, thiosemicarbazide or amidrazone moieties in position 3 [270–275]. Thiadiazino-fused quinolones 88a and compounds derived from displacement of fluorine atoms in positions 8 and 10 with cycloalkylimines are of great interest as promising compounds exhibiting not only antibacterial but also other types of biological activity [276, 277]. Synthesis of tetracyclic quinolones 89, in which the thiadiazine fragment is fused with both the pyridine and triazole rings has been described [278]. Activity of compounds 89 with R = H, Me against Gram-positive and Gram-negative bacteria is comparable with that of ofloxacin. Another core structure close to ofloxacin is 1,2,4-oxadiazino[i,j]-annelated fluoroquinolone 90 which was obtained by cyclization of 3-[1-(hydroxyiminoethyl)amino] acrylate [279]. The synthesis of tetracyclic fluoroquinolones 91 has been reported [280, 281]. The structure of novel pentacyclic fluoroquinolones 92 (Scheme 41), obtained by cyclization of ethyl 3-(benzazol-2-yl)hydrazino-2-polyfluorobenzoyl acrylates, was elucidated by X-ray crystallography [282–284].

Structure of fluoroquinolones 91, 92
As a rule, cyclizations of 1-substituted 8-fluoroquinolones have an advantage in comparison with annelation of the pyridine ring to a benzazine moiety, thus allowing one to vary annelated fragments to a greater extent. However, the synthesis of levofloxacin is an exception, since the scheme suggesting to obtain first the optically active benzoxazine, as the key intermediate, followed by annelation of the pyridone fragment proved to be a more successful one.
2.2.7 Tetracyclic [a,i,j]-Annelated Fluoroquinolones
Several examples of tetracyclic [a,i,j]-annelated fluoroquinolones are available in the literature. In particular, compounds 93 and 94, bearing 3-aminopyrrolidine and (1S,4S)-5-methyl-2,5-diazabicyclo[2.2.1]heptane fragments, respectively are considered to be rather promising because they both exceed ofloxacin in antibacterial activity (Scheme 42) [285, 286].

Structure of tetracyclic fluoroquinolones 93, 94
3 Other Types of Biological Activity of Fluoroquinolones
During the last decades compounds of the fluoroquinolone family proved to be not only effective inhibitors of bacterial enzymes; their antineoplastic [287], antiviral [41] (including concerning HIV [288]), anti-diabetic [289] and other types [290, 291] of biological activity have been intensively elucidated.
3.1 Anticancer Activity
Some representatives of the fluoroquinolone family, especially polycyclic compounds, are capable of inhibiting topoisomerase II, the key enzyme for replication DNA, and this is why they are promising for development of antineoplastic drugs [172, 292, 293]. In particular, a profound antineoplastic activity is demonstrated by quinobenzoxazines 95–97 (Scheme 43) [293–298]. Fluoroquinolone 95 (R’ = Н) is more active towards some tumor cells than such antineoplastic drugs, as adriamicin, camptotecin and etoposide [299]. Relationships between the nature of substituents in the amino fragment and the benzene ring of compounds 95–96 and their abilities to suppress the growth of tumor cells have been studied. Compounds with R’ = Н and R = Cl, NO2 were shown to inhibit not only topoisomerase II, but also topoisomerase I [280, 299–301]. Amides 97 proved to suppress effectively the growth of HCT-116 cells, IC50 values 0,03–0,4 μM [295].

Structure of fluoroquinolones 95–97
Further steps to modify the structure quinobenzoxazines 95 involve annelation of the benzene rings to the benzoxazine fragment, as illustrated by the synthesis of benzo- and dibenzoderivatives 98 and 99 (Scheme 44) [299, 302]. Research studies on activity of pentacyclic derivatives 98 towards a number of tumor cells have shown that R-isomers are much more active, than S-isomers (Table 7). Also it has been revealed that a molecular target for fused fluoroquinolones 99 is the site of DNA capable of forming the quadruplex [303]. It has been shown that R-isomer 99 is characterized by a strong linkage with G-quadruplex and a low influence on topoisomerase II, while the S-isomer 99 has a strong linkage with topoizomerase II and a low interaction with G-quadruplex [296].

Structure of fluoroquinolones 98, 99
The data of biological tests on activity of compound 100 (drug QQ58), as an intercalator of DNA [304] confirmed that this compound inhibits human telomerase (IC50 28 μМ); in organisms it is transformed into qarfloxacin which is linked with DNA G-quadruplexes [300, 304–306]. Polynuclear fluoroquinolones, containing the amide fragment, for example 101 (Scheme 45), have been shown to inhibit effectively the HeLa (mammalian cancer) growth (IC50 0,1–0,2 μМ) [303, 307, 308].

Structure of fluoroquinolones 100, 101
Other fused fluoroquinolones, derivatives of benzazolotriazino[i,j]-annelated quinolon-6-carboxylic acids 92 have shown anticancer activity [309]. Biological tests on 9 types of tumors revealed that annelation of 1-methylbenzimidazo fragment to the triazine ring is more effective for suppression of cell growth, than that of the benzothiazole ring. An increase in numbers of fluorine atoms in the benzene rings of quinoline or benzazole fragments enhance antineoplastic action of pentacyclic derivatives; acids suppress growth of cells more strongly, than the corresponding ethyl esters. The biggest effect on melanoma has been observed in vivo experiments for fluoroquinolone 92d (Scheme 46, Fig. 2) [310].

Structure of fused fluoroquinolones 92

Cells growth suppression for derivatives 92а-d
Derivatives of levofloxacin 103 (Scheme 47), bearing in position 3 a lipophilic fragment, or the benzothiazole fragment instead of the carboxyl group, proved to exhibit antineoplastic activity (Table 8) [311]. The highest level of activity against glioblastoma has been observed for the ester 103а.

Structure of fluoroquinolones 103–105
Antineoplastic activity of fluorine-containing derivatives of 1,3,4-oxa(thia)-diazine[6,5,4-i,j]quinolon-6-carboxylic acids 104, 105 has been studied on cultures of 60 lines of cancer cells for nine groups, such as leukemia, lung cancer, colon cancer, CNS cancer, melanoma, ovarian cancer, renal cancer, prostate cancer, mammalian cancer [309, 310]. In the series of thiadiazinoquinolines the highest effect on antineoplastic activity gas been observed for compounds 105а and 105b bearing such pharmacophoric fragment, as N,N-dimethyl-1,3-diaminopropane. In case of compound 105b the full death of nearly all tumor cells MCF7 and SF-268 (more than 90 %) has been reached. Biological tests of compounds 105а,c,d,f have shown that the presence of a fluorine atom in position 8 facilitates suppression of cell growth. Also a high activity of compound 105а towards leukemia has been established [309, 310].
Not only [i,j]-annelated fluoroquinolones, but also polycyclic fluoroquinolones, in which an additional ring is annelated to [c]- or [h]-sides proved to possess antineoplastic action. Research studies on antineoplastic activity of 5-cyclopropyl-6,8-difluoro-7-(2,6-dimethyl-4-pyridinyl)-5H-pyrazolo[4,3-c]quinolin-3(2Н)-ones 106 have shown that derivatives containing the cyclohexyl group in position 2 are the most effective inhibitors of topoisomerase II of HeLa cells (mammalian cancer), while the dimethylaminocyclohexyl compound has shown the best data on cytotoxicity towards Р388 (leukemia) cells (Table 9) [312]. 6-Fluoro-4-oxopyridino[2,3-a]-carbazol-3-carboxylic acids 107 inhibit MCF-7 (breast cancer) and A549 (lung cancer), activity of 107b towards MCF-7 is twice higher, than that of ellipticine (Scheme 48) [198].

Structure of fluoroquinolones 106, 107
Also a number of bicyclic fluoroquinolones are capable of suppressing the growth of tumor cells. Incorporation of pyrrolo[2,1-c][1,4]benzodiazepine fragment in position 1 of fluoroquinolones resulted in compounds 108, which inhibit the growth of HT-29 (colon cancer) cells and А549 (lung cancer) up to 80 % [313]. Derivatives of 1-phenylsubstituted fluoroquinolones 109 suppress the growth of Solo205 (carcinoma) cells (IC50 values 2–20 nМ) [314]. 3-Benzimidazolyl fluoroquinolone 110 and its analogues (Scheme 49), including [i,j]-oxazino annelated compounds, proved to suppress the growth of tumor KV, А2780 and Bel7404 cells [315].

Structure of fluoroquinolones 108–110
Rather high antineoplastic activity of ciprofloxacin derivatives, containing a substituent in position 4 of the piperazine fragment has been shown [302]. Elucidation of the “structure-activity” relationships for 1-(2-thiazolyl)-6-fluoro-1,4-dihydro-4-oxo-1,8-naphthyridin-3-carboxylic acids has shown that several compounds of this series exhibit activity, comparable with the well-known drug etoposide [316–318]. Also the data on activity of amides of 7-substituted 1-(2-thia-zolyl)- and 1-(2-benzothiazolyl)-1,8-naphthyridin-4-on-3-carboxylic acids have been reported [319]. Ethyl 1-(4-cyano-2,3,5,6-tetrafluorophenyl)-6,7,8-trifluoro-4-oxo-1,4-dihydroquinolin-3-carboxylate proved to inhibit the phosphorylation process of transcription STAT3 activator that plays an important role for cancer therapy [320].
3.2 Tuberculostatic Activity
Being effective inhibitors of DNA-gyrase of mycobacteria some derivatives of fluoroquinolones are important for therapy of rifampicin-resistant tuberculosis [321]. In particular, values of minimum inhibitory concentrations against M. tuberculosis for a number of elucidated fluoroquinolones proved to be in the range from 0,12 to 128 μg/ml (Table 10) [322, 323].
An important synthetic approach for development of fluoroquinolones which are active against Mycobacterium tuberculosis appears to be introduction of isoniazide and pyrazinamide residues into the piperazine fragment in position 7. Indeed, 1-tert-butyl substituted fluoroquinolones 111 and 1-cyclopropyl-5-amino-fluoroquinolones 112 proved to exhibit a high activity towards Mycobacterium tuberculosis in vivo [38]. The minimum inhibitory concentration against M. tuberculosis H37Rv for compound 113b is 0,78 μg/ml (Scheme 50) [324]. Also quinolones, bearing residues of hydrazides of substituted benzoic acids, which can be regarded as isosters of isoniazide, proved to be active compounds (MIC 0,5 μg/ml for multiresistant M. tuberculosis А8 241) [325].

Structure of fluoroquinolones 111–113
1-Cyclopropyl-8-methoxyquinolones 114 are active against Mycobacterium tuberculosis, its multi-resistant strains, as well as Mycobacterium smegmatis [326]. Derivative 115 possesses tuberculostatic activity against Meningitis tuberculosis Н 37 R v (MIC 0,16–0,35 μg/ml) [327]. 1-[(6′-Fluoro-1′,4′-dihydro-7-(4”-methyl-1”-piperazinyl)-1′-ethyl-4′-oxo-3′-quinolylamido)-3-iminomethyl]-rifampicin 116 proved to exhibit a considerable tuberculostatic activity (Scheme 51) [328].

Structure of fluoroquinolones 114–116
1-(4′-Amino-2′-fluoro)phenyl substituted fluoroquinolones 117 (R = H, Me) inhibit the growth of M. tuberculosis [329]. Incorporation of aminoester or polyethyleneamino fragments has been suggested to increase their ability to penetrate through cellular membranes. Indeed, fluoroquinolones 118 have been established to possess a high specific activity against mycobacteria and a low toxicity [330]. Tuberculostatic activity of derivatives 118 (R = H; X, Y = 0; n = 4) proved to be five times higher than that of pefloxacin (Scheme 52).

Structure of compounds 117, 118
Several compounds [331] of the benzothiazolo[3,2-a]quinolone-6-carboxylic acids 119 family (Scheme 53) exhibit high tuberculostatic activity relative to multi-resistant strain of M. tuberculosis (Table 11).

Structure of fluoroquinolones 119–121
Ofloxacin and its analogs are promising drugs for tuberculosis treatment. Ofloxacin (daily dose 300–800 mg) and levofloxacin (250–500 mg a day) in combination with p-aminosalicylic acid, cycloserine, or ethionamid are effective for the treatment of multi-resistant strains of tuberculosis. On using of these fluoro-quinolones, a relatively high concentration in cells is reached, that increasing their antibacterial activity [38]. Derivatives of ofloxacin, containing the nitro group in position 8, e.g. 120 proved to possess a high tuberculostatic activity [332]. Also compounds showing tuberculostatic activity have been found among oxadiazino-quinolines 121 and thiadiazinoquinolines 105 (MIC 0,2–0,4 μg/ml) [276, 277].
3.3 Antiviral Activity
Fluoroquinolones 122, bearing the (triazolylmethyl)phenyl fragment in position 1 and an aryl substituent in position 4 of piperazine, are capable of protecting the HIV-infected cells from a virus-induced destruction (IC50 0,25–0,7 μМ). They appear to be a new structural type of effective drugs for treatment and prevention of viral diseases caused by HIV retroviruses [333]. Fluoroquinolones 123 with 4-(2′-pyridinyl)-1-piperazine fragment in position 7, inhibit reverse transcriptase of HIV-1 [334]. 8-Difluoromethoxy- and 8-trifluoromethylcarboxylic acids 124 inhibit replication of HIV-1, while CF3- derivatives are more active against HIV-1 than the corresponding difluoromethoxy compounds (Scheme 54, Table 12) [335–338].

Structure of fluoroquinolones 122–124
[i,j]-Annelation of the oxazine ring is favorable for exhibiting of antiviral activity, but does not lead to such promising compounds, as 8-methoxy- and difluoro-methoxy derivatives [339]. Fluoroquinolone 125c is more active against the virus HIV-1, than thiazeto derivative 126 [336]. Values IC50 3,7 μМ for 125а and 1,7 μМ for 125b have been found, while values EC50 0,074 μg/ml for 125c and 0,4 μg/ml for 126 have been obtained. Also a number of tricyclic fluoroquinolones 127 proved to possess a high activity (ЕС50 0,008−2.3 μg/ml) (Scheme 55) [340]. Also effective compounds against HIV-1 have been discovered in the series of the Mannich bases of norfloxacin [341].

Structure of fluoroquinolones 125–127
Fluoroquinolone 128 bearing the (2-hydroxyethoxy)methyl fragment at N-1 is active against herpes virus HSV-1 (EC50 2,30 μМ), however the level of its activity is lower than that of acyclovir (EC50 1,09 μМ) [41]. 8-Trifluoro-methylquinolones 124 have been reported to suppress human cytomegalovirus [342]. Fluoroquinolones 129, containing the sulphamidomethyl group in a piperazine fragment, are active against influenza H1N1, H3N2 and H5N1 viruses [343]. Tricyclic fluoroquinolones 130, 131 were found to possess a high activity against hepatitus B virus (IC50 0,1 μМ) (Scheme 56) [344, 345]. Ciprofloxacin and levofloxacin are recommended for treatment of patients after transplantation surgery operations in order to prevent the disease caused by poliomavirus BK [346].

Structure of fluoroquinolones 128–131
3.4 Other Types of Biological Activity
Some fluoroquinolones appear to be active against fungi and parasites. For instance, the Mannich derivatives of norfloxacin 132 demonstrate a considerable antifungal activity against Histoplasma capsulatum. One of compounds of this family is more active than clotrimazole towards Microsporum audouinii, while other derivatives surpass clotrimazole in relation to Cryptococcus neoformans or Microsporum gypsum. From all derivatives 132 which have been studied (Scheme 57), compound with R = Br, X = N, R1 = NH2, Y-Z = CH, A = COMe, R2-R3 = OMe proved to exhibit the highest antifungal activity (MIC for Cryptococcus neoformans and Microsporum audouinii 0,6 μg/ml) [341].

Structure of fluoroquinolones 132
Moxifloxacin, gatifloxacin, trovafloxacin, and grepafloxacin belong to a new generation of fluoroquinlones, showing anti-parasitic activity against Toxoplasma gondii and Plasmodium falciparum which cause such severe diseases as toxoplasmosis and malaria, respectively. These fluoroquinolones are targeting at the DNA-gyrase, located in a top layer of parasites [347]. For example, the IC50 value for trovafloxacin against Toxoplasma gondii is 0,96 μM. The data on activity of fluoroquinolones 133 against parasites (Coccidia) [348], and activity of 7-(3′-azabicyclo[3.1.0]hexyl)quinolones 134 in relation to plasmodium have recently been reported (Scheme 58) [149].

Structure of fluoroquinolones 133–135
Some fluoroquinolones have been shown to exhibit cardiovascular, hypertensive, and antitrombocyte activities. For instance, compound 135 inhibits aggregation of trombocytes [349]. According to the recently published data, 5-amonofluoroquinolones 136 and 137 are active as glicogensyntase-kinase-3β inhibitors (GSK, serine-treonine-proteinkinase) [265]. Bi- and tricyclic fluoroquinolones, bearing the fragment of N-(2-pyridinyl)ethylenediamine appear to be promising GSK inhibitors (Table 13) [265]. 138, their 8-fluoro- and 5,8-difluoroderivatives proved to be selective allosteric modulators of М1 receptor, activation of which is important for therapy of the Alzheimer’s disease (Scheme 59) [350–353].

Structure of fluoroquinolones 136–138
4 Structure and Spectral Characteristics
The structure of fluoroquinolones has been elucidated in crystals and solutions. The data on X-ray crystallography analysis of fluoroquinolines are available in the literature for both quinolones [89, 123, 354, 355], and their polycyclic [204, 231, 232, 270, 271, 282, 283, 356] condensed systems.
The 1Н, 13С and 19 F NMR spectra for the series of fluoroquinolines have been registered and analyzed. 1Н, 13С NMR spectra of fluoroquinolones bearing rather complicated optically active fragments, including heteronuclear correlation experiments, have been discussed in the literature [164–168]. Elucidation of NMR 19 F spectra of compounds 12 has revealed long-range coupling constants 7 J F-F between the trifluoromethyl group and fluorine atom in position 8, which are realized through space due to vicinity of interacting spins [39]. The 19 F NMR spectra of benzimidazo [2′,3′:3,4]-1,2,4-triazino[5,6,1-i,j]quinoline ring system 92 demonstrate unusual through space 1H-19 F and 19 F-19 F spin-spin interactions with coupling constants 7 J(F1, F11) = 3.5–4.0 Hz and 6 J(F1, Н12) = 2.0–3.0 Hz (Scheme 60) [284].

Long-range coupling constants in compounds 12, 92
5 Complexes of Fluoroquinolones with Metals
Due to the presence of the carboxyl and β − oxo groups, as well as azaheterocyclic fragments, fluoroquinolones have a profound ability to form metal-chelates, and other ionic structures. It is known that complexes with metals may enhance activity of fluoroquinolones due to a better solubility and endocellular accumulation [357, 358]. The crystal structures of a number of metal complexes, results of their thermal analysis, IR and NMR spectra of complexes and their bioactivity have been considered [359]. In the recently published review article [360] the data concerning the structure and properties of metal complexes of fluoroquinolones, and their interaction with DNA have been analyzed. Also physical and chemical characteristics, as well as pharmacokinetic data and antibacterial properties of fluoroquinolones complexes with a variety of metals have been reviewed [361].
The Cu(II)-complex of ciprofloxacin was shown to possess a high activity against Mycobacterium tuberculosis than the parent compound [362]. An enhanced solubility of metal complexes in lipids facilitates their transport into bacteria cells, while an easily proceeding reduction of metal leads to the formation of Cu(I) and activation of oxygen which kills mycobacteria. Authors came to a conclusion that redox-active metal complexes are very promising compounds for development of highly active antitubercular drugs. Indeed, the minimum inhibitory concentration for enrofloxacin complex Cu(erx)2(H2O) against E. coli и P. aeruginosa is 0.125 μg/ml, while the same index for the parent enrofloxacin is 1.0 μg/ml [363]. Antibacterial activity of N-propyl norfloxacin (pr-norf) complex with CuCl2 and phenanthroline (phen) [Cu(pr-norf)(phen)Cl] has been was reported [364]. For instance, the formation of sparfloxacin (sflx) (Scheme 61) dimeric complex with Cu(II) [Cu2(sflx)2] and mononuclear complex with phenanthroline [Cu(phen)(sflx)H2O] has been shown (Figs. 3 and 4) [365].

Structure of sparfloxacin (sflx)

Structure of complex Cu2(sflx)2 (Reproduced with permission of Elsevier [365])

Structure of complex Cu(phen)(sflx)H2O (Reproduced with permission of Elsevier [365])
Antiproliferative effect of sparfloxacin and its metal complexes against hormone independent BT20 breast cancer cell line has been studied (Fig. 5) [365]. Coordination of sparfloxacin with copper in the form of dimeric complex Cu2(sflx)2 has been established to diminish the value of inhibitor concentration IC50 (μΜ) in approximately ten times. These data are in agreement with a hypothesis that biological activity of fluoroquinolones is in many respects caused by their ability for metal chelate formation. Antitumor activity of moxifloxacin-copper complexes against breast cancer cell lines has also been described [366].

Suppression of cells growth by sparfloxacin and its metal complexes, a- spfx, b-Cu2(sflx)2, c-[Cu(bpy)(sflx)H2O], d-[Cu(phen)(sflx)H2O], e-[Cu(df)(sflx)H2O]; bpy = bipyridine, phen = phenanthroline, df = 4,5-diazafluoren-9-one (Reproduced with permission of Elsevier [365])
Complex of norfloxacin [Fe(nf)2(H2O)2]Cl3 · 6H2O was shown to exhibit a higher antibacterial activity than the parent norfloxacin against E. coli and Bacillus dysenteriae bacteria [367]. Also it is worth noting that antimicrobial activity of cobalt complexes of ciprofloxacin is less, than that of copper complexes [368].
The reaction of ciprofloxacin (cfH) with metal salts in the presence of aromatic polycarboxylate ligands (or under basic conditions) has been found to give original metal–cfH complexes, for example, [Ba2(cf)2(1,4-bdc)(H2O)2]∙H2O and [Mn(cfH)(1,3-bdc)] (bdc = benzenedicarboxylate). The structure of [Ba2(cf)2(1,4-bdc)(H2O)2]∙ H2O consists of unique two-dimensional arm-shaped layers (Fig. 6), while the second complex contains double-chain-like ribbons constructed from [Mn2(cfH)2(CO2)2] dimers and 1,3-bdc (Fig. 7) [369].

Structure of complex [Ba2(cf)2(1,4-bdc)(H2O)2]∙H2O (Reproduced with permission of Wiley [369])

Structure of complex, [Mn(cf)(1,3-bdc)] (Reproduced with permission of Wiley [369])
Supramolecular structure of cadmium complexes of ciprofloxacin [Cd2(cf)2(bptc)(H2O)2]∙8H2O is shown in Fig. 8 [369]. Two units are connected together by μ3-O atoms of carboxylic groups from cf ligands in an edge-sharing mode to form [M2(cfH)2(H2O)2] dimers.

Supramolecular structure of ciprofloxacin complex, [Cd2(cf)2(bptc)(H2O)2]∙8H2O (bptc = 3,3′,4,4′-benzophenontetracarboxylate) (Reproduced with permission of Wiley [369])
Complexes of norfloxacin with zinc(II), such as [Zn(nf)2] · 4H2O and [Zn(H2O)2(nf)2](NO3)2, were found to exhibit a strong blue fluorescent emission [370]. The complex of Zn(II) with enrofloxacin and pyridine, as the second N-donative ligand, [Zn(erx)2(py)2] · 6H2O · MeOH has been obtained (Fig. 9). Such complexes were found to interact with CT-DNA, thus demonstrating their ability to bind with DNA. According to the data obtained by using the UV spectroscopic titration technique, the binding strength for Zn(orx)2(py)2 corresponds to the highest K b value [371].

Structure of complex [Zn(erx)2(py)2] · 6H2O · MeOH (Reproduced with permission of Elsevier [371])
The formation of ofloxacin complexes with magnesium has been studied by using NMR 1Н and 2D 1H-13C HSQC methods [372]. Behavior of coordinative compounds of ciprofloxacin, levofloxacin and lomefloxacin with Al(III) in water solutions has been elucidated by NMR 1Н and 13С spectroscopy [373]. Tetrakis[4-(3-carboxy-1-ethyl-6-fluoro-4-hydroxonio-1,4-dihydro-7-quinolyl)-1-methyl-piperazin-1-ium] di-μ2-chlorido-bis[tetrachloridobismuthate(III)] tetrachloride octahydrate, (C17H22FN3O3)4[Bi2Cl10]Cl4 · 8H2O, is composed of edge-shared centrosymmetric dinuclear [Bi2Cl10]4−anions, Cl−anions, dihydrogen pefloxacinium cations and water molecules. The BiIII coordination polyhedron is a distorted octahedron [374].
One of the modern trend in the chemistry of fluoroquinolones is the formation of Pd(II) and Pt(II) complexes with a number of fluoroquinolones, such as ciprofloxacin, levofloxacin, ofloxacin, sparfloxacin and gatifloxacin [375, 376]. Two examples are given below Scheme 62.

Pd(II) and Pt(II) complexes of fluoroquinolones
A great deal of complexes derived from enoxacin, norfloxacin, lomefloxacin, fleroxacin, ofloxacin, rufloxacin, gatifloxacin and sparfloxacin and their luminescence properties of Tb3+– and Eu3+–complexes have been investigated (Fig. 10) [377]. Complexes of Tb3+–enoxacin, Tb3+–norfloxacin, Tb3+– lomefloxacin and Tb3+–fleroxacin were shown to display a relatively strong emission intensity compared with Tb3+–ofloxacin, Tb3+–rufloxacin, Tb3+–gatifloxacin and Tb3+– sparfloxacin. Quite weak peaks with unique characters of Eu3+ at 590 and 617 nm have been observed in the luminescence spectra of Eu3+–enoxacin, however no luminescence of Eu3+ could be detected when Eu3+ was added to other fluoroquinolones. The distinct changes in emission intensities for Tb3+–fluoroquinolone and Eu3+–fluoroquinolone complexes might originate from different energy gaps between the triplet levels of fluoroquinolones and the excited levels of Ln3+. Thus, research studies in the field of complexes of fluoroquinolones with metals are aimed at obtaining of biologically active coordination compounds, and also to use of complex formation for quantitative analysis of fluoroquinolones.

Emission spectra of Tb3+–complexes of some fluoroquinolones (Reproduced with permission of Elsevier [377])
In conclusion it is worth noting that despite the successes reached in area of synthesis, studying of biological activity and application of fluoroquinolones, tasks of design of new structures, development of synthetic approaches, modifications of existing drugs by means of incorporation of substituents into positions 1–8 as well as annelation of additional rings to quinolone fragment continue to remain actual. Not less important studying of structure–activity relations among fluoroquinolones as in process of accumulation of such material all new dependences of antibacterial activity on positions and the nature of the substituents in a fluoroquinolone fragment become clear. The increasing attention is given to the synthesis of optically active isomers among fluoroquinolones and to their use as medicines. Fluoroquinolones are known to be not only antibacterial drugs, but also as compounds exhibiting other types of biological activity. Development of novel anticancer and antiviral agents in the series of fluoroquinolones is in progress. Researches in the field of metalocomplexes of fluoroquinolonecarboxylic acids directed to elucidation of “structure – bioactivity” relations and cation roles in interaction of fluoroquinolones with DNA are developed. Studying of complex formation of fluoroquinolones plays a crucial role for obtaining the fullest data on pharmacokinetic interaction of fluoroquinolones with other drugs.
References
Andriole T (ed) (1988) The quinolones. Academic Press, New York
Wolfson J, Hooper D (eds) (1989) Quinolone antimicrobial agents. American Society for Microbiology, Washington, DC
Siporin C, Heifetz C, Damaglia J (1990) The new generation of quinolones. Marcel Dekker Inc., New York
Mokrushina G, Alekseev S, Charushin V, Chupakhin O (1991) Zhurnal Vsesoyuznogo Khimicheskogo obschestva im. D.I. Mendeleeva 36:447–455
Chu D, Fernandes P (1991) Recent developments in the field of quinolone antibacterial agents. Adv Drug Res 21:39–144
Fadeeva N, Shul’gina M, Glushkov G (1993) Molecular and biological features of antibacterial action of derivatives 4-quinolon-3-carboxylic acids. Pharm Chem J 27:4–9
Mokrushina G, Charushin V, Chupakhin O (1995) Relationship between structure and antibacterial activity in the fluoroquinolone series of compounds. Pharm Chem J 29:590–606
Padeyskaya E, Yakovlev V (1995) Quinolones. Bioinform, Moscow
Andriole T (1998) The quinolones, 2nd edn. Academic Press, New York
Mokrushina G, Nosova E, Lipunova G, Charushin V (1999) Polycyclic fluoroquinolones. Russ J Org Chem 35:1447–1463
Hooper D, Rubinstein E (eds) (2003) Quinolone antibacterial agents. ASM Press, Washington, DC
Shams W, Evans M (2005) Guide to selection of fluoroquinolones in patients with lower respiratory tract infections. Drugs 65:949–991
Keam S, Croom K, Keating G (2005) Gatifloxacin: a review of its use in the treatment of bacterial infections in the US. Drugs 65:695–724
Bouzard D (1993) In: Krohn R, Kirst H, Maag H (eds) Antibiotics and antiviral compounds. Wiley, Weinheim
Hamada Y, Watanabe T, Umezu K (1999) Preparation of quinolinecarboxylic acid esters. JP Patent 11147875, 2 Jan 1999
Stankovic S, Mitov S, Stanojovic C (2003) A process for synthesis of antibiotic fluoroquinolinic acid derivatives. WO Patent 10144, 6 Feb 2003
Maslennikov E, Strunin B, Kalashnik V, Gusejnov F, Khaev E, Kovalev V (2003) Cyclocondensation method for preparing ethyl-6-fluoro-7-chloro-1,4-dihydro-4-oxo-3-quinolinecarboxylate from diethyl-2-(3-chloro-4-fluoroanilinometylenecarboxylate acid-Et ester. RU Patent 2206564, 20 June 2003
Chupakhin O, Charushin V, Rusinov V, Mokrushina G, Kotovskaya S, Baskakova Z, Kolmakova T (1996) Method for production of 1-ethyl-6-fluoro-7-(piperazinyl-1)-4-oxo-1,4-dihydro-3-quinolinecarboxylic acid. RU Patent 2054005, 10 Feb 1996
Azev Yu, Alekseev S, Charushin V, Rusinov V, Chupakhin O (1996) Process for preparing ethyl ester derivatives of 7-fluoro-1,4-dihydro-4-oxo-3-quinolinecarboxylic acid. RU Patent 2052454, 20 Jan 1996
Chupakhin O, Charushin V, Mokrushina G, Kotovskaya S, Karpenko I, Karpin I, Petrova G, Sidjrjv E, Nefedov O, Volchkov N, Lipkind M, Shajdurov V, Zabolotskich V, Shipilov A, Tolstikov G, Gruzdev V, Navashin S, Fomina I (1992) Preparation of 1-ethyl-6-fluoro-7-(4-methylpiperazinyl)-4-oxo-1,4-dihydro-3-quinolinecarboxylic acid. SU Patent 1766921, 7 Oct 1992
Dzhemilev U, Tolstikov G, Nefedov O, Chupakhin O, Charoshin V, Navashin S, Dokichev V, Sultanov S, Gruzdev V, Zverev V (1993) Preparation of ethyl 6,7-difluoro-1,4-dihydro-4-oxo-3-quinolinecarboxylate. SU Patent 1786028, 7 Jan 1993
Richardson T, Shanbhag V, Adair K, Smith S (1998) Synthesis of 7-benzoxazol-2-yl and 7-benzothiazol-2-yl-6-fluoroquinolones. J Heterocycl Chem 35:1301–1304
Kumar N, Bhandari P (1997) A new process for the preparation of 1,4-dihydro-1-alkyl-6-fluoro-4-oxo-7-(1-piperazinyl)quinoline-3-carboxylic acid derivatives. IN Patent 178696, 10 Feb 1997
Shin H, Chang J, Lee K (2005) One-pot four-step process for preparing 7-chloro-1-cyclopropyl-6-fluoro-4-oxo-1,4-dihydro-1,8-naphthyridine-3-carboxylic acid using DMF dialkyl acetals. WO Patent 40164, 6 May 2005
Gomez C, Villasante Prieto A, Francisko P (2005) One-pot process for preparing gatifloxacin. WO Patent 47260, 26 May 2005
Iki M, Ikemoto T, Sato T (2002) Process for preparing quinolinecarboxylic acid esters and 1,8-naphthyridinecarboxylic acid esters. JP Patent 155081, 28 May 2002
Lee T, Park N, Khoo J, Song S, An J (2004) Preparation of quinolonecarboxylate derivatives. WO Patent 56781, 8 July 2004
Wang Y, Chen R, Dong Z, Ben S, Nan H, Yu B, Zhao C (2002) Preparation of quinolone carboxylic acids. CN Patent 1338455, 6 Mar 2002
Randall J (2004) Preparation of quinoline derivatives as antibiotic intermediates using silylating agents for cyclization of ethoxy-substituted aromatic intermediates. WO Patent 13103, 14 Mar 2004
Muto M, Miura M, Kitagawa Y (2004) Process for the production of optically active quinolinecarboxylic acid and intermediates therefor. WO Patent 108680, 16 Dec 2004
Vales M, Lokshin V, Pepe G, Samat A, Guglielmetti R (2001) Enaminones acylation: competitive formation of quinolin-4-one and isoquinolin-1-one derivatives. Synthesis 2419–2426
Nishimura Y, Minamida A, Matsumoto K (1988) Synthesis and antibacterial activity of enoxacin analogues with a variant at position 1. Chem Pharm Bull 36:1223–1226
Ptaszynska K, Winiarski J, Zadelek S, Biedrzycki M, Dziegielewski K, Lewandowska B, Nowakowska K, Michalowska J (2004) Preparation alkyl-N-(2-fluoroethyl)-6,8-difluoro-7-(4-methyl-1-piperazinyl)-4-oxo-3-quinoline-carboxylate. Pol Patent 187824, 29 Oct 2004
Yoshikazu A, Kazuhiko I, Fujio I, Masaki H, Takayoshi I (2005) Synthesis and antibacterial activity of 1-(2-fluorovinyl)-7-substituted-4-quinolone-3-carboxylic acid derivatives, conformationally restricted analogues of fleroxacin. J Med Chem 48:3194–3202
Ritsumosa M, Sadahiro S (1994) Preparation of quinolinecarboxylic acids as intermediates for microbicides. JP Patent 0673013, 15 Mar 1994
Takemura M, Takahashi H, Sugita K, Miyauchi R (1998) Preparation of substituted cyclobutylamine derivatives as antibacterial agents. WO Patent 54169, 3 Dec 1998
Sheu J, Chen Y, Fang K, Wang T, Tzeng C, Peng C (1998) Synthesis and antibacterial activity of 1-(substituted-benzyl)-6-fluoro-1,4-dihydro-4-oxoquinoline-3-carboxylic acids and their 6,8-difluoro analogs. J Heterocycl Chem 35:955–964
Shindikar A, Viswanathan C (2005) Novel fluoroquinolones: design, synthesis, and in vivo activity in mice against Mycobacterium tuberculosis H 37 Rv. Bioorg Med Chem Lett 15:1803–1806
Aizikovich A, Nikonov M, Kodess M, Korotayev V, Charushin V, Chupakhin O (2000) Novel 1-trifluoromethyl substituted 1,2-ethylenediamines and their use for the synthesis of fluoroquinolones. Tetrahedron 56:1923–1927
Hanessian S, Saladino R, Nunez Y (1996) On the binding site of quinolone antibacterials. An attempt to probe the shen model. Bioorg Med Chem Lett 6:2333–2338
Lucero B, Gomez C, Frugulhetti I, Faro L, Alvarenga L, de Souza M, de Souza T, Ferreira V (2006) Synthesis and anti-HSV-1 activity of quinolonic acyclovir analogues. Bioorg Med Chem Lett 16:1010–1013
Masoudi A, Iman A (2003) Synthesis and reactions of some New 6,7-dihaloquinolones bearing mercapto groups. Phosphorus Sulfur Silicon Relat Elem 178:2393–2402
Zheng H, Liu J, Zhang P (2010) One-pot synthesis and antimicrobial activity of novel quinolone heterocyclic derivatives. J Heterocycl Chem 47:1411–1414
Hong W, Lee K (2006) Baylis-Hillman route to several quinolone antibiotic intermediates. Synthesis 963–968
Dumas J, Khire U, Lasch S, Nagarathnam D, Scott W (2004) Preparation of quinolinecarboxylic acid derivatives and methods for use in treating cancer. WO Patent 80465, 21 Feb 2004
Khire U, Liu X, Nagaratham D, Wood J, Wang L, Wang L, Liu D, Zhao J, Guernon L, Zhang L (2005) Quinolinecarboxylic acid derivatives for treatment of hyperproliferative conditions, their preparation and pharmaceutical compositions. WO Patent 97752, 20 Oct 2005
Kuramoto Y, Ohshita Y, Yoshida J, Yazaki A, Shiro M, Koike T (2003) A novel antibacterial 8-chloroquinolone with a distorted orientation of the N1-(5-amino-2,4-difluorophenyl) group. J Med Chem 46:1905–1917
Gordeev M, Hackbarth C, Barbachyn M, Banitt L, Gage J, Luehr G, Gomez M, Trias J, Morin S, Zurenko G, Parker C, Evans J, White R, Patel D (2003) Novel oxazolidinone–quinolone hybrid antimicrobials. Bioorg Med Chem Lett 13:4213–4216
Yoon S, Chung Y, Lee C, Oh Y, Choi D, Kim N, Lim J, Jin Y, Lee D, Lee W (1997) Synthesis, pharmacokinetics, and biological activity of a series of new pyridonecarboxylic acid antibacterial agents bearing a 5-fluoro-2-pyridyl group or a 3-fluoro-4-pyridyl group at N-1. J Heterocycl Chem 34:1021–1027
Mealy N, Castaner J (2002) Quinolone antibacterial. Drug Future 27:1033–1038
Yoon S, Yong H, Lee C, Oh Yo, Choi D, Kim N (1995) Novel quinolinecarboxylic acid derivatives. WO Patent 5373, 23 Feb 1995
Yoshikazu A, Ichiro A, Kazuhiko I, Iinuma F, Hosaka N, Ishizaki T (2005) Synthesis and antibacterial activity of the 4-quinolone-3-carboxylic acid derivatives having a trifluoromethyl group as a novel N-1 substituent. J Med Chem 48:3443–3446
Jung J, Jung Y, Park O (2001) Synthesis of 4-hydroxyquinolin-2(1H)-one analogues and 2-substituted quinolone derivatives. J Heterocycl Chem 38:61–67
Jung J, Oh S, Kim W, Park W, Kong J, Park O (2003) Synthesis and biological properties of 4-substituted quinolin-2(1H)-one analogues. J Heterocycl Chem 40:617–623
Rao V, Wentrup C (2002) Synthesis of fluorinated 2-phenyl-4-quinolones from pyrrole-2,3-diones. J Chem Soc Perkin Trans I 1232–1235
Saloutin V, Bazyl’ I, Skryabina Z, Aleksandrov G, Chupakhin O (1995) Crystalline hydrogen-bonded adducts of dimethyl sulphoxide and 7-hydroxypolyfluoroquinolones (chromones). J Fluorine Chem 74:15–18
Naik P, Chimatadar S, Nandibewoor S (2009) Kinetics and oxidation of fluoroquinoline antibacterial agent, norfloxacin, by alkaline permanganate: a mechanistic study. Ind Eng Chem Res 48:2548–2553
Pucci M, Ackerman M, Thanassi J, Shoen C, Cynamon M (2010) In vitro antituberculosis activities of ACH-702, a novel isothiazoloquinolone, against quinolone-susceptible and quinolone-resistant isolates. Antimicrob Agents Chemother 54:3478–3484
Molina-Torres C, Ocampo-Candiani J, Rendon A, Pucci M, Vera-Cabrera L (2010) In vitro activity of a new isothiazoloquinolone, ACH-702, against Mycobacterium tuberculosis and other mycobacteria. Antimicrob Agents Chemother 54:2188–2193
Marks K, Malik M, Mustaev A, Hiasa H, Drlika K, Kerns R (2011) Synthesis and evaluation of 1-cyclopropyl-2-thioalkyl-8-methoxy fluoroquinolones. Bioorg Med Chem Lett 21:4585–4588
Kondo H, Sakamoto F, Kawakami K, Tsukamoto G (1988) Studies on prodrugs. 7. Synthesis and antimicrobial activity of 3-formylquinolone derivatives. J Med Chem 31:221–225
Tanaka K, Houghton T, Kang T, Dietrich E, Delorme D, Ferreira S, Caron L, Viens F, Arhin F, Sarmiento I, Lehoux D, Fadhil I, Laquerre K, Liu J, Ostiguy V, Poirier H, Moeck G, Parr T, Far A (2008) Bisphosphonated fluoroquinolone esters as osteotropic prodrugs for the prevention of osteomyelitis. Bioorg Med Chem 16:9217–9229
Patel N, Patel A, Chauhan H (2007) Synthesis of amide derivatives of quinolone and their antimicrobial studies. Indian J Chem 46B:126–134
Patel N, Patel S, Patel J, Patel J, Corgamwala Y (2011) Synthesis and antibacterial activity of thioureido amide of fluoroquinolone. Int J Biol Chem 5:37–45
Srivastava S, Srivastava SK, Shukla A, Chauhan P, Puri S, Bhaduri A, Pandey V (1999) Synthesis and methemoglobin toxicity of the amides of 6/7 mono or disubstituted quinolone. Bioorg Med Chem Lett 9:25–30
Al-Soud Y, Al-Masoudi N (2003) A New class of dihaloquinolones bearing N’-aldehydoglycosylhydrazides, mercapto-1,2,4-triazole, oxadiazoline and α-amino ester precursors: synthesis and antimicrobial activity. J Brazilian Chem Soc 14:790–796
Patel N, Patel S (2009) Synthesis and antimicrobial activity of 2-phenyl-3-{1-cyclopropyl-6-fluoro-7-[4-methylpiperazin-1-yl]-4-quinolone}carboxamido-3-thiazolidin-4-ones. Pharm Chem J 43:305–309
Sharad S, Ganesh M, Sunil G, Charnsingh G (2010) Green synthesis and biological evaluation of some novel azoles as antimicrobial agents. Bioorg Med Chem Lett 20:7200–7204
Obanin G, Fokin A, Ya B, Ryzhkov O, Skryabina Z, Saloutin V, Chupakhin O (2000) Synthesis of N-substituted 2-(5,6,7,8-tetrafluoro-4-oxo-1,4-dihydroquinolin 3-yl)glyoxalic acids. Russ Chem Bull 49:1231–1236
Fokin A, Burgart Y, Ryzhkov O, Saloutin V (2001) Reactions of 1-aryl(alkyl)-3-ethoxalyl-5,6,7,8-tetrafluoro-1,4-d-hydrocinnolin(quinolin)-4-ones with aromatic dinucleophiles. Russ Chem Bull 50:689–692
Clark R, Wang S, Ma Z, Weitzberg M, Motter C, Tufano M, Wagner R, Gu Y, Dandliker P, Lerner C, Chovan L, Cai Y, Black-Schaefer C, Lynch L, Kalvin D, Nilius A, Pratt S, Soni N, Zhang T, Zhang X (2004) Novel inhibitors of bacterial protein synthesis: structure–activity relationships for 1,8-naphthyridine derivatives incorporating position 3 and 4 variants. Bioorg Med Chem Lett 14:3299–3302
Iones R, Barry A, Thomsberry C (1989) Antimicrobial activity of Ro 23-9424, a novel ester-linked codrug of fleroxacin and desacetylcefotaxime. Antimicrob Agents Chemother 33:944–950
Al-Hajjar F (2002) Preparation of 1-cyclopropyl-6-fluoro-7-(piperazin-1-yl)quinolin-4-ones and derivatives as antibiotics. Eur Patent 1245566, 2 Oct 2002
Kerns R, Rybak M, Kaatz G, Vaka F, Cha R, Grucz R, Diwadkar V (2003) Structural features of piperazinyl-linked ciprofloxacin dimers required for activity against drug-resistant strains of Staphylococcus aureus. Bioorg Med Chem Lett 13:2109–2112
Park C, Lee J, Jung H, Kim M, Lim S, Yeo H, Choi E, Yoon E, Kim K, Cha J, Kim S, Chang D, Kwon D, Li F, Suh Y (2007) Identification, biological activity, and mechanism of the anti-ischemic quinolone analog. Bioorg Med Chem 15:6517–6521
Nguyen S, Ding X, Butler M, Tashjian T, Peet N, Bowlin T (2011) Preparation and antibacterial evaluation of decarboxylated fluoroquinolones. Bioorg Med Chem Lett 21:5961–5963
Vysokov V, Charushin V, Afanasyeva G, Chupakhin O (1993) The synthesis of fluorinated 4H-1,4-benzothiazine-2-carboxylic acid 1,1-dioxides – thionated analogues of Pefloxacin. Mendeleev Commun 3:159–160
Guo H, Qi J (2003) Preparation of 7-(aminomethyl-azaspiroheptyl)-quinoline-carboxylic acid derivatives as bactericides. WO Patent 14108, 20 Feb 2003
Saito T, Jouno T, Tani Y, Akiba T (2001) Process for producing quinolinecarboxylic acids and intermediates thereof. WO Patent 62734, 30 Aug 2001
Guo H, Liu J, Wang Y (2004) Preparation of 5-amino-1-cyclopropyl-6-fluoro-8-methoxy-1,4-dihydroquinolin-4-one-3-carboxylic acid derivatives as antibacterial agents. CN Patent 1491944, 28 Apr 2004
Akiba T, Kitagawa Yu, Muto M (2003) Improved preparation of 5-acylamino-4-oxo-quinoline-3-carboxylic acids as bactericides and their intermediates. JP Patent 160567, 3 June 2003
Griffin J, Judice J (1999) Novel multi-binding therapeutic agents that modulate enzymatic processes. Patent 64037, 16 12 WO. 1999
Demuth T, White R (1997) 5-(N-Heterosubstituted amino)quinolone antimicrobials. US Patent 5646163, 8 July 1997
Hansen T, Gu Y, Rehm T, Dandliker P, Chovan L, Bui M, Nilius A, Beutel B (2005) Synthesis and antibacterial activity of 5-methoxy- and 5-hydroxy-6-fluoro-1,8-naphthyridone-3-carboxylic acid derivatives. Bioorg Med Chem Lett 15:2716–2719
Takahashi H, Mijauchi R, Itoh M, Takemura M, Hayakawa I (2002) Preparation of dehalogenoquinolinecarboxylic acid derivatives. WO Patent 40478, 25 May 2002
Cecchetti V, Fravolini A, Terni P Pagella P, Tabardini O (1993) 6-Aminopiperazinylquinolones and analogs, their synthesis and their use as antibacterial agents. Eur Patent 531958, 17 Mar 1993
Cecchetti V, Fravolini A, Palumbo M, Sissi C, Tabarrini O, Terni P, Xin T (1996) Potent 6-desfluoro-8-methylquinolones as new lead compounds in antibacterial chemotherapy. J Med Chem 39:4952–4957
Lawrence L, Wu P, Fan L, Gouveia K, Card A, Casperson M, Denbleyker K, Barrett J (2001) The inhibition and selectivity of bacterial topoisomerases by BMS-284756 and its analogues. J Antimicrob Chemother 48:195–201
Miyauchi R, Kawakami K, Ito M, Matsuhashi N, Ohki H, Inagaki H, Takahashi H, Takemura M (2009) Design, synthesis and biological evaluations of novel 7-[3-(1-aminocycloalkyl)pyrrolidin-1-yl]-6-desfluoro-8-methoxyquinolones with potent antibacterial activity against multi-drug resistant gram-positive bacteria. Bioorg Med Chem 17:6879–6889
Ruzic M, Pucelj J, Tomsic Z, Makuc S, Brne P, Barut M, Strancar A (2005) Process for preparing ciprofloxacin by contacting it with a novel support. WO Patent 75430, 18 Aug 2005
Niddam H, Dolitzky B, Pilarski G, Sterimbaum G (2004) Synthesis of gatifloxacin. WO Patent 69825, 19 Sep 2004
Mody S, Mehata B, Patel M, Shrikhande A, Mahajan R (1999) An improved process for the preparation of 1-ethyl-6,8-difluoro-1,4-dihydro-7-(3-methyl-1-piperazinyl)-4-oxoquinoline-3-carboxylic acid and its salts. IN Patent 177148, 5 July 1999
Berthon-Cedille L, Leguern M (2008) Process for the preparation of fluoroquinolone-3-carboxylic acids via amination of alkyl fluoro(haloquinolone)carboxylates with amines. US Patent 54643, 28 Nov 2008
Himmler T, Jaetsch T, Hallenbach W, Rast H, Wetzstein H, Heinen E, Pirro F, Scheer M, Stegemann M, Stupp H (1998) Preparation of 7-(3-vinylpiperazin-1-yl)quinolinecarboxylic acid as antibacterials. DE Patent 19651687, 1 Jan 1998
Liu B, Yang C, Xu G, Zhu Y, Cui J, Wu X, Xie Y (2005) Syntheses of quinolone hydrochloride enantiomers from synthons (R)- and (S)-2-methylpiperazine. Bioorg Med Chem 13:2451–2458
Mulvihill M, Shaber S (2004) Chemical modification of drugs into labile derivatives with enhanced properties. US Patent 254182, 16 Dec 2004
Md-Saleh S, Chilvers E, Kerr K, Milner S, Snelling A, Weber J, Thomas G, Duhme-Klair A, Routledge A (2009) Synthesis of citrate–ciprofloxacin conjugates. Bioorg Med Chem Lett 19:1496–1498
Hayakama I, Atarashi S, Kimura E (1990) Preparation of 7-(azaspiroalkanyl)quinolonecarboxylates and analogs as bactericides. RU Patent 2094432, 7 Mar 1990
Rameshkumar N, Ashokkumar M, Subramanian E, Llavarasan R, Sridhar S (2003) Synthesis of 6-fluoro-1,4-dihydro-4-oxoquinoline-3-carboxylic acid derivatives as potential antimicrobial agents. Eur J Med Chem 38:1001–1004
Yun S, Jung Y, Lee S, Lee J (1997) Antimicrobial quinoline derivatives and process for the preparation thereof. KR Patent 9703501, 18 Mar 1997
Li Y, Lu R, Yang A, Zhang Y (2004) Synthesis of novel fluoroquinolone compounds. Heterocycl Commun 10:447–450
Foroumadi A, Emami S, Hassanzadeh A, Rajaee M, Sokhanvar K, Moshafi M, Shafiee A (2005) Synthesis and antibacterial activity of N-(5-benzylthio-1,3,4-thiadiazol-2-yl) and N-(5-benzylsulfonyl-1,3,4-thiadiazol-2-yl)piperazinyl quinolone derivatives. Bioorg Med Chem Lett 15:4488–4492
Foroumadi A, Mansouri S, Kiani Z, Rahmani A (2003) Synthesis and in vitro antibacterial evaluation of N-[5-(5-nitro-2-thienyl)-1,3,4-thiadiazole-2-yl] piperazinyl quinolones. Eur J Med Chem 38:851–854
Chatterjee N, Bharat S, Naik S (1996) A process for the preparation of β-lactan antibiotic linked fluoroquinolones as hybrid antibacterial agents. IN Patent 180479, 11 Apr 1996
Zhi C, Wright G (2003) Preparation of uracils and related compounds as antibacterials that inhibit bacterial DNA polymerase III C and type II bacterial topoisomerase. US Patent 181719, 25 Sep 2003
Zhi C, Long Z, Manikowski A, Comstock J, Xu W, Brown N, Tarantino P, Karsten J, Holm A, Dix E, Wright G, Barnes M, Butler M, Foster K, LaMarr W, Bachand B, Bethell R, Cadilhac C, Charron S, Lamothe S, Motorina I, Storer R (2006) Hybrid antibacterials. DNA polymerase−topoisomerase inhibitors. J Med Chem 49:1455–1465
Zhi C. Wright G (2002) Preparation of uracils and related compounds as antibacterials that inhibit bacterial DNA polymerase III C and type II bacterial topoisomerase. WO Patent 102792, 27 Dec 2002
Darehkordi A, Javanmiri M, Ghazi S, Assar S (2011) Synthesis of N-aryl-2,2,2-trifluoroacetimidoyl piperazinylquinolone derivatives and their antibacterial evaluations. J Fluorine Chem 132:263–268
Hutschiterlen G, Specklin J, Baeschlin D, Lochev H, Sigwalt C (2004) Preparation and use of oxazolidinone-quinolinone and oxazolidinone-naphthyridinone hybrid antibiotics for the treatment of anthrax and other infections. WO Patent 96221, 12 Jan 2004
Ellsworth E, Hutchings K, Murphy S, Powell S, Sciotti R, Tran T (2005) Synthesis of azetidinyl quinolones as antibacterial agents. WO Patent 26146, 24 Mar 2005
Kato N, Iwasaki N, Azuma T (2000) Preparation of antibacterial 5-amino-8-methyl-7-pyrrolidinylquinoline-3-carboxylic acids and their intermediates. JP Patent 247970, 12 Sep 2000
Takahashi H, Ruroyanagi J, Miyauchi R, Nagamochi M, Takemura M, Hayakawa I (2005) Preparation of quinoline compounds containing pyrrolidine moiety as antibacterial agents. WO Patent 111015, 24 Nov 2005
Takemura M, Takahashi H, Ohki H, Kimura K, Miyauchi R, Takeda T (1998) Preparation of cis-substituted fluoromethylpyrrolidine derivatives of 1,4-dihydro-4-oxoquinoline-3-carboxylic acid as antibacterial agents. WO Patent 58923, 30 Dec 1998
Ellsworth E, Tayler C, Murphy S, Ranckhorst M, Starr J, Hutchings K, Limberakis C, Hoyer D (2005) Preparation of quinoline antibacterial agents. WO Patent 49602, 2 June 2005
Kim B (2001) A process for preparation of pyrrolidino-quinolinecarboxylic acid derivatives (e.g. gemifloxacin) with improved filtration. WO Patent 68649, 20 Sep 2001
Hong С, Kim Y, Lee Y, Kwak J (1998) Methyloxime-substituted aminopyrrolidine: a new surrogate for 7-basic group of quinolone. Bioorg Med Chem Lett 8:221–226
Hong C, Kim Y, Kim S, Chang J, Choi H, Nam D, Kim A, Lee J, Park K (1998) Preparation of quinoline (or naphthyridine)-3-carboxylic acids such as 7-(4-aminomethyl-3-mrthyloxyiminopyrrolidin-1-yl)-1-cyclopropyl-6-fluoro-4-oxo-1,4-dihydro-1,8-naphthyridine-3-carboxylic acid as antibacterials. US Patent 5776944, 7 July 1998
Choi H, Choi S, Nam D, Choi B (2003) Improved two-step process for preparing acid salts of gemifloxacin via Schiff-base protected intermediates. WO Patent 87100, 23 Oct 2003
Choi D, Shin J, Yang J, Yoon S, Jung Y (2004) Syntheses and biological evaluation of new fluoroquinolone antibacterials containing chiral oxiimino pyrrolidin. Bioorg Med Chem Lett 14:1273–1277
Lv K, Liu M, Feng L, Sun L, Sun Y, Wei Z, Guo H (2012) Synthesis and antibacterial activity of naphthyridone derivatives containing mono/difluoro-methyloxime pyrrolidine scaffolds. Eur J Med Chem 47:619–625
Nakayama T (2004) Preparation of intermediates for antibacterial quinoline-carboxylic acids. JP Patent 244380, 14 Feb 2004
Naoki O, Toshifumi A (2003) Process for producing antibacterial quinolone-carboxylic acid derivatives. WO Patent 97634, 10 Sep 2003
Kimura Y, Atarashi S, Kawakami K, Sato K, Hayakawa I (1994) Fluorocyclopropyl)quinolones. 2. Synthesis and stereochemical structure-activity relationships of chiral 7-(7-amino-5-azaspiro[2.4]heptan-5-yl)-1-(2-fluorocyclopropyl)quinolone antibacterial agents. J Med Chem 37:3344–3352
Yoon S, Chung Y, Lee C, Oh Y, Kim N, Lim J, Jin Y (1999) Preparation and antibacterial activity of quinolone carboxylic acid derivatives. WO Patent 00393, 12 Jan 1999
Feng L, Liu M, Wang S, Chai Y, Lv K, Shan G, Cao J, Li S, Guo H (2011) Synthesis of naphthyridone derivatives containing 8-alkoxyimino-1,6-dizaspiro[3.4]octane scaffolds. Tetrahedron 67:8264–8270
Petersen U, Schenke T, Krebs A, Grohe K, Schriewer M, Haller I, Metzger K, Endermann R, Zeiler H (1997) Preparation of 7-(1-pyrrolidinyl)-3-quinolonecarboxylic acids and naphthyridine-3-carboxylic acids as antimicrobial agents and feed additives. US Patent 5607942, 4 Mar 1997
Inagaki H, Miyauchi S, Miyauchi R, Kawato H, Ohki H, Matsuhashi N, Kawakami K, Takahashi H, Takemura M (2003) Synthesis and structure−activity relationships of 5-amino-6-fluoro-1-[(1R,2S)-2-fluorocyclopropan-1-yl]-8-methylquinolonecarboxylic acid antibacterials having fluorinated 7-[(3R)-3-(1-aminocyclopropan-1-yl)pyrrolidin-1-yl] substituents. J Med Chem 46:1005–1015
Inagaki H, Takeda T, Miyauchi R, Kawakami K, Takahashi H, Takemura M (2004) Practical synthesis of DQ-113, a new quinolone antibacterial agent, by using the intramolecular Horner-Wadsworth-Emmons reaction. Heterocycles 63:699–706
Muto M, Kitagawa Y (2004) Process for preparation of quinolinone derivatives. WO Patent 113321, 29 Dec 2004
Asahina Y, Takei M (2005) Preparation of quinolonecarboxylic acid derivatives as antibacterial agents. Eur Patent 1666477, 24 Mar 2005
Ellsworth E, Murphy S (2005) Preparation of quinolone derivatives as antibacterial agents. WO Patent 111030, 24 Nov 2005
Park T, Lee S, Han Ch (2002) Preparation of pyridinyl (pyrrolidinyl) quinolone carboxylates as antimicrobials. US Patent 130302, 20 Nov 2002
Asahina J, Takei M (2005) Preparation of quinolonecarboxylic acid derivatives as antibacterial agents. WO Patent 26147, 24 Mar 2005
Ellsworth E, Sciotti R, Stark J (2005) Preparation of pyrrolidinylquinolones as antibacterials. WO Patent 26165, 24 Mar 2005
Hubschwerlen C, Specklin J, Surivet J, Baeschlin D (2005) Preparation of oxazolidinones-quinolinones as hybrid antibiotics. WO Patent 23801, 17 Mar 2005
De Souza N, Patel M, Deshpande P, Agarwal S, Sreenivas K, Nair S, Chugh Ya, Shukla M (2003) Preparation of chiral, broad-spectrum antimicrobial 7-substituted piperidino quinolone carboxylic acids derivatives effective against multidrug-resistant bacteria. US Patent 216568, 20 Nov 2003
Hilty P, Hubschwerlen C, Thomas A (2001) Expeditious solution phase synthesis of fluoroquinolone antibacterial agents using polymer supported reagents. Tetrahedron Lett 42:1645–1646
Ganapati Reddy P, Baskaran S (2001) Microwave assisted amination of quinolone carboxylic acids: an expeditious synthesis of fluoroquinolone antibacterials. Tetrahedron Lett 42:6775–6777
Deshpande V, Ravindvanathan T (2000) An improved process for the preparation of ciprofloxacin. IN Patent 184650, 16 Nov 2000
Hu X, Kim N, Gray J, Almstead J, Seibel W, Ledoussal B (2003) Discovery of (3S)-amino-(4R)-ethylpiperidinyl quinolones as potent antibacterial agents with a broad spectrum of activity and activity against resistant pathogens. J Med Chem 46:3655–3661
Ledaussal B, Alnstead J, Grey J, Hu X (1999) Preparation of quinolones as antimicrobials. US Patent 6329391, 25 Mar 1999
Bowers GE, Macielag MJ, Xu X, Paget S, Weidner W (2005) Preparation of 7-(alkylidene-substituted-heterocyclic amino) quinolones and naphthyridones as bactericides. WO Patent 33108, 14 Apr 2005
Chai Y, Liu M, Wang B, You X, Feng L, Zhang Y, Cao J, Guo H (2010) Synthesis and in vitro antibacterial activity of novel fluoroquinolone derivatives containing substituted piperidines. Bioorg Med Chem Lett 20:5195–5198
Chai Y, Wang B, Liu M, Yi H, Sun L, You X, Guo H (2011) Design, synthesis and in vitro antibacterial activity of 7-(4-alkoxyimino-3-aminomethylpiperidin-1-yl)fluoroquinolone derivatives. Bioorg Med Chem Lett 21:3377–3380
Zhang Y, Li G, Liu M, You X, Feng L, Lv K, Cao J, Guo H (2011) Synthesis and in vitro antibacterial activity of 7-(3-alkoxyimino-5-amino/methylaminopiperidin-1-yl)fluoroquinolone derivatives. Bioorg Med Chem Lett 21:928–931
Huang X, Zhang A, Chen D, Jia Z, Li X (2010) 4-Substituted 4-(1H-1,2,3-triazol-1-yl)piperidine: novel C7 moieties of fluoroquinolones as antibacterial agents. Bioorg Med Chem Lett 20:2859–2863
Chiu C, Lewin T (1999) Process for preparing naphthyridones and intermediates. Eur. Patent 930297, 21 July1999
Okada H, Chiba K, Nakada K (1997) Preparation of pyridonecarboxylic acids and their use as antibacterial agents against Helibacter. JP Patent 9208578, 12 Aug 1997
Anquetin G, Rouquayrol M, Mahmoudi N, Santillana-Hayat M, Gozalbes R, Greiner J, Farhati K, Derouin F, Guedj R, Vierling P (2004) Synthesis of new fluoroquinolones and evaluation of their in vitro activity on Toxoplasma gondii and Plasmodium spp. Bioorg Med Chem Lett 14:2773–2776
Vilsmaier E, Goerz T (1998) Diastereoselective syntheses of N-protected derivatives of 1α,5α,6β-6-amino-3-azabicyclo[3.1.0]hexane; a route to trovafloxacin 6β-diastereomer. Synthesis 1998(5):739–744
Ota N, Shirono T, Akiba T (2003) Process for preparing of quinolinecarboxylic acid derivatives. JP Patent 96075, 3 Apr 2003
De Souza N, Patel M, Gupta S, Upadhyay D, Shukla M, Chaturvedi N, Bhawsar S, Nair S, Jafri M, Khozakiwala H (2002) Preparation and use of quinolone and naphthyridine derivatives as inhibitors of cellular efflux pumps of microbes. WO Patent 9758, 7 Feb 2002
Hagen S, Josyula V, Venkata N (2005) Preparation of substituted quinolones and derivatives there of as antibacterial agents. WO Patent 26161, 24 Mar 2005
Huang X, Chen D, Wu N, Zhang A, Jia Z, Li X (2009) The synthesis and biological evaluation of a novel series of C7 non-basic substituted fluoroquinolones as antibacterial agents. Bioorg Med Chem Lett 19:4130–4133
Norris T, Braish T, Butters M, DeVries K, Hawkins J, Massett S, Rose P, Santafianos D, Sklavounos C (2000) Synthesis of trovafloxacin using various (1α,5α,6α)-3-azabicyclo[3.1.0]hexane derivatives. J Chem Soc Perkin Trans 1 2000:1615–1622
Norris T (2000) Preparation of trovafloxacin and analogs. Eur. Patent 976749, 2 Feb 2000
Inagaki H, Takahashi H, Takemura M (2004) Synthesis and antibacterial activity of novel 6-fluoro-1-[(1R,2S)-2-fluorocyclopropan-1-yl]-4-oxoquinoline-3-carboxylic acids bearing cyclopropane-fused 2-amino-8-aza-bicyclo[4.3.0]nonan-8-yl substituents at the C-7 position. Bioorg Med Chem Lett 14:5193–5198
Himmer T, Rast H (2000) Semihydrochloride of 8-cyano-1-cyclopropyl-7-(1S,6S-2,8-diazabicyclo[4.3.0]nonan-8-yl)-6-fluoro-1,4-dihydro-4-oxo-3-quinoline-carboxylic acid. DE Patent 19854357, 31 May 2000
Himmer T, Hallenbach W, Rast H (2000) Crystal modification A of 8-cyano-1-cyclopropyl-7-(1S,6S-2,8-diazabicyclo[4.3.0]nonan-8-yl)-6-fluoro-1,4-dihydro-4-oxo-3-quinolinecarboxylic acid. DE Patent 19854356, 31 May 2000
Himmer T, Hallenbach W, Rast H (2000) Crystal modification B of 8-cyano-1-cyclopropyl-7-(1S,6S-2,8-diazabicyclo[4.3.0]nonan-8-yl)-6-fluoro-1,4-dihydro-4-oxo-3-quinoline-carboxylic acid. DE Patent 19854355, 31 May 2000
Guo H, Liu J (2005) Preparation of quinolonecarboxylic acid derivative as antibiotics. WO Patent 103048, 3 Nov 2005
Matzke M, Petersen U, Jaetsch T, Bartel S, Schenke T, Himmler T, Baasner B, Werling H, Scharler K, Labischinski H (1998) Preparation of 7-(2-oxa-5,8-diazabicyclo[4.3.0]non-8-yl)quinolone- and naphthyridinecarboxylic acid derivatives for therapy of Helicobacter pylori infections and associated gastroduodenal illnesses. DE Patent 19652239, 18 June 1998
Bhushan L, Bhushan L, Kumar S (2005) Preparation of quinolones as novel antiinfective compounds. WO Patent 19224, 3 Mar 2005
Nagibina N, Sidorova L, Klyuev N, Carushin V, Chupakhin O (1997) Application of 1,3-dipolar cycloaddition methodology for the synthesis of novel fluoroquinolones. Russ J Org Chem 33:1468–1475
Nagibina N, Charushin V, Sidorova L, Klyuev N (1998) Molecular rearrangement of 1,2,3-triazolines – adducts of 7-azido-6-fluoroquinolone-4 with alkenes. Russ J Org Chem 34:434–446
Mochulskaya N, Charushin V, Sidorova L, Chupakhin O, Tkachev A (2000) Azomethinoxide fragment in the structure modification of fluoroquinolones. Russ J Org Chem 36:1800–1808
Mochulskaya N, Sidorova L, Charushin V (2002) Three-component cyclization of hydroxylamino-substituted quinoline with reactive methylene compounds and formaldehyde: new method for the synthesis of 7-(isoxazolidin-2-yl)-6-fluoroquinolones. Russ Chem Bull 51:2106–2108
Leyva S, Leyva E (2007) Thermochemical reaction of 7-azido-1-ethyl-6,8-difluoroquinolone-3-carboxylate with heterocyclic amines. An expeditious synthesis of novel fluoroquinolone derivatives. Tetrahedron 63:2093–2097
McPherson J, Runner R, Buxton T, Hartmann J, Farcasiu D, Bereczki L, Roth E, Tollas S, Ostorhazi E, Rozgonyi F, Herczegh P (2012) Synthesis of osteotropic hydroxybisphosphonate derivatives of fluoroquinolone antibacterials. Eur J Med Chem 47:615–618
Yasumichi F, Masanori T, Yoshkazu A, Sato T, Kurasaki H, Ebisu H, Takei M, Fukuda H (2008) Preparation of mutilin derivatives containing heterocyclic aromatic carboxylic acid moiety at 14-position. Eur Patent 2149571, 27 Nov 2008
Elmore S, Cooper C, Schultz C, Hutchinson D, Donner P, Green B, Anderson D, Xie Q, Dinges J, Lynch L (2001) Quinoline- and naphthyridinecarboxylic acid antibacterials. WO Patent 32655, 10 May 2001
Zhang X, Mu F, Robinson B, Wang P (2010) Concise route to the key intermediate for divergent synthesis of C7-substituted fluoroquinolone derivatives. Tetrahedron Lett 51:600–601
Zang Z, Zhou W (2005) Arylation of nitromethane: masked nucleophilic formylation of fluoroquinolones. Tetrahedron Lett 46:3855–3858
Zang Z, Zhou W, Yu A (2004) Synthesis and antibacterial activity of 7-(substituted)aminomethyl quinolones. Bioorg Med Chem Lett 14:393–395
Zhu B, Marinelli B, Goldschmidt R, Foleno B, Hilliard J, Bush K, Macielag M (2009) Synthesis and antibacterial activity of 7-(1,2,3,4-tetrahydropyrrolo[1,2-a]pyrazin-7-yl) quinolones. Bioorg Med Chem Lett 19:4933–4936
Takemura M, Kimura Y, Takahashi H, Ishida Y (1998) Preparation and formulation of aminocyclopropylpyrrolidinylquinolone derivatives as bactericides. Eur Patent 0919553, 22 Jan 1998
Tang X, Tang X (2004) Preparation of gatifloxacin hydrobromide and application antibacterial agents. CN Patent 1548435, 24 Nov 2004
Ravikumar K, Sridhar B (2006) Moxifloxacinium chloride–water–methanol (2/1/1), a novel antibacterial agent. Acta Crystallogr C 62:478–482
Keating G, Scott L (2004) Moxifloxacin: a review of its use in the management of bacterial infections. Drugs 64:2347–2377
Xiao Y, Yong D, Li L, Liang Q, Chang Ya, Chen Yu, Lu X, Ye Z (2003) Process for the preparation of gatifloxacin. CN Patent 1461748, 17 Dec 2003
Lee E, Chris L, Bentlej T (2005) Preparation of quinolone antibacterial agents. WO Patent 26145, 24 Mar 2005
De Souza N, Patel M, Deshpande P, Agarwal S, Gupte S, Upadhyay D, Bhawsar S, Beri R, Sreenivas K, Nair S, Sheela C, Shukla M, ChughY, Shetty N, Yeole R, Reddy M (2002) Preparation of chiral broad-spectrum antimicrobial 7-substituted piperidino-quinolinecarboxylic acid derivatives. WO Patent 85886, 31 Oct 2002
Deshpande P, Bhavsar S, Chugh Y, Yeole R, De Souza N, Patel M (2005) Novel polymorphs of racemic dextrorotatory and levorotatory enantiomers of 1-cyclopropyl-6-fluoro-8-methoxy-7-(4-amino-3,3-dimethylpiperidin1-yl)-1,4-dihydro-4-oxoquinoline-3-carboxylic acid and hydrochloride and mesylate salts. WO Patent 66154, 21 July 2005
Takahashi H, Hagiwara T, Hayakawa I (2002) Preparation of fluoroquinoline drug with reduced effect on the heart. WO Patent 76458, 3 Oct 2002
Schriewer M, Grohe K, Krebs A, Petersen U, Schenke T, Haller I, Metzger K, Endermann R, Zeiler H (1992) Antibacterial 5-alkylquinolinecarboxylic acids. US Patent 5140033, 18 Aug 1992
Takemura M, Kimura Y, Takahashi H, Kimura K, Miyauchi S, Ohki H, Sugita K, Miyauchi R (2000) Preparation of cis-substitutedaminocycloalkylpyrrolidine derivatives of 1,4-dihydro-4-oxo-quinoline-3-carboxylic acids as antimicrobial drugs. US Patent 6121285, 19 Sep 2000
Gehring R, Mohrs K, Heilmann W, Diehl H (1997) Preparation of 8-methoxyquinolonecarboxylates. DE Patent 19751948, 24 Nov 1997
Ochi K, Shimizu H (1993) Preparation of 6-fluoro-7-(heterocyclic amino)-3-quinolonecarboxylic acid derivatives as intermediates for antimicrobial agents. US Patent 5869661, 2 May 1993
Takahashi H, Miyauchi R, Takemura M (2005) Preparation of 8-cyanoquinolone-carboxylic acid derivatives as antibacterial agents. WO Patent 30752, 7 Apr 2005
Bartel S, Jaetsch T, Himmler T (1997) 8-Cyano-1-cyclopropyl-7-(2,8-diazabicyclo[4.3.0]nonan-8-yl)-6-fluoro-1,4-dihydro-4-oxo-3-quinoline-carboxylic acid derivatives. WO Patent 31001, 28 Aug 1997
Stepanchikova A, Lagunin A, Filimonov D, Poroikov V (2003) Prediction of biological activity spectra for substances: evaluation on the diverse sets of drug-like structures. Curr Med Chem 10:225–233
Lagunin A, Zakharov A, Filimonov D, Poroikov V (2007) A new approach to QSAR modelling of acute toxicity. SAR QSAR Environ Res 18:285–298
Lei B, Xi L, Li J, Liu H, Yao X (2009) Global, local and novel consensus quantitative structure-activity relationship studies of 4-(phenylaminomethylene) isoquinoline-1, 3 (2H, 4H)-diones as potent inhibitors of the cyclin-dependent kinase 4. Anal Chim Acta 644:17–24
Li X, Zhu Z, Cheng X, Yang X (2007) Quantitative structure-pharmacokinetic/pharmacodynamic relationship for fluoroquinolones. Chem Pharm J 41:23–28
Wagman A, Wentland M (2007) In: Taylor J, Triggle D (eds) Comprehensive medicinal chemistry II. Elsevier, Oxford
Bryskier A (2005) In: Bryskier A (ed) Antimicrobial agents. ASM Press, Washington, DC
Dalhoff A, Schmitz F (2003) In vitro antibacterial activity and pharmacodynamics of new quinolones. Eur J Clin Microbiol Infect Dis 22:203–207
Al-Trawneh S, Zahra J, Kamal M, El-Abadelah M, Zani F, Incerti M, Cavazzoni A, Alfieri R, Petronini P, Vicini P (2010) Synthesis and biological evaluation of tetracyclic fluoroquinolones as antibacterial and anticancer agents. Bioorg Med Chem Lett 18:5873–5884
Emami S, Shafiee A, Foroumadi A (2006) Structural features of new quinolones and relationship to antibacterial activity against gram-positive bacteria. Mini-Rev Med Chem 6:375–386
Boteva A, Krasnykh O (2009) The methods of synthesis, modification and biological activity of 4-quinolones. Chem Het Comp 45:757–785
Chu D (1985) Preparation of benzoxazoloquinolines as antibacterial agents ZA Patent 02802, 27 Nov 1985
Lipunova G, Nosova E, Vasil’eva P, Charushin V (2003) Fluorinated benzimidazo[1,2-a]quinolones. Russ Chem Bull 52:457–460
Saloutin V, Burgart Y, Chupakhin O (2002) Fluorinated tricarbonyl compounds. UrO RAN, Ekaterinburg
Barrett D, Sasaki H, Kinoshita T, Sakane K (1996) A novel [3 + 2] annulation: synthesis and X-ray crystallographic structure of a novel tetrahydropyrazolo[1,5-a]quinoline, an intermediate towards new tricyclic quinolone antibacterials. J Chem Soc Chem Commun 61–62
Barrett D, Sasaki H, Kinoshita T, Tsutsumi H, Sakane K (1996) Alkylation of 1-[N -(hydroxymethyl)- N -methylamino]-4-quinolones. An improved preparation of intermediates for novel potent tricyclic quinolone antibacterial agents. Bull Chem Soc Jp 69:1371–1375
Tsoi E, Charushin V, Nosova E, Lipunova G, Tkachev A (2001) New approach to [a]-fused fluoroquinolones: the synthesis of 5-oxo-1,2,3,3a,4,5-hexahydropyrrolo[1,2-a]quinolines. Mendeleev Commun 11:53–55
Edmont D, Marot C, Chenault J (2002) Synthesis of novel fused tricyclic quinolones: 4a,5-dihydro-1H-[1;2,4]triazino[1,6-a]quinoline-2,4,6(3H)-triones. J Heterocycl Chem 39:1161–1167
Edmont D, Chenault J (2003) 8-Fluoro-4-hydroxy-1H-[1,2,4]triazino[4,5-a]-quinoline-1,6(2H)-dione: synthesis and reactivity. J Heterocycl Chem 40:789–793
Edmont D, Chenault J (2001) A convenient selective N-alkylation of 4-Oxo-1,4-dihydro-2-quinoline carboxylic acid. Synlett 6:833–837
Azev Y, Shorshnev S, Gabel’ D, Dul’ks T (2003) Intramolecular thermal condensation of 3-acetyl-5-oxopyrazolo[1,5-a]quinoline-4-ethylcarboxylate: a simple pathway to the new tetracyclic system containing fluoroquinolone fragment. Pharm Chem J 37:327–328
Gao Y (2004) Preparation of prulifloxacin from 3,4-difluoroaniline and 3-hydroxy-2-butanone. CN Patent 1478781, 3 Mar 2004
Segawa J, Kazuno K, Matsuoka M, Amimoto I, Ozaki M, Matsuda M, Tomii Y, Kitano M, Kise M (1995) Studies on pyridonecarboxylic acids. IV. Synthesis and antibacterial activity evaluation of S-(-)- and R-(+)-6-fluoro-1-methyl-4-oxo-7-(1-piperazinyl)-4H-[1,3]thiazeto-[3,2-a]quinoline-3-carboxylic acids. Chem Pharm Bull 43:1238–1240
Segawa J, Kazuno K, Matsuoka M, Shiranase I, Ozaki M, Matsuda M, Tomii Y, Kitano M, Kise M (1995) Studies on pyridonecarboxylic acids. III. Synthesis and antibacterial activity evaluation of 1,8-disubstituted 6-fluoro-4-oxo-7-piperazinyl-4H-[1,3]thiazeto[3,2-a]quino-line-3-carboxylic acid derivatives. Chem Pharm Bull 43:63–70
Petersen U, Matzke M, Jaetsch T, Schenke T, Himmler T, Bartel S, Baasner B, Werling H, Schaller K, Labischinski H, Endermann R (1998) Use of 7-(1-aminomethyl-2-oxa-7-azabicyclo[3.3.0.]oct-7-yl)quinolonecarboxylates, naphthyridinones and related compounds for Helibacter pylori infection therapy and associated gastroduodenalillinesses. DE Patent 19652219, 18 June 1998
Matsuoka M, Segawa J, Makita Y (1997) Studies on pyridonecarboxylic acids. V. A practical synthesis of ethyl 6,7-difluoro-1-methyl-4-oxo-4H-[1,3]thiazeto[3,2-a]quinoline-3-carboxylate, a key intermediate for the new tricyclic quinolone, prulifloxacin (NM441) and versatile new syntheses of the 2-thioquinoline skeleton. J. Heterocycl Chem 34:1773–1779
Keam S, Perry C (2004) Prulifloxacin. Drugs 64:2221–2234
Matsuoka M, Segawa J, Aminito I, Masui Y, Tomii Y, Kitano M, Kise M (1999) Synthesis and antibacterial activity of novel 7-substituted 6-fluoro-1-methylene-4-oxo-4H-[1,3]thiazeto[3,2-a]quinoline-3-carboxylic acid derivatives. Heterocycles 51:2915–2930
Petersen U, Schenke T, Saetsch T, Bartel S, Bremm K, Endermann R, Metzger K (1994) Preparation of quinolone and naphthyridine carboxylic acid-derivative antibiotics. DE Patent 4427530 4 Aug 1994
Cecchetti V, Cruciani G, Filipponi E, Fravolini A, Tabarrini O, Xin T (1997) Synthesis and antibacterial evaluation of [1,3]benzothiazino[3,2-a]quinoline- and [3,1]benzothiazino[1,2-a]quinoline-6-carboxylic acid derivatives. Bioorg Med Chem 5:1339–1344
Wiles J, Song Y, Wang Q, Lucien E, Hashimoto A, Cheng J, Marlor C, Ou Y, Podos S, Thanassi J, Thoma C, Deshpande M, Pucci M, Bradbury B (2006) Biological evaluation of isothiazoloquinolones containing aromatic heterocycles at the 7-position: in vitro activity of a series of potent antibacterial agents that are effective against methicillin-resistant Staphylococcus aureus. Bioorg Med Chem Lett 16:1277–1281
Wiles J, Hashimoto A, Thanassi J, Cheng J, Incarvito C, Deshpande M, Pucci M, Bradbury B (2006) Isothiazolopyridones: synthesis, structure, and biological activity of a new class of antibacterial agents. J Med Chem 49:39–42
Bradbury B, Deshphande M, Pucei M, Wang Q, Wiles J, Song M, Hashimoto A, Lucien E (2005) Preparation of isothiazoloquinolones and related compounds as antiinfective agents. WO Patent 19228, 3 Mar 2005
Wiles J, Wang Q, Lucien E, Hashimoto A, Song Y, Cheng J, Marlor C, Ou Y, Podos S, Thanassi J, Thoma C, Deshpande M, Pucci M, Bradbury B (2006) Isothiazoloquinolones containing functionalized aromatic hydrocarbons at the 7-position: synthesis and in vitro activity of a series of potent antibacterial agents with diminished cytotoxicity in human cells. Bioorg Med Chem Lett 16:1272–1276
Hashimoto A, Pais G, Wang Q, Lucien E, Incarvito C, Deshpande M, Bradbury B, Wiles J (2007) Practical synthesis and molecular structure of a potent broad-spectrum antibacterial isothiazoloquinolone. Org Process Res Dev 11:389–398
Wang Q, Lucien E, Hashimoto A, Pais G, Nelson D, Song Y, Thanassi J, Marlor C, Thoma C, Cheng J, Podos S, Ou Y, Deshpande M, Pucci M, Buechter D, Bradbury B, Wiles J (2007) Isothiazoloquinolones with enhanced antistaphylococcal activities against multidrug-resistant strains: effects of structural modifications at the 6-, 7-, and 8-positions. J Med Chem 50:199–210
Kim H, Wiles J, Wang Q, Pais G, Lucien E, Hashimoto A, Nelson D, Thanassi J, Podos S, Deshpande M, Pucci M, Bradbury B (2011) Exploration of the activity of 7-pyrrolidino-8-methoxyisothiazoloquinolones against methicillin-resistant Staphylococcus aureus (MRSA). J Med Chem 54:3268–3282
Kawamura K, Michara S, Nukii S, Uchida I (2003) Preparation of 1-methyl-1,4-dihydro-9H-pyrazolo[4,3-b]-quinoline-9-one derivatives as protein kinase C inhibitors. JP Patent 55376, 26 Feb 2003
Fujita M, Egawa H, Kataoka M, Miyamoto T, Nakano J, Matsumoto J (1995) Imidazo- and triazoloquinolones as antibacterial agents. Synthesis and structure-activity relationships. Chem Pharm Bull 43:2123–2132
Fujita M, Egawa H, Miyamoto T, Nakano J, Matsumoto J (1996) 5-Alkoxyimidazoquinolones as potential antibacterial agents. Synthesis and structure-activity relationships. Chem Pharm Bull 44:987–990
Cooper C, Tufano M, Donner P, Chu D (1996) The synthesis and in vitro antibacterial activity of conformationally restricted quinolone antibacterial agents. Bioorg Med Chem 4:1307–1315
Yusuf M, Monther A, Khanfar A, Shuheil M, Ei-Abadelah M, Boese R (2006) Heterocycles [h]fused onto 4-oxoquinolines. Part I. Synthesis of 6-Oxo-6,9-dihydro[1,2,5]oxadiazolo[3,4-h]quinoline-7-carboxylic acid N-oxide. Heterocycles 68:1163–1172
Al-Qawasmeh R, Zahra J, Zani F, Vicini P, Boese R, El-Abadelah M (2009) Synthesis and antibacterial activity of 9-cyclopropyl-4-fluoro-6-oxo-6,9-dihydro-[1,2,5]thiadiazolo[3,4-h]-quinoline-7-carboxylic acid and its ethyl ester. Arkivoc 2009(12):322–336
Al-Dweik M, Zahra J, Khanfar M, El-Abadelah M, Zeller K, Voelter W (2009) Heterocycles [h]-fused to 4-oxoquinoline-3-carboxylic acid. Part VII: synthesis of some 6-oxoimidazo[4,5-h]quinoline-7-carboxylic acids and esters. Monatsh Chem 140:221–228
Sidorenko S (2006) Levofloxacin nowadays. Antibiot Chemother 51:28–37
Tunitskaya V, Khomutov А, Kochetkov S, Kotovskaya S, Charushin V (2011) Inhibiting DNA-gyrase by levofloxacin and other derivatives of fluoroheterocycles. Acta Nat 3:98–104
Dorgan R (1997) Preparation of pyrido[3,2,1-i,j]-1,3,4-benzoxadiazines as antibacterial agents. WO Patent 26261, 24 July 1997
Tanba H, Imai E, Mao S (2004) Preparation of optically active tricyclic compounds without forming diastereomers. JP Patent 99494, 2 Apr 2004
Wang B, Wang J (2002) Preparation of levofloxacin CN Patent 1357547, 10 Jan 2002
Wang J, Wang B (2002) The process comprises substituting 2,4,5-trifluoro-3-nitrobenzoyl fluoride with Cl2 at 190–195 °C for 16-18 h to obtain 3-chloro-2,4,5-trifluorobenzoyl fluoride substituting. CN Patent 1357548, 10 Jan 2002
Shirato S (2007) Preparation of 3S-9,10-difluoro-2,3-dihydro-3-methyl-7-oxo-7H-pyrido[1,2,3-de][1,4]benzoxazine-6-carboxylic acid. JP Patent 210914, 23 Aug 2007
Patel M, Gupte S, Chugh Y, Saoji D, Agarwal S, deSouza N, Khorakiwala H (2000) Antibacterial optically pure benzoquinolizinecarboxylic acid derivatives processes, compositions and methods of treatment. WO Patent 68229, 16 Nov 2000
Yang Y, Ji R, Chen K (1999) A practical stereoselective synthesis of (S)-(-)-ofloxacin. Chin J Chem 17:539–544
Kim S, Kang S, Seo H, Kim J, Na H (2006) Cloning and characterization of ofloxacin ester-enantioselective lipase, and use for levofloxacin production. KR Patent 0109105, 19 Oct 2006
Lee S, Min B, Hwang S, Koo Y, Lee C, Song S, Oh S, Min S, Lin S, Kim D (2001) Enantioselective production of levofloxacin by immobilized porcine liver esterase. Biotechnol Lett 23:1033–1037
Lee S, Min B, Seong S, Oh S, Lim S, Kim S, Kim D (2001) Polyacrylamide gel immobilization of porcine liver esterase for the enantioselective production of levofloxacin. Biotec Bioprocess Eng 6:179–182
Kang S, Park S, Kim Y, Kim Y (1997) An improved synthesis of levofloxacin. Heterocycles 45:137–145
Lee B, Shin S (2002) Process for preparation of alkyl 2-(2,3,5-trifluoro-4-(4-methyl-1-piperazinyl)) benzoyl-3(S)-((1-hydroxyprop-2-yl)amino)acrylate KR Patent 0026961, 13 Apr 2002
Lee B, Shin S (2001) 248. Ethyl 2-2,3,5-trifluoro-4-(4-methyl-1-piperazinyl)benzoyl-3(S)-(1-hydroxyprophy-2-ylamino)acrylate and method for manufacturing the same. KR Patent 0018722, 15 Mar 2001
Lee B, Shin S (2001) Method for manufacturing ethyl 2,3,5-trifluoro-4-(4-methyl-1-piperazinyl)benzoylacetate. KR Patent 0018721, 15 Mar 2001
Lee B, Shin S (2008) Process for preparation of levofloxacin WO Patent 077643, 7 Mar 2008
Kawakami K, Atarashi S, Kimura Y, Takemura M, Hayakawa I (1998) Synthesis and antibacterial activity of novel pyridobenzoxazine analogues. Chem Pharm Bull 46:1710–1715
Park Y, Lee H, Kim M, Kim K (2000) Preparation of (-)-pyrido-benzoxazine carboxylates from (+)-ethyl-2-(4-chloro-5-fluoro-2-halo-3-nitrobenzoyl)-3-[(1-hydroxypropy-2(S)-amino)]acrylate. WO Pat 50428, 31 Aug 2000
Kim Y, Kang S, Park S (1999) Method for the preparation of 9-fluoro-7-oxo-7H-pyrido[1,2,3-de][1,4]benzoxazine-6-carboxylic acid derivatives. US Patent 5952494, 14 Sep 1999
Adrio J, Carretero J, Ruano J, Pallares A, Vicioso M (1999) An efficient synthesis of ofloxacin and levofloxacin from 3,4-difluoroaniline. Heterocycles 51:1563–1572
Sato K, Takayanagi Y, Okano K, Nakayama K, Imura A, Iton M, Yagi T, Kobayashi Y, Nagai T (2001) Process for the preparation of benzoxazine derivatives and intermediates therefor. Eur Patent 1211254, 12 Feb 2001
Chupakhin O, Krasnov V, Levit G, Charushin V, Korolyova M, Tzoi E, Lee H, Park Y, Kim M, Kim K (2000) Preparation of (S)-benzoxazines and racemization of (R)-benzoxazines. JP Patent 178265, 17 June 2000
Charushin V, Krasnov V, Levit G, Korolyova M, Kodess M, Chupakhin O, Kim M, Lee H, Park Y, Kim K (1999) Kinetic resolution of (±)-2,3-dihydro-3-methyl-4H-1,4-benzoxazines with (S)-naproxen. Tetrahedron Asymmetry 10:2691–2695
Krasnov V, Levit G, Bukrina I, Andreeva I, Sh L, Sadretdinova L, Korolyova M, Kodess M, Charushin V, Chupakhin O (2003) Kinetic resolution of (±)-2,3-dihydro-3-methyl-4H-1,4-benzoxazine, (±)-2-methyl-1,2,3,4-tetrahydroquino-line and (±)-2-methylindoline using N-tosyl-(S)-prolyl chloride. Tetrahedron Asymmetry 14:1985–1989
Krasnov V, Levit G, Kodess M, Charushin V, Chupakhin O (2004) N-phthaloyl-(S)-alanyl chloride as a chiral resolving agent for the kinetic resolution of heterocyclic amines. Tetrahedron Asymmetry 15:859–864
Krasnov V, Levit G, Andreeva I, Grishakov A, Charushin V, Chupakhin O (2002) Kinetic resolution of (±)-2-methyl-1,2,3,4-tetrahydroquinoline and (±)-2-methylindoline. Mendeleev Commun 12:27–29
Potemkin V, Krasnov V, Levit G, Bartashevich E, Andreeva I, Kuzminsky M, Anikin N, Charushin V, Chupakhin O (2004) Kinetic resolution of (±)-2,3-dihydro-3-methyl-4H-1,4-benzoxazine in the reaction with (S)-naproxen chloride: a theoretical study. Mendeleev Commun 14:69–71
Rueping M, Stoeckel M, Sugiono E, Theissmann T (2010) Asymmetric metal-free synthesis of fluoroquinolones by organocatalytic hydrogenation. Tetrahedron 66:6565–6568
Takemura M, Takahashi H, Kawakami K (1996) Preparation of pyridobenzoxazine derivatives as antibacterial agents. WO Patent 13370, 27 Sep 1996
Han C, Lee J, Lobkovsky E, Porco J (2005) Catalytic ester − amide exchange using group (IV) metal alkoxide − activator complexes. J Am Chem Soc 127:10039–10044
Cociorva O, Li B, Nomanbhoy T, Li Q, Nakamura A, Nakamura K, Nomura M, Okada K, Seto S, Yumoto K, Liyanage M, Zhang M, Aban A, Leen B, Szardenings A, Rosenblum J, Kozarich J, Kohno Y, Shreder K (2011) Synthesis and structure–activity relationship of 4-quinolone-3-carboxylic acid based inhibitors of glycogen synthase kinase-3β. Bioorg Med Chem Lett 21:5948–5951
Takamura M, Ohki H (2005) Preparation of pyridobenzoxazine derivatives as antibacterial agents. WO Patent 73238, 11 Aug 2005
Jefferson E, Swayze E, Osgood S, Miyaji A, Risen L, Blyn L (2003) Antibacterial activity of quinolone–macrocycle conjugates. Bioorg Med Chem Lett 13:1635–1638
Asahina Y, Takei M (2003) Preparation of 10-(3-cyclopropylaminomethyl-1-pyrrolidinyl)pyridobenzoxazinecarboxylic acid derivatives effective against resistant bacteria. WO Patent 78439, 25 Sep 2003
Bortolaso R, Stivanello M (2002) Process for the preparation of marbofloxacin via benzyl ether intermediates. IT Patent 1313683, 9 Sep 2002
Lipunova G, Nosova E, Charushin V, Sidorova L, Chasovskikch O (1998) 1,3,4-Oxa(thia)diazino [i, j]-annelated quinolines: a new type of key intermediate in the synthesis of tricyclic fluoroquinolones. Mendeleev Commun 8:131–133
Lipunova G, Sidorova L, Nosova E, Perova N, Charushin V, Aleksandrov G (1999) Derivatives of 1,3,4-thiadiazino[6,5,4-i, j]quinoline –new heterocyclic system. Zhurn Org Khim (Russ J Org Chem) 35:1729–1735
Lipunova G, Nosova E, Charushin V, Chasovskikh O (2001) Synthesis of fluorinated 1,3,4-oxadiazino[6,5,4-i, j]quinolines. Chem Heterocycl Compd 37:1278–1288
Nosova E, Sidorova L, Lipunova G, Mochul’skaya N, Chasovskikh O, Charushin V (2002) Synthesis of new fluorinated derivatives of quinolinecarboxylic acids. Chem Heterocycl Compd 38:922–928
Lipunova G, Nosova E, Mochul’skaya N, Andreiko A, Chasovskikh O, Charushin V (2002) 1,2,4-Triazino[5,6,1-i, j]quinolines: a new type of tricyclic analogs of fluoroquinolones. Russ Chem Bull 51:663–667
Nosova E, Lipunova G, Sidorova L, Charushin V (2001) New derivatives of 1,3,4-thiadiazino[6,5,4-i, j]quinoline. Russ J Org Chem 37:1169–1176
Nosova E, Lipunova G, Charushin V (2001) Synthesis and antibacterial activity of 1,3,4-thia(oxa)diazino[6,5,4-i, j]quinoline derivatives. Pharm Chem J 35:599–601
Lipunova G, Nosova E, Kravchenko M, Sidorova L, Tsoi E, Mokrushina G, Chasovskikh O, Charushin V (2004) Fluorinated quinolones possessing antituberculous activity. Pharm Chem J 38:597–601
Hu G, Zhang Z, Huang W (2004) Synthesis and antibacterial activity of new tetracyclic triazolothiadiazino fluoroquinolones. Chin Chem Lett 15:23–25
Miao H, Ceccetti V, Tabarrini O, Fravolini A (2000) New 1,8-peri-annelated tricyclic quinolone antibacterials. J Heterocycl Chem 37:297–302
Kwon Y, Na Y (2006) Study on the synthesis and cytotoxicity of new quinophenoxazine derivatives. Chem Pharm Bull 54:248–251
Schwaebe M, Nagasawa J, Haggach M (2008) Preparation of fused pyridone hydrazides as anticancer drugs. WO Patent 131134, 30 Oct 2008
Lipunova G, Mokrushina G, Nosova E, Rusinova L, Charushin V (1997) Novel pentacyclic fluoroquinolones. Mendeleev Commun 7:109–111
Nosova E, Lipunova G, Mokrushina G, Chasovskikh O, Rusinova L, Charushin V (1998) Novel pentacyclic fluoroquinolones. Zhurn Org Khim (Russ J Org Chem) 34:436–441
Charushin V, Nosova E, Lipunova G, Kodess M (2001) Fused fluoroquinolones: synthesis and 1H and 19 F NMR studies. J Fluorine Chem 110:25–28
Wang E, Zhang X, Wu W (2003) Preparation of tetracyclic fluoroquinolonecarboxylates as antibacterial agents. CN Patent 1425668, 25 June 2003
Wang E, Zhang X, Wu B, Wu W (2003) Preparation of tetracyclic fluoroquinolonescarboxylates as antibacterial agents. CN Patent 1425669, 25 June 2003
Shaharyar M, Ali M, Abdullah M (2007) Synthesis and antiproliferative activity of 1-[(sub)]-6-fluoro-3-[(sub)]-1, 3,4-oxadiazol-2-yl-7-piperazino-1, 4-dihydro-4-quinolinone derivatives. Med Chem Res 16:292–299
Tabarrini O, Massari S, Daelemans D, Stevens M, Manfroni G, Sabatini S, Balzarini J, Cecchetti V, Pannecouque C, Fravolini A (2008) Structure − activity relationship study on anti-HIV 6-desfluoroquinolones. J Med Chem 51:5454–5458
Edmont D, Rocher R, Plisson C, Chenault J (2000) Synthesis and evaluation of quinoline carboxyguanidines as antidiabetic agents. Bioorg Med Chem Lett 10:1831–1834
Srivastava S, Chauhan P, Bhaduri A, Fatima N, Chatterjee R (2000) Quinolones: novel probes in antifilarial chemotherapy. J Med Chem 43:2275–2279
Dixit S, Mishra N, Sharma M, Singh S, Agarwal A, Awasthi S, Bhasin V (2012) Synthesis and in vitro antiplasmodial activities of fluoroquinolone analogs. Eur J Med Chem 51:52–59
Anderson V, Osheroff N (2001) Type II topoisomerases as targets for quinolone antibacterials turning Dr. Jekyll into Mr. Hyde. Curr Pharm Des 7:337–353
Zeng Q, Kwok Y, Kerwin S, Mangold G, Hurley L (1998) Design of new topoisomerase II inhibitors based upon a quinobenzoxazine self-assembly model. J Med Chem 41:4273–4278
Whitten J, Schwaebe M, Siddiqui-Jain A, Moran T (2005) Preparation of substituted quinobenzoxazine analogs as antitumor agents. US Patent 200585468, 21 Apr 2005
Whitten J, Pierre F, Schwaebe M (2006) Quinobenzoxazine analogs binding to G quartet structure in DNA and their preparation, pharmaceutical compositions, pharmacokinetics and use for treatment of proliferative diseases. WO Patent 113509, 26 Oct 2006
Kim M, Duan W, Gleason-Guzman M, Hurley L (2003) Design, synthesis, and biological evaluation of a series of fluoroquinoanthroxazines with contrasting dual mechanisms of action against topoisomerase II and G-quadruplexes. J Med Chem 46:571–583
Kwok Y, Zeng Q, Hurley L (1999) Structural insight into a quinolone-topoisomerase II-DNA complex. J Biol Chem 274:17226–17235
Schwaebe M, Ryckman D, Nagasawa J, Pierre F, Vialettes A, Haddach M (2011) Facile and efficient generation of quinolone amides from esters using aluminum chloride. Tetrahedron Lett 52:1096–1100
Kang D, Kim J, Jung M, Lee E, Jahng Y, Kwon Y, Na Y (2008) New insight for fluoroquinophenoxazine derivatives as possibly new potent topoisomerase I inhibitor. Bioorg Med Chem Lett 18:1520–1524
Duan W, Rangan A, Vankayalapati H, Kim M, Zeng Q, Sun D, Han H, Fedorov O, Nishioka D, Rha S, Lzbicka E, Von Hoff D, Hurley L (2001) Design and synthesis of fluoroquinophenoxazines that interact with human telomeric G-quadruplexes and their biological effects. Mol Cancer Ther 1:103–120
Kwok Y, Sun D, Clement J, Hurley L (1999) The quinobenzoxazines: relationship between DNA binding and biological activity. Anti-Cancer Drug Des 14:443–447
Azema J, Guidetti B, Dewelle J, Calve B, Mijatovic T, Korolyov A, Vaysse J, Malet-Martino M, Martino M, Kiss R (2009) 7-((4-Substituted)piperazin-1-yl) derivatives of ciprofloxacin: synthesis and in vitro biological evaluation as potential antitumor agents. Bioorg Med Chem 17:5396–5407
Whitten J, Schwaebe M, Siddiqui-J, Moran T (2004) Preparation of substituted quinolene analogs as antitumor agents. WO Patent 91504, 16 Feb 2004
Qin Y, Hurley L (2008) Structures, folding patterns, and functions of intramolecular DNA G-quadruplexes found in eukaryotic promoter regions. Biochimie 90:1149–1171
Kelland R (2005) Overcoming the immortality of tumour cells by telomere and telomerase based cancer therapeutics – current status and future prospects. Eur J Cancer 41:971–979
Parkinson G, Lee M, Neidle S (2002) Crystal structure of parallel quadruplexes from human telomeric DNA. Nature 417:876–880
Whitten J, Pierre F, Regan C, Schwaebe M, Yiannikouros G, Jung M (2006) Preparation of fused quinolone analogs which inhibit cell proliferation and/or induce cell apoptosis. US Patent 0063761, 6 Apr 2006
Hurley L, Guzman M (2007) Combination cancer chemotherapy. WO Patent 137000, 29 Nov 2007
Lipunova G, Nosova E, Mokrushina G, Sidorova L, Charushin V (2000) Antitumor activity of the fluorinated derivatives of condensed quinolines and quinazolines. Pharm Chem J 34:19–22
Lipunova G, Nosova E, Sidorova L, Charushin V (2011) Synthesis and antitumor activity of fluorinated derivatives of [i, j]-annelated quinolones. Pharm Chem J 45:208–210
Korolyov A, Dorbes S, Azema J, Guidetti B, Danel M, Lamoral-Theys D, Gras T, Dubois J, Kiss R, Martino R, Malet-Martino M (2010) Novel lipophilic 7H-pyrido[1,2,3-de]-1,4-benzoxazine-6-carboxylic acid derivatives as potential antitumor agents: improved synthesis and in vitro evaluation. Bioorg Med Chem 18:8537–8548
Wentland M, Aldous S, Gruett M, Perni R, Powles R, Danz D, Klingbeil K, Peverly A, Robinson R, Corbett T, Rake J, Coughlin S (1995) The antitumor activity of novel pyrazoloquinoline derivatives. Bioorg Med Chem Lett 5:405–410
Kamal A, Devaiah V, Reddy K, Kumar M (2005) Synthesis and biological activity of fluoroquinolone-pyrrolo[2,1-c][1,4]benzodiazepine conjugates. Bioorg Med Chem 13:2021–2029
Khire U, Liu X, Nagarathnam D, Wood J, Wang L, Liu D, Zhao J, Guernon L, Zhang L (2005) Quinolonecarboxylic acid derivatives for treatment of hyperproliferative conditions, their preparation and pharmaceutical compositions. WO Patent 097752, 20 Oct 2005
Qidong Y, Xungui H, Zhiyu L (2004) Preparation of quinolone derivatives as antitumor agents. CN Patent 1473827, 11 Feb 2004
Tomita K, Tsuzuki Y, Shibamori K, Tashima M, Kajikawa F, Sato Y, Kashimoto S, Chiba K, Hino K (2002) Synthesis and structure − activity relationships of novel 7-substituted 1,4-dihydro-4-oxo-1-(2-thiazolyl)-1,8-naphthyridine-3-carboxylic acids as antitumor agents. Part 1. J Med Chem 45:5564–5575
Tsuzuki Y, Tomita K, Shibamori K, Sato Y, Kashimoto S, Chiba K (2004) Synthesis and structure − activity relationships of novel 7-substituted 1,4-dihydro-4-oxo-1-(2-thiazolyl)-1,8-naphthyridine-3-carboxylic acids as antitumor agents. Part 2. J Med Chem 47:2097–2109
Tsuzuki Y, Tomita K, Sato Y, Kashimoto S, Chiba K (2004) Synthesis and structure–activity relationships of 3-substituted 1,4-dihydro-4-oxo-1-(2-thiazolyl)-1,8-naphthyridines as novel antitumor agents. Bioorg Med Chem Lett 14:3189–3193
Whitten J, Schwaebe M, Moran T (2004) Preparation of heterocyclic-substituted 1,4-dihydro-4-oxo-1,8-naphthyridine analogs. WO Patent 91627, 28 Jan 2004
Xu J, Cole D, Chang C, Ayyad R, Asselin M, Hao W, Gibbons J, Jelinsky S, Saraf K, Park K (2008) Inhibition of the signal transducer and activator of transcription-3 (STAT3) signaling pathway by 4-Oxo-1-phenyl-1,4-dihydroquinoline-3-carboxylic acid esters. J Med Chem 51:4115–4121
Bryskier A, Lowther J (2005) Antituberculoses agents. In: Bryskier A (ed) Antimicrobial agents. ASM Press, Washington, DC
Aubry A, Pan X, Fisher M, Jarlier V, Cambau E (2004) Mycobacterium tuberculosis DNA gyrase: interaction with quinolones and correlation with antimycobacterial drug activity. Antimicrob Agents Chemother 48:1281–1288
Artico M, Nai A, Sbardella G, Massa S, Musiu C, Lostia S, Demontis F, Colla P (1999) Nitroquinolones with broad-spectrum antimycobacterial activity in vitro. Bioorg Med Chem Lett 9:1651–1656
Imramovsky A, Polanc S, Vinsova J, Kocevar M, Jampilek J, Reckova Z, Kaustova J (2007) A new modification of anti-tubercular active molecules. Bioorg Med Chem 15:2551–2559
Vavrikova E, Polanc S, Kocevar M, Horvati K, Bosze S, Stolarikova J, Vavrova K, Vinsova J (2011) New fluorine-containing hydrazones active against MDR-tuberculosis. Eur J Med Chem 46:4937–4945
Senthilkumar P, Dinakaran M, Banerjee D, Devakaram R, Yogeeswari P, China A, Nagaraja V, Sriram D (2008) Synthesis and antimycobacterial evaluation of newer 1-cyclopropyl-1,4-dihydro-6-fluoro-7-(substituted secondary amino)-8-methoxy-5-(substituted)-4-oxoquinoline-3-carboxylic acids. Bioorg Med Chem 16:2558–2569
Sharma K, Fernandes P (2006) Synthesis and biological activity of substituted quinolones derived from 6-fluoro-3-carbethoxy-1H-quinolin-4-one. Ind J Heterocycl Chem 15:253–258
Lakhina V, Zinchenko E, Yarotskij S, Charushin V, Chupakhin O, Tsoj E, Shorshnev S (1997) Antibiotic of rifamycin order showing antibacterial and antimycobacterial antituberculosis activity. RU Patent 2098419, 10 Dec 1997
Sheu J, Chen Y, Tzeng C, Hsu S, Fang K, Wang T (2003) Synthesis, and antimycobacterial and cytotoxic evaluation of certain fluoroquinolone derivatives. Helv Chim Acta 86:2481–2489
Fedorova O, Rusinov G, Mordovskoj G, Zueva M, Kravchenko M, Ovchinnikova I, Chupakhin O (1997) Synthesis and tuberculostatic activity of podands with fluoroquinoline fragment. Khim Farm Zhurn (Chem Pharm J) 31:21–23
Dinarakan M, Senthilkumar P, Yogeeswari P, China A, Nagaraja V, Sriram D (2008) Antimycobacterial activities of novel 2-(sub)-3-fluoro/nitro-5,12-dihydro-5-oxobenzothiazolo[3,2-a]quinoline-6-carboxylic acid. Bioorg Med Chem 16:3408–3418
Dinarakan M, Senthilkumar P, Yogeeswari P, China A, Nagaraja V, Srieam D (2008) Novel ofloxacin derivatives: synthesis, antimycobacterial and toxicological evaluation. Bioorg Med Chem Lett 18:1229–1236
Bartel S, Kleefeld G, Schulze T, Paessens A, Neumann R, Reefschlaeger J, Streissle G (1994) Quinolone- and naphthyridinecarboxylic acids. DE Patent 4303657, 11 Aug 1994
Ceccetti V, Parolin C, Moro S, Pecere T, Filipponi E, Calistri A, Tabarrini O, Gatto B, Palumbo M, Fravolini A, Palu G (2000) 6-aminoquinolones as new potential anti-HIV agents. J Med Chem 43:3799–3802
Witvrouw M, Daelemans D, Pannecouque C (1998) Broad-spectrum antiviral activity and mechanism of antiviral action of the fluoroquinolone derivative K-12. Antivir Chem Chemother 9:403–411
Kimura T, Katsube T (1993) Preparation of aminoquinolone derivatives as anti-HIV agents. US Patent 5519016, 1 Dec 1993
Kimura T, Katsube T, Nishigaki T (1997) Preparation of trifluoromethyl(piperazinyl)quinolinecarboxylic acids as anti-HIV agents. JP Patent 09249568, 22 Sep 1997
Ohmine T, Katsube T, Tsuzaki Y, Kazui M, Kobayashi N, Komai T, Hagihara M, Nishigaki T, Iwamoto A, Kimura T, Kashiwase H, Yamashita M (2002) Anti-HIV-1 activities and pharmacokinetics of new arylpiperazinyl fluoroquinolones. Bioorg Med Chem Lett 12:739–742
Hagihara M, Kashiwase H, Katsube T, Kimura T, Komai T, Momota K, Ohmine T, Nishigaki T, Kimura S, Shimada K (1999) Synthesis and anti-HIV activity of arylpiperazinyl fluoroquinolones: a new class of anti-HIV agents. Bioorg Med Chem Lett 9:3063–3068
Tomoahi K, Toshinoro O, Hidekiho F, Masako T, Toshinoro N, Yoshinaki K, Tetsushi N (1998) Preparation and formulation of pyridobenzoxazinecarboxylic acid derivatives as virucides. WO Patent 33835, 7 Apr 1998
Pandeya S, Srirama D, Nathb G, DeClercqc E (2000) Synthesis, antibacterial, antifungal and anti-HIV activities of norfloxacin mannich bases. Eur J Med Chem 35:249–255
Ishimura M, Furukawa H, Katsube T (1999) Preparation of fluoroquinolones as anticytomegalovirus agents. WO Patent 42106, 26 Aug 1999
Selvam P, Rathore P, Karthikumar S, Velkumar K, Palanisamy P, Vijayalakhsmi S, Witvroum M (2009) Synthesis and antiviral studies of novel N-sulphonamidomethyl piperazinyl fluoroquinolones. Ind J Pharm Sci 71:432–436
Schneider S, Ruppelt M, Schriewer M, Schulze T, Neumann R (1993) 9-fluoro-7-oxo-7H-pyrido(1,2,3-de)(1,4)benzoxazine carboxylic acids and esters, and their use as antiviral agents. Patent EP 563734, 6 Oct 1993
Schneider S, Bartel S, Ruppelt M, Sriewer M, Schulze T, Neumann R (1993) 7-oxo-7H-pyrido(1,2,3-de)(1,4)benz-oxazinecarboxylic acids and esters and their use as antiviral agents. Patent EP 563732, 6 Oct 1993
Gabardi S, Waikar S, Martin S, Roberts K, Chen J, Borgi L, Sheashaa H, Dyer C, Malek S, Tullius S, Vadivel N, Grafals M, Abdi R, Najafian N, Milford E, Chandraker A (2010) Evaluation of fluoroquinolones for the prevention of BK viremia after renal transplantation. Clin J Am Soc Nephrol 5:1298–1304
Anquetin G, Greiner J, Vierling P (2005) Synthesis of mono- and di-substituted 2,4,5-trifluorobenzoic acid synthons, key precursors for biologically active 6-fluoroquinolones. Tetrahedron 61:8394–8404
Abdul-Rahman S (1999) Preparation of quinolonecarboxylates as bactericides and paraziticides. US Patent 6,967,205, 15 Nov 1999
Watanuki S, Kogo Y, Moritomo H, Tsukamoto I, Kaga D, Okuda T, Hirayama F, Moritani Y, Takasaki J (2005) Preparation of quinolone derivatives as platelet aggregation inhibitors WO Patent 009971, 3 Feb 2005
Yang F, Shipe W, Bunda J, Nolt M, Wisnoski D, Zhao Z, Barrow J, Ray W, Ma L, Wittmann M, Seager M, Koeplinger K, Hartman G, Lindsley C (2010) Parallel synthesis of N-biaryl quinolone carboxylic acids as selective M1 positive allosteric modulators. Bioorg Med Chem Lett 20:531–536
Kuduk S, DiMarco C, Cofre V, Pitts D, Ray W, Ma L, Wittmann M, Seager M, Koeplinger K, Thompson C, Hartman G, Bilodeau M (2010) Pyridine containing M1 positive allosteric modulators with reduced plasma protein binding. Bioorg Med Chem Lett 20:657–661
Kuduk S, DiMarco C, Cofre V, Pitts D, Ray W, Ma L, Wittmann M, Veng L, Seager M, Koeplinger K, Thompson C, Hartman G, Bilodeau M (2010) N-heterocyclic derived M1 positive allosteric modulators. Bioorg Med Chem Lett 20:1334–1337
Kuduk S, DiMarco C, Cofre V, Ray W, Ma L, Wittmann M, Seager M, Koep[linger K, Thompson C, Hartman G, Bilodeau M (2011) Fused heterocyclic M1 positive allosteric modulators. Bioorg Med Chem Lett 21:2769–2772
Toffoli P, Rodier N (1987) Méthanesulfonate de péfloxacinium (péflacine DCI). Acta Crystallogr Sect C 43:1745–1748
Turel I, Leban I, Zupancic M, Bukovec P, Gruber K (1996) An adduct of magnesium sulfate with a member of the quinolone family (Ciprofloxacin). Acta Crystallogr Sect C 52:2443–2445
Hashimoto K, Fujita N, Tanaka T, Kido M (1995) 6-ethyl-9-fluoro-6,7-dihydro-8-(4-hydroxy-piperidino)-5-methyl-1-oxo-1H,5H-benzo[ij]quinoli-zine-2-carboxylic acid. Acta Crystallogr Sect C 51:519–521
Anacona J, Toledo C (2002) Synthesis and antibacterial activity of metal complexes of ciprofloxacin. Trans Met Chem 26:228–236
Jakics E, Iyobe S, Hirai K, Fukuda H, Hashimoto H (1992) Occurrence of the nfxB type mutation in clinical isolates of Pseudomonas aeruginosa. Antimicrob Agents Chemother 36:2562–2567
Turel I (2002) The interactions of metal ions with quinolone antibacterial agents. Coord Chem Rev 232:27–47
Lipunova G, Nosova E, Charushin V (2009) Metal complexes of fluoroquinolonecarboxylic acids. Russ Chem J 53:74–85
Serafin A, Stanczak A (2009) The complexes of metal ions with fluoroquinolones. Russ J Coord Chem 35:83–97
Saha D, Padhye S, Anson C, Powell A (2002) Hydrothermal synthesis, crystal structure, spectroscopy, electrochemistry and antimycobacterial evaluation of the copper (II) ciprofloxacin complex: [Cu(cf)2(BF4)2] · 6H2O. Inorg Chem Commun 5:1022–1027
Efthimiadou E, Sanakis Y, Katsarou M (2006) Antibacterial activity of enrofloxacine metallocomplexes. J Inorg Biochem 100:1378–1388
Katsarou M, Efthimiadou E, Psomas G, Karaliota A, Vourloumis D (2008) Novel copper(II) complex of N-propyl-norfloxacin and 1,10-phenanthroline with enhanced antileukemic and DNA nuclease activities. J Med Chem 51:470–478
Shingnapurkat D, Bucther R, Afrabiasi Z, Sinn E, Ahmed F, Sarkar F, Padhye S (2007) Neutral dimeric copper–sparfloxacin conjugate having butterfly motif with antiproliferative effects against hormone independent BT20 breast cancer cell line. Inorg Chem Commun 10:459–462
Patitungkho S, Absule S, Dandawate P, Padhye S, Ahmad A, Sarkar F (2011) Synthesis, characterization and anti-tumor activity of moxifloxacin–copper complexes against breast cancer cell lines. Bioorg Med Chem Lett 21:1802–1806
Gao F, Yang P, Xie J, Wang H (1995) Norfloxacin metallocomplexes: structure and antibacterial activity. J Inorg Chem 60:61–67
Jimenez-Garrido N, Perello L, Ortiz R (2005) Cobalt and copper complexes of ciprofloxacine. J Inorg Biochem 99:677–689
Xiao D, Wang E, An H, Su Z, Li Y, Gao L, Sun C, Xu L (2005) Rationally designed, polymeric, extended metal–ciprofloxacin complexes. Chem Eur J 11:6673–6686
Chen Z, Xiong R, Zhang J, Chen X, Xue Z, You X (2001) 2D molecular square grid with strong blue fluorescent emission: a complex of norfloxacin with zinc(II). Inorg Chem 40:4075–4077
Tarushi A, Psomas G, Raptopoulou C, Psycharis V, Kessissoglou D (2009) Structure and DNA-binding properties of bis(quinolonato)bis(pyridine)zinc(II) complexes. Polyhedron 28:3272–3278
Drevensek P, Kosmrlj J, Giester G (2006) Spectral study of ofloxacine coordination. J Inorg Biochem 100:1755–1763
Sakai M, Hara A, Anjo S, Nakamura M (1999) Al (III) complexes of fluoroquinoline formation: NMR study. J Pharm Biomed Anal 18:1057–1067
Polishchuk A, Gerasimenko A, Gayvoronskaya K, Karaseva E (2008) Tetrakis(dihydrogen pefloxacinium) di-μ2-chlorido-bis-[tetrachloridobismuthate(III)] tetrachloride octahydrate. Acta Crystal E 64:m931–m932
Vieira L, deAlmeida M, Lourenco M, Bezerra A, Fontes A (2009) Synthesis and antitubercular activity of palladium and platinum complexes with fluoroquinolones. Eur J Med Chem 44:4107–4111
Vieira L, deAlmeida M, de Abreu H, Duarte H, Grazul R, Fontes A (2009) Platinum(II) complexes with fluoroquinolones: synthesis and characterization of unusual metal–piperazine chelates. Inorg Chim Acta 362:2060–2064
Sun C, Ping H, Zhang M, Li H, Guan F (2011) Spectroscopic studies on the lanthanide sensitized luminescence and chemiluminescence properties of fluoroquinolone with different structure. Spectrochim Acta A Mol Biomol Spectrosc 82:375–382
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Charushin, V.N., Nosova, E.V., Lipunova, G.N., Chupakhin, O.N. (2014). Fluoroquinolones: Synthesis and Application. In: Nenajdenko, V. (eds) Fluorine in Heterocyclic Chemistry Volume 2. Springer, Cham. https://doi.org/10.1007/978-3-319-04435-4_3
Download citation
DOI: https://doi.org/10.1007/978-3-319-04435-4_3
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-04434-7
Online ISBN: 978-3-319-04435-4
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)