
Abstract
The invention of telescopes and microscopes about 400 years ago revolutionized our perception of the world, extending our sense of seeing. Extending it further and further has since been the driving force for major scientific developments. Local probe techniques extend our sense of touching into the micro- and nanoworld and in this way provide complementary new insight into these worlds we see with microscopic techniques. Furthermore, touching things is an essential prerequisite to manipulating things, and the ability to feel and manipulate single molecules and atoms for sure marks another of these revolutionizing steps in our relation to the world in which we live.
Local probes are small-sized objects, such as the very end of sharp tips, which interact with a sample, or better, the surface of a sample at selected positions. Proximity to or contact with the sample is required to have a high spatial resolution. This, in principle, is an old idea that appeared in the literature from time to time in context with bringing a source of electromagnetic radiation in close contact with a sample (Synge, Philos Mag 6:356, 1928; O’Keefe, J Opt Soc 46:359, 1956; Ash and Nicolls, Nature 237:510, 1972). It found no resonance and therefore was not pursued until the early 1980s. Nanoscale local probes require atomically stable tips and high-precision manipulation devices. The latter are based on mechanical deformations of spring-like structures by piezoelectric, electrostatic, or magnetic forces to ensure continuous and reproducible displacements with precision down to the picometer level. They also require very good vibration isolation. The resolution that can be achieved with local probes is given mainly by the effective probe size, its distance from the sample, and the distance dependence of the interaction between the probes and the sample measured. The last can be considered creating an effective aperture by selecting a small feature of the overall geometry of the probe tip, which then corresponds to the effective probe. One of the great advantages of local probes is that they can work in any environment; this way, they provide the possibility to study live biological processes similar to optical microscopy, but at a resolution similar to electron microscopy (EM).
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
10.1 Introduction
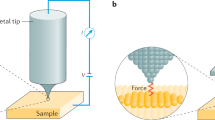
The atomic force microscope (AFM) belongs to the large family of instruments called local or scanning probe microscopes. All of these instruments are based on the principle of measuring the interaction of a small tip structure with a sample surface at close distances. In the case of the first instrument of the family, the scanning tunneling microscope (STM), developed in the early 1980s by Binnig and Rohrer [4] at the IBM Research Laboratory in Zurich, the interacting tip is a single atom (see Fig. 10.1). This is possible because of the very short-range electronic interaction used with a decay length on the size of atoms. This interaction between the tip and surface, which allows electrons to cross a small gap before an electrical contact is made, is related to the quantum-mechanical process of electron tunneling reflected by the name. Such a tunneling current through a vacuum gap is in the range of pico- to nanoamps and drops by an order of magnitude if the distance changes from 0.4 to 0.5 nm. With atoms being the same size, it is possible that the main contribution to this tunneling current is related to the interaction of the foremost tip atom with a single atom at the surface closest to this tip atom. If the surface and the tip are moved with respect to one another at a constant average distance, the variations seen in the current reflect the topography of the surface with atomic resolution if the surface is homogeneous. In the case of different types of atoms present at the surface, the signal measured is a mixture of topography and differences between the atom species in their electronic interaction with the tip atom.

STM imaging with atomic resolution. The tip is scanned at a constant height over the surface, and the current measured between the tip and surface is modulated by the distribution of surface atoms
With this incredible resolution, the most important part of such a microscope is the mechanism that causes the tip to approach the surface and moves it across with the necessary precision and stability. The mechanism Binnig and Rohrer came up with is based on the piezoelectric effect discovered in 1880 by the brothers Pierre and Jacques Curie. They noticed that mechanical stress affects the polarization of crystalline structures like quartz and that this can be used in reverse with an electric field applied across a quartz crystal to change its size. In the case of quartz, a very high electric field is necessary for only a small change in size. In the 1950s, ceramic materials such as perovskites were discovered, which change their size in the micrometer range with only a few hundred volts applied. Three stacks of ceramic discs were used in the first STMs to position the tip in all three dimensions with the necessary precision that allowed a precise approach of the tip to the surface and scanning it across. Meanwhile, more often a ceramic tube with an electrode on the inside and a segmented electrode on the outside is used, which allows tip movement in all three directions in a controlled way by applying a voltage between the inner electrode and all outside electrodes to approach and retract the tip and between the inner and individual outer electrodes to bend the tube sideways to do the scanning (Fig. 10.2).

(a) An electric field applied across a quartz crystal interacts with the crystalline structure through its polarization, which affects the crystal structure and leads to a change in size. (b) The piezotube changes its length if a voltage difference between the inside electrode and all four outer electrodes is applied. The tube bends sideways if a voltage difference between the inside electrode and one segment of the outside electrodes is applied. In this way, a tip mounted in the direction of the z-coordinate can approach a surface very precisely and be scanned across with the same precision, which is mainly related to the stability of the voltage source used
To prevent the tip from crashing into the surface while scanning, due to roughness of the sample surface or when mechanical vibrations of the instruments occur, the electronics that controls the movement of the tip includes a “feedback loop” that limits the tip–surface interaction by moving the tip away from the surface if necessary to reduce the current below an upper limit. The feedback can keep the interaction constant during scanning if it is fast enough, which means the tip will follow the contour of a sample with homogeneous electronic properties. This imaging mode is called constant current imaging. The maximum speed of the feedback control, mainly determined by mechanical resonances, is the main limitation in scanning probe microscopy that determines how fast an image (picture) of an area with a certain size can be produced. Faster imaging can be done in so-called constant-height mode, but this is only possible on sample surfaces that are flat at the atomic scale to avoid the tip’s crashing into the sample at protrusions.
10.2 Atomic Force Microscopy
Due to the electronic interaction measured in the case of the STM, both the tip and the surface have to be conducting. This significantly limits the materials that can be investigated. In 1986, Binnig, together with Quate and Gerber, overcame this limitation with the invention of the next member of the probe microscope family, called the atomic force microscope (AFM) [5]. The idea that allows also the investigation of nonconducting materials was to use the “van der Waals” forces between atoms as a type of interaction present in all types of atoms. The corresponding interaction potential (Lennard–Jones potential) has a very weak attractive regime between 0.7 and .35 nm and becomes strongly repulsive with a 1/r 12 distance dependence below 0.35 nm. A microscope using these interactions between the tip and the surface atoms needs to detect forces in the nano- to pico-Newton range, thus asking for a very sensitive force-detection system. The most straightforward way to measure forces is by using a spring, which can be characterized by its spring constant connecting the spring extension (or bending in the case of a cantilever) and force by Hooke’s law: F = −kΔx. With such a linear force–distance relationship, a spring with a stiffness of 1 N/m needs a precision in the distance measurement in the nanometer range to achieve a force resolution in the nano-Newton range. The STM, invented just five years before by Binnig and collaborators, can easily measure distances with such a precision; therefore, it was the obvious choice to be used in the first AFM to detect the bending of a small cantilever with a tiny piece of diamond as a tip at the end. A schematic of the first AFM instrument is provided in Fig. 10.3.

Schematic of the first AFM from the IBM patent application in 1986 (Adapted from Ref. [6]). In (a) the cantilever is not drawn to scale; the dimensions are shown in (b). A: sample, B: AFM diamond tip, C: STM tip, D: metal cantilever as sample for the STM detection, E: modulating piezo, F: viton vibration insulation
Initially, there was doubt about whether the AFM could achieve a true atomic resolution and whether the van der Waals interaction of one tip atom with just one surface atom on crystalline surfaces is actually measured. Some researchers suggested that clusters of atoms on both sides interact with one another and that the structures seen are interference patterns created while scanning across the surface that have more or less a relationship to its atomic structure. It was much later that Giessibl could show that under certain conditions, even atomic substructures can be imaged with an AFM [7]. However, the normal case, especially on molecular structures, turned out to be that more than one atom of the tip interacts due to various types of surface forces with a surface area of a corresponding size and in this way reduces the AFM resolution to the nanometer range.
An important development providing the basis for the successful use of the AFM in biology was the replacement of the STM detection with an optical detection scheme (Fig. 10.4), as this allowed measurements even in salt-containing solutions. In 1988, Meyer and Amer demonstrated that a reflected laser beam at the tip of a cantilever observed in some distance with a split photodiode can provide a sensitivity similar to that of the STM detection [8]. The standard detection scheme in most commercial AFMs is based meanwhile on a diode laser focused at the end of the cantilever directly above its tip. The bending of the cantilever changes the direction of the reflected laser beam and thus its position on the detector. The change in the intensity distribution between the two halves of the split diode leads to a change in the electric signals produced by both halves. If a sensitive amplifier is used to measure the signal difference between the two halves, it is possible to detect movements of the cantilever in the range below 1 nm, which, with a small cantilever spring constant of 0.01 N/m, leads to a force sensitivity in the range of 10 pN.

Optical detection scheme showing the laser beam deflected by the cantilever toward the quadrant photo detector
The cantilevers used are micro-machined from silicon or silicon nitride wafers with a length between 50 and 250 μm and a thickness between 0.5 and 2 μm, leading to spring constants between 100 and 0.01 Ν/m (Fig. 10.5). The tips are made by different procedures and in some cases have a roof-shaped end due to the crystalline structure of the material used. In such a case, the resolution depends not only on the length of the ridge, but also on its tilt and the scanning direction with respect to the direction of the ridge. A so-called oxide-sharpening technique, which leads to final tip radii of only a few nanometers at a total tip length of 5–10 μm, makes the smallest tip radii. The shape of cantilevers quite often is triangular, to provide a higher stability against lateral forces, which can become quite high due to friction effects while scanning the tip across a surface. These lateral or friction forces twist the cantilever sideward and lead to a movement of the laser beam on the detector perpendicular to the normal deflection, which can be measured by using a quadrant photodiode instead of a split one (Fig. 10.4).

EM image of an AFM cantilever from below, with the tip visible at the end as a small 5-μm square
With a device that can detect the bending of the cantilever, it becomes difficult to move the tip, which is usually done in an STM. Therefore, many AFMs have the sample mounted on the piezotube. As the sample normally has a larger mass than the cantilever, the system’s resonance frequency decreases in such a setup, decreasing the scan speed. However, with a specially designed laser optic, the problem can be overcome and a scanning head can be made that can be placed over a fixed sample. Nevertheless, usual imaging speeds are in the range of minutes; the efforts to build fast-scanning AFMs are still ongoing, mainly based on reducing the cantilever size.
10.2.1 Application Modes
As in the case of an STM, it is important to control the tip interaction while scanning with the AFM in order not to destroy the tip or sample due to surface roughness or external vibrations. The electronic feedback in the case of an AFM tries to keep the force between the tip and the sample within limits. This way, if the reaction time of the feedback is fast compared to the scanning speed, the tip will follow the surface contour, applying the same force to the sample everywhere when working in the repulsive van der Waals regime. This mode is called constant-force mode. The feedback normally is created as fast as the system resonances allow when weighted linear, differential, and integral components of the signal are being used. Nevertheless, over slopes, the feedback with reasonable scan speeds always becomes too slow and characteristic image artifacts occur depending on whether the tip is not retracted or pushed down fast enough. For very flat samples, a constant-height mode is also possible, with forces between the tip and the sample changing during scanning, as higher sample structures will push up the cantilever tip in this case. The tip is kept in contact due to an average preadjusted loading force. In this mode, the cantilever’s spring constant and the corresponding resonance frequency determine the reaction time and, therefore, the maximal scan speed:
where F is the resonance frequency, k is the spring constant, and m is the mass of the cantilever.
If a tip with a certain interaction force is scanned over a sample, lateral forces act on the tip due to the friction between the tip and the sample. Measuring various lateral friction forces is a way to characterize inhomogeneous samples, where different components have different frictions. For such friction-mode measurements, the AFM has to be equipped with a quadrant photodiode instead of a split one, as mentioned earlier. If adjusted in the right way, the quadrant diode detects in one direction the normal bending of the cantilever and perpendicular its sideward twisting due to the lateral friction force. Friction and high forces on rough surfaces, together with a slow feedback, easily destroy soft samples, resulting very often in parts of the sample sticking to the tip. A way to reduce this risk is to use the AFM in a dynamic mode. In this mode, the cantilever is vibrating close to its natural resonance frequency driven by a small piezo mounted where the cantilever is fixed. With small oscillation amplitudes in the ångstrom range in the dynamic AFM mode, force gradients are measured, allowing measurements even in the narrow and shallow attractive range of the van der Waals forces.
Some instruments drive the cantilever oscillation with a magnetic coating above the tip, which allows for much better control of the tip motion in this dynamic mode, especially if the cantilever works in a fluid chamber. If a piezo is used, it has to be mounted outside the fluid chamber to avoid electrical shorting. In this case, the vibrating piezo excites acoustic waves, which drive the cantilever in a more indirect way that makes the control of the actual tip motion difficult.
Working in air under ambient conditions has a serious issue with water, as all materials have thin layers of water present on the outer surface, with the water thickness depending on the humidity, the temperature, as well as the actual material. During the approach of the cantilever tip to the surface, the water layer on the tip and that on the surface coalesce at one point during the approach and form a water meniscus. The forces generated by this meniscus are in the micro-Newton range; scanning this meniscus across a surface will wipe away everything that does not adhere well to the surface. With high enough amplitudes of the cantilever oscillations, in the so-called tapping mode, the water meniscus between the tip and the surface will break during each retraction cycle and no lateral forces of the meniscus will be produced.
In dynamic mode, the amplitude and phase changes of the vibrating cantilever reflect elastic and inelastic tip–sample interactions and can be used to characterize these material properties at the nanometer scale (Fig. 10.6). Images of inhomogeneous samples created using either the amplitude or the phase changes nicely depict distributions of different materials. Unfortunately, with the cantilever immersed into a liquid, the viscosity leads to a strong damping of the cantilever oscillations and a significant broadening of its resonance frequency curve, reducing the resolution in both the amplitude and the phase-shift signal. With a special type of feedback using a 90° phase-shifted input signal, it is possible to restore to some extend the so called quality factor of the cantilever, which was greatly reduced by the damping of the liquid.

Principle of a dynamic AFM that uses an oscillating cantilever to measure tip interactions by amplitude and phase changes of the oscillation driven with a feedback using a phase-shifted signal to enhance the cantilever’s quality factor, which is significantly reduced in liquid due to the viscosity
10.2.2 Working in Liquid
The already-mentioned possibility to work with an AFM in liquid and especially in salt solutions extends the possibility of light microscopy to study the time course of processes from the micro- into the nanometer range. For biological applications, this opened a new era of studies on cellular structures and their dynamics. But as mentioned earlier, the cantilever motion in liquids is damped by the liquid’s viscosity, and with the maximum speed the cantilever can be moved, the maximal imaging speed for high-resolution imaging is reduced. The viscous drag directly affects the mechanical properties of the cantilever immersed into a medium. How the mechanical properties of a cantilever change with increasing viscosity is shown in Fig. 10.7, where the glycol concentration in water is increased systematically, leading to a significant increase in viscosity. The resonance curve is substantially broadened, and its maximum shifted from nearly 24 kHz at a 20 % glycol concentration down to 7 kHz at an 80 % concentration. These resonance curves were recorded as the difference between a cantilever moving in liquid driven by thermal fluctuations and the same cantilever in contact with the liquid chamber wall. This measuring protocol enables one to extract the instrument’s mechanical and electrical noise (from the signal) to a level where the resonance curve excited by thermal fluctuations even in a very viscous medium can be measured.

Changes to the frequency spectrum of a cantilever were measured while the ratio of the concentration of glycol to water is increased from 20 % (a) to 80 % (d) in 20 % increments
The response of a mechanical cantilever to time-dependent external forces is described by a differential equation that characterizes the cantilever by its mass and its spring constant and the surrounding medium by its viscosity η, leading to a velocity-dependent damping γ:
-
F = time-dependent driving force,
-
m = moving mass,
-
γ = velocity-dependent damping,
-
k = spring constant.
With this equation, we can derive the maximum of the resonance curve of the cantilever as
To describe the way the resonance maximum changes with changing viscosity, as depicted in Fig. 10.8, the viscosity η and other parameters have to change to produce the monotonous decrease in the resonance frequency with increasing viscosity. There is no reason to assume that the spring constant of the cantilever may change, as it is only related to the material properties of the cantilever itself, which do not change. Another possibility is to assume that some mass of the medium is added to the cantilever mass. When a cantilever moves in a medium, some of the medium moves with the cantilever and some of the medium flows around these moving components to make way in front and to fill the space left behind.

Schematic of the different mass components necessary to describe the motion of a cantilever in a viscous medium
Therefore, to account for the hydrodynamic effects, we need to add two additional components of mass into the equation (see Fig. 10.9):

Fit to the maximum values of the resonance curves at different mixtures of water and glycol, which allows the values of m 1 and m 2 to be determined
-
m = cantilever mass,
-
m 1 = fluid mass moving with the cantilever,
-
m 2 = fluid mass moving in the opposite direction.
With this equation, we can fit the observed changes of f max and calculate the different mass components. Assuming that the shape of the cantilever is causing only secondary effects, we can approximate the actual rectangular cantilever with a cylinder of the same mass and length, which allows us to solve the differential equation. The results show that in pure water twice the mass of the cantilever moves with the cantilever, and a little bit less than half the mass moves in the opposite direction. With an addition of 80 % glycol, and the corresponding high viscosity, 140 times the mass of the cantilever is moved together with it, but only 20 % more than in pure water is moving in the opposite direction. In the case of a cantilever with a diameter of 6 μm, the layer of liquid moving with it changes from 2.4 μm in pure water to 80 μm if 80 % glycol is added. In the latter case with 80 % glycol, the layer thickness of the liquid moving against the cantilever is 200 nm, and in pure water it is 10 nm. This calculation shows that in water the hydrodynamic effects are limited to a range below 3 μm around the cantilever. With an actual tip length of 5 μm for most commercial cantilevers, this will not influence the sample interaction.
10.3 Applications in Biological Systems
Interactions of molecular structures are traditionally characterized by binding constants, on and off rates, and corresponding binding energies. The relevant energies for single binding events range from thermal energy up to some hundred times that of the thermal energy when covalent bonding is involved. Besides covalent bonds, hydrogen bonds, with energies between 4 and 16 kT, play an important role for macromolecular structures and their spatial organization. At interfaces of these structures, coulomb and dipole forces determine the interaction with the aqueous environment and thus between molecules. An important question for the biological function of these molecular interactions is their distance dependence in the actual environment. This question can be addressed using AFM force spectroscopy on the single-molecule level, giving a new, more physical view of the molecular interactions in terms of forces and their distance dependence.
An important aspect of measuring forces at the molecular level is the dependence of the force value on the timescale in which the force is applied. This connection can be understood by the deformation to the molecular interaction potentials caused by an applied force. This deformation leads to an effective lowering of the binding energy if the force tries to pull the molecular structures apart. Consider thermal fluctuations with a pulling force applied: It becomes more likely that the bond will break during a certain observation time. The off rate for the biotin–avidin binding, for instance, at room temperature is on the order of six months. If a small force of about 80 pN is applied, the lowering of the binding potential reduces the off rate to about 9 s. Doubling this force further decreases the lifetime of the binding by three orders of magnitude. Unfortunately, these experiments cannot be modeled with “molecular dynamic” computer simulations, as the timeframe of these simulations with the available computer power is limited to the pico- up to the nanosecond time range. Simulations of the rupture forces of biotin–avidin at this fast timescale lead to calculated forces of 600 pN. For AFM measurements completed at the time scale of 100 ms, the actual measured forces are 100–200 pN [9].
10.3.1 Intermolecular Forces
Protein adsorption is a very important topic for many biomedical and biotechnological applications. For instance, many chromatographic separations, such as hydrophobic, displacement, and ion-exchange chromatography, are based on differences in the binding affinities of proteins to surfaces. In addition, in vitro cell cultures require cell-surface adhesion, which is mediated by a sublayer of adsorbed proteins. The molecular basis of cell adhesion is, in general, an important process in tissue development, tissue engineering, and tissue tolerance to implants.
Protein adsorption to a surface is a net result of various complex interactions between and within all components, including the chemistry and topology of the solid surface, the protein itself, and the medium in which it takes place. The interaction forces involved include dipole and induced dipole moments, hydrogen bond formation, and electrostatic forces. All inter- and intramolecular forces contribute to a decrease in the Gibbs free energy during adsorption, defining the binding energy.
An important question for protein-adsorption processes is the reversibility of the adsorption process. One approach to this question is an analysis of the time course of adsorption. As the adsorption is a multiple-step process in most cases, this relates to the more specific question of until which step is the process reversible? The most common way to quantify adsorption is by using the adsorption isotherm, characterizing at a constant temperature the number of molecules adsorbed in relation to the steady-state concentration of the same molecules in the bulk solution. Adsorption isotherms provide a convenient method for determining whether or not an adsorption process is reversible. Reversibility is commonly observed with the adsorption of small molecules on solids, but only rarely in the case of more complex random coil polymers.
Proteins are polymers, but globular proteins do not have a random coil structure. The native state of these proteins in aqueous solution is highly ordered; most of the polypeptide backbone has little or no rotational freedom. Therefore, significant denaturing processes have to occur to form numerous contacts with any surface. Structural rearrangements may occur in a way that the internal stability of globular proteins prevents them from completely unfolding on a surface into a loose “loop and tail” type of structure. Thus, a subtle balance between intermolecular and intramolecular forces determines the number of protein-segment to surface contacts formed at steady state.
The thermodynamic description of the protein-adsorption process is based on the laws of irreversible thermodynamics [10]. The process is strongly time-dependent; some of the involved steps of molecular rearrangement are remarkably slow and probably lead to significant binding of proteins only on the timescale of seconds. With different timescales for the various interactions, the adsorption process can be divided into fast steps—which can be reversible—and slow steps, where protein structure rearrangements can occur controlled by the adhering surface and the rest of the environment. The latter processes are irreversible in most cases.
With the AFM, it is possible to measure the adhesion forces established by single proteins, such as protein A and tubulin molecules, within contact times of milliseconds to seconds. Protein A and tubulin are both globular proteins and can be seen as examples of different types of protein binding. Protein A is often used to bind antibodies to solid substrates, and tubulin forms the filament structure of microtubules as part of the cell’s cytoskeleton. The molecules can be attached to the cantilever tip and then brought into contact with different solid surfaces, which might be covered with layers of other molecular structures. To study implant compatibilities, bare metal surfaces such as gold or titanium are of interest. In addition, the optically transparent indium tin-oxide (ITO) is of relevance for the development of interfaces between biological molecules and electro-optic devices.
The procedure for measuring adhesion forces between proteins and surfaces using an AFM needs to define the approach speed, maximal applied force, contact time, retraction speed, retraction distance, and time away from the surface (see Fig. 10.10). With these parameters fixed, one can measure the interaction force by approaching gold, titanium, or ITO surfaces, for instance, with protein-coated tips. It turns out that for the first contact, there is a specific interaction characteristic for the different molecules and the different metals [11]. With the observed reproducibility of a certain value for an adhesion force within a series of measurements, it has to be assumed that in these experiments a well-defined interaction between a specific amino acid group located at the surface of the protein and the metal determines the first step of the adhesion process. The experiments performed by Eckert et al. [11] demonstrated that this technique, combined with an adequate preparation procedure, can be used to measure these first steps in the protein-adhesion process at the single-molecule level. Furthermore, these experiments demonstrated that the technique can be used to study the dependence of these interactions on the environment.

AFM approach and retraction scheme for molecular adhesion measurements. The image sequence to the left shows a cantilever-tip approach/retract cycle with a molecule linked to the tip by a molecular tether. The top right image depicts the changes during the cycle in the vertical position of the cantilever base; it is bending, which corresponds to the force acting on the cantilever tip. The bottom right image shows the movement of the cantilever tip during the approach. The jump indicates an adhesion force pulling the cantilever toward the surface due to attractive van der Waals forces. The following linear change corresponds to the cantilever response while pushed against the surface; its slope reflects the cantilever spring constant
10.3.2 Intramolecular Forces
The modular structure of proteins in natural fibers and the cytoskeleton seems to be a general strategy for resistance against mechanical stress. One of the most abundant modular proteins in the cytoskeleton is spectrin. In erythrocytes, spectrin molecules are part of a two-dimensional network that provides the red blood cells with their special elastic features. The basic constituent of spectrin subunits is a structural repeat, which has 106 amino acids forming three antiparallel α-helices with a left-handed coiled-coil structure. Helical linkers of 10–20 amino acids most likely connect the repeats. The mechanical properties of several modular proteins have been investigated by AFM so far. The first study was done on titin [12], which exhibits a β-sheet secondary structure. The experiments have demonstrated that the elongation events observed during stretching of single proteins may be attributed to the unfolding of individual domains; experiments with optical tweezers have corroborated these results [13, 14]. These studies suggest that single domains unfold one at a time in an all-or-none fashion when subjected to directional mechanical stress.
A major technological step forward in protein-folding studies with the AFM was the development of a double-tip detection system. The concept uses one tip that stays in contact with the surface during the force and distance measurements, to stabilize the distance between the second force-measuring tip and the surface. This distance normally has drift speeds of several nanometers per second in AFM due to the long mechanical connection between the cantilever and the sample holder. The double-tip scheme (Fig. 10.11) allows long-term stabilization of this distance with a fraction-of-an-ångstrom precision over minutes and makes so-called force-clamp measurements over such timescales possible. With this technique, it became possible to perform unfolding and refolding experiments on extremely small protein structures such as spectrin, with only four repeats [15]. In these experiments (Fig. 10.12), the proteins were attached to clean, freshly prepared gold surfaces. While the clean tip of the second force-measuring cantilever approaches such a surface, a single molecule can become attached to its tip, and forces in the range of pico to nano-Newtons can be applied to the molecular structure. With forces of about 80 pN, the molecules are stretched at a speed of 3 nm/s to more than 10 times their folded length, reaching almost the total contour length. The force–extension curves show characteristic sawtooth-like patterns. The reaction coordinate of unfolding is imposed by the direction of pulling, and unfolding events occurring in a single protein can be studied this way. Each peak in the bottom trace of Fig. 10.12 can be attributed to a breakage of a main stabilizing connection of a folded structure.

Schematic of double-cantilever AFM with one lever stabilizing the other cantilever’s tip and surface distance, which allows it to be used exclusively measure forces

Stretching spectrin repeats between a gold surface and a cantilever tip
Recombinant DNA techniques were used in the experiments to construct chains of repeats from identical spectrin domains. The method extends the monomer (R16) at both ends, so that the polymeric protein product contains a 13-residue linker between the consecutive R16 units, with two cysteine residues at the C-terminal end. The force–extension relationship was measured on a four-repeat construct fixed to the gold surface with the cysteine residues introduced at the C-terminal end. In the sawtooth-like pattern of the force–extension curves (Fig. 10.12, bottom), each peak represents an unfolding event of one subunit. After each peak, the force drops back as additional length of the protein chain unfolds, is made available, and starts to extend. The maximal extension in these experiments is variable because the polymer is not always picked up at the end, and sometimes the tip can be attached to the surface at multiple locations. Nevertheless, the observed extensions never exceeded the contour length of four repeats.
The so-called worm-like chain (WLC) model describes the measured force–extension curves well. It predicts the isothermal restoring forces of a flexible polymer, acting as an entropic spring during extension. It also defines a persistence length to the polymer chain, which turns out to be 0.6 nm for the spectrin chain. Two consecutive force peaks are spaced by a distance that represents the gain in length produced by a single unfolding event. These distances were measured for several hundred unfolding events. The histogram (Fig. 10.13) of the measured distances reveals a length distribution with two statistically relevant peaks. When the histogram is fitted with two Gaussian distributions, the maxima are at 15.5 and 31 nm. The force distributions associated with the short and long elongation events can also clearly be separated. The mean force for the 15.5- and 31-nm elongation events are 60 and 80 pN, respectively, at a pulling speed of 3 nm/ms.

Distribution of elongation length observed by stretching the α-helical spectrin with the AFM. The left peak relates to partial unfolding, as shown in Fig. 10.14, and the right peak relates to the total unfolding of the structure
These experiments demonstrated for the first time at the single-protein level that α-helical domains can unfold in several discrete steps in a much more complex way than previously suggested by molecular dynamic simulations. The appearance of different elongation events suggests that at least one intermediate state for the spectrin repeat exists between the folded state and the completely unfolded state (see Fig. 10.14). In thermodynamic studies, such an intermediate state has not been found. A statistical analysis of the AFM force curves shows that the short elongation events are half the length of the total unfolding length of one domain. The molecular pathway of unfolding of a spectrin domain can be simulated based on the AFM measurements and shows that the first steps involve a partial opening of the bundle and the loss of the secondary structure at the repeats connecting helices. The central part reorganizes around a hydrophobic core resembling a shorter version of the repeat structure.

Normal spectrin repeat folding of the three α-helices forming a slightly twisted “N” in space, with connection possibilities to other repeats at both ends. Below the stable intermediate structure is shown with the connecting a-helices extended, but with the structure around the hydrophobic core still stable
Protein-folding and -unfolding pathways are very complex. However, by applying a force, the unfolding pathways can become very strongly restricted. The results with only two possible unfolding pathways can be explained along this line, as outlined in Fig. 10.15. One leads directly from the folded to the unfolded state, the other first to an intermediate state. The intermediate state is conceptually available along both pathways, but each pathway by itself leads from the native folded state 0 either to the partially unfolded state 1 or to the completely unfolded state 2. The relative difference in the height of the free energy barrier along either of the two pathways determines whether state 1 or state 2 is attained. The advantage of such a modeling approach is that only one free parameter is needed for differentiating between the two pathways.

Energy scheme for the pathway leading to an intermediate state, and the energy scheme that shows how the process leads directly to the completely unfolded state when the force is applied faster and the energy landscape is further deformed
In the native folded state, the protein is in state 0 at the bottom of the potential well. The mechanical stress applied by the AFM tip not only decreases the barrier height for thermally activated unfolding but also reduces the options of the protein to follow either path 1 or 2 during unfolding (Fig. 10.15). A certain protein will follow only one path, leading to the observed bimodal probability distribution with 35 % and 65 % probability for path 1 and 2, respectively.
For such a simple model, a Monte Carlo simulation can be used to test the reaction kinetics simultaneously for the short and long elongation events, which means paths 1 and 2, respectively. The kinetics can be characterized by two parameters: the width of the first barrier, and an effective so-called attempt frequency related to the barrier height as an exponential multiplication factor normalized by the thermal energy. In such a simulation, there is no reason to assume a change in the width of the first barrier, but the attempt frequency should vary and needs to be adjusted to agree with the relative difference in barrier height. The force and elongation histograms obtained from 5,000 simulations reproduce the observed general features well, with a barrier width of 0.4 nm and a difference in the barrier height between both pathways of twice the thermal energy.
The experiments shed new light on the mechanical behavior of spectrin, which is an essential cytoskeletal protein with unique elastic properties. They demonstrated that the unfolding of spectrin repeats occurs in a stepwise fashion during stretching with stable intermediates. The stable stretched states under the applied forces show, on the one hand, that protein structures are determined by the environment and, on the other hand, how the elastic properties of spectrin come about. Actually, spectrin behaves within a certain force range as a quantized but perfectly elastic structure: Applied forces stretch the molecule to a defined length; after the forces are turned off, it goes back to the original state. These details about the unfolding of single domains revealed by precise AFM measurements show that force spectroscopy can be used to determine forces that stabilize protein structures and also to analyze the energy landscape and the transition probabilities between different conformational states.
10.3.3 AFM and Optical Microscopy
From the very beginning, the AFM was thought to be an ideal tool for biological research. Imaging living cells under physiological conditions and studying dynamic processes at the plasma membrane were envisioned although it was clear that such experiments are quite difficult, as the AFM cantilever is by far more rigid than cellular structures. Therefore, the way cells are supported becomes quite important. In addition, a parallel optical observation is necessary to control the cantilever-tip approach to distinct cellular features. In order to address these problems, the IBM Physics Group in Munich started the development of a special AFM built into an inverted optical microscope [16]. This instrument finally provided the first reproducible images of the outer membrane of a living cell held by a pipette in its normal growth medium. A conventional piezotube scanner moved this pipette. The detection system for the cantilever movement was, in principle, the normal optical-detection scheme, but a glass fiber was used as a light source very close to the cantilever to avoid optical distortions of the light while going through the water surface. This setup allowed a very fast scanning speed of up to a picture per second for imaging cells in constant-height mode. This is possible because the parts moving in the liquid are very small and—quite different from the imaging of cells adsorbed to a microscope slide—no significant excitation of disturbing waves or convection in the liquid occurs. It is possible to keep the cell alive and well for days within such a liquid cell. This makes studies of live activities and kinematics possible in addition to the application of other cell physiology–measuring techniques. With this step in the development of scanning probe instruments, the capability of optical microscopy to investigate the dynamics of biological processes of cell membranes under physiological conditions was extended into the nanometer range. With imaging rates of up to one frame per second, structures as small as 10 nm could be resolved and their dynamics studied. This made it possible to visualize processes such as the binding of labeled antibodies, endo- and exocytosis, pore formation, and the dynamics of surface and membrane cytoskeletal structures in general.
With the cantilever’s integrated tip, forces in the range from pico- to nano-Newtons are applied to the investigated cell; the mechanical properties of cell surface structures therefore dominate the imaging process. On the one hand, this fact mixes topographic and elastic properties of the sample in the images; on the other hand, it provides additional information about the underlying membrane’s cytoskeleton and its dynamics in various situations during the life of a cell. To separate the elastic and topographic properties, we need further information. The additional information can be provided by using AFM modulation techniques. The pipette AFM concept is well suited for such modulation measurements, as the convection or excitation of waves in the solution caused by movement of the pipette is negligible and the modulation can be done very quickly. Nevertheless, for a thorough analysis of a cell wall elasticity map, we would have to record pixel by pixel a complete frequency spectrum of the cantilever response to derive image data in various frequency regimes. This would take too long for a highly dynamic system like a living cell; therefore, such analyses are restricted to certain small areas. Experiments performed on living cells with this setup showed in some cases a certain weak mechanical resonance in the regime of several kHz. In 2007, Cross and coworkers rediscovered this [17] and used it to characterize cancer cells. It might lead to new methods of medical diagnosis at the cellular level and to new techniques in drug development.
Figure 10.16 shows the schematic arrangement of the first AFM set up to study live cells. It was built on a highly stable sample holder stage within an inverted optical microscope. The sample area can be observed from below through a planar surface defined by a glass plate with a magnification of 600×–1200× and from above by a stereo microscope with a magnification of 40×–200×. The illumination is from the top through the less well-defined surface of the aqueous solution. In order not to block the illumination, the manipulator for the optical fiber and for the micropipette point toward the focal plane at a 45° angle. The lever is mounted in a fixed position within the liquid slightly above the glass plate tilted also by 45°. A single-mode optical fiber is used to avoid beam distortions at the water surface by bringing the end as close as possible to the lever within the liquid. To do so requires removing the last several millimeters of the fiber’s protective jacket. The minimum distance is determined by the diameter of the fiber cladding and the geometry of the lever. In the original experiments, fibers for 633-nm light with a nominal cladding diameter of 125 μm and levers with a length of 200 μm have been used. Holding the fiber at a 45° angle with respect to the lever means that the fiber core can be brought as close as 150 μm to the lever. A 4-μm-diameter core has a numerical aperture of 0.1, and the light emerging from the fiber therefore expands with an apex angle of 6°. For the geometry given above, the smallest spot size achievable is 50 μm, approximately the size of the triangular region at the end of the cantilever. Due to the construction of the mechanical pieces holding the pipette and the fiber, the closest possible distance of the position-sensitive quadrant photodetector to the lever is about 2 cm. This implies a minimum spot size of 2.1 mm, within the 3-mm × 3-mm borders of the detector. Using a 2-mW HeNe laser under normal operational conditions produces a displacement sensitivity of better than 0.01 nm, with a signal-to-noise ratio of 10, which compares well with the sensitivity of other optical detection techniques. The advantage of this method is that there are no lenses and there is no air–liquid or air–solid interface for the incoming light beam to cross. Only the outgoing light crosses the water–air interface, as the detector cannot be immersed in the water, but this does not influence the information about the changes in the cantilever’s bending.

Schematic of the first AFM built in1988 to image live cells under physiological conditions [16]
The pipette used for holding the cell in the setup was made from a 0.8-mm borosilicate glass capillary, which was pulled to about a 2–4-μm opening in three pulling steps. It is mounted on the piezotube scanner and coupled to a fine and flexible Teflon tube, through which the pressure in the pipette can be adjusted by a piston or water pump. The pipette is fixed at an angle that allows imaging of the cell without the danger of touching the pipette with the lever. All these components are located in a 50-ml container. The glass plate above the objective of the optical microscope forms the bottom of this container.
After several microliters of the cell suspension have been added, a single cell can be sucked onto the pipette and fixed there by maintaining low pressure in the pipette. Other cells are removed through the pumping system. The cell fixed at the pipette’s end is placed close to the AFM lever by a rough approach with screws and finally positioned by the piezo scanner. When the cell is in close contact with the lever’s tip, scanning of the capillary with the cell attached leads to position-dependent deflections of the lever. The levers used were micro-fabricated silicon nitrite triangles with a length of 200 μm and a spring constant of 0.12 N/m. The forces that can be applied with these levers and the sensitivity of the detection system can be as good as 0.1 nN.
With such forces acting on the plasma membrane of a cell, the stiffness of the structures dominates the images. Therefore, the scaffolds of the cortical layer of actin filaments and actin-binding proteins, which are cross-linked into a three-dimensional network and closely connected to the surface membrane, are prominent in the images. The relatively stable arrangements of the actin filaments are responsible for their relatively persistent structure. However, these surface actin filaments are not permanent. During phagocytosis or cell movement, rapid changes of their shape can be observed. These changes depend on the transient and regulated polymerization of cytoplasmic free actin or the depolarization during the breakdown of the actin filament complex. The timescale of cell surface changes on a larger scale observed with the AFM was about 1–2 h at room temperature. Except for small structures some 10 nm in size, everything is quite stable on this timescale. More and larger structures are rearranged within 10–15 min after these periods of stability.
In a series of experiments, Hörber and coworkers studied the membrane dynamics after infection of the cells with pox viruses [18]. The first effects were observed seconds to minutes after adding the virus solution to the chamber. After this time, the cell surface became smooth and so soft that the tip tended to penetrate the cell surface even with a loading force far below 1 nN. After a few minutes, the cells became rigid again; with stable imaging, the same structures as before became visible. During the first hour after infection, no significant variation in the membrane structures was observed. It is known that within 4–8 h, viruses are reproduced inside the cell and are passed through the cell membrane via exocytosis [19]. However, approximately 2.5 h after infection, with the AFM a series of processes started at the membrane. Single clear protrusions became visible, which were growing in size. The objects quickly disappeared and the original structure of the membrane was restored. Such processes sometimes occurred several times in the same area and lasted about 90 s for small protrusions of about 20 nm and up to 10 min for larger ones (up to 100 nm in size). Each process proceeded distinctly and apparently independently of the others, was never observed with uninfected cells, and was never observed before 2 h after infection.
The fact that the growing protrusions abruptly disappeared makes it likely that the exocytosis of particles related to the starting virus reproduction was observed, but not viruses. The first progeny viruses should appear 4–8 h after infection and are clearly bigger than the structures observed. Nevertheless, after 2–3 h, the early stage of virus reproduction is finished and the final virus assembly just begins. The observation of protrusions after this characteristic timespan supports the assumption that they are related to the exocytosis of protein agglomerates originating from the virus assembly.
Significantly more than 6 h after infection, even more dramatic changes at the cell membrane can be observed. Large protrusions, with cross sections of 200–300 nm, grow out of the membrane near deep folds. These events occur much less frequently than those observed after 2 h. These large protrusions also abruptly disappear, leaving behind small scars at the cell surface. Considering the timing and their size, it is very likely that these protrusions are now progeny viruses exiting the cell after the assembly is finished. With approximately 20–100 virus particles exiting an infected cell, and with roughly 1/40 of the cell surface accessible to the AFM tip, the observation of one or two of those events should be possible. In the experiments, the number of processes exhibiting the right size and timing observed varied between none and two. During the first experiment of 46 h of continuous observation of a single infected cell, such events were seen 19 and 35 h after infection. Stokes could show with electron microscopy that individual viruses sometimes exit the cell at the end of microvilli [19]. Such a process might have been observed in one case with the AFM when the cantilever tip was pushed up by the growing microvillus and the release of a hard structure with the right size for a viral particle was detected. The release of other hard structures of the right size occurred in flatter regions, but the number of microvilli observed always increased significantly after infection. The striking similarity between the EM and AFM images shown in Fig. 10.17 makes us believe that indeed the exocytosis of a progeny virus through the membrane of a living infected cell was observed and that the AFM can be used to study such processes.

EM images of viral particles at the end of microvilli structures on the left taken from the publication by Stokes et al. (Adapted from Ref. [19]) compare well with processes observed by AFM [18] showing the process of microvillus development from the lower right, with the release of a particle at the end before the last image on the upper left
These experiments make two advantages of the AFM obvious: First, it provides a resolution similar to that of the EM without the need for fixation and coating of the sample; and, second, it can produce a movie of a biological process under natural conditions for the cells studied similar to what optical microscopy can provide at a lower resolution.
10.3.4 AFM Application in Electrophysiology
The method of fixing a cell to a pipette and scanning it across an AFM cantilever tip is flexible enough to allow the integration of and combination with well-established cell physiological techniques of manipulation and investigation of single cells. The structures observed with the AFM can be related to known features of membranes, but more important than just the imaging of structures with this technique is the possibility of observing their time evolution. In this way, the AFM can provide information that brings us closer to understanding not only the “being” of these structures, but also their “becoming.”
10.3.4.1 Single Cells and Excised Membrane Patches
In principle, the AFM setup with the cell fixed on the end of a pipette allows simultaneous patch-clamp measurements for studying ion channels in the membrane of whole cells. Therefore, a logical step in the development of combinations of the AFM technique with established cell biological techniques was a combined patch-clamp /AFM setup that could be used to investigate excised membrane patches for single-ion-channel recording (see Fig. 10.18) [20]. In particular, the study of mechanosensitive ion channels in the membrane, which become activated by mechanical stress in the membrane, is an obvious application for a force-measuring device such as an AFM. The importance of such ion channels is very obvious in our senses of touching and hearing. During the development that started in 1991, a new type of patch-clamp setup was developed that was much more stable to satisfy the needs of AFM applications. It was designed to accommodate the usual electrophysiological and optical components besides the AFM. The chamber—where a constant flow of buffer solution guarantees the right conditions for the experiments—consists of two optical transparent plastic plates, one at the top and one at the bottom, keeping the water inside just by its surface tension. In this way, this type of flow cell is accessible from two sides. The chamber is mounted on an xyz-stage together with the optical detection of the AFM lever movement and a double-barrel application pipette. The latter was integrated to use the setup also for standard patch-clamp measurements, with the application of chemicals and different kinds of ions at different concentrations to characterize the sample further. The patch-clamp pipette in this setup is again mounted on a piezotube scanner fixed within the field of view of an inverse optical microscope necessary to control the approach of the pipette to the cell and to the AFM cantilever tip. In patch-clamp experiments, small membrane pieces are excised from a cell inside the chamber, which can contain none, one, or a few of the ion channels to be studied. This allows the measurement of currents through a single ion channel that is only open on the timescale of micro- to milliseconds, resulting in currents in the picoampere range.

Schematic of the AFM/patch-clamp setup (top) with an image of the cantilever pipette approach controlled through an optical microscope (left)
In such a setup, with a membrane patch at the end of the pipette, the AFM can image the tip of the pipette with the patch on top. Furthermore, changes in the patch according to the changing pressure in the patch pipette can be monitored, along with the reaction of the patch to the change of the electric potential across the membrane [20]. In the images, cytoskeleton structures are clearly seen excised together with the membrane. On these stabilizing structures of the membrane and on the rim of the pipette, resolutions down to 10–20 nm can be achieved, showing reproducible structures and changes that can be induced by the application of force.
Voltage-sensitive ion channels change shape in electrical fields, leading eventually to the opening of the ion-permeable pore. To investigate the size of this electromechanical transduction, the AFM setup was used in studies of cells from a cancer cell line (HEK 293) that were kept at a certain membrane potential (voltage-clamped) [21]. Cells transfected with Shaker K+ ion channels were used as controls. In these control cells, movements of 0.5–5 nm normal to the plane of the membrane were measured, tracking a ±10-mV peak-to-peak AC carrier stimulus to frequencies above 1 kHz with a phase shift of 90–120°, as expected of a displacement current. The movement was outward with depolarization, and the amplitude of the movement only weakly influenced the holding potential. In contrast, cells transfected with a non-inactivating mutant of Shaker K+ channels showed movements that were sensitive to the holding potential, decreasing with depolarization between −80 and 0 mV.
Further control experiments used open or sealed pipettes and cantilever placements just above the cells. The results suggested that the observed movement is produced by the cell membrane rather than by artificial movement of the patch pipette and/or acoustic or electrical interaction of the membrane and the AFM tip. The large amplitude of the movements and the fact that they also occurred in control cells with a low density of voltage-sensitive ion channels imply the presence of multiple electromechanical motors. These experiments open up a route to exploit the voltage-dependent movements as a source of contrast for imaging membrane proteins with an AFM.
10.3.4.2 Tissue Sections
Cochlear hair cells of the inner ear are responsible for the detection of sound (Fig. 10.19). They encode the magnitude and time course of an acoustic stimulus as an electric receptor potential, which is generated by a still-unknown interaction of cellular components. In the literature, different models for the mechanoelectrical transduction of hair cells are discussed [22, 23]. All hypotheses have in common that a force applied to the so-called hair bundle—which are specialized stereocilia at the apical end of the cells—in the positive direction toward the tallest stereocilia opens transduction channels, whereas negative deflection closes them. For a better understanding of the transduction process, it is important to know what elements of the hair bundle contribute to the opening of transduction channels and to study their mechanical properties. The morphology of the hair cells is described precisely by scanning and transmission electron microscopy. Unfortunately, this method is restricted to fixed and dehydrated specimen. Therefore, the development of an AFM setup to enable studies of cochlear hair cells in physiological solution was thought to provide further information on the dynamic properties of the system.

EM images of the mammalian hair cells of the inner ear with the V-shaped bundles of stereocilia. The right image shows a close-up where the lateral linkers between the stereocilia are marked by arrows
To control the approach to the tissue section, the AFM was again built into a patch-clamp setup onto an upright differential interference contrast (DIC) light microscope [24]. A water-immersion objective was used to achieve a high resolution of about 0.5 μm even on organotypic cell cultures with a thickness of about 300 μm. With this setup, it is possible to see ciliary bundles of inner and outer cochlear hair cells extracted from rats several days old and to approach these structures in a controlled way with the AFM cantilever tip. AFM images can be obtained by measuring the local force interaction between the AFM tip and the specimen surface while scanning the tip. The question of whether or not morphological artifacts occurred at the hair bundles during AFM investigation can be clarified by preparing the cell cultures for the electron microscope directly after the AFM measurements. Forces up to 1.5 nN applied in the direction of the stereocilia axis did not change the structure of the hair bundles. The resolution achieved with the AFM in this situation is high enough to image the tips of individual stereocilia and the typical shape of the ciliar bundle of inner and outer hair cells.
From AFM scan traces, the position of the individual stereocilium as well as its stiffness can be derived. The force interaction in the excitatory direction shows a stiffness increase with bending from 0.73 × 10−3 N/m, reaching a steady-state level at about 2 × 10−3 N/m. The mean stiffness value of stereocilia in the excitatory direction, calculated at the steady-state level, was determined to be (2.5 ± 0.6) × 10−3 N/m and (3.1 ± 1.5) × 10−3 N/m in the inhibitory direction. Taking into account the standard deviation of 0.6 × 10−3 N/m, the average force constant showed only a weak dependence on the position of the stereocilium in the excitatory direction. However, some stereocilia located at the center had an exceptionally high stiffness, and some located at the outer region revealed a very low stiffness. In the inhibitory direction, the mean stiffness in general is slightly higher, with a standard deviation 2.5 times above the excitatory direction. The higher stiffness might be explained by a direct contact between the taller and the adjacent shorter stereocilia, while in the excitatory direction, shorter stereocilia are pulled by elastic tip links. These links connect the rows of taller and shorter stereocilia [23]. The high standard deviation might be an effect of the variation in the spatial interaction between taller and shorter stereocilia. Depending on the angle between the scan direction and the direction defined by the centers of adjacent taller and shorter stereocilia, mechanical compliance can vary in a wide range. For the excitatory direction, two stiffness distributions are distinguishable. One group of values is located around 2.2 × 10−3 N/m, and a smaller one around 3.1 × 10−3 N/m, which corresponds to stereocilia located in the central region of the investigated hair bundles. Comparing the stiffness to the corresponding AFM images, one can find a correlation between the arrangement and stiffness of stereocilia. Those standing a little bit apart from their neighbors clearly show a higher stiffness, with a value of 3.2 × 10−3 N/m. A possible hypothesis may be that lateral links connecting these stereocilia with their direct neighbors are oriented in the direction of the exerted force. This will allow the transmission of additional force along these links to their neighbors.
The hypothesis above implies that all stereocilia standing closer display much less interaction with their neighbors mediated by side links. It can be directly tested by another AFM experiment. The force transmission between adjacent stereocilia in this experiment is examined by using a lock-in amplifier for detecting the transmitted forces stimulated by a glass fiber touching individual pairs of stereocilia. AFM imaging again allows an exact localization of stereocilia and the stimulating glass fiber. The AFM force signal is detected by a lock-in amplifier in phase with the vertical oscillation of the AFM cantilever at 357 Hz. The lateral force transmitted by lateral links can be calculated from the output signal and related to the stereocilium’s position. Only that half of the hair bundle can be taken into account, where the fiber is in contact with the directly stimulated stereocilium across its entire width but doesn’t touch the nearest adjacent stereocilium at all. During the experiment, the arrangement of stereocilia and stimulating fibers was regularly controlled by AFM imaging. Force interaction between the stereocilia and the AFM tip leads to displacements of stereocilia from 0 nm to about 250 nm. Therefore, the relative displacement between the directly stimulated stereocilium and the stereocilium pushed by the AFM tip is expected to result in the continuous stretching of lateral links located in between. This allows detection of transmitted forces by lateral links for different states of stretching. For us to be able to compare the results more accurately, the forces have to be normalized with respect to the corresponding maximum force detected at the directly stimulated stereocilium. Normalized forces rapidly decrease from the directly stimulated to the first adjacent stereocilium. During the fiber tip’s approach to the stereocilium, the relative position is detected, but not the preload force. Obviously, forces transmitted by lateral links rapidly decrease from a directly stimulated stereocilium to an adjacent stereocilium in the experiments done with rats at postnatal age day 4. This result supports the hypothesis of a weak interaction between stereocilia by lateral links.
In these experiments, the noise level made it difficult to distinguish between slight couplings by lateral links from mechanically independent stereocilia. Besides the modulation signal at 357 Hz, the same AFM curves of the identical outer hair cells contain information about the stiffness of stereocilia as described above. This information can be used to detect the mechanical effect of the touching glass fiber on the stiffness of adjacent stereocilia. If lateral links contribute to the stiffness measured at individual stereocilia, one would expect to see an increase in the stiffness of adjacent stereocilia compared to data shown for stereocilia investigated without the fiber attached. Stiffness data were derived only for the excitatory direction, where the AFM tip displaces the stereocilia toward the fiber tip. The results of these experiments show that not only does the stiffness of the directly touched stereocilium increase, but also the stiffness of the five stereocilia next to it increases. The mean stiffness in the excitatory direction turns out to be (4.8 ± 1.8) × 10−3 N/m, which is about 1.9 times higher than the mean stiffness of stereocilia not touched by the fiber.
In the experiments described, the AFM allows local stiffness measurements on the level of individual stereocilia. The results represent the local elastic properties of the directly touched stereocilium and its nearest neighbors. It can be seen that stiffness depends on the orientation of links with regard to the direction of the stimulus.
A stimulating fiber had only little mechanical effect on adjacent stereocilia if not in direct contact with the fiber. For a partial decoupling of the tectorial membrane from hair bundles of outer hair cells, it therefore can be assumed that only stereocilia still in contact with the tectorial membrane and their nearest neighbors are displaced by an incoming mechanical stimulus. Lateral links may not compensate for loss in contact with the tallest stereocilia of the outer hair cells. Decoupling of the tectorial membrane following exposure to pure tones at high sound-pressure levels is supposed to protect hair cells and to avoid damage [25]. A strong interaction between the tectorial membrane and hair bundles of the outer hair cells seems to be essential for the efficacy of the cochlear amplifier and the transduction of sound into an electrical signal.
In the experiments described above, the AFM was used only to examine the elastic properties of stereocilia on hair cells, similar to all the micromechanical measurements that already have been performed on the entire stereocilia bundles of sensory hair cells using thin glass fibers directly attached to the bundle or by using fluid jets. The receptor potential or transduction current in these experiments was measured in response to the displacement of stereocilia. To study the kinetics of a single transduction channel over the whole range of its open probability requires a technique that allows stimulating a single stereocilium [26]. As shown in the previous section, the AFM is such a technique, allowing a force to be exerted very locally to an individual stereocilium. To patch the hair cells with a pipette necessitates removing the supporting cells with a cleaning pipette. After cleaning, a patch pipette filled with intracellular solution (concentrations in mM: KCl 135, MgCl2 3.5, CaCl2 0.1, EGTA 5, HEPES 5, ATP 2.5; at pH 7.4) can be attached to the lateral wall of the outermost row of outer hair cells. This so-called cell-attached configuration is the precursor of the whole-cell configuration where the microelectrode is in direct electrical contact with the inside of the cell. For low-noise measurements of single ion channels, the seal resistance should typically be in the range >1 GΩ. A pulse of suction applied to the pipette breaks the patch, creating a hole in the plasma membrane, and provides access to the cell interior. During recording, the electrical resistance between the inside of the pipette and the hair cell should be very small. Many voltage-activated K+ ion channels are embedded in the lipid membrane of outer hair cells. The opening and closing of these channels increases the background noise level during transduction current measurements. The current response of outward-rectifying K+ ion channels can be controlled by applying 10-mV voltage steps across the cell membrane (progressively increasing from −100 to +40 mV). The outward currents mainly correspond to potassium ion currents of voltage-gated K+ channels. During transduction current measurements, the holding potential of the hair cell is set to −80 mV, corresponding to the reversal potential of K+ ion channels. After forming a seal, the AFM tip is moved to the top of the corresponding hair bundle under optical control. It then is used to successively displace each stereocilium within a hair bundle as described before, but in contrast to the force-transmission measurements, a sinusoidal voltage is now added to the normal AFM scan signal, modulating the AFM tip in horizontal directions with an amplitude of 190 nm at 98 Hz. In this way, the hair bundle is slightly displaced several times while interacting with the lateral side of the AFM tip. The tip is repeatedly scanning along the same line while approaching the hair bundle. As expected, an inward current cannot be detected until a stereocilium is displaced in the excitatory direction. In the experiments described above, a weak transmission of force from the directly stimulated stereocilium to adjacent stereocilia was detected, implying that only a few channels are opened. The results of the elasticity measurements are nicely complemented by the electrophysiological results of experiments where the tip of the AFM cantilever is repeatedly scanned across the same stereocilium of an outer hair cell from the medial turn of a postnatal rat at day 3. With applied horizontal forces of up to 800 pN resulting in stereocilia displacements of about 350 nm in the excitatory direction and 250 nm in the inhibitory direction, the AFM tip opens up transduction channels in the excitatory direction for about 90–130 ms. The current amplitudes reflect the displacements and show for small forces the expected characteristics of single-channel currents. With these experiments, Langer and colleagues [26–34] demonstrated for the first time single-channel recording of mechanosensitive ion channels (Fig. 10.20).

AFM force signal (bottom) and ion-current response (top) while stimulating a single stereocilium with the cantilever tip
10.4 Conclusion
Since its invention in 1986, the AFM was continuously developed to suit many different applications. In biological research, it provides high-resolution, three-dimensional imaging not only of single molecules and macromolecular assemblies, but also of intact, living cells in physiological solutions and gives a detailed view of surface features of large biological structures such as grains and seeds. In addition to imaging, the AFM has the capability to simultaneously measure biophysical properties, such as the stiffness of molecular structures, but it also provides the means to analyze binding interactions down into the range of only a few pico-Newtons and to analyze the effect of the environment (pH, salt, temperature, etc.) on the interaction. The implications of this are far-reaching for many biomedical applications, including the development of new drugs, targeted drug delivery, biocompatible materials for implants, as well as in vitro and in vivo sensors, and in the agro-food area for quality control.
For the study of many biological samples, a parallel optical observation is essential to control the approach to the sample and to have available certain standard controls for the actual sample. However, an AFM built on top of an optical microscope loses part of its stability. A major technological breakthrough in this respect was the development of a multiple-detection scheme using two cantilevers in parallel to separate force and distance measurement. This development allows long-term stabilization of the distance between the cantilever tip and the surface with picometer precision, thereby enabling so-called force-clamp measurements. With such an approach, it is possible to perform unfolding and refolding experiments of even small protein structures, for instance. The details about the unfolding of single-protein domains revealed by precise AFM measurements have demonstrated that force spectroscopy can be used to determine the forces that stabilize protein structures and to analyze the energy landscape and the transition probabilities between different conformational states. In this way, the AFM can make essential contributions to the understanding of the connection between protein structure and function.
The AFM used as an imaging tool and as a force-measuring tool simultaneously allows the localization of molecular structures and the determination of their biophysical properties such as elasticity. Unfortunately, the AFMs that are currently available commercially are still quite large and bulky, expensive, and difficult to use, requiring a heavy vibration-stabilization platform. The future development of a mass-producible “micro-”AFM with a stability and price that enable field applications will extend the application range dramatically not only in biomedical and bioengineering laboratories, but also in many industrial areas, especially for quality control. Such a development will automatically include a much higher imaging speed, allowing the analysis of processes in the millisecond range. It will also eliminate many operational problems, which then can be fully managed by software developments.
The future AFM will be (1) compact, (2) rapid-scanning, (3) user-friendly, (4) mass-producible and, consequently, cheap.
References
Synge EH. A suggested method for extending microscopic resolution into the ultra-microscopic region. Philos Mag. 1928;6:356.
O’Keefe JA. Resolving power of visible light. J Opt Soc. 1956;46:359.
Ash EA, Nicolls G. Super-resolution aperture scanning microscope. Nature. 1972;237:510.
Binnig G, Rohrer H, Gerber C, Weibel E. Tunneling through a controllable vaccuum gap. Appl Phys Lett. 1982;40:178.
Binnig G, Quate CF, Gerber C. Atomic force microscope. Phys Rev Lett. 1986;56:930.
Atomic force microscope and method for imaging surfaces with atomic resolution, issued to Gerd K. Binnig of IBM’s Zurich Research Laboratory in 1988, U.S. Patent 4724318.
Giessibl FJ, Hembacher S, Bielefeldt H, Mannhardt J. Subatomic features on the silicon(111)-(7×7) surface observed by atomic force microscopy. Science. 2000;289(5478):422.
Meyer E, Amer NM. Novel optical approach to atomic force microscopy. Appl Phys Lett. 1988;53:2400.
Florin E-L, Moy VT, Gaub HE. Adhesion forces between individual ligand-receptor pairs. Science. 1994;264:415.
Haynes CA, Norde W. Globular proteins at solid-liquid interfaces. Colloids Surf B Biointerfaces. 1994;2:517–66.
Eckert R, Jeney S, Hörber JKH. Understanding intercellular interactions and cell adhesion: lessons from studies on protein-metal interactions. Cell Biol Int. 1998;21:707.
Rief M, Gautel M, Oesterhelt F, Fernandez JM, Gaub HE. Reversible Unfolding of Individual Titin Immunoglobulin Domains by AFM. Science. 1997;276:1109.
Kellermayer MSZ, Smith SB, Granzier HL, Bustamante C. Folding-unfolding transitions in single titin molecules characterized with force-measuring laser tweezers. Science. 1997;276:1112.
Tskhovrebova L, Trinick J, Sleep JA, Simmons RM. Elasticity and unfolding of single molecules of the giant muscle protein titin. Nature. 1997;387:308.
Altmann SM, Grunberg RG, Lenne PF, Ylanne J, Raae A, Herbert K, Saraste M, Nilges M, Hörber JKH. Pathways and intermediates in forced unfolding of spectrin repeats. Structure. 2002;10:1085–96.
Häberle W, Hörber JKH, Binnig G. Force microscopy on living cells. J Vac Sci Technol. 1991;B9:1210.
Cross SE, Yu-Sheng Jin, Jianyu Rao, Gimzewski JK. Nanomechanics of human metastatic cancer cells in clinical pleural effusions. Nat Nanotechnol. 2007;2:780–3.
Hörber JKH, Häberle W, Ohnesorge F, Binnig G, Liebich HG, Czerny CP, Mahnel H, Mayr A. Investigation of living cells in the nanometer regime with the scanning force microscope. Scanning Microsc. 1992;6:919.
Stokes GV. High-voltage electron microscope study of the release of vaccinia virus from whole cells. J Virol. 1976;18:636.
Hörber JKH, Mosbacher J, Häberle W, Ruppersberg P, Sakmann B. A look at membrane patches with a scanning force microscope. Biophys J. 1995;68:1687.
Mosbacher J, Langer M, Hörber JKH, Sachs F. Voltage-dependent membrane displacements measured by atomic force microscopy. J Gen Physiol. 1998;111:65–74.
Corey DP, Hudspeth AJ. Analysis of the microphonic potential of the bullfrog’s sacculus. J Neurosci. 1983;3:962.
Furness DN, Hackney CM, Benos DJ. The binding site on cochlear stereocilia for antisera raised against renal Na channels is blocked by amiloride and dihydrostreptomycin. Hear Res. 1996;93:136.
Langer MG, Öffner W, Wittmann H, Flösser H, Schaar H, Häberle W, Pralle A, Ruppersberg JP, Hörber JKH. A scanning force microscope for simultaneous force and patch-clamp measurements on living cell tissues. Rev Sci Instrum. 1997;68:2583.
Adler HJ, Poje CP, Saunders JC. Recovery of auditory function and structure in the chick after two intense pure tone exposures. Hear Res. 1993;71:214.
Langer MG, Koitschev A, Haase H, Rexhausen U, Hörber JKH, Ruppersberg JP. Mechanical stimulation of individual stereocilia of living cochlear hair cells by atomic force microscopy. Ultramicroscopy. 2000;82:269.
Goldsbury C, Scheuring S. Curr Protoc Protein Sci. 2002;17.7.1–17
Hörber JKH, Miles M. Science. 2003;302:1002–5.
Braga PC, Ricci D. Atomic force microscopy: biomedical methods and applications. Totowa: Humana Press; 2004.
Connell SDA, Smith DAM. Mol Membr Biol. 2006;23(1):17–28.
Forman JR, Clarke J. Curr Opin Struct Biol. 2007;17:58.
Yang H, Wang Y, Lai S, An H, Li Y, Chen F. J Food Sci. 2007;72(4):65–5.
Gonçalves RP, Buzhysnskyy N, Scheuring S. J Bioenerg Biomembr. 2008;40:133–8.
Liu H, Fu S, Zhu JY, Li H, Zhan H. Enzyme Microb Technol. 2009;45:274–81.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Hoerber, J.K.H. (2014). Atomic Force Microscopy: Applications in the Field of Biology. In: Smentkowski, V. (eds) Surface Analysis and Techniques in Biology. Springer, Cham. https://doi.org/10.1007/978-3-319-01360-2_10
Download citation
DOI: https://doi.org/10.1007/978-3-319-01360-2_10
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-01359-6
Online ISBN: 978-3-319-01360-2
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)