
Abstract
The importance of iron to the metabolism of the mycobacteria was gradually appreciated during the first half of the last century. Frank Winder working in Dublin in the 1950s and 1960s was the first to establish the absolute amounts of iron needed for growth and, from his work, it was then possible to investigate the consequences of iron deficiency and subsequently how iron was solubilized and transported into mycobacterial cells. Parallel with this work, was the discovery by Alan Snow, at ICI Ltd, UK, of the mycobactins. These are essential growth factors for Mycobacterium paratuberculosis and their role in iron binding was then pivotal to elucidating the main aspects of iron uptake. However, mycobactins, being wholly intracellular materials, were unable to act as external siderophores for the solubilization of iron; this role was then found to be carried out by the exochelins discovered by the author of this review. The exochelins were of two types: those from the non-pathogenic mycobacteria were water-soluble pentapeptides whereas those from pathogenic species were modifications of mycobactin and were then named as the carboxymycobactins. The interdependency of these materials and others is then unraveled in this review. The review focuses mainly on the research work carried out over the last century leaving the present work on iron uptake to be covered in other reviews in this monograph.
“Mycobacteria are nothing more than E. coli wrapped up in a fur coat”—Frank Winder.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
2.1 Introduction
In writing this review I have attempted to cover as much as possible of the early research work of how our understanding of iron metabolism has developed over the past six or seven decades as this material now receives little, if any, mention in current research papers or reviews. However, there are many nuggets of valuable information tucked away in many of these early papers that should not be forgotten as they can help to identify aspects of the topic that still need investigation and explanation.
Iron metabolism does not, though, have a long history. The real beginnings only start in the 1950s through the work of Frank Winder. He was the pioneer in this area as he was the first to appreciate that, in order to study how iron was metabolized, it was necessary to know how much iron was needed by the mycobacteria to grow. It was then essential to prepare culture medium with as much iron as possible being removed from it; this then provided the first culture media that were genuinely, and knowingly, iron deficient. Thus, Winder’s research group in Dublin, Ireland, was then the first group to be engaged on understanding the consequences of iron deprivation in mycobacteria. This deprivation of iron in the growth medium was the essential first step in understanding how iron was assimilated into the bacteria. Now, many laboratories throughout the world are actively pursuing a variety of aspects of the subject but these still have a long way to go before the problems of iron metabolism, both in vitro and in vivo, could be considered as being solved.
The importance of iron in the growth and multiplication of the tubercle bacillus, and other pathogenic mycobacteria has gradually been appreciated over the past three or four decades as it has been increasingly realized that iron-deficient growth conditions are the ‘natural’ state for pathogenic bacteria to be in when they are causing infections in a host animal. Thus, from the early work of Frank Winder from the 1950s and into the 1970s has developed a crucial understanding of how mycobacteria are able to acquire the iron that is essential for their growth and multiplication when causing tuberculosis and related mycobacterial diseases.
The other pioneer in this field was Alan Snow who was involved in the discovery of the mycobactins that were subsequently to become central to the iron metabolism story. However, the involvement of the mycobactins with iron only came in the 1960s, some 20 years after the initial descriptions of the molecule. By this time, interest in mycobactins, as possible target molecules for the design of rational anti-tuberculosis agents, had faded and with only one or two groups then pursing the biochemical puzzles that had been opened up by the discovery of these unique microbial siderophores. Interestingly, however, these early thoughts of synthesizing novel mycobactin antagonists are now being re-visited as the need for novel chemotherapeutic agents for the treatment of tuberculosis becomes ever-more urgent.
I hope, therefore, that this early history of the subject will be of interest to readers. I have placed much less emphasis on developments over the past 10–15 years as, although a lot is now happening in many laboratories, these aspects are mainly focusing on filling in the details of individual parts of the overall picture and are, in any case, covered elsewhere in this monograph. Solving the problems of iron metabolism is a little like doing a jigsaw puzzle: in this case, however, one is never sure that one has all the pieces available to provide the final picture. One tries to provide a coherent and comprehensible view of iron metabolism in the mycobacteria from the pieces that can be found. But how many more are still missing? In spite of us thinking that we now know what the final picture is going to look like, there is clearly still a long way to go!
2.2 The Trouble with Iron
Iron is an essential trace element for almost all living cells. The only exceptions appear to be some lactobacilli and the spirochete, Borrelia burgdorferi, that is the causative agent of Lyme disease. Iron is needed as an essential co-factor in many enzymes and is also the critical metal ion in all haem compounds, including all the cytochromes that carry out essential functions in energy metabolism and also are components of several key enzymes. Iron, however, is unique amongst the nutrients needed for cell growth in that is insoluble at neutral pH values. However, this needs to be qualified as iron exists in two states: the reduced ferrous form and the oxidized ferric form. It is the latter form that is insoluble and, although ferrous salts are water-soluble, they quickly oxidize to the ferric form, a reaction which is accelerated if the iron is in a chelated form. Although the solubility of ferric iron at pH 7 has usually been stated to be 10−18 M, more recent measurements give this as about 10−9 to 10−10 M [1, 2]. This revised lower value arises because it is now appreciated that the principal ionic species that exists in aqueous solution is \( {\text{Fe}}({\text{OH}})_{2}^{+} \) and not Fe(OH)3 as previously thought. Even though this revised value is a billion times higher than the earlier value, it still results in iron being effectively insoluble as 10−9 M corresponds to 56 pg/ml. This then effectively renders iron as being unavailable to cells. Specific mechanisms have therefore evolved so that cells may acquire iron from the environment and also hold it within themselves in a usable form. These mechanisms differ between animals, plants and microorganisms.
For microbial pathogens, solving the problem of iron acquisition is essential. If they cannot acquire iron from the sources of iron inside the host that they have infected, then they will be unable to grow and thus cause disease. As pathogens do cause disease, we can obviously conclude that all pathogens must have evolved mechanisms for iron acquisition. The principal sources of iron within an animal are: transferrin, ferritin, haemoglobin and haem-containing proteins.
Transferrin is the principle iron transporting protein in the blood. There are related proteins of lactoferrin, found in milk and other extracellular fluids and secretions, and ovaferrin (formerly known as conalbumin) that is found in eggs. These are large proteins (~80 kDa) but only have two binding sites for ferric iron.
Ferritin is a protein (~50 kDa) comprising 24 identical subunits that form a hollow sphere into which up to 4,000 atoms of Fe(III) can be stored. This is the principal form of iron storage in animal cells. A similar form of it, bacterioferritin, exists in bacteria, including mycobacteria, for the same function.
In haemoglobin and other haem containing molecules iron is tightly bound as the ferrous ion and therefore, for metal release, the molecule must be degraded. This occurs principally with haemolytic bacteria which therefore excludes most pathogenic mycobacteria.
To achieve release of iron from transferrin and ferritin requires that pathogenic bacteria, including mycobacteria, must either attack the protein itself by secreting various proteases, use a ferric reductase that would generate ferrous ions that might then be directly assimilated, or, alternatively, use molecules of very high iron binding strength that can then, literally, strip the iron out of the molecules. The materials that can do this are known as siderophores and those relevant to the mycobacteria will be covered in this review and also elsewhere in this monograph.
Thus, we can conclude that all mycobacteria, whether pathogenic or saprophytic, require iron and that the acquisition of iron from whatever source requires specific mechanisms for achieving this.
2.3 Iron as an Essential Nutrient for Mycobacteria
In the early days of mycobacteriology, iron was ‘guessed’ to be an essential minor trace element and workers, such as Sauton [3], in devising appropriate growth medium for the cultivation of mycobacteria, recommended that iron be added to the culture medium at 10 μg/ml. Obviously, this was an empirical amount but later workers [4–6] were able to confirm that iron was indeed required to achieve full growth of various mycobacteria. Edson and Hunter [4] indicated that, for M. phlei, 3.75 μg Fe/ml was needed for full growth but Turian [6] revised this to just 1 μg/ml. Clearly, the amount of iron added to the medium would depend on the amounts of iron which were adventitiously included by other ingredients of the medium. Nor should the presence of iron in the water or in the glassware being used for cultivation be ignored. Thus, to establish what amounts of iron might be necessary for growth it was first necessary to prepare culture medium that with as little iron as possible. This was first appreciated by Frank Winder who then pioneered a series of in-depth studies on the role of iron in the metabolism of the mycobacteria.
Frank Winder (Fig. 2.1) carried out all his work on the mycobacteria at Trinity College, Dublin, Republic of Ireland, starting from about 1953 and continuing to his death in 2007 at the age of 79. Initially, Winder’s work was done in the Laboratories of the Medical Research Council of Ireland and then later in the Department of Biochemistry also within the College. In a seminal paper in which the conditions of iron deficient (and also zinc deficient) growth of a mycobacterium (Mycobacterium smegmatis) were first described, Winder and Denneny [7] observed that when some, but not all, cultures of the bacterium were grown in modified Proskauer and Beck medium to which no iron or other trace elements had been added (simply because the formulation of this medium did not include addition of an iron or zinc salt), the cells prematurely ceased growth, failed to form the usual pellicle and became elongated with a low level of DNA. The cells had, in fact, been cultivated in a medium that was accidentally deficient in both iron and zinc. In addition, the tubes being used for the cultivations had been recycled a number of times thereby exhausting adventitious metal ions from the glassware itself. When both iron and zinc were subsequently included in the medium, full and normal growth of M. smegmatis was restored including restoration of DNA synthesis.

Frank Gerald Augustine Winder (1928–2007)
This discovery then opened up the door to a major study of iron metabolism in the mycobacteria by Winder and his associates that also included the author of this review (see Fig. 2.2) who joined his team as a post-doctoral fellow in 1960 and worked in the MRC of Ireland Laboratories until 1964. Winder and O’Hara [8, 9] then described the effects of both iron and zinc deficiencies on the composition of M. smegmatis; some of the key findings are summarized in Table 2.1. However, before these studies could begin, it was necessary to devise a simple protocol for the removal of the trace metal ions from the medium otherwise there would be an inadequate basis on which to conduct the subsequent studies. The procedure used, which was derived from the work of Donald et al. [10], was to autoclave 2 L of medium in a 5 L flask with 1 % (w/v) high-grade, activated alumina. When the flask came out of the autoclave, the contents was immediately shaken thoroughly and then, when cool, filtered through Whatman number 542 filter paper which was considered to be the highest quality, ash-free paper then available. The first 50 ml of the filtered medium was discarded as this has come into contact with measuring cylinder, the filter funnel and the filter paper itself. The remaining medium was then dispensed in 100 ml lots into cleaned 250 ml conical flasks. Cleaning the flasks necessitated devising another strategy. Initially flasks were cleaned with chromic acid although this procedure was soon replaced by filling the flasks with alcoholic KOH and leaving overnight. This was followed by washing them in distilled water then standing for another night in 2 M HNO3. The flasks were finally thoroughly rinsed in distilled water and allowed to drain upside down before being filled with medium the next day. The procedure was extremely tedious and time-consuming and services of a dedicated technician were then needed to do all the preparatory work.

Colin Ratledge (1936–)
Winder and O’Hara [11] also carried out some significant analytical work on the cellular content of iron in the mycobacterial cells. Under the most stringent iron deficient growth conditions, M. smegmatis contained 64 μg Fe/g cell dry weight suggesting that this was the lowest possible concentration needed for the cells to function. Obviously, as the mycobacteria have an essential requirement for iron, the cells would not be able to grow if there was, literally, no iron in the medium. They could not synthesize the various cytochromes and iron-containing enzymes that are vital for cell metabolism and growth. If iron was not limiting, then the iron content of the cells rose to 224 μg/g cell dry wt. Values for the zinc content of the cells were simultaneously calculated as 11 and 43 μg/g cell dry weight, respectively. It was evident, however, that iron-deficiently growing cells were adapting to allow some growth to occur but clearly major changes were occurring within the metabolic pathways to minimize the detrimental effects of iron deficiency.
One of the main effects of iron deficiency on metabolism appeared to be a decrease in the DNA to protein ratio [8]. This was subsequently attributed to there being a considerable increase in the activity of an ATP-dependent DNAase [12, 13] and a DNA polymerase [14]. Further work on the DNAase [15, 16] considered that it was involved in recombination repair and possibly in excision repair. Interestingly, how the increased activities of both these enzymes then correlated with the original discovery, that there were low concentrations of DNA in iron-deficient cells, was resolved by Winder and Barber [17] who reported that hydroxyurea could induce the same effects as iron deficiency, including cell elongation. However, one cause of DNA degradation might be in the lowered activity of ribonucleotide reductase which, in E. coli, is known to contain iron as an essential co-factor [18] and would therefore lead to an alteration in the pool of nucleotides. This possibility, though, does not appear to have been followed up in M. smegmatis and the general consensus was that the decrease in DNA during iron deficient growth was a secondary but not a primary effect.
The paper by Winder and Barber [17] and a subsequent one by MacNaughton and Winder [19] were the last papers that Frank would write on aspects of his work connected to iron deficiency in the mycobacteria. He then, with respect to his continuing interest in the mycobacteria, concentrated on trying to unravel the mechanism of action of isoniazid (INH) as one of the more potent anti-TB compounds then available. This work had also begun in the 1960s and was carried out in parallel with the iron deficiency studies.
It was not, though, surprising that Winder and O’Hara [9] reported considerable decreases in activity of many iron-containing enzymes. These included various cytochromes. These findings were subsequently confirmed by McCready and Ratledge [20] also working with M. smegmatis. Non-haem iron in the cells dropped to ~0.2 nmol/g CDW from ~5 nmol/g CDW and cytochromes a and b could not be detected. However, cytochrome c was scarcely affected, as were the flavoproteins, indicating that these iron-containing components were, to some extent, protected from the severest ravages of iron deficiency. Presumably these components have very high affinities for iron and thus can acquire iron even when it was available in the smallest concentrations inside the cell and against competition from other iron-dependent cytochromes and enzymes.
Iron deficiency therefore produces a major diminution of most components in the respiratory chain and this, in turn, will inevitably cause a decline in activity of glycolytic enzymes as the final oxidation of pyruvate, via the tricarboxylic acid cycle and its linkage to oxidative phosphorylation that involves many cytochromes, would be seriously impaired by iron deficiency. Thus, it is not unexpected to find a general down-regulation of many enzyme activities in the central pathways of metabolism simply as a consequence of there being diminished energy (ATP) production.
It was apparent from these experiments of the 1960s and 1970s that iron deficiency in mycobacteria was causing the cells to become ‘anaemic’, somewhat equivalent to the condition that seen in humans and other animals. Cells became ‘lethargic’: they had a diminished supply of energy, failed to grow properly and failed to carry out normal metabolism. They also become noticeably much paler than cells grown with a surfeit of iron. McCready and Ratledge [20] and McCready [21] found that the content of porphyrins in iron deficient cells was adversely affected: in M smegmatis, coproporphyrin III was less than 25 μmol/g CDW after iron deficient growth compared to over 200 μmol/g CDW in cells grown iron sufficiently. It is then this absence of porphyrin that accounts for the very pale appearance (the ‘anaemic’ condition) of the iron-deficient cells. This low content of porphyrin was later also observed in iron-deficiently grown M. avium [Barclay and Ratledge, unpublished work in the 1980s] and may then be a general explanation for most mycobacteria being much paler when grown without adequate amounts of iron. This phenomenon, which is not seen with other bacteria, must be caused by repression of the biosynthesis of the porphyrin nucleus due to lack of iron in the cells. This makes metabolic sense. Why synthesize something that cannot be converted into the end-product: haem? Therefore stopping the synthesising of the precursor of haem is a sensible metabolic strategy under iron deficient conditions.
But this then poses a major problem to the cells: if iron then becomes available to the cells for whatever reason, what are the cells going to do with this iron if it cannot be immediately converted into haem because there are no precursor molecules of porphyrin available? Up-regulation of porphyrin biosynthesis cannot be immediate. The cells must therefore have some means of acquiring the iron and holding it in a form which can then be mobilized as porphyrins begin to be re-synthesized. This aspect of iron metabolism then is considered in the next section of this review.
Such is the need to scavenge whatever iron might be available from the environment, so as to rescue the cells from their ‘anaemia’, that it is therefore not surprising to observe that much metabolic effort is expended by the iron-deficient mycobacterium to acquire iron from its environment. Iron deficiency, however, is not just a man-made artificial construct for laboratory cultivation experiments. There is good reason now to consider that iron deficiency is the normal status of pathogenic mycobacterium within the animal tissue which it is infecting. Pathogenic mycobacteria therefore must overcome the natural defenses of the infected host animal that seeks to withhold iron from the invading bacteria. Unless a pathogen, and not just a mycobacterium, can acquire iron from its host, it will not be able to grow and thus become pathogenic. Gaining iron is therefore possibly the first step for an invading bacterium to achieve in order to grow in vivo. Thus, the mechanisms of iron acquisition by mycobacteria are of prime concern if we are to understand anything about the pathogenicity of these bacilli.
2.4 Early Discoveries of the Major Components of Iron Acquisition by Mycobacteria
2.4.1 The Mycobactins
The first clues about how iron might assimilated by mycobacteria came very indirectly from the initial observations by Twort and Ingram [22–24] when they were attempting to cultivate the mycobacterium that was the causative agent of Johne’s disease in cattle. However, it would be more than 50 years before it would be appreciated how these early observations, and the subsequent discoveries arising from them, fitted in with iron metabolism to unravel a major and unique feature of mycobacterial metabolism.
Johne’s disease in cattle causes chronic enteritis and was found to be caused by a mycobacterium that was then called M. johnei [25] but was re-named as M. paratuberculosis. This name is therefore used in the remainder of this review although more recent taxonomic work has re-classified the bacillus yet again, as mentioned below. The organism, very importantly for the iron assimilation story, could not be cultivated in ordinary laboratory medium but Twort and Ingram [22–24] found that by supplementing the egg-based medium they were using with dry, killed human tubercle bacilli they could then achieve good growth of this previously uncultivatable Mycobacterium species. Animal tissues and extracts were ineffective. They subsequently found that other killed mycobacteria could also support growth: these included M. phlei, M. smegmatis, and M. butryicum as well as other less-well defined mycobacteria. Also extracts from the killed mycobacteria prepared using organic solvents were equally successful in promoting growth. The conclusion was reached that M. paratuberculosis lacked the ability to synthesize some essential growth factor but that this material was synthesized by several competent mycobacteria that could be grown in laboratory culture medium. As was subsequently acknowledged by Snow [26], Twort could be considered to be “… a true pioneer in this field as the concept of vitamins and growth factors were only dimly recognized at that time”. Yet here was a scientist proposing a radical concept of a transferable growth factor, available from some microbial sources but not synthesized by the host microorganism. This was exactly the same concept that lay behind the discovery of many of the vitamins essential for human metabolism but this was a novel concept for achieving growth of a microorganism.
Twort’s findings, however, were not developed for another 30 years. In the meanwhile, cultivation of M. paratuberculosis, which was, and continues to be, a considerable veterinary problem, in laboratory media was routinely achieved by adding a simple extract prepared from cells of M. phlei which, of course, is a non-pathogenic species. Cultivation of Johne’s bacillus did not, therefore, require a growth factor that was confined to pathogenic mycobacteria. A saprophytic mycobacteria could do just as well and was obviously less hazardous to grow and extract.
We now must move to the 1940s for the development of Twort’s observations and to the laboratories of one of the major industrial chemical companies in the UK: Imperial Chemical Industries (ICI) Ltd at their pharmaceutical research laboratories at Blackley near Manchester and later at Wilmslow and finally at Alderley Edge in Cheshire. An account of the thinking that went on in ICI for Twort’s discoveries to become a priority research program has been given by Snow [26]. The person who initially took up the baton was J. Francis who pointed out in 1945, (cited by Snow [26]) that a specific growth factor for M. paratuberculosis was being synthesized by M. tuberculosis. As no other microorganism, other than another species of mycobacteria, could produce such a compound, then this compound must be unique to the mycobacteria. In addition, as Twort had found that extracts of animal tissues, including those from cattle, could not support growth of Johne’s bacillus, this growth factor was not being synthesized by animals.
As there was no chemotherapeutic treatment for tuberculosis in 1945, Francis’s reasoning was clear: M. tuberculosis and other mycobacteria were synthesizing a specific growth factor not found in humans. If this compound could be identified then it opened up the opportunity of designing an appropriate antagonist that would then, hopefully, be specifically inhibitory to the tubercle bacillus. Such inhibitors should not though affect the infected human as there was no suggestion that the missing growth factor for M. paratuberculosis was synthesized in animals. ICI Ltd obviously considered that if this aspiration could be realized then it could be an extremely lucrative project and it is little wonder that major efforts were expended to attain the goals.
GA Snow (see Fig. 2.3), always known as Alan, joined the team in the late 1940s and was to become the major driver of the entire project. He has written that the initial work on isolating and purifying the growth factor was undertaken by J. Madinaveitia and H.M. Macturk working with the initial project leader, J. Francis [27]. They used massive quantities of M. phlei: in all some 50–60 kg dry cells were used and the growth factor was eventually isolated and purified. It was given the name mycobactin.

G Alan Snow (with kind permission of The Biochemical Society, UK)
The work, though, was extremely difficult. It was hindered by there being no adequate assay for the growth factor as its structure was unknown and, although it could bind to metal ions, this was not regarded of any particular significance. Indeed, in the first full-length paper describing the isolation of mycobactin by Francis et al. [28], more attention was given to the copper complex of mycobactin and this was, in fact, the complex of mycobactin that was studied in detail. Iron binding received only one mention in this seminal paper: “Ferric salts react with mycobactin to give an intense reddish purple colour”. How these workers missed the significance of iron binding to mycobactin, or failed to appreciate that this would provide a simple assay for quantifying mycobactin, now seems strange because the preparations of unchelated mycobactin readily form the red ferric mycobactin complex, having a tenacity for iron binding is so strong that the mycobactin readily stripped iron from water, glassware and any materials containing trace amounts of iron. However, as Snow was using such large quantities of cells and extracting considerable amounts of mycobactin, the material was only turning a light brown in color as clearly the amount of available iron was relatively small.
The early work on mycobactin was helped considerably by mycobactin adventitiously forming a crystalline aluminium complex [27] during its purification and passage through a column of chromatographic alumina. This allowed the team to carry out some X-ray crystallography giving a suggested molecular weight of about 914. This was subsequently revised to 870 with the formula of C47H75O10N5. To work out the structure of such a large molecule, however, was then a considerable and daunting task. Also, as mentioned above, Snow and his colleagues had found that it was extremely difficult to assay mycobactin during its purification processes. Initially, assays for its presence had to rely on its growth-stimulating properties for M. paratuberculosis which were slow and extremely tedious even with improved techniques [29]. There were, however, suggestions by Antoine et al. [30] and Reich and Hanks [31] that Arthrobacter terregens, which also required a ‘terregens factor’ for growth, might be a more suitable organism for the bioassay of mycobactin as it could be grown in about 3 days or so. This bacterium was, though, much less sensitive to mycobactin than M. paratuberculosis [26] and was also responsive to growth factors other than mycobactin. It therefore does not appear to have been used to any great extent by Snow himself.
The initial major effort that had been put into the pharmacological aspects of the project appears to have dissipated somewhat by the very early 1950s and the original authors of the 1953 paper [28] do not appear, even in the acknowledgements, of the next papers that were published on the structure of mycobactin in 1954 [32, 33]. Perhaps the discovery of streptomycin in 1943 as the first anti-tuberculosis antibiotic followed by its general availability in the late 1940s, together with the arrival of PAS (p-aminosalicyclic acid) as a second anti-TB agent in 1953, may have influenced the senior managers of ICI that the future of anti-tuberculosis treatment would lie with antibiotics and not with problematic, and still to be synthesized, possible antagonists of a still largely uncharacterized mycobactin. But for whatever reason, Alan Snow then was the person who almost single-handedly elucidated the structures not only of the mycobactin from M. phlei but many other ones as well (Fig. 2.4).

General structures of the ferric-mycobactins [26, 64, 67]. The substituents at R1 are usually alkenyl chains with a cis-double bond at C2 (exceptions are for mycobactins M and N, both from M. marinum). There are usually a number of chain lengths, only the major ones are given; † indicates two distinct mycobactins from the same strain, ‡ mycobactins are considered as being equivalent
The initial description of a possible structure for mycobactin form M. phlei was given in the December 1954 issue of the Journal of the Chemical Society [33]. However, there was a problem with working out how one of the hydrolytic products of mycobactin, 2-amino-6-hydroxyaminohexanoic acid, was orientated in the molecule. Two possible structures for it were offered. It was though another 11 years before this issue was resolved. No full-length papers were published by Snow or any other person on the mycobactins from 1954 to 1965 though there was a short preliminary communication made in 1961 to a meeting of the Biochemical Society in the UK [34] concerning the isolation of the mycobactin from M. tuberculosis. But it seems likely that the mycobactin project was now de-prioritized roundabout this time and, in a letter written to Philip D’Arcy Hart at the Medical Research Council Laboratories in London and dated July 29th 1968, Snow himself said that “We have revived some interest in this topic after a lapse of a number of years”. This would suggest that the project had been completely abandoned in the late 1950s and early 1960s. Sometime then in the 1960s, interest in mycobactins must have then re-started but, in all probability, only Alan Snow, with possibly just one or two technical assistants, would have been engaged on the project for most of the remainder of the program.
The structure of mycobactin from M. phlei was published in January 1965 [35] and given the name mycobactin P. This was to distinguish it from mycobactin T which Snow [34] had briefly described earlier. Snow commented in letter dated August 12th 1955, to Philip D’Arcy Hart with whom he had considerable correspondence, that the team
“… have done quite a number of experiments on Myco. tuberculosis. However, we have met considerable difficulties in this research. In the first place, we have grown our tubercle bacillus on a medium similar to that used for the growth of phlei with beef infusion present in the hope of stimulating production of growth factor (i.e. mycobactin). This medium, however, was not a suitable one for growth of large quantities of the tubercle bacillus, and it took us many months to accumulate even a small quantity of the dried organism as starting material.”
Snow went on to say that the methods used to extract the mycobactin from M. phlei “… were quite unsuitable for extraction of the growth factor from tubercle. We have also been severely hampered in our facilities for testing the activity of the concentrates.”
The initial results of the work being done in the 1950s had also produced some problems. Snow commented in the same letter to Philip Hart that the growth factor for M. johnei that had been extracted from M. tuberculosis was similar but not identical to the mycobactin from M. phlei. Thus, the magnitude of this project that was facing Alan Snow cannot be over-stated.
The structure of mycobactin T itself was eventually solved and published in 1965 by Snow [36]. Hough and Rogers [37] were subsequently able to confirm in detail Snow’s structure and stereochemistry of mycobactin P by using X-ray crystallography. The ferric ion was found to lie in a V-shaped cleft with a very strained octahedral configuration involving five oxygens and one nitrogen. The exceptional stability of mycobactin with ferric iron was then explained together with explaining how the iron could be easily released from mycobactin by reduction of ferric to ferrous iron [38–40] where the resultant ferrous ion had little or no affinity to mycobactin and would thus be available for incorporation in apoenzymes and other proteins.
It was only in the two major papers of 1965 by Snow that dealt with the structures of mycobactins [35, 36] was iron binding recognized as a major attribute of them. Snow [35] now appreciated that mycobactin was, in fact, a microbial siderophore—or what were then called ‘sideramines’. He commented that the isolation of the desferri-form was directly attributable to the cultivation medium being used, that was beef infusion broth, having a low content of ionized iron. In other words, and with hindsight, the iron-containing components of the medium would be various haem compounds and also ferritin and transferrin that would withhold iron from bacteria and, therefore, the cells were accidentally being grown iron deficiently. This was confirmed by Norman Morrison [41, 42], at the Johns Hopkins-Leonard Wood Memorial Laboratory, Baltimore, USA, who in a personal communication to Alan Snow, reported that large amounts of mycobactin could be produced by growing M. phlei in a synthetic medium with less than 0.2 μg iron/ml. This then indicated a much easier way to optimize the accumulation of mycobactins in mycobacteria.
Following the descriptions of the structures of the first two mycobactins, P and T, [35, 36] the structures of other mycobactins were elucidated during the remaining years of the 1960s by Snow who had now been joined by a very able technical assistant, Mr. A. J. White. Mycobactins S and H types from, respectively, M. smegmatis and M. thermoresistible [43] were described; this was followed by descriptions of the mycobactins from M. aurum, M. terrae, M. fortuitum and M. marinum that were labeled as types A, R, F and M and N, respectively [44]. The structures of all the mycobactins that were determined by Alan Snow are given in Fig. 2.4. It is probably worth repeating the observations made in this final paper that the mycobactins M and N, both from M. marinum, were inhibitory towards M. tuberculosis and these two mycobactins were distinctively different from the mycobactins from other mycobacteria in having the characteristic long alkyl chain attached to a different part of the molecule (Fig. 2.4). This observation, however, does not appear to have been taken up by any other group looking to realize the potential of designing inhibitors of M. tuberculosis when this work of Snow and colleagues began 25 years earlier. These papers were, in fact, the last significant publications arising from the work at ICI Ltd. Alan Snow, himself, wrote a review of the mycobactins which remains the definitive account of the chemistry and major properties of these iron-binding compounds [26]. It is still quoted in many research papers today. This review contains many details that are still salient today; this includes considerable information on the binding of metal ions to the molecule including, of course, iron. The tenacity of mycobactin for iron is the major feature of the molecule and Snow, on the basis of desferrimycobactin being able to remove the iron from ferric-desferrioxamine B [45], calculated that its stability constant was well in excess of 1030. The mycobactin project at ICI Ltd came to a close at the end of the 1960s with Snow himself then retiring about a decade later. The original objectives of the research, however, had not been fulfilled though considerable interest is still evident to-day in looking at aspects of iron metabolism in the mycobacteria for opportunities to design novel anti-tuberculosis agents. This topic is then re-visited by other contributors in this monograph.
Alan Snow appears to have received many requests for samples of mycobactin P once it was firmly established that this was the growth factor essential for the growth of johne’s bacillus. I have already commented on some correspondence between Alan and Philip D’Arcy Hart at the MRC Laboratories in London. Hart was not only wanting to grow M. johnei, as it was still referred to then, but also the leprosy bacillus, M. leprae, and the rat leprosy bacillus, M. lepraemurium. Also pursuing the same objectives was John Hanks at Johns Hopkins University, Baltimore, who also had received samples of mycobactin from Snow. Unfortunately, in spite of many attempts both in London and Baltimore, the leprosy bacillus in neither location showed any sign of growth in mycobactin-supplemented medium.
The correspondence between Hart and Snow began in 1955 and continued up to 1972 with Snow finally concluding (October 26th, 1972) that “(o)ur own stocks of mycobactin P are quite low now, because we have given so much away but we can still help (you) with small quantities when required (in a good cause!)”. The author of this chapter has been privileged to receive the original letters concerning the mycobactins that had been sent and received by Philip Hart. Hart published one paper dealing with the growth of M. johnei in a mycobactin-containing medium [46]. Philip died in 2006 aged 106 having remained active in research until his final 2 years.
The question of how M. paratuberculosis was able to grow in vivo and acquire iron from the host was not solved until much later although Norman Morrison, working at the Johns Hopkins University, found that M. paratuberculosis could, in fact, grow without mycobactin if the pH of the growth medium was dropped to 5.5, or even to 5.0 as was later reported by Lambrecht and Collins [47]. This, it was suggested, might then mimic the conditions of growth in vivo and might be enough to increase the solubility of free iron to the point where it could now be acquired without the need for mycobactin. A (partial) resolution of this problem came, however, by the demonstration by Barclay and Ratledge [48] that M. paratuberculosis and other mycobactin-dependent strains of Mycobacterium could synthesize the extracellular counterpart to mycobactin, that is carboxymycobactin, and this would then be the way in which iron was acquired by the cells.
2.4.2 Other Mycobactins and Related Lipid-Soluble Siderophores
Once Alan Snow had solved the structure of mycobactin and worked out the main ways in which it could be extracted and characterized, the way was open for other workers to build on this work and examine other mycobacteria and genera related to the Mycobacterium genus for the presence of other iron binding compounds.
The presence of materials similar to mycobactin was noted by Patel and Ratledge [49] in species of Nocardia. The genus of Nocardia is taxonomically closed related to the mycobacteria and both are members of the actinomyces group of bacteria and have similar cell walls with a high content of lipid. It is therefore not too surprising that similar iron-binding compounds would then be found in these species. These were then named nocobactins and the structure of that from N. asteroides, termed nocobactin NA, was then elucidated by Ratledge and Snow [50]. This is shown in Fig. 2.5 where it is labeled as type a. It resembled the structure of mycobactin M (Fig. 2.4) but had a distinctive oxazole ring instead of an oxazoline ring and with a shorter alkyl chain. This type of nocobactin was also detected in N. paraffinae, N. sylvodorifera and N uniformis [51]. Two other types of nocobactin were found: type b (which is then known as nocobactin NB) was from N. brasiliensis, and type c, or nocobactin NC, was from N. caviae and N. phenotolerans. The latter types were similar to mycobactins S and T but had saturated alkyl chains rather than the unsaturated chains of the latter materials.

Structures of nocobactins from Nocardi species [49, 51]. Type a from N. asteroides, N. sylvoderifa, N. paraffinae and N. uniformis (now all considered to be equivalent to N. asteroides) type b from N. brasiliensis and type c from N. caviae and N. phenotolerans. Types b and c are equivalent in structure to the mycobactins (see Fig. 2.4) but type a has its long alkyl chain at R4 and is somewhat equivalent to mycobactins M and N (see Fig. 2.4) but also has an unusual oxazoline ring instead of the more usual oxazole ring
Once it was appreciated that iron deficient growth of a mycobacterium would increase the content of mycobactin by up to a 100-fold, it was then a relative easy matter to develop simpler methods of producing it. In our own work with M. smegmatis, it was possible to produce cells with 10 % (w/w) of mycobactin, a truly massive over-production of a siderophore. Richard Hall, one of my very able graduate students, was also able to achieve good yields of mycobactin by growing cells on agar plates (though highly purified agar had to be used) but there was no need to de-ferrinate the culture medium beforehand [52]. Cells could then be scraped off just one or two plates and then the mycobactin extracted with ethanol in the usual way. Up to 10 mg mycobactin per plate could be attained. It was then possible to analyze the mycobactins very quickly and easily using both thin layer chromatography and the newly arrived technique of high-pressure chromatography (HPLC) [53]. The former technique, with appropriate solvents, separated the mycobactins according to differences in the R2, R3, R4 and R5 nuclear substituents (Fig. 2.4) and HPLC separated them mainly on the basis of differences in the length of the alkyl chain at R1. Bosne et al. [54] subsequently simplified this procedure by adding EDDA as an iron chelating agent into liquid culture medium to produce iron-deficient growth conditions and thus a stimulation of mycobactin production. They used this method to identify 65 strains of M. fortuitum and M. chelonae according to the type of mycobactin being produced [55].
Using their simplified method of cultivation, Hall and Ratledge [56] analyzed the mycobactins of 39 strains of mycobacteria, principally as a means to determine if the mycobactins “…may be useful as a chemotaxonomic marker in the mycobacteria”. They were indeed able to confirm the validity of the hypothesis and found that the mycobactins were strongly conserved molecules showing strong intra-species consistency and could be therefore used as chemotaxonomic characters of high discriminatory power. The structures of the new mycobactins were not determined, however. Using the same method, Leite et al. [57] were able to identify various clinical mycobacterial isolates from their mycobactins and Barclay et al. [58] were able to suggest an even more rapid method of detecting and identifying mycobacteria using 55Fe-labelling of the mycobactins. A list of species producing mycobactin is given in Table 2.2; it is probably reasonable to conclude that most species of mycobacteria will be found to produce a mycobactin but there are some exceptions.
Rich Hall went on to apply his techniques to other groups of mycobacteria, showing equivalence of the mycobactins from M. senegalense, M. farcinogenes and M. fortuitum but a distinction from that from Nocardia farcinica [59]. He also examined the mycobactins from seven strains of armadillo-derived mycobacteria (ADM) [59] that were of interest because of the association of M. leprae with the armadillo and because, but unlike M. leprae, these bacteria could be grown in laboratory medium. The ADM were found to be a heterogeneous group; four of them produced materials that resembled the mycobactins from M. avium-intracellulare-scrofulaceum (MIAS) complex of mycobacteria suggesting that they could be assigned to this taxonomic grouping.
The MIAS group of mycobacteria had earlier been studied by Barclay and Ratledge [61] for their mycobactins which were of particular interest as some freshly isolated strains of M. avium had been reported as being dependent on mycobactin for growth [62]. The presence of mycobactins in strains of M. avium had, though, been initially reported by Ratledge and McCready [63]. Barclay and Ratledge [61] found that only those strains of M. avium that could grow without mycobactin could produce it themselves but three strains, initially unable to grow unless mycobactin was added to the growth medium, were eventually able to grow without it and now produced small quantities themselves. This indicated that mycobactin biosynthesis was being strongly repressed and then slowly reversed during the subsequent adaptation rather than being indicative of a permanent genetic deletion. The structure of mycobactin Av from M. avium and other MIAS strains was subsequently determined by Barclay et al. [64] (Fig. 2.4). In addition, the same type of mycobactin was also isolated from three strains of M. paratuberculosis of that had lost their original dependency on mycobactin and could now produce it themselves. This suggested a taxonomic similarity between M. paratuberculosis and the MIAS complex of mycobacteria and this indeed has now been confirmed by more conventional taxonomic methods [65]. M. paratuberculosis, having started life as Mycobacterium enteriditis chronicae pseudotuberculosae bovis, Johne, or Johne’s bacillus for short, and then becoming M. johnei, is now re-named as M. avium subsp. paratuberculosis.
Barclay et al. [64] found that mycobactin Av differed from the other mycobactins in having two long lipophilic side-chains (Fig. 2.4), one at the usual R1 position and the second one of about 10 carbons in length at the R4 position. They also showed that the mycobactins from M. tuberculosis, M. bovis and M. africanum, were identical molecules and therefore could all be named as mycobactin T.
A mycobactin from M. paratuberculosis, but of slightly different structure to that isolated by Barclay et al. [64], had been isolated and identified 2 years earlier by Richard Merkal [66, 67]. Again this was being produced by a strain that was initially mycobactin-dependent for growth but had subsequently reverted. This was named as mycobactin J (Fig. 2.4) but it was more like the conventional mycobactins in having just a single alkyl chain at the R1 position. The absolute configuration of the structure of mycobactin J was subsequently confirmed by Schwartz and De Voss [68]. Barclay et al. [64], having kindly been sent a sample of mycobactin J and also the production organism, M. paratuberculosis strain NADC 18 (now ATCC 19698) by Richard Merkal, could only find mycobactin J as a minor component at <10 % of the total mycobactins that they extracted from the same strain as used by Merkal himself. The major mycobactin of this species, in our hands, was equivalent to the mycobactin from M. intracellulare M12, which is part of the avium complex of mycobacteria. It now appears that the original culture of M. paratuberculosis strain 18 probably had been taxonomically misnamed [69, 70] and should therefore be regarded as a strain of M. avium.
What the reasons were for mycobactin J being the dominant siderophore when NADC 18 strain was grown in Merkal’s laboratory but not in the UK remains unsolved but clearly there must be some subtle regulatory mechanism that can cause such a shift. Mycobactin J remains the only commercially available mycobactin and, because of its structural similarities to the main mycobactins, is able to stimulate the growth of both a number of isolates and mutants that are being generated with defects in their iron metabolic pathways.
Hall and Ratledge [71] also isolated mycobactin-like materials from three out of 11 species of Rhodococcus: R. bronchialis, R. terreus and R. rubropertinctus and suggested that the latter two species might be equivalent to each other. Although no structural determinations were carried out, the similarity of these new materials to the mycobactins was apparent and they should have been, but were not, named as rhodobactins. Some species of Rhodococcus, including R. rhodochrous, did not, however, produce a ‘rhodobactin’. Some 20 years later, Dhungana et al. [72] isolated a siderophore from R. rhodochrous, albeit from a different strain to that of Hall and Ratledge, with hexadecane as the sole carbon source. This siderophore was named rhodobactin but it was present in the culture medium and was not apparently in the cells. It was therefore distinct in structure to the mycobactins having two catecholate and one hydroxamate moieties for iron chelation instead of having a salicyloyl and two hydroxamate moieties and without a long alkyl chain for lipid solubility. Pedantically, the name ‘rhodobactin’ is therefore incorrect as it would imply similarity to the mycobactins and nocobactins.
2.4.3 Salicylic Acid
The story of the mycobactins and iron metabolism was then taken up by the author of this review. I carried out research work, as post-doctoral fellow, with Frank Winder in Dublin from October 1960 to June 1964 being asked to focus on the metabolic consequences of iron-deficiency in M. smegmatis being used as a model organism for the tubercle bacillus. The first significant finding [73] was the identification of salicylic acid that accumulated up to about 17 μg/ml in the medium of iron deficiently grown cells whereas in iron replete medium only about 0.6 μg salicylic acid/ml was found (see Fig. 2.6). However, a mistake was made in the latter part of this paper when the concentrations of salicylic acid were being given for M. tuberculosis and M. phlei. The ‘salicylic acid’ in the latter species had only been verified by simple paper chromatography where it ran with the same Rf value as authentic salicylic acid. Alan Snow then wrote to us to ask if the detected acid was indeed salicylate; could it not be 6-methylsalicylate? And, indeed, so it turned out to be.

Snow’s concern over the correct identification of salicylic acid arose from his realization that, although salicylate was a common moiety in most mycobactins, in some mycobactins (see Fig. 2.4) and notably in mycobactin P from M phlei, it was 6-methylsalicylate that occurred. It therefore seemed odd to Snow that we had recorded finding salicylate in the extracellular medium of this species as this would imply a metabolic puzzle. However, by using more selective solvent systems for paper chromatography, we then showed that the original conclusion had been too hasty. We had failed to double-check; M. phlei did indeed secrete 6-methylsalicylate into the culture medium and not salicylic acid.
This useful contact with Alan Snow then immediately brought our attention to the mycobactins which, at that point, were just emerging as probably having significant roles in iron metabolism. Although the biochemical connection between salicylic acid and mycobactin was clear, it was far from evident how these two molecules might interact, if at all, to achieve uptake and assimilation of iron into the mycobacteria. However, for a continuation of this work, there was jump of several years as, following my time in Dublin, I then spent the next 3 years working in industry and it was only when I returned to academia to the University of Hull, UK, in late 1967 that interest in the mycobacteria and iron metabolism was re-awakened. The role of salicylic acid in mycobacteria then became one of the major foci of our work and this is described in more detail in the following section.
2.5 Extracellular Siderophores of Mycobacteria
2.5.1 The Problem with Mycobactin
The essential puzzle with mycobactin was not whether or not it was involved with iron metabolism, as clearly it was just judging from the very large increase in its production during iron-deficient growth of any mycobacterium, but how did it actually acquire iron from the environment. Mycobactins all have a long alkyl chain (see Fig. 2.4) that makes them almost completely insoluble in water but easily soluble in organic solvents such as ethanol, methanol or chloroform. Thus, they were cell-associated materials and were not released into the culture medium. It was later shown that mycobactin forms a discrete but discontinuous layer abutting on to the cytoplasmic membrane of the cells and some distance from the outer surface of the cell (Fig. 2.7) [74]. For this work, staining of mycobactin was achieved by using vanadyl ions as they reacted faster with mycobactin than did iron and was also much more specific in what it was able to bind to. (Alan Snow pointed out to me an interesting experiment that could be done by adding a mixed solution of ferric chloride and vanadyl ions in the form of ammonium metavanadate to desferrimycobactin in ethanol. The solution would immediately turn deep blue due to formation of the vanadate complex but then, on standing overnight, the solution became red due to the formation of the ferric complex. This indicated a difference between the rate of the reaction and the stability of the product. Ferric ions although reacting slower than vanadyl ions nevertheless could replace it and form a complex with a much higher affinity which was then the final stable form of the chelate.)

a (magnification ~140,000-fold), and b (magnification ~700,000-fold). Cellular location of mycobactin. Electron micrographs of iron-deficiently grown Mycobacterium smegmatis incubated with 0.1 % ammonium metavanadate for 10 min at 4 °C (The vanadyl ions react very quickly and specifically with desferrimyobactin but are not metabolically removed from the mycobactin). The large black circular areas in the cytoplasm are polyphosphate granules which are also seen in iron-sufficiently grown cells [74]
It was suggested in this paper of Ratledge et al. [74] that the mycobactin was probably intercalated between the cytoplasmic membrane and the peptidoglycan backbone of the cell wall with, perhaps a small amount of it possibly being within the membrane itself. There did not appear to be any of it within the cytoplasm. Thus, what was the function of mycobactin and how was it able to acquire iron from the extracellular medium, or the environment in which it grew within an infected host cell, if it was a wholly intracellular and water-insoluble material? Ivan Kochan [75] working in Miami of Ohio University, suggested that mycobactin could indeed fulfill this role by demonstrating that mycobactin could remove the iron from transferrin but this did not explain how the two molecules might legitimately come into contact. Kochan et al. [76] and later Golden et al. [77] then showed that mycobactin could be solubilized and transferred into the medium if a detergent, such as Tween 80, Triton or lecithin, was added to the medium. But again, to my group (at Hull University), that now included Leo Macham as post-doctoral research assistant and who made some significant contributions in this area, this did not seem a likely condition for pathogenic mycobacteria to experience when growing in vivo. To us, the experimental conditions used by Kochan seemed contrived and unlikely to be realistic. His proposals certainly could not explain how iron was mobilized by mycobacteria growing in simple laboratory culture medium devoid of any detergent. We, therefore, were of the opinion that another iron binding component, that was water-soluble and which would be released by the cells into their surrounding environment, was necessary to explain how iron was solubilized in the medium before being transferred into the cells.
The first and obvious candidate for this was salicylic acid—why else did it occur in increased quantities in the culture medium during iron-deficient growth (Fig. 2.6)? Although 55Fe-labelled salicylate could readily donate the iron to mycobactin [78] this was only if the experiment was carefully constructed to avoid phosphate buffers. When similar experiments were done in the presence of phosphate buffer, no transfer of iron took place as the iron was quickly converted to ferric phosphate that was insoluble and not immediately accessible to the mycobactin [79]. It was confirmed much later by Chipperfield and Ratledge [2] that, indeed, salicylate could not function as chelating agent for iron at neutral pH values.
The failure of salicylate to function as an extracellular siderophore for the solubilization of iron then raised two questions: if salicylate was not the extracellular iron sequestering agent, what was? And, if salicylate did not have this role, then what function did it have, if any, other than being a precursor of mycobactin? The answer to the latter question remains unsolved though salicylate can function as a means of transferring the ferrous ion, being released from mycobactin by ferric-mycobactin reductase [38–40] across the cell membrane and into a receptor porphyrin for the synthesis of haem. This is shown later in Fig. 2.11. It was also found that the mode of action of the anti-tuberculosis agent, PAS = p-aminosalicylic acid, was, contrary to the initial indications that it was acting as an antagonist of p-aminobenzoic acid for the synthesis of folic acid, was in fact acting as an antagonist of salicylic acid for its role in iron metabolism [80, 81]. This has then been confirmed by later work [82–84].
An alternative role for salicylic acid was advanced by Morrison [42] who pointed out that salicylate may be acting as an energy-uncoupling agent as had been indicated earlier by Brodie [85]. This then might tie in with the original observation by Bernheim [86] that salicylate was readily oxidized by M. tuberculosis and therefore might be an important respiratory substrate. The view of Brodie [84, 87] would be that salicylate uncoupled oxidative phosphorylation and, therefore, with insufficient ATP for growth, cells have to increase their respiration rate. But in the iron-deficient cells that over-produce salicylate, the salicylate may be a means of rapidly down-regulating energy production by directly uncoupling oxidative phosphorylation in order to conserve key metabolic processes and not to generate ATP needlessly. This is certainly an intriguing suggestion from Morrison that has never been followed up.
The search for the iron-solubilizing agent that would be present in the culture medium of mycobacteria then began. Clearly the answer to this question was of considerable importance as it would help to explain how pathogenic mycobacteria were able to acquire iron from host tissues and iron-containing cell components. Macham and Ratledge [88] carried out experiments with M. smegmatis and M. bovis BCG both being grown iron deficiently. They showed that there was some material in the cell-free culture filtrates of both organisms that could hold 55Fe in solution at pH 7 in the presence of phosphate ions and could be dialyzed; in other words, it had a molecular size of <10000 Da and was therefore not some form of colloidal iron. These materials were named exochelins. But it quickly became evident that the material from M. smegmatis was quite different from that of M. bovis. The former was completely water-soluble and could not be extracted into any organic solvent, including ethanol, whereas the other exochelin could, when in the ferric-form, be extracted into chloroform [88, 89]. It was also found that the two exochelins had different mechanisms for iron uptake: exochelin from M. smegmatis was taken up by an active (energy-dependent) transport system but the one from M. bovis was by facilitated diffusion and was not energy dependent [90, 91]. Furthermore, the M. smegmatis exochelin could not be taken up by M. bovis but that from M. bovis could be taken up by M. smegmatis. Thus, two distinct types of extracellular siderophores were being produced by the mycobacteria. Other water-soluble exochelins were recovered from other non-pathogens: M. neoaurum [92] and M. vaccae [93]. The latter siderophore was of interest as this species of mycobacteria did not appear to have a mycobactin.
2.5.2 The Water-Soluble Exochelins
It was nearly 20 years after the initial discoveries of the exochelins before their structures were resolved though it was established soon after their initial isolation that exochelin MS from M. smegmatis was probably a pentapeptide with three N-epsilon-hydroxyornithines providing the chelating centre. Various research groups in the UK had been approached by the author for assistance in trying to work out the structures and to see how the ornithine residues, together with a beta-alanine and an allo-threonine, were assembled. But none had been able to complete the work until the author approached the group headed by Dudley Williams in the Department of Chemistry at the University of Cambridge to help solve the structure of the water-soluble exochelins. The structure of the chloroform-soluble exochelins was solved by collaboration with a research group at Glaxo Research Laboratories.
The structure of the exochelin from M. smegmatis, then named as exochelin MS, was determined by a PhD student, Gary Sharman working under Dudley Williams at Cambridge University. Exochelin MS was an ornithinyl siderophore with three hydroxamate groups that provided the iron chelating center (Fig. 2.8a) [94]. Further, but unpublished, work of the author indicated that the exochelin from M. vaccae was probably similar if not identical to this exochelin. The structure of the exochelin from M. neoarum, called exochelin MN, was different [95] but was still based on a peptide backbone as seen with exochelin MS (Fig. 2.8b). It had an unusual 2-hydroxyhistidine residue as part of its iron chelating center.

Although the structure of exochelin MS was not elucidated until 1995, quite a lot of its properties and function had been worked out beforehand. Its uptake, see above, was by an active transport process requiring the input of energy (i.e., ATP was involved at some point of the mechanism) [90]. It was produced in a growth-related manner and could readily solubilize iron from not only inorganic forms of insoluble iron, such as ferric hydroxide and ferric phosphate, but also from ferritin the storage form of iron found in all animals [88, 89]. The involvement of mycobactin in the uptake of iron into M. smegmatis was not immediately apparent when small concentrations of ferric-exochelin were used; however, when higher concentrations were used, a second uptake process became evident. This was a slower process and was not inhibited by energy poisons and other agents; it was deduced to be by a process involving transfer to mycobactin itself [90] and could be explained by the mycobactin preventing a sudden over-load of iron into the cells. Cells that were iron-deficient had very low contents of porphyrins (Table 2.1) and thus the key precursors of haem synthesis were not instantly available to utilize the iron. But even though iron could not be immediately used and incorporated into cell components, a mechanism of iron storage was necessary that would then serve as a ‘pantry’ of iron. This then began to shape the view that mycobactin was an intracellular store of iron and that it acquired iron only when there was a sudden availability of it to the cells (see Fig. 2.11). Of interest was the finding that an exochelin may be involved in iron uptake into the leprosy bacillus, M. leprae. Somewhat fortuitously, Hall et al. [96] had isolated the exochelin from M. neoarum simply because this species was taxonomically related to M. vaccae [97] and which, in turn, had been suggested might be related to M. leprae [98]. Iron metabolism in M. neoarum therefore might be worth investigating. Richard Hall showed that, by using 55Fe-labelled exochelin MN, the iron was taken up by cells of M. leprae isolated from armadillo livers but the process was not one of active transport (as was with its uptake into M. neoarum) and appeared to be by facilitated diffusion [96]. The process though was specific in that there was no transfer of iron when chelated to exochelin MS. However, another exochelin, this time isolated from an armadillo-derived Mycobacterium (ADM) and which could be grown in the laboratory, also could donate iron to M. leprae [92]. Examination of the two exochelins—from M. neoaurum and Mycobacterium ADM8563—showed that they had similar properties and were possibly identical. The presence of a chloroform-soluble carboxymycobactin was not investigated in this work but it would not have been present in the preparations used because these had been purified by ion exchange chromatography which would have excluded the carboxymycobactins. The rate of uptake of iron into M. leprae was, as might have been expected, very slow but, as exochelin-mediated iron uptake did not occur in ADM cells that had been grown iron sufficiently, it was concluded that this result might indicate that M. leprae had been growing iron-deficiently in its host animal (the armadillo) in order for iron uptake to have taken place at all. It was also suggested that one of these exochelins might be useful additions to any growth medium that might be being developed for the possible growth of M. leprae in the laboratory. This however was never additionally studied and the cultivation of M. leprae in culture medium still remains a distant prospect.
2.5.3 The Carboxymycobactins
The name ‘carboxymycobactin’ was not given to the extracellular siderophores that had been isolated from pathogenic species of mycobacteria, including M. tuberculosis, M. bovis and M. avium and related species, before their structures had been established which was not until 1995. Up to that date, they were referred to as the chloroform-soluble exochelins. Perhaps, with hindsight an alternative name to ‘exochelin’ might have been used to avoid confusion with the obviously different water-soluble exochelins that were described in the previous section. But we (as all the work had been done in the author’s laboratory) simply had no idea what might be their structures. To call the material ‘exomycobactin’, as was belatedly suggested by another group, presupposed we knew it was related to mycobactin itself. For all we knew, it could have been based on a completely different type of structure. What made matters worse, was that initial analysis of the siderophores from the pathogenic species showed that they were composed of a variety of materials. Barclay and Ratledge [48, 99] analyzed the exochelins from species of mycobacteria belonging to the tuberculosis group, including both H37Rv (virulent) and H37Ra (avirulent) strains of M. tuberculosis, and to the avium groups using both high performance thin layer chromatography (HPTLC) and high performance liquid chromatography (HPLC). Multiple spots or peaks were revealed; in some cases upwards of 15 individual compounds could be seen. This was completely puzzling and suggested that, as there was no large single entity, determining the structures might be a long and daunting task if each component had to be isolated and purified. If only we had used a less discriminating technique, such as ordinary thin layer chromatography, we might have seen just a single spot on the chromatograms that would then have encouraged us to have the material examined without delay. But a multiple of spots on HPTLC and peaks with HPLC suggested there might be a multiple of structures.
It was, however, quite clear to us from very early in our experiments with the chloroform-soluble exochelins that they were of significance in the uptake of iron into pathogenic mycobacteria. Perhaps the most important work on their role came from the work of Raymond Barclay who was a post-doctoral research assistant working with me. Raymond showed that the bacteriostatic effects of serum (containing the iron-withholding protein of transferrin) towards the growth of M. avium and M. paratuberculosis in laboratory medium could be reversed not only by mycobactin but also by the exochelins that these bacteria were producing [48]. This was strong evidence in favor of these siderophores being of major importance in the development of mycobacterial infections in animals. Our much earlier work following the initial discovery of the exochelins had already established their ability to extract the iron from animal ferritin to support growth of M. bovis var. BCG [100]. Finding out what was the structure or structures of these siderophores then became major of pre-occupation in our work. But would be nearly another 10 years before the problem was finally solved.
The breakthrough in determining the structure of the chloroform-soluble exochelins came in collaboration between researchers at Glaxo Research Laboratories at Stevenage near London. As soon as the first high resolution NMR spectrum of the purified exochelin from M. avium was obtained, Steve Lane and his colleagues were immediately able to suggest a strong similarity in structure to mycobactin but with a variation: the long alkyl chain of mycobactin was now shorter and, instead of terminating in a methyl group, now terminated in a carboxy group (Fig. 2.9). As there was now competition to publish the structure of these extracellular siderophores, an instant decision was made to call the material ‘carboxymycobactin’ as this seemed to be an accurate descriptor of the molecule. The structure was then published by Lane et al. [101] with the paper being submitted on February 15th 1995. In this paper, mention was made that a preliminary examination of the related materials from M. tuberculosis and M. bovis had revealed that these were also carboxymycobactins of similar structures. A group led by Marcus Horwitz in California, USA, simultaneously published (their paper was submitted on February 21st, a week after the paper of Lane et al. [101]) the structure of the related material that they had isolated from M. tuberculosis [102, 103]. In this case, however, the terminating group of the alkyl chain was not a carboxy group but was the methyl ester of this group. Possibly the esterification reaction may have occurred during the late growth phase of the cultures that had been used whereas for the work of Lane et al. [101, 104], culture filtrates had been prepared from cells in their active phase of growth during which iron solubilization and uptake should be at their maximum.

Structure of the carboxymycobactins (originally termed the chloroform-soluble exochelins) from Mycobacterium avium, M. bovis BCG and M. tuberculosis [101] where n = 2–9. Related siderophores have been reported in M. smegmatis [53, 104]. Related molecules but with a terminal methyl ester group on the acyl chain from M. tuberculosis and M. avium were found [102, 103]
It is clear that all the siderophores obtained earlier from culture filtrates of pathogenic mycobacteria and had been termed ‘chloroform-soluble exochelins’ were, in fact, carboxymycobactins. A list of species and strains that produce carboxymycobactins is given in Table 2.3. Of considerable importance and interest was the finding that all (13 out of 13) strains of M. paratuberculosis that had been examined by Barclay and Ratledge [99] produced a carboxymycobactin but all of them still required mycobactin for growth. However, it was ruled out that the added mycobactin was being converted into the extracellular product as the amount of carboxymycobactin recovered was much greater than the amount of mycobactin that had been added as a growth factor into the culture medium. Also there was no observable conversion of 14C-labelled carboxymycobactin into mycobactin itself when fed to cultures of M. bovis [100].
This discovery then provides a partial explanation as to how M. paratuberculosis and other mycobactin-dependent species are able to grow in vivo. They simply produce carboxymycobactin, an extracellular siderophore that then acquires the iron from host tissues and transfers the iron to the bacterial cells. There must then be a slow transfer of iron into the pathogenic bacteria without the participation of mycobactin, though how this occurs is still not clear. Evidently, however, when the bacteria are isolated from an infected animal, mycobactin becomes essential for the uptake process to be complete and for the bacteria to grow in vitro. Homuth et al. [105] took an alternative view: they suggested, on the basis of their finding of an extracellular ferric reductase in cultures of M. paratuberculosis that could reduce the iron in transferrin and lactoferrin, that this may be an alternative means of iron acquisition in this bacterium in vivo. The possibility of the direct uptake of iron from transferrin had been suggested earlier by Lambrecht and Collins [106]. But, if this were the case, one would then have expected extracts of animal tissues to have supported growth of this bacterium in laboratory culture medium but clearly this does not happen [22–24]. Further work on this aspect of iron metabolism in M. paratuberculosis and other mycobactin-dependent strains is therefore still needed to give some vital clues as to how it is accomplished.
The only species of Mycobacterium that failed to produce detectable amounts of carboxymycobactin was M. microti which also appeared not to produce a mycobactin [99]. This species therefore is an oddity amongst the mycobacteria in producing neither mycobactin nor a carboxymycobactin and clearly would warrant further investigation as to what is its mechanism of iron acquisition. It is, however, a genuine mycobacterium in that it is the causative organism of tuberculosis in voles and other rodents but has also been associated with some isolated cases of TB in humans [107]. Some investigation into iron metabolism in this species may therefore be warranted.
What came as a complete surprise during this phase of our work was the finding of a carboxymycobactin in culture filtrates of M. smegmatis grown iron deficiently [108]. The presence of exochelin MS in this species had appeared to be sufficient to account for iron uptake in the saprophyte so the presence of a second, and alternative, method of iron acquisition was unexpected. However, the amounts of carboxymycobactin (10–25 μg/ml) that were produced were, at most, only 10 % of the total siderophores. Also it was produced later in the growth of the cells indicating that the exochelin-dependent route of iron assimilation was probably the major one. It was also noted that the amount of carboxymycobactin was some 20 times higher when glycerol was used as a carbon source instead of glucose. The structure of this carboxymycobactin was subsequently determined [104] and was found to resemble the structure of mycobactin S with a family of short carboxylic acids attached to the mycobactin nucleus. Some eight variants of the molecule were identified [6, 104] and this could then partly explain why Barclay and Ratledge [61, 99] found such a plethora of iron-binding molecules when they first examined the carboxymycobactins from pathogenic mycobacteria by high resolution chromatographic techniques.
2.6 Putting it All Together
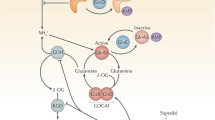
How the various components of iron metabolism come together to present a coherent picture of iron uptake and transport in mycobacteria still has a long way to go. But we are getting there. It is not, however, the remit of this chapter to go into the details of how the various components enumerated above dovetail together but, for the sake of completeness, I have included the very first model that was proposed in 1975 for iron transport in the mycobacteria (Fig. 2.10) that does not distinguish between the water-soluble exochelins of the saprophytic strains and the chloroform-soluble ones of the pathogens [100]. A more detailed model proposed in 1999 is given in Fig. 2.11. In this model, besides the major routes of iron uptake via the exochelins and carboxymycobactins, the uptake of iron chelated to citrate was worked out by Ann Messenger [93] and assimilation of iron by direct binding of a mycobacterial cell to transferrin or lactoferrin was proposed by Lambrecht and Collins [106] probably involving an extracellular ferric reductase as identified by Homuth et al. [105]. This enzyme could also work with ferritin and ferric ammonium citrate. Iron storage within the cytoplasm of the cell is by bacterioferritin [109–111] but only when the iron is in excess of immediate metabolic requirements. Bacterioferritin receives the iron being released by ferrimycobactin after passage through the cell membrane [112]. Iron would then be released from bacterioferritin on demand from the cell probably by an internal ferric-reductase.

An early model proposed to explain iron uptake and transport in the mycobacteria [98]

Possible mechanisms for iron uptake in mycobacteria as further elucidated during the 1980s and 1990s [113]. FeRex an extracellular ferric reductase, FeR ferric-mycobactin reductase, My mycobactin, Rep receptor protein for exochelin, FxuA, FxuB etc. ferric-exochelin uptake proteins, ExiT exochelin transport protein, FxbA, FxbB, etc. ferric-exochelin biosynthesis proteins, Bfr bacterioferritin, ? unknown mechanisms but the uptake of ferric-carboxymycobactin is now known to involve an ABC transporter
It will then be of doubtless interest to the reader to compare this model given in Fig. 2.11 to the ones then described in detail in the other chapters of this monograph. When all is known and understood, these early beginnings of iron acquisition may, hopefully, still prove to have been modeled along the right lines but there is still much to be elucidated in this fascinating aspect of mycobacterial metabolism.
References
Boukhalfa H, Crumbliss AL (2002) Chemical aspects of siderophore mediated iron transport. Biometals 15:325–339
Chipperfield JR, Ratledge C (2000) Salicylate is not a bacterial siderophore: a theoretical study. Biometals 13:165–168
Sauton A (1912) Sur la nutrition minerale du bacilli tuberculeux. CR Acad Sci (Paris) 155:860–861
Edson NL, Hunter GJE (1943) Respiration and nutritional requirements of certain members of the genus Mycobacterium. Biochem J 37:563–571
Goth A (1945) The antitubercular activity of aspergillic acid and its probable mode of action. J Lab Clin Med 30:899–905
Turian G (1951) Action plasmogene du fer chez les Mycobacteries. Le bacilli de la fleole, indicateur du fer. Helv Chim Acta 34:917–920
Winder F, Denneny J (1959) Effect of iron and zinc on nucleic acid and protein synthesis in Mycobacterium smegmatis. Nature 184:742–743
Winder FG, O’Hara C (1962) Effect of iron deficiency and of zinc deficiency on the composition of Mycobacterium smegmatis. Biochem J 82:98–102
Winder FG, O’Hara C (1964) Effects of iron deficiency and of zinc deficiency on the activities of some enzymes in Mycobacterium smegmatis. Biochem J 90:122–126
Donald C, Passey BI, Swaby RJ (1952) A comparison of methods for removing trace metals from microbiological media. J Gen Microbiol 7:211–220
Winder FG, O’Hara C (1966) Levels of iron and zinc in Mycobacterium smegmatis grown under conditions of trace metal limitation. Biochem J 100:38P
Winder FG, Coughlan MP (1969) A nucleoside triphosphate-dependent deoxyribonucleic acid-breakdown system in Mycobacterium smegmatis and the effect of iron limitation on the activity of this system. Biochem J 111:679–687
Winder FG, Coughlan MP (1971) Comparison of the effects of carbon, nitrogen and iron limitation on the growth and on the RNA and DNA content of Mycobacterium smegmatis. Irish J Med Sci 140:16–25
Winder FG, McNulty MS (1970) Increased DNA polymerase activity accompanying decreased DNA content in iron-deficient Mycobacterium smegmatis. Biochim Biophys Acta 209:578–580
Winder FG, Lavin MF (1971) Partial purification and properties of a nucleoside triphosphate-dependent deoxyribonuclease from Mycobacterium smegmatis. Biochim Biophys Acta 247:542–561
Winder FG, Sastry PA (1971) The formation of a long-lived complex between an ATP-dependent deoxyribonuclease and DNA. FEBS Lett 17:27–30
Winder FG, Barber DS (1973) Effects of hydroxyurea, nalidixic acid and zinc limitation on DNA polymerase and ATP-dependent deoxyribonuclease activities of Mycobacterium smegmatis. J Gen Microbiol 76:189–196
Ehrenberg A, Reichard P (1972) Electron spin resonance of the iron-containing protein B2 from ribonucleotide reductase. J Biol Chem 247:3485–3488
MacNaughton AW, Winder FG (1977) Increased DNA polymerase and ATP-dependent deoxyribonuclease activities following DNA damage in Mycobacterium smegmatis. Mol Gen Genet 150:301–308
McCready KA, Ratledge C (1978) Amounts of iron, haem and related compounds in Mycobacterium smegmatis grown in various concentrations of iron. Biochem Soc Trans 6:421–423
McCready KA (1980) Studies on iron metabolism in Mycobacterium smegmatis and other mycobacteria. PhD thesis, University of Hull, UK
Twort FW, Ingram GLY (1912) A method for isolating and cultivating the Mycobacterium enteritidis chronicae pseudotuberculosae bovis, Johne, and some experiments on the preparation of a diagnostic vaccine for pseudo-tuberculosis enteritis of bovines. Proc R Soc Lond B Biol Sci 84:517–530
Twort FW, Ingram GLY (1913) A monograph on Johne’s disease (enteritis chronicae pseudotuberculosa bovis). Baliere, Tindall and Cox
Twort and Ingram (1914) Further experiments on the biology of Johne’s bacillus. Zentr Bakteriol Parasitenk Abt 1 Org 73:277–283
Johne HA, Frothingham L (1895) Ein eigentuemlicher Fall von Tuberculose beim Rind. Dtsch Ztschr Tiermed Pathol 21:438–454
Snow GA (1970) Mycobactins: iron-chelating growth factors from mycobacteria. Bacteriol Rev 34:99–125
Francis J, Madinaveitia J, Macturk HM Snow GA (1949) Isolation from acid-fast bacteria of a growth-factor for Mycobacterium johnei and of a precursor of phthiocol. Nature 163:365–366
Francis J, Macturk HM, Madinaveitia J Snow GA (1953) Mycobactin, a growth factor for Mycobacterium johnei. I isolation from Mycobacterium phlei. Biochem J 55:596–607
Wheater DWF, Snow GA (1966) Assay of the mycobactins by measurement of the growth of Mycobacterium johnei. Biochem J 100:47–49
Antoine AD, Morrison NE, Hanks JH (1964) Specificity of improved methods for mycobactin bioassay by Arthrobacter terregens. J Bacteriol 88:1672–1677
Reich CV, Hanks JH (1964) Use of Arthrobacter terregens for bioassay of mycobactin. J Bacteriol 87:1317–1320
Snow GA (1954a) Mycobactin. A growth factor for Mycobacterium johnei. II Degradation and identification of fragments. J Chem Soc 2588–2596
Snow GA (1954b) Mycobactin. A growth factor for Mycobacterium johnei. III Degradation and tentative structure. J Chem Soc 4080–4093
Snow GA (1961) An iron-containing growth factor from Mycobacterium tuberculosis. Biochem J 81:hn 4P
Snow GA (1965) The structure of mycobactin P, a growth factor for Mycobacterium johnei, and the significance of its iron complex. Biochem J 94:160–165
Snow GA (1965) Isolation and structure of mycobactin T, a growth factor from Mycobacterium tuberculosis. Biochem J 94:166–175
Hough E, Rogers D (1784) The crystal structure of ferrimycobactin P, a growth factor for the mycobacteria. Biochem Biophys Res Commun 57:73–77
Brown KA, Ratledge C (1974) Iron transport in Mycobacterium smegmatis: ferrimycobactin reductase [NAD(P)H:ferrimycobactin oxido-reductase], the enzyme releasing iron from its carrier. FEBS Lett 53:262–266
McCready KA, Ratledge C (1979) Ferrimycobactin reductase activity from Mycobacterium smegmatis. J Gen Microbiol 113:67–72
Ratledge C (1971) Transport of iron by mycobactin in Mycobacterium smegmatis. Biochem Biophys Res Commun 45:856–862
Morrison NE (1965) Circumvention of the mycobactin requirement of Mycobacterium paratuberculosis. J Bacteriol 89:762–767
Morrison NE (1995) Mycobacterium leprae: iron nutrition: bacterioferritin, mycobactin, exochelin and intracellular growth. Int J Lepr 63:86–91
White AJ, Snow GA (1969) Isolation of mycobactins from various mycobacteria. The properties of mycobactins S and H. Biochem J 111:785–792
Snow GA, White AJ (1969) Chemical and biological properties of mycobactins isolated from various mycobacteria. Biochem J 115:1031–1045
Snow GA (1969) Metal complexes of mycobactin P and of desferrisideramines. Biochem J 115:119–205
D’Arcy Hart P (1958) A mycobactin-containing liquid medium for the study of Mycobacterium johnei. J Pathol Bacteriol 76:205–210
Lambrecht RS, Collins MT (1992) Mycobacterium paratuberculosis factors that influence mycobactin dependence. Diagn Microbiol Infect Dis 15:239–246
Barclay R, Ratledge C (1986) Participation of iron on the growth inhibition of pathogenic strains of Mycobacterium avium and M. paratuberculosis in serum. Zentralbl Bakteriol Mikrobiol Hyg (Ser A) 262:189–194
Patel PV, Ratledge C (1973) Isolation of lipid-soluble compounds that bind ferric ions from Nocardia species. Biochem Soc Trans 1:886–888
Ratledge C, Snow GA (1974) Isolation and structure of nocobactin NA, a lipid-soluble iron-binding compound from Nocardia asteroides. Biochem J 139:407–413
Ratledge C, Patel PV (1976) Lipid-soluble, iron-binding compounds in Nocardia and related organisms. In: Goodfellow M, Brownell GH, Serrano JA (eds) The biology of the Nocardiae. Academic Press, London
Hall RM, Ratledge C (1982) A simple method for the production of mycobactin, the lipid-soluble siderophore from mycobacteria. FEMS Microbiol Lett 15:133–136
Ratledge C, Ewing DE (1978) The separation of the mycobactins from Mycobacterium smegmatis by using high-pressure liquid chromatography. Biochem J 175:853–857
Bosne S, Papa F, Clavel-Seres S, Rastogi N (1993) A simple and reliable EDDA method for mycobactin production in mycobacteria: optimal conditions and use in mycobacterial speciation. Curr Microbiol 26:353–358
Bosne S, Levy-Frebault VV (1992) Mycobactin analysis as an aid for the identification of Mycobacterium fortuitum and Mycobacterium chelonae subspecies. J Clin Microbiol 30:1225–1231
Hall RM, Ratledge C (1984) Mycobactins as chemotaxonomic characters for some rapidly growing mycobacteria. J Gen Microbiol 130:1883–1892
Leite CQF, Barreto AMW, Leite SRA (1995) Thin-layer chromatography of mycobactins and mycolic acids for the identification of clinical mycobacteria. Rev Microbiol 26:192–196
Barclay R, Furst V, Smith I (1992) A simple and rapid method for the detection and identification of mycobacteria using mycobactin. J Med Microbiol 37:286–290
Hall RM, Ratledge C (1985) Equivalance of mycobactins from Mycobacterium senegalense, Mycobacterium farcinogenes and Mycobacterium fortuitum. J Gen Microbiol 131:1691–1996
Hall RM, Ratledge C (1985) Mycobactins in the classification and identification of armadillo-derived mycobacteria. FEMS Microbiol Lett 28:243–247
Barclay RM, Ratledge C (1983) Iron-binding compounds of Mycobacterium avium, M. intracellulare, M. scrofulaceum, and mycobactin-dependent M. paratuberculosis and M. avium. J Bacteriol 153:1138–1146
Matthews PRJ, McDiarmid A, Collins P, Brown A (1977) The dependence of some strains of Mycobacterium avium on mycobactin for initial and subsequent growth. J Med Microbiol 11:53–57
Ratledge C, McCready KA (1977) Mycobactins from Mycobacterium avium. Int J Syst Bacteriol 27:288–289
Barclay R, Ewing DF, Ratledge C (1985) Isolation, identification, and structural analysis of the mycobactins of Mycobacterium avium, Mycobacterium intracellulare, Mycobacterium scrofulaceum and Mycobacterium paratuberculosis. J Bacteriol 164:896–903
Thorel MF, Krichevsky M, Levy-Frebault VV (1990) Numerical taxonomy of mycobactin-dependent mycobacteria, emended description of Mycobacterium avium, and description of Mycobacterium avium subsp. paratuberculosis subsp. nov., and Mycobacterium avium subsp. silvaticum subsp. nov. Int J Syst Bacteriol 40:254–260
Merkal RS, McCullough WG (1982) A new mycobactin, mycobactin J from Mycobacterium paratuberculosis. Curr Microbiol 7:333–335
McCullough WG, Merkal RS (1982) Structure of mycobactin. J Curr Microbiol 7:337–341
Schwartz BD, De Voss JJ (2001) Structure and absolute configuration of mycobactin. J Tetrahedron Lett 42:3653–3655
Collins DM, De Lisle GW (1986) Restriction endonuclease analysis of various strains of Mycobacterium paratuberculosis isolated from cattle. Am J Vet Res 10:2226–2228
Whipple DL, Le Febvre RB, Andrews RE et al (1987) Isolation and analysis of restriction endonuclease digestive patterns of chromosomal DNA from Mycobacterium paratuberculosis and other Mycobacterium species. J Clin Microbiol 25:1511–1515
Hall RM, Ratledge C (1986) Distribution and application of mycobactins for the characterization of species within the genus Rhodococcus. J Gen Microbiol 132:853–856
Dhungana S, Michalczyk R, Boukhalfa H et al (2007) Purification and characterization of rhodobactin: a mixed ligand siderophore from Rhodococcus rhodochrous strain OFS. Biometals 20:853–867
Ratledge C, Winder FG (1962) The accumulation of salicylic acid by mycobacteria during growth on an iron-deficient medium. Biochem J 84:501–506
Ratledge C, Patel PV, Mundy J (1982) Iron transport in Mycobacterium smegmatis: the locaction of mycobactin by electron microscopy. J Gen Microbiol 128:1559–1565
Kochan I, Pellis NR, Golden CA (1971) Mechanism of tuberculostasis in mammalian serum. Infect Immun 3:553–558
Kochan I, Cahall DL, Golden CA (1971) Employment of tuberculostasis in serum-agar medium for the study of production and activity of mycobactin. Infect Immun 4:130–137
Golden CA, Kochan I, Spriggs DR (1974) Role of mycobactin in the growth and virulence of tubercle bacilli. Infect Immun 9:34–40
Ratledge C, Marshall BJ (1972) Iron transport in Mycobacterium smegmatis: the role of mycobactin. Biochim Biophys Acta 279:58–74
Ratledge C, Macham LP, Brown KA et al (1974) Iron transport in Mycobacterium smegmatis: a restricted role for salicylic acid in the extracellular environment. Biochim Biophys Acta 372:39–51
Brown KA, Ratledge C (1972) Inhibition of mycobactin formation in Mycobacterium smegmatis by p-aminosalicylate: a new proposal for the mode of action of p-aminosalicylate. Am Rev Respir Dis 106:774–776
Brown KA, Ratledge C (1975) The effect of p-aminosalicylic acid on iron transport and assimilation in mycobacteria. Biochim Biophys Acta 385:207–220
Adilakshmi T, Ayling PD, Ratledge C (2000) Mutational analysis of a role for salicylic acid in iron metabolism of Mycobacterium smegmatis. J Bacteriol 182:264–271
Nagachar N, Ratledge C (2010) Roles of trpE2, entC and entD in salicylic acid biosynthesis in Mycobacterium smegmatis. FEMS Microbiol Lett 308:159–165
Nagachar N, Ratledge C (2010) Knocking out salicylate biosynthesis genes in Mycobacterium smegmatis induces hypersensitivity to p-aminosalicylate (PAS). FEMS Microbiol Lett 311:193–199
Brodie AF (1967) Microbial phosphorylating preparations: Mycobacterium. Methods Enzymol 10:157–169
Bernheim F (1951) Metabolism of aromatic compounds by mycobacteria. Adv Tuberc Res 5:5–39
Brodie AF, Gray CT (1957) Bacterial particles in oxidative phosphorylation. Science 125:534–537
Macham LP, Ratledge C (1975) A new group of water-soluble iron-binding compounds from mycobacteria: the exochelins. J Gen Microbiol 89:379–382
Macham LP, Stephenson MC, Ratledge C (1977) Iron transport in Mycobacterium smegmatis: the isolation, purification and function of exochelin MS. J Gen Microbiol 101:41–49
Stephenson MC, Ratledge C (1979) Iron transport in Mycobacterium smegmatis: uptake of iron from ferriexochelin. J Gen Microbiol 110:193–202
Stephenson MC, Ratledge C (1980) Specificity of exochelins for iron transport in three species of mycobacteria. J Gen Microbiol 116:521–523
Hall RM, Ratledge C (1987) Exochelin-mediated iron acquisition by the leprosy bacillus, Mycobacterium leprae. J Gen Microbiol 133:193–199
Messenger AJM, Hall RM, Ratledge C (1986) Iron uptake processes in Mycobacterium vaccae R877R, a mycobacterium lacking mycobactin. J Gen Microbiol 132:845–852
Sharman GJ, Williams DH, Ewing DF et al (1995) Isolation, purification and structure of exochelin MS, the extracellular siderophore from Mycobacterium smegmatis. Biochem J 305:187–196
Sharman GJ, Williams DH, Ewing DF et al (1995) Determination of the structure of exochelin MN, the extracellular siderophore from Mycobacterium neoaurum. Chem Biol 2:553–561
Hall RM, Wheeler PR, Ratledge C (1983) Exochelin-mediated iron uptake into Mycobacterium leprae. Int J Lepr 51:490–494
Goodfellow M, Wayne LG (1982) Taxonomy and nomenclature. In: Ratledge C, Stanford J (eds) The biology of the mycobacteria, vol 1. Academic Press, London
Stanford JL, Rook GAW (1976) Taxonomic studies on the leprosy bacillus. Int J Lepr 44:216–221
Barclay R, Ratledge C (1988) Mycobactins and exochelins of Mycobacterium tuberculosis, M. bovis, M. africanum and other related species. J Gen Microbiol 134:771–776
Macham LP, Ratledge C, Nocton JC (1975) Extracellular iron acquisition by mycobacteria: role of the exochelins and evidence against the participation of mycobactin. Infect Immun 12:1242–1251
Lane SJ, Marshall PS, Upton RJ et al (1995) Novel extracellular mycobactins, the carboxymycobactins from M. avium. Tetrahedron Lett 36:4129–4132
Gobin J, Moore CH, Reeve JR, Wong DK et al (1995) Iron acquisition by Mycobacterium tuberculosis: isolation and characterization of a family of iron-binding exochelins. Proc Nat Acad Sci USA 92:5189–5193
Wong DK, Gobin J, Horwitz MA, Gibson BW (1996) Characterization of exochelins from Mycobacterium avium: evidence for saturated and unsaturated and for acid and ester forms. J Bacteriol 178:6394–6398
Lane SJ, Marshall PS, Upton RJ, Ratledge C (1998) Isolation and characterization of carboxymycobactins as the second extracellular siderophores in Mycobacterium smegmatis. Biometals 11:13–20
Homuth M, Valentin-Weigand P, Rohle M et al (1998) Identification and characterization of a novel extracellular ferric reductase from Mycobacterium paratuberculosis. Infect Immun 66:710–716
Lambrecht RS, Collins MT (1993) Inability to detect mycobactin in mycobacteria-infected tissues suggests and alternative iron acquisition mechanism by mycobacteria in vivo. Microb Pathog 14:229–238
Emmanuel FX, Seagar AL, Doig C et al (2007) Human and animal infections with Mycobacterium microti, Scotland. Emerg Infect Dis 13:1924–1927
Ratledge C, Ewing M (1996) The occurrence of carboxymycobactin, the siderophore of pathogenic mycobacteria, as a second extracellular siderophore in Mycobacterium smegmatis. Microbiology 142:2207–2212
Brooks BW, Yong NM, Watson DC et al (1991) Mycobacterium paratuberculosis antigen D: characterization and evidence that it is bacterioferritin. J Clin Microbiol 29:1652–1658
Inglis NF, Stevenson K, Hosie AH et al (1994) Complete sequence of the gene encoding the bacterioferritin subunit of Mycobacterium avium subspecies silvaticum. Gene 150:205–206
Pessolani MCV, Smith DR, Rivoire B et al (1994) Purification, characterization, gene sequence, and significance of a bacterioferritin from Mycobacterium leprae. J Exp Med 180:319–327
Matzanke BF, Bohnke R, Mollmann U et al (1997) Iron uptake and intracellular metal transfer in mycobacteria mediated by xenosiderophores. Biometals 10:193–203
Ratledge C (1999) Iron metabolism. In: Ratledge C, Dale J (eds) Mycobacteria: molecular biology and virulence, Blackwell, Oxford, pp 260–286
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 The Author(s)
About this chapter
Cite this chapter
Ratledge, C. (2013). A History of Iron Metabolism in the Mycobacteria. In: Byers, B. (eds) Iron Acquisition by the Genus Mycobacterium. SpringerBriefs in Molecular Science(). Springer, Heidelberg. https://doi.org/10.1007/978-3-319-00303-0_2
Download citation
DOI: https://doi.org/10.1007/978-3-319-00303-0_2
Published:
Publisher Name: Springer, Heidelberg
Print ISBN: 978-3-319-00302-3
Online ISBN: 978-3-319-00303-0
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)