
Abstract
Cystic fibrosis is a common autosomal recessive disease caused by mutations in the CFTR gene that encodes an anion channel expressed in epithelia and other cell types. While the disease affects multiple organ systems, it is progressive pulmonary disease, characterized by airway infection and inflammation, that is life limiting. The origins of the lung disease associated with loss of CFTR function are complex and likely multifactorial. Current research is defining how loss of CFTR anion channel activity alters the volume and composition of respiratory secretions and thereby impacts host defenses. Here we review the current understanding of the defect in innate immunity that characterizes the airway disease in cystic fibrosis. Advances in cystic fibrosis basic science research and the development of new animal models of disease are shedding new light on the causes of lung disease and may lead to new, more targeted therapies.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Cystic Fibrosis
- Cystic Fibrosis Transmembrane Conductance Regulator
- Airway Epithelium
- Secretory Leukocyte Protease Inhibitor
- Submucosal Gland
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Introduction
Cystic fibrosis (CF) is an autosomal recessive disease caused by mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene (Rommens et al. 1989). CFTR encodes an anion channel regulated by nucleotides and phosphorylation. Over 1,500 disease-associated CFTR mutations have been reported. The most common mutation is a three-base deletion in exon 10 resulting in the loss of a phenylalanine residue at position 508 (ΔF508), present on ~70 % of mutant alleles (Kerem et al. 1989). In addition to primary or secondary CFTR-associated changes that lead to pulmonary disease manifestations, polymorphisms in other genetic loci may influence the CF phenotype (Garred et al. 1999; Henry et al. 2001; Salvatore et al. 2002). Several candidate modifier genes have been proposed for CF including mannose-binding protein (Garred et al. 1999), HBD-1 and HBD-2 (Salvatore et al. 2002), alpha 1-antitrypsin enhancer (Henry et al. 2001), and HLA class II (Aron et al. 1999). A recent genome-wide association study identified two new modifier loci for CF (Wright et al. 2011).
CF is a multiorgan system disease affecting the gastrointestinal tract (liver, gall bladder, small and large intestine, pancreas), sweat glands, reproductive system, sinuses, and respiratory tract. While the signs and symptoms of the disease have been recognized for centuries, it was only in the 1930s that Fanconi (Fanconi et al. 1936) and Andersen (1938) recognized and characterized the disorder as a distinct pathologic entity and noted its genetic basis. Although loss of CFTR function causes disease in many tissues and cell types, it is the effect on the respiratory tract that is most life limiting. More than 90 % of people with CF die of progressive lung disease associated with chronic bacterial infection and inflammation within the airways (Rowe et al. 2005). CF lung disease is associated with the eventual chronic colonization of the airways with large numbers of bacteria, notably Haemophilus influenzae, Staphylococcus aureus, and Pseudomonas aeruginosa (Rosenfeld et al. 2001). It is increasingly recognized that CF-associated airway infections are polymicrobial and involve biofilm formation (Bjarnsholt et al. 2009; Sibley and Surette 2011; Singh et al. 2000a). Remarkably, these infections are confined to the respiratory tract and spread to other organs is extremely rare.
In this chapter, we will focus our attention on the host defense problem within the airways. In CF, bacteria grow in regions of the lung that are normally sterile. The clinical course of CF lung disease correlates with the acquisition of bacterial infection and its progression. Whatever the cause underlying this propensity for infection, it is lung specific. These features indicate that loss of CFTR activity impairs the innate defenses of the lung. The precise link between loss of CFTR function in the airways and the host defense defect remains an area of intense study and scientific debate. Here we review our current understanding of CF lung disease.
2 Overview of CF Lung Disease
The loss of CFTR anion channel activity has a profound impact on the function of many organs, most notably those lined by epithelia and involved in the elaboration of secretions at mucosal surfaces (Welsh et al. 2001). In addition, there is growing evidence that CFTR may be important to the function of non-epithelial cell types, including alveolar macrophages (Di et al. 2006; Zhang et al. 2010), neutrophils (Painter et al. 2006, 2008, 2010), lymphocytes (Bubien 2001; Bubien et al. 1990; Mueller et al. 2011), smooth muscle cells (Robert et al. 2004, 2005; Vandebrouck et al. 2006), neurons (Rogan et al. 2010), and others. Here we will confine our focus to the impact of loss of CFTR function on the onset and progression of lung disease.
Lung disease in infants and preschool-aged children with CF can be remarkably asymptomatic at its earliest stages. A series of bronchoscopy and bronchoalveolar lavage (BAL) studies helped document that infants with CF may have significant inflammation and bacterial infection in the face of no respiratory symptoms or signs (Armstrong et al. 2005; Balough et al. 1995; Khan et al. 1995). Early laboratory evidence of lung disease includes the presence of neutrophils, proinflammatory cytokines, and culturable bacteria such as H. influenzae and S. aureus in BAL fluid (Khan et al. 1995; Muhlebach et al. 1999). High-resolution chest CT scans are among the most sensitive early radiologic measures of disease and may demonstrate inhomogeneity of aeration, subsegmental atelectasis, bronchial wall thickening, and airway obstruction in healthy-appearing young children. As the disease progresses, bronchial dilatation and bronchiectatic changes are observed in the airways. These findings indicated that the respiratory tract host defenses of children with CF are compromised early on in their ability to eradicate bacteria encountered by inhalation or microaspiration. While aggressive, early treatments have slowed the rate of progression of lung disease, CF remains a serious chronic disease. Lung transplant is currently the only option available for patients with advanced disease.
While studies of the host defense defect associated with CF usually focus on bacterial infections, there is evidence that people with CF may have problems in their ability to tolerate infections by respiratory viruses. One prospective study of infants and children with CF reported an increased morbidity associated with respiratory syncytial virus infections (Abman et al. 1988). Hiatt and coworkers found that compared to non-CF subjects, infants with CF were more likely to develop lower respiratory tract infections associated with hospitalization and reduction in lung function (Hiatt et al. 1999). In contrast, Ramsey and colleagues studied school-aged children with CF prospectively and did not identify any significant adverse effect of respiratory viral infections on pulmonary function compared with age-matched non-CF controls (Ramsey et al. 1989). In experiments using cultured primary CF and non-CF airway epithelia, Erzurum et al. noted an increase in parainfluenza type III replication in CF cells (Zheng et al. 2003). This increase in virus replication was associated with reduced nitric oxide synthase 2 (NOS2) and 2′,5′-oligoadenylate synthetase (OAS) 1 induction in response to virus or interferon gamma. The investigators linked these reductions in antiviral defenses to an impaired activation of signal transducer and activator of transcription (STAT)1. Recently, Sutanto and coworkers studied primary cells from CF and non-CF subjects and noted that human rhinovirus 1B replicated to higher levels in CF respiratory epithelia (Sutanto et al. 2011). This finding was also associated with a reduced apoptotic response and increased release of IL-8 in the CF epithelia. These studies are intriguing as some antimicrobial peptides and proteins exhibit antiviral properties (Daher et al. 1986), and signaling mediated by type I and type III interferons may also promote antibacterial defenses (Li et al. 2008). In addition, intracellular pathogen sensing mediated by NLRP1 and NLRP3 inflammasomes may influence both antiviral and antibacterial host defense responses (Cassel and Sutterwala 2010; Guarda et al. 2011; Poeck et al. 2010; Poeck and Ruland 2011; Strunk et al. 2011). Further studies are needed to determine if mutations in CFTR also cause a defect in antiviral innate immunity.
While our discussion focuses on how loss of CFTR function results in the primary manifestations of CF lung disease, it is important to understand that disease progression results in secondary complications that further compromise airway defenses. As airway bacterial infection in children with CF evolves from intermittent infection to chronic colonization, the host mounts an impressive response. Predominant findings include the release of proinflammatory cytokines by epithelia and immune effector cells that include IL-1, TNF-alpha, IL-6, 1L-17 (Decraene et al. 2010; Dubin and Kolls 2011; McAllister et al. 2005; Tan et al. 2011), and 1L-23 (Decraene et al. 2010; McAllister et al. 2005) and neutrophil chemoattractants such as IL-8, IL-17, and CCL20 (Armstrong et al. 2005; Balough et al. 1995; Khan et al. 1995; McAllister et al. 2005; Muhlebach et al. 1999) and the secretion of pathogen-specific antibodies (Doring et al. 1988). Intense neutrophilic infiltration of the airways ensues, and the associated inflammatory responses include the release of enzymes such as neutrophil elastase, myeloperoxidase, cathepsins, and others. In addition, airway pathogens can release proteases and other inflammatory stimuli that gradually lead to disease progression. These secondary inflammatory responses contribute to the destruction of airway tissue and stimulate proliferative responses in the airway epithelium (Leigh et al. 1995) with associated remodeling, such as goblet cell metaplasia (Bedrossian et al. 1976; Davis and Dickey 2008; Groneberg et al. 2002). This burden of proteases causes further secondary compromise in antimicrobial defenses by directly degrading host defense proteins such as lactoferrin (Britigan et al. 1993), defensins (Taggart et al. 2003), SLPI (Weldon et al. 2009), and elafin (Guyot et al. 2008). In addition, the protease-rich environment of the CF airways can cleave TLR-2 and TLR-4, which may further impair innate immune signaling (Greene et al. 2004). These persistent host responses to polymicrobial infection add to the difficulties in defining the host defense defects directly linked to loss of CFTR function and distinguishing them from those that arise as a consequence of the host inflammatory response.
3 Innate Immune Defenses in the Airways
The airways face a daily burden of inhaled or aspirated bacteria, viruses, and other potentially damaging particulates. To protect against these threats, the epithelium of the respiratory tract has evolved multiple mechanisms to prevent microbial infection and minimize tissue damage in response to infectious agents. At the most basic level, the epithelium protects the airways from such insults by serving as a physical barrier between the environment and the underlying tissue. However, the defensive capacity of this mucosal surface extends far beyond its simple barrier function. The conducting airways are lined by a pseudostratified columnar epithelium consisting of ciliated and non-ciliated surface cells, mucin-producing goblet cells, and a progenitor cell type termed basal cells. The conducting airway epithelium also possesses submucosal glands, which supply bulk liquid secretion, additional mucins, and other molecules with antimicrobial, anti-inflammatory, or other host defense functions to the airway surface. Together, the surface airway epithelia and submucosal glands are responsible for generating the airway surface liquid (ASL)—a mixture of secreted proteins and peptides with innate immune functions, as well as lipids, surfactants, and electrolytes important for ASL volume homeostasis. The ASL is organized into two compartments: an aqueous phase near the cell surface that bathes the cilia (the sol or periciliary layer) and a layer of hydrated mucus (the gel layer) that rests atop the sol phase (Fig. 1). Inhaled microbes and other particles are trapped in this mucus layer, which, propelled by the coordinated beating of the cilia, slides along the top of the periciliary fluid layer in a process known as mucociliary clearance. In this way, particles are swept up and out of the airways to the nasopharynx, where they are eliminated by swallowing. Similarly, cough clearance also removes particles from the airway lumen (Bartlett et al. 2008a).

The airway surface liquid (sol) and mucus (gel) layers provide an optimal environment for the function of secreted host defense factors. (A) Scanning electron microscope image of osmium and perfluorocarbon fixed cultured well-differentiated non-CF primary human airway epithelial cells. Black arrowhead indicates gel layer; white arrowhead indicates sol layer. This sentence refers to fig. 1B. (B) Note ciliated cells (c), goblet cells (g), and basal cells (b). Light microscopy image of osmium and perfluorocarbon fixed newborn non-CF pig tracheal epithelium stained with toluidine blue. Black arrowhead indicates gel layer; white arrowhead indicates sol layer. Scale bars in both images indicate 10 μm
In addition to these physical and mechanical defense mechanisms, the airway epithelium actively interfaces with the environment and constitutively or inducibly secretes an array of innate immune effector molecules into the ASL that sense and respond to microbial threats (Table 1). This “chemical shield” includes cationic peptides with broad-spectrum antimicrobial activity, such as the beta-defensins (Pazgier et al. 2006; Schutte and McCray 2002; Singh et al. 1998), CCL20 (Starner et al. 2003), and the human cathelicidin LL-37 (Bals et al. 1998b), as well as the prototypic antimicrobial protein lysozyme (Fleming 1922; Fleming and Allison 1922), which kills bacteria by degrading peptidoglycan in the bacterial cell wall. There are also innate immune molecules that combat bacteria in ways that do not involve direct killing, such as iron sequestration by lactoferrin (Masson et al. 1966; Oram and Reiter 1968) or binding of bacterial siderophores (iron-chelating molecules) by the neutrophil gelatinase-associated lipocalin (NGAL, or lipocalin-2) (Goetz et al. 2002). Many of these molecules are multifunctional. For example, the collectins surfactant protein A (SP-A) and surfactant protein D (SP-D) bind microbes and act as opsonins (Kuan et al. 1992; Tenner et al. 1989; van Iwaarden et al. 1990, 1991, 1994) and also help to modulate inflammation through their interactions with phagocytes and inflammatory-signaling molecules (Murakami et al. 2002; Sano et al. 1999; Sato et al. 2003). The abundant secreted protein PLUNC (palate, lung, nasal epithelium clone) possesses potent surfactant activity and is proposed to contribute to airway epithelial defenses through antimicrobial as well as anti-biofilm effects (Chu et al. 2007; Gakhar et al. 2010; Lukinskiene et al. 2011; McGillivary and Bakaletz 2010; Zhou et al. 2008). Airway secretions also contain proteins with anti-inflammatory functions, such as the protease inhibitors elafin and secretory leukocyte protease inhibitor (SLPI) (Butler et al. 2006; Henriksen et al. 2004; Sallenave 2010). Porter and coworkers demonstrated that airway epithelia secrete lipids that exert direct antimicrobial activity and synergize with host defense proteins (Do et al. 2008).
In addition to polypeptide- and lipid-based defenses, the secreted enzyme lactoperoxidase (LPO) contributes to an oxidative host defense system. In this system, LPO uses H2O2 produced by the dual oxidases DUOX1 and DUOX2 (members of the NOX gene family) to catalyze the oxidation of the secreted anion thiocyanate (SCN−) in the ASL. This reaction generates the antibacterial product hypothiocyanate (OSCN−), which is toxic to several relevant airway pathogens (Conner et al. 2002; Forteza et al. 2005; Gerson et al. 2000; Moskwa et al. 2007; Wijkstrom-Frei et al. 2003). In all cases, these defensive molecules may be either constitutively expressed or induced in response to various inflammatory stimuli. This redundancy in the number of host defense factors that act by multiple mechanisms is further augmented by their ability to act together in synergistic or additive manners (Singh et al. 2000b).
A third arm of lung host defense involves the resident phagocytic cells, including alveolar macrophages and neutrophils that may be recruited to sites of airway infection and inflammation. In addition to engulfing and destroying bacteria, both macrophages and neutrophils release a variety of potent antimicrobial factors to augment the epithelial responses to invading microorganisms. Macrophages and neutrophils store and secrete many of the same innate immune factors released by airway epithelia, such as alpha- and beta-defensins (Ganz et al. 1985; Garcia et al. 2001a), LL-37 (Cowland et al. 1995), lysozyme (Hiatt et al. 1952), lactoferrin (Masson et al. 1969), NGAL (Kjeldsen et al. 1993), and PLUNC (Bartlett et al. 2008b), as well as phagocyte-specific products such as the anti-inflammatory bactericidal/permeability-increasing protein (BPI) (Weiss et al. 1978). Macrophages and neutrophils actively communicate with airway epithelia by releasing and responding to cytokines and other molecules involved in amplification and/or regulation of inflammatory responses.
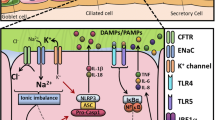
In airway epithelia, as well as in macrophages and neutrophils, many innate immune responses are mediated by Toll-like receptors (TLRs), a family of pattern recognition receptors that evolved to recognize a variety of pathogen-associated molecular patterns (PAMPs). Engagement of a TLR typically initiates a signaling cascade that results in an immune response tailored to the invading organism. TLRs respond by activating the transcription of numerous cytokine genes, including TNF, IL-6, and IL-12 as well as a variety of chemokine genes (Akira and Takeda 2004). TLRs also induce proforms of IL-1β and IL-18, which are then processed and secreted by NOD-like receptor-mediated caspase-1 activity. Additional intracellular receptors also coordinate epithelial innate immune responses to pathogens, including the NOD-like receptors, RIG-I, and MDA5 (Andrejeva et al. 2004; Yoneyama et al. 2004). Reactive oxygen species (ROS) are among the most evolutionarily conserved pathway of responses to infection or injury and are triggered by all danger-associated molecular patterns (DAMPs) and PAMPs. A ROS-dependent pathway triggers the inflammasome complex formation in myeloid cells like macrophages (Cruz et al. 2007).
4 How Does Loss of CFTR Function Alter Airway Host Defenses?
It has been challenging to explain the complex pathogenesis of CF lung disease. While it is well established that CF is caused by absent or reduced CFTR anion channel activity, it has been difficult to draw a direct line from this molecular defect to the varied CF disease manifestations. In the case of CF lung disease, the critical question is: How does the loss of anion channel activity cause defective innate immunity in the airways? This question has given rise to a number of hypotheses, which touch on multiple components of mucosal innate immunity. We stress that these hypotheses are not necessarily mutually exclusive and that it is possible, even likely, that the primary pathogenesis of CF lung disease is multifactorial.
4.1 Altered Na+ and Cl− Transport in CF
CFTR is a Cl− channel that also conducts HCO −3 , SCN−, I−, and other anions (Anderson et al. 1991a, b). For this reason, considerable effort has focused on identifying links between altered electrolyte transport by airway epithelia and the functions of the innate immune system. As epithelia use Cl− secretion and Na+ absorption to coordinately regulate the volume of secretions, it is important to understand both Cl− and Na+ transport and osmotically coupled liquid movement across the airway epithelium in CF patients. Several groups have investigated how CF may alter the volume or composition of respiratory secretions.
One hypothesis posits that CFTR activity is essential for the regulation of ASL volume and mucus hydration and that absence of CFTR causes dehydration of the ASL and reduced mucociliary clearance (Boucher 2004, 2007). This model rests on the concept that the depth of the ASL periciliary layer is tightly regulated to ensure a height of ~7 μm (Matsui et al. 1998). This ~7 μm depth, the approximate length of a cilium, is thought to be optimal for efficient ciliary beating and movement of the mucus layer. Early studies assessing the nasal and airway transepithelial voltage (Vt) in CF subjects indicated that the CF epithelium had a lower (lumen-negative) Vt and also exhibited a greater reduction in nasal Vt after application of amiloride, an inhibitor of the epithelial Na+ channel (ENaC), relative to non-CF epithelia (Knowles et al. 1981, 1983a, b). Based on this observation, it was suggested that Na+ hyperabsorption and concomitant liquid absorption reduces ASL volume in the CF airways. This depletion of ASL would then lead to dehydration of the mucus layer, resulting in thick, sticky, adherent mucus that is difficult to move, ultimately impairing mucociliary clearance in the CF airways. In support of this hypothesis, evidence for altered ASL volume homeostasis, thickened mucus, and reduced mucociliary transport has been reported using cultured airway epithelia from human CF and non-CF patients (Matsui et al. 1998). In a study of bronchial tissue biopsies from human CF and non-CF subjects, CF samples showed a trend toward reduced ASL depth, although this did not reach statistical significance (Griesenbach et al. 2011). In the same study, a comparison of the periciliary liquid height of the nasal epithelium in wild-type and CFTR-null mice revealed a significant decrease in samples from the CFTR-null animals (Griesenbach et al. 2011).
Data from recent experiments in cultured CF and non-CF epithelia provide an alternative interpretation for the increased nasal Vt observed in people with CF. Itani and coworkers studied cultured primary well-differentiated human airway epithelia and failed to find evidence for increased Na+ conductance in CF epithelia (Itani et al. 2011). Under basal or cAMP-stimulated conditions, the transepithelial conductance of CF epithelia was reduced compared with non-CF epithelia, and the reduction could be accounted for solely by the loss of the CFTR channel conductance. The addition of amiloride resulted in greater decreases in Vt and short-circuit current in CF epithelia than non-CF epithelia. However, amiloride caused similar reductions in conductance and Na+ absorption in cells from both genotypes, indicating that the effects of amiloride were due to a loss of Cl− conductance. These results suggest that loss of anion conductance is the critical defect in electrolyte transport in CF epithelia.
Inhibition of Na+ transport in the respiratory tract as a therapy has been investigated in CF subjects given the ENaC inhibitor amiloride by inhalation. A multicenter randomized double-blind placebo-controlled clinical trial investigated the efficacy of thrice-daily inhaled amiloride on pulmonary function of CF patients over a 6-month period. The study failed to demonstrate significant benefit (Pons et al. 2000).
A second hypothesis proposes that changes in ASL ionic strength might adversely affect the activity of innate immune effector proteins. In this model, loss of CFTR-dependent Cl− transport results in increased ASL NaCl concentrations due to an inability of the surface epithelium to modify ASL composition by absorption of Cl− and Na+. A predicted outcome of this scenario is a reduction in the activity of salt-sensitive antimicrobial factors. This hypothesis emphasizes the central importance of antimicrobials, particularly cationic peptides and proteins, in protecting the airways from microbes (Cole et al. 1999, 2002). It is known that the activity levels of numerous antimicrobial factors, including lysozyme, lactoferrin, alpha- and beta-defensins, and others, are diminished in solutions of increased ionic strength (Goldman et al. 1997; Porter et al. 1997; Singh et al. 1998, 2000b; Travis et al. 1999; Valore et al. 1998). In experiments using cultured well-differentiated human primary airway epithelia derived from CF and non-CF donors, Smith and colleagues found that CF epithelia exhibited a killing defect when a small bacterial inoculum was applied directly to the apical surface (Smith et al. 1996). However, when in vitro killing assays were performed using ASL that had been removed from the apical surface of the cultures using water as a diluent, there was no significant difference in killing between CF and non-CF-derived samples. This result implied that it was unlikely that CF ASL lacked a critical bactericidal factor, and suggested instead that the activity of a bactericidal factor or factor(s) was inhibited by some aspect of the ASL environment in the CF epithelia. To test the hypothesis that the NaCl concentration influences antimicrobial activity in CF ASL, the authors replaced the ASL of both CF and non-CF cultures with solutions containing either high (182 mM) or low (92 mM) Cl− concentrations and repeated the bacterial killing assays. Bacterial killing was significantly reduced in non-CF cells that received the high salt solution, while killing activity was restored in CF cells under the low salt conditions (Smith et al. 1996).
While this hypothesis remains a subject of debate, the idea that loss of CFTR function alters the ASL milieu in a manner that impairs the activity of host defense factors remains attractive. A confounding factor has been the difficulty of accurately measuring ASL salt concentrations in vivo. Some investigators reported that Cl− concentrations are indeed elevated in CF (Gilljam et al. 1989; Joris et al. 1993; Kozlova et al. 2006a, b; Vanthanouvong et al. 2006; Zabner et al. 1998), while others found no significant difference in NaCl concentrations between CF and non-CF (Caldwell et al. 2002; Grubb et al. 2002; Jayaraman et al. 2001a, b; Knowles et al. 1997). If a difference in the concentration of ASL NaCl is not validated in CF, the experimental findings of the study of Smith and colleagues (1996) continue to point to a compositional change in CF ASL that negatively impacts the function of host defense factors.
4.2 Altered SCN− Transport in CF
This hypothesis builds from the observation that CFTR conducts thiocyanate (SCN−) (Tabcharani et al. 1993). While many secreted host defense proteins and peptides are well characterized, the recognition of an airway epithelial oxidative microbicidal system is recent. This oxidative system consists of two H2O2-generating enzymes of airway epithelia, dual oxidases (DUOX)1 and 2, along with a pseudohalide anion (thiocyanate, SCN−), and the enzyme lactoperoxidase (LPO) (Conner et al. 2002; Forteza et al. 2005; Gerson et al. 2000; Moskwa et al. 2007; Wijkstrom-Frei et al. 2003). The DUOX enzymes generate H2O2 into the apical extracellular space where H2O2 reacts with SCN− in a LPO-catalyzed reaction to form the antibacterial molecule OSCN− (H2O2 + SCN− → OSCN−) (Fig. 2). Both LPO and SCN− are highly concentrated in the airway surface liquid. SCN− is secreted apically by airway epithelia and accumulates in the ASL in concentrations of approximately 400–460 μM (Lorentzen et al. 2011; Wijkstrom-Frei et al. 2003). The DUOX/LPO/SCN− system can generate sufficient OSCN− to eliminate bacteria in vitro and in vivo (Conner et al. 2007; Moskwa et al. 2007). SCN− secretion is reduced in CF cells and tissues (Conner et al. 2007; Moskwa et al. 2007), leading to the hypothesis that diminished SCN− (and therefore reduced OSCN−) availability impairs defenses against airway pathogens. In support of this, Moskwa and coworkers reported that OSCN−-mediated killing of Staphylococcus aureus was inhibited on the apical surface of cultured CF airway epithelia (Moskwa et al. 2007). The relative contribution of this system to normal airway defenses in vivo, and how relevant its inactivation may be to CF pathogenesis, is currently unresolved. In a recent study, Lorentzen and colleagues found that the concentrations of SCN− were variable, and not significantly different, in the nasal secretions of humans with and without CF, suggesting that the bacterial killing activity of OSCN− was unlikely to be different between genotypes (Lorentzen et al. 2011). However, lung function correlated positively with SCN− levels in the CF subjects in this study. These findings suggest that while oxidative defenses are important for overall lung health, CFTR activity may not be the only determinant of ASL SCN− levels; the authors cite SCN− transport by alternative ion channels as potential compensating mechanisms in the CF airways (Lorentzen et al. 2011). One candidate for alternative SCN− transport, the sodium-independent chloride/iodide transporter pendrin (also known as SLC26A4), is expressed by human airway epithelia and is upregulated in response to IL-4 (Pedemonte et al. 2007) and to viral infections (Nakagami et al. 2008). Therefore, it is possible that, in some individuals, increased pendrin expression might partially compensate for loss of CFTR activity in inflamed CF airways.

Model of the oxidative host defense system at the apical side of airway epithelia. DUOX enzymes are the H2O2-generating cytochromes in the apical membrane of airway epithelia. LPO is secreted by submucosal glands. SCN− is utilized by LPO for OSCN− generation
4.3 Altered HCO −3 Transport in CF
It has been long recognized that loss of CFTR function results in the acidification of pancreatic secretions and that CF pancreatic secretions fail to properly alkalinize in response to secretagogues (Gaskin et al. 1982; Kopelman et al. 1988). CFTR expressed within the airways also transports bicarbonate (HCO −3 ) and thereby helps buffer ASL (Fischer and Widdicombe 2006; Smith and Welsh 1992). In the airways, loss of HCO −3 secretion via CFTR is predicted to result in a diminished capacity to alkalinize respiratory secretions. A number of studies have assessed ASL pH using in vitro systems and in vivo models. There is evidence that secretions from submucosal glands derived from human CF nasal tissues are hyperacidified relative to non-CF secretions (Song et al. 2006) and that cultured CF human bronchial epithelia acidify their ASL more rapidly than do non-CF cells (Coakley et al. 2003). In studies comparing CFTR-null and wild-type mice, Jayaraman and coworkers noted no significant differences in the in vivo airway pH as measured using a tracheal window preparation (Jayaraman et al. 2001b). Reductions in ASL pH could negatively influence innate immunity by several mechanisms [reviewed in (Coakley and Boucher 2001; Poschet et al. 2002)] including inhibiting the activity of antimicrobials (Dorschner et al. 2006; Lehrer et al. 1983; Selsted et al. 1985), altering the viscosity of secretions (Bhaskar et al. 1991; Holma 1985), and decreasing ciliary beat frequency (Clary-Meinesz et al. 1998).
4.4 CF-Associated Changes in Airway Submucosal Gland Function and Secreted Mucins
Alterations in airway submucosal gland physiology are also implicated in the innate immune defects in CF. Submucosal glands are responsible for the secretion of liquid and mucins, such as the gel-forming mucin MUC5B (Groneberg et al. 2002), as well as numerous proteins and peptides with antimicrobial, anti-inflammatory, and other host defense functions (Wine and Joo 2004). A submucosal gland is formed by invagination of the airway epithelial surface to form a single collecting duct, into which multiple mucous tubules empty (Wine and Joo 2004). The more proximal portions of these tubules are lined by mucous cells, responsible for mucin production, while the distal acini of the tubules contain serous cells that secrete liquid, electrolytes, and various protein and peptide components of ASL. CFTR is expressed primarily in the serous acini (Engelhardt et al. 1992), where its anion channel activity is thought to play a key role in liquid secretion. As major sites of mucus production, the submucosal glands contribute to the quantity and quality of mucus in the airways and are studied for their possible involvement in generating the abnormal mucus that is a hallmark of CF lung disease.
Several investigators hypothesize that liquid secretion by serous cells is reduced in CFTR-deficient submucosal glands, leading to altered hydration of secreted mucins and the ASL. The resulting mucus is predicted to be unusually viscous and would impede the efficient mechanical clearance of particles from the airways. Such changes in mucus rheologic properties could also lead to obstruction of submucosal gland ducts. In support of this hypothesis, there is abundant evidence that liquid secretion in response to secretory signals is reduced when CFTR function is impaired. A number of studies confirmed that CF glands fail to secrete in response to forskolin and vasoactive intestinal peptide (VIP), agonists that stimulate secretion by elevating intracellular cAMP levels (Choi et al. 2009; Joo et al. 2002, 2006, 2010; Lee and Foskett 2010). Similarly, secretion in CF glands is impaired in response to the neuropeptide substance P (Choi et al. 2009; Joo et al. 2010) and to carbachol when administered in combination with other agonists (Choi et al. 2007). In keeping with these data, studies of secretions collected from individual submucosal glands suggest that viscosity is significantly elevated in secretions from CF individuals (Jayaraman et al. 2001a; Salinas et al. 2005).
Defective HCO −3 secretion is also hypothesized to impact mucin release and viscosity (Quinton 2008). One model posits that, early in the process of mucus gel formation and extrusion from the submucosal glands, polyanionic mucin molecules are packed together into condensed mucin granules. To maintain this structure, the mucin molecules are highly cross-linked and their charges neutralized by a “shield” of Ca2+ ions. HCO −3 , which effectively chelates Ca2+, can sequester these Ca2+ ions, thereby releasing the cross-links between mucins and allowing the mucins to rapidly expand as they are released from submucosal glands and/or goblet cells. In this way, HCO −3 may be intimately involved in the swelling and hydration of the ASL mucus layer. This model is supported by the demonstration that adding HCO −3 to mucus gels increases mucus dispersal in vitro (Chen et al. 2010a). Therefore, in CF glands and tissues, the absence of CFTR-mediated HCO −3 secretion may result in mucus granules that de-condense inappropriately, giving rise to viscous, under-hydrated mucus that impairs mucociliary clearance (Chen et al. 2010a).
In addition to the impact of impaired secretory responses of CF glands on the hydration status of mucins, it is suggested that this phenomenon could affect the abundance of secreted antimicrobials and other host defense factors from gland serous cells. While there is currently little direct evidence for this, Wine and colleagues reported that a number of known innate immune molecules, including lysozyme, NGAL (lipocalin-2), HSC-71, and the protease inhibitors alpha 1-antitrypsin and alpha 1-antichymotrypsin, are secreted by Calu-3 cells (a serous cell model) in response to forskolin stimulation (Joo et al. 2004). This observation leads to the prediction that secretion of antimicrobials may be reduced in the absence of CFTR. Additionally, when this observation is considered with the above-mentioned defects in liquid secretion and mucus hydration, it is tempting to speculate that in CF glands, secreted innate immune molecules may become “trapped” within viscous polyanionic mucus and are therefore less likely to be properly presented to the airway lumen.
Increased mucin production by the surface epithelium may also contribute to impaired mucociliary clearance in CF. Airway surface goblet cells secrete the gel-forming mucin MUC5AC and, to a lesser extent, MUC5B (Groneberg et al. 2002; Hovenberg et al. 1996). Goblet cell metaplasia is a response to chronic inflammation and is commonly seen in CF lung disease (Bedrossian et al. 1976; Davis and Dickey 2008; Groneberg et al. 2002). Therefore, the increased viscous secretions associated with CF are derived in part from the surface epithelium. In keeping with this, Derichs and colleagues reported that the viscosity was increased in both the mucus and the periciliary liquid layers of the ASL from cultured CF bronchial epithelia, which do not possess submucosal glands (Derichs et al. 2011). This raises the possibility that increased mucin production from goblet cells may also impact innate immunity by providing more binding sites for cationic antimicrobials and other molecules that normally associate with the polyanionic mucin polymers in the airways (Felgentreff et al. 2006), making those antimicrobial factors less available to interact with their target pathogens.
5 New Animal Models of CF Lung Disease
A limitation in advancing knowledge of the molecular basis of CF lung disease has been the lack of animal models that recapitulate key features of lung and other organ disease pathology. The mouse models with CFTR-null alleles and human CFTR mutations available since the early 1990s have contributed greatly to disease understanding but do not develop spontaneous lung disease similar to humans with CF. Recently, several groups used somatic cell targeting of the CFTR gene, followed by nuclear transfer and cloning to develop novel models in pigs (Klymiuk et al. 2011; Rogers et al. 2008a, b) and ferrets (Sun et al. 2008, 2010). These new animal models recapitulate key features of CF disease (Klymiuk et al. 2011; Rogers et al. 2008b; Sun et al. 2010). At birth, the airways of CFTR targeted pigs are free of inflammation but manifest a bacterial host defense defect without the secondary consequences of infection (Rogers et al. 2008b; Stoltz et al. 2010). Pigs with targeted CFTR genes spontaneously develop hallmark features of CF including airway inflammation, remodeling, mucus, and infection within months of birth (Fig. 3). Their lungs contain multiple bacterial species (Gram-negative and Gram-positive), suggesting an equal opportunity host defense defect. While the lungs of newborn pigs show no inflammation, they are less often sterile than wild-type littermate controls. Moreover, after intrapulmonary bacterial challenge with Staphylococcus aureus, CF pigs fail to eradicate bacteria as effectively as wild-type pigs (Stoltz et al. 2010). These results suggest that impaired bacterial elimination is the pathogenic event initiating a cascade of inflammation and pathology.

Airway disease in a pig model of cystic fibrosis. Airways from a CFTR+/+ (left panel, H&E stain) and a CFTR−/− pig (middle and right, PAS and H&E, respectively), bar = 90 μm. CFTR−/− pig is infected with the Gram-negative organism Bordetella bronchiseptica. Note the increased mucosal thickening and mucin expression in the epithelium with mucocellular lumen obstruction (middle panel). A CF pig airway is obstructed with neutrophils and mucopurulent debris (right panel)
Studies of CF pig submucosal glands have shown that the model exhibits responses to agonists qualitatively similar to glands from human patients with CF (Joo et al. 2010). CF pig glands produce almost no liquid in response to cAMP agonists and reduced volumes in response to all other stimuli except carbachol. Furthermore, glands from newborn CF pigs, like human CF glands, exhibit a reduced secretory response to substance P (Joo et al. 2010). Thus, CF glands have a reduced ability to respond to important secretagogues. These findings, combined with the hypoplastic submucosal glands of the newborn CF airways (Meyerholz et al. 2010), raise the possibility that the gland contributions to ASL are altered in their volume and composition in CF.
Recent studies in newborn CF pigs provide further support for the idea that an altered ASL environment is a primary cause of impaired innate immunity in the lung. In vitro and in vivo experiments showed that ASL pH was lower in CF pigs, and this reduction in pH reduced the antimicrobial activity of ASL (Pezzulo et al. 2012). Interestingly, a more acidic ASL pH also diminished bacterial killing in wild-type pigs, while increasing ASL pH increased antimicrobial activity in the ASL of pigs with CF. These results in newborn animals support the concept that reductions in ASL pH due to loss of CFTR-dependent HCO −3 secretion impair the activity of endogenous antimicrobial proteins and peptides, allowing for the onset of airway infection.
Experiments in the CF pig model also yielded some unexpected findings. CFTR targeted pigs exhibit morphological abnormalities of the newborn trachea and large airways, including tracheal cartilage alterations, a decrease in submucosal gland mass, and more noticeable smooth muscle bundles in the posterior trachea (Meyerholz et al. 2010). Interestingly, retrospective analysis of chest CT scans from children with CF revealed that the tracheas of CF subjects were less circular than the non-CF control individuals (Meyerholz et al. 2010), echoing the tracheal abnormalities observed in the CF piglets. These results are contrary to the dogma that the “lungs of children with CF are normal at birth,” and raise the possibility that loss of CFTR function perturbs a developmental program in the lung resulting in structural abnormalities. Such changes could also contribute to the lung disease phenotype.
In a series of experiments using cultured airway epithelia and freshly excised nasal and tracheal tissues from CFTR−/− pigs, as well as in vivo electrophysiological measurements, Chen et al. reported that airway epithelia from newborn CF pigs displayed a reduced Cl− conductance without an increase in Na+ conductance (Chen et al. 2010b). The authors implicate the loss of the CFTR-dependent Cl− conductance, rather than increased Na+ conductance, as the explanation for the greater reduction in Vt in response to amiloride observed in CF epithelia relative to non-CF epithelia. Measurements of the periciliary liquid depth in fixed tracheal tissue from pigs indicated no significant difference in ASL depth between the CF and non-CF airways at birth. These data suggest that ASL/mucus dehydration due to Na+ hyperabsorption in CF airways may not be a primary event in CF pathogenesis, although they do not rule out the possibility that perturbations in Na+ transport may affect airway immunity as the disease progresses.
CFTR-null ferrets also develop multiorgan system disease, and neonatal animals manifest a pulmonary host defense defect in the airways associated with colonization by bacteria (Sun et al. 2010). Early results also indicate that adult CF ferrets develop a lung disease phenotype with similarities to human CF, including bacterial colonization (John Engelhardt, personal communication). Continued studies of animal models that reproduce key phenotypic features of human CF lung disease are likely to provide further insights into the links between loss of CFTR function and the initial pathologic events.
6 Future Directions
With recent advances in CF basic science research and the development of new animal models, the field is poised to make further breakthroughs in the understanding of disease pathogenesis, including a better grasp of the molecular basis of the airway host defense defect. Advancements in these areas are critical to the development of new pulmonary specific or systemic therapies to prevent the onset or slow the rate of disease progression.
References
Abman SH, Ogle JW, Butler-Simon N, Rumack CM, Accurso FJ (1988) Role of respiratory syncytial virus in early hospitalizations for respiratory distress of young infants with cystic fibrosis. J Pediatr 113(5):826–830
Agerberth B, Charo J, Werr J, Olsson B, Idali F, Lindbom L, Kiessling R, Jornvall H, Wigzell H, Gudmundsson GH (2000) The human antimicrobial and chemotactic peptides LL-37 and alpha-defensins are expressed by specific lymphocyte and monocyte populations. Blood 96(9):3086–3093
Akira S, Takeda K (2004) Toll-like receptor signalling. Nat Rev Immunol 4(7):499–511
Andersen DH (1938) Cystic fibrosis of the pancreas and its relation to celiac disease: a clinical and pathological study. Am J Dis Child 56:344–399
Anderson MP, Gregory RJ, Thompson S, Souza DW, Paul S, Mulligan RC, Smith AE, Welsh MJ (1991a) Demonstration that CFTR is a chloride channel by alteration of its anion selectivity. Science 253:202–205
Anderson MP, Rich DP, Gregory RJ, Smith AE, Welsh MJ (1991b) Generation of cAMP-activated chloride currents by expression of CFTR. Science 251(4994):679–682
Andrejeva J, Childs KS, Young DF, Carlos TS, Stock N, Goodbourn S, Randall RE (2004) The V proteins of paramyxoviruses bind the IFN-inducible RNA helicase, mda-5, and inhibit its activation of the IFN-beta promoter. Proc Natl Acad Sci USA 101(49):17264–17269
Andresen E, Lange C, Strodthoff D, Goldmann T, Fischer N, Sahly H, Branscheid D, Heine H (2011) S100A7/psoriasin expression in the human lung: unchanged in patients with COPD, but upregulated upon positive S. aureus detection. BMC Pulm Med 11:10
Armstrong DS, Hook SM, Jamsen KM, Nixon GM, Carzino R, Carlin JB, Robertson CF, Grimwood K (2005) Lower airway inflammation in infants with cystic fibrosis detected by newborn screening. Pediatr Pulmonol 40(6):500–510
Arnold RR, Brewer M, Gauthier JJ (1980) Bactericidal activity of human lactoferrin: sensitivity of a variety of microorganisms. Infect Immun 28(3):893–898
Aron Y, Polla BS, Bienvenu T, Dall’ava J, Dusser D, Hubert D (1999) HLA class II polymorphism in cystic fibrosis. A possible modifier of pulmonary phenotype. Am J Respir Crit Care Med 159(5 Pt 1):1464–1468
Balough K, McCubbin M, Weinberger M, Smits W, Ahrens R, Fick R (1995) The relationship between infection and inflammation in the early stages of lung disease from cystic fibrosis. Pediatr Pulmonol 20(2):63–70
Bals R, Wang X, Wu Z, Freeman T, Bafna V, Zasloff M, Wilson JM (1998a) Human beta-defensin 2 is a salt-sensitive peptide antibiotic expressed in human lung. J Clin Invest 102(5):874–880
Bals R, Wang X, Zasloff M, Wilson JM (1998b) The peptide antibiotic LL-37/hCAP-18 is expressed in epithelia of the human lung where it has broad antimicrobial activity at the airway surface. Proc Natl Acad Sci USA 95(16):9541–9546
Bartlett JA, Fischer AJ, McCray PB Jr (2008a) Innate immune functions of the airway epithelium. Contrib Microbiol 15:147–163
Bartlett JA, Hicks BJ, Schlomann JM, Ramachandran S, Nauseef WM, McCray PB Jr (2008b) PLUNC is a secreted product of neutrophil granules. J Leukoc Biol 83(5):1201–1206
Bedrossian CW, Greenberg SD, Singer DB, Hansen JJ, Rosenberg HS (1976) The lung in cystic fibrosis. A quantitative study including prevalence of pathologic findings among different age groups. Hum Pathol 7(2):195–204
Bhaskar KR, Gong DH, Bansil R, Pajevic S, Hamilton JA, Turner BS, LaMont JT (1991) Profound increase in viscosity and aggregation of pig gastric mucin at low pH. Am J Physiol 261(5 Pt 1):G827–G832
Bingle CD, Bingle L (2000) Characterisation of the human plunc gene, a gene product with an upper airways and nasopharyngeal restricted expression pattern. Biochim Biophys Acta 1493(3):363–367
Bjarnsholt T, Jensen PO, Fiandaca MJ, Pedersen J, Hansen CR, Andersen CB, Pressler T, Givskov M, Hoiby N (2009) Pseudomonas aeruginosa biofilms in the respiratory tract of cystic fibrosis patients. Pediatr Pulmonol 44(6):547–558
Boucher RC (2004) New concepts of the pathogenesis of cystic fibrosis lung disease. Eur Respir J 23(1):146–158
Boucher RC (2007) Evidence for airway surface dehydration as the initiating event in CF airway disease. J Intern Med 261(1):5–16
Britigan BE, Hayek MB, Doebbeling BN, Fick RB Jr (1993) Transferrin and lactoferrin undergo proteolytic cleavage in the Pseudomonas aeruginosa-infected lungs of patients with cystic fibrosis. Infect Immun 61(12):5049–5055
Bubien JK (2001) CFTR may play a role in regulated secretion by lymphocytes: a new hypothesis for the pathophysiology of cystic fibrosis. Pflugers Arch 443(Suppl 1):S36–S39
Bubien JK, Kirk KL, Rado TA, Frizzell RA (1990) Cell cycle dependence of chloride permeability in normal and cystic fibrosis lymphocytes. Science 248(4961):1416–1419
Butler MW, Robertson I, Greene CM, O’Neill SJ, Taggart CC, McElvaney NG (2006) Elafin prevents lipopolysaccharide-induced AP-1 and NF-kappaB activation via an effect on the ubiquitin-proteasome pathway. J Biol Chem 281(46):34730–34735
Caldwell RA, Grubb BR, Tarran R, Boucher RC, Knowles MR, Barker PM (2002) In vivo airway surface liquid Cl- analysis with solid-state electrodes. J Gen Physiol 119(1):3–14
Candiano G, Bruschi M, Pedemonte N, Musante L, Ravazzolo R, Liberatori S, Bini L, Galietta LJ, Zegarra-Moran O (2007) Proteomic analysis of the airway surface liquid: modulation by proinflammatory cytokines. Am J Physiol Lung Cell Mol Physiol 292(1):L185–L198
Cassel SL, Sutterwala FS (2010) Sterile inflammatory responses mediated by the NLRP3 inflammasome. Eur J Immunol 40(3):607–611
Chen EY, Yang N, Quinton PM, Chin WC (2010a) A new role for bicarbonate in mucus formation. Am J Physiol Lung Cell Mol Physiol 299(4):L542–L549
Chen JH, Stoltz DA, Karp PH, Ernst SE, Pezzulo AA, Moninger TO, Rector MV, Reznikov LR, Launspach JL, Chaloner K, Zabner J, Welsh MJ (2010b) Loss of anion transport without increased sodium absorption characterizes newborn porcine cystic fibrosis airway epithelia. Cell 143(6):911–923
Chertov O, Michiel DF, Xu L, Wang JM, Tani K, Murphy WJ, Longo DL, Taub DD, Oppenheim JJ (1996) Identification of defensin-1, defensin-2, and CAP37/azurocidin as T-cell chemoattractant proteins released from interleukin-8-stimulated neutrophils. J Biol Chem 271(6):2935–2940
Choi JY, Joo NS, Krouse ME, Wu JV, Robbins RC, Ianowski JP, Hanrahan JW, Wine JJ (2007) Synergistic airway gland mucus secretion in response to vasoactive intestinal peptide and carbachol is lost in cystic fibrosis. J Clin Invest 117(10):3118–3127
Choi JY, Khansaheb M, Joo NS, Krouse ME, Robbins RC, Weill D, Wine JJ (2009) Substance P stimulates human airway submucosal gland secretion mainly via a CFTR-dependent process. J Clin Invest 119(5):1189–1200
Chu HW, Thaikoottathil J, Rino JG, Zhang G, Wu Q, Moss T, Refaeli Y, Bowler R, Wenzel SE, Chen Z, Zdunek J, Breed R, Young R, Allaire E, Martin RJ (2007) Function and regulation of SPLUNC1 protein in Mycoplasma infection and allergic inflammation. J Immunol 179(6):3995–4002
Clary-Meinesz C, Mouroux J, Cosson J, Huitorel P, Blaive B (1998) Influence of external pH on ciliary beat frequency in human bronchi and bronchioles. Eur Respir J 11(2):330–333
Coakley RD, Boucher RC (2001) Regulation and functional significance of airway surface liquid pH. JOP 2(4 Suppl):294–300
Coakley RD, Grubb BR, Paradiso AM, Gatzy JT, Johnson LG, Kreda SM, O'Neal WK, Boucher RC (2003) Abnormal surface liquid pH regulation by cultured cystic fibrosis bronchial epithelium. Proc Natl Acad Sci USA 100(26):16083–16088
Cole AM, Dewan P, Ganz T (1999) Innate antimicrobial activity of nasal secretions. Infect Immun 67(7):3267–3275
Cole AM, Kim YH, Tahk S, Hong T, Weis P, Waring AJ, Ganz T (2001) Calcitermin, a novel antimicrobial peptide isolated from human airway secretions. FEBS Lett 504(1–2):5–10
Cole AM, Liao HI, Stuchlik O, Tilan J, Pohl J, Ganz T (2002) Cationic polypeptides are required for antibacterial activity of human airway fluid. J Immunol 169(12):6985–6991
Conner GE, Salathe M, Forteza R (2002) Lactoperoxidase and hydrogen peroxide metabolism in the airway. Am J Respir Crit Care Med 166(12 Pt 2):S57–S61
Conner GE, Wijkstrom-Frei C, Randell SH, Fernandez VE, Salathe M (2007) The lactoperoxidase system links anion transport to host defense in cystic fibrosis. FEBS Lett 581(2):271–278
Corbin BD, Seeley EH, Raab A, Feldmann J, Miller MR, Torres VJ, Anderson KL, Dattilo BM, Dunman PM, Gerads R, Caprioli RM, Nacken W, Chazin WJ, Skaar EP (2008) Metal chelation and inhibition of bacterial growth in tissue abscesses. Science 319(5865):962–965
Cornish CJ, Devery JM, Poronnik P, Lackmann M, Cook DI, Geczy CL (1996) S100 protein CP-10 stimulates myeloid cell chemotaxis without activation. J Cell Physiol 166(2):427–437
Cowland JB, Johnsen AH, Borregaard N (1995) hCAP-18, a cathelin/pro-bactenecin-like protein of human neutrophil specific granules. FEBS Lett 368(1):173–176
Cowland JB, Sorensen OE, Sehested M, Borregaard N (2003) Neutrophil gelatinase-associated lipocalin is up-regulated in human epithelial cells by IL-1 beta, but not by TNF-alpha. J Immunol 171(12):6630–6639
Cruz CM, Rinna A, Forman HJ, Ventura AL, Persechini PM, Ojcius DM (2007) ATP activates a reactive oxygen species-dependent oxidative stress response and secretion of proinflammatory cytokines in macrophages. J Biol Chem 282(5):2871–2879
Daher KA, Selsted ME, Lehrer RI (1986) Direct inactivation of viruses by human granulocyte defensins. J Virol 60(3):1068–1074
Davis CW, Dickey BF (2008) Regulated airway goblet cell mucin secretion. Annu Rev Physiol 70:487–512
Decraene A, Willems-Widyastuti A, Kasran A, De Boeck K, Bullens DM, Dupont LJ (2010) Elevated expression of both mRNA and protein levels of IL-17A in sputum of stable Cystic Fibrosis patients. Respir Res 11:177
Derichs N, Jin BJ, Song Y, Finkbeiner WE, Verkman AS (2011) Hyperviscous airway periciliary and mucous liquid layers in cystic fibrosis measured by confocal fluorescence photobleaching. FASEB J 25(7):2325–2332
Di A, Brown ME, Deriy LV, Li C, Szeto FL, Chen Y, Huang P, Tong J, Naren AP, Bindokas V, Palfrey HC, Nelson DJ (2006) CFTR regulates phagosome acidification in macrophages and alters bactericidal activity. Nat Cell Biol 8(9):933–944
Do TQ, Moshkani S, Castillo P, Anunta S, Pogosyan A, Cheung A, Marbois B, Faull KF, Ernst W, Chiang SM, Fujii G, Clarke CF, Foster K, Porter E (2008) Lipids including cholesteryl linoleate and cholesteryl arachidonate contribute to the inherent antibacterial activity of human nasal fluid. J Immunol 181(6):4177–4187
Doring G, Albus A, Hoiby N (1988) Immunologic aspects of cystic fibrosis. Chest 94(2 Suppl):109S–115S
Dorschner RA, Lopez-Garcia B, Peschel A, Kraus D, Morikawa K, Nizet V, Gallo RL (2006) The mammalian ionic environment dictates microbial susceptibility to antimicrobial defense peptides. FASEB J 20(1):35–42
Dubin PJ, Kolls JK (2011) IL-17 in cystic fibrosis: more than just Th17 cells. Am J Respir Crit Care Med 184(2):155–157
Elsbach P, Weiss J, Franson RC, Beckerdite-Quagliata S, Schneider A, Harris L (1979) Separation and purification of a potent bactericidal/permeability-increasing protein and a closely associated phospholipase A2 from rabbit polymorphonuclear leukocytes. Observations on their relationship. J Biol Chem 254(21):11000–11009
Engelhardt JF, Yankaskas JR, Ernst SA, Yang Y, Marino CR, Boucher RC, Cohn JA, Wilson JM (1992) Submucosal glands are the predominant site of CFTR expression in the human bronchus. Nat Genet 2(3):240–248
Fanconi G, Uehlinger E, Knauer C (1936) Das Coeliakie-syndrom bei angeborener zystischer Pankreasfibromatose und Bronchiektasien (Celiac syndrome with congenital cystic fibromatosis of the pancreas and bronchiectasis). Wien Med Wchnschr 86:753–756
Felgentreff K, Beisswenger C, Griese M, Gulder T, Bringmann G, Bals R (2006) The antimicrobial peptide cathelicidin interacts with airway mucus. Peptides 27(12):3100–3106
Fiedler MA, Kaetzel CS, Davis PB (1991) Sustained production of secretory component by human tracheal epithelial cells in primary culture. Am J Physiol 261(4 Pt 1):L255–L261
Fischer H, Widdicombe JH (2006) Mechanisms of acid and base secretion by the airway epithelium. J Membr Biol 211(3):139–150
Fleming A (1922) On a remarkable bacteriolytic element found in tissues and secretions. Proc R Soc (Lond) 93:306–319
Fleming A, Allison VD (1922) Observations on a bacteriolytic substance ("lysozyme") found in secretions and tissues. Br J Exp Pathol 3:252–260
Fluckinger M, Haas H, Merschak P, Glasgow BJ, Redl B (2004) Human tear lipocalin exhibits antimicrobial activity by scavenging microbial siderophores. Antimicrob Agents Chemother 48(9):3367–3372
Forteza R, Salathe M, Miot F, Forteza R, Conner GE (2005) Regulated hydrogen peroxide production by Duox in human airway epithelial cells. Am J Respir Cell Mol Biol 32(5):462–469
Gakhar L, Bartlett JA, Penterman J, Mizrachi D, Singh PK, Mallampalli RK, Ramaswamy S, McCray PB Jr (2010) PLUNC is a novel airway surfactant protein with anti-biofilm activity. PLoS One 5(2):e9098
Ganz T, Selsted ME, Szklarek D, Harwig SS, Daher K, Bainton DF, Lehrer RI (1985) Defensins. Natural peptide antibiotics of human neutrophils. J Clin Invest 76(4):1427–1435
Garcia JR, Jaumann F, Schulz S, Krause A, Rodriguez-Jimenez J, Forssmann U, Adermann K, Kluver E, Vogelmeier C, Becker D, Hedrich R, Forssmann WG, Bals R (2001a) Identification of a novel, multifunctional beta-defensin (human beta-defensin 3) with specific antimicrobial activity. Its interaction with plasma membranes of Xenopus oocytes and the induction of macrophage chemoattraction. Cell Tissue Res 306(2):257–264
Garcia JR, Krause A, Schulz S, Rodriguez-Jimenez FJ, Kluver E, Adermann K, Forssmann U, Frimpong-Boateng A, Bals R, Forssmann WG (2001b) Human beta-defensin 4: a novel inducible peptide with a specific salt-sensitive spectrum of antimicrobial activity. FASEB J 15(10):1819–1821
Garred P, Pressler T, Madsen HO, Frederiksen B, Svejgaard A, Hoiby N, Schwartz M, Koch C (1999) Association of mannose-binding lectin gene heterogeneity with severity of lung disease and survival in cystic fibrosis. J Clin Invest 104(4):431–437
Gaskin KJ, Durie PR, Corey M, Wei P, Forstner GG (1982) Evidence for a primary defect of pancreatic HCO3-secretion in cystic fibrosis. Pediatr Res 16(7):554–557
Gerson C, Sabater J, Scuri M, Torbati A, Coffey R, Abraham JW, Lauredo I, Forteza R, Wanner A, Salathe M, Abraham WM, Conner GE (2000) The lactoperoxidase system functions in bacterial clearance of airways. Am J Respir Cell Mol Biol 22(6):665–671
Gilljam H, Ellin A, Strandvik B (1989) Increased bronchial chloride concentration in cystic fibrosis. Scand J Clin Lab Invest 49(2):121–124
Glaser R, Harder J, Lange H, Bartels J, Christophers E, Schroder JM (2005) Antimicrobial psoriasin (S100A7) protects human skin from Escherichia coli infection. Nat Immunol 6(1):57–64
Goetz DH, Holmes MA, Borregaard N, Bluhm ME, Raymond KN, Strong RK (2002) The neutrophil lipocalin NGAL is a bacteriostatic agent that interferes with siderophore-mediated iron acquisition. Mol Cell 10(5):1033–1043
Goldman MJ, Anderson GM, Stolzenberg ED, Kari UP, Zasloff M, Wilson JM (1997) Human beta-defensin-1 is a salt-sensitive antibiotic in lung that is inactivated in cystic fibrosis. Cell 88(4):553–560
Gottsch JD, Eisinger SW, Liu SH, Scott AL (1999) Calgranulin C has filariacidal and filariastatic activity. Infect Immun 67(12):6631–6636
Greene CM, McElvaney NG, O’Neill SJ, Taggart CC (2004) Secretory leucoprotease inhibitor impairs Toll-like receptor 2- and 4-mediated responses in monocytic cells. Infect Immun 72(6):3684–3687
Griesenbach U, Soussi S, Larsen MB, Casamayor I, Dewar A, Regamey N, Bush A, Shah PL, Davies JC, Alton EW (2011) Quantification of periciliary fluid height in human airway biopsies is feasible, but not suitable as a biomarker. Am J Respir Cell Mol Biol 44(3):309–315
Groneberg DA, Eynott PR, Oates T, Lim S, Wu R, Carlstedt I, Nicholson AG, Chung KF (2002) Expression of MUC5AC and MUC5B mucins in normal and cystic fibrosis lung. Respir Med 96(2):81–86
Grubb BR, Chadburn JL, Boucher RC (2002) In vivo microdialysis for determination of nasal liquid ion composition. Am J Physiol Cell Physiol 282(6):C1423–C1431
Guarda G, Braun M, Staehli F, Tardivel A, Mattmann C, Forster I, Farlik M, Decker T, Du Pasquier RA, Romero P, Tschopp J (2011) Type I interferon inhibits interleukin-1 production and inflammasome activation. Immunity 34(2):213–223
Guignard F, Mauel J, Markert M (1995) Identification and characterization of a novel human neutrophil protein related to the S100 family. Biochem J 309(Pt 2):395–401
Guyot N, Butler MW, McNally P, Weldon S, Greene CM, Levine RL, O’Neill SJ, Taggart CC, McElvaney NG (2008) Elafin, an elastase-specific inhibitor, is cleaved by its cognate enzyme neutrophil elastase in sputum from individuals with cystic fibrosis. J Biol Chem 283(47):32377–32385
Harder J, Bartels J, Christophers E, Schroder JM (2001) Isolation and characterization of human beta -defensin-3, a novel human inducible peptide antibiotic. J Biol Chem 276(8):5707–5713
Hartshorn KL, Crouch EC, White MR, Eggleton P, Tauber AI, Chang D, Sastry K (1994) Evidence for a protective role of pulmonary surfactant protein D (SP-D) against influenza A viruses. J Clin Invest 94(1):311–319
Hartshorn KL, Crouch E, White MR, Colamussi ML, Kakkanatt A, Tauber B, Shepherd V, Sastry KN (1998) Pulmonary surfactant proteins A and D enhance neutrophil uptake of bacteria. Am J Physiol 274(6 Pt 1):L958–L969
Heilborn JD, Nilsson MF, Kratz G, Weber G, Sorensen O, Borregaard N, Stahle-Backdahl M (2003) The cathelicidin anti-microbial peptide LL-37 is involved in re-epithelialization of human skin wounds and is lacking in chronic ulcer epithelium. J Invest Dermatol 120(3):379–389
Henriksen PA, Hitt M, Xing Z, Wang J, Haslett C, Riemersma RA, Webb DJ, Kotelevtsev YV, Sallenave JM (2004) Adenoviral gene delivery of elafin and secretory leukocyte protease inhibitor attenuates NF-kappa B-dependent inflammatory responses of human endothelial cells and macrophages to atherogenic stimuli. J Immunol 172(7):4535–4544
Henry MT, Cave S, Rendall J, O’Connor CM, Morgan K, FitzGerald MX, Kalsheker N (2001) An alpha1-antitrypsin enhancer polymorphism is a genetic modifier of pulmonary outcome in cystic fibrosis. Eur J Hum Genet 9(4):273–278
Hiatt RB, Engle C, Karush K (1952) The role of the granulocyte as a source of lysozyme in ulcerative colitis. J Clin Invest 31(7):721–726
Hiatt PW, Grace SC, Kozinetz CA, Raboudi SH, Treece DG, Taber LH, Piedra PA (1999) Effects of viral lower respiratory tract infection on lung function in infants with cystic fibrosis. Pediatrics 103(3):619–626
Hiemstra PS, Maassen RJ, Stolk J, Heinzel-Wieland R, Steffens GJ, Dijkman JH (1996) Antibacterial activity of antileukoprotease. Infect Immun 64(11):4520–4524
Holma B (1985) Influence of buffer capacity and pH-dependent rheological properties of respiratory mucus on health effects due to acidic pollution. Sci Total Environ 41(2):101–123
Hovenberg HW, Davies JR, Carlstedt I (1996) Different mucins are produced by the surface epithelium and the submucosa in human trachea: identification of MUC5AC as a major mucin from the goblet cells. Biochem J 318(Pt 1):319–324
Iovine NM, Elsbach P, Weiss J (1997) An opsonic function of the neutrophil bactericidal/permeability-increasing protein depends on both its N- and C-terminal domains. Proc Natl Acad Sci USA 94(20):10973–10978
Isemura S, Saitoh E, Ito S, Isemura M, Sanada K (1984) Cystatin S: a cysteine proteinase inhibitor of human saliva. J Biochem 96(4):1311–1314
Itani OA, Chen JH, Karp PH, Ernst S, Keshavjee S, Parekh K, Klesney-Tait J, Zabner J, Welsh MJ (2011) Human cystic fibrosis airway epithelia have reduced Cl- conductance but not increased Na+ conductance. Proc Natl Acad Sci USA 108(25):10260–10265
Jayaraman S, Joo NS, Reitz B, Wine JJ, Verkman AS (2001a) Submucosal gland secretions in airways from cystic fibrosis patients have normal [Na(+)] and pH but elevated viscosity. Proc Natl Acad Sci USA 98(14):8119–8123
Jayaraman S, Song Y, Vetrivel L, Shankar L, Verkman AS (2001b) Noninvasive in vivo fluorescence measurement of airway-surface liquid depth, salt concentration, and pH. J Clin Invest 107(3):317–324
Jia HP, Schutte BC, Schudy A, Linzmeier R, Guthmiller JM, Johnson GK, Tack BF, Mitros JP, Rosenthal A, Ganz T, McCray PB Jr (2001) Discovery of new human beta-defensins using a genomics-based approach. Gene 263(1–2):211–218
Joo NS, Irokawa T, Wu JV, Robbins RC, Whyte RI, Wine JJ (2002) Absent secretion to vasoactive intestinal peptide in cystic fibrosis airway glands. J Biol Chem 277(52):50710–50715
Joo NS, Lee DJ, Winges KM, Rustagi A, Wine JJ (2004) Regulation of antiprotease and antimicrobial protein secretion by airway submucosal gland serous cells. J Biol Chem 279(37):38854–38860
Joo NS, Irokawa T, Robbins RC, Wine JJ (2006) Hyposecretion, not hyperabsorption, is the basic defect of cystic fibrosis airway glands. J Biol Chem 281(11):7392–7398
Joo NS, Cho HJ, Khansaheb M, Wine JJ (2010) Hyposecretion of fluid from tracheal submucosal glands of CFTR-deficient pigs. J Clin Invest 120(9):3161–3166
Joris L, Dab I, Quinton PM (1993) Elemental composition of human airway surface fluid in healthy and diseased airways. Am Rev Respir Dis 148(6 Pt 1):1633–1637
Kerem B, Rommens JM, Buchanan JA, Markiewicz D, Cox TK, Chakravarti A, Buchwald M, Tsui LC (1989) Identification of the cystic fibrosis gene: genetic analysis. Science 245(4922):1073–1080
Khan TZ, Wagener JS, Bost T, Martinez J, Accurso FJ, Riches DW (1995) Early pulmonary inflammation in infants with cystic fibrosis. Am J Respir Crit Care Med 151(4):1075–1082
King AE, Critchley HO, Sallenave JM, Kelly RW (2003) Elafin in human endometrium: an antiprotease and antimicrobial molecule expressed during menstruation. J Clin Endocrinol Metab 88(9):4426–4431
Kjeldsen L, Johnsen AH, Sengelov H, Borregaard N (1993) Isolation and primary structure of NGAL, a novel protein associated with human neutrophil gelatinase. J Biol Chem 268(14):10425–10432
Klymiuk N, Mundhenk L, Kraehe K, Wuensch A, Plog S, Emrich D, Langenmayer MC, Stehr M, Holzinger A, Kroner C, Richter A, Kessler B, Kurome M, Eddicks M, Nagashima H, Heinritzi K, Gruber AD, Wolf E (2011) Sequential targeting of CFTR by BAC vectors generates a novel pig model of cystic fibrosis. J Mol Med (Berl) 90(5):597–608. doi:10.1007/s00109-011-0839-y
Knowles M, Gatzy J, Boucher R (1981) Increased bioelectric potential difference across respiratory epithelia in cystic fibrosis. N Engl J Med 305(25):1489–1495
Knowles M, Gatzy J, Boucher R (1983a) Relative ion permeability of normal and cystic fibrosis nasal epithelium. J Clin Invest 71(5):1410–1417
Knowles MR, Stutts MJ, Spock A, Fischer N, Gatzy JT, Boucher RC (1983b) Abnormal ion permeation through cystic fibrosis respiratory epithelium. Science 221(4615):1067–1070
Knowles MR, Robinson JM, Wood RE, Pue CA, Mentz WM, Wager GC, Gatzy JT, Boucher RC (1997) Ion composition of airway surface liquid of patients with cystic fibrosis as compared with normal and disease-control subjects. J Clin Invest 100(10):2588–2595
Koczulla R, von Degenfeld G, Kupatt C, Krotz F, Zahler S, Gloe T, Issbrucker K, Unterberger P, Zaiou M, Lebherz C, Karl A, Raake P, Pfosser A, Boekstegers P, Welsch U, Hiemstra PS, Vogelmeier C, Gallo RL, Clauss M, Bals R (2003) An angiogenic role for the human peptide antibiotic LL-37/hCAP-18. J Clin Invest 111(11):1665–1672
Kopelman H, Corey M, Gaskin K, Durie P, Weizman Z, Forstner G (1988) Impaired chloride secretion, as well as bicarbonate secretion, underlies the fluid secretory defect in the cystic fibrosis pancreas. Gastroenterology 95(2):349–355
Korfhagen TR, Bruno MD, Ross GF, Huelsman KM, Ikegami M, Jobe AH, Wert SE, Stripp BR, Morris RE, Glasser SW, Bachurski CJ, Iwamoto HS, Whitsett JA (1996) Altered surfactant function and structure in SP-A gene targeted mice. Proc Natl Acad Sci USA 93(18):9594–9599
Kozlova I, Nilsson H, Henriksnas J, Roomans GM (2006a) X-ray microanalysis of apical fluid in cystic fibrosis airway epithelial cell lines. Cell Physiol Biochem 17(1–2):13–20
Kozlova I, Vanthanouvong V, Johannesson M, Roomans GM (2006b) Composition of airway surface liquid determined by X-ray microanalysis. Ups J Med Sci 111(1):137–153
Kuan SF, Rust K, Crouch E (1992) Interactions of surfactant protein D with bacterial lipopolysaccharides. Surfactant protein D is an Escherichia coli-binding protein in bronchoalveolar lavage. J Clin Invest 90(1):97–106
Larrick JW, Hirata M, Balint RF, Lee J, Zhong J, Wright SC (1995) Human CAP18: a novel antimicrobial lipopolysaccharide-binding protein. Infect Immun 63(4):1291–1297
Lee KC, Eckert RL (2007) S100A7 (Psoriasin)–mechanism of antibacterial action in wounds. J Invest Dermatol 127(4):945–957
Lee RJ, Foskett JK (2010) cAMP-activated Ca2+ signaling is required for CFTR-mediated serous cell fluid secretion in porcine and human airways. J Clin Invest 120(9):3137–3148
Lehrer RI, Selsted ME, Szklarek D, Fleischmann J (1983) Antibacterial activity of microbicidal cationic proteins 1 and 2, natural peptide antibiotics of rabbit lung macrophages. Infect Immun 42(1):10–14
Lehrer RI, Ganz T, Szklarek D, Selsted ME (1988) Modulation of the in vitro candidacidal activity of human neutrophil defensins by target cell metabolism and divalent cations. J Clin Invest 81(6):1829–1835
Leigh MW, Kylander JE, Yankaskas JR, Boucher RC (1995) Cell proliferation in bronchial epithelium and submucosal glands of cystic fibrosis patients. Am J Respir Cell Mol Biol 12(6):605–612
LeVine AM, Gwozdz J, Stark J, Bruno M, Whitsett J, Korfhagen T (1999a) Surfactant protein-A enhances respiratory syncytial virus clearance in vivo. J Clin Invest 103(7):1015–1021
LeVine AM, Kurak KE, Wright JR, Watford WT, Bruno MD, Ross GF, Whitsett JA, Korfhagen TR (1999b) Surfactant protein-A binds group B streptococcus enhancing phagocytosis and clearance from lungs of surfactant protein-A-deficient mice. Am J Respir Cell Mol Biol 20(2):279–286
LeVine AM, Whitsett JA, Gwozdz JA, Richardson TR, Fisher JH, Burhans MS, Korfhagen TR (2000) Distinct effects of surfactant protein A or D deficiency during bacterial infection on the lung. J Immunol 165(7):3934–3940
Li XL, Ezelle HJ, Kang TJ, Zhang L, Shirey KA, Harro J, Hasday JD, Mohapatra SK, Crasta OR, Vogel SN, Cross AS, Hassel BA (2008) An essential role for the antiviral endoribonuclease, RNase-L, in antibacterial immunity. Proc Natl Acad Sci USA 105(52):20816–20821
Lindahl M, Stahlbom B, Tagesson C (1999) Newly identified proteins in human nasal and bronchoalveolar lavage fluids: potential biomedical and clinical applications. Electrophoresis 20(18):3670–3676
Lindahl M, Stahlbom B, Tagesson C (2001) Identification of a new potential airway irritation marker, palate lung nasal epithelial clone protein, in human nasal lavage fluid with two-dimensional electrophoresis and matrix-assisted laser desorption/ionization-time of flight. Electrophoresis 22(9):1795–1800
Lorentzen D, Durairaj L, Pezzulo AA, Nakano Y, Launspach J, Stoltz DA, Zamba G, McCray PB Jr, Zabner J, Welsh MJ, Nauseef WM, Banfi B (2011) Concentration of the antibacterial precursor thiocyanate in cystic fibrosis airway secretions. Free Radic Biol Med 50(9):1144–1150
Lukinskiene L, Liu Y, Reynolds SD, Steele C, Stripp BR, Leikauf GD, Kolls JK, Di YP (2011) Antimicrobial activity of PLUNC protects against Pseudomonas aeruginosa infection. J Immunol 187(1):382–390
Marra MN, Wilde CG, Griffith JE, Snable JL, Scott RW (1990) Bactericidal/permeability-increasing protein has endotoxin-neutralizing activity. J Immunol 144(2):662–666
Masson P, Heremans JF, Prignot J (1965) Immunohistochemical localization of the iron-binding protein lactoferrin in human bronchial glands. Experientia 21(10):604–605
Masson PL, Heremans JF, Prignot JJ, Wauters G (1966) Immunohistochemical localization and bacteriostatic properties of an iron-binding protein from bronchial mucus. Thorax 21(6):538–544
Masson PL, Heremans JF, Schonne E (1969) Lactoferrin, an iron-binding protein in neutrophilic leukocytes. J Exp Med 130(3):643–658
Matsui H, Grubb BR, Tarran R, Randell SH, Gatzy JT, Davis CW, Boucher RC (1998) Evidence for periciliary liquid layer depletion, not abnormal ion composition, in the pathogenesis of cystic fibrosis airways disease. Cell 95(7):1005–1015
McAllister F, Henry A, Kreindler JL, Dubin PJ, Ulrich L, Steele C, Finder JD, Pilewski JM, Carreno BM, Goldman SJ, Pirhonen J, Kolls JK (2005) Role of IL-17A, IL-17 F, and the IL-17 receptor in regulating growth-related oncogene-alpha and granulocyte colony-stimulating factor in bronchial epithelium: implications for airway inflammation in cystic fibrosis. J Immunol 175(1):404–412
McCray PB Jr, Bentley L (1997) Human airway epithelia express a beta-defensin. Am J Respir Cell Mol Biol 16(3):343–349
McGillivary G, Bakaletz LO (2010) The multifunctional host defense peptide SPLUNC1 is critical for homeostasis of the mammalian upper airway. PLoS One 5(10):e13224
Meyerholz DK, Stoltz DA, Namati E, Ramachandran S, Pezzulo AA, Smith AR, Rector MV, Suter MJ, Kao S, McLennan G, Tearney GJ, Zabner J, McCray PB Jr, Welsh MJ (2010) Loss of cystic fibrosis transmembrane conductance regulator function produces abnormalities in tracheal development in neonatal pigs and young children. Am J Respir Crit Care Med 182(10):1251–1261
Mihaila A, Tremblay GM (2001) Human alveolar macrophages express elafin and secretory leukocyte protease inhibitor. Z Naturforsch C 56(3–4):291–297
Moskwa P, Lorentzen D, Excoffon KJ, Zabner J, McCray PB Jr, Nauseef WM, Dupuy C, Banfi B (2007) A novel host defense system of airways is defective in cystic fibrosis. Am J Respir Crit Care Med 175(2):174–183
Mueller C, Braag SA, Keeler A, Hodges C, Drumm M, Flotte TR (2011) Lack of cystic fibrosis transmembrane conductance regulator in CD3+ lymphocytes leads to aberrant cytokine secretion and hyperinflammatory adaptive immune responses. Am J Respir Cell Mol Biol 44(6):922–929
Muhlebach MS, Stewart PW, Leigh MW, Noah TL (1999) Quantitation of inflammatory responses to bacteria in young cystic fibrosis and control patients. Am J Respir Crit Care Med 160(1):186–191
Murakami S, Iwaki D, Mitsuzawa H, Sano H, Takahashi H, Voelker DR, Akino T, Kuroki Y (2002) Surfactant protein A inhibits peptidoglycan-induced tumor necrosis factor-alpha secretion in U937 cells and alveolar macrophages by direct interaction with toll-like receptor 2. J Biol Chem 277(9):6830–6837
Murthy AR, Lehrer RI, Harwig SS, Miyasaki KT (1993) In vitro candidastatic properties of the human neutrophil calprotectin complex. J Immunol 151(11):6291–6301
Nakagami Y, Favoreto S Jr, Zhen G, Park SW, Nguyenvu LT, Kuperman DA, Dolganov GM, Huang X, Boushey HA, Avila PC, Erle DJ (2008) The epithelial anion transporter pendrin is induced by allergy and rhinovirus infection, regulates airway surface liquid, and increases airway reactivity and inflammation in an asthma model. J Immunol 181(3):2203–2210
Nisapakultorn K, Ross KF, Herzberg MC (2001a) Calprotectin expression in vitro by oral epithelial cells confers resistance to infection by Porphyromonas gingivalis. Infect Immun 69(7):4242–4247
Nisapakultorn K, Ross KF, Herzberg MC (2001b) Calprotectin expression inhibits bacterial binding to mucosal epithelial cells. Infect Immun 69(6):3692–3696
Niyonsaba F, Iwabuchi K, Someya A, Hirata M, Matsuda H, Ogawa H, Nagaoka I (2002) A cathelicidin family of human antibacterial peptide LL-37 induces mast cell chemotaxis. Immunology 106(1):20–26
Nordahl EA, Rydengard V, Nyberg P, Nitsche DP, Morgelin M, Malmsten M, Bjorck L, Schmidtchen A (2004) Activation of the complement system generates antibacterial peptides. Proc Natl Acad Sci USA 101(48):16879–16884
Oram JD, Reiter B (1968) Inhibition of bacteria by lactoferrin and other iron-chelating agents. Biochim Biophys Acta 170(2):351–365
Painter RG, Valentine VG, Lanson NA Jr, Leidal K, Zhang Q, Lombard G, Thompson C, Viswanathan A, Nauseef WM, Wang G, Wang G (2006) CFTR Expression in human neutrophils and the phagolysosomal chlorination defect in cystic fibrosis. Biochemistry (Mosc) 45(34):10260–10269
Painter RG, Bonvillain RW, Valentine VG, Lombard GA, LaPlace SG, Nauseef WM, Wang G (2008) The role of chloride anion and CFTR in killing of Pseudomonas aeruginosa by normal and CF neutrophils. J Leukoc Biol 83(6):1345–1353
Painter RG, Marrero L, Lombard GA, Valentine VG, Nauseef WM, Wang G (2010) CFTR-mediated halide transport in phagosomes of human neutrophils. J Leukoc Biol 87(5):933–942
Pazgier M, Hoover DM, Yang D, Lu W, Lubkowski J (2006) Human beta-defensins. Cell Mol Life Sci 63(11):1294–1313
Pedemonte N, Caci E, Sondo E, Caputo A, Rhoden K, Pfeffer U, Di Candia M, Bandettini R, Ravazzolo R, Zegarra-Moran O, Galietta LJ (2007) Thiocyanate transport in resting and IL-4-stimulated human bronchial epithelial cells: role of pendrin and anion channels. J Immunol 178(8):5144–5153
Pezzulo AA, Tang XX, Hoegger MJ, Abou Alaiwa MH, Ramachandran S, Moninger TO, Karp PH, Wohlford-Lenane CL, Haagsman HP, van Eijk M, Banfi B, Horswill AR, Stoltz DA, McCray PB Jr, Welsh MJ, Zabner J (2012) Reduced airway surface pH impairs bacterial killing in the porcine cystic fibrosis lung. Nature 487(7405):109–113
Phalipon A, Cardona A, Kraehenbuhl JP, Edelman L, Sansonetti PJ, Corthesy B (2002) Secretory component: a new role in secretory IgA-mediated immune exclusion in vivo. Immunity 17(1):107–115
Poeck H, Ruland J (2011) From virus to inflammation: mechanisms of RIG-I-induced IL-1beta production. Eur J Cell Biol 91(1):59–64
Poeck H, Bscheider M, Gross O, Finger K, Roth S, Rebsamen M, Hannesschlager N, Schlee M, Rothenfusser S, Barchet W, Kato H, Akira S, Inoue S, Endres S, Peschel C, Hartmann G, Hornung V, Ruland J (2010) Recognition of RNA virus by RIG-I results in activation of CARD9 and inflammasome signaling for interleukin 1 beta production. Nat Immunol 11(1):63–69
Pons G, Marchand MC, d’Athis P, Sauvage E, Foucard C, Chaumet-Riffaud P, Sautegeau A, Navarro J, Lenoir G (2000) French multicenter randomized double-blind placebo-controlled trial on nebulized amiloride in cystic fibrosis patients. The Amiloride-AFLM Collaborative Study Group. Pediatr Pulmonol 30(1):25–31
Porter EM, van Dam E, Valore EV, Ganz T (1997) Broad-spectrum antimicrobial activity of human intestinal defensin 5. Infect Immun 65(6):2396–2401
Poschet J, Perkett E, Deretic V (2002) Hyperacidification in cystic fibrosis: links with lung disease and new prospects for treatment. Trends Mol Med 8(11):512–519
Quinton PM (2008) Cystic fibrosis: impaired bicarbonate secretion and mucoviscidosis. Lancet 372(9636):415–417
Ramsey BW, Gore EJ, Smith AL, Cooney MK, Redding GJ, Foy H (1989) The effect of respiratory viral infections on patients with cystic fibrosis. Am J Dis Child 143(6):662–668
Redl B, Wojnar P, Ellemunter H, Feichtinger H (1998) Identification of a lipocalin in mucosal glands of the human tracheobronchial tree and its enhanced secretion in cystic fibrosis. Lab Invest 78(9):1121–1129
Robert R, Thoreau V, Norez C, Cantereau A, Kitzis A, Mettey Y, Rogier C, Becq F (2004) Regulation of the cystic fibrosis transmembrane conductance regulator channel by beta-adrenergic agonists and vasoactive intestinal peptide in rat smooth muscle cells and its role in vasorelaxation. J Biol Chem 279(20):21160–21168
Robert R, Norez C, Becq F (2005) Disruption of CFTR chloride channel alters mechanical properties and cAMP-dependent Cl- transport of mouse aortic smooth muscle cells. J Physiol 568(Pt 2):483–495
Rogan MP, Reznikov LR, Pezzulo AA, Gansemer ND, Samuel M, Prather RS, Zabner J, Fredericks DC, McCray PB Jr, Welsh MJ, Stoltz DA (2010) Pigs and humans with cystic fibrosis have reduced insulin-like growth factor 1 (IGF1) levels at birth. Proc Natl Acad Sci USA 107(47):20571–20575
Rogers CS, Hao Y, Rokhlina T, Samuel M, Stoltz DA, Li Y, Petroff E, Vermeer DW, Kabel AC, Yan Z, Spate L, Wax D, Murphy CN, Rieke A, Whitworth K, Linville ML, Korte SW, Engelhardt JF, Welsh MJ, Prather RS (2008a) Production of CFTR-null and CFTR-DeltaF508 heterozygous pigs by adeno-associated virus-mediated gene targeting and somatic cell nuclear transfer. J Clin Invest 118(4):1571–1577
Rogers CS, Stoltz DA, Meyerholz DK, Ostedgaard LS, Rokhlina T, Taft PJ, Rogan MP, Pezzulo AA, Karp PH, Itani OA, Kabel AC, Wohlford-Lenane CL, Davis GJ, Hanfland RA, Smith TL, Samuel M, Wax D, Murphy CN, Rieke A, Whitworth K, Uc A, Starner TD, Brogden KA, Shilyansky J, McCray PB Jr, Zabner J, Prather RS, Welsh MJ (2008b) Disruption of the CFTR gene produces a model of cystic fibrosis in newborn pigs. Science 321(5897):1837–1841
Rommens JM, Iannuzzi MC, Kerem B-S, Drumm ML, Melmer G, Dean M, Rozmahel R, Cole JL, Kennedy D, Hidaka N, Zsiga M, Buchwald M, Riordan JR, Tsui L-C, Collins FS (1989) Identification of the cystic fibrosis gene: chromosome walking and jumping. Science 245:1059–1065
Rosenfeld M, Gibson RL, McNamara S, Emerson J, Burns JL, Castile R, Hiatt P, McCoy K, Wilson CB, Inglis A, Smith A, Martin TR, Ramsey BW (2001) Early pulmonary infection, inflammation, and clinical outcomes in infants with cystic fibrosis. Pediatr Pulmonol 32(5):356–366
Rosenthal MD, Gordon MN, Buescher ES, Slusser JH, Harris LK, Franson RC (1995) Human neutrophils store type II 14-kDa phospholipase A2 in granules and secrete active enzyme in response to soluble stimuli. Biochem Biophys Res Commun 208(2):650–656
Rowe SM, Miller S, Sorscher EJ (2005) Cystic fibrosis. N Engl J Med 352(19):1992–2001
Ryckman C, Vandal K, Rouleau P, Talbot M, Tessier PA (2003) Proinflammatory activities of S100: proteins S100A8, S100A9, and S100A8/A9 induce neutrophil chemotaxis and adhesion. J Immunol 170(6):3233–3242
Saitoh H, Masuda T, Shimura S, Fushimi T, Shirato K (2001) Secretion and gene expression of secretory leukocyte protease inhibitor by human airway submucosal glands. Am J Physiol Lung Cell Mol Physiol 280(1):L79–L87
Salinas D, Haggie PM, Thiagarajah JR, Song Y, Rosbe K, Finkbeiner WE, Nielson DW, Verkman AS (2005) Submucosal gland dysfunction as a primary defect in cystic fibrosis. FASEB J 19(3):431–433
Sallenave JM (2010) Secretory leukocyte protease inhibitor and elafin/trappin-2: versatile mucosal antimicrobials and regulators of immunity. Am J Respir Cell Mol Biol 42(6):635–643
Sallenave JM, Shulmann J, Crossley J, Jordana M, Gauldie J (1994) Regulation of secretory leukocyte proteinase inhibitor (SLPI) and elastase-specific inhibitor (ESI/elafin) in human airway epithelial cells by cytokines and neutrophilic enzymes. Am J Respir Cell Mol Biol 11(6):733–741
Sallenave JM, Si Tahar M, Cox G, Chignard M, Gauldie J (1997) Secretory leukocyte proteinase inhibitor is a major leukocyte elastase inhibitor in human neutrophils. J Leukoc Biol 61(6):695–702
Salvatore F, Scudiero O, Castaldo G (2002) Genotype-phenotype correlation in cystic fibrosis: the role of modifier genes. Am J Med Genet 111(1):88–95
Sano H, Sohma H, Muta T, Nomura S, Voelker DR, Kuroki Y (1999) Pulmonary surfactant protein A modulates the cellular response to smooth and rough lipopolysaccharides by interaction with CD14. J Immunol 163(1):387–395
Sano H, Nagai K, Tsutsumi H, Kuroki Y (2003) Lactoferrin and surfactant protein A exhibit distinct binding specificity to F protein and differently modulate respiratory syncytial virus infection. Eur J Immunol 33(10):2894–2902
Sato M, Sano H, Iwaki D, Kudo K, Konishi M, Takahashi H, Takahashi T, Imaizumi H, Asai Y, Kuroki Y (2003) Direct binding of Toll-like receptor 2 to zymosan, and zymosan-induced NF-kappa B activation and TNF-alpha secretion are down-regulated by lung collectin surfactant protein A. J Immunol 171(1):417–425
Schutte BC, McCray PB Jr (2002) [beta]-defensins in lung host defense. Annu Rev Physiol 64:709–748
Selsted ME, Szklarek D, Ganz T, Lehrer RI (1985) Activity of rabbit leukocyte peptides against Candida albicans. Infect Immun 49(1):202–206
Sibley CD, Surette MG (2011) The polymicrobial nature of airway infections in cystic fibrosis: Cangene Gold Medal Lecture. Can J Microbiol 57(2):69–77
Simpson AJ, Maxwell AI, Govan JR, Haslett C, Sallenave JM (1999) Elafin (elastase-specific inhibitor) has anti-microbial activity against gram-positive and gram-negative respiratory pathogens. FEBS Lett 452(3):309–313
Singh PK, Jia HP, Wiles K, Hesselberth J, Liu L, Conway BA, Greenberg EP, Valore EV, Welsh MJ, Ganz T, Tack BF, McCray PB Jr (1998) Production of beta-defensins by human airway epithelia. Proc Natl Acad Sci USA 95(25):14961–14966
Singh PK, Schaefer AL, Parsek MR, Moninger TO, Welsh MJ, Greenberg EP (2000a) Quorum-sensing signals indicate that cystic fibrosis lungs are infected with bacterial biofilms. Nature 407(6805):762–764
Singh PK, Tack BF, McCray PB Jr, Welsh MJ (2000b) Synergistic and additive killing by antimicrobial factors found in human airway surface liquid. Am J Physiol Lung Cell Mol Physiol 279(5):L799–L805
Smith JJ, Welsh MJ (1992) cAMP stimulates bicarbonate secretion across normal, but not cystic fibrosis airway epithelia. J Clin Invest 89(4):1148–1153
Smith JJ, Travis SM, Greenberg EP, Welsh MJ (1996) Cystic fibrosis airway epithelia fail to kill bacteria because of abnormal airway surface fluid. Cell 85(2):229–236
Sohnle PG, Collins-Lech C, Wiessner JH (1991) The zinc-reversible antimicrobial activity of neutrophil lysates and abscess fluid supernatants. J Infect Dis 164(1):137–142
Sohnle PG, Hahn BL, Santhanagopalan V (1996) Inhibition of Candida albicans growth by calprotectin in the absence of direct contact with the organisms. J Infect Dis 174(6):1369–1372
Sonesson A, Ringstad L, Nordahl EA, Malmsten M, Morgelin M, Schmidtchen A (2007) Antifungal activity of C3a and C3a-derived peptides against Candida. Biochim Biophys Acta 1768(2):346–353
Song Y, Salinas D, Nielson DW, Verkman AS (2006) Hyperacidity of secreted fluid from submucosal glands in early cystic fibrosis. Am J Physiol Cell Physiol 290(3):C741–C749
Sorensen O, Arnljots K, Cowland JB, Bainton DF, Borregaard N (1997) The human antibacterial cathelicidin, hCAP-18, is synthesized in myelocytes and metamyelocytes and localized to specific granules in neutrophils. Blood 90(7):2796–2803
Starner TD, Barker CK, Jia HP, Kang Y, McCray PB Jr (2003) CCL20 is an inducible product of human airway epithelia with innate immune properties. Am J Respir Cell Mol Biol 29(5):627–633
Steinbakk M, Naess-Andresen CF, Lingaas E, Dale I, Brandtzaeg P, Fagerhol MK (1990) Antimicrobial actions of calcium binding leucocyte L1 protein, calprotectin. Lancet 336(8718):763–765
Stoltz DA, Meyerholz DK, Pezzulo AA, Ramachandran S, Rogan MP, Davis GJ, Hanfland RA, Wohlford-Lenane C, Dohrn CL, Bartlett JA, Nelson GA, Chang EH, Taft PJ, Ludwig PS, Estin M, Hornick EE, Launspach JL, Samuel M, Rokhlina T, Karp PH, Ostedgaard LS, Uc A, Starner TD, Horswill AR, Brogden KA, Prather RS, Richter SS, Shilyansky J, McCray PB Jr, Zabner J, Welsh MJ (2010) Cystic fibrosis pigs develop lung disease and exhibit defective bacterial eradication at birth. Sci Transl Med 2(29):29ra31
Strunk T, Richmond P, Prosser A, Simmer K, Levy O, Burgner D, Currie A (2011) Method of bacterial killing differentially affects the human innate immune response to Staphylococcus epidermidis. Innate Immun 17(6):508–516
Sun X, Yan Z, Yi Y, Li Z, Lei D, Rogers CS, Chen J, Zhang Y, Welsh MJ, Leno GH, Engelhardt JF (2008) Adeno-associated virus-targeted disruption of the CFTR gene in cloned ferrets. J Clin Invest 118(4):1578–1583
Sun X, Sui H, Fisher JT, Yan Z, Liu X, Cho HJ, Joo NS, Zhang Y, Zhou W, Yi Y, Kinyon JM, Lei-Butters DC, Griffin MA, Naumann P, Luo M, Ascher J, Wang K, Frana T, Wine JJ, Meyerholz DK, Engelhardt JF (2010) Disease phenotype of a ferret CFTR-knockout model of cystic fibrosis. J Clin Invest 120(9):3149–3160
Sutanto EN, Kicic A, Foo CJ, Stevens PT, Mullane D, Knight DA, Stick SM (2011) Innate inflammatory responses of pediatric cystic fibrosis airway epithelial cells: effects of nonviral and viral stimulation. Am J Respir Cell Mol Biol 44(6):761–767
Tabcharani JA, Rommens JM, Hou YX, Chang XB, Tsui LC, Riordan JR, Hanrahan JW (1993) Multi-ion pore behaviour in the CFTR chloride channel. Nature 366(6450):79–82
Taggart CC, Greene CM, Smith SG, Levine RL, McCray PB Jr, O’Neill S, McElvaney NG (2003) Inactivation of human beta-defensins 2 and 3 by elastolytic cathepsins. J Immunol 171(2):931–937
Tan HL, Regamey N, Brown S, Bush A, Lloyd CM, Davies JC (2011) The th17 pathway in cystic fibrosis lung disease. Am J Respir Crit Care Med 184(2):252–258
Tenner AJ, Robinson SL, Borchelt J, Wright JR (1989) Human pulmonary surfactant protein (SP-A), a protein structurally homologous to C1q, can enhance FcR- and CR1-mediated phagocytosis. J Biol Chem 264(23):13923–13928
Territo MC, Ganz T, Selsted ME, Lehrer R (1989) Monocyte-chemotactic activity of defensins from human neutrophils. J Clin Invest 84(6):2017–2020
Tjabringa GS, Ninaber DK, Drijfhout JW, Rabe KF, Hiemstra PS (2006) Human cathelicidin LL-37 is a chemoattractant for eosinophils and neutrophils that acts via formyl-peptide receptors. Int Arch Allergy Immunol 140(2):103–112
Travis SM, Conway BA, Zabner J, Smith JJ, Anderson NN, Singh PK, Greenberg EP, Welsh MJ (1999) Activity of abundant antimicrobials of the human airway. Am J Respir Cell Mol Biol 20(5):872–879
Triebel S, Blaser J, Reinke H, Tschesche H (1992) A 25 kDa alpha 2-microglobulin-related protein is a component of the 125 kDa form of human gelatinase. FEBS Lett 314(3):386–388
Turner J, Cho Y, Dinh NN, Waring AJ, Lehrer RI (1998) Activities of LL-37, a cathelin-associated antimicrobial peptide of human neutrophils. Antimicrob Agents Chemother 42(9):2206–2214
Valore EV, Park CH, Quayle AJ, Wiles KR, McCray PB Jr, Ganz T (1998) Human beta-defensin-1: an antimicrobial peptide of urogenital tissues. J Clin Invest 101(8):1633–1642
van Iwaarden F, Welmers B, Verhoef J, Haagsman HP, van Golde LM (1990) Pulmonary surfactant protein A enhances the host-defense mechanism of rat alveolar macrophages. Am J Respir Cell Mol Biol 2(1):91–98
van Iwaarden JF, van Strijp JA, Ebskamp MJ, Welmers AC, Verhoef J, van Golde LM (1991) Surfactant protein A is opsonin in phagocytosis of herpes simplex virus type 1 by rat alveolar macrophages. Am J Physiol 261(2 Pt 1):L204–L209
Van Iwaarden JF, Pikaar JC, Storm J, Brouwer E, Verhoef J, Oosting RS, van Golde LM, van Strijp JA (1994) Binding of surfactant protein A to the lipid A moiety of bacterial lipopolysaccharides. Biochem J 303(Pt 2):407–411
van’t Hof W, Blankenvoorde MF, Veerman EC, Amerongen AV (1997) The salivary lipocalin von Ebner’s gland protein is a cysteine proteinase inhibitor. J Biol Chem 272(3):1837–1841
Vandal K, Rouleau P, Boivin A, Ryckman C, Talbot M, Tessier PA (2003) Blockade of S100A8 and S100A9 suppresses neutrophil migration in response to lipopolysaccharide. J Immunol 171(5):2602–2609
Vandebrouck C, Melin P, Norez C, Robert R, Guibert C, Mettey Y, Becq F (2006) Evidence that CFTR is expressed in rat tracheal smooth muscle cells and contributes to bronchodilation. Respir Res 7:113
Vanthanouvong V, Kozlova I, Johannesson M, Naas E, Nordvall SL, Dragomir A, Roomans GM (2006) Composition of nasal airway surface liquid in cystic fibrosis and other airway diseases determined by X-ray microanalysis. Microsc Res Tech 69(4):271–276
Weinrauch Y, Elsbach P, Madsen LM, Foreman A, Weiss J (1996) The potent anti-Staphylococcus aureus activity of a sterile rabbit inflammatory fluid is due to a 14-kD phospholipase A2. J Clin Invest 97(1):250–257
Weiss J, Elsbach P, Olsson I, Odeberg H (1978) Purification and characterization of a potent bactericidal and membrane active protein from the granules of human polymorphonuclear leukocytes. J Biol Chem 253(8):2664–2672
Weldon S, McNally P, McElvaney NG, Elborn JS, McAuley DF, Wartelle J, Belaaouaj A, Levine RL, Taggart CC (2009) Decreased levels of secretory leucoprotease inhibitor in the Pseudomonas-infected cystic fibrosis lung are due to neutrophil elastase degradation. J Immunol 183(12):8148–8156
Welsh MJ, Ramsey BW, Accurso F, Cutting GR (2001) Cystic fibrosis. In: Scriver CR, Beaudet AL, Sly WS, Valle D, Childs B, Vogelstein B (eds) The metabolic and molecular basis of inherited disease, vol 3, 8th edn. McGraw-Hill, New York, NY, pp 5121–5189
Wijkstrom-Frei C, El-Chemaly S, Ali-Rachedi R, Gerson C, Cobas MA, Forteza R, Salathe M, Conner GE (2003) Lactoperoxidase and human airway host defense. Am J Respir Cell Mol Biol 29(2):206–212
Wilkinson TS, Dhaliwal K, Hamilton TW, Lipka AF, Farrell L, Davidson DJ, Duffin R, Morris AC, Haslett C, Govan JR, Gregory CD, Sallenave JM, Simpson AJ (2009) Trappin-2 promotes early clearance of Pseudomonas aeruginosa through CD14-dependent macrophage activation and neutrophil recruitment. Am J Pathol 174(4):1338–1346
Wine JJ, Joo NS (2004) Submucosal glands and airway defense. Proc Am Thorac Soc 1(1):47–53
Wright FA, Strug LJ, Doshi VK, Commander CW, Blackman SM, Sun L, Berthiaume Y, Cutler D, Cojocaru A, Collaco JM, Corey M, Dorfman R, Goddard K, Green D, Kent JW Jr, Lange EM, Lee S, Li W, Luo J, Mayhew GM, Naughton KM, Pace RG, Pare P, Rommens JM, Sandford A, Stonebraker JR, Sun W, Taylor C, Vanscoy LL, Zou F, Blangero J, Zielenski J, O'Neal WK, Drumm ML, Durie PR, Knowles MR, Cutting GR (2011) Genome-wide association and linkage identify modifier loci of lung disease severity in cystic fibrosis at 11p13 and 20q13.2. Nat Genet 43(6):539–546
Xu YY, Samaranayake YH, Samaranayake LP, Nikawa H (1999) In vitro susceptibility of Candida species to lactoferrin. Med Mycol 37(1):35–41
Yang D, Chertov O, Bykovskaia SN, Chen Q, Buffo MJ, Shogan J, Anderson M, Schroder JM, Wang JM, Howard OM, Oppenheim JJ (1999) Beta-defensins: linking innate and adaptive immunity through dendritic and T cell CCR6. Science 286(5439):525–528
Yang D, Chen Q, Schmidt AP, Anderson GM, Wang JM, Wooters J, Oppenheim JJ, Chertov O (2000) LL-37, the neutrophil granule- and epithelial cell-derived cathelicidin, utilizes formyl peptide receptor-like 1 (FPRL1) as a receptor to chemoattract human peripheral blood neutrophils, monocytes, and T cells. J Exp Med 192(7):1069–1074
Yang D, Chen Q, Hoover DM, Staley P, Tucker KD, Lubkowski J, Oppenheim JJ (2003) Many chemokines including CCL20/MIP-3alpha display antimicrobial activity. J Leukoc Biol 74(3):448–455
Yoneyama M, Kikuchi M, Natsukawa T, Shinobu N, Imaizumi T, Miyagishi M, Taira K, Akira S, Fujita T (2004) The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat Immunol 5(7):730–737
Yoshida M, Korfhagen TR, Whitsett JA (2001) Surfactant protein D regulates NF-kappa B and matrix metalloproteinase production in alveolar macrophages via oxidant-sensitive pathways. J Immunol 166(12):7514–7519
Zabner J, Smith JJ, Karp PH, Widdicombe JH, Welsh MJ (1998) Loss of CFTR chloride channels alters salt absorption by cystic fibrosis airway epithelia in vitro. Mol Cell 2(3):397–403
Zallen G, Moore EE, Johnson JL, Tamura DY, Barkin M, Stockinger H, Silliman CC (1998) New mechanisms by which secretory phospholipase A2 stimulates neutrophils to provoke the release of cytotoxic agents. Arch Surg 133(11):1229–1233
Zhang Y, Li X, Grassme H, Doring G, Gulbins E (2010) Alterations in ceramide concentration and pH determine the release of reactive oxygen species by Cftr-deficient macrophages on infection. J Immunol 184(9):5104–5111
Zheng S, De BP, Choudhary S, Comhair SA, Goggans T, Slee R, Williams BR, Pilewski J, Haque SJ, Erzurum SC (2003) Impaired innate host defense causes susceptibility to respiratory virus infections in cystic fibrosis. Immunity 18(5):619–630
Zhou HD, Li XL, Li GY, Zhou M, Liu HY, Yang YX, Deng T, Ma J, Sheng SR (2008) Effect of SPLUNC1 protein on the Pseudomonas aeruginosa and Epstein-Barr virus. Mol Cell Biochem 309(1–2):191–197
Acknowledgements
This work was supported by P01 HL-51670, P01 HL-091842, and the Roy J. Carver Charitable Trust. We thank Erin Burnight, Jeydith Gutierrez, and Sateesh Krishnamurthy for critically reviewing the manuscript. We thank Tom Moninger and David Meyerholz for assistance in preparing photomicrographs.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer Basel AG
About this chapter
Cite this chapter
Bartlett, J.A., McCray, P.B. (2013). Cystic Fibrosis and Defective Airway Innate Immunity. In: Hiemstra, P., Zaat, S. (eds) Antimicrobial Peptides and Innate Immunity. Progress in Inflammation Research. Springer, Basel. https://doi.org/10.1007/978-3-0348-0541-4_11
Download citation
DOI: https://doi.org/10.1007/978-3-0348-0541-4_11
Published:
Publisher Name: Springer, Basel
Print ISBN: 978-3-0348-0540-7
Online ISBN: 978-3-0348-0541-4
eBook Packages: MedicineMedicine (R0)