
Abstract
Herein we review the state of knowledge regarding the role of IL-17 in tuberculosis (TB). IL-17 is induced following exposure to mycobacteria in mice and humans, yet its role in protection and the immunopathologic consequences of infection is not defined. It appears that the IL-17 response to mycobacterial infection is dependent on IL-23 and that the induction of this cytokine is dependent upon pathogen-associated molecules on the surface of the mycobacterium. While there is evidence for a protective role, there is also evidence for a pathologic role; it is therefore critical to decipher the impact of IL-17 so that preventive and therapeutic measures can be appropriately tailored.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
There are two main reasons for studying IL-17 and tuberculosis. The first is to determine whether IL-17 is important in disease. The second is that the dynamic interaction between host and this unique pathogen may allow us to identify as yet unknown functions for this cytokine. Therefore, when considering the role of IL-17 in tuberculosis, the ability of this cytokine to mediate and regulate antibacterial activity as well as its ability to impact the inflammatory response should be determined. In addition, the bacterial and host factors that mediate induction and regulation of this cytokine should be addressed.
Tuberculosis is a disease resulting from the interaction of a persistent yet inflammatory intracellular bacterium and a responsive but regulated immune response [1–3]. While cellular immunity is essential to control bacterial growth, the concurrent development of inflammation leads to tissue damage that then promotes dissemination. The acquired cellular response consists of antigen-specific effector T cells and B cells, and while the requirement for T cells in control of infection is clear, the role of B cells and immunoglobulin is less so [3]. From both animal studies and patient data, it appears that CD4 T cells are the primary mediators of protection and that the IL-12/IFN-γ axis is critical to controlling disease [1–3]. CD8 T cells have the ability to protect the host from disease and have unique antibacterial activity, but the absence of these cells can be overcome by the presence of CD4 T cells [1–3]. There is active regulation of the acquired immune response, and it is likely that this serves to limit not only antibacterial activity but also tissue damage during chronic disease [4, 5]. Taking into account the known functions of IL-17, this cytokine could impact the innate, acquired, and inflammatory response during TB.
2 Evidence for a Role of IL-17 in TB
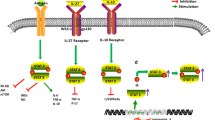
Support for a role of IL-17 in disease is provided by the fact that mRNA and cellular analyses demonstrate that IL-17 is induced following infection of mice with Mycobacterium tuberculosis (Mtb) [6]. More importantly, antigen-specific IL-17-producing cells can be detected in both exposed individuals and TB patients thus providing substantial impetus for investigation of this cytokine [7]. In mice, IL-17 production occurs rapidly upon a high-dose intratracheal challenge with Mycobacterium bovis BCG (BCG), and this response is dependent upon the γδ T cell population [8]. This population is also a major producer of IL-17 in response to Mtb infection in the lung [9]. In the absence of the early γδ T cell-derived IL-17 response to BCG, there is a reduced early neutrophil and reduced later mononuclear response in the lung [8] (see Fig. 1a). In Mtb infection of IL-23-deficient mice, there is very little IL-17 mRNA induction in the lung, and in this model the inflammatory response is altered [6]. It is tempting to speculate that the disrupted granulomatous response previously reported for Mtb-infected γδ T cell-deficient mice [10] may relate to the loss of an early IL-17 response. The presence of IL-17-producing γδ T cells is particularly interesting in light of the demonstration that unrestrained IL-17-producing γδ T cells can lead to tissue damage and death in a fungal model of infection [11]. Indeed, regulation of these IL-17-producing γδ T cells is superoxide dependent [11], and in the absence of superoxide, there is an increase in neutrophil accumulation in the TB granuloma in mice [12] suggesting that the ability of IL-17-producing γδ T cells to recruit neutrophils may also be regulated by superoxide. There are other innate sources of IL-17 in the lung such as invariant NK T cells [13], but the role of this response in TB has not been addressed.

Functions of IL-17 during Mtb infection. (a) Upon mycobacterial infection in the lung, phagocytes respond to the bacteria, generate IL-23, and promote γδ T cells to make IL-17 and thereby mediate recruitment of neutrophils. Absence of this response limits early neutrophil recruitment and alters later mononuclear accumulation. (b) Vaccine-induced memory T cells, expressing the tissue-homing chemokine receptor CCR4, recognize infected lung phagocytes and respond by producing or inducing CXCR9, CXCR10, and CXCR11 in the lung. These chemokines then recruit memory Th1 cells that mediate accelerated control of bacterial growth. (c) The dynamic development of the granuloma is altered in the absence of IL-23 and IL-17; IFN and IL-17 appear to counterregulate the development of a mononuclear or granulocytic inflammation in response to Mtb
3 Evidence for an Antigen-Specific IL-17 Response in TB
Recent thinking posits that IL-17 is an ancient cytokine that acts to bridge the gap between innate and acquired immunity [14, 15]. In TB it appears that IL-17 is present both during the innate and acquired response. This has been shown in mice [6, 9, 16] and more recently in humans [7]. Whether the acquired IL-17 response depends upon the innate IL-17 response is not yet clear. The antigen-specific IL-17-producing T cell response is induced with the same kinetics in the mouse as the IFN-γ-producing antigen-specific response but at about a five- to tenfold lower number of cells [6, 16]. Interestingly, despite the reports of IFN-γ and IL-17 responses counterregulating each other and the fact that the IL-17 response is regulated by IFN-γ following BCG infection [17], the antigen-specific IL-17 response is maintained in the lung during Mtb infection despite the presence of a strong IFN-γ response [6]. The accepted model for induction of IL-17-producing T cells (Th17) has a requirement for TGFβ to initiate the polarization process in both mice and humans [15, 18] and that inflammatory cytokines, such as IL-6, act to direct the cells to a Th17 rather than regulatory, FoxP3-expressing phenotype (Treg) [19]. IL-23 is thought then to promote the release of IL-17 by polarized cells [20] (see Fig. 2). IFN-γ limits the amount of IL-23 made by dendritic cells infected with mycobacteria [17], and this may be one way IFN-γ-producing T cells (Th1) regulate the Th17 response. In mycobacterial models, however, IL-1β in conjunction with IL-23 can drive IL-17 from γδ T cells (and CD4+ cells) without the need for TCR engagement [21, 22]. Interestingly, IFN inhibits IL-1β production by mouse cells but enhances it for human cells [22], and thus, the impact of IFN on IL-17 regulation in mice and humans may differ.

Polarization of Th17 cells during mycobacterial infection. TGFβ drives induction of the transcription factor RORγt, if no inflammation is present, cells will tend toward a regulatory phenotype. If mycobacteria are present, however, IL-6 will induce IL-21 which in turn promotes more RORγt and expression of the IL-23R and thereby responsiveness to IL-23. Naïve cells polarized with these cytokines can then produce both IL-17 and IL-22. Induction of the Th1 transcription factor T-bet in naïve T cells conversely promotes expression of the IL-12Rβ2 molecule which allows the cells to respond to IL-12p70 and produce IFN-γ. The IL-12Rβ1 component of both IL-12R and IL-23R requires different levels of stimulus to be expressed. If IL-27 is present, then STAT1 is activated and this reduces the impact of IL-6 on the polarizing T cells and reduces Th17 development. Both IL-12 and IL-23 appear to be required for long-term persistence of Th1 and Th17 cells respectively during Mtb infection. BCG and Mtb interact with the antigen-presenting cell to define the conditions that pertain during activation of the naïve T cells. In some cases memory T cells and γδT can become IL-17 producers without TCR ligation in the presence of IL-1β and IL-23
It is likely that the nature of phagocyte activation and the local cytokine environment will determine the polarization of T cells both during priming and expression of effector function. These conditions will be defined to some degree by the impact of mycobacterial molecules on phagocyte pattern recognition receptors.
4 When and Where Is IL-17 Expressed During Human Disease?
Following the original demonstration of antigen-specific IL-17-producing CD4+ T cells in PPD-positive individuals [7], many further studies have examined IL-17 production in both active and latent human tuberculosis. In the original publication, IL-17-, IFN-, and IL-22-producing cells were detected in the peripheral blood of patients with active TB; however, only IFN- and IL-22-producing cells were present in the bronchoalveolar lavage; it was suggested that IFN-γ limits the IL-17 response in the lungs [7]. In several recent studies, patients with active TB have been shown to have a stronger IFN response to mycobacterial antigens than do those with latent disease; however, the IL-17 response is less clearly associated with disease state [23, 24]. There is however a tendency for the IL-17 response to antigen to be higher in latent than active TB patients [23, 25] possibly due to the higher IFN response in the active cases inhibiting IL-17. While one study was able to correlate decreased IL-17 levels in serum during treatment with increased risk of death [26], a second study could not correlate IL-17 in plasma during treatment to disease state or response to treatment [27]. In the mouse model, a drop in IL-17 was found to occur during effective treatment [28].
The potential for patients to generate an IL-17 response following exposure or during disease seems to be associated with geographical location and/or the nature of the disease. In one study of a Swiss population, IL-17 was not detected strongly in response to purified protein derivative (PPD) of Mtb; cells that did respond were CD45RA−, CD25−, CCR6+, and CXCR3+ and produced mainly IFN-γ [29]. In contrast, in a South African population, the BCG-reactive cells were CD45RA− cells and those making IL-17 or IL-22 were predominantly CCR7+ and CD27+/−, whereas those making IFN-γ were predominantly CCR7− and CD27− [7]. In Chinese studies of cells from pleural effusions, IL-17 was produced by CCR7+, CD27+, CD4 T cells but only after restimulation [30, 31]. Again in contrast, a South African study of both pleural and pericardial effusions found very low to negligible IL-17 in these effusions although high levels of IL-22 and IFN were detected; a low level of CD4+ cells capable of making IL-17 was also detected, but these did not appear to be specific for mycobacterial antigen [32].
5 Can IL-17 Mediate Protection?
IL-17 acts to regulate granulocyte homeostasis [33], chemokine expression and cell recruitment [34, 35] and to orchestrate germinal center formation [36]. IL-17 is required for protection against Klebsiella pneumoniae [34], Citrobacter rodentium [37], Candida albicans [38], and Mycoplasma pulmonis [39]. IL-17 has also been extensively implicated as a mediator of autoimmune-mediated disease (reviewed in [20]). These myriad functions make it difficult to immediately identify where within the complex interaction between host and bacteria IL-17 is likely to act. In revising this chapter for 2012, I had thought to be able to more definitively answer the question raised in the title; however, the primary role of IL-17 in protection against Mtb remains unclear. Following low-dose aerosol delivery of Mtb to IL-23-deficient mice, there is no difference in the level of bacterial burden within the lung for the first 90 days of infection, this despite the fact that there is very little IL-17 mRNA or Th17 response detected [6]. Similarly in a BCG model, absence of IL-23 or IL-17 does not alter the bacterial burden [40]. Interestingly, in both these studies, IL-23 could mediate protection in the absence of IL-12, but this was associated with the ability of IL-23 to promote IFN-γ responses [6, 40]. Despite this absence of protective activity, it was possible to improve bacterial control during pulmonary Mtb infection if IL-23 was delivered prior to infection [41] suggesting that the natural IL-23/IL-17 response may be improved upon. In later studies we have shown that, although early control is not affected by the absence of IL-23, later control of bacterial growth following a low-dose challenge is lost. Importantly, this loss of protection does not appear to be related to the loss of IL-17, as bacterial growth is identical between wild-type and IL-17RA-deficient mice in this model over a prolonged period [42, 43]. In stark contrast to the data from low-dose aerosol delivery of Mtb, others have shown increased susceptibility to Mtb infection in mice lacking IL-17A when a moderately higher dose is delivered intratracheally [44]. The difference in bacterial growth between the wild-type and IL-17A-deficient mice following intratracheal infection occurs within the first 4 weeks and is associated with a defective inflammatory response. This differential requirement for IL-17 between the aerosol and intratracheal model is intriguing. It may be that the intratracheal model is more inflammatory and that therefore the cells recruited to the lung are different and more dependent upon IL-17 for effective function. In contrast, the aerosol model is known to induce very little inflammation early, and there may therefore be less of a need for early IL-17. The relevance of either model to natural human disease is a matter for discussion, but it is clear that defining why the two models differ so dramatically will be informative in the dissection of the function of IL-17 in lung disease.
One aspect of the role of IL-17 in protection may be related to its impact on dissemination of bacteria from the site of infection. Recent work showed an increase in IL-17 in the IL-10-deficient mouse, and while depletion of IL-17 had no effect on bacterial growth in the lung, it appeared that dissemination of bacteria to the spleen was increased in the absence of IL-17 [45]. Further, in a study of the dissemination of BCG from the skin to the draining lymph node, it was shown that absence of migratory IL-17-producing γδ T cells restricted the accumulation of BCG in the lymph node [46]. It is possible therefore that IL-17 plays a role in defining the cells which mediate dissemination of the bacteria. As the induction of the acquired response depends upon dissemination of bacteria from the lung to the draining lymph node [3], the kinetics of the acquired response may be delayed in the absence of IL-17. While this may not be critical in the low-dose aerosol model, it may be more so in the higher dose intratracheal model.
In a couple of recent reductionist studies, the ability of T cells making IL-17 to mediate protection in mice infected with Mtb was assessed. One group transferred IFN-deficient, antigen-specific, IL-17-polarized T cells into immune-deficient mice and found that these cells could protect more efficiently than unpolarized, IFN-deficient, antigen-specific cells [47]. In a second study, the ability of cells to protect was dependent upon the cells retaining plasticity and thereby maintaining a mixed rather than fully polarized IL-17 phenotype [48]. These data suggest that while IFN is not essential for protection in these transfer models, IL-17 alone is not sufficient to mediate protection. Further studies are clearly warranted.
Whether IL-23 and IL-17 play a protective role in human disease has not yet been clarified. It is apparent however that patients with defective IL12Rb1 gene function are susceptible to mycobacterial disease, that their T cells are defective in their response to both IL-12 and IL-23 [49, 50], and that IL-23 drives efficient IL-17 responses in human T cells [51]. In view of the mouse data, it is likely that the role of IL-17 in protection against TB will be subtle and therefore very difficult to define in humans; however, this should not deter investigation. In this regard, the association between polymorphisms in the IL23R gene and damaging inflammation [52, 53] provides impetus and potentially useful subjects for analysis. As discussed above, there have been several studies on the expression of IL-17 in healthy-exposed, infected, and diseased subjects, and while there is some evidence that circulating IL-17 is seen in the healthier exposed individuals, there is no strong current evidence linking IL-17 to either protection or disease development.
6 How Does IL-17 Impact Vaccine-Induced Protection?
Despite the absence of a defined role in controlling bacterial burden, the absence of IL-23, and thereby IL-17, does result in reduced protection in a model of immunological memory in mice (see Fig. 1b). In this model, the accelerated memory Th1 response induced by a subunit vaccine is lost in the absence of IL-23, and this is associated with the absence of an IL-17-producing CD4+ memory population in the lungs of vaccinated IL-23-deficient mice [16]. The expression of IL-17 mRNA in vaccinated lungs correlates with the expression of the chemokines CXCL9, CXCL10, and CXCL11, and in the absence of this response, the accelerated accumulation of CXCR3-expressing IFN-γ-producing memory cells fails to occur, and thus, vaccine-induced protection is lost [16]. The vaccine-induced IL-17-producing cells were also shown to be positive for the tissue-homing chemokine receptor CCR4 and capable of populating normal lung tissue. This suggested that memory cells induced by vaccination could populate noninflamed tissue and provide a surveillance function [16]. Further, in the absence of CCR4 using a BCG model, it was found that both IFN and IL-17-producing cells failed to be maintained and that recall granulomatous responses were defective [54].
When IL-23 is added to a DNA vaccine regimen, it is capable of increasing both IFN-γ and IL-17 responses and protection to a degree equivalent to IL-12 [55, 56]. In view of the downregulatory activity of IL-27 with regard to IL-17 cellular responses [57, 58], it is not surprising that this cytokine fails to improve vaccine-induced protection [55]. In contrast to these studies, BCG vaccination did not require IL-23 to be protective against a subsequent BCG challenge [40]; however, as this was a systemic challenge, the need for a surveillance cell would be less evident. It is also the case however that the cross regulation of Th17 and Th1 cells is more apparent following BCG infection; thus, a substantial Th17 response cannot be detected following subcutaneous or systemic delivery of BCG unless the IL-12/IFN-γ axis is absent [17, 56]. A recent study of the mechanism of IL-17 induction during BCG vaccination has shown that BCG induces prostaglandin E2 and that this drives IL-10 which in turn limits the IFN response to BCG [59]. At the same time, however, BCG also induces IL-23 which promotes IL-17 production which in turn limits the IL-10 thereby promoting an overall IFN response; in the absence of IL-10, there is no need for IL-17 to promote the IFN response [59]. Thus, expression of IL-17 during BCG vaccination may promote the IFN response. Studies with genetically manipulated BCG have shown increased protection, and this is associated with an increased IL-17 response to the genetically manipulated BCG compared to the wild-type BCG [60].
Human and animal vaccine studies have increasingly measured IL-17 responses after vaccination. In the bovine model, a viral boost of BCG vaccination showed increased efficacy, and this was associated with an increased antigen-specific IL-17 response prior to challenge [61]. Boosting BCG-vaccinated people with virus-delivered antigen results in multifunctional antigen-specific cells with a preference for IL-17 in adolescents compared to children [62]. A higher dose of the virally delivered antigen promotes more IL-17-producing cells; however, previous exposure may limit the IL-17 response [63]. Detailed analysis of the cells responding to the virally delivered antigen suggests that the IL-17 response may be limited by regulatory T cells that act to limit levels of the inflammatory extracellular ATP [64].
Extensive studies using the whole-blood assay have measured the level of IL-17 to mycobacterial antigens in vaccinated individuals. Vaccination does result in an increased IL-17 production in infants [65], and while age at vaccination could impact size of the IL-17 response, this response evened out as infants aged [66]. The efficacy of BCG differs geographically, and so the whole-blood assay was used to compare responses in UK and Malawian infants receiving BCG. It was found that while the Malawian children had a greater general inflammatory response including IL-17, the UK infants had stronger type 1 responses; it was suggested that these different biosignatures may explain why BCG is more effective in the UK [67]. However, in an extensive study on the potential for a biosignature to predict protective efficacy of BCG vaccination, no difference in the cytokine profile (IL-2, IFN, TNF, or IL-17) or cell types was associated with the development of tuberculosis in vaccinated individuals [68]. It would seem therefore that the role of IL-17 in vaccine studies in humans is still undefined.
7 How Could IL-17 Impact Inflammation?
Dissecting the role of such a pluripotent entity as IL-17 in a chronic disease like TB is difficult. A major confounding factor is that the granulomatous structure, even within the tractable mouse model, is a constantly developing entity [69]. The ability of IL-17 to impact granulocyte homeostasis and accumulation could be a major factor in granuloma formation. IL-17 is likely a mediator of stress-induced granulopoiesis acting via G-CSF [70]; however, in the absence of IL-23 and IL-17, there is a modest decrease in the inflammatory response early in infection [6] (see Fig. 1c). This is in contrast to the impact of IL-17 and IL-23 on the inflammatory response to fungal infection via the mucosal route wherein IL-23 and IL-17 downregulate the protective Th1 response and increase the pyogranulomatous nature of the inflammation [71]. Further, IL-23 and IL-17 improve survival and reduce the killing activity of neutrophils in this fungal model [71]. The potential for neutrophils to impact granuloma formation during Mtb infection [72] and the causal connection between early and enhanced neutrophil recruitment with susceptibility to TB [73] prompted us to examine the role of IL-17 and IL-23 in the Koch phenomenon. We found that repeated delivery of mycobacterial antigen drove an IL-23-dependent IL-17-response in the lungs of Mtb-infected mice that did not impact bacterial control but did increase the pyogranulomatous nature of the lesions [74] (Fig. 1c). In two other recent studies, the impact of IFN in regulating the inflammatory action of IL-17 has been highlighted. In the first study, the absence of the IFNγR on nonhematopoietic cells was found to limit the anti-inflammatory activity of these cells, and this resulted in enhanced IL-17 responses which compromised immunity [75]. In the second study, the absence of IFN in memory cells did not impact their ability to control bacteria but did limit their ability to limit neutrophil recruitment and thereby tissue damage [76] (Fig. 1c). These data demonstrate that IL-17 is associated with the potential for a more damaging inflammatory response to Mtb infection. In this regard, an intriguing paper from Argentina shows that patients with multidrug-resistant tuberculosis and high bacterial burdens had higher levels of IL-17 [77]. This is in contrast to the lack of apparent IL-17 in pleural and pericardial tubercular effusions (see discussion above).
An important aspect of the Mtb granuloma that is often dismissed is the accumulation of B cells in secondary lymphoid structures in both mice and humans [78–80]. Indeed, an altered granulomatous response [81] and reduced protection can be seen following Mtb infection of B cell-deficient mice [82]. The ability of IL-17 to mediate germinal centers [36] may impact the B cell response, as recent studies have identified IL-17 mRNA associated with the B cell follicles in human tuberculosis lesions in the lung [83]. In our recent studies, we have found that the absence of IL-17RA does not greatly inhibit the development of B cell follicles but that IL-23 and CXCL13 seem to be critical for these structures and for T cell recruitment to the granulomatous site [42, 84].
8 Induction and Regulation of the IL-17 Response
IL-23 is a key mediator of IL-17 secretion in memory T cells [85] and γδ T cells [9], and in its absence there is very little IL-17 produced in response to Mtb infection [6]. Mycobacteria can induce IL-23 in dendritic cells [9, 17], and this induction can be regulated by IFN-γ, at least for BCG [17]. Recent studies have implicated dectin-1 in the induction of IL-23 and as a promoter of IL-17-producing CD4 T cells during fungal infection [86]. During mycobacterial infection, dectin-1 has been implicated as a mediator of macrophage [87] and dendritic cell [88] activation with a specific role for dectin-1 in induction of IL-12p40, a subunit of IL-23. Recently, dectin-1 ligation by β-glucan or Mtb on monocyte-derived dendritic cells was reported to result in IFN and IL-17 production in a mixed lymphocyte reaction, whereas ligation of DC-SIGN or the mannose receptor resulted in an IFN response [89, 90]. The role of dectin-1 in stimulating the phagocytic response to fungal bodies is dependent on developmentally regulated display of β-glucan moieties [91, 92]. It is possible therefore that the expression of β-glucan moieties on different mycobacterial species will regulate the amount of IL-23 and therefore the amount of IL-17 they induce. One could hypothesize therefore that while the Th1 response can regulate the Th17 in BCG infection, it is possible that a greater induction of IL-23 by Mtb allows the Th17 response in TB to overcome Th1 regulation [6, 17]. The importance of the early cytokine milieu in defining polarization is highlighted by the fact that the requirement for induction of the il12rb1 gene (a component of both the IL-12 and IL-23 receptor) in naïve T cells is lower for the induction of IL-17-producing cells than it is for IFN-γ-producing cells [93].
Although it is technically difficult to assess the very earliest events during low-dose aerosol infection directly in vivo, the response of CD4 T cells and dendritic cells to vaccination with trehalose dimycolate (TDM, or cord factor) has been studied. TDM is the primary inflammatory mediator of mycobacteria, and alterations in its structure affect the granulomatous response in the lung [94]. In vaccine studies it has been shown that delivery of an IAb-restricted peptide in the presence of TDM and the detoxified monophosphoryl lipid A (MPL) results in early expression of IL-6, TGFβ, and IL-23 in dendritic cells and IL-17 but not IFN-γ in the CD4 T cells [16]. This early IL-17 corresponds with early expression of the IL-23 receptor but not the IL-12 receptor in the CD4 cells [16]. These data suggest that Th17 cells, while not requiring IL-23 to become polarized [18], are able to respond to IL-23 very early during activation. Indeed in the absence of IL-23, there is a small but reproducible reduction in very early proliferation of CD4 T cells [16]. The initiation of Th17 cells during Mtb infection is less well defined, although the absence of IL-23 does ablate the IL-17 response. It is likely that the balance between IL-6 and TGFβ [19] as well as that between IL-1β and IFN during activation of naïve T cells will define the relative induction of a Th17 or Treg population. The importance of this balance is highlighted by the observed ability of Treg cells to limit bacterial control [4, 95, 96]. The receptor for TDM was recently identified as the macrophage-inducible C-type lectin (Mincle) [97, 98], and this molecule acts through the FcRγ and Syk-CARD9 signaling pathway to promote inflammation as well as both IFN and IL-17 responses to mycobacteria [98]. In human studies, antigen-experienced cells make IL-17 in response to Mtb-activated antigen-presenting cells in an IL-23-dependent manner; it appears also that the production of IL-23 can be induced by NK cells recognizing the infected antigen-presenting cells [99].
9 Conclusions
This chapter does not answer the question raised in the title, as the animal model evidence suggests both positive and negative impacts in the TB disease process and the human data is still equivocal. We know however that this cytokine is induced in and expressed by innate and acquired cells in mice and humans and that the expression of this response is dependent upon IL-23. Induction of the IL-17 response is likely to be dependent upon the counterregulation of IL-1β, IL-12, and IL-23 at the dendritic cell level, and this may be related to the level of cytokine-inducing moieties (such as β-glucans) on the mycobacterial surface. There is conflicting evidence on the role of IL-17 in control of bacterial growth during mycobacterial infection, and this conflict may be resolved with careful analysis of the role of dose and concurrent inflammation during initial infection. There are several areas that should be investigated in order to clarify the role of IL-17 in TB. These are the impact of IL-17 on inflammation and tissue damage, the induction and regulation of the IL-17 response, the role of surface molecules of various mycobacterial species in determining the IL-17 response, the impact of IL-23R polymorphisms on TB in humans, the ability of IL-17-producing memory cells to improve vaccine-induced protection, and the impact of IL-17 and IL-23 on neutrophil and B cell function during TB.
References
Flynn J, Chan J (2001) Immunology of tuberculosis. Annu Rev Immunol 19:93–129
North R, Jung Y (2004) Immunity to tuberculosis. Annu Rev Immunol 22:599–623
Cooper AM (2009) Cell mediated immune responses in tuberculosis. Annu Rev Immunol 27:393–422
Scott-Browne J, Shafiani S, Tucker-Heard G, Ishida-Tsubota K, Fontenot J, Rudensky A, Bevan M, Urdahl K (2007) Expansion and function of Foxp3-expressing T regulatory cells during tuberculosis. J Exp Med 204:2159–2169
Koch M, Tucker-Heard G, Perdue N, Killebrew J, Urdahl K, Campbell D (2009) The transcription factor T-bet controls regulatory T cell homeostasis and function during type 1 inflammation. Nat Immunol 10:595–602
Khader S, Pearl J, Sakamoto K, Gilmartin L, Bell G, Jelley-Gibbs D, Ghilardi N, deSauvage F, Cooper A (2005) IL-23 compensates for the absence of IL-12p70 and is essential for the IL-17 response during tuberculosis but is dispensable for protection and antigen-specific IFN-gamma responses if IL-12p70 is available. J Immunol 175:788–795
Scriba T, Kalsdorf B, Abrahams D-A, Isaacs F, Hofmeister J, Black G, Hassan H, Wilkinson R, Walzl G, Gelderbloem S et al (2008) Distinct, specific IL-17 and IL-22-producing CD4+ T cell subsets contribute to the human anti-mycobacterial immune response. J Immunol 180:1962–1970
Umemura M, Yahagi A, Hamada S, Begum M, Watanabe H, Kawakami K, Suda T, Sudo K, Nakae S, Iwakura Y et al (2007) IL-17-mediated regulation of innate and acquired immune response against pulmonary Mycobacterium bovis bacille Calmette-Guerin Infection. J Immunol 178:3786–3796
Lockhart E, Green A, Flynn J (2006) IL-17 production is dominated by gammadelta T cells rather than CD4 T cells during Mycobacterium tuberculosis infection. J Immunol 177:4662–4669
D’Souza CD, Cooper AM, Frank AA, Mazzaccaro RJ, Bloom BR, Orme IM (1997) An anti-inflammatory role for γδ T lymphocytes in acquired immunity to Mycobacterium tuberculosis. J Immunol 158:1217–1221
Romani L, Fallarino F, De Luca A, Montagnoli C, D’Angelo C, Zelante T, Vacca C, Bistoni F, Fioretti M, Grohmann U et al (2008) Defective tryptophan catabolism underlies inflammation in mouse chronic granulomatous disease. Nature 451:211–215
Cooper AM, Segal BH, Frank AA, Holland SM, Orme IM (2000) Transient loss of resistance to pulmonary tuberculosis in p47phox−/− mice. Infect Immun 68:1231–1234
Michel M, Keller A, Paget C, Fujio M, Trottein F, Savage P, Wong C, Schneider E, Dy M, Leite-de-Moraes M (2007) Identification of an IL-17-producing NK1.1(neg) iNKT cell population involved in airway neutrophilia. J Exp Med 204:995–1001
Weaver C, Hatton R, Mangan P, Harrington L (2007) IL-17 family cytokines and the expanding diversity of effector T cell lineages. Annu Rev Immunol 25:821–852
Stockinger B, Veldhoen M, Martin B (2007) Th17 T cells: linking innate and adaptive immunity. Semin Immunol 19:353–361
Khader S, Bell G, Pearl J, Fountain J, Rangel-Moreno J, Cilley G, Shen F, Eaton S, Gaffen S, Swain S et al (2007) IL-23 and IL-17 in establishment of protective pulmonary CD4+ T cell responses upon vaccination and during Mycobacterium tuberculosis challenge. Nat Immunol 8:369–377
Cruz A, Khader S, Torrado E, Fraga A, Pearl J, Pedrosa J, Cooper A, Castro A (2006) Cutting edge: IFN-γ regulates the induction and expansion of IL-17-producing CD4 T cells during mycobacterial infection. J Immunol 177:1416–1420
Stockinger B, Veldhoen M (2007) Differentiation and function of Th17 T cells. Curr Opin Immunol 19:281–286
Ivanov I, Zhou L, Littman D (2007) Transcriptional regulation of Th17 cell differentiation. Semin Immunol 19:409–417
McGeachy M, Cua D (2007) The link between IL-23 and Th17 cell-mediated immune pathologies. Semin Immunol 19:372–376
Sutton CE, Lalor SJ, Sweeney CM, Brereton CF, Lavelle EC, Mills KH (2009) Interleukin-1 and IL-23 induce innate IL-17 production from gammadelta T cells, amplifying Th17 responses and autoimmunity. Immunity 31:331–341
Masters SL, Mielke LA, Cornish AL, Sutton CE, O’Donnell J, Cengia LH, Roberts AW, Wicks IP, Mills KHG, Croker BA (2010) Regulation of interleukin-1[beta] by interferon-[gamma] is species specific, limited by suppressor of cytokine signalling 1 and influences interleukin-17 production. EMBO Rep 11:640–646
Sutherland JS, Adetifa IM, Hill PC, Adegbola RA, Ota MOC (2009) Pattern and diversity of cytokine production differentiates between Mycobacterium tuberculosis infection and disease. Eur J Immunol 39:723–729
Sutherland JS, de Jong BC, Jeffries DJ, Adetifa IM, Ota MOC (2010) Production of TNF-α, IL-12(p40) and IL-17 can discriminate between active TB disease and latent infection in a West African cohort. PLoS One 5:e12365
Stern J, Keskin D, Romero V, Zuniga J, Encinales L, Li C, Awad C, Yunis E (2009) Molecular signatures distinguishing active from latent tuberculosis in peripheral blood mononuclear cells, after in vitro antigenic stimulation with purified protein derivative of tuberculin (PPD) or Candida: a preliminary report. Immunol Res 45:1–12
Chen Y-C, Chin C-H, Liu S-F, Wu C-C, Tsen C-C, Wang Y-H, Chao T-Y, Lie C-H, Chen C-J, Wang C-C et al (2011) Prognostic values of serum IP-10 and IL-17 in patients with pulmonary tuberculosis. Dis Markers 31:101–110
Djoba Siawaya JF, Beyers N, Van Helden P, Walzl G (2009) Differential cytokine secretion and early treatment response in patients with pulmonary tuberculosis. Clin Exp Immunol 156:69–77
de Steenwinkel J, de Knegt G, ten Kate M, Verbrugh H, Ottenhoff T, Bakker-Woudenberg I (2011) Dynamics of interferon-gamma release assay and cytokine profiles in blood and respiratory tract specimens from mice with tuberculosis and the effect of therapy. Eur J Clin Microbial Infect Dis 31:1195–1201
Acosta-Rodriguez E, Rivino L, Geginat J, Jarrossay D, Gattorno M, Lanzavecchia A, Sallusto F, Napolitani G (2007) Surface phenotype and antigenic specificity of human interleukin 17-producing T helper memory cells. Nat Immunol 8:639–646
Qiao D, Yang BY, Li L, Ma JJ, Zhang XL, Lao SH, Wu CY (2011) ESAT-6- and CFP-10-specific Th1, Th22 and Th17 cells in tuberculous pleurisy may contribute to the local immune response against Mycobacterium tuberculosis infection. Scand J Immunol 73:330–337
Wang T, Lv M, Qian Q, Nie Y, Yu L, Hou Y (2011) Increased frequencies of T helper type 17 cells in tuberculous pleural effusion. Tuberculosis 91:231–237
Matthews K, Wilkinson KA, Kalsdorf B, Roberts T, Diacon A, Walzl G, Wolske J, Ntsekhe M, Syed F, Russell J et al (2011) Predominance of interleukin-22 over interleukin-17 at the site of disease in human tuberculosis. Tuberculosis 91:587–593
Stark M, Huo Y, Burcin T, Morris M, Olson T, Ley K (2005) Phagocytosis of apoptotic neutrophils regulates granulopoiesis via IL-23 and IL-17. Immunity 22:285–294
Ye P, Rodriguez FH, Kanaly S, Stocking KL, Schurr J, Schwarzenberger P, Oliver P, Huang W, Zhang P, Zhang J et al (2001) Requirement of interleukin-17 receptor signalling for lung CXC chemokine and granulocyte colony-stimulating factor expression, neutrophil recruitment, and host defense. J Exp Med 194:519–527
Kolls J, Linden A (2004) Interleukin-17 family members and inflammation. Immunity 21:467–476
Hsu H, Yang P, Wang J, Wu Q, Myers R, Chen J, Yi J, Guentert T, Tousson A, Stanus A et al (2007) Interleukin 17-producing T helper cells and interleukin 17 orchestrate autoreactive germinal center development in autoimmune BXD2 mice. Nat Immunol 9:166–175
Mangan P, Harrington L, O’Quinn D, Helms W, Bullard D, Elson C, Hatton R, Wahl S, Schoeb T, Weaver C (2006) Transforming growth factor-beta induces development of the T(H)17 lineage. Nature 441:231–234
Huang W, Na L, Fidel P, Schwarzenberger P (2004) Requirement of interleukin-17A for systemic anti-Candida albicans host defense in mice. J Infect Dis 190:624–631
Wu Q, Martin R, Rino J, Breed R, Torres R, Chu H (2007) IL-23-dependent IL-17 production is essential in neutrophil recruitment and activity in mouse lung defense against respiratory Mycoplasma pneumoniae infection. Microbes Infect 9:78–86
Chackerian A, Chen S, Brodie S, Mattson J, McClanahan T, Kastelein R, Bowman E (2006) Neutralization or absence of the interleukin-23 pathway does not compromise immunity to mycobacterial infection. Infect Immun 74:6092–6099
Happel K, Lockhart E, Mason C, Porretta E, Keoshkerian E, Odden A, Nelson S, Ramsay A (2005) Pulmonary interleukin-23 gene delivery increases local T-cell immunity and controls growth of Mycobacterium tuberculosis in the lungs. Infect Immun 73:5782–5788
Khader SA, Guglani L, Rangel-Moreno J, Gopal R, Fallert Junecko BA, Fountain JJ, Martino C, Pearl JE, Tighe M, Lin YY et al (2011) IL-23 is required for long-term control of Mycobacterium tuberculosis and B cell follicle formation in the infected lung. J Immunol 187:5402–5407
Aujla SJ, Chan YR, Zheng M, Fei M, Askew DJ, Pociask DA, Reinhart TA, McAllister F, Edeal J, Gaus K et al (2008) IL-22 mediates mucosal host defense against gram-negative bacterial pneumonia. Nat Med 14:275–281
Okamoto YY, Umemura M, Yahagi A, O’Brien R, Ikuta K, Kishihara K, Hara H, Nakae S, Iwakura Y, Matsuzaki G (2010) Essential role of IL-17A in the formation of a mycobacterial infection-induced granuloma in the lung. J Immunol 184:4414–4422
Redford P, Boonstra A, Read S, Pitt J, Graham C, Stavropoulos E, Bancroft G, O’Garra A (2010) Enhanced protection to Mycobacterium tuberculosis infection in IL-10-deficient mice is accompanied by early and enhanced Th1 responses in the lung. Eur J Immunol 40:2200–2210
Sumaria N, Roediger B, Ng LG, Qin J, Pinto R, Cavanagh LL, Shklovskaya E, de St Groth BF, Triccas JA, Weninger W (2011) Cutaneous immunosurveillance by self-renewing dermal γδ T cells. J Exp Med 208:505–518
Wozniak TM, Saunders BM, Ryan AA, Britton WJ (2010) Mycobacterium bovis BCG-specific Th17 cells confer partial protection against Mycobacterium tuberculosis infection in the absence of gamma interferon. Infect Immun 78:4187–4194
Curtis MM, Rowell E, Shafiani S, Negash A, Urdahl KB, Wilson CB, Way SS (2010) Fidelity of pathogen-specific CD4+ T cells to the Th1 lineage is controlled by exogenous cytokines, interferon-γ expression, and pathogen lifestyle. Cell Host Microbe 8:163–173
Filipe-Santos O, Bustamante J, Chapgier A, Vogt G, de Beaucoudrey L, Feinberg J, Jouanguy E, Boisson-Dupuis S, Fieschi C, Picard C et al (2006) Inborn errors of IL-12/23- and IFN-gamma-mediated immunity: molecular, cellular, and clinical features. Semin Immunol 18:347–361
Hoeve M, de Boer T, Langenberg D, Sanal O, Verreck F, Ottenhoff T (2003) IL-12 receptor deficiency revisited: IL-23-mediated signaling is also impaired in human genetic IL-12 receptor beta1 deficiency. Eur J Immunol 33:3393–3397
Hoeve M, Savage N, de Boer T, Langenberg D, de Waal Malefyt R, Ottenhoff T, Verreck F (2006) Divergent effects of IL-12 and IL-23 on the production of IL-17 by human T cells. Eur J Immunol 36:661–670
Cargill M, Schrodi S, Chang M, Garcia V, Brandon R, Callis K, Matsunami N, Ardlie K, Civello D, Catanese J et al (2007) A large-scale genetic association study confirms IL12B and leads to the identification of IL23R as psoriasis-risk genes. Am J Hum Genet 80:273–290
Duerr R, Taylor K, Brant S, Rioux J, Silverberg M, Daly M, Steinhart A, Abraham C, Regueiro M, Griffiths A et al (2006) A genome-wide association study identifies IL23R as an inflammatory bowel disease gene. Science 314:1461–1463
Stolberg VR, Chiu B-C, Schmidt BM, Kunkel SL, Sandor M, Chensue SW (2011) CC chemokine receptor 4 contributes to innate NK and chronic stage T helper cell recall responses during Mycobacterium bovis infection. Am J Pathol 178:233–244
Wozniak T, Ryan A, Triccas J, Britton W (2006) Plasmid interleukin-23 (IL-23), but not plasmid IL-27, enhances the protective efficacy of a DNA vaccine against Mycobacterium tuberculosis infection. Infect Immun 74:557–565
Wozniak T, Ryan A, Britton W (2006) Interleukin-23 restores immunity to Mycobacterium tuberculosis infection in IL-12p40-deficient mice and is not required for the development of IL-17-secreting T cell responses. J Immunol 177:8684–8692
Stumhofer J, Laurence A, Wilson E, Huang E, Tato C, Johnson L, Villarino A, Huang Q, Yoshimura A, Sehy D et al (2006) Interleukin 27 negatively regulates the development of interleukin 17-producing T helper cells during chronic inflammation of the central nervous system. Nat Immunol 7:937–945
Batten M, Li J, Yi S, Kljavin N, Danilenko D, Lucas S, Lee J, de Sauvage F, Ghilardi N (2006) Interleukin 27 limits autoimmune encephalomyelitis by suppressing the development of interleukin 17-producing T cells. Nat Immunol 7:929–936
Gopal R, Lin Y, Obermajer N, Slight S, Nuthalapati N, Ahmed M, Kalinski P, Khader SA (2012) IL-23-dependent IL-17 drives Th1-cell responses following Mycobacterium bovis BCG vaccination. Eur J Immunol 42:364–373
Desel C, Dorhoi A, Bandermann S, Grode L, Eisele B, Kaufmann SHE (2011) Recombinant BCG ΔureC hly+ induces superior protection over parental BCG by stimulating a balanced combination of type 1 and type 17 cytokine responses. J Infect Dis 204:1573–1584
Vordermeier HM, Villarreal-Ramos B, Cockle PJ, McAulay M, Rhodes SG, Thacker T, Gilbert SC, McShane H, Hill AVS, Xing Z et al (2009) Viral booster vaccines improve Mycobacterium bovis BCG-induced protection against bovine tuberculosis. Infect Immun 77:3364–3373
Scriba TJ, Tameris M, Mansoor N, Smit E, van der Merwe L, Isaacs F, Keyser A, Moyo S, Brittain N, Lawrie A et al (2010) Modified vaccinia Ankara-expressing Ag85A, a novel tuberculosis vaccine, is safe in adolescents and children, and induces polyfunctional CD4+ T cells. Eur J Immunol 40:279–290
de Cassan SC, Pathan AA, Sander CR, Minassian A, Rowland R, Hill AVS, McShane H, Fletcher HA (2010) Investigating the induction of vaccine-induced Th17 and regulatory T cells in healthy, Mycobacterium bovis BCG-immunized adults vaccinated with a new tuberculosis vaccine, MVA85A. Clin Vaccine Immunol 17:1066–1073
Griffiths KL, Pathan AA, Minassian AM, Sander CR, Beveridge NER, Hill AVS, Fletcher HA, McShane H (2011) Th1/Th17 cell induction and corresponding reduction in ATP consumption following vaccination with the novel Mycobacterium tuberculosis vaccine MVA85A. PLoS One 6:e23463
Lalor MK, Smith SG, Floyd S, Gorak-Stolinska P, Weir RE, Blitz R, Branson K, Fine PE, Dockrell HM (2010) Complex cytokine profiles induced by BCG vaccination in UK infants. Vaccine 28:1635–1641
Burl S, Adetifa UJ, Cox M, Touray E, Ota MO, Marchant A, Whittle H, McShane H, Rowland-Jones SL, Flanagan KL (2010) Delaying bacillus Calmette-Guérin vaccination from birth to 4 1/2 months of age reduces postvaccination Th1 and IL-17 responses but leads to comparable mycobacterial responses at 9 months of age. J Immunol 185:2620–2628
Lalor MK, Floyd S, Gorak-Stolinska P, Ben-Smith A, Weir RE, Smith SG, Newport MJ, Blitz R, Mvula H, Branson K et al (2011) BCG vaccination induces different cytokine profiles following infant BCG vaccination in the UK and Malawi. J Infect Dis 204:1075–1085
Kagina BMN, Abel B, Scriba TJ, Hughes EJ, Keyser A, Soares A, Gamieldien H, Sidibana M, Hatherill M, Gelderbloem S et al (2010) Specific T cell frequency and cytokine expression profile do not correlate with protection against tuberculosis after bacillus Calmette-Guérin vaccination of newborns. Am J Respir Crit Care Med 182:1073–1079
Rhoades ER, Frank AA, Orme IM (1997) Progression of chronic pulmonary tuberculosis in mice aerogenically infected with virulent Mycobacterium tuberculosis. Tuber Lung Dis 78:57–66
Aujla S, Dubin P, Kolls J (2007) Th17 cells and mucosal host defense. Semin Immunol 19:377–382
Zelante T, De Luca A, Bonifazi P, Montagnoli C, Bozza S, Moretti S, Belladonna M, Vacca C, Conte C, Mosci P et al (2007) IL-23 and the Th17 pathway promote inflammation and impair antifungal immune resistance. Eur J Immunol 37:2695–2706
Seiler P, Aichele P, Bandermann S, Hauser A, Lu B, Gerard N, Gerard C, Ehlers S, Mollenkopf H, Kaufmann S (2003) Early granuloma formation after aerosol Mycobacterium tuberculosis infection is regulated by neutrophils via CXCR3-signaling chemokines. Eur J Immunol 33:2676–2686
Keller C, Hoffmann R, Lang R, Brandau S, Hermann C, Ehlers S (2006) Genetically determined susceptibility to tuberculosis in mice causally involves accelerated and enhanced recruitment of granulocytes. Infect Immun 74:4295–4309
Cruz A, Fraga A, Fountain J, Rangel-Moreno J, Torrado E, Saraiva M, Pereira D, Randall T, Pedrosa J, Cooper A et al (2010) Pathological role of interleukin 17 in mice subjected to repeated BCG vaccination after infection with Mycobacterium tuberculosis. J Exp Med 207:1609–1616
Desvignes L, Ernst JD (2009) Interferon-γ-responsive nonhematopoietic cells regulate the immune response to Mycobacterium tuberculosis. Immunity 31:974–985
Nandi B, Behar SM (2011) Regulation of neutrophils by interferon-γ limits lung inflammation during tuberculosis infection. J Exp Med 208:2251–2262
Basile JI, Geffner LJ, Romero MM, Balboa L, Sabio y García C, Ritacco V, García A, Cuffré M, Abbate E, López B et al (2011) Outbreaks of Mycobacterium tuberculosis MDR strains induce high IL-17 T-cell response in patients with MDR tuberculosis that is closely associated with high antigen load. J Infect Dis 204:1054–1064
Ulrichs T, Kosmiadi G, Trusov V, Jörg S, Pradl L, Titukhina M, Mishenko V, Gushina N, Kaufmann S (2004) Human tuberculous granulomas induce peripheral lymphoid follicle-like structures to orchestrate local host defence in the lung. J Pathol 204:217–228
Tsai M, Chakravarty S, Zhu G, Xu J, Tanaka K, Koch C, Tufariello J, Flynn J, Chan J (2006) Characterization of the tuberculous granuloma in murine and human lungs: cellular composition and relative tissue oxygen tension. Cell Microbiol 8:218–232
Kahnert A, Höpken U, Stein M, Bandermann S, Lipp M, Kaufmann S (2007) Mycobacterium tuberculosis triggers formation of lymphoid structure in murine lungs. J Infect Dis 195:46–54
Bosio C, Gardner D, Elkins K (2000) Infection of B cell-deficient mice with CDC 1551, a clinical isolate of Mycobacterium tuberculosis: delay in dissemination and development of lung pathology. J Immunol 164:6417–6425
Maglione P, Xu J, Chan J (2007) B cells moderate inflammatory progression and enhance bacterial containment upon pulmonary challenge with Mycobacterium tuberculosis. J Immunol 178:7222–7234
Zhang M, Wang Z, Graner MW, Yang L, Liao M, Yang Q, Gou J, Zhu Y, Wu C, Liu H et al (2011) B cell infiltration is associated with the increased IL-17 and IL-22 expression in the lungs of patients with tuberculosis. Cell Immunol 270:217–223
Khader S, Rangel-Moreno J, Fountain J, Martino C, Reiley W, Pearl J, Winslow G, Woodland D, Randall T, Cooper A (2009) In a murine tuberculosis model, the absence of homeostatic chemokines delays granuloma formation and protective immunity. J Immunol 183:8004–8014
Aggarwal S, Ghilardi N, Xie M, de Sauvage FJ, Gurney AL (2002) Interleukin-23 promotes a distinct CD4+ T cell activation state characterised by the production of IL-17. J Biol Chem 278:1910–1914
Leibundgut-Landmann S, Groß O, Robinson M, Osorio F, Slack E, Tsoni S, Schweighoffer E, Tybulewicz V, Brown G, Ruland J et al (2007) Syk- and CARD9-dependent coupling of innate immunity to the induction of T helper cells that produce interleukin 17. Nat Immunol 8:630–638
Yadav M, Schorey J (2006) The beta-glucan receptor dectin-1 functions together with TLR2 to mediate macrophage activation by mycobacteria. Blood 108:3168–3175
Rothfuchs A, Bafica A, Feng C, Egen J, Williams D, Brown G, Sher A (2007) Dectin-1 interaction with Mycobacterium tuberculosis leads to enhanced IL-12p40 production by splenic dendritic cells. J Immunol 179:3463–3471
Zenaro E, Donini M, Dusi S (2009) Induction of Th1/Th17 immune response by Mycobacterium tuberculosis: role of dectin-1, mannose receptor, and DC-SIGN. J Leukoc Biol 86:1393–1401
van de Veerdonk F, Teirlinck A, Kleinnijenhui J, Jan Kullberg B, van Creval R, van der Meer J, Joosten L, Netea M (2010) Mycobacterium tuberculosis induces IL-17A responses through TLR4 and dectin-1 and is critically dependent on endogenous IL-1. J Leukoc Biol 88(2):227–232
Hohl T, Van Epps H, Rivera A, Morgan L, Chen P, Feldmesser M, Pamer E (2005) Aspergillus fumigatus triggers inflammatory responses by stage-specific beta-glucan display. PLoS Pathog 1:e30
Steele C, Rapaka R, Metz A, Pop S, Williams D, Gordon S, Kolls J, Brown G (2005) The beta-glucan receptor dectin-1 recognizes specific morphologies of Aspergillus fumigatus. PLoS Pathog 1:e42
Kano S, Sato K, Morishita Y, Vollstedt S, Kim S, Bishop K, Honda K, Kubo M, Taniguchi T (2008) The contribution of transcription factor IRF1 to the interferon-gamma-interleukin 12 signaling axis and T(H)1 versus T(H)-17 differentiation of CD4(+) T cells. Nat Immunol 9:34–41
Rao V, Fujiwara N, Porcelli S, Glickman M (2005) Mycobacterium tuberculosis controls host innate immune activation through cyclopropane modification of a glycolipid effector molecule. J Exp Med 201:535–543
Kursar M, Koch M, Mittrücker H, Nouailles G, Bonhagen K, Kamradt T, Kaufmann S (2007) Cutting edge: regulatory T cells prevent efficient clearance of Mycobacterium tuberculosis. J Immunol 178:2661–2665
Shafiani S, Tucker-Heard G, Kariyone A, Takatsu K, Urdahl KB (2010) Pathogen-specific regulatory T cells delay the arrival of effector T cells in the lung during early tuberculosis. J Exp Med 207:1409–1420
Ishikawa E, Ishikawa T, Morita Y, Toyonaga K, Yamada H, Takeuchi O, Kinoshita T, Akira S, Yoshikai Y, Yamasaki S (2009) Direct recognition of the mycobacterial glycolipid, trehalose dimycolate, by C-type lectin Mincle. J Exp Med 206:2879–2888
Schoenen H, Bodendorfer B, Hitchens K, Manzanero S, Werninghaus K, Nimmerjahn F, Agger E, Stenger S, Andersen P, Ruland J et al (2010) Cutting edge: Mincle is essential for recognition and adjuvanticity of the mycobacterial cord factor and its synthetic analog trehalose-dibehenate. J Immunol 184:2756–2760
Paidipally P, Periasamy S, Barnes PF, Dhiman R, Indramohan M, Griffith DE, Cosman D, Vankayalapati R (2009) NKG2D-dependent IL-17 production by human T cells in response to an intracellular pathogen. J Immunol 183:1940–1945
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer Basel
About this chapter
Cite this chapter
Cooper, A.M. (2013). Is IL-17 Required to Control Tuberculosis?. In: Quesniaux, V., Ryffel, B., Padova, F. (eds) IL-17, IL-22 and Their Producing Cells: Role in Inflammation and Autoimmunity. Progress in Inflammation Research. Springer, Basel. https://doi.org/10.1007/978-3-0348-0522-3_14
Download citation
DOI: https://doi.org/10.1007/978-3-0348-0522-3_14
Published:
Publisher Name: Springer, Basel
Print ISBN: 978-3-0348-0521-6
Online ISBN: 978-3-0348-0522-3
eBook Packages: MedicineMedicine (R0)