
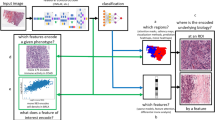
Abstract
Weakly supervised classification of whole slide images (WSIs) in digital pathology typically involves making slide-level predictions by aggregating predictions from embeddings extracted from multiple individual tiles. However, these embeddings can fail to capture valuable information contained within the individual cells in each tile. Here we describe an embedding extraction method that combines tile-level embeddings with a cell-level embedding summary. We validated the method using four hematoxylin and eosin stained WSI classification tasks: human epidermal growth factor receptor 2 status and estrogen receptor status in primary breast cancer, breast cancer metastasis in lymph node tissue, and cell of origin classification in diffuse large B-cell lymphoma. For all tasks, the new method outperformed embedding extraction methods that did not include cell-level representations. Using the publicly available HEROHE Challenge data set, the method achieved a state-of-the-art performance of 90% area under the receiver operating characteristic curve. Additionally, we present a novel model explainability method that could identify cells associated with different classification groups, thus providing supplementary validation of the classification model. This deep learning approach has the potential to provide morphological insights that may improve understanding of complex underlying tumor pathologies.
Access provided by Autonomous University of Puebla. Download conference paper PDF
Similar content being viewed by others


Keywords
1 Introduction
Accurate diagnosis plays an important role in achieving the best treatment outcomes for people with cancer [1]. Identification of cancer biomarkers permits more granular classification of tumors, leading to better diagnosis, prognosis, and treatment decisions [2, 3]. For many cancers, clinically reliable genomic, molecular, or imaging biomarkers have not been identified and biomarker identification techniques (e.g., fluorescence in situ hybridization) have limitations that can restrict their clinical use. On the other hand, histological analysis of hematoxylin and eosin (H&E)-stained pathology slides is widely used in cancer diagnosis and prognosis. However, visual examination of H&E-stained slides is insufficient for classification of some tumors because identifying morphological differences between molecularly defined subtypes is beyond the limit of human detection.
The introduction of digital pathology (DP) has enabled application of machine learning approaches to extract otherwise inaccessible diagnostic and prognostic information from H&E-stained whole slide images (WSIs) [4, 5]. Current deep learning approaches to WSI analysis typically operate at three different histopathological scales: whole slide-level, region-level, and cell-level [4]. Although cell-level analysis has the potential to produce more detailed and explainable data, it can be limited by the unavailability of sufficiently annotated training data. To overcome this problem, weakly supervised and multiple instance learning (MIL) based approaches have been applied to numerous WSI classification tasks [6,7,8,9,10]. However, many of these models use embeddings derived from tiles extracted using pretrained networks, and these often fail to capture useful information from individual cells. Here we describe a new embedding extraction method that combines tile-level embeddings with a cell-level embedding summary. Our new method achieved better performance on WSI classification tasks and had a greater level of explainability than models that used only tile-level embeddings.
2 Embedding Extraction Scheme
Transfer learning using backbones pretrained on natural images is a common method that addresses the challenge of using data sets that largely lack annotation. However, using backbones pretrained on natural images is not optimal for classification of clinical images [11]. Therefore, to enable the use of large unlabeled clinical imaging data sets, as the backbone of our neural network we used a ResNet50 model [12]. The backbone was trained with the bootstrap your own latent (BYOL) method [13] using four publicly available data sets from The Cancer Genome Atlas (TCGA) and three data sets from private vendors that included healthy and malignant tissue from a range of organs [14].
2.1 Tile-Level Embeddings
Following standard practice, we extracted tiles with dimensions of 256 × 256 pixels from WSIs (digitized at 40 × magnification) on a spatial grid without overlap. Extracted tiles that contained artifacts were discarded (e.g., tiles that had an overlap of >10% with background artifacts such blurred areas or pen markers). We normalized the tiles for stain color using a U-Net model for stain normalization [15] that was trained on a subset of data from one of the medical centers in the CAMELYON17 data set to ensure homogeneity of staining [16].
To create the tile-level embeddings, we used the method proposed by [17] to summarize the convolutional neural network (CNN) features with nonnegative matrix factorization (NMF) for K = 2 factors. We observed that the feature activations within the last layer of the network were not aligned with the cellular content. Although these features may still have been predictive, they were less interpretable, and it was more difficult to know what kind of information they captured. Conversely, we observed that the self-supervised network captured cellular content and highlighted cells within the tiles (Fig. 1). Therefore, the tile-level embeddings were extracted after dropping the last layer (i.e., dropping three bottleneck blocks in ResNet50) from the pretrained model.

A visualization of the output features of the backbone for a typical input tile (left), from the last layer (middle), and from the second to last layer (right) of the pretrained CNN summarized using NMF with K = 2 factors. Resolution: 0.25 μm/pixel.
2.2 Cell-Level Embeddings
Tiles extracted from WSIs may contain different types of cells, as well as noncellular tissue such as stroma and blood vessels and nonbiological features (e.g., glass). Cell-level embeddings may be able to extract useful information, based on the morphological appearance of individual cells, that is valuable for downstream classification tasks but would otherwise be masked by more dominant features within tile-level embeddings.
We extracted deep cell-level embeddings by first detecting individual cellular boundaries using StarDist [18] and extracting 32 × 32-pixel image crops centered around each segmented nucleus to create cell-patch images. We then used the pre-trained ResNet50 model to extract cell-level embeddings in a similar manner to the extraction of the tile-level embeddings. Since ResNet50 has a spatial reduction factor of 32 in the output of the CNN, the 32 × 32-pixel image had a 1:1 spatial resolution in the output. To ensure the cell-level embeddings contained features relevant to the cells, prior to the mean pooling in ResNet50 we increased the spatial image resolution to 16 × 16 pixels in the output from the CNN by enlarging the 32 × 32-pixel cell-patch images to 128 × 128 pixels and skipping the last 4-layers in the network.
Because of heterogeneity in the size of cells detected, each 32 × 32-pixel cell-patch image contained different proportions of cellular and noncellular features. Higher proportions of noncellular features in an image may cause the resultant embeddings to be dominated by noncellular tissue features or other background features. Therefore, to limit the information used to create the cell-level embeddings to only cellular features, we removed portions of the cell-patch images that were outside of the segmented nuclei by setting their pixel values to black (RGB 0, 0, 0). Finally, to prevent the size of individual nuclei or amount of background in each cell-patch image from dominating over the cell-level features, we modified the ResNet50 Global Average Pooling layer to only average the features inside the boundary of the segmented nuclei, rather than averaging across the whole output tensor from the CNN.
2.3 Combined Embeddings
To create a combined representation of the tile-level and cell-level embeddings, we first applied a nuclei segmentation network to each tile. Only tiles with ≥ 10 cells per tile, excluding any cells which overlapped the tile border, were included for embedding extraction. For the included tiles, we extracted the tile-level embeddings as described in Sect. 2.1 and for each detected cell we extracted the cell-level embeddings as described in Sect. 2.2. We then calculated the mean and standard deviation of the vectors of the cell-level embeddings for each tile and concatenated those to each tile-level embedding. This resulted in a combined embedding representation with a total size of 1536 pixels (1024 + 256 + 256).
In addition to the WSI classification results presented in the next sections, we also performed experiments to compare the ability of combined embeddings and tile-level embeddings to predict nuclei-related features that were manually extracted from the images and to identify tiles where nuclei had been ablated. The details and results of these experiments are available in supplementary materials and provide further evidence of the improved ability to capture cell-level information when using combined embeddings compared with tile-level embeddings alone.
3 WSI Classification Tasks
For each classification task we compared different combinations of tile-level and cell-level embeddings using a MIL framework. We also compared two different MIL architectures to aggregate the embeddings for WSI-level prediction.
The first architecture used an attention-MIL (A-MIL) network [19] (the code was adapted from a publicly available implementation [20]). We trained the network with a 0.001 learning rate and tuned the batch size (48 or 96) and bag sample size (512, 1024, or 2048) for each classification task separately. When comparing the combined embedding extraction method with the tile-level only embeddings, parameters were fixed to demonstrate differences in performance without additional parameter tuning.
Transformer (Xformer) was used as the second MIL architecture [21]. We used three Xformer layers, each with eight attention heads, 512 parameters per token, and 256 parameters in the multi-layer perceptron layers. The space complexity of the Xformer was quadratic with the number of tokens. While some WSIs had up to 100,000 tiles, we found, in practice, that we could not fit more than 6000 tokens in the memory. Consequently, we used the Nyströformer Xformer variant [22] since it consumes less memory (the code was adapted from a publicly available implementation [23]). This Xformer has two outputs, was trained with the Adam optimizer [24] with default parameters, and the loss was weighted with median frequency balancing [25] to assign a higher weight to the less frequent class. Like A-MIL, the batch and bag sample sizes were fixed for each classification task. During testing a maximum of 30,000 tiles per slide were used. The complete flow for WSI classification is shown in Fig. 2. The models were selected using a validation set, that was a random sample of 20% of the training data. All training was done using PyTorch version 1.12.1 (pytorch.org) on 8 NVIDIA Tesla V100 GPUs with Cuda version 10.2.

Schematic visualization of the classification pipeline based on combined embeddings. Tile-level and cell-level embeddings are extracted in parallel and then concatenated embedding vectors are passed through the MIL model for the downstream task. aMi equals the number of cells in tile i.
3.1 Data
We tested our feature representation method in several classification tasks involving WSIs of H&E-stained histopathology slides. The number of slides per class for each classification task are shown in Fig. 3.

Class distributions in the data used for WSI classification tasks. Numbers in the bars represent the number of WSIs by classification for each task.
For breast cancer human epidermal growth factor receptor 2 (HER2) prediction, we used data from the HEROHE Challenge data set [26]. To enable comparison with previous results we used the same test data set that was used in the challenge [27]. For prediction of estrogen receptor (ER) status, we used images from the TCGA-Breast Invasive Carcinoma (TCGA-BRCA) data set [28] for which the ER status was known. For these two tasks we used artifact-free tiles from tumor regions detected with an in-house tumor detection model.
For breast cancer metastasis detection in lymph node tissue, we used WSIs of H&E-stained healthy lymph node tissue and lymph node tissue with breast cancer metastases from the publicly available CAMELYON16 challenge data set [16, 29]. All artifact-free tissue tiles were used.
For cell of origin (COO) prediction of activated B-cell like (ABC) or germinal center B-cell like (GCB) tumors in diffuse large B-cell lymphoma (DLBCL), we used data from the phase 3 GOYA (NCT01287741) and phase 2 CAVALLI (NCT02055820) clinical trials, hereafter referred to as CT1 and CT2, respectively. All slides were H&E-stained and scanned using Ventana DP200 scanners at 40× magnification. CT1 was used for training and testing the classifier and CT2 was used only as an independent holdout data set. For these data sets we used artifact-free tiles from regions annotated by expert pathologists to contain tumor tissue.
4 Model Classification Performance
For the HER2 prediction, ER prediction, and metastasis detection classification tasks, combined embeddings outperformed tile-level only embeddings irrespective of the downstream classifier architecture used (Fig. 4).

Model performance using the Xformer and A-MIL architectures for the breast cancer HER2 status, breast cancer ER status, and breast cancer metastasis detection in lymph node tissue classification tasks. Error bars represent 95% confidence intervals computed by a 5000-sample bias-corrected and accelerated bootstrap.
In fact, for the HER2 classification task, combined embeddings obtained using the Xformer architecture achieved, to our knowledge, the best performance yet reported on the HEROHE Challenge data set (area under the receiver operating characteristic curve [AUC], 90%; F1 score, 82%).
For COO classification in DLBCL, not only did the combined embeddings achieve better performance than the tile-level only embeddings with both the Xformer and A-MIL architectures (Fig. 5) on the CT1 test set and CT2 holdout data set, but they also had a significant advantage versus tile-only level embeddings in respect of the additional insights they provided through cell-level model explainability (Sect. 4.1).

Model performance using the Xformer and A-MIL architectures for the COO in DLBCL classification task. Error bars represent 95% confidence intervals computed by a 5000-sample bias-corrected and accelerated bootstrap.
4.1 Model Explainability
Tile-based approaches in DP often use explainability methods such as Gradient-weighted Class Activation Mapping [30] to highlight parts of the image that correspond with certain category outputs. While the backbone of our model was able to highlight individual cells, there was no guaranteed correspondence between the model activations and the cells. To gain insights into cell-level patterns that were very difficult or impossible to obtain from tile-level only embeddings, we applied an explainability method that assigned attention weights to the cellular average part of the embedding.
Cellular Explainability Method. The cellular average embedding is \(\frac{1}{N}\mathop \sum \limits_{i = 0}^{N - 1} e_{ij} \) where \(e_{ij} \in R^{256}\) is the cellular embedding extracted from every detected cell in the tile \(j \left( {i \in \left\{ {1, 2, \ldots , N_{j} } \right\}} \right)\) where Nj is the number of cells in the tile j. This can be rewritten as a weighted average of the cellular embeddings \(\mathop \sum \limits_{i = 0}^{N - 1} e_{{ij{ }}} Sigmoid \left( {w_{i} } \right)/\mathop \sum \limits_{i = 0}^{N - 1} Sigmoid{ }\left( {w_{i} } \right)\) where \(w_{i} \in R^{256}\) are the per cell attention weights that if initialized to 0 result in the original cellular average embedding. The re-formulation does not change the result of the forward pass since \(w_{i}\) are not all equal. Note that the weights are not learned through training but calculated per cell at inference time to get the per cell contribution. We computed the gradient of the output category (of the classification method applied on top of the computed embedding) with respect to the attention weights \(w_{i}\): \(grad_{{i{ }}} = \partial Score_{i} /\partial w_{i} \) and visualized cells that received positive and negative gradients using different colors.
Visual Example Results. Examples of our cellular explainability method applied to weakly supervised tumor detection on WSIs from the CAMELYON16 data set using A-MIL are shown in Fig. 6. Cells with positive attention gradients shifted the output towards a classification of tumor and are labeled green. Cells with negative attention gradients are labeled red. When reviewed by a trained pathologist, cells with positive gradients had characteristics previously associated with breast cancer tumors (e.g., larger nuclei, more visible nucleoli, differences in size and shape). Conversely, negative cells had denser chromatin and resembled other cell types (e.g., lymphocytes). These repeatable findings demonstrate the benefit of using cell-level embeddings and our explainability method to gain a cell-level understanding of both correct and incorrect slide-level model predictions (Fig. 6). We also applied our explainability method to COO prediction in DLBCL.
In this case, cells with positive attention gradients that shifted the output towards a classification of GCB were labeled green and cells with negative attention gradients that shifted the classification towards ABC were labeled red. Cells with positive attention gradients were mostly smaller lymphoid cells with low grade morphology or were normal lymphocytes, whereas cells with negative attention gradients were more frequently larger lymphoid cells with high grade morphology (Fig. 6).

Cellular explainability method applied to breast cancer metastasis detection in lymph nodes and COO prediction in DLBCL. Cells in the boundary margin were discarded.
5 Conclusions
We describe a method to capture both cellular and texture feature representations from WSIs that can be plugged into any MIL architecture (e.g., CNN or Xformer-based), as well as into fully supervised models (e.g., tile classification models). Our method is more flexible than other methods (e.g., Hierarchical Image Pyramid Transformer) that usually capture the hierarchical structure in WSIs by aggregating features at multiple levels in a complex set of steps to perform the final classification task. In addition, we describe a method to explain the output of the classification model that evaluates the contributions of histologically identifiable cells to the slide-level classification. Tile-level embeddings result in good performance for detection of tumor metastases in lymph nodes. However, introducing more cell-level information, using combined embeddings, resulted in improved classification performance. In HER2 and ER prediction tasks for breast cancer we demonstrate that addition of a cell-level embedding summary to tile-level embeddings can boost model performance by up to 8%. Finally, for COO prediction in DLBCL and breast cancer metastasis detection in lymph nodes, we demonstrated the potential of our explainability method to gain insights into previously unknown associations between cellular morphology and disease biology.
References
Neal, R.D., et al.: Is increased time to diagnosis and treatment in symptomatic cancer associated with poorer outcomes? Systematic review. Br. J. Cancer 112(Suppl 1), S92-107 (2015)
Henry, N.L., Hayes, D.F.: Cancer biomarkers. Mol. Oncol. 6(2), 140–146 (2012)
Park, J.E., Kim, H.S.: Radiomics as a quantitative imaging biomarker: practical considerations and the current standpoint in neuro-oncologic studies. Nucl. Med. Mol. Imaging 52(2), 99–108 (2018)
Lee, K., et al.: Deep learning of histopathology images at the single cell level. Front. Artif. Intell. 4, 754641 (2021)
Niazi, M.K.K., Parwani, A.V., Gurcan, M.N.: Digital pathology and artificial intelligence. Lancet Oncol. 20(5), e253–e261 (2019)
van der Laak, J., Litjens, G., Ciompi, F.: Deep learning in histopathology: the path to the clinic. Nat. Med. 27(5), 775–784 (2021)
Shao, Z., et al.: TransMIL: transformer based correlated multiple instance learning for whole slide image classification. In: Advances in Neural Information Processing Systems, vol. 34 pp. 2136–2147 (2021)
Wang, Y., et al.: CWC-transformer: a visual transformer approach for compressed whole slide image classification. Neural Comput. Appl. (2023)
Lu, M.Y., Williamson, D.F.K., Chen, T.Y., Chen, R.J., Barbieri, M., Mahmood, F.: Data-efficient and weakly supervised computational pathology on whole-slide images. Nat. Biomed. Eng. 5(6), 555–570 (2021)
Chen, R.J., et al.: Scaling vision transformers to gigapixel images via hierarchical self-supervised learning. In: Proceedings of the IEEE/CVF Conference on Computer Vision and Pattern Recognition, pp. 16144–16155 (2022)
Kang, M., Song, H., Park, S., Yoo, D., Pereira, S.: Benchmarking self-supervised learning on diverse pathology datasets. In: Proceedings of the IEEE/CVF Conference on Computer Vision and Pattern Recognition (CVPR), pp. 3344–3354 (2023)
He, K., Zhang, X., Ren, S., Sun, J.: Deep residual learning for image recognition. In: Proceedings of the IEEE/CVF Conference on Computer Vision and Pattern Recognition, pp. 770–778 (2016)
Grill, J.-B., et al.: Bootstrap your own latent - a new approach to self-supervised learning. Adv. Neural. Inf. Process. Syst. 33, 21271–21284 (2020)
Abbasi-Sureshjani, S., et al.: Molecular subtype prediction for breast cancer using H&E specialized backbone. In: MICCAI Workshop on Computational Pathology, pp. 1–9 (2021)
Tellez, D., et al.: Quantifying the effects of data augmentation and stain color normalization in convolutional neural networks for computational pathology. Med. Image Anal. 58, 101544 (2019)
Litjens, G., et al.: H&E-stained sentinel lymph node sections of breast cancer patients: the CAMELYON dataset. GigaScience 7(6), giy065 (2018)
Collins, E., Achanta, R., Süsstrunk, S.: Deep feature factorization for concept discovery. In: Ferrari, V., Hebert, M., Sminchisescu, C., Weiss, Y. (eds.) Computer Vision – ECCV 2018, pp. 352–368. Springer International Publishing, Cham (2018)
Schmidt, U., Weigert, M., Broaddus, C., Myers, G.: Cell detection with star-convex polygons. In: Frangi, A.F., Schnabel, J.A., Davatzikos, C., Alberola-López, C., Fichtinger, G. (eds.) MICCAI 2018. LNCS, vol. 11071, pp. 265–273. Springer, Cham (2018). https://doi.org/10.1007/978-3-030-00934-2_30
Ilse, M., Tomczak, J.M., Welling, M.: Attention-based deep multiple instance learning. ArXiv abs/1802.04712 (2018)
Attention-based Deep Multiple Instance Learning. https://github.com/AMLab-Amsterdam/AttentionDeepMIL. Accessed 24 Feb 2023
Vaswani, A., et al.: Attention is all you need. In: Guyon, I., et al. (eds.) Advances in Neural Information Processing Systems (NeurIPS 2017), vol. 31, pp. 5998–6008 (2017)
Xiong, Y., et al.: Nyströmformer: a Nystöm-based algorithm for approximating self-attention. Proc. Conf. AAAI Artif. Intell. 35(16), 14138–14148 (2021)
Nyström Attention. https://github.com/lucidrains/nystrom-attention. Accessed 24 Feb 2023
Kingma, D.P., Ba, J.: Adam: a method for stochastic optimization. In: Proceedings of the 3rd International Conference on Learning Representations (ICLR 2015), abs/1412.6980 (2015)
Eigen, D., Fergus, R.: Predicting depth, surface normals and semantic labels with a common multi-scale convolutional architecture. In: Proceedings of the IEEE International Conference on Computer Vision, pp. 2650–2658 (2015)
HEROHE ECDP2020. https://ecdp2020.grand-challenge.org/. Accessed 24 Feb 2023
Conde-Sousa, E., et al.: HEROHE challenge: predicting HER2 status in breast cancer from hematoxylin-eosin whole-slide imaging. J. Imaging 8(8) (2022)
National Cancer Institute GDC Data Portal. https://portal.gdc.cancer.gov/. Accessed 24 Feb 2023
CAMELYON17 Grand Challenge. https://camelyon17.grand-challenge.org/Data/. Accessed 24 Feb 2023
Selvaraju, R.R., Cogswell, M., Das, A., Vedantam, R., Parikh, D., Batra, D.: Grad-CAM: visual explanations from deep networks via gradient-based localization. In: 2017 IEEE International Conference on Computer Vision (ICCV), pp. 618–626 (2017)
Acknowledgments
We thank the Roche Diagnostic Solutions and Genentech Research Pathology Core Laboratory staff for tissue procurement and immunohistochemistry verification. We thank the participants from the GOYA and CAVALLI trials. The results published here are in part based upon data generated by the TCGA Research Network: https://www.cancer.gov/tcga. We thank Maris Skujevskis, Uwe Schalles and Darta Busa for their help in curating the datasets and the annotations and Amal Lahiani for sharing the tumor segmentation model used for generating the results on HEROHE. The study was funded by F. Hoffmann-La Roche AG, Basel, Switzerland and writing support was provided by Adam Errington PhD of PharmaGenesis Cardiff, Cardiff, UK and was funded by F. Hoffmann-La Roche AG.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
1 Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Copyright information
© 2023 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this paper

Cite this paper
Gildenblat, J., Yüce, A., Abbasi-Sureshjani, S., Korski, K. (2023). Deep Cellular Embeddings: An Explainable Plug and Play Improvement for Feature Representation in Histopathology. In: Greenspan, H., et al. Medical Image Computing and Computer Assisted Intervention – MICCAI 2023. MICCAI 2023. Lecture Notes in Computer Science, vol 14225. Springer, Cham. https://doi.org/10.1007/978-3-031-43987-2_75
Download citation
DOI: https://doi.org/10.1007/978-3-031-43987-2_75
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-031-43986-5
Online ISBN: 978-3-031-43987-2
eBook Packages: Computer ScienceComputer Science (R0)