
Abstract
The side-chain hydroxylation of cholesterol by specific enzymes produces 24(S)-hydroxycholesterol, 25-hydroxycholesterol, 27-hydroxycholesterol, and other products. These enzymatically formed side-chain oxysterols act as intermediates in the biosynthesis of bile acids and serve as signaling molecules that regulate cholesterol homeostasis. Besides these intracellular functions, an imbalance in oxysterol homeostasis is implicated in pathophysiology. Furthermore, growing evidence reveals that oxysterols affect cell proliferation and cause cell death. This chapter provides an overview of the pathophysiological role of side-chain oxysterols in developing human diseases. We also summarize our understanding of the molecular mechanisms underlying the induction of various forms of cell death by side-chain oxysterols.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Side-chain oxysterols
- 24(S)-hydroxycholesterol
- 25-hydroxycholesterol
- 27-hydroxycholesterol
- Endoplasmic reticulum
- Cell death
- Sterol regulatory element-binding protein
- Liver X receptor
- Acyl-CoA:cholesterol acyltransferase
- Integrated stress response
1 Enzymatically Formed Oxysterols
Oxysterols are sterols derived from the oxidation of cholesterol or its precursors and can be generated by autoxidation, enzymatically, or by both processes (Mutemberezi et al. 2016). Cholesterol oxidation can occur on the hydrocarbon rings (A or B) with an additional hydroxyl, epoxide, or ketone group, or on the side chain with a hydroxyl group. Several major oxysterols act as intermediates in the synthesis pathways of bile acids and steroid hormones (Wang et al. 2021). The hydroxylation of the side chain of cholesterol by specific enzymes generates oxysterols, particularly 24(S)-hydroxycholesterol (24S-OHC, also known as cerebrosterol), 25-hydroxycholesterol (25-OHC), 27-hydroxycholesterol (27-OHC, also known as [25R]-26-hydroxycholesterol), and other products (Fig. 10.1). This reaction is mediated by mitochondrial or microsomal cytochrome P450 enzymes, except for 25-OHC, which is produced by cholesterol 25-hydroxylase (CH25H). Side-chain oxysterols serve as physiological key regulators of cholesterol homeostasis and primarily function as signaling molecules for excess cholesterol levels (Griffiths and Wang 2022; Poli et al. 2022). This is due to the increased hydrophilicity of oxysterols with that of cholesterol, which allows them to move more rapidly between the intracellular membrane organelles and facilitates access to their receptors (Dias et al. 2019). While physiological levels of oxysterols are involved in numerous biological processes, an imbalance in oxysterol homeostasis has been implicated in pathophysiology. This chapter focuses on the importance of enzymatically formed oxysterols, especially side-chain oxysterols (24S-OHC, 25-OHC, and 27-OHC), in understanding the regulation of cholesterol homeostasis. We also provide an overview of the pathophysiological role of oxysterol homeostasis imbalance in the development of human diseases and discuss our understanding of the molecular mechanisms by which side-chain oxysterols induce cell death.

Synthesis and structures of enzymatically formed side-chain oxysterols. Oxysterols are formed by enzymatic activity or nonenzymatic reactions (autoxidation). Cytochrome P450 enzymes and cholesterol 25-hydroxylase (CH25H) catalyze the enzymatic formation of side-chain oxysterols
2 The Role of Oxysterols in Cholesterol Homeostasis
To maintain cellular cholesterol homeostasis, cholesterol levels are tightly regulated by a balance in uptake, storage, endogenous biosynthesis, and efflux (Luo et al. 2020). When cellular cholesterol levels become excessive, some of the cholesterol is enzymatically converted to side-chain oxysterols. Besides serving as substrates for bile acid biosynthesis in cholesterol excretion pathways, these oxysterols play essential roles in the feedback loop of cholesterol homeostasis. Because oxysterols can be transported more easily than cholesterol, they can function as important signaling molecules.
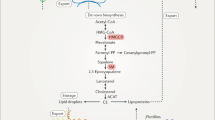
Oxysterols play critical roles in cholesterol homeostasis at the transcriptional and posttranslational levels (Fig. 10.2). One of the primary regulators by which oxysterol regulates cellular cholesterol homeostasis at the transcriptional level is the sterol regulatory element-binding protein 2 (SREBP-2), which is encoded by sterol regulatory element-binding factor 2 (SREBF2) (Brown et al. 2018). The SREBP family comprises three protein subtypes, termed SREBP-1a, -1c, and -2. SREBP-1 isoforms mainly regulate the expression of enzymes involved in fatty acid biosynthesis pathway, whereas SREBP-2 regulates the expression of proteins involved in cholesterol biosynthesis and uptake. When the endoplasmic reticulum (ER) cholesterol levels are high, SREBP-2 resides as an inactive precursor protein within the ER membrane through the formation of a complex with the SREBP cleavage-activating protein (Scap) (Hua et al. 1996) and insulin-induced genes (Insigs) (Yang et al. 2002). Cholesterol causes a conformational change in Scap, which triggers the association with Insigs and the retention of the Scap/SREBP-2 complex in the ER. When the ER cholesterol levels become low, Scap dissociates from the Insigs, allowing the transport of the Scap/SREBP-2 complex from the ER to the Golgi via coat protein complex II (COPII)-mediated vesicles. Two Golgi-resident proteases (Site-1 protease and Site-2 protease) sequentially cleave SREBP-2, releasing an NH2-terminal transcriptionally active mature form, which is transported to the nucleus. The mature form of SREBP-2 binds to SREs in the promoters of target genes and transactivates the genes involved in cholesterol biosynthesis (e.g., 3-hydroxy-3-methylglutaryl coenzyme A reductase [HMGCR] and HMGCS) and low-density lipoprotein receptor (LDLR) for cholesterol uptake (Horton et al. 2003). Whereas Scap senses cholesterol, Insigs sense 25-OHC and other side-chain oxysterols, causing the formation of SREBP-2/Scap/Insig complexes in the ER, thereby preventing SREBP-2 cleavage and the production of its active form (Adams et al. 2004; Radhakrishnan et al. 2007).

(a) The role of side-chain oxysterols in cholesterol homeostasis. Oxysterols are important regulators of cholesterol homeostasis. The transcriptional regulation by side-chain oxysterols is mediated by Insig/Scap/SREBP-2 for cholesterol uptake and biosynthesis or by LXR/RXR for cholesterol efflux. At the posttranslational regulation levels, side-chain oxysterols promote Insig-mediated degradation of HMGCR, which is a key enzyme in cholesterol biosynthesis, or allosteric activation of ACAT for cholesterol storage. (b) Mechanisms of side-chain oxysterol action in cholesterol homeostasis. (1) When cholesterol concentrations in the ER membrane are high, cholesterol binds to Scap, causing the retention of Scap/SREBP-2 in the ER, whereas oxysterols bind to Insigs, triggering their complex formation with Scap/SREBP-2 in the ER. (2) When the sterol levels are low, Scap dissociates from the Insigs, eliciting the transport of SREBP-2 to the Golgi, where the active form of SREBP-2 (mSREBP-2) is generated. mSREBP-2 subsequently induces the transcription of SRE-containing genes in the nucleus, which regulates cholesterol biosynthesis and uptake. (3) Oxysterols also act as ligands for LXR signaling, which regulates associated with cholesterol efflux. (4) Moreover, oxysterols cause the association of Insigs with HMGCR, promoting its proteasomal degradation. (5) Cholesterol and oxysterols induce allosteric ACAT activation, producing sterol esters that are incorporated into LDs for storage
Another transcription factor that controls cholesterol homeostasis through oxysterols is the liver X receptor (LXR) (Wang and Tontonoz 2018). There are two isoforms of LXR in mammals, termed LXRα (NR1H3) and LXRβ (NR1H2), which belong to a subclass of the nuclear receptor superfamily. Side-chain oxysterols are ligands of LXR (Janowski et al. 1996, 1999). Ligand-activated LXR forms heterodimers with retinoid X receptor (RXR), which is the receptor for 9-cis retinoic acid. Activating these transcription factors promotes cholesterol efflux and excretion by upregulating target genes, including ATP-binding cassette transporter A1 (ABCA1), ABCG1, and apolipoprotein E (ApoE).
Side-chain oxysterols also regulate cholesterol homeostasis at the posttranslational level. The mevalonate pathway is pivotal for the biosynthesis of cholesterol and other essential biomolecules, such as coenzyme Q (ubiquinone), dolichols, and isoprenoids (Goldstein and Brown 1990). The ER-resident protein HMGCR is a rate-limiting enzyme in the mevalonate pathway and a target of statins, which are cholesterol-lowering drugs (Schumacher and DeBose-Boyd 2021). HMGCR catalyzes the conversion of HMG-CoA to mevalonic acid. The increase in oxysterol level causes the binding of HMGCR to the Insigs, which initiates HMGCR ubiquitination and proteasomal degradation, thereby slowing cholesterol biosynthesis (Sever et al. 2003). HMGCR is a target gene of SREBP-2, suggesting that side-chain oxysterols regulate HMGCR both at the levels of transcription and enzymatic activity. Because cholesterol does not affect this Insig-mediated degradation of HMGCR, cholesterol synthesis can be inhibited by oxysterols even more potently and more rapidly than cholesterol.
Another posttranslational regulator of cholesterol homeostasis is acyl-CoA:cholesterol acyltransferase (ACAT), which is also known as sterol O-acyltransferase (SOAT). ACAT is an ER transmembrane protein that utilizes long-chain fatty acyl-CoA and cholesterol as substrates to form cholesteryl esters (Chang et al. 2009). Two ACAT isoenzymes, ACAT1 and ACAT2, have been identified in mammals. ACAT1 is expressed in many tissues, whereas ACAT2 is expressed predominantly in the liver and small intestine. Cholesteryl esters are stored in cytoplasmic lipid droplets (LDs) to prevent cytotoxic accumulation of free cholesterol or to serve as the precursors for steroid hormone production. In addition to cholesterol, other sterols that possess the 3β-hydroxy group, including oxysterols (such as 24S-OHC and 27-OHC), plant sterols, and pregnenolone, could be ACAT substrates (Rogers et al. 2015; Takabe et al. 2016). As a posttranslational regulation, both ACAT1 and ACAT2 function as allosteric enzymes activated not only by cholesterol but also by oxysterols (Zhang et al. 2003; Liu et al. 2005), which suggests that an increase in oxysterol levels promotes the conversion of excess cholesterol to cholesteryl esters. Because ACAT1 and ACAT2 do not have SRE in their promoter regions, oxysterol-mediated regulation of ACAT activity modulates cholesterol homeostasis independent of SREBP/Scap/Insigs-mediated regulation.
As noted, cellular cholesterol homeostasis is controlled by the precise regulation of transcriptional and posttranslational mechanisms that are sensitive to oxysterol levels. By controlling the production of nascent proteins, transcriptional regulation can respond efficiently to changes in cellular cholesterol concentrations but on relatively slow time scales of hours. In contrast, posttranslational regulation directly alters protein activities, allowing faster cellular responses to changes in cholesterol levels (Olsen et al. 2012). Notably, the physiological importance of side-chain oxysterols as regulators of cholesterol homeostasis in vivo remains uncertain because several studies have reported that mice with transgenic or knockout genes of side-chain oxysterol-producing enzymes showed modest changes in cholesterol homeostasis (Meir et al. 2002; Rosen et al. 1998; Lund et al. 2003; Björkhem 2009).
3 24S-OHC
3.1 24S-OHC and Brain Disorder
The brain is the most cholesterol-rich organ and contains approximately 25% of the total amount of cholesterol in the body (Björkhem and Meaney 2004). Because the blood–brain barrier prevents cholesterol translocation between the brain and systemic circulation, brain cholesterol levels are not affected by dietary cholesterol levels (Björkhem et al. 1997). In the adult brain, the major source of cholesterol is in situ biosynthesis by astrocytes; this cholesterol is delivered to neurons via ApoE-dependent transportation (Li et al. 2022). Therefore, to maintain a steady-state level of cholesterol in the brain, excess cholesterol is transported outside the brain mainly after its conversion to 24S-OHC by the neuronal cytochrome P450 enzyme cholesterol 24-hydroxylase (CYP46A1) (Russell et al. 2009). 24S-OHC can readily cross the blood–brain barrier by diffusion or using an organic anion transporting polypeptide 2 (Björkhem et al. 2019; Ohtsuki et al. 2007), after which 24S-OHC is transported to the liver, where it is metabolized to bile acids (Björkhem et al. 1997). In addition to their pivotal roles in maintaining brain cholesterol homeostasis, CYP46A1 and 24S-OHC affect higher-order brain functions, such as memory and learning, in a physiological setting (Li et al. 2022).
Growing evidence suggests that dysregulation of cholesterol homeostasis in the brain is associated with several neurodegenerative diseases, including Alzheimer’s disease (AD), Parkinson’s disease (PD), and Huntington’s disease (HD) (Dai et al. 2021; Chang et al. 2017). Moreover, increasing evidence suggests that CYP46A1 and 24S-OHC could be possible biomarkers of neurodegenerative diseases and may have a role in the pathogenesis or progression (Leoni and Caccia 2013; Noguchi et al. 2015; Sodero 2021). For example, polymorphisms in CYP46A1 are associated with AD (Kölsch et al. 2002). Selective expression of CYP46A1 around neuritic plaques has also been reported (Brown 3rd et al. 2004). Production of amyloid-β (Aβ) peptides is inhibited (Brown 3rd et al. 2004; Urano et al. 2013) or promoted by 24S-OHC (Gamba et al. 2014). Measurement of 24S-OHC levels in four different brain areas of healthy controls and patients with AD showed that 24S-OHC existed at an approximate concentration of 20 ng/mg tissue in controls, and its concentration was lower in patients with AD (Heverin et al. 2004). Assuming a volume of 1 μL for 1 mg tissue, the level of 24S-OHC in the brain tissue may be approximately 50 μM. Several studies reported that plasma 24S-OHC levels were lower in patients with AD than in controls (Bretillon et al. 2000; Kölsch et al. 2004), which may be caused by decreased CYP46A1 levels due to neuronal cell death. However, several studies also reported elevated 24S-OHC levels in the plasma (Lütjohann et al. 2000; Zuliani et al. 2011) and cerebrospinal fluid (CSF) (Schönknecht et al. 2002) of patients with AD or mild cognitive impairment. This finding may suggest increased brain cholesterol turnover as a result of the early stage of neurodegeneration. Differences in disease progression between the patients may explain the conflicting findings of these increased and decreased 24S-OHC levels.
In the case of patients with PD, unchanged or lower levels of 24S-OHC in the plasma but significantly increased levels of 24S-OHC in the CSF relative to the levels in the controls were reported (Björkhem et al. 2013). Furthermore, a significant correlation was observed between 24S-OHC levels in the CSF and disease duration. In the case of HD, plasma 24S-OHC levels were lower in patients with HD than in controls at all disease stages (Leoni et al. 2013). Change in 24S-OHC levels in the plasma or CSF was further reported in brain disorders, such as multiple sclerosis (MS) (Leoni et al. 2002; Fellows Maxwell et al. 2019), amyotrophic lateral sclerosis (ALS) (Hartmann et al. 2022), and autism spectrum disorders (ASD) (Grayaa et al. 2018). In the case of drug-free patients with schizophrenia (SZ), 24-OHC levels in plasma were not significantly different between SZ patients and healthy controls (Guidara et al. 2022). In the SZ group, plasma 24-OHC levels were positively correlated with the positive and negative syndrome scale indicating the severity of symptoms. Furthermore, a link exists between CYP46A1 and retinal diseases, such as age-related macular degeneration (AMD) and glaucoma. For example, plasma 24-OHC levels were specifically associated with AMD (Lin et al. 2018). An association between CYP46A1 polymorphism and glaucoma has also been reported (Fourgeux et al. 2009).
Moreover, 24-OHC is a positive allosteric modulator of N-methyl-D-aspartate receptors (NMDARs) (Paul et al. 2013; Sun et al. 2016). Because NMDARs are ionotropic glutamate receptors for excitatory neurotransmission throughout the central nervous system, NMDAR functions are crucial for synaptic plasticity and cognition. In HD neurons and mouse models, CYP46A1 overexpression shows neuroprotective effects (Boussicault et al. 2016, 2018).
Although some conflicting findings remain to be clarified, the quantification of 24S-OHC is important in advancing our understanding of neurological disorders. More neurological status likely needs to be assessed for changes in 24S-OHC levels in the plasma or CSF.
3.2 24S-OHC-Induced Neuronal Cell Death
The types of cell death are broadly divided into accidental cell death (ACD) and regulated cell death (RCD). ACD is an uncontrolled process of cell death. In contrast, RCD is a tightly controlled process regulated by complex signaling pathways to eliminate cells that are no longer needed, damaged, or harmful in a targeted fashion. It plays a significant role in both physiological and pathophysiological processes, such as embryonic development, tissue homeostasis, aging, inflammation, and immunity (Galluzzi et al. 2018). Inappropriate neuronal cell death is involved in many neurodegenerative diseases. Beyond the classically defined apoptosis, growing evidence has revealed various forms of nonapoptotic RCD and new pathways of cell death machinery (Tang et al. 2019).
Because 24S-OHC at higher concentrations has been shown to induce neuronal cell death, it is presumed to be involved in the etiology of neurodegenerative diseases (Noguchi et al. 2014, 2015). Using human neuroblastoma SH-SY5Y cell lines, the cytotoxic mechanism of 24S-OHC has been well investigated. At concentrations higher than 10 μM, 24S-OHC induces cell death in SH-SY5Y cells and primary cortical neuronal cells (Kölsch et al. 1999; Yamanaka et al. 2011). Moreover, 24S-OHC-treated SH-SY5Y cells exhibited neither typical apoptotic features, such as nuclear fragmentation and caspase-3 activation, nor the necrotic feature, ATP depletion. Instead, 24S-OHC-treated cells showed necroptosis-like cell death features. Necroptosis is characterized as a type of RCD that is necrosis-like, caspase-independent, and mediated through a pathway dependent on receptor-interacting serine/threonine kinase 3 (RIPK3) and mixed lineage kinase domain-like (MLKL) and (at least in some settings) the kinase activity of RIPK1 (Yuan et al. 2019). Caspase-8 is known to be the molecular switch for extrinsic apoptosis and necroptosis (Yuan et al. 2016). SH-SY5Y cells lack caspase-8 expression, which is responsible for the absence of apoptotic features in response to 24S-OHC (Yamanaka et al. 2011). Although 24S-OHC-induced cell death is effectively inhibited using a RIPK1 inhibitor or RIPK1 siRNA-mediated knockdown, neither RIPK3 nor MLKL is expressed, suggesting that 24S-OHC induces a necroptosis-like but unconventional type of RCD in SH-SY5Y cells (Vo et al. 2015). When SH-SY5Y cells were differentiated using all-trans-retinoic acid in which caspase-8 expression is induced, the exposure to 24S-OHC induced apoptosis as evidenced by caspase-3 activation (Kölsch et al. 2001; Nakazawa et al. 2017). In caspase-8-expressing human T-lymphoma Jurkat cells, 24S-OHC also induced apoptosis (Yamanaka et al. 2014).
Further studies demonstrated that ACAT1-catalyzed esterification of 24S-OHC at the 3β-hydroxyl group with unsaturated long-chain fatty acids in the ER is responsible for initial key pro-cell death events during 24S-OHC-induced cell death in SH-SY5Y cells (Yamanaka et al. 2014; Takabe et al. 2016) (Fig. 10.3). Consequently, abnormal accumulation of 24S-OHC esters between the two leaflets of the ER membrane bilayer evoked the formation of an LD-like structure coupled with an enlarged ER structure, resulting in the disruption of ER membrane integrity and release of ER luminal chaperone proteins into the cytosol (Urano et al. 2019). In general, a decrease in protein folding capacity in the ER causes unfolded protein response (UPR) to restore the ER homeostasis and maintain the fidelity of protein folding (Hetz 2012). Although UPR is a prosurvival adaptive response, under unresolvable ER stress conditions, the UPR represses the adaptive response and even triggers cell death (Kim et al. 2008; Tabas and Ron 2011). Indeed, 24S-OHC-induced ER dysfunction is accompanied by the activation of pro-death UPR. The ER dysfunction and integrated stress response (ISR) described below are the key signaling pathways for 24S-OHC-induced cell death (Urano et al. 2019, 2022).

Schematic representation of the mechanism proposed for 24S-OHC-induced cell death in SH-SY5Y cells. Under conditions of excess 24S-OHC levels in SH-SY5Y cells, ACAT1 catalyzes 24S-OHC esterification with unsaturated long-chain fatty acid in the ER. Accumulation of 24S-OHC esters within the ER membrane bilayer leads to the formation of an LD-like structure coupled with an enlarged ER structure, resulting in the disruption of ER membrane integrity, which in turn induces the release of ER luminal proteins into the cytosol. ER stress and/or disruption of the ER membrane integrity activates pro-death UPR signaling, including RIDD. ISR activation through PERK and GCN2 phosphorylation causes the phosphorylation of eIF2α and subsequent inhibition of protein synthesis and the formation of SGs. ISR, UPR, and ER dysfunction together lead to disruption of protein homeostasis and ultimately result in the induction of an unconventional type of cell death by 24S-OHC
ISR and UPR constitute overlapping signaling pathways mediated by eukaryotic translation initiator factor 2α (eIF2α) (Hetz et al. 2020; Pakos-Zebrucka et al. 2016). UPR is activated by the accumulation of unfolded/misfolded proteins in the ER. In mammalian cells, the UPR employs three main signaling pathways, including activating transcription factor 6 (ATF6), inositol-requiring enzyme 1 (IRE1), and protein kinase R-like ER kinase (PERK) pathways. Of these, the PERK pathway is responsible for a part of the ISR signaling pathway (Pakos-Zebrucka et al. 2016). ISR is an evolutionarily conserved signaling pathway activated in response to intracellular and extracellular disturbances (Costa-Mattioli and Walter 2020). Disturbance in protein homeostasis is the main cause of ISR signaling activation. Although ISR primarily serves as a prosurvival cell response, severe or prolonged stress leads ISR signaling toward cell death (Liu et al. 2015; Rutkowski et al. 2006). Activated PERK (via autophosphorylation) functions as an eIF2α kinase. In addition to PERK, the ISR sensor relies on three other eIF2α kinases, including general control nonderepressible 2 (GCN2), heme-regulated eIF2α kinase (HRI), and double-stranded RNA-dependent protein kinase R (PKR). GCN2 is activated by amino acid deprivation and UV light exposure, HRI responds to heme deficiency, and PKR is activated during viral infections. Phosphorylated eIF2α sequesters the eIF2, thereby inhibiting the assembly of a 43S translation initiation complex and causing the general attenuation of 5′ cap-dependent protein synthesis (Liu et al. 2015). Moreover, phosphorylation of eIF2α enhances the translation of selective mRNAs, including not only those that encode prosurvival proteins but also those that encode proapoptotic proteins. The global inhibition of protein translation caused by ISR induces the formation of cytoplasmic membraneless ribonucleoprotein-based compartments known as stress granules (SGs) by liquid–liquid phase separation. While SGs sequester mRNAs upon stress and maintain a translation arrest until recovery from stress, persistent or aberrant SG formation is implicated in disease pathology and cell death (Reineke and Neilson 2019).
Intriguingly, 24S-OHC triggers eIF2α phosphorylation and subsequent SG formation (Urano et al. 2022). Inhibition of ISR signaling suppresses 24S-OHC-induced SG formation and cell death, suggesting that ISR and SG formation are involved in 24S-OHC-induced cell death. Moreover, 24S-OHC-induced ISR is activated through PERK and GCN2 activation, which downregulates global protein de novo synthesis. Inhibition of ACAT1 suppresses the activation of both PERK and GCN2, as well as downstream eIF2α activation, suggesting that ACAT1-catalyzed esterification of 24S-OHC is responsible for the activation of ISR. Furthermore, 24S-OHC-induced ER dysfunction has been proposed to drive the activation of PERK signaling, although it is not clear how 24S-OHC causes the activation of GCN2 signaling. Because 25-OHC has also been reported to activate GCN2 in an LXR- and SREBP-independent manner in bone-marrow-derived macrophages (Shibata et al. 2013), oxysterols may have some common machinery for GCN2 activation. In addition to the PERK pathway, the IRE1 pathway via regulated IRE1-dependent decay (RIDD) is also implicated in 24S-OHC-induced pro-death UPR signaling (Urano et al. 2019). RIDD is the mechanism by which the RNase activity of activated IRE1 upon UPR induces posttranscriptional degradation of a subset of ER-localized mRNAs (Hollien and Weissman 2006; Maurel et al. 2014).
From food-derived products, vitamin E homologs α- and γ-tocopherol have been found to significantly suppress 24S-OHC-induced cell death in SH-SY5Y cells (Nakazawa et al. 2017; Kimura et al. 2018). Vitamin E is a family of eight isoforms, namely α-, β-, γ-, and δ-tocopherols and α-, β-, γ-, and δ-tocotrienols. All vitamin E homologs comprise a chromanol ring and an aliphatic side chain. Tocopherols have a saturated side chain, whereas tocotrienols have an unsaturated side chain containing three unsaturated double bonds. Both tocopherols and tocotrienols are potent lipid-soluble antioxidants, but only tocopherols exert inhibitory effects on 24S-OHC-induced cell death in SH-SY5Y cells. Cotreatment with α-tocopherol but not α-tocotrienol effectively suppressed 24S-OHC-induced UPR and ER membrane disruption (Chiba et al. 2023). Neither reactive oxygen species (ROS) generation nor lipid peroxidation was observed in 24S-OHC-treated cells. Therefore, α-tocopherol with a saturated side chain may protect the ER membrane from 24S-OHC-induced disruption through its nonantioxidant activity.
3.3 24S-OHC-Induced Cell Death in Other Cell Types
The cytotoxicity of 24S-OHC has also been examined in other cell types. In human hepatic cells (HepG2 cells), 24S-OHC caused caspase- and ACAT-mediated esterification-independent cell death that was suppressed by both α-tocopherol and α-tocotrienol, suggesting the involvement of free radical-mediated lipid peroxidation in 24S-OHC-induced cell death in hepatic cells (Suzuki et al. 2021). In human keratinocytes (HaCaT cells), 24S-OHC caused a caspase-dependent but ACAT-mediated esterification-independent cell death that was inhibited by α-tocopherol but not α-tocotrienol (Suzuki et al. 2021). In murine oligodendrocytes 158N cells, microglial BV-2, and neuroblastoma N2a cells, 24S-OHC induced a type of cell death termed oxiapoptophagy, which was characterized by oxidative stress and several features of apoptosis and autophagy (Nury et al. 2015, 2021). Furthermore, α-tocopherol prevented 24S-OHC-induced oxiapoptophagy. However, it is unclear whether ACAT-mediated esterification is involved in 24S-OHC-induced oxiapoptophagy.
Interestingly, CYP46A1 was identified as one of the most dramatically dysregulated cholesterol metabolism genes in glioblastoma (Han et al. 2020). A reduction in CYP46A1 expression was associated with an increase in tumor grade and a worse prognosis in patients with glioblastoma. Ectopic expression of CYP46A1 suppressed cell proliferation of glioblastoma cells in vitro and in xenografts by increasing the 24S-OHC levels. Treatment of glioblastoma cells with 24S-OHC increased apoptosis in a dose-dependent manner. Furthermore, restoration of CYP46A1 activity using its activator, efavirenz, inhibits glioblastoma growth by regulating LXR and SREBP-1 activities. These data demonstrated that the CYP46A1/24S-OHC axis is a potential target for glioblastoma therapy.
Together, these findings suggest that 24S-OHC induces different types of cell death through various mechanisms in a cell-type-dependent manner. Considering the involvement of 24S-OHC in neurodegenerative diseases, further studies in a nonproliferative neuronal culture, including iPS cell-derived neuronal culture, stem cell-derived neuronal culture, and the neuron-glia coculture system, are warranted to understand the molecular actions of 24S-OHC.
3.4 Protective or Damage-Promoting Effects of 24S-OHC Against Other Cytotoxic Stimulations
The cytoprotective effects of 24S-OHC have also been demonstrated. 7-Ketocholesterol (7-KC) is an oxysterol generated by the autoxidation of cholesterol. Because 7-KC has been shown to have a highly cytotoxic potential in neuronal cells, it may contribute to the pathogenesis of neurodegenerative diseases. Treatment with 24S-OHC at sublethal concentrations showed a significant reduction in cell death induced by subsequent treatment with 7-KC in both undifferentiated and retinoic acid-differentiated SH-SY5Y cells (Okabe et al. 2013). Cotreatment of 24S-OHC with the RXR ligand promoted the cytoprotective effects of 24S-OHC against 7-KC-induced cell death. Knockdown of LXR or ABCG1 using siRNA significantly diminished 24S-OHC-induced cytoprotective effects. This finding suggests that 24S-OHC at sublethal concentrations induces adaptive responses via the activation of the LXR/RXR pathway, thereby protecting cells from the subsequent 7-KC-induced cytotoxicity. It has also been reported that 24S-OHC at low concentrations protects cells from cell death induced using staurosporine, which is a toxic substance that induces apoptosis (Emanuelsson and Norlin 2012). In addition, a low concentration of 24S-OHC stimulated cellular processes critical to maintaining redox homeostasis and showed a protective action against oxidative stress in human glioblastoma U-87 MG cells (Cigliano et al. 2019).
Regarding the damage-promoting effects of 24S-OHC, it was reported that 24S-OHC at low concentrations but not 27-OHC or 7β-hydroxycholesterol enhanced Aβ binding to human differentiated neuronal cell lines by the upregulation of a multireceptor complex involving CD36 and β1-integrin (Gamba et al. 2011). In addition, 24S-OHC promoted the generation of NADPH oxidase-dependent generation of ROS, resulting in the disruption of the redox equilibrium and potentiation of Aβ42 neurotoxicity.
4 25-OHC
4.1 25-OHC and Related Diseases
In the plasma, 25-OHC has been reported to be a minor side-chain oxysterol compared with 24S-OHC and 27-OHC (Mutemberezi et al. 2016). ER-resident CH25H is a key enzyme for the production of 25-OHC (Cao et al. 2020). Expression of CH25H is found in macrophages and the liver, and it is regulated in response to immune and inflammatory conditions and in an LXR-dependent manner (Liu et al. 2018; Park and Scott 2010). Besides CH25H, other cytochrome P450 enzymes, such as CYP3A4, CYP46A1, and sterol 27-hydroxylase (CYP27A1), and even ROS-induced reactions can generate 25-OHC (Diczfalusy and Björkhem 2011; Honda et al. 2011; Diczfalusy 2013). However, the importance of these reactions in vivo remains unclear.
Growing evidence indicates that 25-OHC is involved in antiviral process and inflammatory immune response (Cyster et al. 2014). CH25H expression is induced in response to toll-like receptor ligands and the subsequent signaling through the interferon receptor/Janus kinase (JAK)/signal transducer and activator of transcription 1 (STAT1) pathway in human macrophages (Park and Scott 2010). The subsequently produced 25-OHC shows antiviral activities against a broad range of enveloped viruses (Blanc et al. 2013; Liu et al. 2013). Furthermore, 25-OHC has been shown to block membrane fusion between the virus and host cell, ultimately inhibiting viral replication, but the exact mechanism may vary depending on the virus and cell type. Recently, 25-OHC has been reported to suppress the replication of severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2), which causes novel coronavirus disease-2019 (COVID-19), by blocking membrane fusion (Zang et al. 2020; Wang et al. 2020). Regarding cytokine production, 25-OHC can have both pro-inflammatory and anti-inflammatory roles (Reboldi et al. 2014).
4.2 25-OHC-Induced Cell Death
It has been reported that 25-OHC induces cell death by different mechanisms in various types of cells. For example, it induces apoptosis in human aortic smooth muscle cells (Ares et al. 1997). Moreover, it induces Ca2+ influx through plasma membrane channels. Using Ca2+ entry blocker effectively inhibited apoptosis, suggesting that Ca2+ plays a crucial role in 25-OHC-induced cell death. Furthermore, 25-OHC induced apoptosis in vascular smooth muscle cells, resulting in the promotion of vascular calcification (Dong et al. 2020). Activation of pro-death UPR signaling, including increases in ATF4 and CCAAT/enhancer-binding protein homologous protein (CHOP) expression, has been implicated in 25-OHC-induced apoptosis and vascular calcification. Another report showed that soluble adenylyl cyclase (sAC) played a key role in the 25-OHC-induced apoptosis of rat vascular smooth muscle cells by controlling protein kinase A (PKA)-dependent phosphorylation as well as mitochondrial translocation of Bax and mitochondrial ROS formation (Appukuttan et al. 2013).
Oxysterol-binding protein-related protein 4L (ORP4L) has been reported to bind 25-OHC and be involved in cell proliferation and survival (Charman et al. 2014). In macrophages, excessive 25-OHC led to the disassembly of the ORP4L/Gαq/11/phospholipase C (PLC)-β3 complex, resulting in disruption of Ca2+ signaling, a decrease of antiapoptotic Bcl-XL expression, and an increase in apoptosis (Zhong et al. 2016). In thioglycolate-elicited peritoneal macrophages, 25-OHC caused ER stress, including CHOP induction and subsequent apoptosis (Sekiya et al. 2014). A deficiency of neutral cholesterol ester hydrolase 1 (NCEH1) promoted the accumulation of the 25-OHC ester in the ER due to its defective hydrolysis, resulting in the augmentation of 25-OHC-induced apoptosis. Moreover, ACAT1 inhibition suppressed NCEH1-dependent augmentation of 25-OHC-induced ER stress and apoptosis. These findings suggest that the esterification of 25-OHC in the ER plays an important role, and ACAT and NCEH1 can exert proapoptotic and antiapoptotic effects, respectively, in 25-OHC-induced macrophage cell death.
Oxysterol-binding protein-related protein 8 (ORP8) is another member of the ORP family and it binds to 25-OHC (Yan et al. 2008). In hepatic cell lines, HepG2 and Huh7, 25-OHC induced apoptosis via the pro-death UPR pathway (Li et al. 2016). ORP8 knockdown diminished 25-OHC-induced ER stress and apoptosis, suggesting that ORP8 mediates the cytotoxicity of 25-OHC. Cotreatment with either α-tocopherol or α-tocotrienol effectively suppressed 25-OHC-induced HepG2 cell death (Suzuki et al. 2021). The accumulation of lipid hydroperoxides was also observed in 25-OHC-treated HepG2 cells, suggesting the involvement of free radical-mediated lipid peroxidation in 25-OHC-induced cell death. In a rat liver tumorigenesis model, antitumor effects of 25-OHC were reported to induce the entry of rat AH136B ascites hepatoma cells into the sub-G1 phase (Yokoyama et al. 1999). Anticancer effects of 25-OHC were also observed in head and neck squamous cell carcinoma cells (You et al. 2020).
UV irradiation has been shown to increase 25-OHC levels but not 24S-OHC or 27-OHC levels in human primary keratinocytes (Olivier et al. 2017). In human keratinocyte HaCaT cells, 25-OHC caused caspase-3-dependent apoptosis (Suzuki et al. 2021) and caspase-1-dependent pyroptosis (Olivier et al. 2017). Pyroptosis is characterized as a type of RCD that is caused by the caspase-1/4/5/11-dependent formation of gasdermin family-mediated pores on the plasma membrane, leading to the release of pro-inflammatory cytokines and cell rupture (Galluzzi et al. 2018; Vandenabeele et al. 2022). Because the P2X7 receptor and caspase-1 were activated by 25-OHC, the P2X7/NLRP3 inflammasome signaling pathway was implicated in 25-OHC-induced pyroptosis in HaCaT cells.
In the CSF and serum of patients with untreated ALS, the 25-OHC levels were higher than those in the control and treatment groups, and the serum 25-OHC levels were associated with the disease severity and progression rate (Kim et al. 2017). Increased expression of CH25H and CYP3A4 mRNA was also observed in the early symptomatic stages of ALS model mice. In the motor neuron-like cell line (NSC-34) expressing human G93A mutant of superoxide dismutase 1, 25-OHC was shown to induce apoptosis. Cotreatment of 22(S)-hydroxycholesterol as an LXR antagonist significantly attenuated cell death, indicating the involvement of the LXR signaling pathway in 25-OHC-induced motor neuron cell death.
In addition to the observation in thioglycolate-elicited peritoneal macrophages (Sekiya et al. 2014), ACAT inhibition also suppressed 25-OHC-induced cell death in SH-SY5Y cells, but not in HepG2 and HaCaT cells (Suzuki et al. 2021). As noted, because UPR/ER stress causes 25-OHC-induced cell death in many types of cells, it is of interest to verify the cytoprotective effects of ACAT inhibition in those cells.
5 27-OHC
5.1 27-OHC and Related Diseases
It has been shown that 27-OHC is the most abundant oxysterol in circulation and an important intermediate in cholesterol catabolism to bile acid biosynthesis (Kim et al. 2022). In the mitochondria, CYP27A1 mediates the enzymatic synthesis of 27-OHC from cholesterol. In the liver, 27-OHC is further catabolized toward bile acids by oxysterol 7α-hydroxylase (CYP7B1). Although CYP27A1 is mainly expressed in the liver, it is also expressed in macrophages, the brain, and the lung, indicating that 27-OHC concentration can be locally regulated in nonhepatic tissues (Kim et al. 2022). Bile acids have been reported to suppress the transcription of CYP27A1 (Chen and Chiang 2003). Mice with CYP27A1 deficiency had normal plasma levels of cholesterol and markedly reduced levels of bile acid synthesis (Rosen et al. 1998) and showed hepatomegaly and hypertriglyceridemia (Repa et al. 2000). Mutations in human CYP27A1 have been linked to a rare autosomal recessive lipid storage disease called cerebrotendinous xanthomatosis, which is characterized by progressive dementia, xanthomatosis, and accelerated atherosclerosis (Cali et al. 1991). The diverse phenotypes observed in patients with cerebrotendinous xanthomatosis demonstrate the importance of CYP27A1 in bile acid biosynthesis, reverse cholesterol transport, and vitamin D3 biosynthesis.
Accumulating evidence indicates that 27-OHC is related to neurodegenerative diseases. This oxysterol is synthesized at low levels in neurons and glial cells (Russell 2000). The peripherally derived 27-OHC crosses the blood–brain barrier via free diffusion (Heverin et al. 2005). In the case of patients with AD, 27-OHC levels in the CSF are elevated in AD and mild cognitive impairment subjects compared with those in the controls (Wang et al. 2016b), which could be attributed to dysfunction of the blood–brain barrier and blood–CSF barrier. Moreover, 27-OHC increased Aβ levels by increasing the levels of β-secretase (Marwarha et al. 2013) and decreasing the levels of the insulin-degrading enzyme (IDE), which is known as an Aβ-degrading enzyme (Zhang et al. 2019). Furthermore, 27-OHC also increased phosphorylated tau levels (Marwarha et al. 2010). In the case of PD, 27-OHC reduced the levels of tyrosine hydroxylase, which is the rate-limiting enzyme in dopamine synthesis, and increased the levels of α-synuclein, which is the major component of Lewy bodies in SH-SY5Y cells (Marwarha et al. 2011). In patients with hereditary spastic paraplegia type 5 (SPG5) with mutations in CYP7B1, a marked accumulation of 27-OHC was observed (Schüle et al. 2010).
It has been established that 27-OHC functions as an endogenous selective estrogen receptor modulator (SERM). SERM is an estrogen receptor ligand that shows agonist or antagonist activity in a cell- and promoter-dependent manner. In the vasculature, 27-OHC acted as an estrogen receptor α antagonist and promoted atherosclerosis progression via pro-inflammatory processes (Umetani et al. 2014). However, 27-OHC also acted as an agonist for estrogen receptor α and promoted the proliferation of cancer cells and tumorigenesis (Kim et al. 2022). The pro-tumor properties of 27-OHC were found to be associated with various cancers, notably breast cancer (Nelson et al. 2013). Furthermore, 27-OHC increased tumor metastasis due to increased myeloid immune cell function and decreased cytotoxic CD8+T lymphocytes (Baek et al. 2017). In addition, it also induced hematopoietic stem cell mobilization and extramedullary hematopoiesis during pregnancy by regulating estrogen receptor α function (Oguro et al. 2017).
5.2 27-OHC-Induced Cell Death
It has been shown that 27-OHC induces apoptotic cell death in different cell types. There are two different types of apoptotic pathways: one is initiated when stress occurs within the cell (the intrinsic pathway) and the other is triggered by the binding of ligands to cell surface death receptors (the extrinsic pathway). It has been reported that 27-OHC activates both intrinsic and extrinsic apoptotic pathways. For example, 27-OHC treatment increased apoptosis via increased ROS levels and ER stress in hematopoietic stem and progenitor cells (Woo et al. 2022). Exogenous 27-OHC treatment also promoted ROS production and apoptosis in leukemia cells. In contrast, 27-OHC was shown to evoke the extrinsic pathway by inducing the production of tumor necrosis factor-α in macrophages (Kim et al. 2013; Umetani et al. 2014).
Other types of cell death processes are also observed in 27-OHC-induced cell death. In colon cancer Caco-2 cells, a high concentration of 27-OHC induced nonapoptotic cell death (Warns et al. 2018). These effects are independent of LXR or estrogen receptor activation and are due to decreased activation of Akt, which is associated with cell proliferation. In cocultured SH-SY5Y cells and astrocyte C6 cells, 27-OHC induced apoptosis associated with the reduction of mitochondrial membrane potential (Wang et al. 2016a) and pyroptosis by causing lysosomal membrane permeabilization and subsequent cathepsin B leakage into the cytosol (Chen et al. 2019). However, it remains unclear how 27-OHC damages mitochondria and lysosomes.
Ferroptosis is a type of programmed cell death that is characterized by iron-dependent peroxidation of polyunsaturated phospholipids in cell membranes (Galluzzi et al. 2018; Yang and Stockwell 2016). Recent work suggests that ferroptosis plays a pivotal role in tumor suppression (Li et al. 2020). In estrogen receptor α-negative cancer cell lines, acute exposure to 27-OHC attenuated cell growth and migration by disrupting lipid metabolism through interfering with SREBPs signaling (Liu et al. 2021). Chronic exposure to 27-OHC led to the selection of 27-OHC-resistant cells, which induced adaptive responses to ferroptosis by upregulating the activity of processes that allowed the cells to withstand lipid oxidative stress. Moreover, 27-OHC-resistant cells were more tumorigenic and metastatic when evaluated in an in vivo model. The enhanced tumorigenic and metastatic activities of 27-OHC-resistant cells were attenuated by inhibition of the phospholipid hydroperoxide-reducing enzyme glutathione peroxidase 4 to increase the sensitivity to ferroptosis. Because circulating 27-OHC levels are elevated in hypercholesterolemia, the possibility that 27-OHC impacts cancer pathogenesis by selecting cells that are resistant to ferroptosis was considered (Liu et al. 2021).
6 Concluding Remarks
Growing evidence indicates the importance of side-chain oxysterols in various pathogeneses. Side-chain oxysterols induce various forms of cell death depending on the hydroxylated site of cholesterol and the cell types; they also exert protective effects against cell death caused by other stresses. These functions are thought to be due to the various physiological activities of side-chain oxysterols at the transcriptional and posttranslational levels. Suppression of oxysterol-induced cell death is considered effective for disease treatment. Inhibitors of side-chain oxysterol-producing enzymes may be therapeutic agents for diseases that are caused by oxysterol-induced cell death. ACAT inhibitors are also promising drugs for cell death caused by the esterification of oxysterols. Thus, further studies of the roles of side-chain oxysterols in various pathogeneses may pave the way for the development of therapeutic reagents for oxysterol-related diseases.
References
Adams CM et al (2004) Cholesterol and 25-hydroxycholesterol inhibit activation of SREBPs by different mechanisms, both involving SCAP and Insigs. J Biol Chem 279:52772–52780. https://doi.org/10.1074/jbc.m410302200
Appukuttan A et al (2013) Oxysterol-induced apoptosis of smooth muscle cells is under the control of a soluble adenylyl cyclase. Cardiovasc Res 99:734–742. https://doi.org/10.1093/cvr/cvt137
Ares MP et al (1997) Ca2+ channel blockers verapamil and nifedipine inhibit apoptosis induced by 25-hydroxycholesterol in human aortic smooth muscle cells. J Lipid Res 38:2049–2061. https://doi.org/10.1016/S0022-2275(20)37135-2
Baek AE et al (2017) The cholesterol metabolite 27 hydroxycholesterol facilitates breast cancer metastasis through its actions on immune cells. Nat Commun 8:864. https://doi.org/10.1038/s41467-017-00910-z
Björkhem I (2009) Are side-chain oxidized oxysterols regulators also in vivo? J Lipid Res 50(Suppl):S213–S218. https://doi.org/10.1194/jlr.r800025-jlr200
Björkhem I, Meaney S (2004) Brain cholesterol: long secret life behind a barrier. Arterioscler Thromb Vasc Biol 24:806–815. https://doi.org/10.1161/01.atv.0000120374.59826.1b
Björkhem I et al (1997) Importance of a novel oxidative mechanism for elimination of brain cholesterol. Turnover of cholesterol and 24(S)-hydroxycholesterol in rat brain as measured with 18O2 techniques in vivo and in vitro. J Biol Chem 272:30178–30184. https://doi.org/10.1074/jbc.272.48.30178
Björkhem I et al (2013) Oxysterols and Parkinson’s disease: evidence that levels of 24S-hydroxycholesterol in cerebrospinal fluid correlates with the duration of the disease. Neurosci Lett 555:102–105. https://doi.org/10.1016/j.neulet.2013.09.003
Björkhem I et al (2019) On the fluxes of side-chain oxidized oxysterols across blood-brain and blood-CSF barriers and origin of these steroids in CSF. J Steroid Biochem Mol Biol 188:86–89. https://doi.org/10.1016/j.jsbmb.2018.12.009
Blanc M et al (2013) The transcription factor STAT-1 couples macrophage synthesis of 25-hydroxycholesterol to the interferon antiviral response. Immunity 38:106–118. https://doi.org/10.1016/j.immuni.2012.11.004
Boussicault L et al (2016) CYP46A1, the rate-limiting enzyme for cholesterol degradation, is neuroprotective in Huntington’s disease. Brain 139:953–970. https://doi.org/10.1093/brain/awv384
Boussicault L et al (2018) CYP46A1 protects against NMDA-mediated excitotoxicity in Huntington’s disease: Analysis of lipid raft content. Biochimie 153:70–79. https://doi.org/10.1016/j.biochi.2018.07.019
Bretillon L et al (2000) Plasma levels of 24S-hydroxycholesterol in patients with neurological diseases. Neurosci Lett 293:87–90. https://doi.org/10.1016/s0304-3940(00)01466-x
Brown J 3rd et al (2004) Differential expression of cholesterol hydroxylases in Alzheimer’s disease. J Biol Chem 279:34674–34681. https://doi.org/10.1074/jbc.M402324200
Brown MS et al (2018) Retrospective on cholesterol homeostasis: the central role of scap. Annu Rev Biochem 87:783–807. https://doi.org/10.1146/annurev-biochem-062917-011852
Cali JJ et al (1991) Mutations in the bile acid biosynthetic enzyme sterol 27-hydroxylase underlie cerebrotendinous xanthomatosis. J Biol Chem 266:7779–7783. https://doi.org/10.1016/S0021-9258(20)89518-0
Cao Q et al (2020) Multiple roles of 25-hydroxycholesterol in lipid metabolism, antivirus process, inflammatory response, and cell survival. Oxid Med Cell Longev 2020:8893305. https://doi.org/10.1155/2020/8893305
Chang TY et al (2009) Acyl-coenzyme A:cholesterol acyltransferases. Am J Physiol Endocrinol Metab 297:E1–E9. https://doi.org/10.1152/ajpendo.90926.2008
Chang TY et al (2017) Cellular cholesterol homeostasis and Alzheimer’s disease. J Lipid Res 58:2239–2254. https://doi.org/10.1194/jlr.r075630
Charman M et al (2014) Oxysterol-binding protein (OSBP)-related protein 4 (ORP4) is essential for cell proliferation and survival. J Biol Chem 289:15705–15717. https://doi.org/10.1074/jbc.M114.571216
Chen W, Chiang JY (2003) Regulation of human sterol 27-hydroxylase gene (CYP27A1) by bile acids and hepatocyte nuclear factor 4alpha (HNF4alpha). Gene 313:71–82. https://doi.org/10.1016/S0378-1119(03)00631-0
Chen S et al (2019) 27-Hydroxycholesterol contributes to lysosomal membrane permeabilization-mediated pyroptosis in co-cultured SH-SY5Y cells and C6 cells. Front Mol Neurosci 12:14. https://doi.org/10.3389/fnmol.2019.00014
Chiba R et al (2023) α-Tocopherol suppresses 24(S)-hydroxycholesterol-induced cell death via inhibition of endoplasmic reticulum membrane disruption. Steroids 189:109136. https://doi.org/10.1016/j.steroids.2022.109136
Cigliano L et al (2019) 24S-hydroxycholesterol affects redox homeostasis in human glial U-87 MG cells. Mol Cell Endocrinol 486:25–33. https://doi.org/10.1016/j.mce.2019.02.013
Costa-Mattioli M, Walter P (2020) The integrated stress response: From mechanism to disease. Science 368:eaat5314. https://doi.org/10.1126/science.aat5314
Cyster JG et al (2014) 25-Hydroxycholesterols in innate and adaptive immunity. Nat Rev Immunol 14:731–743. https://doi.org/10.1038/nri3755
Dai L et al (2021) Cholesterol metabolism in neurodegenerative diseases: molecular mechanisms and therapeutic targets. Mol Neurobiol 58:2183–2201. https://doi.org/10.1007/s12035-020-02232-6
Dias IH et al (2019) Localisation of oxysterols at the sub-cellular level and in biological fluids. J Steroid Biochem Mol Biol 193:105426. https://doi.org/10.1016/j.jsbmb.2019.105426
Diczfalusy U (2013) On the formation and possible biological role of 25-hydroxycholesterol. Biochimie 95:455–460. https://doi.org/10.1016/j.biochi.2012.06.016
Diczfalusy U, Björkhem I (2011) Still another activity by the highly promiscuous enzyme CYP3A4: 25-hydroxylation of cholesterol. J Lipid Res 52:1447–1449. https://doi.org/10.1194/jlr.e017806
Dong Q et al (2020) 25-Hydroxycholesterol promotes vascular calcification via activation of endoplasmic reticulum stress. Eur J Pharmacol 880:173165. https://doi.org/10.1016/j.ejphar.2020.173165
Emanuelsson I, Norlin M (2012) Protective effects of 27- and 24-hydroxycholesterol against staurosporine-induced cell death in undifferentiated neuroblastoma SH-SY5Y cells. Neurosci Lett 525:44–48. https://doi.org/10.1016/j.neulet.2012.07.057
Fellows Maxwell K et al (2019) Oxysterols and apolipoproteins in multiple sclerosis: a 5 year follow-up study. J Lipid Res 60:1190–1198. https://doi.org/10.1194/jlr.m089664
Fourgeux C et al (2009) Primary open-angle glaucoma: association with cholesterol 24S-hydroxylase (CYP46A1) gene polymorphism and plasma 24-hydroxycholesterol levels. Invest Ophthalmol Vis Sci 50:5712–5717. https://doi.org/10.1167/iovs.09-3655
Galluzzi L et al (2018) Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ 25:486–541. https://doi.org/10.1038/s41418-017-0012-4
Gamba P et al (2011) Interaction between 24-hydroxycholesterol, oxidative stress, and amyloid-β in amplifying neuronal damage in Alzheimer’s disease: three partners in crime. Aging Cell 10:403–417. https://doi.org/10.1111/j.1474-9726.2011.00681.x
Gamba P et al (2014) Up-regulation of β-amyloidogenesis in neuron-like human cells by both 24- and 27-hydroxycholesterol: protective effect of N-acetyl-cysteine. Aging Cell 13:561–572. https://doi.org/10.1111/acel.12206
Goldstein JL, Brown MS (1990) Regulation of the mevalonate pathway. Nature 343:425–430. https://doi.org/10.1038/343425a0
Grayaa S et al (2018) Plasma oxysterol profiling in children reveals 24-hydroxycholesterol as a potential marker for Autism Spectrum Disorders. Biochimie 153:80–85. https://doi.org/10.1016/j.biochi.2018.04.026
Griffiths WJ, Wang Y (2022) Cholesterol metabolism: from lipidomics to immunology. J Lipid Res 63:100165. https://doi.org/10.1016/j.jlr.2021.100165
Guidara W et al (2022) Plasma oxysterols in drug-free patients with schizophrenia. J Steroid Biochem Mol Biol 221:106123. https://doi.org/10.1016/j.jsbmb.2022.106123
Han M et al (2020) Therapeutic implications of altered cholesterol homeostasis mediated by loss of CYP46A1 in human glioblastoma. EMBO Mol Med 12:e10924. https://doi.org/10.15252/emmm.201910924
Hartmann H et al (2022) Cholesterol dyshomeostasis in amyotrophic lateral sclerosis: cause, consequence, or epiphenomenon? FEBS J 289:7688–7709. https://doi.org/10.1111/febs.16175
Hetz C (2012) The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat Rev Mol Cell Biol 13:89–102. https://doi.org/10.1038/nrm3270
Hetz C et al (2020) Mechanisms, regulation and functions of the unfolded protein response. Nat Rev Mol Cell Biol 21:421–438. https://doi.org/10.1038/s41580-020-0250-z
Heverin M et al (2004) Changes in the levels of cerebral and extracerebral sterols in the brain of patients with Alzheimer’s disease. J Lipid Res 45:186–193. https://doi.org/10.1194/jlr.m300320-jlr200
Heverin M et al (2005) Crossing the barrier: net flux of 27-hydroxycholesterol into the human brain. J Lipid Res 46:1047–1052. https://doi.org/10.1194/jlr.M500024-JLR200
Hollien J, Weissman JS (2006) Decay of endoplasmic reticulum-localized mRNAs during the unfolded protein response. Science 313:104–107. https://doi.org/10.1126/science.1129631
Honda A et al (2011) Cholesterol 25-hydroxylation activity of CYP3A. J Lipid Res 52:1509–1516. https://doi.org/10.1194/jlr.m014084
Horton JD et al (2003) Combined analysis of oligonucleotide microarray data from transgenic and knockout mice identifies direct SREBP target genes. Proc Natl Acad Sci U S A 100:12027–12032. https://doi.org/10.1073/pnas.1534923100
Hua X et al (1996) Sterol resistance in CHO cells traced to point mutation in SREBP cleavage-activating protein. Cell 87:415–426. https://doi.org/10.1016/s0092-8674(00)81362-8
Janowski BA et al (1996) An oxysterol signalling pathway mediated by the nuclear receptor LXR alpha. Nature 383:728–731. https://doi.org/10.1038/383728a0
Janowski BA et al (1999) Structural requirements of ligands for the oxysterol liver X receptors LXRalpha and LXRbeta. Proc Natl Acad Sci U S A 96:266–271. https://doi.org/10.1073/pnas.96.1.266
Kim I et al (2008) Cell death and endoplasmic reticulum stress: disease relevance and therapeutic opportunities. Nat Rev Drug Discov 7:1013–1030. https://doi.org/10.1038/nrd2755
Kim SM et al (2013) 27-hydroxycholesterol induces production of tumor necrosis factor-alpha from macrophages. Biochem Biophys Res Commun 430:454–459. https://doi.org/10.1016/j.bbrc.2012.12.021
Kim SM et al (2017) 25-Hydroxycholesterol is involved in the pathogenesis of amyotrophic lateral sclerosis. Oncotarget 8:11855–11867. https://doi.org/10.18632/oncotarget.14416
Kim D et al (2022) Pathophysiological role of 27-hydroxycholesterol in human diseases. Adv Biol Regul 83:100837. https://doi.org/10.1016/j.jbior.2021.100837
Kimura Y et al (2018) Tocopherol suppresses 24(S)-hydroxycholesterol-induced cell death via inhibition of CaMKII phosphorylation. Biochimie 153:203–209. https://doi.org/10.1016/j.biochi.2018.07.004
Kölsch H et al (1999) The neurotoxic effect of 24-hydroxycholesterol on SH-SY5Y human neuroblastoma cells. Brain Res 818:171–175. https://doi.org/10.1016/s0006-8993(98)01274-8
Kölsch H et al (2001) Neurotoxicity of 24-hydroxycholesterol, an important cholesterol elimination product of the brain, may be prevented by vitamin E and estradiol-17beta. J Neural Transm 108:475–488. https://doi.org/10.1007/s007020170068
Kölsch H et al (2002) Polymorphism in the cholesterol 24S-hydroxylase gene is associated with Alzheimer’s disease. Mol Psychiatry 7:899–902. https://doi.org/10.1038/sj.mp.4001109
Kölsch H et al (2004) Altered levels of plasma 24S- and 27-hydroxycholesterol in demented patients. Neurosci Lett 368:303–308. https://doi.org/10.1016/j.neulet.2004.07.031
Leoni V, Caccia C (2013) 24S-hydroxycholesterol in plasma: a marker of cholesterol turnover in neurodegenerative diseases. Biochimie 95:595–612. https://doi.org/10.1016/j.biochi.2012.09.025
Leoni V et al (2002) Changes in human plasma levels of the brain specific oxysterol 24S-hydroxycholesterol during progression of multiple sclerosis. Neurosci Lett 331:163–166. https://doi.org/10.1016/s0304-3940(02)00887-x
Leoni V et al (2013) Plasma 24S-hydroxycholesterol correlation with markers of Huntington disease progression. Neurobiol Dis 55:37–43. https://doi.org/10.1016/j.nbd.2013.03.013
Li J et al (2016) Oxysterol binding protein-related protein 8 mediates the cytotoxicity of 25-hydroxycholesterol. J Lipid Res 57:1845–1853. https://doi.org/10.1194/jlr.M069906
Li J et al (2020) Ferroptosis: past, present and future. Cell Death Dis 11:88. https://doi.org/10.1038/s41419-020-2298-2
Li D et al (2022) Brain cell type-specific cholesterol metabolism and implications for learning and memory. Trends Neurosci 45:401–414. https://doi.org/10.1016/j.tins.2022.01.002
Lin JB et al (2018) Oxysterol signatures distinguish age-related macular degeneration from physiologic aging. EBioMedicine 32:9–20. https://doi.org/10.1016/j.ebiom.2018.05.035
Liu J et al (2005) Investigating the allosterism of acyl-CoA:cholesterol acyltransferase (ACAT) by using various sterols: in vitro and intact cell studies. Biochem J 391:389–397. https://doi.org/10.1042/bj20050428
Liu SY et al (2013) Interferon-inducible cholesterol-25-hydroxylase broadly inhibits viral entry by production of 25-hydroxycholesterol. Immunity 38:92–105. https://doi.org/10.1016/j.immuni.2012.11.005
Liu Z et al (2015) Protein kinase R-like ER kinase and its role in endoplasmic reticulum stress-decided cell fate. Cell Death Dis 6:e1822. https://doi.org/10.1038/cddis.2015.183
Liu Y et al (2018) 25-Hydroxycholesterol activates the expression of cholesterol 25-hydroxylase in an LXR-dependent mechanism. J Lipid Res 59:439–451. https://doi.org/10.1194/jlr.m080440
Liu W et al (2021) Dysregulated cholesterol homeostasis results in resistance to ferroptosis increasing tumorigenicity and metastasis in cancer. Nat Commun 12:5103. https://doi.org/10.1038/s41467-021-25354-4
Lund EG et al (2003) Knockout of the cholesterol 24-hydroxylase gene in mice reveals a brain-specific mechanism of cholesterol turnover. J Biol Chem 278:22980–22988. https://doi.org/10.1074/jbc.m303415200
Luo J et al (2020) Mechanisms and regulation of cholesterol homeostasis. Nat Rev Mol Cell Biol 21:225–245. https://doi.org/10.1038/s41580-019-0190-7
Lütjohann D et al (2000) Plasma 24S-hydroxycholesterol (cerebrosterol) is increased in Alzheimer and vascular demented patients. J Lipid Res 41:195–198. https://doi.org/10.1016/S0022-2275(20)32052-6
Marwarha G et al (2010) Leptin reduces the accumulation of Abeta and phosphorylated tau induced by 27-hydroxycholesterol in rabbit organotypic slices. J Alzheimers Dis 19:1007–1019. https://doi.org/10.3233/JAD-2010-1298
Marwarha G et al (2011) The oxysterol 27-hydroxycholesterol regulates α-synuclein and tyrosine hydroxylase expression levels in human neuroblastoma cells through modulation of liver X receptors and estrogen receptors—relevance to Parkinson’s disease. J Neurochem 119:1119–1136. https://doi.org/10.1111/j.1471-4159.2011.07497.x
Marwarha G et al (2013) Gadd153 and NF-κB crosstalk regulates 27-hydroxycholesterol-induced increase in BACE1 and β-amyloid production in human neuroblastoma SH-SY5Y cells. PLoS One 8:e70773. https://doi.org/10.1371/journal.pone.0070773
Maurel M et al (2014) Getting RIDD of RNA: IRE1 in cell fate regulation. Trends Biochem Sci 39:245–254. https://doi.org/10.1016/j.tibs.2014.02.008
Meir K et al (2002) Human sterol 27-hydroxylase (CYP27) overexpressor transgenic mouse model. Evidence against 27-hydroxycholesterol as a critical regulator of cholesterol homeostasis. J Biol Chem 277:34036–34041. https://doi.org/10.1074/jbc.m201122200
Mutemberezi V et al (2016) Oxysterols: From cholesterol metabolites to key mediators. Prog Lipid Res 64:152–169. https://doi.org/10.1016/j.plipres.2016.09.002
Nakazawa T et al (2017) Effect of vitamin E on 24(S)-hydroxycholesterol-induced necroptosis-like cell death and apoptosis. J Steroid Biochem Mol Biol 169:69–76. https://doi.org/10.1016/j.jsbmb.2016.03.003
Nelson ER et al (2013) 27-Hydroxycholesterol links hypercholesterolemia and breast cancer pathophysiology. Science 342:1094–1098. https://doi.org/10.1126/science.1241908
Noguchi N et al (2014) Diverse functions of 24(S)-hydroxycholesterol in the brain. Biochem Biophys Res Commun 446:692–696. https://doi.org/10.1016/j.bbrc.2014.02.010
Noguchi N et al (2015) New aspects of 24(S)-hydroxycholesterol in modulating neuronal cell death. Free Radic Biol Med 87:366–372. https://doi.org/10.1016/j.freeradbiomed.2015.06.036
Nury T et al (2015) Induction of oxiapoptophagy on 158N murine oligodendrocytes treated by 7-ketocholesterol-, 7β-hydroxycholesterol-, or 24(S)-hydroxycholesterol: Protective effects of α-tocopherol and docosahexaenoic acid (DHA; C22:6 n-3). Steroids 99:194–203. https://doi.org/10.1016/j.steroids.2015.02.003
Nury T et al (2021) Oxiapoptophagy: A type of cell death induced by some oxysterols. Br J Pharmacol 178:3115–3123. https://doi.org/10.1111/bph.15173
Oguro H et al (2017) 27-Hydroxycholesterol induces hematopoietic stem cell mobilization and extramedullary hematopoiesis during pregnancy. J Clin Invest 127:3392–3401. https://doi.org/10.1172/jci94027
Ohtsuki S et al (2007) Brain-to-blood elimination of 24S-hydroxycholesterol from rat brain is mediated by organic anion transporting polypeptide 2 (oatp2) at the blood-brain barrier. J Neurochem 103:1430–1438. https://doi.org/10.1111/j.1471-4159.2007.04901.x
Okabe A et al (2013) Adaptive responses induced by 24S-hydroxycholesterol through liver X receptor pathway reduce 7-ketocholesterol-caused neuronal cell death. Redox Biol 2:28–35. https://doi.org/10.1016/j.redox.2013.11.007
Olivier E et al (2017) 25-Hydroxycholesterol induces both P2X7-dependent pyroptosis and caspase-dependent apoptosis in human skin model: New insights into degenerative pathways. Chem Phys Lipids 207:171–178. https://doi.org/10.1016/j.chemphyslip.2017.06.001
Olsen BN et al (2012) Side-chain oxysterols: from cells to membranes to molecules. Biochim Biophys Acta 1818:330–336. https://doi.org/10.1016/j.bbamem.2011.06.014
Pakos-Zebrucka K et al (2016) The integrated stress response. EMBO Rep 17:1374–1395. https://doi.org/10.15252/embr.201642195
Park K, Scott AL (2010) Cholesterol 25-hydroxylase production by dendritic cells and macrophages is regulated by type I interferons. J Leukoc Biol 88:1081–1087. https://doi.org/10.1189/jlb.0610318
Paul SM et al (2013) The major brain cholesterol metabolite 24(S)-hydroxycholesterol is a potent allosteric modulator of N-methyl-D-aspartate receptors. J Neurosci 33:17290–17300. https://doi.org/10.1523/jneurosci.2619-13.2013
Poli G et al (2022) Oxysterols: From redox bench to industry. Redox Biol 49:102220. https://doi.org/10.1016/j.redox.2021.102220
Radhakrishnan A et al (2007) Sterol-regulated transport of SREBPs from endoplasmic reticulum to Golgi: oxysterols block transport by binding to Insig. Proc Natl Acad Sci U S A 104:6511–6518. https://doi.org/10.1073/pnas.0700899104
Reboldi A et al (2014) Inflammation. 25-Hydroxycholesterol suppresses interleukin-1-driven inflammation downstream of type I interferon. Science 345:679–684. https://doi.org/10.1126/science.1254790
Reineke LC, Neilson JR (2019) Differences between acute and chronic stress granules, and how these differences may impact function in human disease. Biochem Pharmacol 162:123–131. https://doi.org/10.1016/j.bcp.2018.10.009
Repa JJ et al (2000) Disruption of the sterol 27-hydroxylase gene in mice results in hepatomegaly and hypertriglyceridemia. Reversal by cholic acid feeding. J Biol Chem 275:39685–39692. https://doi.org/10.1074/jbc.M007653200
Rogers MA et al (2015) Acyl-CoA:cholesterol acyltransferases (ACATs/SOATs): Enzymes with multiple sterols as substrates and as activators. J Steroid Biochem Mol Biol 151:102–107. https://doi.org/10.1016/j.jsbmb.2014.09.008
Rosen H et al (1998) Markedly reduced bile acid synthesis but maintained levels of cholesterol and vitamin D metabolites in mice with disrupted sterol 27-hydroxylase gene. J Biol Chem 273:14805–14812. https://doi.org/10.1074/jbc.273.24.14805
Russell DW (2000) Oxysterol biosynthetic enzymes. Biochim Biophys Acta 1529:126–135. https://doi.org/10.1016/S1388-1981(00)00142-6
Russell DW et al (2009) Cholesterol 24-hydroxylase: an enzyme of cholesterol turnover in the brain. Annu Rev Biochem 78:1017–1040. https://doi.org/10.1146/annurev.biochem.78.072407.103859
Rutkowski DT et al (2006) Adaptation to ER stress is mediated by differential stabilities of pro-survival and pro-apoptotic mRNAs and proteins. PLoS Biol 4:e374. https://doi.org/10.1371/journal.pbio.0040374
Schönknecht P et al (2002) Cerebrospinal fluid 24S-hydroxycholesterol is increased in patients with Alzheimer’s disease compared to healthy controls. Neurosci Lett 324:83–85. https://doi.org/10.1016/s0304-3940(02)00164-7
Schüle R et al (2010) Marked accumulation of 27-hydroxycholesterol in SPG5 patients with hereditary spastic paresis. J Lipid Res 51:819–823. https://doi.org/10.1194/jlr.M002543
Schumacher MM, DeBose-Boyd RA (2021) Posttranslational regulation of HMG CoA reductase, the rate-limiting enzyme in synthesis of cholesterol. Annu Rev Biochem 90:659–679. https://doi.org/10.1146/annurev-biochem-081820-101010
Sekiya M et al (2014) Absence of Nceh1 augments 25-hydroxycholesterol-induced ER stress and apoptosis in macrophages. J Lipid Res 55:2082–2092. https://doi.org/10.1194/jlr.M050864
Sever N et al (2003) Accelerated degradation of HMG CoA reductase mediated by binding of insig-1 to its sterol-sensing domain. Mol Cell 11:25–33. https://doi.org/10.1016/s1097-2765(02)00822-5
Shibata N et al (2013) 25-Hydroxycholesterol activates the integrated stress response to reprogram transcription and translation in macrophages. J Biol Chem 288:35812–35823. https://doi.org/10.1074/jbc.m113.519637
Sodero AO (2021) 24S-hydroxycholesterol: Cellular effects and variations in brain diseases. J Neurochem 157:899–918. https://doi.org/10.1111/jnc.15228
Sun MY et al (2016) Endogenous 24S-hydroxycholesterol modulates NMDAR-mediated function in hippocampal slices. J Neurophysiol 115:1263–1272. https://doi.org/10.1152/jn.00890.2015
Suzuki A et al (2021) Different functions of vitamin E homologues in the various types of cell death induced by oxysterols. Free Radic Biol Med 176:356–365. https://doi.org/10.1016/j.freeradbiomed.2021.10.008
Tabas I, Ron D (2011) Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat Cell Biol 13:184–190. https://doi.org/10.1038/ncb0311-184
Takabe W et al (2016) Esterification of 24S-OHC induces formation of atypical lipid droplet-like structures, leading to neuronal cell death. J Lipid Res 57:2005–2014. https://doi.org/10.1194/jlr.m068775
Tang D et al (2019) The molecular machinery of regulated cell death. Cell Res 29:347–364. https://doi.org/10.1038/s41422-019-0164-5
Umetani M et al (2014) The cholesterol metabolite 27-hydroxycholesterol promotes atherosclerosis via proinflammatory processes mediated by estrogen receptor alpha. Cell Metab 20:172–182. https://doi.org/10.1016/j.cmet.2014.05.013
Urano Y et al (2013) Suppression of amyloid-β production by 24S-hydroxycholesterol via inhibition of intracellular amyloid precursor protein trafficking. FASEB J 27:4305–4315. https://doi.org/10.1096/fj.13-231456
Urano Y et al (2019) 24(S)-Hydroxycholesterol induces ER dysfunction-mediated unconventional cell death. Cell Death Discov 5:113. https://doi.org/10.1038/s41420-019-0192-4
Urano Y et al (2022) Integrated stress response is involved in the 24(S)-hydroxycholesterol-induced unconventional cell death mechanism. Cell Death Discov 8:406. https://doi.org/10.1038/s41420-022-01197-w
Vandenabeele P et al (2022) Pore-forming proteins as drivers of membrane permeabilization in cell death pathways. Nat Rev Mol Cell Biol. https://doi.org/10.1038/s41580-022-00564-w
Vo DK et al (2015) 24(S)-Hydroxycholesterol induces RIPK1-dependent but MLKL-independent cell death in the absence of caspase-8. Steroids 99:230–237. https://doi.org/10.1016/j.steroids.2015.02.007
Wang B, Tontonoz P (2018) Liver X receptors in lipid signalling and membrane homeostasis. Nat Rev Endocrinol 14:452–463. https://doi.org/10.1038/s41574-018-0037-x
Wang H et al (2016a) The cytotoxicity of 27-hydroxycholesterol in co-cultured SH-SY5Y cells and C6 cells. Neurosci Lett 632:209–217. https://doi.org/10.1016/j.neulet.2016.08.056
Wang HL et al (2016b) Cholesterol, 24-hydroxycholesterol, and 27-hydroxycholesterol as surrogate biomarkers in cerebrospinal fluid in mild cognitive impairment and Alzheimer’s disease: a meta-analysis. J Alzheimers Dis 51:45–55. https://doi.org/10.3233/JAD-150734
Wang S et al (2020) Cholesterol 25-Hydroxylase inhibits SARS-CoV-2 and other coronaviruses by depleting membrane cholesterol. EMBO J 39:e106057. https://doi.org/10.15252/embj.2020106057
Wang Y et al (2021) Cholesterol metabolism pathways—are the intermediates more important than the products? FEBS J 288:3727–3745. https://doi.org/10.1111/febs.15727
Warns J et al (2018) 27-hydroxycholesterol decreases cell proliferation in colon cancer cell lines. Biochimie 153:171–180. https://doi.org/10.1016/j.biochi.2018.07.006
Woo SY et al (2022) Role of reactive oxygen species in regulating 27-hydroxycholesterol-induced apoptosis of hematopoietic progenitor cells and myeloid cell lines. Cell Death Dis 13:916. https://doi.org/10.1038/s41419-022-05360-0
Yamanaka K et al (2011) 24(S)-hydroxycholesterol induces neuronal cell death through necroptosis, a form of programmed necrosis. J Biol Chem 286:24666–24673. https://doi.org/10.1074/jbc.M111.236273
Yamanaka K et al (2014) Induction of apoptosis and necroptosis by 24(S)-hydroxycholesterol is dependent on activity of acyl-CoA:cholesterol acyltransferase 1. Cell Death Dis 5:e990. https://doi.org/10.1038/cddis.2013.524
Yan D et al (2008) OSBP-related protein 8 (ORP8) suppresses ABCA1 expression and cholesterol efflux from macrophages. J Biol Chem 283:332–340. https://doi.org/10.1074/jbc.M705313200
Yang WS, Stockwell BR (2016) Ferroptosis: death by lipid peroxidation. Trends Cell Biol 26:165–176. https://doi.org/10.1016/j.tcb.2015.10.014
Yang T et al (2002) Crucial step in cholesterol homeostasis: sterols promote binding of SCAP to INSIG-1, a membrane protein that facilitates retention of SREBPs in ER. Cell 110:489–500. https://doi.org/10.1016/s0092-8674(02)00872-3
Yokoyama S et al (1999) Suppression of rat liver tumorigenesis by 25-hydroxycholesterol and all-trans retinoic acid: differentiation therapy for hepatocellular carcinoma. Int J Oncol 15:565–574. https://doi.org/10.3892/ijo.15.3.565
You JS et al (2020) 25-Hydroxycholesterol induces death receptor-mediated extrinsic and mitochondria-dependent intrinsic apoptosis in head and neck squamous cell carcinoma cells. Anticancer Res 40:779–788. https://doi.org/10.21873/anticanres.14009
Yuan J et al (2016) Roles of caspases in necrotic cell death. Cell 167:1693–1704. https://doi.org/10.1016/j.cell.2016.11.047
Yuan J et al (2019) Necroptosis and RIPK1-mediated neuroinflammation in CNS diseases. Nat Rev Neurosci 20:19–33. https://doi.org/10.1038/s41583-018-0093-1
Zang R et al (2020) Cholesterol 25-hydroxylase suppresses SARS-CoV-2 replication by blocking membrane fusion. Proc Natl Acad Sci U S A 117:32105–32113. https://doi.org/10.1073/pnas.2012197117
Zhang Y et al (2003) Cholesterol is superior to 7-ketocholesterol or 7 alpha-hydroxycholesterol as an allosteric activator for acyl-coenzyme A:cholesterol acyltransferase 1. J Biol Chem 278:11642–11647. https://doi.org/10.1074/jbc.m211559200
Zhang X et al (2019) 27-hydroxycholesterol promotes Aβ accumulation via altering Aβ metabolism in mild cognitive impairment patients and APP/PS1 mice. Brain Pathol 29:558–573. https://doi.org/10.1111/bpa.12698
Zhong W et al (2016) ORP4L facilitates macrophage survival via G-protein-coupled signaling: ORP4L-/- mice display a reduction of atherosclerosis. Circ Res 119:1296–1312. https://doi.org/10.1161/CIRCRESAHA.116.309603
Zuliani G et al (2011) Plasma 24S-hydroxycholesterol levels in elderly subjects with late onset Alzheimer’s disease or vascular dementia: a case-control study. BMC Neurol 11:121. https://doi.org/10.1186/1471-2377-11-121
Acknowledgments
We thank Ren Chiba for his support in making the figures.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Ethics declarations
The authors declare no competing interests.
Funding
This work was supported in part by a JSPS KAKENHI Grant-in-Aid for Scientific Research (C) 16K08254, 19K07093 and 23K10902 to YU.
Rights and permissions
Copyright information
© 2024 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Urano, Y., Noguchi, N. (2024). Enzymatically Formed Oxysterols and Cell Death. In: Lizard, G. (eds) Implication of Oxysterols and Phytosterols in Aging and Human Diseases. Advances in Experimental Medicine and Biology, vol 1440. Springer, Cham. https://doi.org/10.1007/978-3-031-43883-7_10
Download citation
DOI: https://doi.org/10.1007/978-3-031-43883-7_10
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-031-43882-0
Online ISBN: 978-3-031-43883-7
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)