
Abstract
Cardiogenic shock is a life-threatening state of end-organ hypoperfusion secondary to low cardiac output caused by underlying cardiac dysfunction. Cardiogenic shock is associated with significant in-hospital mortality despite advancing therapies. Careful patient evaluation should be made to identify cardiogenic shock as the correct diagnosis, recognizing that acute coronary syndrome is the most prevalent etiology for cardiogenic shock. Once diagnosed, the goal of therapy is to maintain adequate tissue perfusion. Management should be geared toward circulatory support, ventricular unloading, and myocardial perfusion. This chapter describes the pathophysiology, patient evaluation, and management of patients in cardiogenic shock.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
Introduction
Cardiogenic shock (CS) is a life-threatening state of end-organ hypoperfusion secondary to low cardiac output (CO). CS is associated with significant in-hospital mortality and significant healthcare cost. Mortality rates have been documented in excess of 80% despite modern therapies [1, 2].
Over the last several years, there has been a subtle shift in etiology of CS as early identification and treatment of acute coronary syndrome (ACS) have become the standard of care. Myocardial infarction (MI) remains the most prevalent etiology with mortality reported as greater than 35% [1], but CS secondary to advanced heart failure is now commonly seen in the cardiac intensive care unit (CICU) setting around the country [1, 3].
Differentiating Shock
Shock is a state of circulatory failure, which leads to cellular and tissue hypoxia. There are multiple underlying etiologies of shock based on the mechanism of hypoperfusion. The classifications include cardiogenic, distributive, obstructive, hypovolemic, and neurogenic (Table 24.1). In this chapter, we will focus on cardiogenic shock, but it is important to recognize the other causes and that patients may have a combination of more than one type of shock.
Pathophysiology
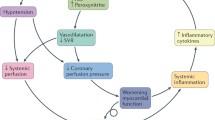
Cardiogenic shock is a condition of decreased myocardial contractility secondary to underlying cardiac dysfunction and hypotension causing hypoperfusion of the myocardium and other end organs. This hypoperfusion causes even more ischemia to cardiac tissue, which further reduces low stroke volume and worsens diastolic filling. This cycle can progress rapidly leading to patient death if not treated.
As cardiac output decreases, intrinsic compensatory mechanisms designed to raise blood pressure cause vasoconstriction and fluid retention. This is reflected as increased systemic vascular resistance (SVR) and elevated pulmonary capillary wedge pressure (PCWP). In CS, these mechanisms become maladaptive and cause an increase in myocardial oxygen requirements, further worsening myocardial dysfunction.
Catecholamines are released by the sympathetic nervous system in an attempt to increase stroke volume by raising the heart rate and constricting blood vessels. Simultaneously, the renin-angiotensin-aldosterone system is activated when the renal system is poorly perfused and attempts to increase blood volume. Fluid is then retained in an attempt to raise blood pressure, which increases both preload and afterload. As the myocardium is stretched, brain natriuretic peptide (BNP) is released and further contributes to the physiologic cycle.
The goal of these intrinsic mechanisms is to increase cardiac output by raising preload, stroke volume, and heart rate. However, if left unchecked, they increase the myocardial workload leading to worsening cardiac output, decreased tissue perfusion, hypotension, ischemia, and ultimately myocardial dysfunction with remodeling [4].
End-organ dysfunction occurs secondary to tissue hypoperfusion. When systemic tissue is hypoperfused, an inflammatory process is triggered. This inflammatory process leads to the release of cytokines and nitric oxide, which cause vasodilation in the microcirculation, further affecting blood pressure and worsening hypoperfusion. As vasodilation occurs, oxygen delivery decreases and ischemia develops [5, 6]. Poor tissue perfusion and hypoxia lead to the development of lactic acidosis.
Clinical Presentation
History and Physical
Past medical history is key to the workup of CS. Myocardial infarction, particularly ST-elevation MI, is the most common cause of CS, and anterior MI is the most likely to develop CS. Any primary cardiac diagnosis that causes myocardial dysfunction can deteriorate to CS. Chronic heart failure (HF) can deteriorate into an acute decompensated state and now accounts for as much as 30% of CS presentations [2]. Other causes of CS include cardiac arrest, valvular heart disease, tamponade, myocarditis, congenital heart disease, hypertrophic cardiomyopathy, refractory ventricular tachycardia, apical ballooning, pulmonary hypertension, and PE [3, 5, 6].
Patients may present with a variety of symptoms and/or feelings that include chest pain, dyspnea, PND/orthopnea, syncope, presyncope, progressive fatigue, and palpitations. Physical exam findings may include pallor, cyanosis, or mottling of the skin. Assessment of the extremities for strength of pulses and temperature can provide an understanding of the patient’s perfusion status. Cardiac auscultation may reveal extra heart sounds, particularly an S3 being indicative of HF, or murmurs. Evaluation of elevated jugular venous pressure, pulsatile liver, significant hepatojugular reflex, ascites, and lower extremity edema may be helpful in determining the patient’s volume status, as well as assessment of the lungs for rales suggestive of pulmonary edema.
Patients with CS may present with symptoms consistent with their underlying pathology, and the physical exam will be dictated by the CS phenotype [2]. Three phenotypes of CS exist. These phenotypes are categorized according to volume status and cardiac output or peripheral exam. Clinically, phenotypes can be broken down into warm or cold and wet or dry (Table 24.2). The first phenotype is described as classic CS. Patients will have evidence of decreased CO, increased SVR, and evidence of increased preload. Euvolemic CS also has evidence of decreased cardiac output and increased SVR, but preload is normal. Mixed or vasodilatory CS is a decrease in CO and increase in preload, but the SVR is normal to low. Lastly, vasodilatory shock which is non-cardiogenic is described as an increase in CO, with decreased preload and afterload.
When there is clinical evidence for CS, assessment of the severity is crucial to understanding the patient’s risk for deterioration and overall prognosis. Clinical evidence of CS may include ashen or mottled appearance, cold and clammy to the touch, elevated lactate (>2.0), rales on physical exam, evidence of organ involvement including transaminitis or rise in creatinine (double in creatine or 50% decrease of GFR), hypotension (systolic BP <90, MAP <60), and altered mental status [7].
The Killip classification assessment can be of value when attempting to determine the patient’s overall clinical picture and mortality risk [8]. This system relies on the physical exam for appropriate classification. Killip Class I was defined as no evidence of heart failure. Class II was defined as heart failure with the presence of an S3 and rales on physical exam. Class III was defined as severe heart failure which included the presence of significant pulmonary edema. Class IV was defined as frank cardiogenic shock.
Identification and management of early stages of CS can prevent further deterioration. The Society for Cardiovascular Angiography and Intervention (SCAI) has developed a classification for CS (Fig. 24.1) [7]. The SCAI classification includes five stages of increased CS severity. Stage A identifies patients at risk. Stage B identifies patients beginning to show signs of deterioration. These patients develop hypotension and/or tachycardia without evidence of hypoperfusion but require intervention to prevent the development of end-organ damage. Stage C is classic CS. The patient has frank evidence of hypoperfusion and requires hemodynamic intervention. Stage D is CS that continues to worsen despite intervention and escalation of therapy. Stage E is refractory CS [7].

SCAI cardiogenic shock stages classifies patients in or at risk for CS according to clinical status. (Permission granted by Naidu et al. [9])
Diagnostic Studies
An electrocardiogram (ECG) should be performed within 10 min of patient arrival [4, 5]. As MI is the most common cause of CS, assessment of ST-segments and T-wave abnormalities is crucial. In addition, ECG can determine rhythm and underlying conduction.
Laboratory workup is a crucial part of evaluating end-organ involvement. Troponins should be drawn at baseline. Troponin I or T is acceptable, although recently institutions have transitioned to high sensitivity troponins. Isolated troponin elevation in the absence of ACS is not specific but is a strong predictor of mortality when significantly elevated. A complete blood count to include hemoglobin and white blood cell count will be important to assess for underlying signs of anemia and infection. Electrolytes, creatinine, and cystatin C for kidney function assessment, liver function tests with INR, LDH, and lactate. Elevated lactate levels are associated with increased mortality in patients with CS [4].
NT proBNP can be helpful for differentiating the etiology of shortness of breath and for prognosis. ACS patients with increased BNP levels are at increased risk of mortality [4].
A point-of-care ultrasound (POCUS) exam of the heart, lungs, and IVC can add valuable information to your physical exam. POCUS is a goal-directed ultrasound and does not take the place of a formal transthoracic echocardiogram (TTE). Figure 24.2 demonstrates an example of the constellation of finding on POCUS in a patient with CS. Formal TTE should be a standard of care and be performed as part of the CS repertoire. A TTE adds valuable information about cardiac structure and function and will further define the direction of care.

(a) B-lines consistent with pulmonary edema present on lung imaging, (b) dilated IVC consistent with increased preload, and (c) dilated left ventricle with a small circumferential pericardial effusion
Chest X-ray should also be performed and can help differentiate infection from pulmonary edema or other etiology during evaluation for CS.
Management
Once CS has been identified, the goal of therapy is to maintain adequate tissue perfusion. Management should be geared toward circulatory support, ventricular unloading, and myocardial perfusion. The underlying cause of CS must be identified and managed while simultaneously providing supportive care. As the most common cause of CS remains ACS, early consideration for reperfusion therapy will be of utmost importance [5].
The cornerstone of treating patients with confirmed or suspected CS is getting the patient to the correct level of care. Patients with CS should be triaged to a setting that offers percutaneous coronary intervention (PCI), mechanical circulatory support (MCS), a Cardiac Intensive Care Unit (CICU), and cardiac transplant capabilities [10].
As patients with CS are commonly volume overloaded and develop pulmonary edema, ensuring an adequate airway and oxygenation is crucial. In the setting of acute decompensated heart failure and CS, noninvasive positive pressure ventilation (NIPPV) is required to optimize ventilation and oxygenation. NIPPV recruits lung tissue resulting in an increase in oxygenation and a decrease in work of breathing. If noninvasive positive pressure ventilation is felt to be inadequate, then consider mechanical ventilation with the goal of lung protection ventilation and oxygenation [10, 11].
Continuous hemodynamic monitoring is an important aspect of managing CS. Using arterial lines for continuous blood pressure management and pulmonary artery catheters (PAC) to titrate medications and guide additional therapy can be helpful. If a PAC is unavailable or there is a contraindication, then a central line or a PICC line can also provide the ability to monitor hemodynamics and give central access for vasoactive medications. There are also minimally invasive hemodynamic monitors available, and noninvasive measures of hemodynamic parameters can be obtained with echocardiography.
Historically, PAC were used regularly in post MI patients. Subsequent literature demonstrated a correlation between PA catheter use and increase in mortality, and routine PAC use is no longer recommended for MI. Despite decline in use, PAC remain the standard for hemodynamic monitoring in the setting of moderate to severe CS. Careful patient assessment and risk evaluation should continue when deciding on monitoring. Complications include bleeding, embolism, infection, pulmonary infarct or hemorrhage, and inaccurate data collection.
Continuous hemodynamic monitoring with PAC can provide real-time feedback for care teams to react and adjust treatments. PAC consist of ports enabled to transduce right atrial pressure, pulmonary arterial pressure with the ability to measure a PCWP tracing, and mixed venous oxygen saturation. From these measurements, CO, SVR, PVR, cardio power output (CPO), and pulmonary artery pulsatility index (PAPi) can be calculated.
CO and cardiac index (CI) can be obtained via thermodilution, which is considered the most accurate, or by Fick calculation. Thermodilution is a procedure performed at the bedside ideally using a dedicated rapid injector. Normal saline is rapidly injected into the RA port of the PAC, and a thermistor monitors the temperature from the RA to the PA. Limitations to the thermodilution method include less reliable readings associated with tricuspid valve regurgitation and ventricular septal defects [12].
A CO and CI calculated by Fick can be done as an alternative. The Fick calculation takes into account oxygen consumption (VO2), height, weight, SaO2 from an ABG, SvO2 from a PAC, hemoglobin, heart rate, and age.
To maintain tissue perfusion, CS management should be focused on increasing CO. CO is improved by increasing the heart rate and the stroke volume. Inotrope support is the first line for increasing stroke volume, improving contractility, and off-loading pressures working against failing ventricles. In the CICU, there are common IV inotropes used regularly for the treatment of CS (Tables 24.3 and 24.4). Each has pros and cons for use, and each should be chosen carefully based on the patient’s clinical picture. Monitoring should be based on signs of end-organ function including lactate, creatinine, urine output, skin temperature, and mottling.
Dobutamine is a fast-acting beta receptor agonist with strong beta-1 stimulation and some beta-2 stimulation. It is a typical first-line IV agent for inotropic support in the treatment of CS [13]. In addition to inotropy, dobutamine causes vasodilation and therefore has some afterload reduction effect. The combination of increased cardiac contractility and decreased afterload improves stroke volume and therefore increases CO. Dobutamine has side effects including increased heart rate and is known to be proarrhythmic. The onset of action of dobutamine can be seen within minutes of initiation. Dosing should be started low and increased as needed. Typical dose initiation is 2.5 μg/kg/min.
Milrinone is a phosphodiesterase inhibitor, which helps to activate beta receptors. This results in an inotropic effect in the heart with beta-1 receptor activation and pulmonary and systemic vasodilation with beta-2 receptor activation. Milrinone has a slower onset of action than dobutamine. In addition, because milrinone is renally cleared, accumulation of the drug can occur and cause worsening side effects including arrhythmias and hypotension.
Studies suggest increased mortality with the use of dopamine in patients with CS [2]. It has both inotropic and vasopressor activity. At low doses (0.5–2 μg/kg/min), the effects are primarily dopaminergic with peripheral vasodilation. Intermediate doses (2–10 μg/kg/min) have primarily beta-1-adrenergic effect with increased cardiac contractility, heart rate, and blood pressure. Doses >10 μg/kg/min have alpha-adrenergic effect with primary vasoconstriction and increased blood pressure.
Epinephrine is considered a second-line medication that acts as both inotrope and vasopressor. At lower doses, epinephrine acts more as an inotrope given strong beta receptor agonist properties. Epinephrine is proarrhythmic and can therefore be problematic in the setting of underlying cardiac dysfunction. In addition, epinephrine is associated with high lactate levels. Dose range is similar to norepinephrine ranging from 0.01 to 0.3 μg/kg/min.
Vasopressor support in the setting of hypotension should be used to support tissue perfusion with the goal to maintain MAP greater than 65 mmHg in conjunction with other therapies. Norepinephrine has been a standard first-line vasopressor agent commonly used to treat hypotensive states like septic shock. Norepinephrine offers vasoconstriction and mild inotrope effect.
Afterload reduction should be considered if tolerated by blood pressure. Afterload reduction can assist with improving CO by decreasing cardiac oxygen demands. If IV afterload reduction is necessary, consider nitroglycerin, clevidipine, or nitroprusside for short-term therapy with plans to transition to an oral agent based on clinical picture including kidney function.
Long-Term Care
Although mortality is high, patients can recover. The patient should be supported while decompensated with plans to intervene and treat their underlying cardiac dysfunction.
Guideline-directed medical therapy (GDMT) for the treatment of heart failure should be considered in patients who have recovered from CS. Beta blockers, ACE inhibitors, and other evidence-based therapies can be initiated when the patient is close to a euvolemic state and weaning off IV inotrope and vasopressor agents. Afterload reduction can be transitioned to an oral regimen based on kidney function and diagnosis. Inotrope support may continue. Some patients remain on long-term inotropes in the outpatient setting as a bridge to transplant or as palliative support for quality of life.
Clinical Pearls
-
Patients with confirmed or suspected CS should be triaged to a setting that offers PCI capabilities and a CICU.
-
Evaluation and treatment of underlying cardiac dysfunction should continue while supporting patients in CS. ACS is the most common etiology of CS and should be ruled out immediately upon presentation.
-
To maintain tissue perfusion, CS management should be focused on increasing CO. Inotrope support is the first line for increasing stroke volume, improving contractility, and off-loading pressures working against failing ventricles.
-
Ongoing risk assessment in the setting of CS is crucial. Risk assessment tools including the SCAI shock stages and Killip classification should be considered for mortality prediction.
-
MCS consideration and cardiac transplant evaluation are warranted in patients with refractory CS.
-
Early involvement of palliative care can assist with goals of care discussions and symptom management and can be particularly useful in the setting of chronic end-stage heart failure.
-
GDMT for the treatment for heart failure should be considered in patients who have recovered from CS.
References
O’Brien C, Beaubien-Souligny W, Amsallem M, Denault A, Haddad F. Cardiogenic shock: reflections at the crossroad between perfusion, tissue hypoxia, and mitochondrial function. Can J Cardiol. 2020;36(2):184–96.
van Diepen S, Katz JN, Albert NM, Henry TD, Jacobs AK, Kapur NK, et al. Contemporary management of cardiogenic shock: a scientific statement from the American Heart Association. Circulation. 2017;136(16):e232–e68.
Brener MI, Rosenblum HR, Burkhoff D. Pathophysiology and advanced hemodynamic assessment of cardiogenic shock. Methodist Debakey Cardiovasc J. 2020;16(1):7–15.
Vahdatpour C, Collins D, Goldberg S. Cardiogenic shock. J Am Heart Assoc. 2019;8(8):e011991.
Bertini P, Guarracino F. Pathophysiology of cardiogenic shock. Curr Opin Crit Care. 2021;27(4):409–15.
Lim HS. Cardiogenic shock: failure of oxygen delivery and oxygen utilization. Clin Cardiol. 2016;39(8):477–83.
Baran DA, Grines CL, Bailey S, Burkhoff D, Hall SA, Henry TD, et al. SCAI clinical expert consensus statement on the classification of cardiogenic shock: this document was endorsed by the American College of Cardiology (ACC), the American Heart Association (AHA), the Society of Critical Care Medicine (SCCM), and the Society of Thoracic Surgeons (STS) in April 2019. Catheter Cardiovasc Interv. 2019;94(1):29–37.
Killip T III, Kimball JT. Treatment of myocardial infarction in a coronary care unit. A two year experience with 250 patients. Am J Cardiol. 1967;20(4):457–64.
Naidu SS, Baran DA, Jentzer JC, Hollenberg SM, van Diepen S, Basir MB, et al. SCAI SHOCK stage classification expert consensus update: a review and incorporation of validation studies: this statement was endorsed by the American College of Cardiology (ACC), American College of Emergency Physicians (ACEP), American Heart Association (AHA), European Society of Cardiology (ESC) Association for Acute Cardiovascular Care (ACVC), International Society for Heart and Lung Transplantation (ISHLT), Society of Critical Care Medicine (SCCM), and Society of Thoracic Surgeons (STS) in December 2021. J Am Coll Cardiol. 2022;79(9):933–46.
Jentzer JC, Tabi M, Burstein B. Managing the first 120 min of cardiogenic shock: from resuscitation to diagnosis. Curr Opin Crit Care. 2021;27(4):416–25.
Alviar CL, Miller PE, McAreavey D, Katz JN, Lee B, Moriyama B, et al. Positive pressure ventilation in the cardiac intensive care unit. J Am Coll Cardiol. 2018;72(13):1532–53.
Argueta EE, Paniagua D. Thermodilution cardiac output: a concept over 250 years in the making. Cardiol Rev. 2019;27(3):138–44.
Jentzer JC, Coons JC, Link CB, Schmidhofer M. Pharmacotherapy update on the use of vasopressors and inotropes in the intensive care unit. J Cardiovasc Pharmacol Ther. 2015;20(3):249–60.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2023 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Bennett, C., Solberg, A. (2023). Cardiogenic Shock. In: Musialowski, R., Allshouse, K. (eds) Cardiovascular Manual for the Advanced Practice Provider. Springer, Cham. https://doi.org/10.1007/978-3-031-35819-7_24
Download citation
DOI: https://doi.org/10.1007/978-3-031-35819-7_24
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-031-35818-0
Online ISBN: 978-3-031-35819-7
eBook Packages: MedicineMedicine (R0)