
Abstract
Air pollution is a silent killer. Considered the largest environmental health risk globally, air pollution has been implicated in the development of cardiovascular and respiratory conditions as well as various cancers. Air pollution poses a serious global threat, as 91% of the world’s population resides in areas that fail to meet WHO air quality guidelines. Air pollution in the form of particulate matter (PM) contributes to increased arrhythmia development. Exposure to unhealthy PM levels coupled with those suffering from arrhythmias necessitates analysis into their shared underlying causes.
Epigenetics is defined as alterations, heritable and acquired, to gene expression with preservation of the original DNA sequence. These modifications naturally occur during development but can be modified by external stimuli, ultimately resulting in differential gene expression in offspring. A handful of modifications constitute the essential pillars of this domain, often working concurrently to induce changes: DNA methylation, chromatin remodeling, histone modification, and RNA-based modifications. This chapter aims to discuss the influence of air pollution on epigenetic mechanisms and how these affect the development of cardiovascular disease, including arrhythmia development.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others
Keywords
1 Introduction: Particulate Matter and Cardiovascular Function
Environmental factors can induce profound physiological and pathophysiological consequences on the cardiovascular system [1, 2]. Obesity, stress, and air pollution, among other external stimuli, exemplify established risk factors for cardiovascular disease (CVD), which continues to be the leading cause of death in the United States. The dynamic interplay between genetics and environmental factors is complex: does one develop heart disease from obesity caused by a poor diet? Or are obese people genetically prone to developing cardiovascular disease via exposure to air pollution? Epigenetics is the balance of these dynamics via mechanisms induced by environmental triggers and those that occur during development.
Particulate matter (PM) is the sum of solid particles and liquid droplets suspended in the air. Sources of particulate matter include emissions from automobiles, smokestacks, construction sites, unpaved roads, and the black carbon and/or elemental carbon from these sources [3]. Many forms of air pollution exist alongside PM, such as carbon monoxide, ozone, oxides of nitrogen, etc. WHO estimates that PM exposure is responsible for the death of nearly seven million people annually. A number of studies have linked PM exposure to stroke, heart disease, several lung diseases, and pneumonia [4,5,6]. Another review by the American Heart Association (AHA) stated that CVD has resulted in over 17 million deaths annually. Of these 17 million, nearly 3.3 million were associated with exposure to ambient air pollution. The AHA has also stated that short-term exposure to PM contributed to the development of CVD and long-term exposure reduced life expectancy [5].
PM penetrates the body primarily through inhalation, where it enters the lungs and gains access to the bloodstream via alveoli. PM10 (coarse PM, diameter of <10 μm) has been experimentally linked, via in vivo and in vitro models, to acute and chronic CVD, whereas PM2.5 (fine PM, diameter of <2.5 μm) and PM0.1 (ultrafine PM, diameter of <0.1 μm) have been shown to penetrate further into the airways, alveoli, and systemic circulation, inducing the development of atherosclerosis, CVD, and provoking acute cardiac episodes and arrhythmias [7]. Recent studies of PM and CVD have demonstrated PM-induced development of cardiomyopathy, myocardial infarction, and hypertension, leading to cardiovascular (CV) dysfunction including arrhythmias, specifically tachycardia [8,9,10,11]. PM exposure is also associated with variations in heart rate (HR) in the form of tachycardia and heart rate variability (HRV). HRV, as its name states, is the variation in between heartbeats within a timeframe, measured by differences in the beat-to-beat interval. Decreased parasympathetic nervous system (PNS) activity exhibits decreased HRV, which is associated with respiratory sinus arrhythmia [12]. Furthermore, research of the underlying mechanisms of PM exposure and its proarrhythmic effects on CVD is lacking. Some studies suggest that PM affects the balance of autonomic control on the heart, increases reactive oxygen species (ROS), and changes the ability of blood to coagulate [11, 13]. This section will review the proarrhythmic effects of PM exposure and its long- and short-term effects.
1.1 Clinical Observations
Clinical examinations of the proarrhythmic effects of PM2.5 have been conducted utilizing data from implantable cardioverter defibrillators (ICD), surveys, and clinical records of patients who experienced heart arrhythmias in correlation with PM concentrations recorded via satellite remote sensing, meteorological, and land-use analyses. To study the short-term effects of exposure to PM2.5, ICDs were used to detect and record episodes of ventricular arrhythmias in a study of over 200 patients. There was a positive association between ventricular arrhythmia events and recorded PM levels; the strongest associations found over a 24-h moving average compared to the mean air pollution concentration for the calendar day and the previous day [9]. Patients with previous episodes of ventricular arrhythmias and other CVDs were more susceptible to ventricular arrhythmias correlated with PM exposure [9]. While short-term studies examine the effects of fluctuating PM emissions associated with certain occupations and shifts in weather patterns and development of CVD, long-term studies usually involve exposure of PM levels comparable to industrialized countries.
Recent studies of long-term PM exposure have demonstrated that exposure to PM2.5 has been associated with the onset of tachycardia. An epidemiological study in China of over ten million reproductive-aged adults exposed to PM investigated this phenomenon over a span of 3 years. This study showed that 16% of participants, mean age of 28 years, were classified as having tachycardia. Older individuals (aged 40–64 years) showed a greater increase in HR than younger individuals (<30 years old) after long-term PM exposure, suggesting that older individuals may be more vulnerable to the effects of PM. This study showed a positive relationship between long-term PM exposure and tachycardia, increases in resting HR, and a 4% increase in ventricular tachycardia [14]. This work is supported by another study, which showed increases in PM by as little as 5 μg/m3 were positively associated with HRV by 4.1% and increased diastolic blood pressure [10]. Thus, individuals exposed to prolonged elevation of PM emission are more susceptible to the development of tachycardia. Older individuals and individuals with a medical history of CVD and arrhythmia are more prone to further development and frequent episodes of arrhythmias.
As an increasingly industrialized society, where the combustion of carbon-based fuel is the primary source of PM, the findings of these epidemiological studies are concerning. If increased exposure to PM caused the development of cardiac pathological defects, a decrease in the input of PM would result in the decrease in future developments of PM-induced CVD and arrhythmia. Implementing higher industry and transportation emission standards would provide a means of limiting emitted PM; however, these standards must be implemented at a global level since air, and its associated PM levels, are not the same between cities and countries. When considering the populations that exhibited increased susceptibility to PM-induced CVD presented above, it is also crucial to further investigate what populations are more vulnerable to the effects of PM. Research focused on specific demographics (i.e., socioeconomic status, education level, race, sex, etc.), and the presence of existing disease should be conducted.
1.2 Experimental Studies
Variations in HR and HRV have shown mixed results when reproduced in animal studies of PM exposure. One study showed that increased exposure to concentrated air particles, analogous to PM, increased heart rate during and one hour after exposure, and caused a small increase in ventricular premature beats, though the latter was not statistically significant. These effects were short lived, which may be the result of a short exposure timeframe or the short window of observation [15].
A long-term in vivo study that investigated PM exposures comparable to those of industrialized countries demonstrated that PM2.5 caused cardiovascular remodeling as well as various features of heart failure [16]. Long-term exposure to fine PM was shown to result in cardiac phenotypes consistent with heart failure, spontaneous arrhythmias, and increased HR. A short-term PM exposure in rats further supported these findings providing evidence that daily exposures to PM were shown to generate ROS up to two-fold of control levels [17]. ROS has been associated with increased risk of arrhythmia via several pathways as discussed below in the Underlying Mechanisms section.
Additionally, rats with metabolic syndrome (MetS) show increased susceptibility to the development of cardiac pathophysiology caused by PM exposure, including arrhythmia development. MetS rats showed altered HRV and increased HR after exposure to PM for 5 h/day over 12 days [18]. These rats, fed a high fructose diet and exposed to PM, exhibited altered cardiac electrophysiology, including a fourfold increase in atrioventricular arrhythmias, a diminished baroreflex response, and a decreased vagal influence on cardiac function, which provides another example of a population more susceptible to the development of arrhythmias. These results are similar to those from an epidemiological study in China, which found that older individuals and those afflicted with comorbidities were more susceptible to CV dysfunction after PM exposure [14].
1.3 Underlying Mechanisms
Recent epidemiological studies in adolescents have found that increased PM exposure caused an abnormal shift from parasympathetic control of the heart to predominant sympathetic control, associated with decreased HRV [11, 12]. As mentioned in the introduction, decreases in HRV are associated with respiratory sinus arrhythmia. Explanations for the imbalance of autonomic control of the heart may be explained by the production of ROS caused by PM exposure [19]. After long-term exposure to PM, the increase in ROS triggers ROS-dependent cell-signaling pathways, which result in an acute increase in blood pressure and a shift toward activation of the sympathetic nervous system, which results in arrhythmia.
PM exposure has also been linked to increased activity of NADPH oxidase, which generates •O2 upon phosphorylation of p47phox, a subunit of NADPH, and binds to p22phox in the cell membrane resulting in increased ROS in the vasculature [20]. ROS in the vasculature results in the activation of inflammatory response via several pathways including focal activity, disruption of cardiac ionic currents, increased cardiac fibrosis, and impaired gap junction function [13]. Focal activity caused by ROS has been shown to prolong action potential duration, and induce early and delayed after depolarization, which facilitates arrhythmia development [13]. ROS disrupts ion currents as a result of residual Na+ flow after the action potential peak, abnormal release, and reuptake of Ca2+ in the sarcoplasmic reticulum, as well as inhibiting KATP channels which prolongs action potential duration.
Other studies have suggested that exposure to PM caused an inflammatory response in the alveoli and release of cytokines, ultimately changing blood coagulation. The ultrafine PM is retained in the interstitial tissue of the lungs where an inflammatory response follows. This retention is likely due to the macrophages’ inability to phagocytize ultrafine PM particles. Changes in blood coagulation have been linked to increased susceptibility to CVD event (i.e., myocardial infarction) [21]. Further research is still needed to fully understand the mechanisms of PM exposure on heart arrhythmias.
2 Epigenetic Modifications Overview
Epigenetic modifications are essential for normal physiological processes. However, there is clear evidence for the role of epigenetic processes in the development of cancers, cardiovascular disease, neurological, and autoimmune disorders. An increased understanding of the mechanisms that drive epigenetics could offer the missing link in developing more effective therapeutic approaches for cardiovascular disease. While epigenetic modifications can be detrimental to cardiovascular health, the epigenome possesses reversible properties that enable it to be reprogrammed. This feature has been established and applied to the development of cancer therapeutics: epigenetic drugs, termed epidrugs, are currently being developed to target the epigenetic modifications responsible for inhibiting tumor suppressor and DNA repair genes [22, 23]. Epidrugs are designed to target enzymes responsible for completing epigenetic modifications including DNA methyltransferases and histone deacetylases. By inhibiting such enzymes, epidrugs prevent unwanted silencing of genes and help to reestablish normal function within the epigenome. Thus, certain epigenetic modifications relevant in the development of disease can be reversed. Mounting evidence suggests the coupling of epigenetic drugs with chemotherapy could provide an effective treatment option for diseases such as cancers. Although still in its infancy, this research suggests that epigenetic modification may be utilized to treat cardiovascular diseases [24].
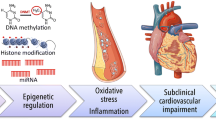
A description of each epigenetic mechanism, providing the foundation for this chapter, is depicted in Fig. 25.1. The subsequent sections will expand on the links between external stimuli, induced epigenetic modifications, and the resulting cardiac dysfunction.

Particulate matter-induced epigenetic modifications of chromosome in cardiomyocyte including DNA methylation, histone modifications, chromatin remodeling, and RNA-based modifications and resulting dysfunction. Figure created with BioRender.com
2.1 DNA Methylation
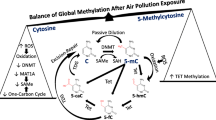
DNA methylation, an essential epigenetic regulator in many eukaryotic organisms, occurs when a methyl (CH3) group is covalently added to the 5-C position of a cytosine on DNA. The execution and maintenance of this modification is determined by a family of three DNA methyltransferase (DNMT) enzymes: DNMT3a, DNMT3b, and DNMT1. Common changes induced by DNA methylation include regulation of gene expression, suppression of transposable elements, genomic imprinting, and X-chromosome inactivation [25, 26]. DNA is highly methylated in vertebrates: ~60–90% of cytosine preceding guanosine (CpG) sites are methylated, with the exception of “islands” of unmethylated areas called CpG islands [27]. Methylation plays a highly regulatory role in gene expression at CpG sites; however, its role changes when CpG islands are the targets of modification.
CpG islands comprise around 1000 CpG-dense base pairs. Under normal conditions, CpG islands lack methylation [28]. These islands are nonrandom; they encompass ~1% of genomic DNA and are located at the promoter regions (5’ N-terminus) of many genes. Housekeeping genes, which act to maintain cellular function and to normalize expression values of variable genes for experimental and computational studies, are among the most common genes possessing CpG islands [29, 30]. Housekeeping gene promoter regions must remain unmethylated to ensure their constitutive expression to maintain normal cell functionality. However, when methylation within CpG islands occurs, access to DNA is decreased, blocking transcription factor binding and gene expression [28]. Methylation of CpG islands and dysregulation of prevalent methylation patterns (60–90% of CpG) potentiate an array of consequences, ranging from developmental disorders to various diseases, including CVD [27, 28, 31].
DNMT1 is the primary methyltransferase responsible for maintaining the covalent attachment between methyl groups and cytosines upon DNA replication. DNMT1 acts to copy the parent strand methylation pattern onto the unmethylated daughter strand, transforming the double-stranded DNA from hemi-methylated to fully methylated. DNMT3a and DNMT3b (de novo methyltransferases) aid in establishing novel patterns of methylation early on in development by adding methyl groups to unmethylated CpG bases [32]. DNA methylation is vital to cell differentiation and embryonic development, and DMNT1 plays a crucial role in subsequent methylation pattern maintenance [32, 33]. Genetic knockout experiments have revealed DMNT1, DMNT2, and DMNT3 are essential to offspring viability, though DMNT2 is not directly involved in the DNA methylation process. DNMT1 inactivation resulted in embryonic lethality, and DMNT3a/DNMT3b inactivation caused death shortly after birth in mice. Reduced or disrupted patterns of these enzymatic catalysts contribute to genomic destabilization, promoting cancer, and disease development [34]. Hypermethylation of CpG island promoter regions exemplifies abnormal DNMT activity, causing transcriptional silencing effects on cellular pathways (mimicking those of mutations/deletions). Hypermethylation of DNA repair genes has also been associated with cancer and disease development [34]. Proper DNA methylation is essential to the growth, development, and maintenance of the human genome.
Methylation can be reversibly modulated by demethylation, accomplished through both an active and passive approach: demethylases are the active enzymatic agents responsible for combating the effects of malfunctioning DNMTs and methylation of CpG islands, often at promoter regions, where hyper/hypomethylation can silence gene expression and result in cancer and disease development [34]. The passive mechanism involves failure to retain methylation patterns during DNA replication and cell division. However, demethylation remains a highly uncharted concept, limiting its current application for therapeutics. Furthermore, the 5C-methyl modification is thought to be highly stable, and it has yet to be determined if breaking the carbon–carbon bond necessary to remove the methyl group from the 5-methylcytosine is possible. Still, cytidine deaminases, elongator complexes, base excision repairs, nucleotide excisions, DNA methyltransferases, and DNA glycosylases are candidate gene families and pathways suspected to play a role in the biochemical mechanisms associated with DNA demethylation [35]. With the increasing potential to reverse methylation of undesirable regions with accuracy, there is hope for detecting and treating diseases caused by epigenetic modifications by offering a directed therapeutic approach [32, 35].
2.2 Chromatin Remodeling
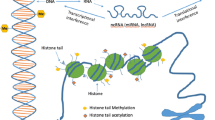
DNA is tightly packaged by wrapping around an octamer of duplicate histone proteins (H2A, H2B, H3, and H4) to form a nucleosome, which is the structural unit of the chromatin complex [36]. This system offers an effective way of housing large amounts of genetic material while dividing the genome between transcriptionally active and inactive/repressed zones [37]. Nucleosomes provide a primary means of genomic compaction and a “signaling hub” for chromatin-templated processes. During chromatin remodeling, ATP-dependent enzymes manipulate the function of nucleosomes, resulting in post-translational modifications [38]. Energy from ATP hydrolysis is used to elicit changes from nucleosome movement to destabilization or restructuring by altering associations between histones and DNA on the nucleosome [26, 39].
There are four classes of ATP-dependent chromatin remodeling complexes that are powered by ATP hydrolysis and function to slide or eject nucleosomes. The complexes manage nucleosome grouping and configuration by communicating with histone chaperones and swapping histones and histone variants [40]. The classes include switching defective/sucrose no-fermenting complexes (SWI/SNF); imitation switch complexes (ISWI); chromodomain, helicase, DNA-binding complexes (CHD); and inositol-requiring 80 complexes (INO80) [26]. The chromatin remodeling complex families contain varied levels of domains, subunits, and accessory subunits, which dictate their target specificity and role in gene expression regulation [40]. Regardless of differences in domain and subunits, the complexes serve a common purpose of altering gene expression at some capacity. Some protein complexes promote access to nucleosomal DNA, while others inhibit access by retightening and reforming the DNA-protein complexes that comprise the nucleosome [36]. The degree that chromatin is condensed modulates transcription, DNA replication, and DNA damage response. Chromatin remodeling complexes function to modulate the state of chromatin and its degree of accessibility, effectively altering gene expression [40]. Furthermore, chromatin remodeling directs protein access to DNA and plays a regulatory role in DNA damage-induced signaling pathways [41]. Irregular functionality of the ATP-dependent chromatin remodeling complexes is linked to disease development, since they are key players in fundamental cellular processes (i.e., transcription, DNA replication, and repair).
2.3 Histone Modifications
Histones are protein structures that organize DNA into nucleosomes as a means of packing a significant amount of genetic information into a condensed structure. Histone modifications provide a means of post-translational control of gene expression by modulating the degree of chromatin compaction. Histone modifications include phosphorylation, ubiquitination, sumoylation, acetylation, deacetylation, ADP-ribosylation, proline isomerization, deamination, methylation, and demethylation [42]. Histone acetylation refers to the introduction of an acetyl group (CH3CO) to a member of a histone tail (commonly lysine). It is the most studied modification and dysregulation that has been implicated in the development of CVD [43]. Acetylation provides greater access to chromosomal domains that are highly compacted and elicits increased access to promoter regions, ultimately activating transcription. Without acetylation, nucleosomes will continue folding into more complex and inaccessible structures. Accessibility is thus a balance of the synergistic actions between acetylation and chromatin remodeling via ATPases and methylation [44]. Histone acetyltransferases (HATs) transfer an acetyl group to histone proteins and play a crucial role in the activation of genes and transcription. Histone deacetylases (HDACs) effectively remove the acetyl group from histone tails, resulting in a reversal of DNA accessibility and an increased level of nucleosomal fiber folding, inducing repression [45]. A disruption of the balance between HAT and HDAC standard functionality can elicit cardiac pathologies, which will be discussed later in this chapter.
Histone methyltransferase (HMT) enzymes use S-adenosylmethionine (SAM) as a cofactor to catalyze the methylation of basic residues (lysines, arginines, and histidines) in histone tails. Individual residues and different residues of the same histone can be methylated to varied degrees. For example, lysine can be mono-, di-, or tri-methylated [46]. The extent of methylation affects the condition of differential gene expression, causing either activation or repression of a methylated gene [47]. Protein recruitment may cause additional chromatin folding or increased access to the DNA [36]. Demethylation refers to the reversal of methylation via removal of methyl group(s) from histone tails. Similar to histone methylation, demethylation can promote or inhibit gene activity [34]. Experimental rat models have associated heart failure development to two histone modifications: trimethylation of histone H3 on lysine-4 or lysine-9, thus revealing the implications few epigenetic modifications can have in cardiovascular disease development [48].
2.4 RNA-Based Modifications
The central dogma of molecular biology in which DNA is transcribed into RNA and RNA is translated into protein oversimplifies the relationship between DNA, RNA, and protein. DNA is transcribed into RNA, which can later be translated into protein; however, this is not always the case. In fact, genomic DNA is predominately transcribed into noncoding RNA (ncRNA), which fails to be translated into proteins. ncRNAs maintain a highly purposeful role in epigenetics and the evolution of disease [49]; ncRNA is divided into small noncoding RNAs (sncRNAs), which includes microRNAs (miRNAs) and long noncoding RNA (lncRNA), greater than 200 nucleotides in length [50]. miRNA—an endogenous form of sncRNA that is ~22 nucleotides long—is the most understood form of sncRNAs to date. miRNA dysregulation has been found to play a major role in the formation of cancer and disease [51,52,53].
The biogenesis of miRNA involves a multistep process beginning with the transcription of miRNAs into long primary transcripts (pri-miRNAs). Next, pri-miRNAs form unique precursor stem loop structures ~77 nucleotides in length (pre-miRNAs) that are cleaved by RNase III nuclease [54]. The transcripts are then exported from the nucleus to the cytoplasm and cleaved by RNase III Dicer, resulting in a final miRNA ~22 nucleotides in length [54]. Upon reaching mature form, one strand of miRNA is organized into an RNA-induced silencing complex (RISC) that can recognize and bind short fragments of mRNA [55]. The seed region of miRNA is between nucleotides 2 and 8 at the 5′ end of the strand. This region is essential for target recognition, functioning to identify the 3′ untranslated region (UTR) of the mRNA to which it binds. Targeting of 3’ UTR regions is widespread, though regions such as 5’ UTR or coding regions may be targeted instead. The normal function of mRNA can effectively be repressed by miRNA binding, and a single miRNA is capable of targeting several mRNAs [56]. High complementarity between the miRNA and target mRNA promotes degradation of target mRNA by a process termed RNA interference. Low complementarity yields translational inhibition of target mRNA, via induced ribosomal alterations, which is highly prevalent in human cells. The interplay between high and low complementarity and their mechanisms offers an effective way of maintaining homeostasis. In addition to interacting with mRNA, organized actions between miRNAs and other epigenetic modifications also exist [56, 57].
The descriptions above detailing each common epigenetic modification provide an overview of their properties, elucidating the highly complex and cooperative nature of epigenetic modifications and their ability to influence cellular function and genetic expression. These mechanisms apply to various systems throughout the body and can be affected by many external triggers, such as air pollution. Although epigenetic changes can induce unfavorable expression, since alterations are not at the DNA sequence level, the possibility to reverse detrimental modifications is within reach. Throughout this chapter, the relationships between epigenetics, arrhythmias, and air pollution in the form of PM exposure will be introduced. First, we will consider the connection between epigenetics and arrhythmias. We will then highlight the effect of PM on arrhythmias and the epigenome. Lastly, we will consider what role other pollutants, including traditional cigarettes, have on the development of arrhythmias.
3 Epigenetics as a Mechanism for Particulate Matter-Induced Cardiovascular Disease
3.1 Role of DNA Methylation in Arrhythmogenesis
Abnormal patterns of DNA methylation have been closely associated with many CVDs, such as arrhythmias [58]. Parental atrial fibrillation (AF) is a risk factor for arrhythmia development, indicating a genetic component [59, 60]. A variety of remodeling changes, such as fibrosis and signaling pathways, lead to physiological changes and cardiac disease conditions, in which AF is a common endpoint [60,61,62]. Additionally, regulation of extracellular matrix proteins, which cause fibrosis that leads to arrhythmias, has been shown to be regulated by DNA methylation [63]. Fibrosis slows the electrical function by jeopardizing the electrical impulse propagation. There are some discrepancies in the literature concerning the uniformity of methylation across the atrium of the heart. Methylation was largely found to be conserved across atrial tissue of coronary bypass surgery patients where after surgery 25% of the patients developed AF, which contrasts starkly with another study involving induced heart failure in rats that found CpG islands to be hypermethylated in the atria with fibrillation [64, 65]. The link between DNA methylation and gene expression in heart disease needs to be further studied before it can be a predictive indicator of gene expression in human populations [66]. DNA methylation has been shown, however, to play a central role in the maintenance of cardiac fibrosis, which contributes to the pathogenesis of AF [62, 63].
SERCA2a, an ATPase that plays an important role in transporting calcium into the sarcoplasmic reticulum, contains CpG islands in the promoter region of its gene [67]. When the expression of SERCA2a is altered, calcium handling becomes abnormal, a lower expression leads to less capacity for calcium handling, leading to a change in contractility and arrhythmia [67]. The methylation of the SERCA2a promoter region is affected by DNMT levels. In one study, DNMT levels in HL-1 cardiomyocytes were increased after introduction of tumor necrosis factor alpha (TNF-α), leading to an enhancement in methylation of SERCA2a, reducing its expression [67]. Activity of SERCA2a and phospholamban can imperatively influence the calcium handling leading to potential risk to developing an arrhythmia as well as heart failure [68]. When cardiac fibrosis was induced via isoproterenol injections in rats, DNMT3a was found to silence RASSF1A via extracellular signal related kinase (ERK1/2) upregulation, which caused increased fibroblast proliferation [69]. Inhibition of this methylation may be a novel treatment for impaired heart function and arrhythmogenesis [67].
The homeobox gene Pitx2c plays a role in AF, and contains CpG islands in its promoter region, allowing it to be silenced through methylation [65]. Heart failure induces Pitx2c promotor hypermethylation, in part due to increased levels of DNMT1, which decreases the protein levels of Pitx2c [65]. Pitx2c promotor methylation in HL-1 cells (atrial cell line) and DNMT are increased when treated with Angiotensin II (Ang II), thereby downregulating the protein levels of Pitx2c and Kir2.1 [65]. When cultured with the Ang II receptor blocker losartan, the effects of hypermethylation on Pitx2c expression in HL-1 cells that had been treated with Ang II is reduced [65]. Thus, Losartan may be beneficial for blocking Ang II receptors which may be a novel therapeutic target [65]. Pitx2c regulates expression of the sodium channel subunit Nav1.5; as well as rectifier channels KCNA3, KCNC4, and Kir2.1; all of these potassium channels play a role in contractility. A study of both mouse and human atria samples that had atrial arrhythmias found Pitx2c to exhibit increased methylation [70]. Dysregulation of Pitx2c promotes susceptibility to AF by modifying calcium handling, altering function of melanocytes, (melanin producing cells that are found in the valves and septum of the heart among other areas of the body) and changing cell-to-cell communication [70].
SUR1 and SUR2 are two gene subunits that form ATP sensitive potassium channels in cardiac myocytes that combine with Kir6.2, a major potassium channel subunit, whose expression is also susceptible to DNA methylation [71]. By using RNAzol on HL-1 cells cultured from atrial myocytes, it was shown that SUR2 can be silenced by CpG methylation, ultimately leading to changes in KATP density and composition, and may eventually lead to dysfunction in the heart [71]. Bisulfate sequencing on these same HL-1 cells has shown that SUR2 is much more prone to methylation versus SUR1 (57.6% vs 0.14%) at CpG sites within their promotor regions [71]. Investigated as a potential treatment, decitabine caused a decrease in SUR1 and increased the unmethylated fraction of SUR2 mRNA and CpG island expression [71]. More avenues to study these epigenetic changes are needed to explore this multi-faceted function, as the targeted silencing of gene expression can imbalance the structural proteins compromising overall function.
3.2 Role of DNA Histone Modifications in Arrhythmogenesis
HDACs are another way in which expression can change epigenetically via post-translational modification. Alteration of HDAC enzymes may cause DNA to be less accessible to transcription factors. Reduced DNA transcription can lead to a range of arrhythmogenic diseases, including long QT syndrome, Brugada syndrome, short QT syndrome, catecholaminergic polymorphic ventricular tachycardia, and arrhythmogenic right ventricular cardiomyopathy [72]. Knockout of HDAC1 and HDAC2 in a mouse model resulted in neonatal lethality, which severely altered growth and function of the hearts by upregulating calcium channel subunits, resulting in cardiac arrhythmias among other cardiac defects [73]. Much like DNA methylation, histone deacetylases are epigenetic modulators and can influence cell proteostasis, prompting investigation of the role of HDACs in the structural remodeling associated with AF and potential therapeutics to abate AF.
HDACs are vital to calcium homeostasis; AF patients have CaMKII-hyperphosphorylated ryanodine receptors, causing sarcoplasmic reticulum leak, sodium-calcium exchanger over-activation, and delayed after-polarizations [74]. In cardiomyocytes found in the pulmonary vein, the use of MPTOE014 (an HDAC inhibitor) modulated the regulatory proteins for calcium homeostasis, helping negate AF inducibility [75]. Tachypacing, (induced rapid pacing) of HL-1 atrial cardiomyocytes resulted in deacetylation, depolymerization, and degradation of α-tubulin by calpain [76]. HDAC6 activated by the tachypacing, leads to this imbalance in α-tubulin which modifies the microtubule structure in cardiomyocytes causing contractile dysfunction [76]. Additionally, inhibition of HDAC6 via tubastatin A was protective in a dog model of AF, suggesting that the TDAC domain of HDAC6 is a potential upstream target to reduce cardiomyocyte remodeling and protect α-tubulin proteostasis [76]. Inhibiting a targeted HDAC may help regulate natural levels of DNA transcription as a means to inhibit adverse cardiac remodeling.
Histone deacetylase inhibitors have been examined as a therapeutic target of fibrosis in chronic conditions. The HDAC inhibitor mocetinostat was shown to be antifibrotic in the hearts of rats with myocardial infarctions, where HDAC1 and HDAC2 were upregulated via left anterior descending coronary artery occlusion [77]. Valproate, an anticonvulsant drug, impairs HDAC6 and HDAC8 when elevated by cardiac hypertrophy, making it a possible inhibitor to treat or prevent cardiac dysfunction [78]. HDAC inhibitors trichostatin A (TSA), valporic acid, and SK-7041 partially reversed hypertrophy in a rat model [78]. In primary mouse myocytes, TSA inhibits Ang-II-induced cardiac hypertrophy response [70]. TSA has also been shown to reduce cardiac hypertrophy in a mouse model of heart failure [79]. In another mouse model, mice with overexpression of homeodomain protein (HopXtg), which induces cardiac hypertrophy through HDAC activity, saw reduced atrial arrhythmia after TSA treatment [80]. It also normalized size distribution, expression of connexin40, and Angiotension II levels between wild type and HopXtg mutant mice [80]. Thus, HDAC inhibition has its therapeutic uses in reversing connexin40 remodeling, atrial fibrosis, and atrial arrhythmia vulnerability where hypertrophy is present.
3.3 Role of miRNA Reprogramming in Arrhythmogenesis
Studies have demonstrated a clear impact of miRNAs on arrhythmogenesis by regulating gene expression related to AF. Many recent studies have investigated the functions of how miRNA contribute to the genesis of AFs and have been implicated largely in fibrosis and ion channel remodeling, two of the main mechanisms that lead to AF [81]. miRNA may be used as therapeutic target or biomarker for AF. Some miRNAs (miRNA1, miRNA133, miRNA208, and miRNA499) are capable of reprogramming fibroblasts, both in vivo and in vitro, to become cardiac myocyte-like [82]. Many fibrosis-regulating miRNA bind to profibrotic targets [83]. Most atrial fibrosis promoting miRNA are down-regulated in AF conditions, except miRNA-21, which is up-regulated [83].
The table below has a list of the known miRNAs and their likely role in altering heart function, potentially contributing to arrhythmia development. Many miRNAs contribute to electrical and structural remodeling in the heart; miRNA1, miRNA26, miRNA106b-25 cluster, miRNA-133, miRNA 208, miRNA328, and miRNA499 all up- or down-regulate genes that affect electrical remodeling. MiRNA21, miRNA26, miRNA29b, miRNA30, miRNA499, and miRNA 590 play a role in dysregulating genes that lead to structural remodeling. In vivo manipulation of miRNA has been investigated in AF: miRNA1, miRNA21, miRNA26, miRNA29b, and miRNA328 have all been manipulated in either mouse, rat, dog, or rabbit and have been shown to reduce atrial remodeling and fibrillation [62]. Looking into the miRNA that lead to AF can give clues to what may be triggering the onset, and potentially how to abate the disease therapeutically. The Table 25.1 outlines the miRNA that have been found to be linked to AF regulation. miRNA targets for expression and the consequences associated with their dysregulation are also presented.
4 Particulate Matter and the Epigenome
The reactive, pro-oxidant nature of PM makes it a prime target for environmentally caused changes to the epigenome. As PM enters the body, it has the ability to readily interact with cell populations from the lung, blood, and vasculature. How PM affects these cells and its relevance to human health is under investigation at the cellular level, in animal-based research, and in clinical cohorts. The highlight of current clinical investigations is based primarily upon the fact that interactions with PM causes changes in the epigenetic layout. Primary evidence for this involves repeated-element methylation and methylation of specific genes during PM exposure. However, emerging work is centered around the adult origins of developmental disease hypothesis. This hypothesis stipulates that the insults (e.g. PM exposure) that occur during germ cell or in utero development affect phenotypes at adulthood, indicating that epigenetic modifications may play an important role.
4.1 In Vivo Modifications to the Epigenome
As the ability for real-time PM exposure monitoring in human subjects is limited, many studies have utilized clinical cohorts and examined the significance of local PM levels on epigenetic markers using patient data. Altered methylation of repeated elements have been correlated with PM or PM markers in several of these studies. Decreased methylation of long interspersed nuclear element −1 (LINE-1), which comprises approximately 17% of the human genome, has been positively associated with black carbon and SO4 levels [88,89,90], and blood levels for several persistent organic pollutants [91]. Similarly, hypomethylation of the transposable element Alu has been found to be associated with PM and its constituents [37, 38, 41], confirming the presence of global changes to the epigenome in humans exposed to PM. How such changes contribute to disease severity, alter development, and change the genetic makeup of offspring is currently under investigation.
Additional studies have examined altered epigenetic modification and function of specific genes with PM exposure. PM exposure in a cohort of children was negatively associated with inducible nitric oxide synthase (iNOS) promoter methylation [92], which was also found in a cohort of foundry workers, where the NOS2A gene had decreased expression following three days of work [93]. This decrease in iNOS promoter methlyation was also found to be PM-associated and was suggested to be of functional relevance in respiratory outcomes due to increased expression after PM exposure [94]. Correlation of PM levels with methylation of specific genes have also been found including Foxp3 [95], TLR-2, ICAM-1 [90], and TLR4 [96]. One study found PM to be associated with genes in the MAPK pathway, but not the Nf-κB pathway [97], demonstrating that PM can cause specific, nonglobal changes to the epigenome. As to whether these changes are adaptations to organic pollutants providing protection or are damaging and contributing to the phenotype found with PM exposure remains undetermined.
4.2 Germline and In Utero Exposure to PM
The Barker hypothesis, or the developmental origins of adult disease hypothesis, was initiated with the observation that those born following periods of in utero stress, such as famine, and thus had reduced birth weight, are at a greater risk for heart disease later in life [98]. Data therein thus implies fetal genetic imprinting, and thus epigenetics is a clear candidate for these outcomes. How PM affects the developmental timeline, and if there are relevant cardiac effects, is thus a target of important study.
Determining how PM affects the developing human and what changes manifest during adulthood are by nature very difficult, thus several studies in animals have been used to further understand the biology and mechanism. Mice were exposed during pregnancy to a relevant level of PM, and then removed from exposure at birth; resulting pups from these mice compared to pups from dams exposed to filtered air had reduced cardiac output, cardiac electrical remodeling and alterations in gene expression of deacetylases and DNA methyltransferases at adulthood [8]. These mice were born with reduced bodyweight and had an impaired cardiac phenotype merely from exposure to PM in utero. Continuing work is also indicating a germline effect, as preconception exposure of mice to PM has been found to cause a decrease in cardiac function of male offspring [99], perhaps due to the fact that hypermethylation of sperm DNA has been shown to occur after PM exposure [100]. Indeed, pups from mice undergoing transplacental exposure of diesel exhaust particles (DEP) have been found to have genetic deletions [101], adding to the evidence that PM causes gene-level alterations during development.
5 Cigarette Smoke and Arrhythmogenic Potential
Several conditions exhibit arrhythmogenic potential including heart failure, endocarditis, heart valve disease, diabetes, and asthma. Furthermore, lifestyles such as consuming alcohol may impact the likelihood of developing arrhythmias [102]. Epidemiological studies examining traditional cigarette smoking demonstrate mixed results on arrhythmogenic risk potential; however, many in the scientific community posit cigarette smoking as a risk factor. The epidemiological evidence linking PM with increased CVD and respiratory disease incidence has been widely published and accepted [103]. Cigarette smoke is comprised of thousands of chemicals, including PM, that are carcinogenic and include tar, benzene, formaldehyde, and arsenic. While the chemical profile of cigarette smoke contains several similar compounds found in PM, key differences arise in the source and concentration of particles. Automobile emissions, industrial wastes, agricultural practices, and roadside dust are all sources of ambient PM. Therefore, due to regional differences in the production of these pollutants, PM profiles are variable and difficult to characterize. Cigarette usage also bears the risk of complications caused by nicotine, an addictive substance associated with many acute cardiovascular responses [104,105,106,107]. Cigarette smoke, however, has a much more consistent compound profile allowing for more standardized and replicable studies. Here, we compare cigarette smoke-induced arrhythmogenesis to particulate matter induced arrhythmia.
5.1 Epidemiology of Atrial Fibrillation and Cigarette Smoking
AF has been the focus of many population-based studies of smoking. These large analyses have produced varying results, complicating our understanding of the role of smoking on AF. The Atherosclerosis Risk in Communities study (ARIC) analyzed participants over an average follow-up period of 13.1 years. In this study, current smokers exhibited a 2.05 relative risk score of developing AF compared to those who never smoked [108]. Benefits to smoking cessation were found. While those who ceased cigarette use still had a relative risk score of 1.32 compared to never smokers, this score is considerably lower than current smokers [108]. Similar trends in data are seen in the Rotterdam and Hordaland studies as both show an increased risk of AF for current smokers [109, 110], however, these studies resulted in different conclusions on the incidence of AF in former smokers. Interestingly, the Rotterdam study found no benefit for former smokers [109] as the relative risk of AF development was nearly identical between former and current smokers. In contrast, the Hordaland study observed former smokers carried no increased risk for AF [111], thus suggesting substantial benefits for cessation. These studies outlined the adverse association of smoking behavior and AF; however, questions regarding the potential benefits of cessation on arrhythmogenesis must be understood more clearly.
Contradictory to the above correlation between smoking and AF, several notable studies of similar design demonstrated no significant risk of AF with smoking habits. In a cohort of 15,000 men and women, current or former smoking status was not found to correlate with higher incidence of AF [110]. In a retrospective analysis of the Framingham Heart Study, this result was further solidified, as no correlation was found between smoking and AF [112]. However, cardiac disease traditionally related to smoking was linked to AF development through a 10-year risk assessment, thus suggesting that any arrhythmogenic potential due to smoking may require long-term exposures to first establish conditions such as heart failure or ventricular hypertrophy. Additionally, the Manitoba Follow-up Study showed similar findings [112, 113]. These results are significant since they question the association of smoking behavior and AF development. Therefore, further studies are warranted to understand the potential correlation and identify whether smoking directly or indirectly leads to AF.
5.2 Epidemiology of Ventricular Arrhythmogenesis and Cigarette Smoking
Ventricular arrhythmias have also been associated with traditional cigarette usage. Several studies have analyzed electrocardiogram (ECG) recordings of chronic heavy smokers and never smokers and found altered ventricular repolarizations with changes in electrocardiographic T wave (Tp-e) to QT ratios (Tp-e/QT), an index of arrhythmogenesis and predictor of future arrhythmia [114]. In an echocardiographic analysis of healthy male and female subjects (24 smokers and 23 nonsmokers), chronic smoking was found to correlate with prolonged Tp-e interval and increases to Tp-e/QT ratio and Tp-e/QTc ratio [115]. These ECG markers indicate global cardiac electrical dysfunction, a possible pathophysiological mechanism that can be targeted for therapeutic development. The evidence for altered electrical instability in arrhythmogenesis was further confirmed by a large study revealing similar results. Comparing 121 smokers to 70 control subjects, the authors found increases in the following intervals of smokers: QTd, cQTd, Tp-e, cTp-e [116]. Additionally, Tp-e/QT and Tp-e/QTc ratios were significantly increased in smokers compared to nonsmokers [116]. The large body of evidence examining the relationship between smoking, ECG abnormalities, and arrhythmogenesis thus provides a clear framework for clinicians to treat and monitor arrhythmia in the early stages of disease.
5.3 Nicotine Exposure on Molecular Mechanisms Associated with Arrhythmogenesis
The most widely studied constituent of tobacco smoke is nicotine, an addictive substance that acts on nicotinic acetylcholine receptors. The hemodynamic responses to nicotine exposure, regardless of the route of administration (intravenous, nasal spray, and ingestion), have been characterized by increases in heart rate and blood pressure [117, 118]. Since the type of nicotine exposure does not significantly impact the effects of nicotine on the body, studies investigating molecular mechanisms altered by nicotine can be easily translated to other exposures. The implications of adverse cardiovascular responses thus extend clinically to the many nicotine delivery products that exist such as cigarettes, e-cigarettes, and nicotine replacement therapy.
In a case–control study examining the role of nicotine on fibrosis, atrial tissue slices from patients (both smokers and nonsmokers) undergoing elective coronary artery bypass grafting were studied for fibrosis [119]. In this analysis, the authors found that pack years, a unit of measurement in cigarette smoke research to identify tobacco exposure, was the only parameter associated with the magnitude of fibrosis [119]. Furthermore, the authors exposed cultured atrial tissue to increasing doses of nicotine and found an increase in collagen III mRNA expression [119]. Fibrosis changes the composition of atrial tissue and subsequently alters cardiomyocyte electrophysiology, leading to atrial arrhythmias like AF [120]. To find a molecular pathway for nicotine-induced fibrosis, researchers analyzed canine atrial fibroblasts incubated with nicotine for 24 hours. These fibroblasts demonstrated an increase in collagen production and upregulation of TGF-β1 and TGF-βRII via downregulation of miR133 and miR590 [121]. The resulting response is an increase in CTGF signaling, a known fibrotic mechanism [121]. In addition to induced cardiac remodeling, nicotine has been shown to directly interact with potassium ion channels. In a study assessing the pharmacologic effects of nicotine on A-type K+ channels in dog ventricular cardiomyocytes, it was discovered that nicotine is a potent blocker of the outward KV4 current [122]. This interaction delays cardiomyocyte repolarization by inhibiting the outward flow of potassium and may contribute to arrhythmogenesis. Nicotine has also been shown to alter sodium current expression in rabbits following in utero exposure. Using a device to continually deliver nicotine subcutaneously, similar to nicotine replacement therapy, researchers found an increase in the INa amplitude of offspring due to upregulated Nav1.5 [123]. Continuing to understand the role of in utero nicotine exposure and its impact on sodium currents, the same group investigated sodium channel response following isoproterenol stimulation, an agonist of β-adrenergic receptors. They found that isoproterenol did not affect INa, suggesting myocyte dysfunction in response to sympathetic activity [124]. This data, paired with alterations in potassium ion flow, shows the potential for nicotine to induce arrhythmogenesis via directly impairing the electrical signaling in the heart. Pharmacological analysis must be further investigated for additional ion channels affected by nicotine to understand changes in cardiac electrical currents. Additionally, since many new nicotine products have entered the market in recent years (e-cigarettes and nicotine replacement therapy), it is imperative to understand the variety of adverse alterations that nicotine may induce on the cardiovascular system.
6 Conclusion
Mounting evidence suggests that global cardiac fibrosis, a preclinical factor associated with arrhythmogenesis, is induced through adverse epigenetic alterations. Analyses of changes in several epigenetic pathways (DNMTs, HDACs, and miRNAs) provide a framework for potential areas of focus. Changes in specific targets due to these mechanisms have previously shown increased fibrosis of cardiac tissue, which creates an environment conducive to development of arrhythmias. However, the precise progression from fibrosis to arrhythmic phenotype is unknown since both cardiac fibrosis and arrhythmia may exist independently of one another.
PM, a pollutant known to be associated with CVD development and mortality, has been shown to alter the epigenome in several experimental models. Research has demonstrated that DNA methylation, particularly hypomethylation, of many genes related to oxidative stress and immune function is changed due to PM exposure. Whether these modifications are adverse or protective adaptations has yet to be discovered. Transient elevations in regional PM concentration correlate with higher incidence of ventricular arrhythmia and overall CVD events. Interestingly, sustained exposure over a long period of time leads to tachycardia. Therefore, the public health implications of PM exposure must not be ignored. Further study into the mechanisms of acute and long-term PM exposure is an important topic, especially in areas where transient changes in PM concentration are common.
Cardiac arrhythmias occur via alterations in the normal conduction sequence of a heartbeat, including tachycardia, bradycardia, or irregular/erratic patterns. Clinically, these conditions can range from life-threatening occurrences to harmless transient episodes. This variation in presentation has caused difficulties in identifying precise pathological mechanisms of arrhythmogenesis. In this chapter, we have discussed how epigenetics is a promising target for arrhythmogenesis research, and how the use of environmental triggers has contributed to our understanding of arrhythmogenesis via epigenetic mechanisms. Elucidating these precise mechanisms will provide targets for epigenetic treatments that are currently being investigated in many diseases including cancer, cardiovascular, neurological, and metabolic diseases.
Abbreviations
- AERP:
-
atrial effective refractory period
- AF:
-
atrial fibrillations
- AHA:
-
American Heart Association
- Ang II:
-
Angiotensin II
- ARIC:
-
Atherosclerosis Risk in Communities
- CHD:
-
chromodomain, helices, DNA-binding complexes
- CpG:
-
cytosine preceding guanosine
- CTGF:
-
connective tissue growth factor
- CV:
-
cardiovascular
- DEP:
-
diesel exhaust particles
- DNMT:
-
DNA methyltransferase
- EGC:
-
electrocardiogram
- ERK:
-
extracellular signal-related kinase
- HATs:
-
histone acetyltransferases
- HDACs:
-
histone deacetylases
- HMTs:
-
histone methyltransferase
- HopXtg:
-
homeodomain protein
- HR:
-
heart rate
- HRV:
-
heart rate variability
- ICD:
-
implantable cardioverter defibrillators
- INO80:
-
inositol-requiring 80 complexes
- iNOS:
-
inducible nitric oxide synthase
- ISWI:
-
imitation switch complexes
- lncRNA:
-
long noncoding RNA
- MetS:
-
metabolic syndrome
- miRNAs:
-
microRNAs
- ncRNA:
-
noncoding RNA
- PM0.1:
-
ultrafine PM, diameter of <0.1 μm
- PM10:
-
coarse PM, diameter of <10 μm
- PM2.5:
-
fine PM, diameter of <2.5 μm
- PNS:
-
parasympathetic nervous system
- pri-miRNAs:
-
primary miRNA transcripts
- RISC:
-
RNA-induced silencing complex
- ROS:
-
reactive oxygen species
- SAM:
-
S-adenosylmethionine
- sncRNAs:
-
small noncoding RNAs
- SWI/SNF:
-
Switching defective/sucrose nonfermenting complexes
- TNF:
-
tumor necrosis factor
- TSA:
-
trichostatin A
- UTR:
-
untranslated region
References
Tanwar V, Katapadi A, Adelstein JM, Grimmer JA, Wold LE. Cardiac pathophysiology in response to environmental stress: a current review. Curr Opin Physiol. 2018;1:198–205.
Gorr MW, Falvo MJ, Wold LE. Air pollution and other environmental modulators of cardiac function. Compr Physiol. 2017;7(4):1479–95.
Particulate Matter (PM) Basics [Internet]. EPA. Environmental Protection Agency; [cited 2021Sep23]. Available from: https://www.epa.gov/pm-pollution/particulate-matter-pm-basics
Shimizu Y. 9 out of 10 people worldwide breathe polluted air, but more countries are taking action [Internet]. World Health Organization. World Health Organization; 2018 [cited 2021 Sep 23]. 2018. Available from: https://www.who.int/news-room/detail/02-05-2018-9-out-of-10-people-worldwide-breathe-polluted-air-but-more-countries-are-taking-action
Brook RD, Franklin B, Cascio W, Hong Y, Howard G, Lipsett M, et al. Air pollution and cardiovascular disease: a statement for healthcare professionals from the expert panel on population and prevention science of the American Heart Association. Circulation. 2004;109(21):2655–71.
Goswami A, Barman J, Rajput K, Lakhlani HN. Behaviour study of particulate matter and chemical composition with different combustion strategies. SAE Technical Paper Series. 2013;
Franklin BA, Brook R, Arden PC. Air pollution and cardiovascular disease. Curr Probl Cardiol. 2015;40(5):207–38.
Tanwar V, Gorr MW, Velten M, Eichenseer CM, Long VP, Bonilla IM, et al. In utero particulate matter exposure produces heart failure, electrical remodeling, and epigenetic changes at adulthood. J Am Heart Assoc. 2017;6(4):e005796.
Rich DQ, Schwartz J, Mittleman MA, Link M, Luttmann-Gibson H, Catalano PJ, et al. Association of short-term ambient air pollution concentrations and ventricular arrhythmias. Am J Epidemiol. 2005;161(12):1123–32.
Cole-Hunter T, de Nazelle A, Donaire-Gonzalez D, Kubesch N, Carrasco-Turigas G, Matt F, et al. Estimated effects of air pollution and space-time-activity on cardiopulmonary outcomes in healthy adults: a repeated measures study. Environ Int. 2018;111:247–59.
Miller JG, Gillette JS, Manczak EM, Kircanski K, Gotlib IH. Fine particle air pollution and physiological reactivity to social stress in adolescence: the moderating role of anxiety and depression. Psychosom Med. 2019;81(7):641–8.
Billman GE. The LF/HF ratio does not accurately measure cardiac sympatho-vagal balance. Front Physiol. 2013;4:4.
Sovari AA. Cellular and molecular mechanisms of arrhythmia by oxidative stress. Cardiol Res Pract. 2016;2016:1–7.
Xie X, Wang Y, Yang Y, Xu J, Zhang Y, Tang W, et al. Long-term exposure to fine particulate matter and tachycardia and heart rate: results from 10 million reproductive-age adults in China. Environ Pollut. 2018;242:1371–8.
Wellenius GA. Cardiac effects of carbon monoxide and ambient particles in a rat model of myocardial infarction. Toxicol Sci. 2004;80(2):367–76.
Wold LE, Ying Z, Hutchinson KR, Velten M, Gorr MW, Velten C, et al. Cardiovascular remodeling in response to long-term exposure to fine particulate matter air pollution. Circulation: Heart Fail. 2012;5(4):452–61.
Gurgueira SA, Lawrence J, Coull B, Murthy GG, González-Flecha B. Rapid increases in the steady-state concentration of reactive oxygen species in the lungs and heart after particulate air pollution inhalation. Environ Health Perspect. 2002;110(8):749–55.
Carll AP, Crespo SM, Filho MS, Zati DH, Coull BA, Diaz EA, et al. Inhaled ambient-level traffic-derived particulates decrease cardiac vagal influence and baroreflexes and increase arrhythmia in a rat model of metabolic syndrome. Part Fibre Toxicol. 2017;14(1):16.
Brook RD, Rajagopalan S, Pope CA, Brook JR, Bhatnagar A, Diez-Roux AV, et al. Particulate matter air pollution and cardiovascular disease. Circulation. 2010;121(21):2331–78.
Ying Z, Kampfrath T, Thurston G, Farrar B, Lippmann M, Wang A, et al. Ambient particulates alter vascular function through induction of reactive oxygen and nitrogen species. Toxicol Sci. 2009;111(1):80–8.
Seaton A, Godden D, MacNee W, Donaldson K. Particulate air pollution and acute health effects. Lancet. 1995;345(8943):176–8.
Fragale A, Romagnoli G, Licursi V, Buoncervello M, Del Vecchio G, Giuliani C, et al. Antitumor effects of epidrug/IFNΑ combination driven by modulated gene signatures in both colorectal cancer and dendritic cells. Cancer Immunol Res. 2017;5(7):604–16.
Nebbioso A, Carafa V, Benedetti R, Altucci L. Trials with ‘epigenetic’ drugs: an update. Mol Oncol. 2012;6(6):657–82.
Miranda Furtado CL, Dos Santos Luciano MC, Silva Santos RD, Furtado GP, Moraes MO, Pessoa C. Epidrugs: targeting epigenetic marks in cancer treatment. Epigenetics. 2019;14(12):1164–76.
Chen T. Mechanistic and functional links between histone methylation and DNA methylation. Progr Mol Biol Translat Sci. 2011;101:335–48.
Duygu B, Poels EM, Costa Martins PA. Genetics and epigenetics of arrhythmia and heart failure. Front Genetics. 2013;4:219.
Ng H-H, Adrian B. DNA methylation and chromatin modification. Curr Op Gen Develop. 1999;9(2):158–63.
Moore LD, Le T, Fan G. DNA methylation and its basic function. Neuropsychopharmacology. 2012;38(1):23–38.
Caiafa P, Zampieri M. DNA methylation and chromatin structure: the puzzling CPG Islands. J Cell Biochem. 2005;94(2):257–65.
Eisenberg E, Levanon EY. Human housekeeping genes, revisited. Trends Genet. 2013;29(10):569–74.
Foy J-P, Pickering CR, Papadimitrakopoulou VA, Jelinek J, Lin SH, William WN, et al. New DNA methylation markers and global DNA hypomethylation are associated with oral cancer development. Cancer Prevent Res. 2015;8(11):1027–35.
Nakao M. Epigenetics: interaction of DNA methylation and chromatin. Gene. 2001;278(1–2):25–31.
Phillips T. The role of methylation in gene expression [Internet]. Nature news. Nature Publishing Group; [cited 2021Sep23]. Available from: https://www.nature.com/scitable/topicpage/the-role-of-methylation-in-gene-expression-1070/
Jin B, Robertson KD. DNA methyltransferases, DNA damage repair, and cancer. Exp Med Biol. 2013;754:3–29.
Carey N, Marques CJ, Reik W. DNA demethylases: a new epigenetic frontier in drug discovery. Drug Disc Today. 2011;16(15–16):683–90.
Alberts B, Johnson A, Lewis J, Morgan D, Raff MC, Roberts K, et al. Chromosomal DNA and its packaging in the chromatin fiber. In: Molecular biology of the cell. 4th ed. New York, NY: Garland Science; 2002.
Narlikar GJ, Sundaramoorthy R, Owen-Hughes T. Mechanisms and functions of ATP-dependent chromatin-remodeling enzymes. Cell. 2013;154(3):490–503.
McGinty RK, Tan S. Nucleosome structure and function. Chem Rev. 2014;115(6):2255–73.
Dong J, Gao Z, Liu S, Li G, Yang Z, Huang H, et al. Slide, the protein interacting domain of imitation switch remodelers, binds DDT-domain proteins of different subfamilies in chromatin remodeling complexes. J Integ Plant Biol. 2013;55(10):928–37.
Aydin ÖZ, Vermeulen W, Lans H. ISWI chromatin remodeling complexes in the DNA damage response. Cell Cycle. 2014;13(19):3016–25.
Luijsterburg MS, van Attikum H. Chromatin and the DNA damage response: the cancer connection. Mol Oncol. 2011;5(4):349–67.
Zhang P, Torres K, Liu X, Liu C-gong E, Pollock R. An overview of chromatin-regulating proteins in cells. Curr Prot Pept Sci. 2016;17(5):401–10.
Wang Y, Miao X, Liu Y, Li F, Liu Q, Sun J, et al. Dysregulation of histone acetyltransferases and deacetylases in cardiovascular diseases. Oxidative Med Cell Longev. 2014;2014:1.
Fry CJ, Peterson CL. Chromatin remodeling enzymes: Who's on first? Curr Bio. 2001;11(5):R185–97.
Eberharter A, Becker PB. Histone acetylation: a switch between repressive and permissive chromatin. EMBO Rep. 2002;3(3):224–9.
Greer EL, Shi Y. Histone methylation: a dynamic mark in health, disease and inheritance. Nature Rev Genet. 2012;13(5):343–57.
Santos-Rosa H, Schneider R, Bannister AJ, Sherriff J, Bernstein BE, Emre NC, et al. Active genes are tri-methylated at K4 of histone H3. Nature. 2002;419(6905):407–11.
Kaneda R, Takada S, Yamashita Y, Choi YL, Nonaka-Sarukawa M, Soda M, et al. Genome-wide histone methylation profile for heart failure. Genes Cells. 2009;14(1):69–77.
Diamantopoulos MA, Tsiakanikas P, Scorilas A. Non-coding RNAs: the riddle of the transcriptome and their perspectives in cancer. Ann Transl Med. 2018;6(12):241.
Hombach S, Kretz M. Non-coding RNAs: classification, biology and functioning. Adv Exp Med Biol. 2016;937:3–17.
Peng Y, Croce CM. The role of microRNAs in human cancer. Signal Transduct Target Ther. 2016;1:15004.
Calin GA, Dumitru CD, Shimizu M, Bichi R, Zupo S, Noch E, et al. Frequent deletions and down-regulation of micro- RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc Natl Acad Sci U S A. 2002;99(24):15524–9.
Klein U, Lia M, Crespo M, Siegel R, Shen Q, Mo T, et al. The DLEU2/miR-15a/16-1 cluster controls B cell proliferation and its deletion leads to chronic lymphocytic leukemia. Cancer Cell. 2010;17(1):28–40.
Lee Y, Ahn C, Han J, Choi H, Kim J, Yim J, et al. The nuclear RNase III Drosha initiates microrna processing. Nature. 2003;425(6956):415–9.
Meltzer PS. Small RNAs with big impacts. Nature. 2005;435(7043):745–6.
Bianchi M, Renzini A, Adamo S, Moresi V. Coordinated actions of microRNAs with other epigenetic factors regulate skeletal muscle development and adaptation. Internat J Mol Sc. 2017;18(4):840.
Ferguson LR. RNA silencing: mechanism, biology and responses to environmental stress. Mutation Res. 2011;714(1–2):93–4.
Zhi H, Ning S, Li X, Li Y, Wu W, Li X. A novel reannotation strategy for dissecting DNA methylation patterns of human long intergenic non-coding RNAs in cancers. Nucleic Acids Res. 2014;42(13):8258–70.
Fox CS, Parise H, D’Agostino RB, Lloyd-Jones DM, Vasan RS, Wang TJ, et al. Parental atrial fibrillation as a risk factor for atrial fibrillation in offspring. J Am Med Assoc. 2004;291(23):2851–5.
Ellinor PT, Shin JT, Moore RK, Yoerger DM, MacRae CA. Locus for atrial fibrillation maps to chromosome 6q14–16. Circulation. 2003;107(23):2880–3.
Wakili R, Voigt N, Kääb S, Dobrev D, Nattel S. Recent advances in the molecular pathophysiology of atrial fibrillation. J Clin Invest. 2011;121(8):2955–68.
Nattel S, Harada M. Atrial remodeling and atrial fibrillation. J Am Coll Cardiol. 2014;63(22):2335–45.
Tao H, Yang J-J, Shi K-H, Deng Z-Y, Li J. DNA methylation in cardiac fibrosis: new advances and perspectives. Toxicology. 2014;323:125–9.
Ma B, Wilker EH, Willis-Owen SA, Byun H-M, Wong KC, Motta V, et al. Predicting DNA methylation level across human tissues. Nucleic Acids Res. 2014;42(6):3515–28.
Kao Y-H, Chen Y-C, Chung C-C, Lien G-S, Chen S-A, Kuo C-C, et al. Heart failure and angiotensin II modulate atrial Pitx2c promotor methylation. Clin Exp Pharmacol Physiol. 2013;40(6):379–84.
Zhong H, Kim S, Zhi D, Cui X. Predicting gene expression using DNA methylation in three human populations. PeerJ. 2019;7:e6757.
Kao Y-H, Chen Y-C, Cheng C-C, Lee T-I, Chen Y-J, Chen S-A. Tumor necrosis factor-α decreases sarcoplasmic reticulum Ca2+-ATPase expressions via the promoter methylation in cardiomyocytes. Crit Care Med. 2010;38(1):217–22.
Frank KF, Bölck B, Brixius K, Kranias EG, Schwinger RH. Modulation of SERCA: implications for the failing human heart. Basic Res Cardiol. 2002;97(Suppl 1):I72-8.
Tao H, Yang J-J, Chen Z-W, Xu S-S, Zhou X, Zhan H-Y, et al. DNMT3A silencing RASSF1A promotes cardiac fibrosis through upregulation of ERK1/2. Toxicology. 2014;323:42–50.
Kirchhof P, Kahr PC, Kaese S, Piccini I, Vokshi I, Scheld H-H, et al. PITX2C is expressed in the adult left atrium, and reducing Pitx2c expression promotes atrial fibrillation inducibility and complex changes in gene expression. Circulation: Cardiovasc Genet. 2011;4(2):123–33.
Fatima N, Schooley JF, Claycomb WC, Flagg TP. Promoter DNA methylation regulates murine SUR1 (ABCC8) and SUR2 (ABCC9) expression in HL-1 cardiomyocytes. PLoS One. 2012;7:e41533.
Monteforte N, Napolitano C, Priori SG. Genetics and arrhythmias: diagnostic and prognostic applications. Revist Españ Cardiol (English Edition). 2012;65(3):278–86.
Montgomery RL, Davis CA, Potthoff MJ, Haberland M, Fielitz J, Qi X, et al. Histone deacetylases 1 and 2 redundantly regulate cardiac morphogenesis, growth, and contractility. Genes Dev. 2007;21(14):1790–802.
Terentyev D, Belevych AE, Terentyeva R, Martin MM, Malana GE, Kuhn DE, et al. miR-1 overexpression enhances Ca2+ release and promotes cardiac arrhythmogenesis by targeting PP2A regulatory subunit B56alpha and causing CaMKII-dependent hyperphosphorylation of RyR2. Circ Res. 2009;104(4):514–21.
Lkhagva B, Chang S-L, Chen Y-C, Kao Y-H, Lin Y-K, Chiu CT-H, et al. Histone deacetylase inhibition reduces pulmonary vein arrhythmogenesis through calcium regulation. Internat. J Cardiol. 2014;177(3):982–9.
Zhang D, Wu C-T, Qi XY, Meijering RAM, Hoogstra-Berends F, Tadevosyan A, et al. Activation of histone deacetylase-6 induces contractile dysfunction through derailment of α-tubulin proteostasis in experimental and human atrial fibrillation. Circulation. 2014;129(3):346–58.
Nural-Guvener HF, Zakharova L, Nimlos J, Popovic S, Mastroeni D, Gaballa MA. HDAC class I inhibitor, Mocetinostat, reverses cardiac fibrosis in heart failure and diminishes CD90+ cardiac myofibroblast activation. Fibrogenesis Tissue Repair. 2014;7:10.
Kee HJ, Bae EH, Park S, Lee KE, Suh SH, Kim SW, et al. HDAC inhibition suppresses cardiac hypertrophy and fibrosis in DOCA-salt hypertensive rats via regulation of HDAC6/HDAC8 enzyme activity. Kidney Blood Press Res. 2013;37(4–5):229–39.
Kook H, Lepore JJ, Gitler AD, Lu MM, Wing-Man Yung W, Mackay J, et al. Cardiac hypertrophy and histone deacetylase–dependent transcriptional repression mediated by the atypical homeodomain protein Hop. J Clin Invest. 2003;112(6):863–71.
Iyer A, Chan V, Brown L. The DOCA-salt hypertensive rat as a model of cardiovascular oxidative and inflammatory stress. Curr Cardiol Rev. 2010;6(4):291–7.
van den Berg NW, Kawasaki M, Berger WR, Neefs J, Meulendijks E, Tijsen AJ, et al. MicroRNAs in atrial fibrillation: from expression signatures to functional implications. Cardiovasc Drugs Ther. 2017;31(3):345–65.
Jayawardena TM, Finch EA, Zhang L, Zhang H, Hodgkinson CP, Pratt RE, et al. MicroRNA induced cardiac reprogramming in vivo evidence for mature cardiac myocytes and improved cardiac function. Circ Res. 2015;116(3):418–24.
Nattel S. Molecular and cellular mechanisms of atrial fibrosis in atrial fibrillation. JACC: Clin Electrophysiol. 2017;3(5):425–35.
Luo X, Pan Z, Shan H, Xiao J, Sun X, Wang N, et al. MicroRNA-26 governs profibrillatory inward-rectifier potassium current changes in atrial fibrillation. J Clin Invest. 2013;123(5):1939–51.
Qi X-Y, Huang H, Ordog B, Luo X, Naud P, Sun Y, et al. Fibroblast inward-rectifier potassium current upregulation in profibrillatory atrial remodeling. Circ Res. 2015;116(5):836–45.
Duisters RF, Tijsen AJ, Schroen B, Leenders JJ, Lentink V, van der Made I, et al. miR-133 and miR-30 regulate connective tissue growth factor: implications for a role of microRNAs in myocardial matrix remodeling. Circ Res. 2009;104(2):170–8.
Zhang X, Jing W. Upregulation of miR-122 is associated with cardiomyocyte apoptosis in atrial fibrillation. Mol Med Rep. 2018;18(2):1745–51.
Baccarelli A, Wright RO, Bollati V, Tarantini L, Litonjua AA, Suh HH, et al. Rapid DNA methylation changes after exposure to traffic particles. Am J Resp Crit Care Med. 2009;179(7):572–8.
Madrigano J, Baccarelli A, Mittleman MA, Wright RO, Sparrow D, Vokonas PS, et al. Prolonged exposure to particulate pollution, genes associated with glutathione pathways, and DNA methylation in a cohort of older men. Environ Health Perspect. 2011;119(7):977–82.
Bind M-A, Baccarelli A, Zanobetti A, Tarantini L, Suh H, Vokonas P, et al. Air pollution and markers of coagulation, inflammation, and endothelial function. Epidemiology. 2012;23(2):332–40.
Rusiecki JA, Baccarelli A, Bollati V, Tarantini L, Moore LE, Bonefeld-Jorgensen EC. Global DNA hypomethylation is associated with high serum-persistent organic pollutants in Greenlandic inuit. Environ Health Perspect. 2008;116(11):1547–52.
Salam MT, Byun H-M, Lurmann F, Breton CV, Wang X, Eckel SP, et al. Genetic and epigenetic variations in inducible nitric oxide synthase promoter, particulate pollution, and exhaled nitric oxide levels in children. J Aller Clin Immunol. 2012;129(1):232.
Tarantini L, Bonzini M, Apostoli P, Pegoraro V, Bollati V, Marinelli B, et al. Effects of particulate matter on genomic DNA methylation content and inos promoter methylation. Environ Health Perspect. 2009;117(2):217–22.
Breton CV, Salam MT, Wang X, Byun H-M, Siegmund KD, Gilliland FD. Particulate matter, DNA methylation in nitric oxide synthase, and childhood respiratory disease. Environ Health Perspect. 2012;120(9):1320–6.
Nadeau K, McDonald-Hyman C, Noth EM, Pratt B, Hammond SK, Balmes J, et al. Ambient air pollution impairs regulatory T-cell function in asthma. J Aller Clin Immunol. 2010;126(4):845.
Bellavia A, Urch B, Speck M, Brook RD, Scott JA, Albetti B, et al. DNA hypomethylation, ambient particulate matter, and increased blood pressure: findings from controlled human exposure experiments. J Am Heart Assoc. 2013;2(3):e000212.
Carmona JJ, Sofer T, Hutchinson J, Cantone L, Coull B, Maity A, et al. Short-term airborne particulate matter exposure alters the epigenetic landscape of human genes associated with the mitogen-activated protein kinase network: a cross-sectional study. Environ Health. 2014;13:1.
Barker DJP. The fetal origins of coronary heart disease. Eur Heart J. 1997;18(6):883–4.
Tanwar V, Adelstein JM, Grimmer JA, Youtz DJ, Katapadi A, Sugar BP, et al. Preconception exposure to fine particulate matter leads to cardiac dysfunction in adult male offspring. J Am Heart Assoc. 2018;7(24):e010797.
Yauk C, Polyzos A, Rowan-Carroll A, Somers CM, Godschalk RW, Van Schooten FJ, et al. Germ-line mutations, DNA damage, and global hypermethylation in mice exposed to particulate air pollution in an urban/industrial location. Proc Natl Acad Sci U S A. 2008;105(2):605–10.
Reliene R, Hlavacova A, Mahadevan B, Baird WM, Schiestl RH. Diesel exhaust particles cause increased levels of DNA deletions after transplacental exposure in mice. Mutat Res. 2005;570(2):245–52.
George A, Figueredo VM. Alcohol and arrhythmias: a comprehensive review. J Cardiovasc Med. 2010;11(4):221–8.
Dominici F, Peng RD, Bell ML, Pham L, McDermott A, Zeger SL, et al. Fine particulate air pollution and hospital admission for cardiovascular and respiratory diseases. J Am Med Ass. 2006;295(10):1127.
Benowitz NL, Gourlay SG. Cardiovascular toxicity of nicotine: implications for nicotine replacement therapy. J Am Coll Cardiol. 1997;29(7):1422–31.
Kerr DMI, Brooksbank KJM, Taylor RG, Pinel K, Rios FJ, Touyz RM, et al. Acute effects of electronic and tobacco cigarettes on vascular and respiratory function in healthy volunteers: a cross-over study. J Hyperten. 2019;37(1):154–66.
Akbarzadeh MA, Yazdani S, Ghaidari ME, Asadpour-Piranfar M, Bahrololoumi-Bafruee N, Golabchi A, et al. Acute effects of smoking on QT dispersion in healthy males. ARYA Atherosclerosis. 2014;10:89–93.
Alshehri AM, Azoz AM, Shaheen HA, Farrag YA, Khalifa MA, Youssef A. Acute effects of cigarette smoking on the cardiac diastolic functions. J Saudi Heart Assoc. 2013;25(3):173–9.
Chamberlain AM, Agarwal SK, Folsom AR, Duval S, Soliman EZ, Ambrose M, et al. Smoking and incidence of atrial fibrillation: results from the atherosclerosis risk in communities (ARIC) study. Heart Rhythm. 2011;8(8):1160–6.
Heeringa J, Kors JA, Hofman A, van Rooij FJA, Witteman JCM. Cigarette smoking and risk of atrial fibrillation: the Rotterdam Study. Am Heart J. 2008;156(6):1163–9.
Stewart S. Population prevalence, incidence, and predictors of atrial fibrillation in the Renfrew/Paisley study. Heart. 2001;86(5):516–21.
Zuo H, Nygård O, Vollset SE, Ueland PM, Ulvik A, Midttun Ø, et al. Smoking, plasma cotinine and risk of atrial fibrillation: the Hordaland Health Study. J Int Med. 2017;283(1):73–82.
Schnabel RB, Sullivan LM, Levy D, Pencina MJ, Massaro JM, D'Agostino RB, et al. Development of a risk score for atrial fibrillation (Framingham Heart Study): a community-based cohort study. Lancet. 2009;373(9665):739–45.
Krahn AD, Manfreda J, Tate RB, Mathewson FAL, Cuddy TE. The natural history of atrial fibrillation: incidence, risk factors, and prognosis in the Manitoba follow-up study. Am J Med. 1995;98(5):476–84.
Gupta P, Patel C, Patel H, Narayanaswamy S, Malhotra B, Green JT, et al. TP-E/Qt ratio as an index of arrhythmogenesis. J Electrocardiol. 2008;41(6):567–74.
İlgenli T, Tokatlı A, Akpınar O, Kılıçaslan F. The effects of cigarette smoking on the TP-e interval, TP-E/qt ratio and TP-E/QTC ratio. Adv Clin Exp Med. 2015;24(6):973–8.
Taşolar H, Ballı M, Bayramoğlu A, Otlu YÖ, Çetin M, Altun B, et al. Effect of smoking on TP-e interval, TP-e/QT and TP-e/qtc ratios as indices of ventricular arrhythmogenesis. Heart Lung Circ. 2014;23(9):827–32.
Felber Dietrich D, Schwartz J, Schindler C, Gaspoz J-M, Barthelemy J-C, Tschopp J-M, et al. Effects of passive smoking on heart rate variability, heart rate and blood pressure: an observational study. International J Epidemiol. 2007;36(4):834–40.
Pope CA, Eatough DJ, Gold DR, Pang Y, Nielsen KR, Nath P, et al. Acute exposure to environmental tobacco smoke and heart rate variability. Environ Health Perspect. 2001;109(7):711–6.
Goette A, Lendeckel U, Kuchenbecker A, Bukowska A, Peters B, Klein HU, et al. Cigarette smoking induces atrial fibrosis in humans via nicotine. Heart. 2007;93(9):1056–63.
Burstein B, Nattel S. Atrial fibrosis: mechanisms and clinical relevance in atrial fibrillation. J Am Coll Cardiol. 2008;51(8):802–9.
Shan H, Zhang Y, Lu Y, Zhang Y, Pan Z, Cai B, et al. Downregulation of miR-133 and miR-590 contributes to nicotine-induced atrial remodelling in canines. Cardiovasc Res. 2009;83(3):465–72.
Wang H, Shi H, Zhang L, Pourrier M, Yang B, Nattel S, et al. Nicotine is a potent blocker of the cardiac A-type K+ channels. Circulation. 2000;102(10):1165–71.
Ton AT, Biet M, Delabre J-F, Morin N, Dumaine R. In-utero exposure to nicotine alters the development of the rabbit cardiac conduction system and provides a potential mechanism for sudden infant death syndrome. Arch Toxicol. 2017;91(12):3947–60.
Biet M, Ton AT, Delabre J-F, Morin N, Dumaine R. In utero exposure to nicotine abolishes the postnatal response of the cardiac sodium current to isoproterenol in newborn rabbit atrium. Heart Rhythm. 2019;16(4):494–501.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2023 Springer Nature Switzerland AG
About this chapter

Cite this chapter
O’Piela, D.R., Grimmer, J.A., Schwieterman, N.A., Mears, M.J., Gorr, M.W., Wold, L.E. (2023). Epigenetic Influences of Air Pollution-Induced Cardiac Arrhythmias. In: Tripathi, O.N., Quinn, T.A., Ravens, U. (eds) Heart Rate and Rhythm. Springer, Cham. https://doi.org/10.1007/978-3-031-33588-4_25
Download citation
DOI: https://doi.org/10.1007/978-3-031-33588-4_25
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-031-33587-7
Online ISBN: 978-3-031-33588-4
eBook Packages: MedicineMedicine (R0)