
Abstract
As a key component of the DNA Damage Response, the Ataxia telangiectasia and Rad3-related (ATR) protein is a promising druggable target that is currently widely evaluated in phase I-II-III clinical trials as monotherapy and in combinations with other rational antitumor agents, including immunotherapy, DNA repair inhibitors, chemo- and radiotherapy. Ongoing clinical studies for this drug class must address the optimization of the therapeutic window to limit overlapping toxicities and refine the target population that will most likely benefit from ATR inhibition. With advances in the development of personalized treatment strategies for patients with advanced solid tumors, many ongoing ATR inhibitor trials have been recruiting patients based on their germline and somatic molecular alterations, rather than relying solely on specific tumor subtypes. Although a spectrum of molecular alterations have already been identified as potential predictive biomarkers of response that may sensitize to ATR inhibition, these biomarkers must be analytically validated and feasible to measure robustly to allow for successful integration into the clinic. While several ATR inhibitors in development are poised to address a clinically unmet need, no ATR inhibitor has yet received FDA-approval. This chapter details the underlying rationale for targeting ATR and summarizes the current preclinical and clinical landscape of ATR inhibitors currently in evaluation, as their regulatory approval potentially lies close in sight.
Carolina Salguero and Christian Valladolid contributed equally.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- DNA damage response (DDR)
- Ataxia telangiectasia and Rad3-related protein (ATR)
- ATR inhibitors
- PARP inhibitors
- Ataxia telangiectasia-mutated (ATM) deficiency
- AT-rich interactive domain-containing protein 1A (ARID1A) deficiency
14.1 Introduction
Preserving genomic integrity is pivotal for cell survival; consequently, cells rely on a network of complex signaling pathways to facilitate faithful DNA replication and maintain genomic stability [1]. Increased proliferation rates are associated with genomic instability via the accumulation of DNA damage in the form of DNA double strand breaks (DSBs) or DNA single strand breaks (SSBs) caused by a variety of events, including replication stress induced by stalling of replication forks [2,3,4]. These events distort genetic material due to subsequent fusion of DSBs and shortening of telomeres, which can result in translocations, gene amplification, and gene mutations [5,6,7].
When DNA damage or replication stress is sensed, cells are prevented entry into mitosis by activating DNA Damage Response (DDR) pathways at varying phases within the cell cycle [8]. DDR signaling pathways orchestrate tightly regulated kinase cascades to resolve DNA damage and replication stress by pausing the cell cycle and initiating repair [9, 10]. Ataxia telangiectasia and Rad3-related protein (ATR) is a key PI3K-related kinase within the DDR that senses replication stress and regulates checkpoints within the cell cycle’s synthesis (S) and gap 2/mitosis (G2/M) phases to preserve genomic stability [1, 10,11,12].
Notably, replication stress and genomic instability are hallmarks of cancer cells, making them dependent on protective DDR pathways for survival [10, 12,13,14]. As such, targeting ATR in cancer medicine is an attractive therapeutic approach to circumvent cancer cell survival by exploiting their dependence on ATR-driven processes.
14.2 ATR Acts as a Gatekeeper of DNA Damage Repair
Endogenous and exogenous sources of DNA damage lead to a wide variety of adducts including DSBs, SSBs, base damage, bulky adducts and base mismatches [1]. Cells have evolved complex DNA repair mechanisms designed to specifically repair these types of damage and maintain genome stability. Repair of DNA DSBs is of particular importance as it is estimated that a single unrepaired DNA DSB can initiate cell death, highlighting the critical role of DDR pathways in cell survival [16]. DSBs are repaired by a number of mechanisms: the best characterized being the error-free homologous recombination repair (HRR) pathway, the highly efficient -but error prone- nonhomologous end joining (NHEJ) pathway, and the error prone microhomology mediated end-joining pathway (regarded as a backup pathway to HRR) [15].
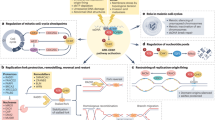
Repair by HRR is initiated by an upstream activator of DDR, the ataxia telangiectasia-mutated (ATM) PI3K-related kinase, which upon sensing DSBs triggers a cascade of events that include cell cycle arrest, repair and apoptosis [17, 18]. During S or G2-phases, exposure of single-stranded DNA can occur as an intermediate of HRR at areas of resected DNA and also at stressed replication forks. These single-stranded DNA regions quickly become coated with the high-affinity ssDNA binding protein, replication protein A (RPA), which protects against DNA degradation. The coating of single-stranded DNA by RPA recruits ATR/ATR-interacting protein (ATRIP) complexes to sites of damage [1, 16]. Following localization to sites of damage, ATR is activated by either topoisomerase II binding protein (TopBP1) or Ewing tumour-associated antigen 1 (ETAA1) [17]. Moreover, recruitment of TopBP1 is mediated by Rad17, which loads the 9-1-1 (Rad9-Hus1-Rad1) complex onto chromatin, binds to TopBP1, and results in ATR activation [4]. Once activated, ATR proceeds to phosphorylate a series of downstream targets; however, its activation of checkpoint kinase 1 (CHK1) is integral to its regulation of cell cycle checkpoints (Fig. 14.1) [16].

ATR activation, inhibition and modulation of antitumor immunity. ATR is activated in response to replication stress, single-stranded DNA, and increased R-loops. ATR activation triggers a kinase cascade of CHK1 and WEE1, resulting in checkpoint activation and cell cycle arrest for DNA repair. ATR inhibition induces inappropriate mitotic entry that culminates in mitotic catastrophe and the release of DNA fragments into the cytosol, which in turn activate the cGAS-STING pathway and a type I IFN response. DDR may also be a means of increasing tumor mutational burden and therefore the generation of neoantigens
ATR commands control over the S and G2/M checkpoints by phosphorylating and activating CHK1 [1, 18]. Active CHK1 kinase in turn phosphorylates and inactivates the cell division cycle 25A/25C (CDC25A/CDC25C) phosphatase proteins, leading to their respective degradation [1]. Degradation of CDC25A thereby renders CDK2 and its associated complexes inactive by removing an inhibitory phosphorylation present on CDK2 [19]. Consequently, progression to S phase is interrupted, preventing DNA replication and promoting DNA repair [16]. ATR-mediated activation of CHK1 also interrupts the G2/M checkpoint in a Wee1-like protein kinase (WEE1) dependent manner. Active CHK1 phosphorylates and stabilizes WEE1, enhancing its activity toward CDK1 [19]. Both CDK1 and CDK2 remain in inactive states induced by WEE1’s inhibitory phosphorylation [19]. Inactivation of CDC25C by CHK1 prevents the removal of the inhibitory phosphorylation on CDK1, which halts the G2/M checkpoint to allow time for post-replicative DNA repair and prevent replication of unrepaired DNA [16].
Additionally, ATR also plays a role in regulating replication forks through multiple mechanisms [1]. One mechanism involves ATR-mediated fork remodeling: ATR phosphorylates the helicase SWI/SNF-related, matrix associated, actin dependent regulator of chromatin, subfamily-A-like 1 (SMARCAL1), promoting maintenance of fork stability and fork restart in cooperation with RAD51 and zinc-finger RANBP2-type containing 3 (ZRANB3) [20]. Another known mechanism underscores the importance of the ATR-CHK1 axis in resolving replication stress during the formation of R-loops, which are RNA–DNA hybrid transcription intermediates that induce genome instability. Here, the ATR-CHK1 pathway is activated by R-loop induced reversed replication forks. Upon activation, ATR protects the genome by regulating the activity of the MUS81 endonuclease, preventing excess nucleolytic degradation of reversed forks. Active ATR also suppresses R-loop accumulation and enables replication recovery, while promoting arrest of the cell cycle at the G2/M-phase [21]. Resolution of replication stress triggers ATR to resume HRR activities by promoting fork reversal and restart, a process involving the recruitment of BRCA2 and RAD51 to sites of damage [11, 22].
In addition to its role in HRR, ATR has further roles in DNA repair through its involvement in the inter-strand crosslink repair (ICLR) and nucleotide excision repair (NER) pathways. ICLR removes toxic inter-strand DNA crosslinks (lesions involving both strands of DNA that can result in replication fork-stalling and inhibition of transcription). The presence of ICLs activates ATR and requires ATR-mediated phosphorylation of Fanconi Anemia proteins, which are key players in mediating ICLR [23, 24]. Lastly, NER is a critical mechanism that repairs a wide variety of DNA lesions caused by chemical agents or environmental factors (particularly UV radiation) [25]. In this repair mechanism, ATR phosphorylates and stabilizes Xeroderma pigmentosa group A (XPA), thereby recruiting the protein to sites of damage during the S-phase of the cell-cycle and aiding in the activation of the NER pathway [26]. As a key player in multiple processes, ATR is a master regulator of DNA repair and more broadly in the replication stress response.
14.3 ATR Signaling Fosters Cancer Cell Survival
Activation of DDR in normal cells can either resolve DNA damage and/or replication stress to promote cell survival, or it can trigger programmed cell death when DNA damage cannot be removed, inhibiting tumorigenesis and preventing inheritance of DNA mutations in daughter cells [28,29,30,31]. Since DDR is often dampened in cancer cells, these cells present increased DNA damage that is tolerated due to a simultaneous amelioration of unrepaired DNA damage response. In this way, DDR is used by cancer cells as a decoy mechanism to shield against cell death and allow genomically unstable cells to traverse the cell-cycle unscathed, making cancer cells dependent on DDR for survival. That is, tolerance of replication stress is crucial for tumor viability, and oncogene-induced dysregulation of DNA replication generates high levels of replication stress in cancer cells [27,28,29]. Furthermore, most cancers also feature defective G1 checkpoints, largely due to p53 signaling loss [30], rendering cancel cells dependent on S- and G2-phase checkpoints, which are ATR-regulated processes [30].
Frequent mutations in DDR genes also increase dependency on ATR signaling [28, 31]. In these situations, cancer cells rely on ATR to respond to and resolve replication stress and repair DNA damage in order to bypass cell death [27]. Given that ATR function is conserved in the vast majority of cancers, it is has emerged as a favorable target in cancer medicine [32]. ATR inhibition in normal cycling cells, with intact cell-cycle checkpoints, leads to moderate cytotoxicity due to replication fork stalling and collapse; however, as cancer cells have high replication stress, they are more dependent on ATR for survival and as a result are more sensitive to its inhibition than normal cells [27, 28]. Furthermore, in-vitro data suggests that chronic use of ATR inhibitors impairs the cell’s ability to repair damage by HRR while also impacting the availability of necessary HRR proteins, such as TopBP1, BRCA1, and RAD51 [28]. This targeted approach has culminated in the development of many clinical studies aimed at evaluating the clinical efficacy of ATR inhibition as both a monotherapy in certain DDR defective cancer backgrounds and in combinational approaches [19, 33].
14.4 Early Development of ATR Inhibitors
The development of potent and selective ATR kinase inhibitors has been closely related to (1) the availability of well-characterized assays that permit accurate measurements of selective kinase activity, (2) the availability of structural and functional insights to guide a drug design strategy that maximizes selectivity, and (3) the development of screening tools and biomarkers that can identify suitable patients. Notably, the first generation of small molecule ATR inhibitors struggled to find balance between potency and selectivity to reach clinical usage. For instance, while Schisandrin B, an active ingredient of the magnolia berry (Schisandra chinensis), inhibited ATR kinase activity at high concentrations leading to off-target effects and toxicity, the small molecule NU6087 demonstrated moderate selectivity over ATM homologs, but did not display selectivity over the wider kinase family [34]. With the development of a cell-screening assay that measured ATP-dependent phosphorylation of H2AX as a more accurate quantification of ATR kinase activity in experimental conditions, the small molecule ETP-46464 was selected from a library of compounds based on its increased selectivity over other ATR homologs. Although poor pharmacological properties in mice prevented ETP-46464 from advancing to clinical studies, the discovery of this compound provided proof of concept for a more reliable biomarker of replication stress that accounts for double stand breaks and has become the standard marker for quantifying DNA damage [35]. Based on recent advances in the development of well-characterized assays, as well as new insights in the structure–function relationship of ATR, many pharmaceutical companies have taken on the challenge to design and develop potent and selective ATP competitive inhibitors of ATR with the most advanced targeted therapies described here.
14.5 ATR Inhibitors in the Clinic
14.5.1 Berzosertib (M6620/VX-970/VE-822)
Shortly after the development of the assay that measured ATR-dependent phosphorylation of γH2AX, a high throughput screen that combined structure–activity relationship with homology modeling led to the discovery of VE-821, a selective inhibitor with 600-fold selectivity for ATR over ATM, DNA-PK, mTOR, and PI3K [36]. In addition to increased selectivity, VE-821 also showed strong inhibition of CHK1 phosphorylation in cellular models of ATM and/or p53 deficiency [37]. In-vitro experiments showed that VE-821 sensitized ovarian cancer cells to DNA damaging agents such as cisplatin and gemcitabine, and the effects of gemcitabine were potentiated when combined with VE-821 in pancreatic cancer cells [38]. It was also observed that VE-821 could further sensitize BRCA1-depleted cells to DNA damaging agents [44]. Such synergistic effects appear to be stronger with DNA-damaging agents, such as cisplatin and carboplatin, since DNA crosslinking triggers early activation of ATR and the DDR machinery. Interestingly, p53-deficient cancer cell lines were shown to be more sensitive to the combination of VE-821 and cisplatin than normal cell lines, and significant synergistic activity was observed in ATM-deficient cell lines [43]. These results were further confirmed by treating ATM-proficient cells with the triple combination of VE-821, cisplatin, and a highly selective ATM inhibitor (KU-55933) [37]. Taken together, these results suggested that cancer cells with defective ATM signaling are more reliant on ATR; hence, demonstrating a synthetic lethal interaction between the S-phase specific ATR and the ATM-p53 pathway mediating the G1 checkpoint [39]. However, it must be noted that recent studies have shown that ATM mutations and p53 status are not enough to predict clinical benefit to ATR inhibition, and mutations in other DDR genes—such as PTEN, XRCC1, BRCA1, BRCA2, and ARID1A—may promote synthetic lethality with ATR inhibitors [40].
Based on promising pre-clinical data, VE-822, an optimized analog of VE-821 with increased potency and selectivity for ATR, became the first inhibitor to enter clinical trials labeled as VX-970, later named M6620 and berzosertib [41]. Berzosertib alone was found to sensitize multiple lung cancer cell lines to a wide variety of DNA-damaging chemotherapeutic agents (cisplatin, oxaliplatin, gemcitabine, etoposide, and the active metabolite of irinotecan, SN38), and the combination of berzosertib and cisplatin showed sustained tumor regression in non-small cell lung cancer (NSCLC) patient-derived xenograft models [42]. A recent CRISPR-Cas9 screen suggested that the ATR-CHK1 pathway has the potential for synthetic lethality in small cell lung cancer (SCLC) [43]. In that study, the combination of berzosertib with cisplatin displayed greater synergistic activity in different SCLC cell lines and primary lung fibroblasts when compared to treatment with the combination of cisplatin and etoposide. Interestingly, while SCLC cell-derived xenografts showed that the combination of berzosertib with cisplatin inhibited tumor growth, other studies showed that pediatric solid tumor xenografts treated with berzosertib and cisplatin displayed a larger event free survival relative to those treated with cisplatin monotherapy [44, 45]. Together, these studies were the first to confirm the clinical potential of berzosertib as a chemo-sensitizer of DNA damaging agents in lung cancer patients, as well as in pediatric solid tumors, setting the stage for several other studies that also showed the clinical potential of berzosertib in combination with cisplatin in other cancer types, including colon cancer, triple negative breast cancer (TNBC), and esophageal tumors, amongst others [44,45,46,47].
Berzosertib monotherapy has already advanced to a phase II clinical trial investigating antitumor activity in molecularly selected solid tumors, leiomyosarcoma and osteosarcoma (NCT03718091). Although clinical trials are currently studying the combination of berzosertib with radiotherapy, chemo-radiotherapy agents, PARP inhibitors, VEGF inhibitors, as well as with anti-PD-L1 antibodies, such as avelumab, the most common strategy for berzosertib treatment combinations in registered clinical trials appears to be with DNA damaging agents such as cisplatin, carboplatin, gemcitabine, topotecan, irinotecan, and paclitaxel, amongst others (Table 14.1).
The first-in-human trial of berzosertib in combination with cytotoxic chemotherapy agents in patients with advanced solid tumors started with a lead-in safety phase of berzosertib monotherapy, followed by three dose escalation arms aimed to determine the safety profile and recommended phase 2 dose (RP2D) of the combinations of berzosertib with (1) cisplatin, (2) gemcitabine with and without cisplatin, and (3) irinotecan. This trial also included 3 expansion cohorts to further elucidate preliminary anti-tumor activity for the combination of berzosertib and gemcitabine in NSCLC patients harboring p53 mutations and/or loss of ATM expression, the combination of berzosertib and cisplatin in TNBC patients with germline (g) BRCA wild type status, and the combination of berzosertib and cisplatin or carboplatin in patients with platinum-resistant advanced SCLC.
Recent results from two dose escalation arms of this first-in-human trial demonstrated preliminary antitumor activity for berzosertib when combined with gemcitabine and/or cisplatin [48]. That is, most patients who received berzosertib in combination with cisplatin (73.1%), and those who received berzosertib in combination with gemcitabine (68.7%) or berzosertib in combination with gemcitabine and cisplatin (71.0%) achieved disease control with partial response (PR) or stable disease (SD) as their best response per RECIST v1.1 [48]. Interestingly, all patients who received prior platinum-based chemotherapy, and had experienced disease progression, achieved PR when treated with berzosertib in combination with cisplatin. Since ATR inhibition can disrupt DNA replication fork stability and homologous recombination repair (the two major mechanisms of PARP inhibitor resistance), preliminary results from this trial suggest that berzosertib inhibition may contribute to re-sensitizing solid tumors to cisplatin [49].
The RP2D for the combination of berzosertib and cisplatin was determined as 140 mg/m2 of berzosertib (administered on days 2 and 9), and 75 mg/m2 of cisplatin administered every 3 weeks (Q3W) on day 1. This RP2D was generally well tolerated, and the safety profile of this combination was consistent with that of cisplatin alone. Importantly, while the human equivalent dose required for berzosertib target engagement was estimated from preclinical models to be ~60 mg/m2, results from the first-in-human trial show that dosing berzosertib at 140 mg/m2 induces a reduction in serine 345-phosphorylated CHK1, without evidence of PK interactions in a range of malignancies, including ovarian, breast, thyroid, and pancreatic cancers [50]. In a similar manner, the RP2D combination of berzosertib and gemcitabine, which is currently being evaluated in patients with advanced NSCLC in an expansion arm of this trial, was established as 210 mg/m2 of berzosertib (administered on days 2 and 9), and 1000 mg/ m2 of gemcitabine, administered Q3W on days 1 and 8. Yet, the dose escalation for berzosertib in combination with both gemcitabine and cisplatin was terminated after two patients experienced febrile neutropenia or neutropenia as dose limiting toxicities (DLTs). Taken together, results from the first two arms of the first-in-human trial of berzosertib demonstrate that a tolerable safety toxicity profile is observed when berzosertib is combined with either gemcitabine or cisplatin, but not when combined with both agents [48].
The pharmacokinetic (PK) profile of a berzosertib monotherapy lead-in was determined across the dose range of 18–210 mg/m2 (n = 30), and it was characterized by biphasic decline with a moderate-to-high clearance, a high distribution volume, and an apparent terminal half-life of approximately 17 hours [48]. While the PK characteristics of berzosertib in combination with either gemcitabine or cisplatin were consistent with the corresponding doses of berzosertib monotherapy, the collective PK data from these two arms suggest that pre-administration of cisplatin 24 hours before berzosertib administration does not affect the PK profile of berzosertib.
Another trial that is currently investigating the combination of berzosertib and gemcitabine is a multicenter, randomized, phase II study that recently published preliminary efficacy and safety data suggesting that this combination provides clinical benefit to platinum-resistant high-grade serous ovarian cancer (HGSOC) patients. At the cutoff date of publication, 70 patients had been randomly assigned to either receive treatment with the berzosertib and gemcitabine combination (n = 34) and achieved a median profession-free survival (PFS) of 22.9 weeks (90%, CI 17.9–72.0), or they were assigned to receive treatment with gemcitabine alone (n = 36) and achieved a median PFS of 14.7 weeks (90%, CI 9.7–36.7). Yet, while the combination of berzosertib and gemcitabine displayed a promising PFS with a hazard ratio of 0.57 (90%, CI 0.33–0.98), a higher objective response was observed for the group of patients who received treatment with gemcitabine alone. According to the authors, discrepancies between ORR and PFS are not uncommon in platinum-resistant ovarian cancer patients [51]. A sub-analysis of the patient population based on the length of the platinum-free interval also showed that patients who are treated with the berzosertib and gemcitabine combination, and who have had a platinum-free interval of 3 months or less, have a 30% increase in median PFS (27.7 weeks compared to 18.6 weeks in patients with intervals larger than 3 months). Since the PFS benefit observed for patients with a platinum-free interval of 3 months or less may be related to the enrichment for biomarkers of replicative stress, the authors followed up on this finding with further correlative assays [51]. Interestingly, results from follow-up studies using the same replication stress signature show that the combination of berzosertib and gemcitabine benefited more patients with tumors displaying low replication stress (RS-low) in contrast to patients with high replication stress tumors (who appeared to receive a greater benefit from the increase of replication stress caused by gemcitabine monotherapy [52]. Based on these results, it is suggested that increasing replication stress in RS-loss with gemcitabine concomitant with ATR inhibition by berzosertib is necessary for lethality [52].
Finally, a few clinical trials have published results about the preliminary efficacy and safety profile of the combination of berzosertib with topotecan in patients with lung cancers. A proof-of-concept phase I clinical trial that investigated the combination of berzosertib with topotecan in patients with platinum-resistant small cell lung cancer (SCLC) showed that 60% (3/5) of the patients treated achieved a PR or prolonged SD lasting ≥6 months, and the combination seemed to be well tolerated with no additive toxicity observed [54]. Yet, shortly after the interim results of the DDRiver SCLC 250 phase II trial investigating the combination of berzosertib with topotecan in platinum-resistant SCLC patients reported an objective response rate of 36% (9/25) and a median duration of response of 6.4 months, the trial was discontinued based on a low probability of meeting the primary objective [55, 56]. Further results from ongoing clinical trials are needed to demonstrate whether treating patients with advanced cancers, whose tumors are undergoing high replicative stress, with the combination of berzosertib and DNA-damaging chemotherapeutic agents may potentially help overcome platinum and/or PARP inhibitor resistance.
14.5.2 Ceralasertib (AZD6738)
Ceralasertib is a potent and selective ATR inhibitor with a promising preclinical data package showing efficacy in DDR-deficient settings [57]. Early preclinical studies showed that ceralasertib increases γH2AX phosphorylation, while inhibiting phosphorylation downstream of CHK1 in a variety of ATM-deficient cell lines and inducing accumulation of unrepaired DNA damage and cell death in ATM/p53-deficient leukemia cells [58, 59]. Recently, a growth inhibition assay assessing the sensitivity of 276 cancer cell lines to ceralasertib reported that cell lines harboring CCNE1 amplification or ARD1A, ATRX, and SETD2 mutations were associated with sensitivity. At first sight, cancer cell lines harboring ATM mutations were not associated with sensitivity; yet, upon stratifying the cancer cells based on ATM expression levels, it was shown that complete absence of ATM function is significantly associated with sensitivity to ceralasertib [60]. This finding, along with previous observations of antitumor responses from patients harboring ATM loss-of-function, supports the idea that patient selection for ATR inhibitors should consider biallelic deleterious mutations and ATM-null expression [61].
Preliminary results from the dose escalation and expansion monotherapy arms of the PATRIOT phase I clinical trial reported that ceralasertib was better tolerated when administered at an intermittent schedule of 2-weeks-on/2-weeks-off because only 20% of the patients experienced grade ≥3 treatment related adverse events (TRAEs) compared to 67% of patients when ceralasertib was administered in a continuous schedule (NCT02223923). Although it was previously shown that ceralasertib monotherapy in-vivo only induces significant tumor control/stasis and that the synergistic effects resulting in tumor regression are pronounced when ceralasertib is combined with DNA damaging agents or certain targeted small molecules, preliminary antitumor activity of the ceralasertib monotherapy arms of the PATRIOT trial show that 7% of patients achieved PR and 48% of them achieved SD as best overall response [60, 62, 63].
In-vivo, the combination of ceralasertib and cisplatin induced significant tumor reduction in HER2-positive breast cancer cells, as well as tumor regression in ATM-deficient lung cancer xenograft models and synergistic effects in ATM-deficient NSCLC cell lines [64, 65]. Results from a phase I trial investigating the combination of ceralasertib and carboplatin in advanced solid tumor patients reported that 2 (6%) of patients with low ATM or SLFN11 expression achieved PR as best response by RECIST v1.1, while 53% patients (including two unconfirmed PRs) achieved SD for ≥35 days (NCT02264678) [66]. Although no association between ATM and SLFN11 expression level and antitumor activity was reported, likely due to the sample size, these findings support the notion that further investigations on the interaction between ATR and loss of ATM function are needed. In contrast, a phase I clinical trial investigating the safety and antitumor activity of the ceralasertib and paclitaxel combination in advanced solid tumors (enriched for melanoma patients) reported one patient achieved complete response (CR), while 21% achieved PR and 32% achieved SD. Even though the ORR for the entire population was 22.6% (95% CI, 12.5–35.5), an ORR of 33.3% (95% CI, 10.8–51.8) was reported for the subset of melanoma patients resistant to PD1/L1 treatment [67].
Although both phase I trials studying the combinations of ceralasertib with chemotherapy agents reported that the combinational strategies are safe and well tolerated, thrombocytopenia, neutropenia and anemia were reported as the most common grade ≥3 TRAEs, with schedule limiting consequences observed with the combination of ceralasertib and carboplatin. Since toxicity may be one of the major challenges in the implementation of ATR inhibitor combinations with DNA damaging agents and other targeted small molecules, the success of clinical trials investigating ceralasertib combinations depends on the optimization of the dose scheduling sequences and targeted genetic tumor aberrations. For instance, recent in-vivo studies suggest that to achieve tumor regressions, concurrent dosing for the ceralasertib and irinotecan combination should be extended at least one day, while a few days of ceralasertib dosing should be included after concurrent dosing with carboplatin [60]. In a similar fashion, the ATRiUM phase I clinical trial is investigating the safety and antitumor activity of ceralasertib with either intermittent or continuous gemcitabine dosing in advanced solid tumors, particularly in patients with advanced pancreatic ductal adenocarcinoma with ATM-loss-of-function [68, 69]. In all, results from an ongoing phase II trial investigating ceralasertib monotherapy in advanced solid tumors (enriching for mCRPC with low ATM expression), as well as results from the remaining arms of the PATRIOT phase I clinical trial and the ATRiUM phase I are required to further assess the clinical efficacy of ceralasertib monotherapy and in combination with chemotherapy agents in a molecularly targeted population.
Although synergistic effects in-vivo were observed when combining ceralasertib with either PARP or WEE1 inhibitors, only the PARP inhibitors and ceralasertib combination has successfully reached phase II clinical trials. Out of the six clinical trials that are currently investigating the combination of ceralasertib and olaparib in the advanced cancer setting, one phase I study reported on safety and preliminary antitumor efficacy, as well as established the RP2D of the ceralasertib and olaparib combination in patients with advanced solid tumors, and two Phase II trials have presented contrasting preliminary results based on patient selection (Table 14.2) [63, 70, 71]. Briefly, results from one of the first modular phase I clinical trials to test the combination of ceralasertib and olaparib established a concurrent RP2D of ceralasertib at 160 mg QD on days 1–7 and olaparib at 300 mg twice daily (BID) on days 1–28, with thrombocytopenia and neutropenia defined as dose limiting toxicities [72]. Within the module that tested the dose escalation of ceralasertib and olaparib, antitumor responses were observed in patients with advanced breast, ovarian, prostate, pancreatic, and ampullary cancer. Interestingly, while some of the responding tumors had BRCA1/2 mutations, antitumor responses were independent of ATM status [72]. Such results are in accordance with recent preclinical studies suggesting that the combination of ceralasertib and olaparib in a concurrent schedule induces tumor regression in TNBC BRCA-wild type and BRCA2-mutated xenograft models [60], and the development of a phase II clinical trial currently recruiting patients to investigate the combination of ceralasertib and olaparib in advanced germline BRCA mutated breast cancer (NCT04090567).
Although differences in study design preclude us from direct comparisons, preliminary antitumor activity from two phase II clinical trials investigating the combination of ceralasertib and olaparib seemed to be influenced by the selection of targeted genetic tumor aberrations. That is, the phase II clinical trial investigating the clinical benefit of this combination in patients with advanced solid tumors with or without ARID1A-deficiency (defined as lack of expression of BAF250a by IHC) reported an ORR of 20% for patients with ARID1A-deficiency, including two patients that achieved sustained CRs, while no objective responses were observed in the cohort of patients with active ARID1A function [73]. In contrast, the phase II clinical trial investigating signals of activity of the ceralasertib and olaparib combination in patients with HGSOC reported no partial or complete responses in a PARP naïve, genetically unselected, platinum-resistant cohort of 12 patients. Nonetheless, 75% of the patients in that trial achieved SD as best overall response by RECIST v1.1 and 27% of the patients achieved ≥ 50% decrease in CA-125, most of them harboring tumors with somatic BRCA1 mutations [74].
As mentioned by investigators of the HGSOC phase II trial, it is likely that more responses may have been achieved by focusing the patient population to ovarian cancer patients harboring tumors with BRCA1/2 mutations and/or CCNE1 copy number amplification [74]. Taking it all together, the ceralasertib and olaparib combination appears to be well tolerated, but it is necessary to continue optimizing patient selection strategies based on the selection of genetic aberrations that induce synthetic lethality in different types of cancer types. Such concept seems to be reflected in recent preliminary results from the HUDSON trial, a phase II multidrug, biomarker selected umbrella study investigating the combination of multiple targeted small molecules with durvalumab, including ceralasertib for NSCLC patients who progressed after anti-PD-1/PD-L1 and platinum therapy (NCT03334617). Although no correlation was found between ATM biomarker status and clinical responses by RECIST 1.1, the HUDSON trial reported an improved ORR (11.1%) and longer PFS (7.43 months) for patients whose tumors harbored ATM mutations or low protein expression -when compared to an ORR of 8.3% and PFS of 4.96 months for NSCLC patients with acquired resistance to prior immunotherapy, regardless of ATM status [75].
By comparing gene expression profiles in paired blood samples from patients with controlled disease and patients whose disease progressed with the ceralasertib monotherapy run-in, the HUDSON trial showed increases in an antigen presentation gene signature and decreases in exhausted T-cell and NK-cell signatures, supporting a model in which ceralasertib also has an active role in the immune activation caused by the combination of ceralasertib with durvalumab [77]. These findings are also in agreement with results from a phase II clinical trial investigating the clinical activity of the ceralasertib and durvalumab combination in advanced gastric cancer patients, which reported (1) significantly longer PFS for patients whose tumors harbored ATM-deficiency and/or HRD-deficiency when compared to patients with active ATM function and HRD-proficient (5.60 months versus 1.65 months, HR 0.13., 95% CI 0.045–0.39, p < 0.001), as well as, (2) upregulation of the innate immune response, (3) activation of intratumoral lymphocytes, and (4) increase of tumor reactive CD8+ T-cells in patients who responded to treatment [76].
Finally, a phase III clinical trial (LATIFY, NCT05450692) will compare the clinical benefit of the ceralasertib and durvalumab combination versus docetaxel monotherapy in NSCLC patients who progressed after anti-PD-1/PD-L1 and platinum therapy. This is based on the finding that NSCLC patients with primary resistance to immunotherapy only responded to the combination of ceralasertib and durvalumab in the HUDSON trial [75].
14.5.3 Elimusertib (BAY1895344)
By evaluating the molecular interactions of available ATR inhibitors within the binding pocket of an ATR homology model created using the crystal structure of a PI3K kinase and performing a high-throughput screen, Bayer identified a lead compound that was further optimized to reduce potential off-target toxicity [78]. BAY1895344, also called elimusertib, is a potent and selective ATR inhibitor shown to increase γH2AX phosphorylation in HT-29 cells and inhibit cell proliferation in a variety of cancer cell lines, including different lymphoma cells and cell lines harboring mutations that affect the ATM pathway, Elimusertib induced stronger antitumor activity than ceralasertib and berzosertib in a lymphoma cell line-derived xenograft (CDX) model, with antitumor activity also observed in ovarian, prostate and colorectal CDX models harboring DDR defects [40]. In addition, elimusertib treatment inhibited neuroblastoma cell growth and induced strong tumor growth inhibition in neuroblastoma xenograft and ALK-driven genetically modified mice models [79]. Interestingly, RNA-seq data from mice who achieved tumor size decrease after elimusertib treatment revealed expression of inflammatory response and immune tumor infiltration, suggesting that ATR inhibition by elimusertib positively impacts the tumoral immune response [79].
Synergistic antitumor efficacy for the combination of elimusertib and DNA-damaging treatments was observed in colorectal cancer cells treated with elimusertib and cisplatin, as well as in colorectal xenograft models treated with elimusertib and radiation therapy [40]. In contrast, antagonistic interactions were observed with the combination of elimusertib and docetaxel [40].
Treatment with elimusertib and olaparib displayed strong antitumor efficacy and a tolerable profile in a TNBC xenograft model and delayed tumor growth in a PARP inhibitor resistant prostate cancer xenograft model [40]. In a similar manner, synergistic antitumor activity was also observed with sequential dosing of anti-PD-1/PD-L1 antibodies and elimusertib in immunocompromised and lymphoma mice models [40]. Taken together, preclinical studies suggest that combining elimusertib with certain DNA damaging agents, as well as with DDR and checkpoint inhibitors, may result in synergistic antitumor activity when compared to the respective singe-agent treatments.
Further studies are currently being conducted to determine the precise combination schedules that are safe and well-tolerated in humans. For instance, results from the dose escalation of the first-in-human trial of elimusertib in patients with advanced solid tumors determined that intermittent dosing of 40 mg BID 3 days on/4 days off is the maximum tolerated dose (MTD) of single-agent elimusertib, with pharmacodynamic data showing on-treatment tumor increases in γH2AX levels (NCT03188965) [80]. The most frequently observed adverse events (AE) in the dose escalation was grade 3 anemia, likely due to limited differentiation and expansion of erythrocyte precursors that are sensitive to replication stress [81]. Based on the safety results from the first elimusertib monotherapy trial, combinational strategies with chemotherapy agents may induce overlapping hematologic toxicity and dose escalations should be approached with caution.
Nonetheless, this trial provided proof-of-concept for the clinical antitumor activity of elimusertib: 4 patients achieved PRs, while 8 achieved SD with a median duration of response of 11.25 months and resulting in 69% disease control rate in patients treated at MTD or above [80]. More importantly, 3 of the 4 patients that achieved PRs had tumors with low ATM expression by IHC, with two of them harboring deleterious ATM mutations. Albeit a small sample size, an ORR of 33.3% was reported for the subgroup of patients with ATM protein loss, and an ORR of 37.5% was calculated for the subgroup of patients harboring ATM deleterious mutations [80]. Within the responders for this trial, the investigators noted one heavily pretreated ovarian cancer patient who had received 9 chemotherapy lines, as well as prior PARP inhibitor and immunotherapy, achieved SD for more than year [80]. The clinical benefit observed for this PARP-resistant ovarian cancer patient harboring a BRCA1 deleterious mutation seem to suggest that PARP inhibitor resistance may be mediated by protection of the DNA replication fork by ATR, opening the possibility of expanding ATR inhibitor treatments to PARP inhibitor-resistant patient population and providing clinical rationale for a phase I clinical trial that investigates the combination of elimusertib and niraparib in patients with advanced ovarian cancer and other solid tumors (Table 14.3, NCT04267939) [80].
14.5.4 Gartisertib (M4344/VX-803)
As an ATP-competitive inhibitor, gartisertib is a selective ATR inhibitor with 100-fold selectivity over a wide range of kinases and strong potency demonstrated by suppression of ATR-driven checkpoint kinase-1 (CHK1) phosphorylation in a prostate cancer cell line, as well as by induction of γH2AX phosphorylation in a small-cell lung cancer cell line [82]. Remarkably, sensitivity assays and gene expression analysis of a variety of cancer lines showed that cancer cells with higher replication stress and high neuroendocrine expression signatures are highly sensitive to gartisertib treatment, suggesting that those genomic signatures may be useful for patient selection and as biomarkers of response [82].
As a single-agent, gartisertib was shown to suppress proliferation in prostate cancer cells at a lower concentration and at a higher rate than berzosertib and ceralasertib, and it was shown to induce tumor stasis and tumor regression in ALT mice models [83, 84]. A variety of preclinical models also demonstrated synergistic effects of different gartisertib combination strategies. For instance, the combination of gartisertib and TOP1 inhibitors showed synergistic antitumor activity in several small-cell lung cancer cell lines and cell-derived mouse xenografts, as well as in prostate cancer patient-derived tumor organoids [85]. In addition, combining gartisertib with DNA damaging agents such as gemcitabine and cisplatin, as well as with PARP inhibitors such as talazoparib, displayed synergy at noncytotoxic concentrations in a small-cell lung cancer cell line.
14.5.5 Camonsertib (RP-3500)
Camonsertib, developed by Repare Therapeutics and recently licensed to Roche, is a highly selective ATR inhibitor that demonstrated potent single-agent efficacy by a dose-dependent inhibition of CHK1 phosphorylation and induction of γH2AX, DNA-PK and KAP1 phosphorylation in-vivo [86]. Camonsertib monotherapy induced significant tumor growth inhibition in an ATM-deficient colorectal xenograft model and also induced complete tumor regression in a gastric xenograft model [86]. Unlike other ATR inhibitors, tumor growth inhibition with minimal hematological adverse effects was observed with intermittent camonsertib treatment in ATM-deficient mouse models [86]. In line with preclinical data suggesting that intermittent camonsertib dosing schedules with dose holidays of at least 4 consecutive days allow for reticulocyte regeneration to avoid hematological toxicities in-vivo, recent preliminary data from the TRESR phase I/IIa clinical trial investigating the safety and preliminary efficacy of camonsertib showed a significant reduction of grade 3 anemia in advanced cancer patients (NCT04497116) [87]. In this study, 14.5% of patients treated with intermittent camonsertib dosing experienced grade 3 anemia, compared with 65.7% of patients who experienced grade 3 anemia after intermittent elimusertib treatment [87, 88]. Preliminary data from the TRESR trial also showed clinical activity across different tumor types, with meaningful clinical benefit in 49% of evaluable patients and an ORR of 25% [87]. Aligned with preclinical data, clinical activity was observed in CRPC patients whose tumors harbored ATM and CDK12 mutations, ovarian cancer with BRCA1 and RAD51C mutations, as well as breast cancer, melanoma, and HNSCC patients with tumors harboring BRCA1 and BRCA2 mutations. Notably, 37 patients whose tumors harbored relevant genomic mutations achieved molecular responses in ctDNA, suggesting that ctDNA responses may predict clinical benefit [87].
Intermittent concomitant rather than sequential administration of camonsertib and PARP inhibitors in different ATM and BRCA1 deficient models led to stronger synergistic antitumor activity without increases in hematological toxicity [86]. Along with the additional modules of the TRESR clinical trial that are currently investigating the combination of camonsertib and talazoparib, the ATTACC phase I/IIa clinical trial investigating the safety and preliminary efficacy of camonsertib in combination with either olaparib or niraparib is currently recruiting patients (NCT04972110).
14.5.6 M1774
Building on learnings from berzosertib, Merck KGaA developed M1774 as a potent and selective ATR inhibitor that has demonstrated antitumor activity in PDX models. The modular DDRiver Solid Tumors 301 clinical trial is currently investigating the safety and tolerability and preliminary efficacy of M1774 in patients with advanced solid tumors harboring selected mutations, including deleterious mutations in ATM, ARID1A, ATRX and/or DAXX (NCT04170153) [89]. Recent results from the dose escalation of this trial suggested an MTD of 180 mg QD continuous dosing and a recommended dose for expansion of 180 mg 2 weeks on/1 week off, with modulation of γH2AX in peripheral blood mononuclear cells achieved in doses starting at 130 mg QD [89]. While the DDRiver 301 trial is currently recruiting patients for two dose expansion modules in biomarker selected cohorts and food effects cohort, it is also recruiting patients in a module investigating the safety and tolerability of the combination of M1174 and niraparib. In addition, a recent clinical trial investigating the safety and tolerability of M1774 in combination with a DDR inhibitor or an immune checkpoint inhibitor has recently started to recruit patients (NCT05396833).
14.5.7 ART0380
ART0380, licensed by Artios Pharma Ltd from The University of Texas MD Anderson Cancer Center and ShangPharma Innovation, demonstrated target engagement by γH2AX and pKAP1 modulation in-vivo and, is currently being investigated as monotherapy and in combination with gemcitabine or irinotecan in a modular phase I/IIa clinical trial for advanced solid tumor patients (NCT04657068). In order to measure target engagement, Artios has developed an assay in normal peripheral blood mononuclear cells (PMBCs) and in circulating tumor cells (CTCs) [90]. Although interim results for this trial have not been presented thus far, Artios recently mentioned in a press release that upon treatment with single-agent ART0380, modulation of γH2AX in patient blood samples is observed at a larger magnitude in CTCs than in PBMCs, and that based on preliminary results from the dose escalation phase of the trial, ART0380 has a safe and tolerable profile. Therefore, the intermittent dose escalation ART0380 has progressed to the dose expansion phase in patients with ATM-deficient tumors.
14.6 ATR and PARP Inhibitor Combination Strategies
Synthetic lethal strategies for cancer treatment, where cell death is induced by targeting proteins or pathways that are redundant in normal cells but not cancer cells, are showing clinical promise. Inhibitors of PARP1 (Poly (ADP)-ribose polymerase 1, a key DDR enzyme) are prime examples of anti-cancer therapeutics capable of harnessing the synthetic lethal mechanism and have revolutionized the field of cancer therapeutics. Seminal work led by multiple teams in the early 2000s identified HR-deficient BRCA1/2-mutated cancers as selective targets for PARP inhibitor-induced lethality [91,92,93]. Today, several PARP inhibitors are FDA-approved for the treatment of BRCA1/2-mutated cancers, including in multiple settings of ovarian cancer, metastatic breast cancer, pancreatic cancer, and advanced castration-resistant prostate cancer (CRPC) [94,95,96,97]. Unfortunately, PARP inhibitor resistance is ubiquitous in the clinic. Acquired PARP inhibitor resistance can occur following prolonged treatment, whereas primary PARP inhibitor resistance is observed in many patients with BRCA1/2-mutated cancers and fail to respond at treatment initiation [98]. One strategy to overcome PARP inhibitor resistance is to develop rational combination treatments to sensitize cells to PARP inhibitors.
Growing evidence suggests that ATR inhibition may help to overcome PARP inhibitor resistance [99, 100]. The ATR gene was identified as a mediator of PARP inhibitor sensitivity in a synthetic lethal siRNA screen [92]. DNA DSBs that are produced following exposure to PARP inhibitors renders cells dependent on ATR for DNA repair [8]. As such, exposure to an ATR inhibitor disables ATR-mediated repair pathways and promotes cell death. Additionally, a known mechanism of PARP inhibitor resistance involves restored replication fork stabilization that may involve ATR, as well as other DDR proteins, such as CHK1 and WEE1 [99]. Preclinical studies have demonstrated that ATR inhibition leads to replication fork collapse that produces irreparable DNA DSBs [101, 102]. Building on this, the rationale for the combination of PARP and ATR inhibitors was demonstrated in another preclinical study in which PARP inhibitor resistant cells exhibited enhanced sensitivity in response to dual ATR and PARP inhibition in ovarian cancer patient-derived xenograft (PDX) models [103]. Furthermore, there are multiple ongoing clinical trials currently evaluating this drug combination, with at least 10 active studies taking place world-wide at the time of publication (Table 14.4).
With an expansive landscape of trials evaluating ATR and PARP inhibitor combinations, it is important to understand the tolerability and clinical efficacy of this approach. Overlapping toxicities stemming from combined ATR and PARP inhibition may be an issue for this drug combination. One example of such toxicity was reported in a dose-finding phase I trial in which ceralasertib was combined with the olaparib PARP inhibitor, which resulted in dose limiting toxicities (DLTs) in the form of thrombocytopenia and neutropenia that restricted continuous concurrent dosing of these agents [104].
Nonetheless, promising clinical activity produced by this drug combination was reported in a separate phase II trials evaluating a cohort of patients with recurrent ovarian cancer who had progressed on prior PARP inhibitor treatment [105]. In a cohort of thirteen patients, the reported objective response rate (ORR) was 46% across six patients who had achieved radiologic PR [105]. Of these patients, 69% had germline BRCA mutations, 23% had somatic BRCA mutations, and 8% had other homologous recombination deficient mutations [105]. Although no patient discontinued treatment due to toxicity, reported adverse events included thrombocytopenia, anemia, and neutropenia, with dose reductions reported for both ceralasertib and olaparib [105]. Interestingly, this same study reported no objective responses in a cohort of PARP inhibitor naïve patients with platinum-resistant ovarian cancer [106]. Enrichment of therapeutic responses in the cohort of patients with past PARP inhibitor exposure further supports the notion that combined ATR and PARP inhibitor strategies may be key to overcome PARP inhibitor resistance in the clinic [106].
14.7 ATR and Immune-Checkpoint Inhibitor Combination Strategies
An emerging body of preclinical and clinical evidence supports the immunomodulatory role of ATR inhibitors in the tumor microenvironment. For instance, a recent single DNA fiber analysis after ATR inhibition showed induction of chromatin bridge formation and chromosome lagging, which in turn accelerated mitotic entry and further activated the cyclic GMP-AMP synthase-stimulator of interferon genes (cGAS-STING) tumor sensing axis [107]. Since genotoxic stress also induces the release of cytosolic DNA fragments that activate the cGAS-STING pathway, the combination of ATR inhibition with chemotherapeutics seems a rational combination to activate the innate immune response [111, 112].
It was also recently shown that treating prostate cancer cell lines with elimusertib induced S-phase DNA damage, activation of cGAS-STING signaling, as well as upregulation of CCL5 (chemokine ligand 5) and CXCL10 (C-X-C motif chemokine ligand 10) expression that culminated in activation of innate immunity [108, 109]. This is further supported by the increase in activated cGAS-STING and TBK1 levels, CD8+ T-cell infiltration, reduction of regulatory T-cell infiltration, and T-cell exhaustion observed in immunocompetent hepatocellular carcinoma mouse models treated with the triple combination of radiation, followed by ceralasertib and PD-L1 inhibition [110]. In addition, shortly after treatment with ceralasertib there was a modest increase in the intratumoral concentration of IFN-γ and proliferating CD8+ T-cells that was accompanied by a reduction of the PD-L1 tumor upregulation induced by radiation. At later time points, the combination of ceralasertib and radiation induced an increase in infiltrating CD8+ T-cells, as well as production of INF-γ and tumor necrosis factor α [110]. Similar results were obtained by studying the combination of ceralasertib and radiation on immunocompetent mouse models of HPV-driven cancer, where a signature of type I and II IFN gene expression and modulation of cytokine gene expression (including CCL3 and CXCL10) were associated with treatment. Interestingly, increased antigen presentation and levels of major histocompatibility complex class I were also observed in vivo with the combination of ceralasertib and radiation [111]. Taken together, results from multiple preclinical studies suggest that the combination of radiation and ATR inhibitors stimulates IFN response and triggers antigen presentation.
ATR inhibition has also been shown to suppress upregulation of the natural killer group 2D (NKG2D) cell surface ligand that binds to activated CD8+ T-cells to trigger pro-inflammatory cytokine production [112]. It has also been suggested that ATM/ATR/CHK1 signaling upregulation leads to transcriptional activation of PD-L1 via the signal transducer and activators of transcription STAT1 and STAT3 and the IFN regulatory factor (IRF1) pathway [113]. In fact, an increase in PD-L1 expression, accompanied by increased infiltrating macrophages and reduced infiltrating CD3+ T-cells, was observed in ATR deficient melanoma models (Fig. 14.1) [114]. Remarkably, such preclinical data is supported by results from the phase I clinical trial investigating elimusertib monotherapy, where paired tumor samples from patients with PD-L1 positive tumors revealed upregulation of PD-L1 [80]. Interestingly, patients with metastatic melanoma that were previously resistant to PD-L1 inhibitors achieved durable responses when treated with the combination of ceralasertib and paclitaxel [115]. In this combinational trial, interlukin-12 fluctuations were also observed in patients that received clinical benefit suggesting activation of the innate immune response [115].
14.8 Candidate Biomarkers of ATR Sensitization
Therapeutic biomarkers are used as indicators of disease prognosis and predictive measures of treatment response [116]. Emerging data from various preclinical and clinical studies that evaluating ATR inhibitors as monotherapy or in combination strategies have identified candidate predictive biomarkers that may indicate sensitivity to ATR inhibition. Here, we summarize key genetic biomarkers and discuss their role in defining target patient populations that may respond best to ATR inhibitors.
ATM is a DDR kinase that senses and repairs dsDNA breaks and whose mutation may confer dependency on the ATR-CHK1 axis, offering an exploitable target for ATR inhibitors [117]. Although ATM is frequently mutated in cancer, the functional impact of many ATM variants is not well established [118]. Furthermore, there is significant overlap between ATM and ATR signaling pathways, as supported by various preclinical and clinical studies evaluating various cancer types including hematological and solid tumors [8, 12, 119]. Clinical responses have been reported from phase I studies of ATR inhibitors specifically in patients with ATM aberrations, including ATM deleterious mutations or protein loss [12, 16, 120]. Although ATM is frequently mutated in cancer, the functional impact of many ATM variants is not well established [118]. However, a large proportion of ATM mutations derive from missense variants, which can lead to a reduction in ATM protein expression levels [31]. This highlights the potential use of immunohistochemistry (IHC) analysis as a clinical tool to probe ATM expression levels and identify those who could benefit from ATR inhibition [31]. Pilie et al., further demonstrated the utility of IHC to understand ATM mutation annotations reported as variants of unknown significance (VUS), in which IHC analysis reported loss of protein in up to 25% of ATM VUS mutations, thus clarifying their functional impact [118]. This study also identified ATM loss of protein in patient tumor samples without identified ATM mutations, which points to the involvement of other mechanisms such as epigenetic or post-translational loss [118].
Another widely evaluated biomarker of ATR inhibitor sensitivity is p53, which plays a prominent role in G1 checkpoint control and whose loss comprises a high proportion of cancer cases [31]. Although there is preclinical data to support p53’s role as a predictive biomarker, the data remains inconsistent. For instance, Toledo et al., showed that cells with defective p53 had augmented replication stress in response to ATR inhibitors compared to cells with wildtype p53 [35]. A similar finding was reported in Kwok et al., in which treatment with the ATR inhibitor AZD6738 resulted in selective toxicity in p53 defective xenografts and cell lines [121]. In contrast, another study showed no increase in sensitivity to single agent ATR inhibition with VE-821 in p53 mutant cell lines compared to matched wildtype p53 cells [122]. Although Dillion et al., reported radio-sensitization by AZD6738 to single radiation fractions in a panel of cell lines, the narrow sensitivity range to AZD6738 was independent of p53 status [123]. Cumulatively, despite strong rationale supporting the use of ATR inhibitors to treat p53 deficient tumors, the conflicting data suggests further studies are necessary to assess its utility as a predictive biomarker of response. Although data is still pending, multiple clinical trials are underway to evaluate ATR inhibitors as monotherapy or in combination strategies in patients with solid tumors harboring TP53 mutations [48, 124].
A link between ATR sensitivity and deficiency of the BAF complex component AT-Rich Interactive Domain-containing protein 1A (ARID1A) was established in a large-scale genetic screen reported in Williamson et al. [125]. In this study, both in-vitro and in-vivo models were used to demonstrate wide-ranged genomic instability and cell death in ARID1A mutant cancer cell lines and tumors in response to ATR inhibition [125]. The clinical significance of this finding is highlighted by the fact that up to 7% of all cancers are associated with ARID1A loss and the frequency of loss is increased in certain cancers, for example, ARID1A loss is reported in up to 50% of clear cell ovarian carcinoma cases [126]. Further support for ATR inhibition in the setting of ARID1A loss was demonstrated in Tsai et al., in which an accumulation of R-loop formation was identified as a driver of replication stress in an ovarian cancer line with ARID1A knockout [126]. Translation of these data to the clinical setting has also produced compelling results. Antitumor activity was observed in patients with ARID1A-deficient solid tumors treated with the single-agent ATR inhibitor ceralasertib, including two patients that achieved RECIST-confirmed complete responses [127]. In addition, treatment with M6620 monotherapy resulted in a RECIST-confirmed complete response after 16 cycles in a patient with metastatic colorectal cancer with ARID1A mutation and IHC confirmed loss of both ARID1A and ATM, with a reported progression free survival of 29 months at their last assessment [128]. The use of cell-free DNA (cfDNA) as an indicator of treatment response was also evaluated in this study, which revealed declining levels of allele frequencies for ARID1A, among other identified mutations, to undetectable levels after 9 cycles of treatment with M6620 compared to baseline [128].
Targeting deficiencies in homologous recombination DNA repair (HRR) also offers a potential opportunity for ATR inhibition [129, 130]. For example, Krajewska et al., demonstrated sensitivity of the breast cancer cell line MCF-7 to ATR inhibitors upon inactivation of RAD51 in the HRR pathway [129]. Other studies have since further elucidated the major role ATR plays in regulating homologous recombination processes. For instance, Kim et al., showed that increased ATR signaling promotes the capacity of HRR in cancer cells by regulating the abundance of homologous recombination factors [28]. In support of this, a phase I trial of the Repare ATR inhibitor RP-3500 monotherapy observed multiple clinical responses in ovarian cancer patients with PARP-inhibitor resistant cancers that harbored actionable BRCA1 and RAD51C mutations [87, 117]. Other responses described in this study included patients with homologous recombination deficiency (HRD), with molecular alterations in ATM, BRCA2 and RAD51B/C [87]. ATR inhibition in HRD-cancers is largely under clinical investigation via multiple trials that are actively recruiting patients with deleterious mutations in HRR genes.
ATR deficiency has also been shown to confer a strong synthetic lethal response with many other DDR genes as well as with inducers of DNA replication stress [29]. For example, ATR inhibition is synthetic lethal in cells with genetic defects in genes such as APOBEC3A and B as well as with overexpression of cyclin E1 (CCNE1) and with c-MYC amplifications [28, 131,132,133]. In addition to those mentioned above, molecular defects in DDR genes such as ERCC1, XRCC1, CHK1, and FANCD2, and even accumulation of R-loops, all have been shown to produce a synthetic lethal effect in response to ATR inhibition [8, 28, 29]. With so many potential synthetic lethal partners possible, results from ongoing preclinical and clinical studies will be instrumental in identifying biomarkers and factors associated with therapeutic response as ATR inhibitors appear poised to enter the clinic in the coming years.
14.9 Concluding Remarks
This chapter provides a rationale for targeting ATR and summarizes the current landscape of ATR inhibitors in clinical evaluation. As a key component of the DDR, ATR is a promising druggable target that is being widely evaluated in phase I, II and III clinical trials as monotherapy and in combinations with other agents, including DNA repair inhibitors, chemo- and radiotherapy, and immunotherapy. Regardless of the approach taken, ongoing clinical studies must address optimization of the therapeutic window for this drug class. A predominantly reported toxicity across ATR inhibitors trials is myelosuppression, which is a mechanism-based toxicity that ultimately limits the therapeutic window in both monotherapy and combination approaches [134]. This carries key implications particularly for combination strategies due to potentiating of overlapping toxicities that may deepen myelosuppression and reduce drug tolerability. Proposed rational combination strategies should limit overlapping toxicity, which may be achieved by coordinating intermittent dosing schedules to facilitate tissue recovery. Another prevalent challenge is refining the target patient population most likely to benefit from ATR inhibition. Molecular technology advances and companion diagnostics have opened the door to precision oncology and the opportunity to offer personalized treatment strategies to patients [135]. Today, many clinical studies are designed on the basis of mutational status, which has led to the approval of several tumor-agnostic drugs [136]. Interestingly, many ongoing ATR inhibitor trials are recruiting patients based on molecular alteration rather than relying solely on tumor-type. A spectrum of molecular alterations have already been identified as potential predictive biomarkers that may sensitize to ATR inhibition; however, to be clinically efficacious, the biomarkers must be sensitive and easy to measure to allow for successful integration into the clinic. In closing, although several ATR inhibitors in development are poised to address a clinically unmet need, no ATR inhibitor has received FDA-approval for cancer indications thus far. We eagerly await the results from ongoing clinical studies as FDA-approval of ATR inhibitors lies close in sight.
References
Ngoi NYL, Peng G, Yap TA (2021) A Tale of Two Checkpoints: ATR Inhibition and PD-(L)1 Blockade. Annu Rev Med 73(1):1–20
Helleday T (2018) Targeting the DNA damage response for anti-cancer therapy. Canc Drug Disc Dev 1–9
Alhmoud JF, Woolley JF, Moustafa A-EA, Malki MI (2020) DNA damage/repair management in cancers. Cancers 12(4):1050
Mazouzi A, Velimezi G, Loizou JI (2014) DNA replication stress: causes, resolution and disease. Exp Cell Res 329(1):85–93
Luo J, Solimini NL, Elledge SJ (2009) Principles of cancer therapy: oncogene and non-oncogene addiction. Cell 138(4):807
Bartkova J, Hořejší Z, Koed K, Krämer A, Tort F, Zieger K et al (2005) DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature 434(7035):864–870
Gorgoulis VG, Vassiliou L-VF, Karakaidos P, Zacharatos P, Kotsinas A, Liloglou T et al (2005) Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature 434(7035):907–913
Sundar R, Brown J, Russo AI, Yap TA (2017) Targeting ATR in cancer medicine. Curr Prob Cancer 41(4):302–315
Branzei D, Foiani M (2010) Maintaining genome stability at the replication fork. Nat Rev Mol Cell Bio 11(3):208–219
Ngoi NYL, Pham MM, Tan DSP, Yap TA (2021) Targeting the replication stress response through synthetic lethal strategies in cancer medicine. Trends Cancer 7(10):930–957
Berti M, Vindigni A (2016) Replication stress: getting back on track. Nat Struct Mol Biol 23(2):103–109
Bradbury A, Hall S, Curtin N, Drew Y (2020) Targeting ATR as Cancer Therapy: A new era for synthetic lethality and synergistic combinations? Pharmacol Therapeut. 207:107450
Baillie KE, Stirling PC (2020) Beyond kinases: targeting replication stress proteins in cancer therapy. Trends Cancer 7(5):430–446
Lecona E, Fernandez-Capetillo O (2018) Targeting ATR in cancer. Nat Rev Cancer 18(9):586–595
Wang X, Wang L, Huang Y, Deng Z, Li C, Zhang J et al (2022) A plant-specific module for homologous recombination repair. Proc Natl Acad Sci 119(16):e2202970119
Rundle S, Bradbury A, Drew Y, Curtin NJ (2017) Targeting the ATR-CHK1 axis in cancer therapy. Cancers 9(5):41
Bass TE, Cortez D (2019) Quantitative phosphoproteomics reveals mitotic function of the ATR activator ETAA1. J Cell Biol 218(4):1235–1249
Zou L, Elledge SJ (2003) Sensing DNA damage through ATRIP Recognition of RPA-ssDNA complexes. Science 300(5625):1542–1548
Butler LR, Gilad O, Brown EJ (2018) Targeting the DNA damage response for anti-cancer therapy. Canc Drug Disc Dev 11–33
Couch FB, Bansbach CE, Driscoll R, Luzwick JW, Glick GG, Bétous R et al (2013) ATR phosphorylates SMARCAL1 to prevent replication fork collapse. Gene Dev 27(14):1610–1623
Matos DA, Zhang J-M, Ouyang J, Nguyen HD, Genois M-M, Zou L (2020) ATR protects the genome against R loops through a MUS81-triggered feedback loop. Mol Cell 77(3):514-527.e4
Sørensen CS, Hansen LT, Dziegielewski J, Syljuåsen RG, Lundin C, Bartek J et al (2005) The cell-cycle checkpoint kinase Chk1 is required for mammalian homologous recombination repair. Nat Cell Biol 7(2):195–201
Wang LC, Gautier J (2010) The Fanconi anemia pathway and ICL repair: implications for cancer therapy. Crit Rev Biochem Mol 45(5):424–439
Sirbu BM, Cortez D (2013) DNA damage response: three levels of DNA repair regulation. Csh Perspect Biol 5(8):a012724
Auclair Y, Rouget R, Drobetsky EA (2009) ATR kinase as master regulator of nucleotide excision repair during S phase of the cell cycle. Cell Cycle 8(12):1865–1871
Lee T-H, Park J-M, Leem S-H, Kang T-H (2014) Coordinated regulation of XPA stability by ATR and HERC2 during nucleotide excision repair. Oncogene 33(1):19–25
Fokas E, Prevo R, Hammond EM, Brunner TB, McKenna WG, Muschel RJ (2014) Targeting ATR in DNA damage response and cancer therapeutics. Cancer Treat Rev 40(1):109–117
Kim D, Liu Y, Oberly S, Freire R, Smolka MB (2018) ATR-mediated proteome remodeling is a major determinant of homologous recombination capacity in cancer cells. Nucleic Acids Res 46(16):8311–8325
Kantidze OL, Velichko AK, Luzhin AV, Petrova NV, Razin SV (2018) Synthetically lethal interactions of atm, ATR, and DNA-PKcs. Trends Cancer 4(11):755–768
Qiu Z, Oleinick NL, Zhang J (2018) ATR/CHK1 inhibitors and cancer therapy. Radiother Oncol 126(3):450–464
Weber AM, Ryan AJ (2015) ATM and ATR as therapeutic targets in cancer. Pharmacol Therapeut. 149:124–138
Hurley PJ, Wilsker D, Bunz F (2007) Human cancer cells require ATR for cell cycle progression following exposure to ionizing radiation. Oncogene 26(18):2535–2542
Mei L, Zhang J, He K, Zhang J (2019) Ataxia telangiectasia and Rad3-related inhibitors and cancer therapy: where we stand. J Hematol Oncol 12(1):43
Nishida H, Tatewaki N, Nakajima Y, Magara T, Ko KM, Hamamori Y et al (2009) Inhibition of ATR protein kinase activity by schisandrin B in DNA damage response. Nucleic Acids Res 37:5678–5689. Available from <Go to ISI>://WOS:000271569100009
Toledo LI, Murga M, Zur R, Soria R, Rodriguez A, Martinez S et al (2011) A cell-based screen identifies ATR inhibitors with synthetic lethal properties for cancer-associated mutations. Nat Struct Mol Biol 18(6):721–U124. Available from <Go to ISI>://WOS:000291308000014
Charrier JD, Durrant SJ, Golec JMC, Kay DP, Knegtel RMA, MacCormick S et al (2011) Discovery of potent and selective inhibitors of ataxia telangiectasia mutated and Rad3 related (ATR) protein kinase as potential anticancer agents. J Med Chem 54:2320–2330. Available from <Go to ISI>://WOS:000289215700028
Reaper PM, Griffiths MR, Long JM, Charrier JD, MacCormick S, Charlton PA et al (2011) Selective killing of ATM- or p53-deficient cancer cells through inhibition of ATR. Nat Chem Biol 7:428–430. Available from <Go to ISI>://WOS:000292252100008
Huntoon CJ, Flatten KS, Hendrickson AEW, Huehls AM, Sutor SL, Kaufmann SH et al (2013) ATR inhibition broadly sensitizes ovarian cancer cells to chemotherapy independent of BRCA status. Cancer Res 73:3683–3691. Available from <Go to ISI>://WOS:000320380300020
Kastan MB, Zhan QM, Eldeiry WS, Carrier F, Jacks T, Walsh WV et al (1992) A mammalian-cell cycle checkpoint pathway utilizing P53 and Gadd45 Is defective in ataxia-telangiectasia. Cell 71:587–597. Available from <Go to ISI>://WOS:A1992JY67600007
Wengner AM, Siemeister G, Lucking U, Lefranc J, Wortmann L, Lienau P et al (2020) The novel ATR inhibitor BAY 1895344 is efficacious as monotherapy and combined with DNA damage-inducing or repair-compromising therapies in preclinical cancer models. Mol Cancer Ther 19(1):26–38. Available from <Go to ISI>://WOS:000505667900003
Knegtel R, Charrier JD, Durrant S, Davis C, O’Donnell M, Storck P et al (2019) Rational design of 5-(4-(Isopropylsulfonyl)phenyl)-3-(3-(4-((methylamino)methyl)phenyl)isoxazol-5-yl)pyrazin-2-amine (VX-970,M6620): optimization of intra- and intermolecular polar interactions of a new ataxia telangiectasia mutated and Rad3-related (ATR) kinase inhibitor. J Med Chem 62:5547–5561. Available from <Go to ISI>://WOS:000471834500020
Hall AB, Newsome D, Wang Y, Boucher DM, Eustace B, Gu Y et al (2014) Potentiation of tumor responses to DNA damaging therapy by the selective ATR inhibitor VX-970. Oncotarget 5(14):5674–5685
Nagel R, Avelar AT, Aben N, Proost N, Ven M van de, van der Vliet J et al (2019) Inhibition of the replication stress response is a synthetic vulnerability in SCLC that acts synergistically in combination with cisplatin. Mol Cancer Ther 18:762–770. Available from <Go to ISI>://WOS:000462996800004
Kurmasheva RT, Kurmashev D, Reynolds CP, Kang M, Wu J, Houghton PJ et al (2018) Initial testing (stage 1) of M6620 (formerly VX-970), a novel ATR inhibitor, alone and combined with cisplatin and melphalan, by the Pediatric Preclinical Testing Program. Pediatr Blood Cancer 65(2):e26825
Leszczynska KB, Dobrynin G, Leslie RE, Ient J, Boumelha AJ, Senra JM et al (2016) Preclinical testing of an Atr inhibitor demonstrates improved response to standard therapies for esophageal cancer. Radiother Oncol 121(2):232–238
Combes E, Andrade AF, Tosi D, Michaud HA, Coquel F, Garambois V et al (2019) Inhibition of Ataxia-telangiectasia mutated and RAD3-related (ATR) overcomes oxaliplatin resistance and promotes antitumor immunity in colorectal cancer. Cancer Res 79:2933–2946. Available from <Go to ISI>://WOS:000470291600015
Tu XY, Kahila MM, Zhou Q, Yu J, Kalari KR, Wang LW et al (2018) ATR inhibition is a promising radiosensitizing strategy for triple-negative breast cancer. Mol Cancer Ther 17:2462–2472. Available from <Go to ISI>://WOS:000448888000017
Middleton MR, Dean E, Evans TRJ, Shapiro GI, Pollard J, Hendriks BS et al (2021) Phase 1 study of the ATR inhibitor berzosertib (formerly M6620, VX-970) combined with gemcitabine +/− cisplatin in patients with advanced solid tumours. Brit J Cancer 125(4):510–59. Available from <Go to ISI>://WOS:000655068600003
Yazinski SA, Comaills V, Buisson R, Genois MM, Nguyen HD, Ho CK et al (2017) ATR inhibition disrupts rewired homologous recombination and fork protection pathways in PARP inhibitor-resistant BRCA-deficient cancer cells. Gene Dev 31(3):318–332. Available from <Go to ISI>://WOS:00039579610001
Yap TA, O’Carrigan B, Penney MS, Lim JS, Brown JS, Luken MJD et al (2020) Phase I trial of first-in-class ATR inhibitor M6620 (VX-970) as monotherapy or in combination with carboplatin in patients with advanced solid tumors. J Clin Oncol 38(27):3195–+. Available from <Go to ISI>://WOS:000574579100010
Konstantinopoulos PA, Cheng SC, Hendrickson AEW, Penson RT, Schumer ST, Doyle LA et al (2020) Berzosertib plus gemcitabine versus gemcitabine alone in platinum-resistant high-grade serous ovarian cancer: a multicentre, open-label, randomised, phase 2 trial. Lancet Oncology 21:957–9568. Available from <Go to ISI>://WOS:000545328900033
Konstantinopoulos PA, da Costa AABA, Gulhan D, Lee EK, Cheng S-C, Hendrickson AEW et al (2021) A replication stress biomarker is associated with response to gemcitabine versus combined gemcitabine and ATR inhibitor therapy in ovarian cancer. Nat Commun 12(1):5574
Shapiro GI, Wesolowski R, Devoe C, Lord S, Pollard J, Hendriks BS et al (2021) Phase 1 study of the ATR inhibitor berzosertib in combination with cisplatin in patients with advanced solid tumours. Brit J Cancer 125:520–57. Available from <Go to ISI>://WOS:000655068600001
Thomas A, Redon CE, Sciuto L, Padiernos E, Ji JP, Lee MJ et al (2018) Phase I study of ATR Inhibitor M6620 in combination with topotecan in patients with advanced solid tumors. J Clin Oncol 2018;36:1594–+. Available from <Go to ISI>://WOS:000434262900008
Thomas A, Takahashi N, Rajapakse VN, Zhang XH, Sun YL, Ceribelli M et al (2021) Therapeutic targeting of ATR yields durable regressions in small cell lung cancers with high replication stress. Cancer Cell 39:566–+. Available from <Go to ISI>://WOS:000640027300015
Merck_KGaA (2022). Merck KGaA, Darmstadt, Germany, advances development programs in oncology focusing on novel mechanisms and pathways. Cited 27 Dec 2022. Available from https://www.emdgroup.com/en/news/development-projects-in-oncology-03-06-2022.html
Foote KM, Nissink JWM, McGuire T, Turner P, Guichard S, Yates JWT et al (2018) Discovery and Characterization<Go to ISI>://WOS:000451496300005 of AZD6738, a potent inhibitor of ataxia telangiectasia mutated and Rad3 related (ATR) kinase with application as an anticancer agent. J Med Chem 61:9889–9907. Available from
Jones CD, Blades K, Foote KM, Guichard SM, Jewsbury PJ, McGuire T et al (2013) Abstract 2348: Discovery of AZD6738, a potent and selective inhibitor with the potential to test the clinical efficacy of ATR kinase inhibition in cancer patients. Cancer Res 73(8_Supplement):2348–2348
Kwok M, Davies N, Agathanggelou A, Smith E, Oldreive C, Petermann E et al (2016) ATR inhibition induces synthetic lethality and overcomes chemoresistance in TP53- or ATM-defective chronic lymphocytic leukemia cells. Blood 127(5):582–595
Wilson Z, Odedra R, Wallez Y, Wijnhoven PWG, Hughes AM, Gerrard J et al (2022) ATR inhibitor AZD6738 (ceralasertib) exerts antitumor activity as a monotherapy and in combination with chemotherapy and the PARP inhibitor olaparib. Cancer Res 82:1140–1152. Available from <Go to ISI>://WOS:000772155800001
Sundar R, Brown J, Russo AI, Yap TA (2017) Targeting ATR in cancer medicine. Curr Prob Cancer 41(4):302–315. Available from https://www.ncbi.nlm.nih.gov/pubmed/28662958
Dillon M, Guevara J, Mohammed K, Smith SA, Dean E, McLellan L et al (2019) A phase I study of ATR inhibitor, AZD6738, as monotherapy in advanced solid tumours (PATRIOT part A, B). Ann Oncol 30:165–+. Available from <Go to ISI>://WOS:000491295501267
Guichard SM, Brown E, Odedra R, Hughes A, Heathcote D, Barnes J et al (2013) The pre-clinical in vitro and in vivo activity of AZD6738: A potent and selective inhibitor of ATR kinase. Cancer Res 73. Available from <Go to ISI>://WOS:000331220602050
Vendetti FP, Lau A, Schamus S, Conrads TP, O’Connor MJ, Bakkenist CJ (2015) The orally active and bioavailable ATR kinase inhibitor AZD6738 potentiates the anti-tumor effects of cisplatin to resolve ATM-deficient non-small cell lung cancer in vivo. Oncotarget 6(42):44289–44305
Kim H, Min A, Im S, Jang H, Lee KH, Lau A et al (2017) Anti-tumor activity of the ATR inhibitor AZD6738 in HER2 positive breast cancer cells. Int J Cancer 140(1):109–119
Yap TA, Krebs MG, Postel-Vinay S, El-Khouiery A, Soria JC, Lopez J et al (2021) Ceralasertib (AZD6738), an oral ATR kinase inhibitor, in combination with carboplatin in patients with advanced solid tumors: a phase I study. Clin Cancer Res 27:5213–5224. Available from: https://www.ncbi.nlm.nih.gov/pubmed/34301752
Kim ST, Smith SA, Mortimer P, Loembe AB, Cho H, Kim KM et al (2021) Phase I study of ceralasertib (AZD6738), a novel DNA damage repair agent, in combination with weekly paclitaxel in refractory cancer. Clin Cancer Res 27:4700–4709. Available from: https://www.ncbi.nlm.nih.gov/pubmed/33975862
Paula BH de, Basu B, Mander A, Khan J, Bundi P, Goodwin R et al (2021) ATRiUM: a first-in-human dose escalation phase I trial of ceralasertib (AZD6738) and gemcitabine as combination therapy. Cancer Res 81. Available from <Go to ISI>://WOS:000680263501296
Dunlop CR, Wallez Y, Johnson TI, Fernandez SBD, Durant ST, Cadogan EB et al (2020) Complete loss of ATM function augments replication catastrophe induced by ATR inhibition and gemcitabine in pancreatic cancer models. Brit J Cancer. 123:1424–1436. Available from Available from: <Go to ISI>://WOS:000554842400004
Bukhari AB, Lewis CW, Pearce JJ, Luong D, Chan GK, Gamper AM (2019) Inhibiting Wee1 and ATR kinases produces tumor-selective synthetic lethality and suppresses metastasis. J Clin Investig 129:1329–1344. Available from <Go to ISI>://WOS:000460125800037
Jin J, Fang HH, Yang F, Ji WF, Guan N, Sun ZJ et al (2018) Combined Inhibition of ATR and WEE1 as a novel therapeutic strategy in triple-negative breast cancer. Neoplasia 20:478–488. Available from <Go to ISI>://WOS:000430688400007
Krebs MG, Lopez J, El-Khoueiry A, Bang YJ, Postel-Vinay S, Abida W et al (2018) Phase I study of AZD6738, an inhibitor of ataxia telangiectasia Rad3-related (ATR), in combination with olaparib or durvalumab in patients (pts) with advanced solid cancers. Cancer Res 78(13_Supplement):CT026–CT026. Available from <Go to ISI>://WOS:000468818900025
Aggarwal R, Umetsu S, Dhawan M, Grabowsky J, Carnevale J, Howell M et al (2021) Interim results from a phase II study of the ATR inhibitor ceralasertib in ARID1A-deficient and ARID1A-intact advanced solid tumor malignancies. Ann Oncol 32:S583–S583. Available from <Go to ISI>://WOS:000700527700488
Shah PD, Wethington SL, Pagan C, Latif N, Tanyi J, Martin LP et al (2021) Combination ATR and PARP Inhibitor (CAPRI): a phase 2 study of ceralasertib plus olaparib in patients with recurrent, platinum-resistant epithelial ovarian cancer. Gynecol Oncol. 163(2):246–253. Available from <Go to ISI>://WOS:000714728500005
Besse B, Awad M, Forde P, Thomas M, Park K, Goss G et al (2021) HUDSON: an open-label, multi-drug, biomarker-directed, phase II platform study in patients with NSCLC, who progressed on anti-PD(L)1 therapy. J Thorac Oncol 16:S118–S119. Available from <Go to ISI>://WOS:000631349600099
Kwon M, Kim G, Kim R, Kim KT, Kim ST, Smith S et al (2022) Phase II study of ceralasertib (AZD6738) in combination with durvalumab in patients with advanced gastric cancer. J Immunother Cancer 10. Available from <Go to ISI>://WOS:000821480200001
Hernandez M, Besse B, Awad M, Forde P, Thomas M, Park K et al (2021) Immuno-modulatory effects of ceralasertib in combination with durvalumab in NSCLC with progression on anti-PD(L)1 treatment (HUDSON). J Thorac Oncol 16:S350–S350. Available from <Go to ISI>://WOS:000631349600531
Lucking U, Wortmann L, Wengner AM, Lefranc J, Lienau P, Briem H et al (2020) Damage incorporated: discovery of the potent, highly selective, orally available ATR inhibitor BAY 1895344 with favorable pharmacokinetic properties and promising efficacy in monotherapy and in combination treatments in preclinical tumor models. J Med Chem 63:7293–7325. Available from <Go to ISI>://WOS:000550753700043
Szydzik J, Lind DE, Arefin B, Kurhe Y, Umapathy G, Siaw JT et al (2021) ATR inhibition enables complete tumour regression in ALK-driven NB mouse models. Nat Commun 12. Available from <Go to ISI>://WOS:000722322900021
Yap TA, Tan DSP, Terbuch A, Caldwell R, Guo C, Goh BC et al (2021) First-in-human trial of the oral ataxia telangiectasia and RAD3-related (ATR) inhibitor BAY 1895344 in patients with advanced solid tumors. Cancer Discov 11(1):80–91. Available from <Go to ISI>://WOS:000607017700021
Austin WR, Armijo AL, Campbell DO, Singh AS, Hsieh T, Nathanson D et al (2012) Nucleoside salvage pathway kinases regulate hematopoiesis by linking nucleotide metabolism with replication stress. J Exp Med 209:2215–2228. Available from <Go to ISI>://WOS:000311295600008
Jo U, Murai Y, Takebe N, Thomas A, Pommier Y (2021) Precision Oncology with drugs targeting the replication stress, ATR, and schlafen 11. Cancers 13(18):4601
Jo U, Senatorov IS, Zimmermann A, Saha LK, Murai Y, Kim SH et al (2021) Novel and highly potent ATR inhibitor M4344 kills cancer cells with replication stress, and enhances the chemotherapeutic activity of widely used DNA damaging agents. Mol Cancer Ther 20(8):1431–1441
Zenke FT, Zimmermann A, Dahmen H, Elenbaas B, Pollard J, Reaper P et al (2019) Abstract 369: Antitumor activity of M4344, a potent and selective ATR inhibitor, in monotherapy and combination therapy. Cancer Res 79(13_Supplement):369
Jo U, Senatorov IS, Zimmermann A, Saha LK, Murai Y, Kim SH et al (2021) Novel and highly potent ATR inhibitor M4344 kills cancer cells with replication stress, and enhances the chemotherapeutic activity of widely used DNA damaging agents. Mol Cancer Ther 20:1431–1441. Available from <Go to ISI>://WOS:000680862700011
Roulston A, Zimmermann M, Papp R, Skeldon A, Pellerin C, Dumas-Bérube É et al (2021) RP-3500: A novel, potent, and selective atr inhibitor that is effective in preclinical models as a monotherapy and in combination with PARP inhibitors. Mol Cancer Ther 21(2):245–256
Yap T, Lee E, Spigel D, Fontana E, Højgaard M, Lheureux S et al (2021) Abstract CC04-01: first-in-human biomarker-driven phase I TRESR trial of ataxia telangiectasia and Rad3-related inhibitor (ATRi) RP-3500 in patients (pts) with advanced solid tumors harboring synthetic lethal (SL) genomic alterations. Mol Cancer Ther 20(12_Supplement):CC04-01–CC04-01
Zhang Y, Hreiki J, Wilkinson G, Ploeger B. Alternative dosing schedules for therapeutic window optimization for the ataxia telangiectasia and Rad3-related pathway (Atr) inhibitor elimusertib in patients with advanced solid tumors: M&S-based exploration using phase 1 data. Clin Pharmacol Ther 111:S41–S41. Available from <Go to ISI>://WOS:000752207700140
Yap TA, Tolcher AW, Plummer ER, Becker A, Fleuranceau-Morel P, Goddemeier T et al (2021) A first-in-human phase I study of ATR inhibitor M1774 in patients with solid tumors. J Clin Oncol 39:TPS3153–TPS3153. Available from: https://ascopubs.org/doi/abs/10.1200/JCO.2021.39.15_suppl.TPS3153
Patel M, Moore KN, Piscitello D, Majithiya J, Luzarraga MR, Millward H et al (2022) Abstract LB520: a pharmacodynamic platform using liquid biopsy to support dose selection for the ATR inhibitor ART0380 (IACS-030380). Cancer Res 82:LB520–LB520. Available from: https://doi.org/10.1158/1538-7445.AM2022-LB520
McCabe N, Lord CJ, Tutt AN, Martin N, Smith GCM, Ashworth A (2005) BRCA2-deficient CAPAN-1 cells are extremely sensitive to the inhibition of poly (ADP-ribose) polymerase: an issue of potency. Cancer Biol Ther 4(9):934–936
McCabe N, Turner NC, Lord CJ, Kluzek K, Białkowska A, Swift S et al (2006) Deficiency in the repair of DNA damage by homologous recombination and sensitivity to poly(ADP-ribose) polymerase inhibition. Cancer Res 66(16):8109–8115
Farmer H, McCabe N, Lord CJ, Tutt ANJ, Johnson DA, Richardson TB et al (2005) Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 434(7035):917–921
Sachdev E, Tabatabai R, Roy V, Rimel BJ, Mita MM (2019) PARP Inhibition in cancer: an update on clinical development. Target Oncol 14(6):657–679
Topatana W, Juengpanich S, Li S, Cao J, Hu J, Lee J et al (2020) Advances in synthetic lethality for cancer therapy: cellular mechanism and clinical translation. J Hematol Oncol 13(1):118
Brown TJ, Reiss KA (2021) PARP inhibitors in pancreatic cancer. Cancer J 27(6):465–475
Tripathi A, Balakrishna P, Agarwal N (2020) PARP inhibitors in castration-resistant prostate cancer. Cancer Treat Res Commun. 24:100199
Li X, Heyer W-D (2008) Homologous recombination in DNA repair and DNA damage tolerance. Cell Res 18(1):99–113
Haynes B, Murai J, Lee J-M (2018) Restored replication fork stabilization, a mechanism of PARP inhibitor resistance, can be overcome by cell cycle checkpoint inhibition. Cancer Treat Rev 71:1–7
Turner NC, Lord CJ, Iorns E, Brough R, Swift S, Elliott R et al (2008) A synthetic lethal siRNA screen identifying genes mediating sensitivity to a PARP inhibitor. Embo J 27(9):1368–1377
Sanjiv K, Hagenkort A, Calderón-Montaño JM, Koolmeister T, Reaper PM, Mortusewicz O et al (2016) Cancer-specific synthetic lethality between ATR and CHK1 kinase activities. Cell Rep 17(12):3407–3416
Toledo LI, Altmeyer M, Rask M-B, Lukas C, Larsen DH, Povlsen LK et al (2014) ATR prohibits replication catastrophe by preventing global exhaustion of RPA. Cell 156(1–2):374
Kim H, George E, Ragland RL, Rafail S, Zhang R, Krepler C et al (2017) Targeting the ATR/CHK1 axis with PARP inhibition results in tumor regression in BRCA-mutant ovarian cancer models. Clin Cancer Res 23(12):3097–3108
Krebs MG, Lopez J, El-Khoueiry A, Bang Y-J, Postel-Vinay S, Abida W et al (2018) Abstract CT026: Phase I study of AZD6738, an inhibitor of ataxia telangiectasia Rad3-related (ATR), in combination with olaparib or durvalumab in patients (pts) with advanced solid cancers. Cancer Res 78(13_Supplement):CT026–CT026
Wethington SL, Shah PD, Martin LP, Tanyi JL, Latif NA, Morgan MA et al (2021) Combination of PARP and ATR inhibitors (olaparib and ceralasertib) shows clinical activity in acquired PARP inhibitor-resistant recurrent ovarian cancer. J Clin Oncol 39(15_suppl):5516–5516
Shah PD, Wethington SL, Pagan C, Latif N, Tanyi J, Martin LP et al (2021) Combination ATR and PARP Inhibitor (CAPRI): a phase 2 study of ceralasertib plus olaparib in patients with recurrent, platinum-resistant epithelial ovarian cancer. Gynecol Oncol 163(2):246–253
Schoonen PM, Kok YP, Wierenga E, Bakker B, Foijer F, Spierings DCJ et al (2019) Premature mitotic entry induced by ATR inhibition potentiates olaparib inhibition-mediated genomic instability, inflammatory signaling, and cytotoxicity in BRCA2-deficient cancer cells. Mol Oncol. 13:2422–2440. Available from <Go to ISI>://WOS:000491106400001
Parkes EE, Walker SM, Taggart LE, McCabe N, Knight LA, Wilkinson R et al (2017) Activation of STING-dependent innate immune signaling by S-phase-specific DNA damage in breast cancer. J Natl Cancer Inst 109. Available from https://www.ncbi.nlm.nih.gov/pubmed/27707838
Pilie P, Tang Z, Park S, Wu C, Dong ZY, Yap T et al (2019) Inhibitors of Ataxia-Telangiectasia Related (ATR) protein lead to innate immune pathway activation and enhanced response to immune therapy in prostate cancer. Journal for Immunotherapy of Cancer. 7. Available from <Go to ISI>://WOS:000496473200135
Sheng HL, Huang Y, Xiao YZ, Zhu ZR, Shen MY, Zhou PT et al (2020) ATR inhibitor AZD6738 enhances the antitumor activity of radiotherapy and immune checkpoint inhibitors by potentiating the tumor immune microenvironment in hepatocellular carcinoma. J Immunother Cancer 8. Available from <Go to ISI>://WOS:000553971800001
Dillon MT, Bergerhoff KF, Pedersen M, Whittock H, Crespo-Rodriguez E, Patin EC et al (2019) ATR inhibition potentiates the radiation-induced inflammatory tumor microenvironment. Clin Cancer Res 25:3392–3403. Available from <Go to ISI>://WOS:000470293000021
Gasser S, Orsulic S, Brown EJ, Raulet DH (2005) The DNA damage pathway regulates innate immune system ligands of the NKG2D receptor. Nature 436:1186–1190. Available from: https://www.ncbi.nlm.nih.gov/pubmed/15995699
Sato H, Niimi A, Yasuhara T, Permata TBM, Hagiwara Y, Isono M et al (2017) DNA double-strand break repair pathway regulates PD-L1 expression in cancer cells. Nat Commun 8:1751. Available from https://www.ncbi.nlm.nih.gov/pubmed/29170499
Chen CF, Ruiz-Vega R, Vasudeva P, Espitia F, Krasieva TB, de Feraudy S et al (2017) ATR mutations promote the growth of melanoma tumors by modulating the immune microenvironment. Cell Rep 18:2331–2342. Available from: https://www.ncbi.nlm.nih.gov/pubmed/28273450
Lee J, Kim ST, Smith S, Mortimer PG, Loembe B, Hong J et al (2020) Results from a phase I, open-label study of ceralasertib (AZD6738), a novel DNA damage repair agent, in combination with weekly paclitaxel in refractory cancer (NCT02630199). J Clin Oncol 38. Available from <Go to ISI>://WOS:000560368301399
Louie AD, Huntington K, Carlsen L, Zhou L, El-Deiry WS (2021) Integrating molecular biomarker inputs into development and use of clinical cancer therapeutics. Front Pharmacol 12:747194
Ngoi NYL, Westin SN, Yap TA (2022) Targeting the DNA damage response beyond poly(ADP-ribose) polymerase inhibitors: novel agents and rational combinations. Curr Opin Oncol 34(5):559–569
Pilie PG, Gheeya JS, Kyewalabye K, Goswamy RV, Wani KM, Le H, et al. Identifying functional loss of ATM gene in patients with advanced cancer. J Clin Oncol 38(15_suppl):3629–3629
Rafiei S, Fitzpatrick K, Liu D, Cai M-Y, Elmarakeby HA, Park J et al (2020) ATM loss confers greater sensitivity to ATR inhibition than PARP inhibition in prostate cancer. Cancer Res 80(11):2094–2100
Yap TA, Tan DSP, Terbuch A, Caldwell R, Guo C, Goh BC et al (2021) First-in-human trial of the oral ataxia telangiectasia and RAD3-related (ATR) inhibitor BAY 1895344 in patients with advanced solid tumors. Cancer Discov 11(1):80–91
Kwok M, Davies N, Agathanggelou A (2016) ATR inhibition induces synthetic lethality and overcomes chemoresistance in TP53- or ATM-defective chronic lymphocytic leukemia cells (vol 127, p 582, 2016). Blood 127:2647–2647. Available from <Go to ISI>://WOS:000378334400022
Middleton FK, Pollard JR, Curtin NJ (2018) The impact of p53 dysfunction in ATR inhibitor cytotoxicity and chemo- and radiosensitisation. Cancers 10(8):275
Dillon MT, Barker HE, Pedersen M, Hafsi H, Bhide SA, Newbold KL et al (2017) Radiosensitization by the ATR inhibitor AZD6738 through generation of acentric micronuclei. Mol Cancer Ther 16(1):25–34
Das S, Whisenant J, Doyle A, Allegra CJ, Berlin J (2019) A phase II study of M6620 and irinotecan in TP53 mutant gastric and gastroesophageal junction (GEJ) adenocarcinoma patients (pts) [NCT03641313]. J Clin Oncol 37(4_suppl):TPS175–TPS175
Williamson CT, Miller R, Pemberton HN, Jones SE, Campbell J, Konde A et al (2016) ATR inhibitors as a synthetic lethal therapy for tumours deficient in ARID1A. Nat Commun 7(1):13837
Tsai S, Fournier L-A, Chang EY, Wells JP, Minaker SW, Zhu YD et al (2021) ARID1A regulates R-loop associated DNA replication stress. Plos Genet 17(4):e1009238
Aggarwal R, Umetsu S, Dhawan M, Grabowsky J, Carnevale J, Howell M et al (2021) 512O Interim results from a phase II study of the ATR inhibitor ceralasertib in ARID1A-deficient and ARID1A-intact advanced solid tumor malignancies. Ann Oncol 32:S583
Yap TA, O’Carrigan B, Penney MS, Lim JS, Brown JS, Luken MJ de M et al (2020) Phase I trial of first-in-class ATR inhibitor M6620 (VX-970) as monotherapy or in combination with carboplatin in patients with advanced solid tumors. J Clin Oncol 38(27):3195–3204
Krajewska M, Fehrmann RSN, Schoonen PM, Labib S, de Vries EGE, Franke L et al (2015) ATR inhibition preferentially targets homologous recombination-deficient tumor cells. Oncogene 34(26):3474–3481
Toh M, Ngeow J (2021) Homologous recombination deficiency: cancer predispositions and treatment implications. Oncol 26(9):e1526–e1537
Buisson R, Lawrence MS, Benes CH, Zou L (2017) APOBEC3A and APOBEC3B activities render cancer cells susceptible to ATR inhibition. Cancer Res 77(17):4567–4578
Savva C, Souza KD, Ali R, Rakha EA, Green AR, Madhusudan S (2019) Clinicopathological significance of ataxia telangiectasia-mutated (ATM) kinase and ataxia telangiectasia-mutated and Rad3-related (ATR) kinase in MYC overexpressed breast cancers. Breast Cancer Res Tr 175(1):105–115
Kok YP, Llobet SG, Schoonen PM, Everts M, Bhattacharya A, Fehrmann RSN et al (2020) Overexpression of Cyclin E1 or Cdc25A leads to replication stress, mitotic aberrancies, and increased sensitivity to replication checkpoint inhibitors. Oncogenesis 9(10):88
Ngoi N, Lin HY, Dumbrava EE, Fu S, Karp DD, Naing A et al (2022) Baseline predictors of hematological toxicity in patients with advanced cancer treated with ATR inhibitors in phase I/II clinical trials. J Clin Oncol 40(16_suppl):3111–3111
Fernandez-Rozadilla C, Simões AR, Lleonart ME, Carnero A, Carracedo Á (2021) Tumor profiling at the service of cancer therapy. Frontiers Oncol 10:595613
Fountzilas E, Tsimberidou AM, Vo HH, Kurzrock R (2022) Clinical trial design in the era of precision medicine. Genome Med 14(1):101
Acknowledgements
Timothy A. Yap is supported by MD Anderson Cancer Center Support grant (NIH/NCI P30 CA016672), The National Cancer Institute (NIH/NCI R01-CA255074), the US Department of Defense Ovarian Cancer Research Program (OC200482), and the V Foundation Clinical Scholar Program (VC2020-001). Helen M.R. Robinson, Graeme C.M. Smith are employed at Artios Pharma.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2023 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Salguero, C., Valladolid, C., Robinson, H.M.R., Smith, G.C.M., Yap, T.A. (2023). Targeting ATR in Cancer Medicine. In: Yap, T.A., Shapiro, G.I. (eds) Targeting the DNA Damage Response for Cancer Therapy. Cancer Treatment and Research, vol 186. Springer, Cham. https://doi.org/10.1007/978-3-031-30065-3_14
Download citation
DOI: https://doi.org/10.1007/978-3-031-30065-3_14
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-031-30064-6
Online ISBN: 978-3-031-30065-3
eBook Packages: MedicineMedicine (R0)