
Abstract
In diabetes, low-density lipoproteins (LDL) are subjected to multiple enzymatic and non-enzymatic modifications that increase their atherogenicity and induce immunogenicity. Modified forms of LDL were capable of inducing endothelial dysfunction and vascular inflammation through activation of innate and acquired immunity, thus contributing to the development and progression of diabetic complications. The immunogenicity of the modified forms of LDL results in induction of self-antibodies specific to them, and these antibodies react with modified forms of LDL forming circulating immune complexes. Those circulating immune complexes exhibit prominent immune modulatory properties that influence not only inflammation but also endothelial dysfunction and clotting. Compared to freely circulating modified forms of LDL that after eliciting epigenetic cell reprogramming, induce inflammation, foam cell formation, endothelial dysfunction, and thrombi formation, modified LDL immune complexes have a more robust atherogenic, pro-inflammatory, and pro-thrombogenic potential and play a crucial role in the development of complications in diabetes. The possible mechanisms for the development of complications, mainly macrovascular disease in diabetes, are also discussed. Various lipid components of the immune complexes may serve not only as diagnostic but also as essential predictive markers of cardiovascular events, retinopathy and nephropathy. This predictive value has been supported by recent research that has shown that ox-LDL-containing immune complexes are most likely a causal biomarker for the development of macrovascular disease in type 1 diabetes.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Modified low-density lipoprotein
- Modified LDL-immune complexes
- Foam cell formation
- Atherosclerosis
- Endothelial dysfunction
- Thrombi formation
- microRNAs
- Macrovascular disease
- Retinopathy
- Nephropathy
Introduction
Hyperglycemia and hyperlipidemia in diabetes lead to overproduction of reactive oxygen species (ROS). Oxidative stress contributes to modification of lipoproteins which is a critical factor to initiate endothelial dysfunction and activate pathogenic pathways that lead to the development and progression of complications in diabetes [1, 2]. Hyperglycemia plays a key role by inducing mitochondrial overproduction of reactive oxygen species (e.g., superoxide anion, hydrogen peroxide, and others), which, in turn, will lead to a variety of modifications of proteins, enzymes, and other substrates, including the formation of advanced glycation end-products (AGE) and oxidation [1, 3, 4].
Lipoproteins can be modified as a consequence of oxidation and glycation. Endothelial cells, monocytes/macrophages, lymphocytes, and smooth muscle cells (SMC) are all able to enhance the rate of oxidation of low-density lipoprotein (LDL). Reactive oxygen species and sulfur-centered radicals initiate metal ion-dependent lipid peroxidation resulting in the generation of aldehydes that interact with lysine residues in ApoB-100. Myeloperoxidase, a heme enzyme secreted by activated macrophages, is able to catalyze lipid peroxidation independently of free metal ions. Oxidation of arachidonic acid, usually secondary to oxidative stress, prostaglandin synthesis by endothelial cells (EC) and platelet activation, lead to the formation of aldehydes that interact with the lysine residues of ApoB100 causing its aggregation, and the resulting modification is generally referred to as malondialdehyde (MDA)-modified LDL [5].
Modified forms of LDL induce endothelial dysfunction and vascular inflammation. Inflammation derives from modified LDL-induced activation of the innate immune system and from the induction of antibodies against the different LDL modifications that lead to the formation of circulating immune complexes that exhibit strong immunomodulatory properties, leading to a robust atherogenic and pro-inflammatory response. LDL-containing IC serve as a predictive biomarker of macrovascular disease in diabetes.
The Pathogenic Role of Modified Forms of LDL
The pathogenic role of modified LDL in the development and progression of atherosclerosis is well established. It has been investigated from two different angles: the direct pro-atherogenic effect of modified forms of LDL [3, 6] and the consequences of the immune response directed against neo-epitopes resulting from lipoprotein modification [7]. Both types of effects have been extensively characterized in the case of oxidized LDL (oxLDL) and of advanced glycation end products-modified LDL (AGE-LDL).
Modified lipoproteins stimulate the release of pro-inflammatory mediators and can affect epigenetic mechanisms leading to the reprogramming of cells such as endothelial cells and monocytes. For instance, oxLDL induces the transformation of macrophages into foam cells, but that only occurs after an epigenetic reprogramming of monocytes. Exposure of reprogrammed monocytes to oxLDL leads to an enhanced response to TLR 2 and 4 as well as to upregulation of CD36 and SRA [8]. Another way to induce an epigenetic reprogramming of monocytes is via a set of mobile small regulatory elements, the microRNAs (miRNAs), which are small endogenous non-coding RNA molecules that regulate post-transcriptional gene expression. MicroRNAs are able to silence gene expression via binding to complementary miRNA recognition elements (MREs) in the 3′ and 5′ untranslated regions of their target mRNAs. To better assess the role of miRNAs in the development of atherosclerosis and other complications in diabetes, miRNAs that regulate cholesterol homeostasis, endothelial cell homeostasis, and the inflammatory response are being carefully studied, but well-validated knowledge in this field is still not available, although many promising results are starting to emerge [9,10,11,12].
As well as inducing the transformation of macrophages into foam cells, a hallmark of the atherosclerotic process, due to its uptake by macrophages via receptor-mediated pathways [3, 13, 14], oxidized LDL can also present oligopeptides to the cell-mediated immune system, leading to activation of T helper 1 cells (Th1 cells) in the vascular wall. As a consequence of their activation, Th-1 cells release, among others, interferon-γ and TNF that activate macrophages and induce the release of chemokines that attract more T cells to the area. The process becomes self-perpetuating, resulting in a chronic inflammatory reaction [15, 16]. Furthermore, oxidized phospholipids generated during LDL oxidation may also activate inflammatory cells through their interaction with TLR4 [17, 18], and oxLDL containing oxidized phospholipids can mediate the uptake of oxLDL by scavenger receptors and it can also be taken up by oxLDL-IC opsonized after interaction with Fc receptors. The differences observed when macrophages are incubated with copper-oxidized LDL versus highly oxidized MDA-LDL could result from differences in the content of oxidized phospholipids in those two forms of oxidized LDL [19, 20].
In addition, oxLDL has chemotactic effects on monocytes [21], enhancing monocyte adhesion to EC in culture [22, 23] as well as the expression of vascular cell adhesion molecule 1 (VCAM 1) and intercellular adhesion molecule 1 (ICAM 1) by human aortic endothelial cells induced by tumor necrosis factor (TNF) [24] and of ICAM-1 in resting human endothelial vein cells [25], thus contributing to the migration of monocytes into the vessel wall. Also high concentrations of oxLDL are cytotoxic and experimental data suggests that oxLDL can injure vascular cells, both endothelial cells and smooth muscle cells (SMC) [26, 27]. Multiple microRNAs and epigenetic modifications also have been described as influencing endothelial and SMC dysfunction [12, 28, 29]. OxLDL induces enhanced synthesis of growth factors including platelet-derived growth factor-AA (PDGF-AA) and PDGF receptors in SMC, as well as of granulocyte-monocyte colony-stimulating factor, macrophage colony-stimulating factor (M-CSF), and granulocyte colony-stimulating factor in aortic endothelial cells from humans and rabbits [30]. In addition, oxidized LDL may affect fibrinolysis by inhibiting the secretion of tissue plasminogen activator (tPA) by human endothelial cells [31] and stimulating the secretion of plasminogen activator inhibitor (PAI)-1 [31]. Thus, oxLDL is unable to stimulate the endothelium-dependent activation of fibrinolysis and may promote a chronic pro-thrombotic state.
The endothelial dysfunction and chronic inflammation induced by oxLDL are extremely relevant to the development of atherosclerosis and other complications in diabetes. Our group has found a positive association between the levels of inflammatory and endothelial dysfunction biomarkers and diabetic retinopathy [32], nephropathy [33], and subclinical atherosclerosis [34].
These pro-inflammatory effects are the result of the activation of a variety of functional pathways. Oxidized LDL has been shown to activate a variety of cell types expressing CD36 and other scavenger receptors and to contribute to the generation of reactive oxygen species (ROS) [35]. On macrophages, the interaction of oxLDL and CD36 (mediated by oxidized phospholipids) results in activation of the src family members Fyn/Lyn, and of several components of the MAP kinase pathway, including MKKK, MKK, FAK, and MAPK (JNK) [14]. The activation of these kinases and associated proteins, such as Vav, is associated with foam cell formation as well as with unregulated actin polymerization and loss of cell polarity causing a migration defect and the trapping of activated cells in the atheromatous lesions [14]. Recently, it was demonstrated that exposure of monocyte-derived macrophages to cytokines and oxLDL through binding to CD36, oxLDL significantly increases production of pro-thrombotic microparticles expressing tissue factor, via a caspase 3/7 dependent pathway [36]. In platelets, the same signaling events lead to enhanced platelet reactivity and enhanced formation of thrombi [37]. It has also been reported that ligation of CD36 by oxLDL leads to the formation of a toll-like receptor heterodimer (TLR-4–TLR-6) that, in turn, activates MyD88 and nuclear factor kappa B (NFkB), a critical step in inducing the synthesis and release of pro-inflammatory cytokines [38]. The balance of pro- and anti-inflammatory mediators, together with resolvins [39], agents that promote the resolution of inflammation, is responsible for atherosclerotic lesion progression or regression
The advanced glycation end-product modified LDL, AGE-LDL, as well as other AGE-modified proteins have also been shown to have pro-inflammatory properties [40, 41]. AGE-modified proteins will impact endothelial cells eliciting increased permeability and pro-coagulant activity [42] as well as overexpression of VCAM-1 [43]. AGEs also contribute to fibroblast proliferation and T lymphocyte activation, which results in the release of increased amounts of interferon-γ that will activate monocytes and macrophages, inducing in turn the release of pro-inflammatory cytokines and chemokines [42], thus creating the conditions for a chronic inflammatory reaction in the arterial wall. The predominant impact of AGE/RAGE in the pathogenesis of oxidative stress in cardiovascular diseases and diabetes has been extensively discussed [44], and the impact of AGE in the atherosclerotic process associated with diabetes was confirmed in streptomycin-induced diabetic ApoE−/− mice [45]. Administration of soluble forms of AGE receptors (RAGE) resulted in reduction of vascular permeability and reduced the progression of atheromatous lesions [45].
The Adaptive Immune Response Elicited by Modified LDL
The pro-inflammatory properties of modified LDL appear to be considerably enhanced as a consequence of their immunogenicity. The immunogenicity of modified LDL was first reported by Steinbrecher et al. based on the immunization of laboratory animals with modified lipoproteins [46]. Of all the modified forms of LDL, oxLDL has been studied in greatest detail from the immunological point of view. Steinbrecher as well as Palinski et al. characterized its immunogenic epitopes [47, 48]. Furthermore, human autoantibodies to oxLDL were the first to be purified and characterized [49,50,51]. Immune complexes (IC) containing modified LDL have been isolated from the peripheral blood of patients with diabetes, cardiovascular disease, and healthy individuals [52, 53]. Both oxidized LDL and corresponding antibodies have been isolated from atheromatous human tissue [49, 54]. Thus, it seems reasonable to use circulating IC as an indicator of the IC that are deposited in the vessel wall. The formation of LDL-IC in circulation is likely to be inconsequential, but those IC formed in the vessel wall will result in enhanced phagocytosis and increased presentation of peptides derived from modified LDL to T helper cells, which is a critical step in the perpetuation vascular inflammation, as described above.
In several studies, we have consistently found that the predominant isotype of modified LDL antibodies is IgG [50, 51, 55,56,57]. This is a significant finding because IgG antibodies are pro-inflammatory [50, 51, 55,56,57]. As reported by our group, predominance of circulating IgG antibodies with higher avidity over IgM antibodies in isolated oxLDL-IC is associated with parameters indicative of deteriorating renal function in the type 1 diabetes Diabetes Control and Complications Trial/Epidemiology of Interventions and Complications (DCCT/EDIC) cohort [57]. We observed significant positive associations of IgG oxLDL antibody concentration in isolated IC with serum creatinine and the urinary albumin excretion rate, as well as a negative correlation with the estimated glomerular filtration rate. IgM oxLDL antibody concentrations did not show any correlation with those parameters [57]. This study, however, was based on a small group of patients with type 1 diabetes. Later we studied a much larger population of 905 patients with type 2 diabetes [58], and this study shows the predominance of IgG over IgM oxLDL antibodies in isolated immune complexes and also shows that high levels of AGE-LDL as well as of IgG antibodies, but not IgM antibodies, reacting with MDA-LDL lysine epitopes in circulating IC, predict the development of macroalbuminuria in patients with type 2 diabetes. Several groups have reported data suggesting that IgM antibodies to oxidized phospholipids and oxidized LDL have protective effects in relation to the development of atherosclerosis [59,60,61,62,63,64], although whether this protective effect extends to antibodies recognizing that modified peptides seem questionable based on data published by Fredrickson and co-workers [65]. If a predominant IgM response has protective effects against the development of atherosclerosis, it is difficult to see how that information can be translated into the clinical setting.
The Composition of Circulating Modified LDL Immune Complexes and Diabetes Complications
Besides studying the pathogenic role of modified LDL antibodies [57, 58, 66,67,68,69], we developed methodology that allows the measurement of modified forms of LDL and the corresponding antibodies involved in IC formation through the isolation and fractionation of circulating IC [53, 57, 70]. This is an important methodological improvement over the direct assay of modified LDL or their corresponding antibodies in serum or plasma samples, as most modified LDL in the circulation is associated with the corresponding antibodies, and measurements of either component of the circulating complexes are inaccurate due to the mutual saturation of antigen and antibody binding sites [53, 56, 70].
In contrast with the conflicting data generated by studies of modified LDL or antibodies to modified LDL [56, 71], data generated in clinical studies carried out on the DCCT/EDIC cohort with our assay have shown that high levels of oxLDL and AGE-LDL in isolated and fractionated IC are associated with increased risk for developing diabetic nephropathy [72]. Also in the DCCT/EDIC cohort, using coronary artery calcification (CAC) indices and carotid intima-media thickness (IMT) as end-points indicative of cardiovascular disease progression, we also found that increased levels of oxLDL and of AGE-LDL in circulating IC are associated with the development of coronary calcification [73] and with increased levels and progression of carotid IMT [74,75,76]. The levels of MDA-LDL in isolated IC showed a significant but weaker correlation with increased carotid IMT [74,75,76,77]. Recently, our group have demonstrated that the levels of AGE-LDL, oxLDL, and MDA-LDL in circulating IC isolated from plasma collected at entry into the DCCT/EDIC study predicted CVD outcomes in people with type 1 diabetes occurring over a 25-year period, even after adjustment for other risk factors including LDL-C levels [78]. When subsequent measurements of these IC were incorporated over time, adjustments by other risk factors mainly LDL-C attenuated the predictive value of the baseline levels and only oxLDL-IC remained independently associated with the risk of all major adverse cardiac and cerebrovascular events, myocardial infarction (MI), and coronary artery disease (CAD). Our data strongly points to a causal association of modified LDL-IC with the development and progression of atherosclerosis. Supporting this concept are our studies showing that F(ab′)2 fragments of anti-oxidized LDL IgG attenuate vascular inflammation and atherosclerosis in a diabetic LDL receptor deficient mice [79]. Our results in type 1 diabetes differ from those obtained in patients with type 2 diabetes (the Veterans Affairs Diabetes Trial (VADT) cohort), in whom the levels of oxLDL and AGE-LDL in circulating IC are not significantly associated with the occurrence of acute events, but high concentrations of MDA-LDL in IC are strong predictors of acute events, especially myocardial infarction (MI) [80]. In agreement with our data, Holvoet et al. reported in two separate studies a link between high levels of oxLDL and established CAD, and between elevated plasma MDA-LDL levels and plaque instability [81, 82].
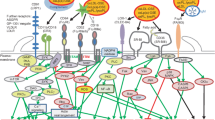
The correlation between MDA-LDL levels and plaque instability is particularly significant because it has been well established that atherosclerotic plaque rupture is a critical event triggering thrombus formation, arterial luminal obstruction, and subsequent acute coronary syndromes [83]. Plaques that are prone to rupture consist of a larger intimal lesion with abundant macrophages and foam cells and a thinned fibrous cap [84]. Necropsy studies have demonstrated that atherosclerosis in people with diabetes is more extensive and accelerated than that in non-diabetic subjects [85]. Furthermore, studies have also shown that atherosclerotic lesions in diabetic patients were more vulnerable as they had larger intimal lesions and increased macrophage infiltration as compared to those in non-diabetic patients [86]. Analysis of gene expression in atherosclerotic plaques showed that when compared to stable plaques, vulnerable plaques have higher expression of matrix metalloproteinases (MMPs) with collagenase activity, which contribute to the thinning of the fibrous cap, causing plaque instability and rupture [87]. Among the metalloproteinases, MMP-9 has been the object of considerable interest in recent years and according to some studies is an independent risk factor for atherothrombotic events [88, 89]. MMP-9 synthesis and release can be induced through TLR-4 stimulation, usually involving bacterial endotoxins [17] but also by minimally modified LDL [90]. The association of circulating MDA-LDL and IC-associated MDA- LDL with plaque instability/acute CV events raises interesting questions such as whether IC containing different modified forms of LDL may lead to distinct gene regulation and cell reprogramming. MDA-LDL-IC seems to lead to plaque instability by inducing macrophage apoptosis and/or increased synthesis of matrix metalloproteinases, such as MMP-9 [91]. OxLDL-IC, in contrast, induce the release of pro-inflammatory cytokines [66] and promote collagen synthesis by smooth muscle cells [92], and therefore are more likely to contribute to atheroma progression without a significant effect on plaque stability (Fig. 13.1).

Diagrammatic representation of the different effects of immune complexes prepared with human copper-oxidized malondialdehyde-modified LDL and the corresponding human antibodies reported by several groups (see text). While both types of immune complexes induce the release of pro-inflammatory cytokines, MDA-LDL-IC are pro-apoptotic while oxLDL-IC are anti-apoptotic and induce the release of proliferation and growth factors by macrophages and smooth muscle cells, and only oxLDL-IC induce collagen synthesis by smooth muscle cells
Considerable interest has been raised by the accumulation of apoptotic macrophages around the necrotic core of vulnerable plaques [91]. A variety of pro-apoptotic insults has been proposed to play a significant role in the evolution of atheromas, including oxidative stress, endoplasmic reticulum (ER) stress, accumulation of non-esterified (free) cholesterol, and effects of pro-inflammatory cytokines released by activated macrophages [91]. Accumulation of free cholesterol in macrophages in combination with signals delivered through scavenger receptors or with interferon-γ, known to be released by activated T lymphocytes in atheromas [16, 93], leads to serine phosphorylation of STAT-1 which is a critical element in the induction of apoptosis secondary to ER stress [94]. The apoptotic macrophages in atheromas are ingested by functional macrophages (efferocytosis). Efferocytosis in early lesions seems to result in suppression of inflammation, while in advanced lesions is associated with enhanced inflammation [91]. This evolution appears to be a result of defective efferocytosis, allowing the apoptotic cells to undergo necrosis, resulting in the accumulation of cell fragments that promote inflammation and plaque instability [91].
Pathogenic Mechanisms of Modified LDL-IC
We have published extensive data proving that oxLDL-IC are more potent activators of human macrophages than oxLDL [66, 67, 95, 96]. The uptake of IC prepared with native or copper-oxidized LDL by human monocyte-derived macrophages is primarily mediated by Fcγ receptors, primarily FcγRI [97,98,99], and it has been shown that the binding of oxLDL antibody blocks the interaction of oxLDL with CD36 [100], so scavenger receptors are not involved in the process. The dependency of the vascular inflammatory process on the activation of phagocytic cells via Fcγ receptors has been demonstrated in double-knockout (DKO) mice generated by crossing apolipoprotein E-deficient mice [apoE(−/−)] with FcγR γ-chain-deficient mice [gamma(−/−)] [101]. The progression of atherosclerosis in the DKO mice is significantly reduced in comparison with apoE(−/−) mice. For MDA-LDL-IC and AGE- LDL-IC, FcγRI is also involved, but possible involvement of scavenger receptors or receptors for AGE-modified proteins has not been excluded. One fundamental property of LDL-IC is their ability to deliver large concentrations of free and esterified cholesterol to macrophages [67, 97, 102]. The intracellular accumulation of free cholesterol is a known inducer of ER stress, which is believed to be the prime stimulus for the chain of events that results in modification of LDL and atheroma formation. However, experimental studies have shown that ER stress usually protects against apoptosis [91]. In fact, both oxLDL at concentrations not exceeding 75 μg/mL and oxLDL-IC prevent macrophage apoptosis [99, 103]. Whether the anti-apoptotic effect of oxLDL is a consequence of the induction of ER stress is not clear, because in addition to the enhanced generation of reactive oxygen and nitrogen species [104], several other mechanisms seem to be involved, including the release of M-CSF mediated by the activation of a PI3K-dependent pathway, upregulation of the anti-apoptotic Bcl-XL gene by NFkB activation, activation of sphingosine kinase, which causes the levels of anti-apoptotic sphingosine-1-phosphate to increase, and inhibition of acid sphingomyelinase, which prevents pro-apoptotic ceramide generation [103, 105]. The anti-apoptotic effect is more pronounced with oxLDL-IC [99, 106] and is not unique to oxLDL-IC, because it has also been reproduced with KLH-anti-KLH IC [99]. However, there are significant differences between oxLDL-IC and other IgG-containing IC. Only oxLDL-IC can induce foam cell formation, and the magnitude of the pro-inflammatory response induced in human macrophages is greater with oxLDL-IC than with KLH-IC, for example [66].
While oxLDL cell signaling is mediated by scavenger receptors, oxLDL-IC deliver activating signals via Fcγ receptors. The cross-linking of Fcγ receptors by IC induces phosphorylation of immunoreceptor tyrosine-based activation motifs (ITAMs) by kinases of the Src family, leading to activation of the Syk pathway [107, 108]. Activation of Syk triggers the mitogen-activated protein kinase (MAPK) signaling cascade, which includes ERK1/2, p38 MAPK, and c-Jun N-terminal kinase (JNK). MAPK activation is also essential for Fc-mediated activation of NFκB [109]. Following the general rule, oxLDL-IC primarily engage FcγRI and induce the activation of the MAPK pathway [110], which is responsible for the expression of pro-inflammatory gene products. In addition, cross-linking of FcγRs by oxLDL-IC activates PI3K and c-Akt [99]. Activated c-Akt promotes cell survival by at least four different mechanisms: (1) phosphorylating the Bad component of the Bad/Bcl-XL complex which results in its dissociation and cell survival, (2) caspase 9 inactivation, (3) regulation of the expression of transcription factors, and (4) activating IKK kinases which phosphorylate IκB and, as a consequence, release the active form of NFkB, which induces the expression of genes favoring cell survival [111] (Fig. 13.2). The repertoire of oxLDL-IC-induced pro-survival genes is much wider than that induced by oxLDL alone [96]. Also, oxLDL-IC induce HSP70B expression in macrophages. This protein binds to the internalized lipid moiety of oxLDL-IC and prevents its degradation, while at the same time inducing sphingokinase-1 [104, 112].

Diagrammatic representation of the activation pathways triggered by oxLDL-IC through the engagement of FcγRI. Two main pathways are activated, the MAPK pathway which is important for the activation of cell proliferation and cytokine synthesis, and the Akt pathway, which also contributes to the induction of cell proliferation and cytokine synthesis through NFκB activation and also promotes cell survival through the dissociation of the Bad/Bcl-XL complex, blocking the pathway that leads to the activation of caspase 9
In contrast to oxLDL, there is no published information concerning pathways of cell activation triggered by MDA-LDL or MDA-LDL-IC. The association of MDA-LDL with acute coronary syndromes [5, 82] and the association of high levels of MDA-LDL in the circulating IC isolated from patients with type 2 diabetes who had acute cardiovascular disease (CVD) events, mainly MI [80], strongly suggest that MDA-LDL and MDA-LDL-IC have pro-apoptotic activity. The different effects of cellular uptake of oxLDL-IC and MDA-LDL-IC (Fig. 13.1) could be a result of structural differences between MDA-LDL and oxLDL. The extent of MDA-lysine modification is much greater in laboratory produced MDA-LDL than in copper-oxidized LDL [70]. This difference results in the generation of epitopes unique to MDA- LDL, and the fact that MDA-LDL antibodies obtained by immunization of rabbits with laboratory-prepared MDA-LDL react with LDL isolated from IC proves that MDA-LDL with identical epitopes and, therefore, with similar structural characteristics, is generated in vivo. Also, while copper oxidation predominantly results in ApoB fragmentation, MDA modification is associated with ApoB aggregation [113]. Obviously, these differences in ApoB could determine different biological properties of the two forms of modified LDL. For example, it has been reported that the processing of heavily oxidized and aggregated LDL by macrophages is defective [114]. Thus, the uptake of MDA-LDL-IC could result in a variety of conditions that could promote apoptosis, including (1) the release of much higher concentrations of free cholesterol in the cell, (2) intracellular accumulation of aggregated LDL, (3) cytoplasmic release of lipoprotein degradation products and oxidized phosphatidylcholine, which could be transported to the extracellular compartment and then react with scavenger receptors and/or TLRs, delivering signals that would favor the activation of pro-apoptotic pathways.
There is considerable interest in identifying biomarkers indicative of plaque instability. A variety of proteins and enzymes have been proposed as candidates, as reviewed recently by Koenig [115]. Besides MMPs, reactive proteins (CRP), cytokines (IL-6, IL-18), enzymes (glutathione peroxidase, lipoprotein-associated phospholipase A-2 (Lp-PLA2)), myeloperoxidase, chemotactic proteins (monocyte chemotactic protein-1), and modified lipoproteins have been proposed as indicators of plaque instability [5, 81, 82, 89, 116, 117]. Our data suggest that modified forms of LDL can also be useful biomarkers for CVD [72, 73, 75] and plaque vulnerability risk [80].
In conclusion, modified forms of LDL play a key role as a persistent insult leading to chronic vascular inflammation and cell reprogramming. The pro-inflammatory effects of modified LDL are significantly enhanced as a consequence of the formation of immune complexes. In general, modified LDL-IC have pro-inflammatory properties, but both clinical and experimental data suggest that there are differences in the consequences of cellular uptake of IC depending on the predominant type of LDL modification. Furthermore, due to cell reprogramming, secondary to epigenetic factors and microRNAs, a major feature of diabetes since hyperglycemia is highly involved in the process, the expression of receptors involved in innate immunity responses and scavenger receptors, as well as expression of pro-thrombotic and pro-inflammatory mediators, may be considerably affected. These novel findings open a variety of basic and clinical research perspectives, ranging from the study of epigenetic and microRNAs cell reprogramming, the investigation of the molecular mechanisms that are responsible for the different cellular effects of different LDL modifications, to the definition of specific LDL modifications as risk factors able to discriminate between people with different types or degrees of diabetes-associated complications.
References
Giacco F, Brownlee M. Oxidative stress and diabetic complications. Circ Res. 2010;107:1058–70.
Balaban RS, Nemoto S, Finkel T. Mitochondria, oxidants, and aging. Cell. 2005;120:483–95.
Miller YI, Choi SH, Fang L, Tsimikas S. Lipoprotein modification and macrophage uptake: role of pathologic cholesterol transport in atherogenesis. Subcell Biochem. 2010;51:229–51.
Baynes JW, Thorpe SR. Glycoxidation and lipoxidation in atherogenesis. Free Radic Biol Med. 2000;28:1708–16.
Holvoet P. Endothelial dysfunction, oxidation of low-density lipoprotein, and cardiovascular disease. Ther Apher. 1999;3:287–93.
Lopes-Virella MF, Virella G. The role of immune and inflammatory processes in the development of macrovascular disease in diabetes. Front Biosci. 2003;8:s750–68.
Lopes-Virella MF, Virella G. Clinical significance of the humoral immune response to modified LDL. Clin Immunol. 2010;134:55–65.
Rios FJ, Koga MM, Ferracini M, Jancar S. Co-stimulation of PAFR and CD36 is required for oxLDL-induced human macrophages activation. PLoS One. 2012;7(5):e36632.
Prattichizzo F, Giuliani A, Ceka A, Rippo MR, Bonfigli AR, Testa R, Procopio AD, Olivieri F. Epigenetic mechanisms of endothelial dysfunction in type 2 diabetes. Clin Epigenet. 2015;7(1):56. https://doi.org/10.1186/s13148-015-0090-4.
Hergenreider E, Heydt S, Tréguer K, Boettger T, Horrevoets AJ, Zeiher AM, Scheffer MP, Frangakis AS, Yin X, Mayr M, Braun T, Urbich C, Boon RA, Dimmeler S. Atheroprotective communication between endothelial cells and smooth muscle cells through miRNAs. Nat Cell Biol. 2012;14:249–56.
La Sala L, Mrakic-Sposta S, Micheloni S, Prattichizzo F, Ceriello A. Glucose-sensing microRNA-21 disrupts ROS homeostasis and impairs antioxidant responses in cellular glucose variability. Cardiovasc Diabetol. 2018;17(1):105.
Brennan E, Wang B, McClelland A, Mohan M, Marai M, Beuscart O, Derouiche S, Gray S, Pickering R, Tikellis C, de Gaetano M, Barry M, Belton O, Ali-Shah ST, Guiry P, Jandeleit-Dahm KAM, Cooper ME, Godson C, Kantharidis P. Protective effect of let-7 miRNA family in regulating inflammation in diabetes-associated atherosclerosis. Diabetes. 2017;66(8):2266–77.
Parthasarathy S, Printz DJ, Boyd D, Joy L, Steinberg D. Macrophage oxidation of low-density lipoprotein generates a modified form recognized by the scavenger receptor. Arteriosclerosis. 1986;6:505–10.
Silverstein RL, Li W, Park YM, Rahaman SO. Mechanisms of cell signaling by the scavenger receptor CD36: implications in atherosclerosis and thrombosis. Trans Am Clin Climatol Assoc. 2010;121:206–20.
Andersson J, Libby P, Hansson GK. Adaptive immunity and atherosclerosis. Clin Immunol. 2010;134:33–46.
De Boer OJ, van der Wal AC, Verhagen CE, Becker AE. Cytokine secretion profiles of cloned T cells from human aortic atherosclerotic plaques. J Pathol. 1999;188:174–9.
Lundberg AM, Hansson GK. Innate immune signals in atherosclerosis. Clin Immunol. 2010;134(1):5–24.
Crispin JC, Virella G. Chapter 11: Cell mediated immunity. In: Virella G, editor. Medical immunology. 7th ed. CRC Press; 2020. p. 141–58.
Kiechl S, Willeit J, Mayr M, Viehweider B, Oberhollenzer M, Kronenberg F, Wiederman C, Oberthaker S, Xu Q, Wiztum JL, Tsimikas S. Oxidized phospholipids, lipoprotein(a), lipoprotein-associated phospholipase activity, and 10-year cardiovascular outcomes: prospective results from the Bruneck study. Arterioscler Thromb Vasc Biol. 2007;27:1788–95.
Virella G, Wilson K, Elkes J, Hammad SM, Rajab HA, Li Y, Chassereau C, Huang Y, Lopes-Virella M. Immune complexes containing malondialdehyde (MDA) LDL induce apoptosis in human macrophages. Clin Immunol. 2018;187:1–9.
Quinn MT, Parthasarathy S, Fong LG, Steinberg D. Oxidatively modified low density lipoproteins: a potential role in recruitment and retention of monocyte/macrophages during atherogenesis. Proc Natl Acad Sci U S A. 1987;84:2995–8.
Berliner JA, Territo MC, Sevanian A, Raimin S, Kim JA, Bamshad B, Ester-son M, Fogelman AM. Minimally modified low density lipoprotein stimulates monocyte endothelial interactions. J Clin Invest. 1990;85:1260–6.
Kume N, Cybulsky MI, Gimbrone MAJ. Lysophosphatidyl-choline, a component of atherogenic lipoproteins, induces mononuclear leukocyte adhesion molecules in cultured human and rabbit arterial endothelial cells. J Clin Invest. 1992;90:1138–44.
Kahn BV, Parthasarathy SS, Alexander RW, Medford RM. Modified LDL and its constituents augment cytokine-activated vascular cell adhesion molecule-1 gene expression in human vascular endothelial cells. J Clin Invest. 1995;95:1262–70.
Takei A, Huang Y, Lopes-Virella MF. Expression of adhesion molecules by human endothelial cells exposed to oxidized low density lipoprotein. Influences of degree of oxidation and location of oxidized LDL. Atherosclerosis. 2001;154:79–86.
Hessler JR, Morel DW, Lewis LJ, Chisolm GM. Lipoprotein oxidation and lipoprotein-induced cytotoxicity. Arteriosclerosis. 1983;3:215–22.
Henriksen T, Evensen SA, Carlander B. Injury to human endothelial cells in culture induced by LDL. Scand J Clin Lab Invest. 1979;39:361–4.
Schober A, Nazari-Jahantigh M, Weber C. MicroRNA mediated mechanisms of the cellular stress response in atherosclerosis. Nat Rev Cardiol. 2015;12:361–74.
Yan MS, Marsden PA. Epigenetics in the vascular endothelium: looking from a different perspective in the epigenomics era. Arterioscler Thromb Vasc Biol. 2015;35:2297–306.
Rajavashisth TB, Andalibi A, Territo MC. Induction of endothelial cell expression of granulocyte and macrophage colony-stimulating factors by modified LDL. Nature. 1990;344:254–7.
Kugiyama K, Sakamoto T, Musumi I, Sugiyama S, Ohgushi M, Ogawa H, Horiguchi M, Yasue H. Transferrable lipids in oxidized LDL stimulate PAI-1 and inhibit tPA release from endothelial cells. Circ Res. 1993;73:335–43.
Rajab HA, Baker NL, Hunt KJ, Klein R, Cleary PA, Lachin J, Virella G, Lopes-Virella MF, DCCT/EDIC Group of Investigators. The predictive role of markers of Inflammation and endothelial dysfunction on the course of diabetic retinopathy in type 1 diabetes. J Diabetes Complicat. 2015;29:108–14.
Lopes-Virella MF, Baker NL, Hunt KJ, Cleary PA, Klein RL, Virella G, DCCT/EDIC Group of Investigators. Baseline markers of inflammation are associated with progression to macroalbuminuria in type 1 diabetic subjects. Diabetes Care. 2013;36:1–7.
Hunt KJ, Baker NL, Cleary PA, Klein R, Virella G, Lopes-Virella MF, DCCT/EDIC Group of Investigators. Longitudinal association between endothelial dysfunction, inflammation, and clotting biomarkers with subclinical atherosclerosis in type 1 diabetes: an evaluation of the DCCT/EDIC cohort. Diabetes Care. 2015. pii: dc142877.
Li W, Febbraio M, Reddy SP, Yu DY, Yamamoto M, Silverstein RL. CD36 participates in a signaling pathway that regulates ROS formation in murine VSMCs. J Clin Invest. 2010;120:3996–4006.
Marchini JF, Manica A, Crestani P, Dutzmann J, Folco EJ, Weber H, Libby P, Croce K. Oxidized low-density lipoprotein induces macrophage production of prothrombotic microparticles. J Am Heart Assoc. 2020;9(15):e015878.
Silverstein RL. Type 2 scavenger receptor CD36 in platelet activation: the role of hyperlipemia and oxidative stress. Clin Lipidol. 2009;4:767.
Stewart CR, Stuart LM, Wilkinson K, van Gils JM, Deng J, Halle A, Rayner KJ, Boyer L, Zhong R, Frazier WA, Lacy-Hulbert A, El Khoury J, Golenbock DT, Moore KJ. CD36 ligands promote sterile inflammation through assembly of a Toll-like receptor 4 and 6 heterodimer. Nat Immunol. 2010;1:155–61.
Hasturk H, Abdallah R, Kantarci A, Nguyen D, Giordano N, Hamilton J, Van Dyke TE. Resolvin E1 (RvE1) attenuates atherosclerotic plaque formation in diet and inflammation-induced atherogenesis. Arterioscler Thromb Vasc Biol. 2015;35:1123–33.
Vlassara H, Cai W, Crandall J, Goldberg T, Oberstein R, Dardaine V, Peppa M, Rayfield EJ. Inflammatory mediators are induced by dietary glycotoxins, a major risk factor for diabetic angiopathy. Proc Natl Acad Sci U S A. 2002;99:15596–601.
Wendt T, Bucciarelli L, Qu W, Lu Y, Yan SF, Stern DM, Schmidt AM. Receptor for advanced glycation endproducts (RAGE) and vascular inflammation: insights into the pathogenesis of macrovascular complications in diabetes. Curr Atheroscler Rep. 2002;4:228–37.
Vlassara H, Bucala R, Striker L. Pathogenic effects of advanced glycosylation: biochemical, biologic, and clinical implications for diabetes and aging. Lab Investig. 1994;70:138–51.
Schmidt AM, Hori O, Chen JX, Li JF, Crandall J, Zhang J, Cao R, Yan SD, Brett J, Stern D. Advanced glycation endproducts interacting with their endothelial receptor induce expression of vascular cell adhesion molecule-1 (VCAM-1) in cultured human endothelial cells and in mice. A potential mechanism for the accelerated vasculopathy of diabetes. J Clin Invest. 1995;96:1395–403.
Daffu G, del Pozo CH, O’Shea KM, Ananthakrishnan R, Ramasamy R, Schmidt AM. Radical roles for RAGE in the pathogenesis of oxidative stress in cardiovascular diseases and beyond. Int J Mol Sci. 2013;14(10):19891–910.
Bucciarelli LG, Wendt T, Qu W, Lu Y, Lalla E, Rong LL, Goova MT, Moser B, Kislinger T, Lee DC, Kashyap Y, Stern DM, Schmidt AM. RAGE blockade stabilizes established atherosclerosis in diabetic apolipoprotein E-null mice. Circulation. 2002;106(22):2827–35.
Steinbrecher UP, Fisher M, Witztum JL, Curtiss LK. Immunogenicity of homologous low density lipoprotein after methylation, ethylation, acetylation, or carbamylation: generation of antibodies specific for derivatized lysine. J Lipid Res. 1984;25:1109–16.
Steinbrecher UP. Oxidation of human low density lipoprotein results in derivatization of lysine residues of apolipoprotein B by lipid peroxide decomposition products. J Biol Chem. 1987;262:3603–8.
Palinski W, Yla-Herttuala S, Rosenfeld ME, Butler SW, Socher SA, Parthasarathy S, Curtiss LK, Witztum JL. Antisera and monoclonal antibodies specific for epitopes generated during oxidative modification of low density lipoprotein. Arteriosclerosis. 1990;10:325–35.
Yla-Herttuala S, Palinski W, Butler S, Picard S, Steinberg D, Witztum JL. Rabbit and human atherosclerotic lesions contain IgG that recognizes epitopes of oxidized LDL. Arterioscler Thromb. 1994;14:32–40.
Mironova M, Virella G, Lopes-Virella MF. Isolation and characterization of human antioxidized LDL autoantibodies. Arterioscler Thromb Vasc Biol. 1996;16:222–9.
Virella G, Koskinen S, Krings G, Onorato JM, Thorpe SR, Lopes-Virella M. Immunochemical characterization of purified human oxidized low-density lipoprotein antibodies. Clin Immunol. 2000;95:135–44.
Mironova MA, Klein RL, Virella GT, Lopes-Virella MF. Anti-modified LDL antibodies, LDL-containing immune complexes, and susceptibility of LDL to in vitro oxidation in patients with type 2 diabetes. Diabetes. 2000;49:1033–41.
Virella G, Thorpe S, Alderson NL, Derrick MB, Chassereau C, Rhett JM, Lopes-Virella MF. Definition of the immunogenic forms of modified human LDL recognized by human autoantibodies and by rabbit hyperimmune antibodies. J Lipid Res. 2004;45:1859–67.
Yla-Herttuala S. Macrophages and oxidized low density lipoproteins in the pathogenesis of atherosclerosis. Ann Med. 1991;2:561–7.
Virella G, Thorpe SR, Alderson NL, Stephan EM, Atchley DH, Wagner F, Lopes-Virella MF, Group DER. Autoimmune response to advanced glycosylation end-products of human low density lipoprotein. J Lipid Res. 2003;443:487–93.
Virella G, Lopes-Virella MF. Lipoprotein autoantibodies: measurement and significance. Clin Diagn Lab Immunol. 2003;10:499–505.
Virella G, Carter RE, Saad A, Crosswell EG, Game BA, Lopes-Virella MF. Distribution of IgM and IgG antibodies to oxidized LDL in immune complexes isolated from patients with type 1 diabetes and its relationship with nephropathy. Clin Immunol. 2008;127:394–400.
Lopes-Virella MF, Hunt KJ, Baker NL, Virella G, VADT Group of Investigators. High levels of AGE-LDL and of IgG antibodies reacting with MDA-lysine epitopes expressed by oxLDL and MDA-LDL in circulating immune complexes predict macroalbuminuria in patients with type 2 diabetes. J Diabetes Complicat. 2016;30:693–6.
Tsimikas S, Brilakis ES, Lennon RJ, Miller ER, Witztum JL, McConnell JP, Kornman KS, Berger PB. Relationship of IgG and IgM autoantibodies to oxidized low density lipoprotein with coronary artery disease and cardiovascular events. J Lipid Res. 2007;48:425–33.
Frostegard J. Low level natural antibodies against phosphorylcholine: a novel risk marker and potential mechanism in atherosclerosis and cardiovascular disease. Clin Immunol. 2010;134:47–54.
Yamashita T, Freigang S, Eberle C, Pattison J, Gupta S, Napoli C, Palinski W. Maternal immunization programs postnatal immune responses and reduces atherosclerosis in offspring. Circ Res. 2006;99:e51–64.
Su J, Georgiades A, Wu R, Thulin T, de Faire U, Frostegard J. Antibodies of IgM subclass to phosphorylcholine and oxidized LDL are protective factors for atherosclerosis in patients with hypertension. Atherosclerosis. 2006;188:160–6.
Faria-Neto JR, Chyu KY, Li X, Dimayuga PC, Ferreira C, Yano J, Cercek B, Shah PK. Passive immunization with monoclonal IgM antibodies against phosphorylcholine reduces accelerated vein graft atherosclerosis in apolipoprotein E-null mice. Atherosclerosis. 2006;189:83–90.
Karvonen J, Paivansalo M, Kesaniemi YA, Horkko S. Immunoglobulin M type of autoantibodies to oxidized low-density lipoprotein has an inverse relation to carotid artery atherosclerosis. Circulation. 2003;108(17):2107–12.
Fredrikson GN, Hedblad B, Berglund G, Alm R, Nilsson JA, Schiopu A, Shah PK, Nilsson J. Association between IgM against an aldehyde-modified peptide in apolipoprotein B-100 and progression of carotid disease. Stroke. 2007;38:1495–500.
Saad AF, Virella G, Chassereau C, Boackle RJ, Lopes-Virella MF. OxLDL immune complexes activate complement and induce cytokine production by MonoMac 6 cells and human macrophages. J Lipid Res. 2006;47:1975–83.
Virella G, Atchley DH, Koskinen S, Zheng D, Lopes- VM. Pro-atherogenic and pro-inflammatory properties of immune complexes prepared with purified human oxLDL antibodies and human oxLDL. Clin Immunol. 2002;105:81–92.
Fu D, Yu JY, Wu M, Du M, Chen Y, Abdel-Samie SA, Li Y, Chen J, Boulton ME, Ma JX, Lopes-Virella MF, Virella G, Lyons TJ. Immune complex formation in human diabetic retina enhances toxicity of oxidized LDL towards retinal capillary pericytes. J Lipid Res. 2014;55:860–9. [Epub ahead of print].
Virella G, Wilson K, Hammad SM, Rajab HA, Li Y, Chassereau C, Huang Y, Lopes-Virella M. Immune complexes containing malondialdehyde (MDA) LDL induce apoptosis in human macrophages. Clin Immunol. 2018;187:1–9. https://doi.org/10.1016/j.clim.2017.06.010.
Virella G, Derrick MB, Pate V, Chassereau C, Thorpe SR, Lopes-Virella MF. Development of capture assays for different modifications of human low-density lipoprotein. Clin Diagn Lab Immunol. 2005;12:68–75.
Lopes-Virella MF, Virella G, Orchard TJ, Koskinen S, Evans RW, Becker DJ, Forrest KY. Antibodies to oxidized LDL and LDL-containing immune complexes as risk factors for coronary artery disease in diabetes mellitus. Clin Immunol. 1999;90:165–72.
Lopes-Virella MF, Carter RE, Baker NL, Lachin J, Virella G, Group DER. High levels of oxidized LDL in circulating immune complexes are associated with increased odds of developing abnormal albuminuria in Type 1 diabetes. Nephrol Dial Transplant. https://doi.org/10.1093/ndt/gfr454. Accessed 19 Aug 2011 [Epub ahead of print].
Lopes-Virella MF, Baker NL, Hunt KJ, Lachin J, Nathan D, Virella G. Oxidized LDL immune complexes and coronary artery calcification in type 1 diabetes. Atherosclerosis. 2011;214:462–7.
Lopes-Virella MF, McHenry MB, Lipsitz S, Yim E, Wilson PF, Lackland DT, Lyons T, Jenkins AJ, Virella G. Immune complexes containing modified lipoproteins are related to the progression of internal carotid intima-media thickness in patients with type 1 diabetes. Atherosclerosis. 2007;190:359–69.
Lopes-Virella MF, Hunt KJ, Baker NL, Lachin J, Nathan DM, Virella G. The levels of oxidized LDL and AGE-LDL in circulating immune complexes are strongly associated with increased levels of carotid intima-media thickness and its progression in type 1 diabetes. Diabetes. 2011;60:582–9.
Hunt KJ, Baker N, Cleary P, Backlund JY, Lyons T, Jenkins A, Virella G, Lopes-Virella MF, DCCT/EDIC Research Group. Oxidized LDL and AGE-LDL in circulating immune complexes strongly predict progression of carotid artery IMT in type 1 diabetes. Atherosclerosis. 2013;231:315–22.
Lopes-Virella MF, Virella G. Modified LDL immune complexes and cardiovascular disease. Curr Med Chem. 2018; https://doi.org/10.2174/0929867325666180524114429.
Lopes-Virella MF, Bebu I, Hunt KJ, Virella G, Baker NL, Braffet B, Gao X, Lachin JM, DCCT/EDIC Research Group. Immune complexes and the risk of CVD in type 1 diabetes. Diabetes. 2019;68:1853–60.
Li Y, Lu Z, Huang Y, Lopes-Virella MF, Virella G. F(ab′)2 fragments of anti-oxidized LDL IgG attenuate vascular inflammation and atherogenesis in diabetic LDL receptor-deficient mice. Clin Immunol. 2016;173:50–6.
Lopes-Virella MF, Hunt KJ, Baker NL, Moritz T, Virella G, Investigators TVGo. Levels of MDA—LDL in circulating immune complexes predict myocardial infarction in the VADT study in the veterans affairs diabetes trial (VADT). ATVB. 2012;224:526–31.
Holvoet P, Perez G, Zhao Z, Brouwers E, Bernar H, Collen D. Malondialdehyde-modified low density lipoproteins in patients with atherosclerotic disease. J Clin Invest. 1995;95:2611–9.
Holvoet P, Vanhaecke J, Janssens S, Van de Werf F, Collen D. Oxidized LDL and malondialdehyde-modified LDL in patients with acute coronary syndromes and stable coronary artery disease. Circulation. 1998;98:1487–94.
Libby P, Theroux P. Pathophysiology of coronary artery disease. Circulation. 2005;111:3481–8.
Shah PK. Pathophysiology of coronary thrombosis: role of plaque rupture and plaque erosion. Prog Cardiovasc Dis. 2002;44:357–68.
Jarrett RJ. Atherosclerosis, diabetes and obesity. Proc Nutr Soc. 1981;40:209–12.
Moreno PR, Murcia AM, Palacios IF, Leon MN, Bernardi VH, Fuster V, Fallon JT. Coronary composition and macrophage infiltration in atherectomy specimens from patients with diabetes mellitus. Circulation. 2000;102:2180–4.
Galis ZS, Sukhova GK, Lark MW, Libby P. Increased expression of matrix metalloproteinases and matrix degrading activity in vulnerable regions of human atherosclerotic plaques. J Clin Invest. 1994;94:2493–503.
Blankenberg S, Rupprecht HJ, Poirier O, Bickel C, Smieja M, Hafner G, Meyer J, Cambien F, Tiret L. Plasma concentrations and genetic variation of matrix metalloproteinase 9 and prognosis of patients with cardiovascular disease. Circulation. 2003;107:1579–85.
Loftus IM, Naylor AR, Bell PR, Thompson MM. Plasma MMP-9—a marker of carotid plaque instability. Eur J Vasc Endovasc Surg. 2001;21:17–21.
Choi SH, Harkewicz R, Lee JH, Boullier A, Almazan F, Li AC, Witztum JL, Bae YS, Miller YI. Lipoprotein accumulation in macrophages via toll-like receptor-4-dependent fluid phase uptake. Circ Res. 2009;104:1355–63.
Seimon T, Tabas I. Mechanisms and consequences of macrophage apoptosis in atherosclerosis. J Lipid Res. 2009;50(Suppl):S382–7.
Abdelsamie SA, Li Y, Huang Y, Lee M-H, Klein RL, Virella G, Lopes-Virella MF. Oxidized LDL immune complexes stimulate collagen IV production in mesangial cells via Fc gamma receptor I. Clin Immunol. 2011;139:258–66.
de Boer OJ, van der Wal AC, Houtkamp MA, Ossewaarde JM, Teeling P, Becker AE. Unstable atherosclerotic plaques contain T-cells that respond to Chlamydia pneumoniae. Cardiovasc Res. 2000;48:402–8.
Lim WS, Timmins JM, Seimon TA, Sadler A, Kolodgie FD, Virmani R, Tabas I. Signal transducer and activator of transcription-1 is critical for apoptosis in macrophages subjected to endoplasmic reticulum stress in vitro and in advanced atherosclerotic lesions in vivo. Circulation. 2008;117:940–51.
Virella G, Munoz JF, Galbraith GM, Gissinger C, Chassereau C, Lopes-Virella MF. Activation of human monocyte-derived macrophages by immune complexes containing low-density lipoprotein. Clin Immunol Immunopathol. 1995;75:179–89.
Hammad SM, Twal WO, Barth JL, Smith KJ, Saad AF, Virella G, Argraves WS, Lopes-Virella MF. Oxidized LDL immune complexes and oxidized LDL differentially affect the expression of genes involved with inflammation and survival in human U937 monocytic cells. Atherosclerosis. 2009;202(2):394–404.
Lopes-Virella MF, Griffith RL, Shunk KA, Virella GT. Enhanced uptake and impaired intracellular metabolism of low density lipoprotein complexed with anti-low density lipoprotein antibodies. Arterioscl Thromb. 1991;11:1356–67.
Lopes-Virella MF, Binzafar N, Rackley S, Takei A, La Via M, Virella G. The uptake of LDL-IC by human macrophages: predominant involvement of the Fc gamma RI receptor. Atherosclerosis. 1997;135:161–70.
Oksjoki R, Kovanen PT, Lindstedt KA, Jansson B, Pentikainen MO. OxLDL-IgG immune complexes induce survival of human monocytes. Arterioscler Thromb Vasc Biol. 2006;26:576–83.
Nagarajan S. Anti-OxLDL IgG blocks OxLDL interaction with CD36, but promotes FcgammaR, CD32A-dependent inflammatory cell adhesion. Immunol Lett. 2007;108:52–61.
Hernandez-Vargas P, Ortiz-Munoz G, Lopez-Franco O, Suzuki Y, Gallego-Delgado J, Sanjuan G, Lazaro A, Lopez-Parra V, Ortega L, Egido J, Gomez-Guerrero C. Fcgamma receptor deficiency confers protection against atherosclerosis in apolipoprotein E knockout mice. Circ Res. 2006;99:1188–96.
Griffith RL, Virella GT, Stevenson HC, Lopes-Virella MF. Low density lipoprotein metabolism by human macrophages activated with low density lipoprotein immune complexes. A possible mechanism of foam cell formation. J Exp Med. 1988;168:1041–59.
Hundal RS, Gomez-Munoz A, Kong JY, Salh BS, Marotta A, Duronio V, Steinbrecher UP. Oxidized low density lipoprotein inhibits macrophage apoptosis by blocking ceramide generation, thereby maintaining protein kinase B activation and Bcl-XL levels. J Biol Chem. 2003;278:24399–408.
Al Gadban MM, Smith KJ, Soodavar F, Piansay C, Chassereau C, Twal WO, Klein RL, Virella G, Lopes-Virella MF, Hammad SM. Differential trafficking of oxidized LDL and oxidized LDL immune complexes in macrophages: impact on oxidative stress. PLoS One. 2010;5(9):e12534.
Chen JH, Riazy M, Wang SW, Dai JM, Duronio V, Steinbrecher UP. Sphingosine kinase regulates oxidized low density lipoprotein-mediated calcium oscillations and macrophage survival. J Lipid Res. 2010;51:991–8.
Hammad SM, Taha TA, Nareika A, Johnson KR, Lopes-Virella MF, Obeid LM. Oxidized LDL immune complexes induce release of sphingosine kinase in human U937 monocytic cells. Prostaglandins Other Lipid Mediat. 2006;79:126–40.
Tohyama Y, Yamamura H. Protein tyrosine kinase, syk: a key player in phagocytic cells. J Biochem. 2009;145:267–73.
Crowley MT, Costello PS, Fitzer-Attas CJ, Turner M, Meng F, Lowell C, Tybulewicz VL, DeFranco AL. A critical role for Syk in signal transduction and phagocytosis mediated by Fcgamma receptors on macrophages. J Exp Med. 1997;186:1027–39.
Luo Y, Pollard JW, Casadevall A. Fcgamma receptor cross-linking stimulates cell proliferation of macrophages via the ERK pathway. J Biol Chem. 2010;285:4232–42.
Huang Y, Jaffa A, Koskinen S, Takei A, Lopes-Virella MF. Oxidized LDL-containing immune complexes induce Fc gamma receptor I-mediated mitogen-activated protein kinase activation in THP-1 macrophages. Arterioscler Thromb Vasc Biol. 1999;19:1600–7.
Datta SR, Brunet A, Greenberg ME. Cellular survival: a play in three Akts. Genes Dev. 1999;13:2905–27.
Smith KJ, Twal WO, Soodavar F, Virella G, Lopes-Virella MF, Hammad SM. Heat shock protein 70B′ (HSP70B′) expression and release in response to human oxidized low density lipoprotein immune complexes in macrophages. J Biol Chem. 2010;285:15985–93.
Viita H, Narvanen O, Yla-Herttuala S. Different apolipoprotein B breakdown patterns in models of oxidized low density lipoprotein. Life Sci. 1999;65:783–93.
Hoff HF, Zyromski N, Armstrong D, O’Neil J. Aggregation as well as chemical modification of LDL during oxidation is responsible for poor processing in macrophages. J Lipid Res. 1993;34:1919–29.
Koenig W, Khuseyinova N. Biomarkers of atherosclerotic plaque instability and rupture. Arterioscler Thromb Vasc Biol. 2007;27:15–26.
Colley KJ, Wolfert RL, Cobble ME. Lipoprotein associated phospholipase A(2): role in atherosclerosis and utility as a biomarker for cardiovascular risk. EPMA J. 2011;2:27–38.
Pelisek J, Rudelius M, Zepper P, Poppert H, Reeps C, Schuster T, Eckstein HH. Multiple biological predictors for vulnerable carotid lesions. Cerebrovasc Dis. 2009;28:601–10.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2023 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Lopes-Virella, M.F., Virella, G. (2023). The Role of Modified Forms of LDL and Corresponding Autoantibodies in the Development of Complications in Diabetes. In: Jenkins, A.J., Toth, P.P. (eds) Lipoproteins in Diabetes Mellitus. Contemporary Diabetes. Humana, Cham. https://doi.org/10.1007/978-3-031-26681-2_13
Download citation
DOI: https://doi.org/10.1007/978-3-031-26681-2_13
Published:
Publisher Name: Humana, Cham
Print ISBN: 978-3-031-26680-5
Online ISBN: 978-3-031-26681-2
eBook Packages: MedicineMedicine (R0)