
Abstract
This chapter aims at describing the recent advances in the genetics of human tremor. Several human disorders are characterized by tremor as one of the possible symptoms, making it almost impossible to fully describe the genetic basis of each of them within the context of a single book chapter. Essential tremor (ET) and Parkinsonian tremor represent the most common forms of human tremor, and their genetics is fully described within the first sections of this chapter. Following the introduction, this chapter starts with a description of the genetics of Parkinson’s disease (PD) given the great advances in our understanding during the last two decades. PD is characterized by resting tremor, rigidity, bradykinesia, and postural instability as well as several non-motor symptoms. Studies in PD families identified six well-validated causative genes for autosomal dominant or recessive forms of the disease and several genes for atypical parkinsonism (Blauwendraat et al., Lancet Neurol 19(2):170–178, 2020; Day and Mullin, Genes (Basel) 12(7):1006, 2021). Moreover, more than 90 independent genome-wide significant risk variants have been identified through genome-wide association studies (GWASs) for the sporadic (idiopathic) forms of the disease (Nalls et al., Lancet Neurol 18(12):1091–1102, 2019; Foo et al., JAMA Neurol 77(6):746–754, 2020). However, despite the continuous advance in our understanding of the genetics of Parkinsonian tremor, little is still known concerning essential tremor, the most common pathologic tremor in humans. Whole-genome and exome sequencing studies revealed several candidate genes possibly responsible for ET in a small number of families, but they likely represent private variants. A recent GWAS revealed five genome-wide significant loci associated with ET, and the search of ET genes is still ongoing (Jiménez-Jiménez et al., Pharmaceuticals (Basel) 14(6):516, 2021; Liao et al., JAMA Neurol 79(2):185–193, 2022). Tremor is often observed in other diseases, including ataxias and dystonias, and several examples of monogenic forms of these disorders are provided within the text. Moreover, this chapter covers the genetics of familial cortical myoclonic tremor with epilepsy, Roussy–Lévy syndrome, and Wilson disease’s tremor.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Genetics
- Tremor
- Genome-wide association study (GWAS)
- Parkinson’s disease (PD)
- Essential tremor (ET)
- Spinocerebellar ataxia
- Dystonia
3.1 Introduction
This chapter aims at describing the recent advances in the genetics of human tremor. Several human disorders are characterized by tremor as one of the possible symptoms, making it almost impossible to fully describe the genetic basis of each of them within the context of a single book chapter. Essential tremor (ET) and Parkinsonian tremor represent the most common forms of human tremor, and their genetics is fully described within the first sections of this chapter. Following the introduction, this chapter starts with a description of the genetics of Parkinson’s disease (PD) given the great advances in our understanding during the last two decades. PD is characterized by resting tremor, rigidity, bradykinesia, and postural instability as well as several non-motor symptoms. Studies in PD families identified six well-validated causative genes for autosomal dominant or recessive forms of the disease and several genes for atypical parkinsonism (Blauwendraat et al. 2020; Day and Mullin 2021). Moreover, more than 90 independent genome-wide significant risk variants have been identified through genome-wide association studies (GWASs) for the sporadic (idiopathic) forms of the disease (Nalls et al. 2019; Foo et al. 2020). Despite the continuous advance in our understanding of the genetics of Parkinsonian tremor, little is still known concerning essential tremor, the most common pathologic tremor in humans. Whole-genome and exome sequencing studies revealed several candidate genes possibly responsible for ET in a small number of families, but they likely represent private variants. A recent GWAS revealed five genome-wide significant loci associated with ET, and the search of ET genes is still ongoing (Jiménez-Jiménez et al. 2021; Liao et al. 2022). Tremor is often observed in other diseases, including ataxias and dystonias, and several examples of monogenic forms of these disorders are provided within the text. Moreover, this chapter covers the genetics of familial cortical myoclonic tremor with epilepsy, Roussy–Lévy syndrome, and Wilson’s tremor.
3.2 Genetics of Parkinson’s Disease
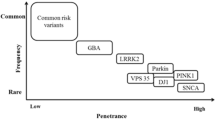
Parkinson’s disease (PD) is the second most common neurodegenerative disorder after Alzheimer’s disease, and advancing age is the major risk factor for this condition that, in industrialized countries, has a reported prevalence of about 1% in people aged more than 60 years and of about 3% in those older than 80 years (Balestrino and Schapira 2020). Indeed, recent estimates suggest that over six million individuals are suffering from the disease worldwide, and because the world’s population is aging the number of affected individuals is expected to more than double by 2040 (Dorsey and Bloem, 2018). The disease is clinically characterized by resting tremor, rigidity, bradykinesia, and postural instability as well as non-motor symptoms such as autonomic insufficiency, cognitive impairment, and sleep disorders. Some improvement can be achieved with levodopa and dopaminergic therapy, but there is currently no treatment that arrests the progression of the disease. Pathologically, PD is characterized by progressive and profound loss of neuromelanin containing dopaminergic neurons in the substantia nigra with the presence of eosinophilic, intracytoplasmic inclusions termed as Lewy bodies (LBs, containing aggregates of α-synuclein as well as other substances), and Lewy neurites in surviving neurons (Thomas and Beal 2011). The majority of PD cases are sporadic (idiopathic PD), likely arising from a combination of polygenic inheritance, environmental exposures, complex gene–environment interactions, and epigenetic dysregulation superimposed on slow and sustained neuronal dysfunction due to aging (Migliore and Coppedè 2009; Coppedè 2021). Familial PD forms account for only 5–15% of the cases, and to date rare variants in over 20 genes have been suggested to cause monogenic PD forms (Blauwendraat et al. 2020). Loci and genes that have been associated with monogenic PD were originally designated as “PARK” loci with a number representing the chronological order of their discovery. However, the relevance of many of these loci is heavily debated and some of them are no longer considered disease-causing ones, so that current recommendations are to use gene names in preference to numbered loci (Blauwendraat et al. 2020; Day and Mullin 2021). Well-established PD genes include autosomal dominant (SNCA, LRRK2, and VPS35) and recessive ones (PRNK, PINK1, and DJ1), as well as other genes leading to atypical parkinsonism (ATP13A2, FBXO7, PLA2G6, and SYNJ1). Large-scale sequencing projects and genome-wide association studies (GWASs) are currently helping to define the genetic landscape of PD (Fig. 3.1). While rare and highly penetrant variants in some genes account for monogenic forms of the disease, sporadic PD is likely resulting from interactions among combinations of more common variants with a smaller effect size, environmental risk factors, and aging. Indeed, more than 90 of such common variants have been identified by PD GWASs (Nalls et al. 2019; Foo et al. 2020). In addition, uncommon but not rare variants of certain genes, such as GBA and LRRK2, exert an intermediate risk for the disease.

The genetic landscape of Parkinson’s disease: The graph shows genes linked to PD, grouped according to the allele frequencies of their variants and the associated risk for the disease. Rare and high-penetrant mutations of SNCA, LRRK2, VPS35, PARKN, PINK1, and DJ1 genes account for monogenic autosomal dominant or recessive PD forms, and rare mutations of ATP13A2, FBXO7, PLA2G6, and SYNJ1 are linked to atypical monogenic parkinsonism. Uncommon but not rare variants in LRRK2 and GBA genes exert an intermediate risk for the disease, and more than 90 common low-risk variants of several genes have been identified through genome-wide association studies
3.2.1 Autosomal Dominant PD
3.2.1.1 SNCA
The a-synuclein gene (SNCA) on 4q21 was the first gene linked to monogenic PD, and alternative gene names are PARK1 or PARK4. A SNCA mutation causing a p.A53T substitution was found to segregate with the disease in an Italian kindred and three unrelated families of Greek origin (Polymeropoulos et al. 1997). Another mutation in the SNCA gene, leading to a p.A30P substitution, was subsequently described in a small German family with PD (Krüger et al. 1998), and a third mutation resulting in a p.E46K substitution, in a Spanish family (Zarranz et al. 2004). A study in a large family identified a triplication of the SNCA gene as causative of PD (Singleton et al. 2003). Individuals from this family had four fully functional copies of SNCA. Other PD families have been subsequently described with SNCA duplication and a disease course less severe of that observed in carriers of SNCA triplication, suggesting the existence of a gene dosage effect (Chartier-Harlin et al. 2004). Particularly, SNCA triplications and the p.E46K mutation are more commonly associated with dementia than the p.A30P mutation and gene duplications. The p.A53T mutation has been associated with dementia and the presence of cortical LBs. Although SNCA has been the first PD gene identified, SNCA missense mutations and multiplications are both extremely rare causes of familial autosomal dominant parkinsonism (Nuytemans et al. 2010). α-Synuclein is expressed throughout the mammalian brain particularly in presynaptic nerve terminals, and mutated α-synuclein has an increased tendency to form aggregates critical to Lewy body formation. These fibrillar aggregates are the major component of LBs in both familial and idiopathic PD, and aggregation of α-synuclein is thought to be a key event in dopaminergic neuronal cell death. The function of α-synuclein under normal physiological conditions is not yet fully elucidated, although there is evidence that implicates SNCA in neurotransmitter release and vesicle turnover at the presynaptic terminals (Abeliovich et al. 2000; Liu et al. 2004). Genetic polymorphisms in the SNCA gene have been consistently associated with PD risk, including a dinucleotide repeat sequence (Rep1) within the promoter region and several single nucleotide polymorphisms (SNPs) at the 3′ end of the gene (Maraganore et al. 2006; Kay et al. 2008; Mata et al. 2011). Moreover, SNCA has been among the genes most significantly associated with PD in GWAS (Pankratz et al. 2009; Satake et al. 2009; Simón-Sánchez et al. 2009; Edwards et al. 2010; Nalls et al. 2019; Foo et al. 2020). Meta-analysis of GWAS reveals that SNCA is a low-risk locus for idiopathic PD (Nalls et al. 2019), and there is evidence suggesting that SNCA alleles associated with increased PD risk are also correlated with higher α-synuclein expression, pointing again to a gene dosage effect (Fuchs et al. 2008). For example, GWASs identified a common SNCA variant in European populations (rs356182) that is associated with an increased risk for PD with an odds ratio of about 1.3 (Blauwendraat et al. 2020).
3.2.1.2 LRRK2
The leucine-rich repeat kinase 2 (LRRK2) gene maps on the PARK8 locus in 12q12 and was the second causal gene linked to autosomal dominant PD (Paisán-Ruíz et al. 2004; Zimprich et al. 2004). LRRK2 encodes the protein dardarin which contains several domains including the catalytic domain of a tyrosine kinase, and whose name is derived from dardara, the Basque word for tremor. The precise physiological role of dardarin is unknown, but the presence of several domains suggests involvement in a wide variety of functions and, as a kinase, LRRK2 is almost certainly involved in signaling cascades, probably relating to cytoskeletal dynamics (Hardy 2010). Recent evidence suggests that LRRK2 is also involved in clathrin-mediated endocytosis (Heaton et al. 2020). All the identified pathogenic mutations occur in predicted functional domains. The most prevalent LRRK2 mutation is a p.G2019S missense mutation occurring in 1–2% of PD patients of European origin, 20% of Ashkenazi Jewish patients, and approximately 40% of Arab Berbers with PD. The penetrance of this mutation is incomplete and variable (15–85%) and influenced by age, environment, and genetic background (Iwaki et al. 2020). Another frequent hotspot of LRRK2 pathogenic mutations is the Arg1441 codon (Nuytemans et al. 2010). A p.G2385R mutation, originally identified as a putative pathogenic mutation in a Taiwanese PD family, was subsequently reported to be a common polymorphism and, probably, one of the most frequent genetic risk factors for PD in Asian populations (Farrer et al. 2007). Large GWASs have confirmed that LRRK2 polymorphisms are well-validated PD risk factors in European and Asian populations (Nalls et al. 2019; Foo et al. 2020). For example, a common noncoding variation (rs76904798) upstream of LRRK2, leading to increased LRRK2 expression, is associated with increased PD risk with an odds ratio of 1.15 (Nalls et al. 2019).
3.2.1.3 VPS35
In 2011, a p.D620N mutation in the VPS35 gene was identified as causative of autosomal dominant late-onset PD in Swiss and Austrian kindreds (Vilariño-Güell et al. 2011; Zimprich et al. 2011). VSP35 is a component of the retromer complex and mediates retrograde transport between endosomes and the trans-Golgi network. Recent studies have demonstrated that VPS35 and the retromer complex influence mitochondrial homeostasis, suggesting that the p.D620N mutation can perturb the maturation of endolysosomes and autophagy as well as membrane receptor recycling, and elicit mitochondrial dysfunction (Sassone et al. 2021).
3.2.2 Autosomal Recessive PD
3.2.2.1 PRKN
Homozygous deletions in PRKN, also known as Parkin or PARK2, were identified in Japanese families as causative of juvenile PD forms (Kitada et al. 1998). Subsequently, several PARK mutations, including missense mutations, frameshift mutations and exonic deletions and insertions, have been observed in PD families (Mata et al. 2004), and PRKN mutations are nowadays regarded as the most common cause of early-onset PD (EOPD) (Jia et al. 2022). Parkin is an ubiquitin E3 ligase preparing target proteins for their degradation mediated by the ubiquitin–proteasome system (Leroy et al. 1998). Moreover, parkin is involved in mitochondrial maintenance, is required for the repair of mitochondrial oxidative DNA damage, might be involved in mitochondrial cytochrome c release, and induces subsequent autophagy of dysfunctional mitochondria (Deng et al. 2008; Narendra et al. 2008; Poole et al. 2008; Rothfuss et al. 2009).
3.2.2.2 PINK1
Several mutations in the PTEN-induced putative kinase 1 gene (PINK-1) on chromosome 1p35-36 (PARK6), encoding a protein which is mitochondrially located and whose loss of function is supposed to render neurons more vulnerable to cellular stress, have been linked to autosomal recessive EOPD (Valente et al. 2004). PINK1 mutations, primarily missense mutations, structural variants, and nonsense mutations, cause mitochondrial deficits contributing to PD pathogenesis, and represent the second most common cause of EOPD (Jia et al. 2022). PINK1 is a kinase with an N-terminal mitochondrial targeting sequence, provides protection against mitochondrial dysfunction and regulates mitochondrial morphology via fission/fusion machinery. PINK1 also acts upstream of parkin in a common pathway. Indeed, studies have described PINK1/parkin function in the maintenance of mitochondrial quality via autophagy (Kawajiri et al. 2011).
3.2.2.3 PARK7
Mutations in PARK7, also known as DJ-1, including exonic deletions and point mutations, have been associated with EOPD (van Duijn et al. 2001; Lockhart et al. 2004). DJ-1 is a mitochondrial protein involved in the protection against oxidative stress, and it was shown that parkin, PINK1, and DJ-1 form a complex to promote ubiquitination and degradation of parkin substrates, including parkin itself (Xiong et al. 2009). Evidence indicates that DJ-1 works in parallel to the PINK1/parkin pathway to maintain mitochondrial function in the presence of an oxidative environment (Thomas et al. 2011). Collectively, PRKN, PINK1, and PARK7 code for proteins required for the ubiquitin-proteasome system and for the maintenance of mitochondria. Their loss of function causes autosomal recessive PD forms that often show early-onset and variable results with respect to Lewy body pathology in the affected brain regions (Jia et al. 2022).
3.2.3 Other Genes Linked to Monogenic PD Forms
Several other genes have been linked to monogenic PD forms but either result in complex or atypical forms of the disease or have less robust evidence for pathogenicity (Blauwendraat et al. 2020). Indeed, the field of PD genetics is in constant flux, with candidates being confirmed, refuted, or newly identified in rapid succession (Wittke et al. 2021). Well-established or high confident PD genes leading to autosomal recessive atypical forms include ATP13A2, FBXO7, PLA2G6, SYNJ1, DNAJC6, and VPS13C (Blauwendraat et al. 2020; Wittke et al. 2021; Jia et al. 2022). Biallelic ATP13A2 mutations have been linked to an autosomal recessive form of early-onset parkinsonism with pyramidal degeneration (Ramirez et al. 2006). A recent analysis of 19 families with ATP13A2 mutations revealed a median age at onset of 14 years and atypical parkinsonism in 83% of the carriers, followed by cognitive decline in 75% of them, and other common signs such as vertical gaze palsy, spasticity/pyramidal signs, mini-myoclonus, and psychotic signs and symptoms in almost 50% (Wittke et al. 2021). Functional studies suggest that ATP13A2 deficiency impairs lysosomal polyamine export (van Veen et al. 2020). The FBXO7 gene encodes for a member of the F-box family of proteins, all of which may have a role in the ubiquitin–proteasome protein-degradation pathway (Shojaee et al. 2008; Di Fonzo et al. 2009) and has been linked to autosomal recessive, juvenile/early-onset parkinsonian-pyramidal syndrome (Di Fonzo et al. 2009). A recent analysis of 26 FBXO7 mutation carriers originating from ten families showed that the mean age at onset was 17 years, atypical parkinsonism signs were present in 92% of them and spasticity/pyramidal signs in 73% (Wittke et al. 2021). The PLA2G6 gene encodes a calcium-independent group VI phospholipase A2 and has been linked to autosomal recessive dystonia-parkinsonism, a disease characterized by levodopa-responsive parkinsonism and dystonia (Paisán-Ruiz et al. 2009, 2010). A recent analysis of 50 patients with PLA2G6-dystonia-parkinsonism revealed a mean age of 26 years old at PD diagnosis; moreover, neuropsychiatric symptoms such as depression, anxiety, or personality changes preceded motor symptoms in almost half of the patients (Vela-Desojo et al. 2022). The SYNJ1 gene codes for polyphosphoinositide phosphatase synaptojanin 1 which has been implicated in synaptic vesicle dynamics, including endocytosis and recycling, and causes autosomal recessive, early-onset parkinsonism (Krebs et al. 2013; Quadri et al. 2013). In addition to parkinsonism, the most commonly reported features in SYNJ1 mutant carriers were dystonia, gait difficulties, cognitive decline, postural instability, hypomimia, and dysarthria/anarthria (Wittke et al. 2021). DNAJC6 encodes for auxilin 1, a protein involved in clathrin-mediated synaptic vesicle endocytosis, and DNAJC6 mutations have been initially described in two families with autosomal recessive juvenile parkinsonism (onset age <11 years), prominent atypical signs, poor or absent response to levodopa, and rapid progression (Edvardson et al. 2012; Koroglu et al. 2013). Subsequently, DNAJC6 mutations have been found also in early-onset PD cases, characterized by symptoms onset in the third-to-fifth decade of life and slow disease progression (Olgiati et al. 2016). The VPS13C gene codes for a member of the VPS13 family of proteins, involved in lipid transport between the endoplasmic reticulum and other organelles, and VPS13C gene mutations have been identified in autosomal recessive, early-onset forms of parkinsonism (Lesage et al. 2016; Rudakou et al. 2020). In addition to the above-described genes causing autosomal recessive EOPD forms, mutations in DCTN1 cause Perry syndrome, a rare autosomal dominant disorder characterized by rapidly progressive early-onset parkinsonism, central hypoventilation, weight loss, insomnia, and depression (Richardson et al. 2020). Several other genes have been linked to typical or atypical parkinsonism, as well as to complex neurological disorders that include parkinsonism as part of their symptoms. For example, POLG mutations may cause variable clinical manifestations, including parkinsonism, and CHCHD2 mutations can cause typical parkinsonism. However, for several of these genes, including LRP10, TMEM230, DNAJC13, EIF4G1, GIGYF2, HTRA2, and UCHL1, the pathogenetic role is still debated, some are no longer considered PD genes or further validation is required prior to be considered PD genes (Blauwendraat et al. 2020; Jia et al. 2022).
3.2.4 Susceptibility Genes
Most of PD occurs as apparently sporadic forms and GWASs have revolutionized our efforts to find loci at which common, normal genetic variability contributes to disease risk. The first GWAS loci for PD were identified in 2009 using data from almost 5000 patients and 9000 controls (Simón-Sánchez et al. 2009). In 2019, a large GWAS including more than 37.000 patients, 18.600 proxy cases (individuals with a first relative with PD) and 1.4 million controls, allowed the identification of 90 independent genome-wide significant risk signals across 78 loci (Nalls et al. 2019). Additional PD loci have been identified in a subsequent GWAS including more than 65.000 cases and almost 1.9 million controls (Foo et al. 2020). Indeed, common variants of small effect size in SNCA, LRRK2, VPS13C, MAPT, RAB29, BST1, GAK, HLA-DRB5, and many other genes have been associated with increased PD risk (Nalls et al. 2019; Blauwendraat et al. 2020; Jia et al. 2022). Notably, the GBA gene is the major genetic risk factor for PD and will be further discussed in the next section.
3.2.4.1 GBA
Mutations in the GBA gene encoding glucocerebrosidase, the enzyme deficient in the lysosomal glycolipid storage disorder Gaucher disease (GD: an autosomal recessive disorder with multisystemic manifestations, including involvement of the liver, spleen, bone marrow, lungs, and nervous system), are associated with the development of PD and other Lewy body disorders (Velayati et al. 2010). The observation that a small subset of GD patients develop parkinsonism with brainstem or diffuse Lewy-related pathology (Tayebi et al. 2003), and that relatives of patients with GD have an increased incidence of parkinsonism (Halperin et al. 2006), led researchers to investigate GBA mutations as a possible risk factor for PD. Several large-scale genetic studies demonstrated that heterozygote GBA variants are the most important genetic risk factor for PD. More than 100 GBA variants have been associated with PD, with an overall odds ratio of about 3.5–6 (Sidransky et al. 2009; Gegg et al. 2022). However, while the odds ratio for “mild” risk variants, such as the common p.N370S variant, is lower than 5, certain high-risk variants, such as the p.L444P one, confer a greater risk for PD. Overall, the estimated lifetime risk of developing PD in heterozygous carriers of a GBA variant ranges from 7.6% at age 50 years to 30% at age 80, and is influenced by other genetic, environmental, and age-related factors. Indeed, GBA variants are the most common genetic risk factor for PD. The accumulation of α-synuclein following loss of glucocerebrosidase activity and subsequent lysosomal dysfunction in neurons is well established. However, the mechanisms by which this occurs warrant further investigation (Gegg et al. 2022).
3.3 Genetics of Essential Tremor
Essential tremor (ET) is one of the most common movement disorders in adults and the most common pathologic tremor in humans. The median disease prevalence is estimated to be 0.4% across all ages, and the mean prevalence 0.67% (Louis and McCreary 2021). However, ET prevalence increases markedly with age, with recent estimates suggesting a 74% increase for every decade increase in age, reaching more than 20% in nonagenarians (Louis and Ferreira 2010; Louis and McCreary 2021). ET shows a bimodal age of onset, with a smaller peak in the second decade of life and a larger peak in the sixth decade (Brin and Koller 1998). Childhood-onset ET is usually hereditary and three times more frequent in males than in females (Ferrara and Jankovic 2009). The disease is characterized by an action tremor with mixed postural and kinetic elements. The postural tremor is commonly seen in the hands and the kinetic tremor is brought out by action, such as writing, eating, or pouring a cup of water (Dalvi and Mercury 2011). ET is a heterogeneous condition with variable clinical expression in affected patients. While the hands are most commonly affected, many patients have a head tremor as well. Approximately 90–95% of the patients have tremor in their upper extremities, 30–34% have a head tremor, 12–20% a voice tremor, and 5–10% a face or trunk tremor. Almost 10% of the patients have a lower limb tremor (Whaley et al. 2007; Dalvi and Mercury 2011). Non-motor symptoms including mild cognitive changes, changes in personality, anxiety, and depression are more frequent in ET patients than in normal age-matched controls (Zesiewicz et al. 2010). According to recent classification criteria, ET is defined as an isolated tremor syndrome manifesting as an action tremor of bilateral upper extremities for a minimum of 3 years duration, in the absence of any other neurological signs such as parkinsonism, ataxia, or dystonia. Tremor involving the voice, head, and lower extremities may or may not be present. ET patients with additional neurological signs (dystonia, rest tremor, impaired tandem gait) are now categorized as “ET plus” (Lenka and Jankovic 2021). The analysis of postmortem ET brains revealed that 75% of them are characterized by cerebellar changes, including loss of Purkinje cells and increase in the number of axonal swellings, termed “torpedoes.” Lewy bodies were observed in the locus coeruleus of the remaining 25% of the brains (Louis et al. 2007). Overall, ET can be considered a cerebellar disorder with pathologic changes affecting either the cerebellum itself or neurons that synapse with Purkinje cells (Dalvi and Mercury 2011). Studies in twins revealed elevated concordance among monozygotic twins, suggesting that the disease has a high heritability (Lorenz et al. 2004). Most of the studies indicate that ET is a familial disorder in 40–50% of the cases, and the disease is often inherited in a manner suggesting an autosomal dominant genetic pattern with incomplete penetrance. A family history of ET appears to correlate with younger age at onset, and first-degree relatives of ET patients have a fivefold increased risk to develop the disease than normal controls. Non-familial “sporadic” ET cases are known and might result from either low-penetrant autosomal dominant loci or from multifactorial inheritance (Deng et al. 2007). Linkage analyses revealed at least four loci for familial ET in Iceland and North American families located at chromosomes 3q13, 2p22-p25, 6p23, and 5q35. However, the causative gene has yet to be identified (Dalvi and Mercury 2011; Jiménez-Jiménez et al. 2021). Whole-genome/exome sequencing approaches have revealed several additional putative genes for ET, but lack of replication in other families suggests that they are likely private variants (Jiménez-Jiménez et al. 2021). GWASs have also identified putative ET loci, but only a recent GWAS including 7.177 ET patients and 475.877 controls revealed five independent genome-wide significant loci that explained approximately 18% of ET heritability (Liao et al. 2022). The results of this GWAS suggest that a portion of ET heritability can be explained by common genetic variants (Liao et al. 2022). Moreover, genetic factors alone do not explain all cases of ET, and several environmental factors including exposure to neurotoxic compounds such as β-carboline alkaloids and ethanol, as well as pesticide and lead exposure, are potential ET risk factors, while smoking and antioxidant intake may be protective (Ong et al. 2019).
3.3.1 Linkage Studies
In 1997, the first ET locus (ETM1) was mapped to chromosome 3q13 in 75 members of 16 Icelandic families (Gulcher et al. 1997). A Ser9Gly variant in the dopamine D3 receptor (DRD3) gene, located in the ETM1 locus, was subsequently associated with disease risk and age at onset (Jeanneteau et al. 2006). Subsequent studies failed to find a significant association of the DRD3 variant with ET, suggesting that it is unlikely to be a causal factor for ET (Lorenz et al. 2009). The ETM2 locus was mapped to a 9.1 cM region on chromosome 2p22-p25 (Higgins et al. 1997) in a large American family of Czech descent. Subsequent studies suggested an association between ET and an A265G substitution in the HS1-binding protein 3 gene (HS1BP3) mapping within the ETM2 locus (Higgins et al. 2005). However, the association with the HS1BP3 gene was not replicated by other investigators (Deng et al. 2005; Shatunov et al. 2005). Linkage to ETM1 and ETM2 loci was not evident in several ET families suggesting genetic heterogeneity in ET. A third ET locus was mapped to chromosome 6p23. Several genes within this locus have been investigated as candidates, but none of them was found to bear pathogenic mutations (Shatunov et al. 2006). No linkage of these three loci with familial ET was found in other studies (Aridon et al. 2008; Novelletto et al. 2011; Zahorakova et al. 2010), suggesting genetic heterogeneity for ET. The analysis of 48 essential tremor patients from five pedigrees revealed another locus on chromosome 5q35 linked to essential tremor, but exome sequencing did not identify a potential causative variant in this region (Hicks et al. 2016).
3.3.2 Whole-Genome and Exome Sequencing Studies
Whole-genome and exome sequencing studies revealed several candidate genes possibly responsible for ET in a small number of families, including FUS (designated as ETM4), HTRA2, TENM4 (designated as ETM5), SORT1, SCN11A, NOTCH2NLC (designated as ETM6), NOS3, KCNS2, HAPLN4, USP46, CACNA1G, SLIT3, CCDC183, MMP10, and GPR151. However, mutations of these genes were found only in singular families, suggesting that they could probably represent private variants (reviewed in Jiménez-Jiménez et al. 2021).
3.3.3 GWASs
The first GWAS in ET identified a sequence variant (rs9652490) of the LINGO1 gene to be a risk factor in European and American populations (Stefansson et al. 2009). Subsequent GWASs identified variants of SLC1A2, STK32B, PPARGC1A, and CTNNA3, as possible ET risk factors, but further GWASs failed to replicate these findings (Thier et al. 2012; Müller et al. 2016; Houle et al. 2017). Recently, a GWAS including 7.177 ET patients and 475.877 control individuals revealed five independent genome-wide significant loci that explained approximately 18% of ET heritability. Functional analyses found significant enrichment in the cerebellar hemisphere, cerebellum, and axonogenesis pathways (Liao et al. 2022). Overall, the genetic etiology of ET remains still largely elusive, and the search of ET genes is still ongoing.
3.4 Tremor in Ataxias
The ataxias are a heterogeneous group of progressive neurodegenerative disorders with ataxia as the leading symptom. Cerebellar ataxias can be divided into acquired, sporadic and hereditary forms (Krygier and Mazurkiewicz-Bełdzińska 2021). Inherited ataxias include autosomal dominant spinocerebellar ataxias (SCAs), autosomal recessive cerebellar ataxias, episodic ataxias (EA), and X-linked ataxias (Manto and Marmolino 2009; Krygier and Mazurkiewicz-Bełdzińska 2021). Tremor is often observed in ataxias (Magrinelli et al. 2020). The aim of this section of this chapter is the discussion of several of the best-known examples of cerebellar ataxias characterized by tremor as one of the symptoms (Table 3.1).
3.4.1 SCA2 and SCA3
Parkinsonism, dystonia, and postural tremor are particularly prevalent in SCAs types 2 (SCA2) and 3 (SCA3), caused by abnormal CAG trinucleotide repeat expansion of ATXN2 and ATXN3 genes, respectively. SCA2 is characterized by a broad group of progressive features, including gait ataxia, postural instability, cerebellar dysarthria, dysmetria, and dysdiachokinesia, as well as non-cerebellar manifestations including slow saccadic eye movements, peripheral neuropathy, cognitive decline, dopamine-responsive parkinsonism, dystonia, and chorea (Krygier and Mazurkiewicz-Bełdzińska 2021). The disease is caused by a CAG repeat expansion of ATNX2, which can expand in families over successive generations resulting in earlier onset age and faster progression. Affected individuals have alleles with 32 or more CAG trinucleotide repeats, resulting in polyglutamine tract expansion in the protein (Lastres-Becker et al. 2008). Machado-Joseph disease (MJD), also known as spinocerebellar ataxia type 3 (SCA3), is the most common form of spinocerebellar ataxia worldwide, caused by CAG trinucleotide repeat expansion in ataxin-3 (ATXN3), coding a conserved and ubiquitous protein known to bind polyubiquitin chains and to function as a deubiquitinating enzyme. Affected individuals have alleles with 52 or more trinucleotide repeats (Matos et al. 2011). The parkinsonian phenotype of SCA2 and SCA3 is often observed in Asians (Lu et al. 2004; Kim et al. 2007).
3.4.2 SCA7
Spinocerebellar ataxia type 7 (SCA7) results from CAG repeat expansion in the ATXN7 gene on chromosome 3p14.1 and is characterized by progressive ataxia and variable age at onset, degree of severity, and rate of progression among and within families. Associated symptoms can include palatal tremor, cone-rod retinal dystrophy, and vision loss (Magrinelli et al. 2020; Krygier and Mazurkiewicz-Bełdzińska 2021).
3.4.3 SCA12
Spinocerebellar ataxia type 12 (SCA12) is a late-onset, autosomal dominant, slowly progressive disorder. Action tremor is the usual presenting sign, often starting in the fourth decade. At disease onset, SCA12 manifests characteristic action tremors in the upper limbs. Disease progression is slow, and patients display varied clinical manifestations, including gait ataxia, abnormal eye movements, parkinsonism, dystonia, hyperreflexia, and psychiatric and cognitive manifestations (Manto 2010; Srivastava et al. 2017; Magrinelli et al. 2020). The disease is caused by a CAG repeat expansion in PPP2R2B, a gene that encodes Bβ, a regulatory subunit of protein phosphatase 2A (PP2A). Alleles with 43 or more CAG repeats are observed in SCA12 patients (Srivastava et al. 2017). The CAG expansion in PPP2R2B correlates with increased Bβ expression and does not result in polyglutamine production. SCA12 may be considered in patients who present with action tremor and later develop signs of cerebellar and cortical dysfunction (O’Hearn et al. 2011).
3.4.4 SCA15 and SCA16
Spinocerebellar ataxia type 15 (SCA15), formerly known as SCA15/SCA16, is rare and slowly progressive dominantly inherited ataxia. Its main distinguishing characteristic is tremor, often affecting the head, which is seen in about half of the affected individuals, and which may be the presenting feature (Storey and Gardner 2012). The disease is due to various deletions of the inositol 1,4,5-triphosphate receptor 1 gene (ITPR1) on chromosome 3. “SCA16” has been shown to be due to an ITPR1 mutation and has now been subsumed into SCA15 (Iwaki et al. 2008).
3.4.5 SCA20
Spinocerebellar ataxia type 20 (SCA20) is a very rare slowly progressive dominantly inherited disorder reported in individuals from a large Anglo-Celtic family from Australia. Its distinguishing clinical features, each present in most affected persons, are palatal tremor, and a form of dysphonia resembling spasmodic dysphonia. The responsible locus was mapped in the pericentric region of chromosome 11 (Knight et al. 2004), but the specific gene(s) underlying SCA20 have not yet been identified (Müller 2021).
3.4.6 SCA27
Spinocerebellar ataxia type 27 (SCA27) is characterized by disease onset in late childhood/early adulthood, and symptoms including slowly progressive cerebellar ataxia, early-onset tremor, orofacial dyskinesia frequently in association with ocular problems, psychiatric symptoms, and cognitive deficits (Müller 2021). The disease results from dominant mutations of the FGF14 gene, on chromosome 13q34 (van Swieten et al. 2003). FGF14 controls channel gating, axonal targeting and phosphorylation in neurons effecting excitability. It is also required for synaptic transmission, plasticity, and neurogenesis (Müller 2021).
3.4.7 Fragile X-Associated Tremor/Ataxia Syndrome
The fragile X-associated tremor/ataxia syndrome (FXTAS) is a late-onset neurodegenerative disorder affecting mainly men over the age of 50 years. The disease is caused by a CGG repeat expansion in the premutation range (55–200) in the fragile X mental retardation 1 (FMR1) gene whose full mutation causes the fragile X syndrome (FXS), the most common cause of inherited mental retardation. Major signs are cerebellar gait ataxia, intention tremor, frontal executive dysfunction, and global brain atrophy. Other frequent findings are parkinsonism, peripheral neuropathy, psychiatric symptoms, and autonomic dysfunction. Affected females have a less severe disease, and some symptoms different from that of men, for example, muscle pain (Leehey 2009).
3.4.8 Others
As discussed in the Introduction of this section tremor is often observed in several ataxias, including other SCAs and recessive ataxias, and not limited to the above detailed examples. Early-onset hypotonia and nonprogressive axial cerebellar ataxia, associated with nystagmus, intention tremor, and dysarthria characterize Cayman ataxia (CA). The name derives from the fact that the disease has been initially found in the Grand Cayman Island. CA is an autosomal recessive disease caused by mutation of ATCAY, which codes for caytaxin, a protein involved in glutamate synthesis and in synaptogenesis of cerebellar granular neurons and Purkinje cells (Hayakawa et al. 2007). More recently, CA resulting from ATCAY mutation has been reported also outside of the Grand Cayman Island, in a Pakistani family (Manzoor et al. 2018). Tremor is also observed in other autosomal recessive ataxias, such as those caused by defects of DNA repair genes (Gueven et al. 2007; Embiruçu et al. 2009). Ataxia with oculomotor apraxia type 1 (AOA1) is a condition characterized by involuntary movements (chorea and dystonia) and/or progressive global ataxia, with dysarthria associated with hands and head tremor. The disease is caused by mutation in the APTX gene which encodes aprataxin, a nuclear protein involved in single-strand DNA repair. Ataxia with oculomotor apraxia type 2 (AOA2) is characterized by global progressive ataxia with onset usually between 8 and 25 years of age. Dystonia, head and postural tremor, chorea, dysphagia, pes cavus, and scoliosis are occasionally seen. The disease is caused by autosomal recessive mutations of SETX encoding senataxin, a DNA/RNA helicase involved in RNA processing and DNA repair. Peripheral neuropathy and movement disorder, as tremor or choreoathetosis, are seen after 5 years of age in ataxia–telangiectasia (AT), a recessive disorder caused by mutations of the ATM gene that encodes for a serine/threonine kinase responsible for DNA repair during the cell cycle (Gueven et al. 2007).
3.5 Familial Cortical Myoclonic Tremor with Epilepsy
Familial cortical myoclonic tremor with epilepsy (FCMTE), also referred to as familial adult myoclonic epilepsy (FAME), benign adult familial myoclonic epilepsy (BAFME) or autosomal dominant cortical myoclonus and epilepsy (ADCME), refers to a clinical entity first described in Japan and subsequently in more than 100 families worldwide and characterized by postural myoclonic tremor of the distal limbs, familial history of epilepsy, a rather benign outcome, and autosomal dominant inheritance (Regragui et al. 2006; van den Ende et al. 2018). Seven loci for FCMTE have been reported (Table 3.2), but the genetic variants underlying the disorder have remained elusive for several years and identified only recently. Intronic pentanucleotide repeat expansions composed of mixed TTTCA/TTTTA repeats in the SAMD12 gene have been identified as the cause of FCMTE1 in Japanese and Chinese populations. SAMD12 codes for the sterile alpha motif domain-containing protein 12, a protein predicted to be involved in transmembrane receptor protein tyrosine kinase signaling pathway (Ishiura et al. 2018; Cen et al. 2018; Zeng et al. 2019; Lei et al. 2019). Subsequently, ATTTC repeat expansions in the first intron of STARD7 was identified as the cause of FCMTE2. STARD7 codes for the protein StAR-related lipid transfer domain containing 7, a phosphatidylcholine-specific lipid transfer protein essential for the maintenance of mitochondrial membrane composition and dynamics (Corbett et al. 2019). Unstable TTTTA/TTTCA expansions in MARCH6 have been associated with FCMTE3; MARCH6 codes for membrane-associated ring-CH-type finger 6, a member of a family of membrane-associated E3 ubiquitin ligases (Florian et al. 2019). FCMTE4 was linked to chromosome 3q26.32-3q28 in a large FCMT family from Thailand, and insertions of the intronic TTTCA repeats in YEATS2, coding a protein involved in histone acetylation, were subsequently identified as the causative mechanism (Yeetong et al. 2019). Pentanucleotide repeat expansions have been also linked to FCMTE6 and FCMTE7, and particularly a TTTCA repeat expansion in the upstream noncoding region of exon 1 of the TNRC6A gene on chromosome 16p12.1 (FCMTE6), and a TTTCA repeat expansion in the RAPGEF2 on chromosome 4q32.1 (FCMTE7) were observed in Japanese families, overall suggesting that pentanucleotide repeat expansions are a common causal mechanism for autosomal dominant cortical myoclonic tremor with epilepsy (Ishiura et al. 2018). One exception is FCMTE5 that was reported in a consanguineous Egyptian family and characterized by onset of seizures in adolescence, followed by the development of cortical myoclonic tremor later in life. The disease showed an autosomal recessive inheritance pattern, patients in this family conformed to the core criteria of FCMTE but some unusual features were also present, and the disease was caused by a homozygous frameshift mutation in the CNTN2 gene (Stogmann et al. 2013).
3.6 Dystonic Tremor
Dystonia is a hyperkinetic movement disorder characterized by sustained muscle contractions, frequently causing twisting and repetitive movements or abnormal postures. According to a consensus statement of the Movement Disorder Society (MDS) expert members, dystonia is defined as “a movement disorder characterized by sustained or intermittent muscle contractions causing abnormal and often repetitive movements, postures, or both. Dystonic movements are typically patterned, twisting, and may be tremulous. Dystonia is often initiated or worsened by voluntary action and associated with overflow muscle activation” (Albanese et al. 2013). It also further classifies dystonia into two main axes: axis 1, clinical characteristics, and axis 2, etiology. Axis 1 takes into consideration age at onset, body distribution, the temporal pattern of the hyperkinetic movement, and associated features, that is, if the dystonia is isolated, combined, or complex. In isolated dystonia, dystonia is the only motor feature except for tremor, in combined dystonia, the dystonia is combined with other movement disorders such as myoclonus, parkinsonism, and others, while complex dystonia is accompanied by neurologic or systemic manifestations beyond movement disorders. Axis 2 defines whether dystonia is inherited, acquired, or idiopathic (di Biase et al. 2022). Inherited dystonias show genetic heterogeneity, including autosomal dominant, autosomal recessive, X-linked, and mitochondrial forms (Müller 2009; di Biase et al. 2022). According to the Human Genome Organization (HUGO) nomenclature, genetic loci for dystonia are named using a DYT prefix followed by a number representing the chronological order of their discovery, that is, DYT1-DYTn. However, the MDS task force has recently proposed a novel nomenclature plan in which pure dystonia is designated with a DYT prefix followed by the gene name (e.g., DYT1 becomes DYT-TOR1A), while dystonia combined with parkinsonism or ataxia would be designated as DYT/PARK or DYT/SCA, respectively, each followed by the gene name (e.g., DYT5a becomes DYT/PARK-GCH1) (Mencacci and Jinnah, 2019). Tremor has been reported as a general manifestation in dystonias. Dystonic tremor manifests as a rhythmic, intermittent, patterned movement in body regions which are primarily affected or not by dystonia (Magrinelli et al. 2020). Recently, Pandey et al. (2021) reviewed the literature to assess the prevalence and clinical characteristics of tremor in different types of primary monogenic dystonia, observing that tremor has been reported in at least 15 different monogenic dystonias and ranges in prevalence according to the different monogenic subtype (Pandey et al. 2021). Table 3.3 shows some of the best-known genes associated with primary monogenic dystonias in which tremor is a common feature.
3.6.1 Dominant Dystonias
Tremor has been observed in several autosomal dominant dystonias. For example, tremor has been reported in 66% of patients with dystonia 24 (DYT24 or DYT-ANO3) an autosomal dominant cranio-cervical dystonia resulting from mutations in the ANO3 gene, coding for anoctamin-3, a transmembrane protein that belongs to a family of calcium-activated chloride channels (Charlesworth et al. 2012; Pandey et al. 2021). Dystonia 6 (DYT6 or DYT-THAP1) is caused by mutations of the transcription factor THAP1, and tremors ranging from mild, asymmetrical, rest, and postural bilateral upper limb to occasional head and lower limb tremors have been reported 18% of the patients (Fuchs et al. 2009; Pandey et al. 2021). Dystonia 25 (DYT 25 or DYT-GNAL) is an autosomal dominant neurologic disorder characterized by adult onset of focal dystonia, usually involving the neck. Tremor is reported to occur in the head and upper limbs and has been reported in 12% of the patients. The disease is caused by mutations of the GNAL gene that encodes a stimulatory alpha-subunit of G proteins with high expression in the basal ganglia (Fuchs et al. 2013; Pandey et al. 2021). Tremor has been also observed in 11% of the patients with dystonia 1 (DYT1 or DYT-TOR1A), an early-onset primary dystonia caused by mutations in the TOR1A gene encoding the protein torsin A, a member of a superfamily of ATPases with chaperone functions (Ozelius et al. 1997; Pandey et al. 2021). Concerning dystonias combined with parkinsonism, postural tremor was observed in 18% of the patients with dystonia 5a (DYT5a or DYT/PARK-GCH1), a rare autosomal dominant dystonia-parkinsonism caused by mutations of GCH1, a gene that codes for GTP-cyclohydrolase I, essential for the synthesis of dopamine (Ichinose et al. 1994; Pandey et al. 2021).
3.6.2 Recessive Dystonias
Bilateral upper limb tremors and occasionally head tremor have been reported in 100% of patients with dystonia 2 (DYT2 or DYT-HPCA), an autosomal recessive dystonia due to mutations in the HPCA gene on 1p35.1 coding for hippocalcin, a member of a family of neuron-specific Ca (2+)-binding proteins found in the retina and brain. However, the analysis included only three patients, so that the complete presence of tremor should be taken with caution (Charlesworth et al. 2015; Pandey et al. 2021). Among combined dystonias, tremor has been reported in 44% of the patients with dystonia 5b (DYT5b or DYT/PARK-TH), caused by mutations of the tyrosine hydroxylase (TH) gene on the short arm of chromosome 11, resulting in lack of the tyrosine hydroxylase enzyme leading to impaired conversion of tyrosine into L-dopa (Verbeek et al. 2007). Tyrosine hydroxylase is a rate-limiting enzyme in dopamine biosynthesis and missense mutations in both alleles of the TH gene cause dopamine-related phenotypes, including dystonia and infantile Parkinsonism. Dystonia 16 (DYT16 or DYT-PRKRA) was observed in seven Brazilian patients and linked to mutations of the gene PRKRA, encoding a protein kinase, interferon-inducible double-stranded RNA-dependent activator. Parkinsonism was observed in four of the seven patients (Camargos et al. 2008), and tremor has been reported in 17% of DYT16 patients. X-linked recessive dystonia or dystonia 3 (DYT3 or DYT-TAF1) is an adult-onset dystonia often accompanied by parkinsonism, resulting from recessive mutations of TAF1 on chromosome Xq13.1 (Makino et al. 2007). Tremor has been reported in 4% of DYT3 patients (Pandey et al. 2021). In addition to the examples described in this chapter, tremor has been observed in other pure or combined dystonias, such as in 64% of the cases of dystonia-parkinsonism resulting from mutations in the SLC6A3 gene, as well as in myoclonus-dystonia resulting from mutations in KCTD17, and other dystonia-parkinsonisms resulting from mutations in SPR and PTS genes, albeit in a limited number of patients (Pandey et al. 2021).
3.7 Roussy–Lévy Syndrome
Charcot–Marie–Tooth (CMT) disease encompasses a genetically heterogeneous group of inherited neuropathies, also known as hereditary motor and sensory neuropathies (HSMN). CMT results from mutations in more than 30 genes expressed in Schwann cells and neurons causing overlapping phenotypes. The classic CMT phenotype reflects length-dependent axonal degeneration characterized by distal sensory loss and weakness, deep tendon reflex abnormalities, and skeletal deformities (Patzkó and Shy 2011). The first cases of CMT associated with ET have been reported more than 30 years ago (Salisachs 1975, 1976). Subsequently, others provided evidence that CMT disease with tremor coincides with the Roussy–Lévy syndrome (Barbieri et al. 1984). The Roussy–Lévy syndrome was first described in 1926 by Roussy and Lévy as a disorder beginning in infancy or childhood and presenting with pes cavus and tendon areflexia, distal limb weakness, tremor in the upper limbs, gait ataxia, and distal sensory loss. In 1998 Auer-Grumbach et al. reported a family with affected members in four generations, showing the clinical signs of Roussy–Lévy syndrome and a partial duplication at chromosome 17p11.2, a genetic defect commonly found in CMT1A patients (the duplication of the PMP22 gene), suggesting a close relation with the CMT syndrome. The PMP22 gene encodes a 22-kDa protein that comprises 2–5% of peripheral nervous system myelin. It is produced primarily by Schwann cells and expressed in the compact portion of essentially all myelinated fibers in the peripheral nervous system (Auer-Grumbach et al. 1998). In members of the original family studied by Roussy and Lévy, Plante-Bordeneuve et al. (1999) identified a heterozygous mutation in the myelin protein zero (MPZ) gene, encoding the major structural protein of peripheral myelin; mutations in this gene are also associated with CMT1B (Plante-Bordeneuve et al. 1999).
3.8 Wilson’s Disease
Wilson’s disease is an inherited autosomal recessive disorder of copper balance leading to hepatic damage and neurological disturbance of variable degree. The hepatic and the neurological form can be distinguished, but many patients present with a mixture of both. An estimate for the disease frequency in most populations is about 17 per million, suggesting a carrier frequency ranging from 1 in 90 to 1 in 122 (Huster 2010; Lorincz 2010). The disease is caused by mutations of the ATP7B gene on chromosome 13q14.3, encoding a copper transporting P-type transmembrane ATPase. Mutations in ATP7B result in abnormal copper metabolism and subsequent toxic accumulation of copper (Thomas et al. 1995). Overall, over 700 ATP7B mutations have been so far identified with only a few of them functionally characterized (Chanpong and Dhawan, 2022). The patient is usually presymptomatic during early life, but the accumulation of copper causes subclinical liver disease. Disease symptoms are highly variable and can manifest between early childhood and the fifth or sixth decade of life, with a peak incidence of around 17 years. Hepatic, neurological, and psychiatric manifestations are observed. Neurological features include dysarthria, dystonia, tremor, parkinsonism, choreoathetosis, ataxia, and subtle cognitive impairment. Tremor is reported in 22–55% of the cases, occurring at rest, upon assumption of a posture, or with action. Clinical signs include asymmetric distal accentuated tremor of the hands, “wing beating” tremor, intention tremor, and sometimes tremor of the trunk and head. Parkinsonism has been reported in 19–62% of the cases. Psychiatric symptoms, including attention deficit, depression, and mood swings, are observed in up to one-third of the patients (Huster 2010; Lorincz 2010; Chanpong and Dhawan, 2022).
3.9 Conclusions
Advances have been obtained in recent years in our understanding of the genetics of PD, both in monogenic and idiopathic forms (Blauwendraat et al. 2020; Jia et al. 2022). Studies in autosomal dominant and recessive forms of the disease, as well as in monogenic atypical forms, have highlighted central roles for protein aggregation and turnover, lysosomal pathways, mitochondrial damage and turnover, endocytosis, and synaptic vesicle trafficking in the pathophysiology of the disease (Jia et al. 2022). In parallel, genome sequencing approaches and large-scale GWASs are increasingly helping to clarify the genetic landscape of the disease that is continuously updated (Blauwendraat et al. 2020; Jia et al. 2022). Less is known concerning the genetics of ET for which some putative gene variants identified in families are likely private ones, and only a recent GWAS provided significant genome-wide signals associated with disease risk (Liao et al. 2022). Genetic heterogeneity, incomplete penetrance, and the fact that some tremor disorders are erroneously referred to as ET because they resemble it at both onset and for many years thereafter are among the most probable reasons explaining why the search of ET genes is still ongoing (Magrinelli et al. 2020). Tremor occurs in several other neurological disorders, such as ataxias, dystonias, and peripheral neuropathies. This chapter provides several examples of hereditary forms of these disorders. Overall, the compromising of several pathways can result in neuronal dysfunction and tremor, and the heterogeneity of the diseases characterized by tremor as one of the possible symptoms is reflected by the heterogeneity of genes and pathways causing such diseases. Mutations in the same gene can cause different diseases, depending on the nature of the mutation itself, making the picture even more complex. Some examples are the MAPT gene, that can cause frontotemporal dementia with parkinsonism or increase the risk for idiopathic PD, or mutations of FRM1 that can lead to either fragile X syndrome or fragile X-associated/tremor ataxia syndrome, depending on the length of the repeated tract. Similarly, as in the case of SNCA and LRRK genes, rare high-penetrant variants are observed in families with monogenic PD, while more common variants confer a slight increase in the risk for idiopathic forms. Overall, this chapter gives a broad overview of human disorders characterized by tremor and of the genetics beyond them.
References
Abeliovich A, Schmitz Y, Fariñas I, Choi-Lundberg D, Ho WH, Castillo PE, Shinsky N, Verdugo JM, Armanini M, Ryan A, Hynes M, Phillips H, Sulzer D, Rosenthal A. Mice lacking alpha-synuclein display functional deficits in the nigrostriatal dopamine system. Neuron. 2000;25(1):239–52.
Albanese A, Bhatia K, Bressman SB, Delong MR, Fahn S, Fung VS, Hallett M, Jankovic J, Jinnah HA, Klein C, Lang AE, Mink JW, Teller JK. Phenomenology and classification of dystonia: a consensus update. Mov Disord. 2013;28(7):863–73.
Aridon P, Ragonese P, De Fusco M, Salemi G, Casari G, Savettieri G. Further evidence of genetic heterogeneity in familial essential tremor. Parkinsonism Relat Disord. 2008;14(1):15–8.
Auer-Grumbach M, Strasser-Fuchs S, Wagner K, Körner E, Fazekas F. Roussy–Lévy syndrome is a phenotypic variant of Charcot-Marie-Tooth syndrome IA associated with a duplication on chromosome 17p11.2. J Neurol Sci. 1998;154(1):72–5.
Balestrino R, Schapira AHV. Parkinson disease. Eur J Neurol. 2020;27(1):27–42.
Barbieri F, Filla A, Ragno M, Crisci C, Santoro L, Corona M, Campanella G. Evidence that Charcot-Marie-Tooth disease with tremor coincides with the Roussy-Levy syndrome. Can J Neurol Sci. 1984;11(4 Suppl):534–40.
Blauwendraat C, Nalls MA, Singleton AB. The genetic architecture of Parkinson’s disease. Lancet Neurol. 2020;19(2):170–8.
Brin MF, Koller W. Epidemiology and genetics of essential tremor. Mov Disord. 1998;13(Suppl 3):55–63.
Camargos S, Scholz S, Simón-Sánchez J, Paisán-Ruiz C, Lewis P, Hernandez D, Ding J, Gibbs JR, Cookson MR, Bras J, Guerreiro R, Oliveira CR, Lees A, Hardy J, Cardoso F, Singleton AB. DYT16, a novel young-onset dystonia-parkinsonism disorder: identification of a segregating mutation in the stress-response protein PRKRA. Lancet Neurol. 2008;7(3):207–15.
Cen Z, Jiang Z, Chen Y, Zheng X, Xie F, Yang X, Lu X, Ouyang Z, Wu H, Chen S, Yin H, Qiu X, Wang S, Ding M, Tang Y, Yu F, Li C, Wang T, Ishiura H, Tsuji S, Jiao C, Liu C, Xiao J, Luo W. Intronic pentanucleotide TTTCA repeat insertion in the SAMD12 gene causes familial cortical myoclonic tremor with epilepsy type 1. Brain. 2018;141(8):2280–8.
Chanpong A, Dhawan A. Wilson disease in children and young adults – state of the art. Saudi J Gastroenterol. 2022;28(1):21–31.
Charlesworth G, Plagnol V, Holmström KM, Bras J, Sheerin UM, Preza E, Rubio-Agusti I, Ryten M, Schneider SA, Stamelou M, Trabzuni D, Abramov AY, Bhatia KP, Wood NW. Mutations in ANO3 cause dominant craniocervical dystonia: ion channel implicated in pathogenesis. Am J Hum Genet. 2012;91(6):1041–50.
Charlesworth G, Angelova PR, Bartolomé-Robledo F, Ryten M, Trabzuni D, Stamelou M, Abramov AY, Bhatia KP, Wood NW. Mutations in HPCA cause autosomal-recessive primary isolated dystonia. Am J Hum Genet. 2015;96(4):657–65.
Chartier-Harlin MC, Kachergus J, Roumier C, Mouroux V, Douay X, Lincoln S, Levecque C, Larvor L, Andrieux J, Hulihan M, Waucquier N, Defebvre L, Amouyel P, Farrer M, Destée A. Alpha-synuclein locus duplication as a cause of familial Parkinson’s disease. Lancet. 2004;364(9440):1167–9.
Coppedè F. One-carbon epigenetics and redox biology of neurodegeneration. Free Radic Biol Med. 2021;170:19–33.
Corbett MA, Kroes T, Veneziano L, Bennett MF, Florian R, Schneider AL, Coppola A, Licchetta L, Franceschetti S, Suppa A, Wenger A, Mei D, Pendziwiat M, Kaya S, Delledonne M, Straussberg R, Xumerle L, Regan B, Crompton D, van Rootselaar AF, Correll A, Catford R, Bisulli F, Chakraborty S, Baldassari S, Tinuper P, Barton K, Carswell S, Smith M, Berardelli A, Carroll R, Gardner A, Friend KL, Blatt I, Iacomino M, Di Bonaventura C, Striano S, Buratti J, Keren B, Nava C, Forlani S, Rudolf G, Hirsch E, Leguern E, Labauge P, Balestrini S, Sander JW, Afawi Z, Helbig I, Ishiura H, Tsuji S, Sisodiya SM, Casari G, Sadleir LG, van Coller R, Tijssen MAJ, Klein KM, van den Maagdenberg AMJM, Zara F, Guerrini R, Berkovic SF, Pippucci T, Canafoglia L, Bahlo M, Striano P, Scheffer IE, Brancati F, Depienne C, Gecz J. Intronic ATTTC repeat expansions in STARD7 in familial adult myoclonic epilepsy linked to chromosome 2. Nat Commun. 2019;10(1):4920.
Dalvi A, Mercury MG. Essential tremor–not just a shake. Dis Mon. 2011;57(3):127–34.
Day JO, Mullin S. The genetics of Parkinson’s disease and implications for clinical practice. Genes (Basel). 2021;12(7):1006.
Deng H, Le WD, Guo Y, Huang MS, Xie WJ, Jankovic J. Extended study of A265G variant of HS1BP3 in essential tremor and Parkinson disease. Neurology. 2005;65(4):651–2.
Deng H, Le W, Jankovic J. Genetics of essential tremor. Brain. 2007;130(Pt 6):1456–64.
Deng H, Dodson MW, Huang H, Guo M. The Parkinson’s disease genes pink1 and parkin promote mitochondrial fission and/or inhibit fusion in Drosophila. Proc Natl Acad Sci U S A. 2008;105(38):14503–8.
di Biase L, Di Santo A, Caminiti ML, Pecoraro PM, Di Lazzaro V. Classification of dystonia. Life (Basel). 2022;12(2):206.
Di Fonzo A, Dekker MC, Montagna P, Baruzzi A, Yonova EH, Correia Guedes L, Szczerbinska A, Zhao T, Dubbel-Hulsman LO, Wouters CH, de Graaff E, Oyen WJ, Simons EJ, Breedveld GJ, Oostra BA, Horstink MW, Bonifati V. FBXO7 mutations cause autosomal recessive, early-onset parkinsonian-pyramidal syndrome. Neurology. 2009;72(3):240–5.
Dorsey ER, Bloem BR. The Parkinson pandemic-a call to action. JAMA Neurol. 2018;75(1):9–10.
Edvardson S, Cinnamon Y, Ta-Shma A, Shaag A, Yim YI, Zenvirt S, Jalas C, Lesage S, Brice A, Taraboulos A, Kaestner KH, Greene LE, Elpeleg O. A deleterious mutation in DNAJC6 encoding the neuronal-specific clathrin-uncoating co-chaperone auxilin, is associated with juvenile parkinsonism. PLoS One. 2012;7(5):e36458.
Edwards TL, Scott WK, Almonte C, Burt A, Powell EH, Beecham GW, Wang L, Züchner S, Konidari I, Wang G, Singer C, Nahab F, Scott B, Stajich JM, Pericak-Vance M, Haines J, Vance JM, Martin ER. Genome-wide association study confirms SNPs in SNCA and the MAPT region as common risk factors for Parkinson disease. Ann Hum Genet. 2010;74(2):97–109.
Embiruçu EK, Martyn ML, Schlesinger D, Kok F. Autosomal recessive ataxias: 20 types, and counting. Arq Neuropsiquiatr. 2009;67(4):1143–56.
Farrer MJ, Stone JT, Lin CH, Dächsel JC, Hulihan MM, Haugarvoll K, Ross OA, Wu RM. Lrrk2 G2385R is an ancestral risk factor for Parkinson’s disease in Asia. Parkinsonism Relat Disord. 2007;13(2):89–92.
Ferrara J, Jankovic J. Epidemiology and management of essential tremor in children. Paediatr Drugs. 2009;11(5):293–307.
Florian RT, Kraft F, Leitão E, Kaya S, Klebe S, Magnin E, van Rootselaar AF, Buratti J, Kühnel T, Schröder C, Giesselmann S, Tschernoster N, Altmueller J, Lamiral A, Keren B, Nava C, Bouteiller D, Forlani S, Jornea L, Kubica R, Ye T, Plassard D, Jost B, Meyer V, Deleuze JF, Delpu Y, Avarello MDM, Vijfhuizen LS, Rudolf G, Hirsch E, Kroes T, Reif PS, Rosenow F, Ganos C, Vidailhet M, Thivard L, Mathieu A, Bourgeron T, Kurth I, Rafehi H, Steenpass L, Horsthemke B, FAME Consortium, LeGuern E, Klein KM, Labauge P, Bennett MF, Bahlo M, Gecz J, Corbett MA, Tijssen MAJ, van den Maagdenberg AMJM, Depienne C. Unstable TTTTA/TTTCA expansions in MARCH6 are associated with familial adult myoclonic epilepsy type 3. Nat Commun. 2019;10(1):4919.
Foo JN, Chew EGY, Chung SJ, Peng R, Blauwendraat C, Nalls MA, Mok KY, Satake W, Toda T, Chao Y, Tan LCS, Tandiono M, Lian MM, Ng EY, Prakash KM, Au WL, Meah WY, Mok SQ, Annuar AA, Chan AYY, Chen L, Chen Y, Jeon BS, Jiang L, Lim JL, Lin JJ, Liu C, Mao C, Mok V, Pei Z, Shang HF, Shi CH, Song K, Tan AH, Wu YR, Xu YM, Xu R, Yan Y, Yang J, Zhang B, Koh WP, Lim SY, Khor CC, Liu J, Tan EK. Identification of risk loci for Parkinson disease in Asians and comparison of risk between Asians and Europeans: a genome-wide association study. JAMA Neurol. 2020;77(6):746–54.
Fuchs J, Tichopad A, Golub Y, Munz M, Schweitzer KJ, Wolf B, Berg D, Mueller JC, Gasser T. Genetic variability in the SNCA gene influences alpha-synuclein levels in the blood and brain. FASEB J. 2008;22(5):1327–34.
Fuchs T, Gavarini S, Saunders-Pullman R, Raymond D, Ehrlich ME, Bressman SB, Ozelius LJ. Mutations in the THAP1 gene are responsible for DYT6 primary torsion dystonia. Nat Genet. 2009;41(3):286–8.
Fuchs T, Saunders-Pullman R, Masuho I, Luciano MS, Raymond D, Factor S, Lang AE, Liang TW, Trosch RM, White S, Ainehsazan E, Hervé D, Sharma N, Ehrlich ME, Martemyanov KA, Bressman SB, Ozelius LJ. Mutations in GNAL cause primary torsion dystonia. Nat Genet. 2013;45(1):88–92.
Gegg ME, Menozzi E, Schapira AHV. Glucocerebrosidase-associated Parkinson disease: Pathogenic mechanisms and potential drug treatments. Neurobiol Dis. 2022;166:105663.
Gueven N, Chen P, Nakamura J, Becherel OJ, Kijas AW, Grattan-Smith P, Lavin MF. A subgroup of spinocerebellar ataxias defective in DNA damage responses. Neuroscience. 2007;145:1418–25.
Gulcher JR, Jónsson P, Kong A, Kristjánsson K, Frigge ML, Kárason A, Einarsdóttir IE, Stefánsson H, Einarsdóttir AS, Sigurthoardóttir S, Baldursson S, Björnsdóttir S, Hrafnkelsdóttir SM, Jakobsson F, Benedickz J, Stefánsson K. Mapping of a familial essential tremor gene, FET1, to chromosome 3q13. Nat Genet. 1997;17(1):84–7.
Halperin A, Elstein D, Zimran A. Increased incidence of Parkinson disease among relatives of patients with Gaucher disease. Blood Cells Mol Dis. 2006;36(3):426–8.
Hardy J. Genetic analysis of pathways to Parkinson disease. Neuron. 2010;68(2):201–6.
Hayakawa Y, Itoh M, Yamada A, Mitsuda T, Nakagawa T. Expression and localization of Cayman ataxia-related protein, Caytaxin, is regulated in a developmental- and spatial-dependent manner. Brain Res. 2007;1129:100–9.
Heaton GR, Landeck N, Mamais A, Nalls MA, Nixon-Abell J, Kumaran R, Beilina A, Pellegrini L, Li Y, International Parkinson Disease Genomics Consortium (IPDGC), Harvey K, Cookson MR. Sequential screening nominates the Parkinson’s disease associated kinase LRRK2 as a regulator of Clathrin-mediated endocytosis. Neurobiol Dis. 2020;141:104948.
Hicks JE, Konidari I, Scott BL, Stajich JM, Ashley-Koch AE, Gilbert JR, Scott WK. Linkage of familial essential tremor to chromosome 5q35. Mov Disord. 2016;31(7):1059–62.
Higgins JJ, Pho LT, Nee LE. A gene (ETM) for essential tremor maps to chromosome 2p22-p25. Mov Disord. 1997;12(6):859–64.
Higgins JJ, Lombardi RQ, Pucilowska J, Jankovic J, Tan EK, Rooney JP. A variant in the HS1-BP3 gene is associated with familial essential tremor. Neurology. 2005;64(3):417–21.
Houle G, Ambalavanan A, Schmouth JF, Leblond CS, Spiegelman D, Laurent SB, Bourassa CV, Grayson C, Panisset M, Chouinard S, Dupré N, Vilariño-Güell C, Rajput A, Girard SL, Dion PA, Rouleau GA. No rare deleterious variants from STK32B, PPARGC1A, and CTNNA3 are associated with essential tremor. Neurol Genet. 2017;3(5):e195.
Huster D. Wilson disease. Best Pract Res Clin Gastroenterol. 2010;24(5):531–9.
Ichinose H, Ohye T, Takahashi E, Seki N, Hori T, Segawa M, Nomura Y, Endo K, Tanaka H, Tsuji S, et al. Hereditary progressive dystonia with marked diurnal fluctuation caused by mutations in the GTP cyclohydrolase I gene. Nat Genet. 1994;8(3):236–42.
Ishiura H, Doi K, Mitsui J, Yoshimura J, Matsukawa MK, Fujiyama A, Toyoshima Y, Kakita A, Takahashi H, Suzuki Y, Sugano S, Qu W, Ichikawa K, Yurino H, Higasa K, Shibata S, Mitsue A, Tanaka M, Ichikawa Y, Takahashi Y, Date H, Matsukawa T, Kanda J, Nakamoto FK, Higashihara M, Abe K, Koike R, Sasagawa M, Kuroha Y, Hasegawa N, Kanesawa N, Kondo T, Hitomi T, Tada M, Takano H, Saito Y, Sanpei K, Onodera O, Nishizawa M, Nakamura M, Yasuda T, Sakiyama Y, Otsuka M, Ueki A, Kaida KI, Shimizu J, Hanajima R, Hayashi T, Terao Y, Inomata-Terada S, Hamada M, Shirota Y, Kubota A, Ugawa Y, Koh K, Takiyama Y, Ohsawa-Yoshida N, Ishiura S, Yamasaki R, Tamaoka A, Akiyama H, Otsuki T, Sano A, Ikeda A, Goto J, Morishita S, Tsuji S. Expansions of intronic TTTCA and TTTTA repeats in benign adult familial myoclonic epilepsy. Nat Genet. 2018;50(4):581–90.
Iwaki A, Kawano Y, Miura S, Shibata H, Matsuse D, Li W, Furuya H, Ohyagi Y, Taniwaki T, Kira J, Fukumaki Y. Heterozygous deletion of ITPR1, but not SUMF1, in spinocerebellar ataxia type 16. J Med Genet. 2008;45(1):32–5.
Iwaki H, Blauwendraat C, Makarious MB, Bandrés-Ciga S, Leonard HL, Gibbs JR, Hernandez DG, Scholz SW, Faghri F, International Parkinson’s Disease Genomics Consortium (IPDGC), Nalls MA, Singleton AB. Penetrance of Parkinson’s disease in LRRK2 p.G2019S carriers is modified by a polygenic risk score. Mov Disord. 2020;35(5):774–80.
Jeanneteau F, Funalot B, Jankovic J, Deng H, Lagarde JP, Lucotte G, Sokoloff P. A functional variant of the dopamine D3 receptor is associated with risk and age-at-onset of essential tremor. Proc Natl Acad Sci U S A. 2006;103(28):10753–8.
Jia F, Fellner A, Kumar KR. Monogenic Parkinson’s disease: genotype, phenotype, pathophysiology, and genetic testing. Genes (Basel). 2022;13(3):471.
Jiménez-Jiménez FJ, Alonso-Navarro H, García-Martín E, Álvarez I, Pastor P, Agúndez JAG. Genomic markers for essential tremor. Pharmaceuticals (Basel). 2021;14(6):516.
Kawajiri S, Saiki S, Sato S, Hattori N. Genetic mutations and functions of PINK1. Trends Pharmacol Sci. 2011;32(10):573–80.
Kay DM, Factor SA, Samii A, Higgins DS, Griffith A, Roberts JW, Leis BC, Nutt JG, Montimurro JS, Keefe RG, Atkins AJ, Yearout D, Zabetian CP, Payami H. Genetic association between alpha-synuclein and idiopathic Parkinson’s disease. Am J Med Genet B Neuropsychiatr Genet. 2008;147B(7):1222–30.
Kim JM, Hong S, Kim GP, Choi YJ, Kim YK, Park SS, Kim SE, Jeon BS. Importance of low-range CAG expansion and CAA interruption in SCA2 parkinsonism. Arch Neurol. 2007;64(10):1510–8.
Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, Yokochi M, Mizuno Y, Shimizu N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature. 1998;392(6676):605–8.
Knight MA, Gardner RJ, Bahlo M, Matsuura T, Dixon JA, Forrest SM, Storey E. Dominantly inherited ataxia and dysphonia with dentate calcification: spinocerebellar ataxia type 20. Brain. 2004;127(Pt 5):1172–81.
Köroğlu Ç, Baysal L, Cetinkaya M, Karasoy H, Tolun A. DNAJC6 is responsible for juvenile parkinsonism with phenotypic variability. Parkinsonism Relat Disord. 2013;19(3):320–4.
Krebs CE, Karkheiran S, Powell JC, Cao M, Makarov V, Darvish H, Di Paolo G, Walker RH, Shahidi GA, Buxbaum JD, De Camilli P, Yue Z, Paisán-Ruiz C. The Sac1 domain of SYNJ1 identified mutated in a family with early-onset progressive Parkinsonism with generalized seizures. Hum Mutat. 2013;34(9):1200–7.
Krüger R, Kuhn W, Müller T, Woitalla D, Graeber M, Kösel S, Przuntek H, Epplen JT, Schöls L, Riess O. Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson’s disease. Nat Genet. 1998;18(2):106–8.
Krygier M, Mazurkiewicz-Bełdzińska M. Milestones in genetics of cerebellar ataxias. Neurogenetics. 2021;22(4):225–34.
Lastres-Becker I, Rüb U, Auburger G. Spinocerebellar ataxia 2 (SCA2). Cerebellum. 2008;7(2):115–24.
Leehey MA. Fragile X-associated tremor/ataxia syndrome: clinical phenotype, diagnosis, and treatment. J Investig Med. 2009;57(8):830–6.
Lei XX, Liu Q, Lu Q, Huang Y, Zhou XQ, Sun HY, Wu LW, Cui LY, Zhang X. TTTCA repeat expansion causes familial cortical myoclonic tremor with epilepsy. Eur J Neurol. 2019;26(3):513–8.
Lenka A, Jankovic J. Tremor syndromes: an updated review. Front Neurol. 2021;12:684835.
Leroy E, Boyer R, Auburger G, Leube B, Ulm G, Mezey E, Harta G, Brownstein MJ, Jonnalagada S, Chernova T, Dehejia A, Lavedan C, Gasser T, Steinbach PJ, Wilkinson KD, Polymeropoulos MH. The ubiquitin pathway in Parkinson’s disease. Nature. 1998;395(6701):451–2.
Lesage S, Drouet V, Majounie E, Deramecourt V, Jacoupy M, Nicolas A, Cormier-Dequaire F, Hassoun SM, Pujol C, Ciura S, Erpapazoglou Z, Usenko T, Maurage CA, Sahbatou M, Liebau S, Ding J, Bilgic B, Emre M, Erginel-Unaltuna N, Guven G, Tison F, Tranchant C, Vidailhet M, Corvol JC, Krack P, Leutenegger AL, Nalls MA, Hernandez DG, Heutink P, Gibbs JR, Hardy J, Wood NW, Gasser T, Durr A, Deleuze JF, Tazir M, Destée A, Lohmann E, Kabashi E, Singleton A, Corti O, Brice A, French Parkinson’s Disease Genetics Study (PDG); International Parkinson’s Disease Genomics Consortium (IPDGC). Loss of VPS13C function in autosomal-recessive parkinsonism causes mitochondrial dysfunction and increases PINK1/Parkin-dependent mitophagy. Am J Hum Genet. 2016;98(3):500–13.
Liao C, Castonguay CE, Heilbron K, Vuokila V, Medeiros M, Houle G, Akçimen F, Ross JP, Catoire H, Diez-Fairen M, Kang J, Mueller SH, Girard SL, Hopfner F, Lorenz D, Clark LN, Soto-Beasley AI, Klebe S, Hallett M, Wszolek ZK, Pendziwiat M, Lorenzo-Betancor O, Seppi K, Berg D, Vilariño-Güell C, Postuma RB, Bernard G, Dupré N, Jankovic J, Testa CM, Ross OA, Arzberger T, Chouinard S, Louis ED, Mandich P, Vitale C, Barone P, García-Martín E, Alonso-Navarro H, JAG A, Jiménez-Jiménez FJ, Pastor P, Rajput A, Deuschl G, Kuhlenbaümer G, Meijer IA, Dion PA, Rouleau GA, 23andMe Research Team. Association of essential tremor with novel risk loci: a genome-wide association study and meta-analysis. JAMA Neurol. 2022;79(2):185–93.
Liu S, Ninan I, Antonova I, Battaglia F, Trinchese F, Narasanna A, Kolodilov N, Dauer W, Hawkins RD, Arancio O. Alpha-synuclein produces a long-lasting increase in neurotransmitter release. EMBO J. 2004;23(22):4506–16.
Lockhart PJ, Lincoln S, Hulihan M, Kachergus J, Wilkes K, Bisceglio G, Mash DC, Farrer MJ. DJ-1 mutations are a rare cause of recessively inherited early onset parkinsonism mediated by loss of protein function. J Med Genet. 2004;41(3):e22.
Lorenz D, Frederiksen H, Moises H, Kopper F, Deuschl G, Christensen K. High concordance for essential tremor in monozygotic twins of old age. Neurology. 2004;62(2):208–11.
Lorenz D, Klebe S, Stevanin G, Thier S, Nebel A, Feingold J, Frederiksen H, Denis E, Christensen K, Schreiber S, Brice A, Deuschl G, Dürr A. Dopamine receptor D3 gene and essential tremor in large series of German, Danish and French patients. Eur J Hum Genet. 2009;17(6):766–73.
Lorincz MT. Neurologic Wilson’s disease. Ann N Y Acad Sci. 2010;1184:173–87.
Louis ED, Ferreira JJ. How common is the most common adult movement disorder? Update on the worldwide prevalence of essential tremor. Mov Disord. 2010;25(5):534–41.
Louis ED, McCreary M. How common is essential tremor? Update on the worldwide prevalence of essential tremor. Tremor Other Hyperkinet Mov (N Y). 2021;11:28.
Louis ED, Faust PL, Vonsattel JP, Honig LS, Rajput A, Robinson CA, Rajput A, Pahwa R, Lyons KE, Ross GW, Borden S, Moskowitz CB, Lawton A, Hernandez N. Neuropathological changes in essential tremor: 33 cases compared with 21 controls. Brain. 2007;130(Pt 12):3297–307.
Lu CS, Chang HC, Kuo PC, Liu YL, Wu WS, Weng YH, Yen TC, Chou YH. The parkinsonian phenotype of spinocerebellar ataxia type 3 in a Taiwanese family. Parkinsonism Relat Disord. 2004;10(6):369–73.
Magrinelli F, Latorre A, Balint B, Mackenzie M, Mulroy E, Stamelou M, Tinazzi M, Bhatia KP. Isolated and combined genetic tremor syndromes: a critical appraisal based on the 2018 MDS criteria. Parkinsonism Relat Disord. 2020;77:121–40.
Makino S, Kaji R, Ando S, Tomizawa M, Yasuno K, Goto S, Matsumoto S, Tabuena MD, Maranon E, Dantes M, Lee LV, Ogasawara K, Tooyama I, Akatsu H, Nishimura M, Tamiya G. Reduced neuron-specific expression of the TAF1 gene is associated with X-linked dystonia-parkinsonism. Am J Hum Genet. 2007;80(3):393–406.
Manto M. Cerebellar disorders. A practical approach to diagnosis and management. Cambridge: Cambridge University Press; 2010.
Manto M, Marmolino D. Cerebellar ataxias. Curr Opin Neurol. 2009;22(4):419–29.
Manzoor H, Brüggemann N, Hussain HMJ, Bäumer T, Hinrichs F, Wajid M, Münchau A, Naz S, Lohmann K. Novel homozygous variants in ATCAY, MCOLN1, and SACS in complex neurological disorders. Parkinsonism Relat Disord. 2018;51:91–5.
Maraganore DM, de Andrade M, Elbaz A, Farrer MJ, Ioannidis JP, Krüger R, Rocca WA, Schneider NK, Lesnick TG, Lincoln SJ, Hulihan MM, Aasly JO, Ashizawa T, Chartier-Harlin MC, Checkoway H, Ferrarese C, Hadjigeorgiou G, Hattori N, Kawakami H, Lambert JC, Lynch T, Mellick GD, Papapetropoulos S, Parsian A, Quattrone A, Riess O, Tan EK, Van Broeckhoven C, Genetic Epidemiology of Parkinson’s Disease (GEO-PD) Consortium. Collaborative analysis of alpha-synuclein gene promoter variability and Parkinson disease. JAMA. 2006;296(6):661–70.
Mata IF, Lockhart PJ, Farrer MJ. Parkin genetics: one model for Parkinson’s disease. Hum Mol Genet. 2004;13 Spec No 1:R127–33.
Mata IF, Yearout D, Alvarez V, Coto E, de Mena L, Ribacoba R, Lorenzo-Betancor O, Samaranch L, Pastor P, Cervantes S, Infante J, Garcia-Gorostiaga I, Sierra M, Combarros O, Snapinn KW, Edwards KL, Zabetian CP. Replication of MAPT and SNCA, but not PARK16-18, as susceptibility genes for Parkinson’s disease. Mov Disord. 2011;26(5):819–23.
Matos CA, de Macedo-Ribeiro S, Carvalho AL. Polyglutamine diseases: the special case of ataxin-3 and Machado-Joseph disease. Prog Neurobiol. 2011;95(1):26–48.
Mencacci NE, Jinnah HA. Naming genes for dystonia: DYT-z or ditzy? Tremor Other Hyperkinet Mov (N Y). 2019:9. https://doi.org/10.7916/tohm.v0.710.eCollection2019
Migliore L, Coppedè F. Genetics, environmental factors and the emerging role of epigenetics in neurodegenerative diseases. Mutat Res. 2009;667(1–2):82–97.
Müller U. The monogenic primary dystonias. Brain. 2009;132(Pt 8):2005–25.
Müller U. Spinocerebellar ataxias (SCAs) caused by common mutations. Neurogenetics. 2021;22(4):235–50.
Müller SH, Girard SL, Hopfner F, Merner ND, Bourassa CV, Lorenz D, Clark LN, Tittmann L, Soto-Ortolaza AI, Klebe S, Hallett M, Schneider SA, Hodgkinson CA, Lieb W, Wszolek ZK, Pendziwiat M, Lorenzo-Betancor O, Poewe W, Ortega-Cubero S, Seppi K, Rajput A, Hussl A, Rajput AH, Berg D, Dion PA, Wurster I, Shulman JM, Srulijes K, Haubenberger D, Pastor P, Vilariño-Güell C, Postuma RB, Bernard G, Ladwig KH, Dupré N, Jankovic J, Strauch K, Panisset M, Winkelmann J, Testa CM, Reischl E, Zeuner KE, Ross OA, Arzberger T, Chouinard S, Deuschl G, Louis ED, Kuhlenbäumer G, Rouleau GA. Genome-wide association study in essential tremor identifies three new loci. Brain. 2016;139(Pt 12):3163–9.
Nalls MA, Blauwendraat C, Vallerga CL, Heilbron K, Bandres-Ciga S, Chang D, Tan M, Kia DA, Noyce AJ, Xue A, Bras J, Young E, von Coelln R, Simón-Sánchez J, Schulte C, Sharma M, Krohn L, Pihlstrøm L, Siitonen A, Iwaki H, Leonard H, Faghri F, Gibbs JR, Hernandez DG, Scholz SW, Botia JA, Martinez M, Corvol JC, Lesage S, Jankovic J, Shulman LM, Sutherland M, Tienari P, Majamaa K, Toft M, Andreassen OA, Bangale T, Brice A, Yang J, Gan-Or Z, Gasser T, Heutink P, Shulman JM, Wood NW, Hinds DA, Hardy JA, Morris HR, Gratten J, Visscher PM, Graham RR, Singleton AB, 23andMe Research Team; System Genomics of Parkinson’s Disease Consortium; International Parkinson’s Disease Genomics Consortium. Identification of novel risk loci, causal insights, and heritable risk for Parkinson’s disease: a meta-analysis of genome-wide association studies. Lancet Neurol. 2019;18(12):1091–102.
Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol. 2008;183(5):795–803.
Novelletto A, Gulli R, Ciotti P, Vitale C, Malaspina P, Blasi P, Pippucci T, Seri M, Cozzolino A, Bilo L, Abbruzzese G, Martinelli P, Bellone E, Barone P, Mandich P. Linkage exclusion in Italian families with hereditary essential tremor. Eur J Neurol. 2011;18(9):e118–20.
Nuytemans K, Theuns J, Cruts M, Van Broeckhoven C. Genetic etiology of Parkinson disease associated with mutations in the SNCA, PARK2, PINK1, PARK7, and LRRK2 genes: a mutation update. Hum Mutat. 2010;31(7):763–80.
O’Hearn E, Holmes SE, Margolis RL. Spinocerebellar ataxia type 12. Handb Clin Neurol. 2011;103:535–47.
Olgiati S, Quadri M, Fang M, Rood JP, Saute JA, Chien HF, Bouwkamp CG, Graafland J, Minneboo M, Breedveld GJ, Zhang J, International Parkinsonism Genetics Network, Verheijen FW, Boon AJ, Kievit AJ, Jardim LB, Mandemakers W, Barbosa ER, Rieder CR, Leenders KL, Wang J, Bonifati V. DNAJC6 mutations associated with early-onset Parkinson’s disease. Ann Neurol. 2016;79(2):244–56.
Ong YL, Deng X, Tan EK. Etiologic links between environmental and lifestyle factors and essential tremor. Ann Clin Transl Neurol. 2019;6(5):979–89.
Ozelius LJ, Hewett JW, Page CE, Bressman SB, Kramer PL, Shalish C, de Leon D, Brin MF, Raymond D, Corey DP, Fahn S, Risch NJ, Buckler AJ, Gusella JF, Breakefield XO. The early-onset torsion dystonia gene (DYT1) encodes an ATP-binding protein. Nat Genet. 1997;17(1):40–8.
Paisán-Ruíz C, Jain S, Evans EW, Gilks WP, Simón J, van der Brug M, López de Munain A, Aparicio S, Gil AM, Khan N, Johnson J, Martinez JR, Nicholl D, Carrera IM, Pena AS, de Silva R, Lees A, Martí-Massó JF, Pérez-Tur J, Wood NW, Singleton AB. Cloning of the gene containing mutations that cause PARK8-linked Parkinson’s disease. Neuron. 2004;44(4):595–600.
Paisán-Ruiz C, Bhatia KP, Li A, Hernandez D, Davis M, Wood NW, Hardy J, Houlden H, Singleton A, Schneider SA. Characterization of PLA2G6 as a locus for dystonia-parkinsonism. Ann Neurol. 2009;65(1):19–23.
Paisán-Ruiz C, Guevara R, Federoff M, Hanagasi H, Sina F, Elahi E, Schneider SA, Schwingenschuh P, Bajaj N, Emre M, Singleton AB, Hardy J, Bhatia KP, Brandner S, Lees AJ, Houlden H. Early-onset L-dopa-responsive parkinsonism with pyramidal signs due to ATP13A2, PLA2G6, FBXO7 and spatacsin mutations. Mov Disord. 2010;25(12):1791–800.
Pandey S, Bhattad S, Dinesh S. Tremor in primary monogenic dystonia. Curr Neurol Neurosci Rep. 2021;21(9):48.
Pankratz N, Wilk JB, Latourelle JC, DeStefano AL, Halter C, Pugh EW, Doheny KF, Gusella JF, Nichols WC, Foroud T, Myers RH, PSG-PROGENI and GenePD Investigators, Coordinators and Molecular Genetic Laboratories. Genomewide association study for susceptibility genes contributing to familial Parkinson disease. Hum Genet. 2009;124(6):593–605.
Patzkó A, Shy ME. Update on Charcot-Marie-Tooth disease. Curr Neurol Neurosci Rep. 2011;11(1):78–88.
Plante-Bordeneuve V, Guiochon-Mantel A, Lacroix C, Lapresle J, Said G. The Roussy-Levy family: from the original description to the gene. Ann Neurol. 1999;46:770–3.
Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanassiadou A, Papapetropoulos T, Johnson WG, Lazzarini AM, Duvoisin RC, Di Iorio G, Golbe LI, Nussbaum RL. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science. 1997;276(5321):2045–7.
Poole AC, Thomas RE, Andrews LA, McBride HM, Whitworth AJ, Pallanck LJ. The PINK1/Parkin pathway regulates mitochondrial morphology. Proc Natl Acad Sci U S A. 2008;105(5):1638–43.
Quadri M, Fang M, Picillo M, Olgiati S, Breedveld GJ, Graafland J, Wu B, Xu F, Erro R, Amboni M, Pappatà S, Quarantelli M, Annesi G, Quattrone A, Chien HF, Barbosa ER, International Parkinsonism Genetics Network, Oostra BA, Barone P, Wang J, Bonifati V. Mutation in the SYNJ1 gene associated with autosomal recessive, early-onset Parkinsonism. Hum Mutat. 2013;34(9):1208–15.
Ramirez A, Heimbach A, Gründemann J, Stiller B, Hampshire D, Cid LP, Goebel I, Mubaidin AF, Wriekat AL, Roeper J, Al-Din A, Hillmer AM, Karsak M, Liss B, Woods CG, Behrens MI, Kubisch C. Hereditary parkinsonism with dementia is caused by mutations in ATP13A2, encoding a lysosomal type 5 P-type ATPase. Nat Genet. 2006;38(10):1184–91.
Regragui W, Gerdelat-Mas A, Simonetta-Moreau M. Cortical tremor (FCMTE: familial cortical myoclonic tremor with epilepsy). Neurophysiol Clin. 2006;36(5–6):345–9.
Richardson D, McEntagart MM, Isaacs JD. DCTN1-related Parkinson-plus disorder (Perry syndrome). Pract Neurol. 2020;20(4):317–9.
Rothfuss O, Fischer H, Hasegawa T, Maisel M, Leitner P, Miesel F, Sharma M, Bornemann A, Berg D, Gasser T, Patenge N. Parkin protects mitochondrial genome integrity and supports mitochondrial DNA repair. Hum Mol Genet. 2009;18(20):3832–50.
Rudakou U, Ruskey JA, Krohn L, Laurent SB, Spiegelman D, Greenbaum L, Yahalom G, Desautels A, Montplaisir JY, Fahn S, Waters CH, Levy O, Kehoe CM, Narayan S, Dauvilliers Y, Dupré N, Hassin-Baer S, Alcalay RN, Rouleau GA, Fon EA, Gan-Or Z. Analysis of common and rare VPS13C variants in late-onset Parkinson disease. Neurol Genet. 2020;6(1):385.
Salisachs P. Unusual motor conduction velocity values in Charcot-Marie-Tooth disease associated with essential tremor: report of a kinship. Eur Neurol. 1975;13(4):377–82.
Salisachs P. Charcot-Marie-Tooth disease associated with “essential tremor”: report of 7 cases and a review of the literature. J Neurol Sci. 1976;28(1):17–40.
Sassone J, Reale C, Dati G, Regoni M, Pellecchia MT, Garavaglia B. The role of VPS35 in the pathobiology of Parkinson’s disease. Cell Mol Neurobiol. 2021;41(2):199–227.
Satake W, Nakabayashi Y, Mizuta I, Hirota Y, Ito C, Kubo M, Kawaguchi T, Tsunoda T, Watanabe M, Takeda A, Tomiyama H, Nakashima K, Hasegawa K, Obata F, Yoshikawa T, Kawakami H, Sakoda S, Yamamoto M, Hattori N, Murata M, Nakamura Y, Toda T. Genome-wide association study identifies common variants at four loci as genetic risk factors for Parkinson’s disease. Nat Genet. 2009;41(12):1303–7.
Shatunov A, Jankovic J, Elble R, Sambuughin N, Singleton A, Hallett M, Goldfarb L. A variant in the HS1-BP3 gene is associated with familial essential tremor. Neurology. 2005;65(12):1995.
Shatunov A, Sambuughin N, Jankovic J, Elble R, Lee HS, Singleton AB, Dagvadorj A, Ji J, Zhang Y, Kimonis VE, Hardy J, Hallett M, Goldfarb LG. Genomewide scans in North American families reveal genetic linkage of essential tremor to a region on chromosome 6p23. Brain. 2006;129(Pt 9):2318–31.
Shojaee S, Sina F, Banihosseini SS, Kazemi MH, Kalhor R, Shahidi GA, Fakhrai-Rad H, Ronaghi M, Elahi E. Genome-wide linkage analysis of a Parkinsonian-pyramidal syndrome pedigree by 500 K SNP arrays. Am J Hum Genet. 2008;82(6):1375–84.
Sidransky E, Nalls MA, Aasly JO, Aharon-Peretz J, Annesi G, Barbosa ER, Bar-Shira A, Berg D, Bras J, Brice A, Chen CM, Clark LN, Condroyer C, De Marco EV, Dürr A, Eblan MJ, Fahn S, Farrer MJ, Fung HC, Gan-Or Z, Gasser T, Gershoni-Baruch R, Giladi N, Griffith A, Gurevich T, Januario C, Kropp P, Lang AE, Lee-Chen GJ, Lesage S, Marder K, Mata IF, Mirelman A, Mitsui J, Mizuta I, Nicoletti G, Oliveira C, Ottman R, Orr-Urtreger A, Pereira LV, Quattrone A, Rogaeva E, Rolfs A, Rosenbaum H, Rozenberg R, Samii A, Samaddar T, Schulte C, Sharma M, Singleton A, Spitz M, Tan EK, Tayebi N, Toda T, Troiano AR, Tsuji S, Wittstock M, Wolfsberg TG, Wu YR, Zabetian CP, Zhao Y, Ziegler SG. Multicenter analysis of glucocerebrosidase mutations in Parkinson’s disease. N Engl J Med. 2009;361(17):1651–61.
Simón-Sánchez J, Schulte C, Bras JM, Sharma M, Gibbs JR, Berg D, Paisán-Ruiz C, Lichtner P, Scholz SW, Hernandez DG, Krüger R, Federoff M, Klein C, Goate A, Perlmutter J, Bonin M, Nalls MA, Illig T, Gieger C, Houlden H, Steffens M, Okun MS, Racette BA, Cookson MR, Foote KD, Fernandez HH, Traynor BJ, Schreiber S, Arepalli S, Zonozi R, Gwinn K, van der Brug M, Lopez G, Chanock SJ, Schatzkin A, Park Y, Hollenbeck A, Gao J, Huang X, Wood NW, Lorenz D, Deuschl G, Chen H, Riess O, Hardy JA, Singleton AB, Gasser T. Genome-wide association study reveals genetic risk underlying Parkinson’s disease. Nat Genet. 2009;41(12):1308–12.
Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, Hulihan M, Peuralinna T, Dutra A, Nussbaum R, Lincoln S, Crawley A, Hanson M, Maraganore D, Adler C, Cookson MR, Muenter M, Baptista M, Miller D, Blancato J, Hardy J, Gwinn-Hardy K. Alpha-synuclein locus triplication causes Parkinson’s disease. Science. 2003;302(5646):841.
Srivastava AK, Takkar A, Garg A, Faruq M. Clinical behaviour of spinocerebellar ataxia type 12 and intermediate length abnormal CAG repeats in PPP2R2B. Brain. 2017;140(1):27–36.
Stefansson H, Steinberg S, Petursson H, Gustafsson O, Gudjonsdottir IH, Jonsdottir GA, Palsson ST, Jonsson T, Saemundsdottir J, Bjornsdottir G, Böttcher Y, Thorlacius T, Haubenberger D, Zimprich A, Auff E, Hotzy C, Testa CM, Miyatake LA, Rosen AR, Kristleifsson K, Rye D, Asmus F, Schöls L, Dichgans M, Jakobsson F, Benedikz J, Thorsteinsdottir U, Gulcher J, Kong A, Stefansson K. Variant in the sequence of the LINGO1 gene confers risk of essential tremor. Nat Genet. 2009;41(3):277–9.
Stogmann E, Reinthaler E, Eltawil S, El Etribi MA, Hemeda M, El Nahhas N, Gaber AM, Fouad A, Edris S, Benet-Pages A, Eck SH, Pataraia E, Mei D, Brice A, Lesage S, Guerrini R, Zimprich F, Strom TM, Zimprich A. Autosomal recessive cortical myoclonic tremor and epilepsy: association with a mutation in the potassium channel associated gene CNTN2. Brain. 2013;136(Pt 4):1155–60.
Storey E, Gardner RJ. Spinocerebellar ataxia type 15. Handb Clin Neurol. 2012;103:561–5.
Tayebi N, Walker J, Stubblefield B, Orvisky E, LaMarca ME, Wong K, Rosenbaum H, Schiffmann R, Bembi B, Sidransky E. Gaucher disease with parkinsonian manifestations: does glucocerebrosidase deficiency contribute to a vulnerability to parkinsonism? Mol Genet Metab. 2003;79(2):104–9.
Thier S, Lorenz D, Nothnagel M, Poremba C, Papengut F, Appenzeller S, Paschen S, Hofschulte F, Hussl AC, Hering S, Poewe W, Asmus F, Gasser T, Schöls L, Christensen K, Nebel A, Schreiber S, Klebe S, Deuschl G, Kuhlenbäumer G. Polymorphisms in the glial glutamate transporter SLC1A2 are associated with essential tremor. Neurology. 2012;79(3):243–8.
Thomas B, Beal MF. Molecular insights into Parkinson’s disease. F1000 Med Rep. 2011;3:7.
Thomas GR, Forbes JR, Roberts EA, Walshe JM, Cox DW. The Wilson disease gene: spectrum of mutations and their consequences. Nat Genet. 1995;9(2):210–7.
Thomas KJ, McCoy MK, Blackinton J, Beilina A, van der Brug M, Sandebring A, Miller D, Maric D, Cedazo-Minguez A, Cookson MR. DJ-1 acts in parallel to the PINK1/parkin pathway to control mitochondrial function and autophagy. Hum Mol Genet. 2011;20(1):40–50.
Valente EM, Abou-Sleiman PM, Caputo V, Muqit MM, Harvey K, Gispert S, Ali Z, Del Turco D, Bentivoglio AR, Healy DG, Albanese A, Nussbaum R, González-Maldonado R, Deller T, Salvi S, Cortelli P, Gilks WP, Latchman DS, Harvey RJ, Dallapiccola B, Auburger G, Wood NW. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science. 2004;304(5674):1158–60.
van den Ende T, Sharifi S, van der Salm SMA, van Rootselaar AF. Familial cortical myoclonic tremor and epilepsy, an enigmatic disorder: from phenotypes to pathophysiology and genetics. A systematic review. Tremor Other Hyperkinet Mov (N Y). 2018;8:503.
van Duijn CM, Dekker MC, Bonifati V, Galjaard RJ, Houwing-Duistermaat JJ, Snijders PJ, Testers L, Breedveld GJ, Horstink M, Sandkuijl LA, van Swieten JC, Oostra BA, Heutink P. Park7, a novel locus for autosomal recessive early-onset parkinsonism, on chromosome 1p36. Am J Hum Genet. 2001;69(3):629–34.
van Swieten JC, Brusse E, de Graaf BM, Krieger E, van de Graaf R, de Koning I, Maat-Kievit A, Leegwater P, Dooijes D, Oostra BA, Heutink P. A mutation in the fibroblast growth factor 14 gene is associated with autosomal dominant cerebellar ataxia [corrected]. Am J Hum Genet. 2003;72(1):191–9.
van Veen S, Martin S, Van den Haute C, Benoy V, Lyons J, Vanhoutte R, Kahler JP, Decuypere JP, Gelders G, Lambie E, Zielich J, Swinnen JV, Annaert W, Agostinis P, Ghesquière B, Verhelst S, Baekelandt V, Eggermont J, Vangheluwe P. ATP13A2 deficiency disrupts lysosomal polyamine export. Nature. 2020;578(7795):419–24.
Vela-Desojo L, Urso D, Osuna-López M, Hoenicka J. Non-motor symptoms in PLA2G6-associated dystonia-parkinsonism: a case report and literature review. J Clin Med. 2022;11(6):1590.
Velayati A, Yu WH, Sidransky E. The role of glucocerebrosidase mutations in Parkinson disease and Lewy body disorders. Curr Neurol Neurosci Rep. 2010;10(3):190–8.
Verbeek MM, Steenbergen-Spanjers GC, Willemsen MA, Hol FA, Smeitink J, Seeger J, Grattan-Smith P, Ryan MM, Hoffmann GF, Donati MA, Blau N, Wevers RA. Mutations in the cyclic adenosine monophosphate response element of the tyrosine hydroxylase gene. Ann Neurol. 2007;62(4):422–6.
Vilariño-Güell C, Wider C, Ross OA, Dachsel JC, Kachergus JM, Lincoln SJ, Soto-Ortolaza AI, Cobb SA, Wilhoite GJ, Bacon JA, Behrouz B, Melrose HL, Hentati E, Puschmann A, Evans DM, Conibear E, Wasserman WW, Aasly JO, Burkhard PR, Djaldetti R, Ghika J, Hentati F, Krygowska-Wajs A, Lynch T, Melamed E, Rajput A, Rajput AH, Solida A, Wu RM, Uitti RJ, Wszolek ZK, Vingerhoets F, Farrer MJ. VPS35 mutations in Parkinson disease. Am J Hum Genet. 2011;89(1):162–7.
Whaley NR, Putzke JD, Baba Y, Wszolek ZK, Uitti RJ. Essential tremor: phenotypic expression in a clinical cohort. Parkinsonism Relat Disord. 2007;13(6):333–9.
Wittke C, Petkovic S, Dobricic V, Schaake S, MDS-endorsed PSP Study Group, Respondek G, Weissbach A, Madoev H, Trinh J, Vollstedt EJ, Kuhnke N, Lohmann K, Dulovic Mahlow M, Marras C, König IR, Stamelou M, Bonifati V, Lill CM, Kasten M, Huppertz HJ, Höglinger G, Klein C. Genotype-phenotype relations for the atypical parkinsonism genes: MDSGene systematic review. Mov Disord. 2021;36(7):1499–510.
Xiong H, Wang D, Chen L, Choo YS, Ma H, Tang C, Xia K, Jiang W, Ronai Z, Zhuang X, Zhang Z. Parkin, PINK1, and DJ-1 form a ubiquitin E3 ligase complex promoting unfolded protein degradation. J Clin Invest. 2009;119(3):650–60.
Yeetong P, Pongpanich M, Srichomthong C, Assawapitaksakul A, Shotelersuk V, Tantirukdham N, Chunharas C, Suphapeetiporn K, Shotelersuk V. TTTCA repeat insertions in an intron of YEATS2 in benign adult familial myoclonic epilepsy type 4. Brain. 2019;142(11):3360–6.
Zahorakova D, Ulmanova O, Kemlink D, Kofrankova M, Roth J, Martasek P, Ruzicka E. No association with the ETM2 locus in Czech patients with familial essential tremor. Neuro Endocrinol Lett. 2010;31(4):549–52.
Zarranz JJ, Alegre J, Gómez-Esteban JC, Lezcano E, Ros R, Ampuero I, Vidal L, Hoenicka J, Rodriguez O, Atarés B, Llorens V, Gomez Tortosa E, del Ser T, Muñoz DG, de Yebenes JG. The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann Neurol. 2004;55(2):164–73.
Zeng S, Zhang MY, Wang XJ, Hu ZM, Li JC, Li N, Wang JL, Liang F, Yang Q, Liu Q, Fang L, Hao JW, Shi FD, Ding XB, Teng JF, Yin XM, Jiang H, Liao WP, Liu JY, Wang K, Xia K, Tang BS. Long-read sequencing identified intronic repeat expansions in SAMD12 from Chinese pedigrees affected with familial cortical myoclonic tremor with epilepsy. J Med Genet. 2019;56(4):265–70.
Zesiewicz TA, Chari A, Jahan I, Miller AM, Sullivan KL. Overview of essential tremor. Neuropsychiatr Dis Treat. 2010;6:401–8.
Zimprich A, Biskup S, Leitner P, Lichtner P, Farrer M, Lincoln S, Kachergus J, Hulihan M, Uitti RJ, Calne DB, Stoessl AJ, Pfeiffer RF, Patenge N, Carbajal IC, Vieregge P, Asmus F, Müller-Myhsok B, Dickson DW, Meitinger T, Strom TM, Wszolek ZK, Gasser T. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron. 2004;44(4):601–7.
Zimprich A, Benet-Pagès A, Struhal W, Graf E, Eck SH, Offman MN, Haubenberger D, Spielberger S, Schulte EC, Lichtner P, Rossle SC, Klopp N, Wolf E, Seppi K, Pirker W, Presslauer S, Mollenhauer B, Katzenschlager R, Foki T, Hotzy C, Reinthaler E, Harutyunyan A, Kralovics R, Peters A, Zimprich F, Brücke T, Poewe W, Auff E, Trenkwalder C, Rost B, Ransmayr G, Winkelmann J, Meitinger T, Strom TM. A mutation in VPS35, encoding a subunit of the retromer complex, causes late-onset Parkinson disease. Am J Hum Genet. 2011;89(1):168–75.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2023 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Coppedè, F. (2023). Advances in the Genetics of Human Tremor. In: Grimaldi, G., Manto, M. (eds) Mechanisms and Emerging Therapies in Tremor Disorders. Contemporary Clinical Neuroscience. Springer, Cham. https://doi.org/10.1007/978-3-031-26128-2_3
Download citation
DOI: https://doi.org/10.1007/978-3-031-26128-2_3
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-031-26127-5
Online ISBN: 978-3-031-26128-2
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)