
Abstract
Abdominal pain is one of the main symptoms of chronic gastrointestinal disorders including inflammatory bowel diseases (IBD) and irritable bowel syndrome (IBS). The pathological mechanisms of chronic visceral pain have not been fully resolved. Pain management remains challenging and a source of frustration for health-care providers as current treatments are limited and lead to many side effects. Growing evidence indicates that functional changes in neuron-glia communication, at different levels of the gut-brain axis, contribute to visceral sensitization. In fact, a key role of spinal glial cells in the development and maintenance of hypersensitivity has been reported in several preclinical models of visceral pain. Here we provide an overview of the current knowledge on the factors, receptors, and transduction signaling pathways responsible for spinal neuron-glia interaction in chronic visceral pain.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
Chronic abdominal pain is one of the most common causes of disability and impaired quality of life for patients with bowel diseases such as inflammatory bowel diseases (IBD) and irritable bowel syndrome (IBS). IBD, including Crohn’s disease and ulcerative colitis, are chronic relapsing and remitting inflammatory diseases associated with abdominal pain (Norton et al. 2017). Pain is reported by over 90% of patients with active IBD, with the expectation that in the majority of patients, symptoms will resolve if the inflammation is under control (Schirbel et al. 2010; Wagtmans 1998). However, many IBD patients (20–60%) continue to experience visceral hypersensitivity (VHS), despite being in endoscopic remission (Farrokhyar et al. 2006; Schirbel et al. 2010). Accordingly, chronic abdominal pain in IBD has often been related to co-existing IBS where abdominal pain is considered a cardinal feature (Schmulson and Drossman 2017). IBS is a functional bowel disorder characterized by abdominal pain, stool irregularities, and bloating (Enck et al. 2016). In comparison with active IBD, IBS is not associated with structural or biochemical abnormalities that are detectable with the current routine diagnostic tools (Enck et al. 2016). Thus, the overlap between quiescent IBD and IBS has proven difficult to tease apart. Some of the chronic abdominal pain in IBD has been reported to be due to IBS-type symptom patterns, with up to 35% of quiescent IBD patients meeting IBS diagnostic criteria (Ozer et al. 2020; Teruel et al. 2016).
Given the complexity of the pathogenesis of chronic abdominal pain under these disease conditions, current pharmacotherapies are quite ineffective and rely mostly on NSAIDs or narcotics that can lead to serious side effects (Zielińska et al. 2019). The establishment of chronic visceral pain results from neuroplasticity in colonic nociceptors first and then along the entire neural axis. Providing functional and metabolic support to neurons, glial cells sense neural activity and respond to fine-tune excitability in the peripheral and central nervous system. Recent work has shown that glial cells in the CNS can contribute to visceral sensitization. Identifying the role of neuronal and non-neuronal cells, particularly glial cells, in the development and persistence of pain, may advance the development of novel and safer treatment modalities to manage chronic abdominal pain.
2 Visceral Pain Circuits
The GI tract is innervated by spinal sensory neurons or nociceptors that have their cell bodies in the dorsal root ganglia (DRGs) along the spinal cord. Primary afferents that innervate the upper GI tract originate from the thoracolumbar region (TL; T10-L2) of the spinal cord (via the splanchnic nerve), while those that innervate the distal colon and rectum originate from the lumbosacral region (LS; L5-S1) (via the pelvic nerve) (Abdullah et al. 2020; Gebhart and Bielefeldt 2016; Grundy et al. 2019). In the dorsal horn of the spinal cord, gut-innervating afferents synapse with interneurons both excitatory and inhibitory and second-order neurons which convey nociceptive signals to supraspinal sites by two ascending spinal pathways: the spinothalamic tract and spinoparabrachial pathway (Gebhart and Bielefeldt 2016; Grundy et al. 2019). The spinoparabrachial pathway is made of superficial dorsal horn neurons (laminae I and II), projecting to affective areas of the brain, including the amygdala, hypothalamus, and periaqueductal gray (PAG). In contrast, the spinothalamic tract originates from the deep dorsal horn (lamina X), relaying visceral sensory input to the thalamus and cortical areas for sensory discrimination and localization (Grundy et al. 2019).
3 Visceral Sensitization
The establishment and maintenance of persistent visceral pain is caused by long-lasting neuroplastic changes that occur in both the peripheral and central nervous system (Brierley and Linden 2014). Under inflammatory conditions, pro-inflammatory lipids, peptides, cytokines, and chemokines not only stimulate immune, epithelial, and stromal cells of the GI tract but also act on gut-innervating primary afferent neurons (Abdullah et al. 2020). Mechanistically, activation of transduction signaling pathways upon inflammation leads to ion channel modulation that in turn induces changes in the electrophysiological properties of colonic sensory neurons, characterized by reduced threshold of activation and enhanced excitability, a process described as sensitization (Basbaum et al. 2009). Among ion channels found in visceral afferent neurons, the transient receptor potential vanilloid 1 (TRPV1) has been implicated in sensing and transducing inflammatory signals (Bourinet et al. 2014; Lapointe et al. 2015). Several studies have demonstrated that peripheral sensitization of TRPV1 mediates VHS in both animal model and IBD/IBS patients (Akbar et al. 2010; Lapointe et al. 2015; Perna et al. 2020; Wouters et al. 2016). Although we have a good understanding of the mechanisms of sensitization at the periphery during intestinal inflammation, little is known about the cells and mediators that drive central sensitization in the spinal cord and the brain (Brierley and Linden 2014). Recent evidence indicates that activation of glial cells could play a central role. Eight major cell types have been identified in the mouse spinal cord (Sathyamurthy et al. 2018). Among them, microglia and astrocytes are activated following nerve injury or inflammation (Gu et al. 2016; Peng et al. 2016; Zhang et al. 2005), highlighting the importance of dorsal root ganglia (DRG) neuron-glial cell interactions in pathological pain.
4 Spinal Glial Cells in VHS
4.1 Microglia Activation in VHS
Microglia, the resident immune-like macrophages of the central nervous system (CNS), make up 10% of the total CNS glial population (Lu 2014). During neurodevelopment, microglia play a central role in the elimination and consolidation of synapses, shaping neural circuit connectivity and regulating subsequent functional activity (Schafer and Stevens 2015). Moreover, microglia control neuronal homeostasis and their activation is associated with a wide range of neurological disorders (Szepesi et al. 2018). Notably, there is a long list of publications on microglia and pain. Over 70% of the publications focused on spinal microglia, showing spinal microglia-neuron cross-talk as a main contributor to the development of chronic pain (Ho et al. 2020). Once activated, they congregate in the dorsal horn of the spinal cord (microgliosis) where they initiate a panel of innate defense mechanisms, including the production of cytokines that contribute to the activation and sensitization of colonic nociceptors (Marinelli et al. 2019). Additionally, microglia are known to express receptors for neuropeptides and neurotransmitters which, upon activation, modulate microglia reactivity (Marinelli et al. 2019). With regard to visceral sensitivity, the role of spinal microglia and the spinal neuro-immune mechanisms underlying visceral sensitization have been barely addressed.
Colitis-induced pain models
In active 2,4,6-trinitrobenzenesulfonic acid solution (TNBS)-induced colitis, Kannampalli et al. first demonstrated that mice with VHS exhibited microglial activation in the lumbosacral (LS) spinal dorsal horn receiving sensory input from the colon (Kannampalli et al. 2014). Notably, Majima et al. showed that microglial reactivity in TNBS-induced colitis model participates to cross-sensitization of bladder afferents (Majima et al. 2018). Using a dextran sodium sulfate (DSS) model of ulcerative colitis, we reported an activation of microglia in the spinal dorsal horn (Basso et al. 2017; Defaye et al. 2021; Huang et al. 2020). This was denoted by an increase in Iba1-labeled cells and a change from ramified to ameboid shape. Interestingly, microgliosis was associated with VHS acutely and post-resolution of inflammation (Defaye et al. 2021; Lucarini et al. 2020), suggesting a putative role of microglia activation in the maintenance of visceral hypersensitivity following disease remission.
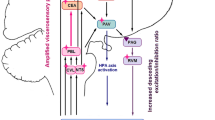
During active colitis, microglial activation is characterized by both microgliosis and increased levels of inflammatory cytokines such as interleukin (IL-1β), IL-6, tumor necrosis factor-α (TNF-α), macrophage inflammatory protein-1 α (MIP-1α or CCL3), brain-derived neurotrophic factor (BDNF), and granulocyte-colony-stimulating factor (G-CSF) (Basso et al. 2017; Huang et al. 2020; Majima et al. 2018). Local administration of the anti-mitotic agent, AraC, that stops microgliosis in the spinal dorsal horn reduced colitis-induced VHS (Kannampalli et al. 2014). Along these lines, inhibiting microglial activation with intrathecal injection of the tetracycline antibiotic minocycline could prevent VHS in TNBS colitis (Kannampalli et al. 2014; Majima et al. 2018). Importantly, intrathecal minocycline was shown to normalize the colitis-induced expression of pro-inflammatory IL-1β and CCL3, as well as the neurogenic and pronociceptive BDNF, in the spinal cord (Majima et al. 2018). Overall, cumulative evidence suggests that colitis-induced activation of microglia leads to the production and release of pro-inflammatory cytokines such as IL-1β and TNF-α, at proximity of synapses that relay nociceptive information from the GI tract. Facilitating synaptic transmission in the spinal dorsal horn by these mediators and neurogenic factors will play a central role in sensitizing nociceptive input and triggering pain-specific neuroplastic changes in the ascending pain pathway (Kawasaki et al. 2008). Accordingly, glutamatergic synapses contribute to the process of central sensitization associated with all forms of pathological pain conditions, and phosphorylation of N-methyl-D-aspartate (NMDA) receptor-NR2B subunit was found to be a surrogate marker of central sensitization in the spinal cord (Huang et al. 2020). Notably, the increase in NR2B phosphorylation was found to persist after resolution of intestinal inflammation and to correlate with hyperalgesia. Whether NMDAR phosphorylation dictates the activation state of microglia in visceral pain models remains to be explored. In the 2,4-dinitrobenzenesulfonic acid (DNBS) model of colitis, inflammation peaks at day 3 and resolves progressively from day 7. Both mice and rat models develop VHS up to 21 days post-administration, and Lucarini et al. reported microglial activation in both dorsal and ventral horn of the lumbar spinal cord during the recovery phase (Lucarini et al. 2020). Interestingly, microglia did not change in density but underwent well-defined morphological alterations (loss of the processes under activation). Using the DSS-induced colitis model, we demonstrated that pro-inflammatory G-CSF activates microglia through G-CSF receptor. We showed that G-CSF drives post-inflammatory pain via cathepsin S-fractalkine-NO signaling axis from spinal microglia and acts on colonic nociceptors that express TRPV1 (Fig. 1) (Basso et al. 2017). Several studies have demonstrated that sensitization of TRPV1-expressing colonic neurons mediates VHS during resolution of colitis, a process that promotes abdominal pain in preclinical models and likely patients with quiescent IBD and IBS (Akbar et al. 2010; Lapointe et al. 2015; Perna et al. 2020; Wouters et al. 2016). We recently investigated whether VHS results from the direct communication between sensitized TRPV1 nociceptors and spinal microglia. Using designer receptors exclusively activated by designer drugs (DREADD) expressed in TRPV1+ nociceptors, we tested whether neuronal activity was indispensable to control spinal microglia activation and VHS. We found that chemogenetic inhibition of TRPV1+ nociceptors could prevent microglial activation in the spinal dorsal horn and subsequent VHS in colitis mice. In contrast, in naïve condition, chemogenetic activation of gut-innervating TRPV1+ nociceptors enhanced microglial activation and associated VHS, in the absence of colitis. We then identified a purinergic signaling mechanism mediated by neuronal ATP and microglial P2Y12 receptor, triggering VHS in colitis. Importantly, inhibition of P2RY12 prevented microglial reactivity and chronic VHS post-colitis. Altogether, these results demonstrated that ATP-releasing TRPV1+ visceral afferents and P2RY12 signaling in the spinal microglia orchestrate the establishment of VHS that persists following remission of colitis (Defaye et al. 2021). Our work strongly suggests that preventing microglial activation or inhibiting P2Y12 receptors centrally could provide therapeutic value to treat both acute and post-inflammatory visceral pain (Fig. 1).

Spinal neuron-glia communication in visceral hypersensitivity. The schematic figure represents the main molecular signaling pathways involved in neuron-glial cell communication. ATP adenosine triphosphate; ADP adenosine diphosphate; G-CSF granulocyte colony-stimulating factor; CX3CL1/FKN fractalkine; CX3CR1 C-X3-C motif chemokine receptor 1; NO nitric oxide; NK1R neurokinin 1 receptor; NMDA receptor N-methyl-D-aspartate receptor; IL-1β interleukin-1β; IL-6 interleukin-6; TNF-α tumor necrosis factor-α; CCL3 C-C motif chemokine ligand 3; BDNF brain-derived neurotrophic factor; TRAF6 TNF receptor-associated factor 6; GFAP glial fibrillary acidic protein; DRG dorsal root ganglion
It will be important to determine whether distinct microglia subpopulations or activation states contribute to different stages of visceral sensitization. This could be done using single-cell transcriptomic analysis in the lumbosacral spinal cord at different stages of colitis. Lastly, while we did not notice any sexual dimorphism in microgliosis under colitis, the activation states and microglia subtypes may be different between males and females (Tansley et al. 2022). Future work will address how transcriptional and functional changes in microglia subpopulations drive VHS. Identifying a VHS-related transcriptional signature of microglia will provide insights into the central mechanisms and potential targets to stop the development and maintenance of VHS in colitis.
Non-inflammatory pain models
IBS-like symptoms, including pain, are often experienced in IBD patients in remission (Enck et al. 2016). Yet, whether similar mechanisms of neuron-microglia interactions contribute to VHS in IBS has been poorly investigated. Using various models of stress-induced IBS, several studies highlighted the role of microglia in non-inflammatory VHS. Saab et al. first showed that stress induced by neonatal irritation (CCI) induced VHS associated with microglial reactivity in the lumbosacral spinal dorsal horn at adulthood (Saab et al. 2006). In addition, chronic psychological stress induced by repetitive water avoidance leads to microglia activation in the lumbar dorsal horn (Bradesi et al. 2009). Both of these studies demonstrated that intrathecal injection of fractalkine (FKN) in naïve animals induces VHS, supporting the hypothesis that FKN could trigger microglial activation and visceral sensitization (Bradesi et al. 2009; Saab et al. 2006). Accordingly, both water avoidance and fractalkine-induced hyperalgesia were blocked by minocycline. Recently, Zhan et al. reported an increase of Iba-1-positive cells in the spinal dorsal horn of acetic acid-treated mice. They found that ulinastatin, a broad-spectrum serine protease inhibitor, reduced acetic acid-induced writhing and microglial activation in the spinal dorsal horn (Zhan et al. 2021).
Importantly, similar to stress, drugs can also trigger neuroinflammation leading to VHS. One example is the use of opioids for functional and chronic gastrointestinal pain, which is not as beneficial as previously assumed. Indeed, opioids can cause unexpected abdominal pain (Farmer et al. 2017; Kong and Burns 2021). When pain is the major symptom of opioid use, this condition is called narcotic bowel syndrome (NBS). NBS is defined by an increase in abdominal pain despite maintaining or increasing doses of narcotics. Agostini et al. showed that chronic morphine-induced VHS is associated with spinal microglia activation in rats (Agostini et al. 2010). Using minocycline, they reduced narcotic-induced hypersensitivity responses to CRD. While it is clear that the mu-opioid receptor is not responsible for morphine-induced microglia activation (Corder et al. 2017), how microglia sense and respond to morphine is still a matter of debate.
4.2 Astrocyte Activation in VHS
Astrocytes make up 20–40% of all of the glial cells in the CNS. Under homeostasis conditions, astrocytes regulate functions, such as synapse formation and CNS homeostasis, by providing neurons with metabolites and growth factors and regulating the balance of ions, fluids, and neurotransmitters (Sofroniew and Vinters 2010). Evidence supports a role of astrocytes in persistent neuropathic pain. After injury, the phenotype, functions, and gene expression profile of spinal astrocytes can undergo significant changes, known as reactive astrogliosis (Li et al. 2019). Only few studies have described the involvement of astrocytes in VHS.
Colitis-induced pain models
Intestinal inflammation induced by TNBS leads to the activation of astrocytes in the lumbosacral spinal dorsal horn as measured by glial fibrillary acidic protein (GFAP) expression, an astrocyte gliosis marker (Sun et al. 2005). The density of GFAP+ astrocytes in the spinal cord was found to decrease comparable to control level at day 28 post-TNBS, when the animals recovered from intestinal inflammation. This indicated that astrocyte activation is inflammation dependent and returns to homeostatic state after remission of colitis. Accordingly, Lucarini et al. reported an activation of astrocytes in both dorsal and ventral horn of the spinal cord in a rat model of DNBS-induced VHS. Astrocytes increased significantly in density (about 30%) and exhibited activated states characterized by expansions of cellular bodies and processes in the dorsal spinal cord (Lucarini et al. 2020) (Fig. 1).
However, the mechanisms underlying astrocyte activation are still unclear. Future works will address whether neuronal subsets and mediators contribute to VHS-associated astrocyte reactivity. Also, as mentioned previously for microglia, different astrocyte subpopulations may contribute to visceral sensitization (Li et al. 2019). It will be important to investigate whether distinct astrocyte subpopulations contribute to different disease stages.
Non-inflammatory pain models
In preclinical models of IBS, spinal astrocyte activation is still controversial. Using the maternal separation model of IBS, Gosselin et al. did not find a global change in spinal astrocytic phenotype (Gosselin et al. 2010). However, an increase of GFAP positive cells in the spinal dorsal horn was reported using neonatal intracolonic acetic acid (AA) model of IBS (Weng et al. 2020; Zhao et al. 2020). In addition, an upregulation of TRAF6 in spinal astrocytes was observed in adult mice in response to neonatal irritation (Weng et al. 2020). Depletion of TRAF6 using small interfering RNA (siRNA) alleviated VHS and reduced the amplitude of spontaneous excitatory postsynaptic currents in the spinal dorsal horn (Weng et al. 2020). Zhao et al. provided insights into the interactions that occur between astrocytes and nociceptors in the spinal cord (Zhao et al. 2020). Previous studies had shown a role of P2Y receptors in stimulating visceral hypersensitivity. P2YR is expressed by 56% to 80% of retrogradely labeled colonic neurons, indicating a P2Y-dependent mechanism of VHS (Hockley et al. 2016). Using the AA model of visceral pain, Zhao et al. found a reduction in spinal expression of IL-6, IL-1β, TNF-α, GFAP, and P2Y1 in response to electroacupuncture. These molecular changes were associated with a decrease in VHS (Zhao et al. 2020). Importantly, intrathecal administration of the astrocyte inhibitor, fluorocitrate, or the P2Y1 receptor antagonist, MRS2179, reduced AA-induced visceral hypersensitivity, raising the hypothesis that electroacupuncture negatively regulates P2Y1 receptor in astrocytes. Finally, as previously described with microglial activation, ulinastatin treatment blocked astrocyte activation (Zhan et al. 2021).
5 Conclusion and Future Perspectives
Children and adults living with chronic visceral pain tend to develop sleep disorder, anxiety, and depression. While efforts have been made to understand the mechanisms and neural circuits of visceral sensation, the pathophysiology of gut pain is very complex and involves many cell types and signaling pathways at the peripheral and central level. Our group and others have demonstrated the importance of both spinal microglia and astrocytes in the transition from acute to chronic visceral pain. Collective results strongly suggest that modulating spinal glial cells via the targeting of purinergic signaling necessary for glial reactivity may provide therapeutic value to treat chronic visceral pain. Although many purinergic blockers are clinically available, it will be important to address their analgesic properties in both IBD and IBS conditions. Lastly, given the growing importance of the role of the microbiome in the gut-brain axis, future work should identify microbial-based molecules as regulators of neuron-glial communication and treatments for visceral pain.
References
Abdullah N, Defaye M, Altier C. Neural control of gut homeostasis [Internet]. Am J Physiol. American Physiological Society. 2020 [cited 2021 Jun 1]:G718–32. Available from: https://pubmed.ncbi.nlm.nih.gov/33026824/
Agostini S, Eutamene H, Cartier C, Broccardo M, Improta G, Houdeau E, et al. Evidence of central and peripheral sensitization in a rat model of narcotic bowel-like syndrome. Gastroenterology [Internet]. Elsevier Inc. 2010;139(2):553–563.e5. Available from: https://doi.org/10.1053/j.gastro.2010.03.046.
Akbar A, Yiangou Y, Facer P, Brydon WG, Walters JRF, Anand P, et al. Expression of the TRPV1 receptor differs in quiescent inflammatory bowel disease with or without abdominal pain. Gut. 2010;59(6):767–74.
Basbaum AI, Bautista DM, Scherrer G, Julius D. Cellular and molecular mechanisms of pain [Internet]. Cell. Elsevier B.V. 2009 [cited 2021 May 3]:267–84. Available from: /pmc/articles/PMC2852643/.
Basso L, Lapointe TK, Iftinca M, Marsters C, Hollenberg MD, Kurrasch DM, et al. Granulocyte-colony–stimulating factor (G-CSF) signaling in spinal microglia drives visceral sensitization following colitis. Proc Natl Acad Sci U S A. 2017;114(42):11235–40.
Bourinet E, Altier C, Hildebrand ME, Trang T, Salter MW, Zamponi GW. Calcium-permeable ion channels in pain signaling. Physiol Rev. 2014;94(1):81–140.
Bradesi S, Svensson CI, Steinauer J, Pothoulakis C, Yaksh TL, Mayer EA. Role of spinal microglia in visceral hyperalgesia and NK1R up-regulation in a rat model of chronic stress. Gastroenterology [Internet]. Gastroenterology. 2009 [cited 2022 Mar 12];136(4). Available from: https://pubmed.ncbi.nlm.nih.gov/19249394/
Brierley SM, Linden DR. Neuroplasticity and dysfunction after gastrointestinal inflammation [Internet]. Nat Rev Gastroenterol Hepatol. Nature Publishing Group. 2014 [cited 2021 May 27]:611–27. Available from: www.nature.com/nrgastro
Corder G, Tawfik VL, Wang D, Sypek EI, Low SA, Dickinson JR, et al. Loss of μ opioid receptor signaling in nociceptors, but not microglia, abrogates morphine tolerance without disrupting analgesia. Nat Med. [Internet]. 2017 Feb 1 [cited 2022 Mar 19];23(2):164–73. Available from: https://pubmed.ncbi.nlm.nih.gov/28092666/
Defaye M, Abdullah NS, Iftinca M, Hassan A, Agosti F, Zhang Z, et al. Gut-innervating TRPV1+ Neurons Drive Chronic Visceral Pain via Microglial P2Y12 Receptor. Cell Mol Gastroenterol Hepatol [Internet]. 2021 [cited 2022 Mar 12];13(4):977–99. Available from: https://pubmed.ncbi.nlm.nih.gov/34954381/
Enck P, Aziz Q, Barbara G, Farmer AD, Fukudo S, Mayer EA, et al. Irritable bowel syndrome. Nat Rev Dis Prim [Internet]. 2016 Mar 24 [cited 2018 Aug 6];2:16014. Available from: http://www.ncbi.nlm.nih.gov/pubmed/27159638
Farmer AD, Gallagher J, Bruckner-Holt C, Aziz Q. Narcotic bowel syndrome. Lancet Gastroenterol Hepatol [Internet]. Elsevier. 2017 May 1 [cited 2022 Mar 12];2(5):361–8. Available from: http://www.thelancet.com/article/S2468125316302175/fulltext
Farrokhyar F, Marshall JK, Easterbrook B, Irvine EJ. 00054725-200601000-00007.Pdf. 2006;12(1):38–46.
Gebhart GF, Bielefeldt K. Physiology of visceral pain. Compr Physiol. 2016;6(4):1609–33.
Gosselin RD, O’Connor RM, Tramullas M, Julio-Pieper M, Dinan TG, Cryan JF. Riluzole normalizes early-life stress-induced visceral hypersensitivity in rats: role of spinal glutamate reuptake mechanisms. Gastroenterology [Internet]. Elsevier Inc. 2010;138(7):2418–25. Available from: https://doi.org/10.1053/j.gastro.2010.03.003.
Grundy L, Erickson A, Brierley SM. Visceral pain [Internet]. Annu Rev Physiol. Annual Reviews Inc. 2019 [cited 2021 May 13]:261–84. Available from: https://doi.org/10.1146/annurev-physiol-020518-.
Gu N, Peng J, Murugan M, Wang X, Eyo UB, Sun D, et al. Spinal microgliosis due to resident microglial proliferation is required for pain hypersensitivity after peripheral nerve injury. Cell Rep [Internet]. The Author(s). 2016;16(3):605–14. Available from: https://doi.org/10.1016/j.celrep.2016.06.018.
Ho IHT, Chan MTV, Wu WKK, Liu X. Spinal microglia-neuron interactions in chronic pain. J Leukoc Biol [Internet]. John Wiley & Sons, Ltd. 2020 Nov 1 [cited 2022 Mar 12];108(5):1575–92. Available from: https://onlinelibrary.wiley.com/doi/full/10.1002/JLB.3MR0520-695R
Hockley JRF, Tranter MM, McGuire C, Boundouki G, Cibert-Goton V, Thaha MA, et al. P2Y receptors sensitize mouse and human colonic nociceptors. J Neurosci [Internet]. 2016 Feb 24 [cited 2022 Mar 12];36(8):2364–76. Available from: https://pubmed.ncbi.nlm.nih.gov/26911685/
Huang Y, Wang C, Tian X, Mao Y, Hou B, Sun Y, et al. Pioglitazone attenuates experimental colitis-associated hyperalgesia through improving the intestinal barrier dysfunction. Inflammation. 2020;43(2):568–78.
Kannampalli P, Pochiraju S, Bruckert M, Shaker R, Banerjee B, Sengupta JN. Analgesic effect of minocycline in rat model of inflammation-induced visceral pain. Eur J Pharmacol [Internet]. NIH Public Access. 2014 Mar 15 [cited 2021 May 14];727(1):87–98. Available from: /pmc/articles/PMC3984928/.
Kawasaki Y, Zhang L, Cheng JK, Ji RR. Cytokine mechanisms of central sensitization: distinct and overlapping role of Interleukin-1β, Interleukin-6, and tumor necrosis factor-α in regulating synaptic and neuronal activity in the superficial spinal cord. J Neurosci [Internet]. Society for Neuroscience. 2008 May 14 [cited 2022 Mar 12];28(20):5189. Available from: /pmc/articles/PMC2408767/.
Kong EL, Burns B. Narcotic Bowel Syndrome. StatPearls [Internet]. StatPearls Publishing. 2021 Aug 11 [cited 2022 Mar 12]; Available from: https://www.ncbi.nlm.nih.gov/books/NBK493207/
Lapointe TK, Basso L, Iftinca MC, Flynn R, Chapman K, Dietrich G, et al. TRPV1 sensitization mediates postinflammatory visceral pain following acute colitis. Am J Physiol. 2015;309(2):G87–99.
Li T, Chen X, Zhang C, Zhang Y, Yao W. Update on reactive astrocyte in CCI. J Neuroimmune Pharmacol [Internet]. 2019:1–13. Available from: https://jneuroinflammation.biomedcentral.com/track/pdf/10.1186/s12974-019-1524-2
Lu CL. Spinal microglia: A potential target in the treatment of chronic visceral pain. J Chinese Med Assoc. No longer published by Elsevier. 2014:3–9.
Lucarini E, Parisio C, Branca JJV, Segnani C, Ippolito C, Pellegrini C, et al. Deepening the mechanisms of visceral pain persistence: an evaluation of the Gut-spinal cord relationship. Cells. 2020;9(8).
Majima T, Funahashi Y, Kawamorita N, Takai S, Matsukawa Y, Yamamoto T, et al. Role of microglia in the spinal cord in colon-to-bladder neural crosstalk in a rat model of colitis. Neurourol Urodyn. 2018;37(4):1320–8.
Marinelli S, Basilico B, Marrone MC, Ragozzino D. Microglia-neuron crosstalk: signaling mechanism and control of synaptic transmission. Semin Cell Dev Biol [Internet]. Elsevier. 2019;94(May):138–51. Available from: https://doi.org/10.1016/j.semcdb.2019.05.017.
Norton C, Czuber-Dochan W, Artom M, Sweeney L, Hart A. Systematic review: interventions for abdominal pain management in inflammatory bowel disease. Aliment Pharmacol Ther [Internet]. John Wiley & Sons, Ltd (10.1111). 2017 Jul 1 [cited 2019 Jun 24];46(2):115–25. Available from: http://doi.wiley.com/10.1111/apt.14108
Ozer M, Bengi G, Colak R, Cengiz O, Akpinar H. Prevalence of irritable bowel syndrome-like symptoms using Rome IV criteria in patients with inactive inflammatory bowel disease and relation with quality of life. Medicine (Baltimore). [Internet]. Wolters Kluwer Health. 2020 May 1 [cited 2022 Mar 12];99(19):e20067. Available from: /pmc/articles/PMC7220554/.
Peng J, Gu N, Zhou L, B Eyo U, Murugan M, Gan WB, et al. Microglia and monocytes synergistically promote the transition from acute to chronic pain after nerve injury. Nat. Commun. [Internet]. Nature Publishing Group; 2016;7(May):1–13. Available from: https://doi.org/10.1038/ncomms12029
Perna E, Aguilera-Lizarraga J, Florens M V., Jain P, Theofanous SA, Hanning N, et al. Effect of resolvins on sensitisation of TRPV1 and visceral hypersensitivity in IBS. Gut [Internet]. BMJ Publishing Group. 2020 Nov 11 [cited 2021 May 13]; Available from: https://gut.bmj.com/content/early/2020/11/10/gutjnl-2020-321530
Saab CY, Wang J, Gu C, Garner KN, Al-Chaer ED. Microglia: a newly discovered role in visceral hypersensitivity? Neuron Glia Biol. 2006;2(4):271–7.
Sathyamurthy A, Johnson KR, Matson KJE, Dobrott CI, Li L, Ryba AR, et al. Massively parallel single nucleus transcriptional profiling defines spinal cord neurons and their activity during behavior. Cell Rep [Internet]. Elsevier B.V. 2018 Feb 20 [cited 2021 May 13];22(8):2216–25. Available from: /pmc/articles/PMC5849084/.
Schafer DP, Stevens B. Microglia function in central nervous system development and plasticity. Cold Spring Harb Perspect Biol [Internet]. Cold Spring Harbor Laboratory Press. 2015 Oct 1 [cited 2022 Mar 12];7(10). Available from: /pmc/articles/PMC4588063/.
Schirbel A, Reichert A, Roll S, Baumgart DC, Büning C, Wittig B, et al. Impact of pain on health-related quality of life in patients with inflammatory bowel disease. World J Gastroenterol. 2010;16(25):3168–77.
Schmulson MJ, Drossman DA. What is new in Rome IV. J Neurogastroenterol Motil [Internet]. 2017 Apr 30 [cited 2018 Aug 6];23(2):151–63. Available from: http://www.ncbi.nlm.nih.gov/pubmed/28274109
Sofroniew MV, Vinters HV. Astrocytes: biology and pathology. Acta Neuropathol [Internet]. Springer. 2010 Jan [cited 2022 Mar 12];119(1):7. Available from: /pmc/articles/PMC2799634/.
Sun YN, Luo JY, Rao ZR, Li L, Duan L. GFAP and Fos immunoreactivity in lumbo-sacral spinal cord and medulla oblongata after chronic colonic inflammation in rats. World J Gastroenterol. Baishideng Publishing Group Inc. 2005 Aug;11(31):4827–32.
Szepesi Z, Manouchehrian O, Bachiller S, Deierborg T. Bidirectional microglia–neuron communication in health and disease. Front Cell Neurosci. Frontiers Media S.A. 2018 Sep 27;12:323.
Tansley S, Uttam S, Ureña Guzmán A, Yaqubi M, Pacis A, Parisien M, et al. Single-cell RNA sequencing reveals time- and sex-specific responses of mouse spinal cord microglia to peripheral nerve injury and links ApoE to chronic pain. Nat Commun [Internet]. Nature Publishing Group. 2022 Dec 1 [cited 2022 Mar 23];13(1). Available from: /pmc/articles/PMC8837774/.
Teruel C, Garrido E, Mesonero F. Diagnosis and management of functional symptoms in inflammatory bowel disease in remission. World J Gastrointest Pharmacol Ther [Internet]. Baishideng Publishing Group Inc. 2016 [cited 2022 Mar 12];7(1):78. Available from: /pmc/articles/PMC4734957/.
Wagtmans MJ. Crohn’s disease in the elderly: a comparison with young adults. J Clin Gastroenterol. 1998;27(2):129–33.
Weng RX, Chen W, Tang JN, Sun Q, Li M, Xu X, et al. Targeting spinal TRAF6 expression attenuates chronic visceral pain in adult rats with neonatal colonic inflammation. Mol Pain. 2020;16:1–9.
Wouters MM, Balemans D, Van Wanrooy S, Dooley J, Cibert-Goton V, Alpizar YA, et al. Histamine receptor H1-mediated sensitization of TRPV1 mediates visceral hypersensitivity and symptoms in patients with irritable bowel syndrome. Gastroenterology [Internet]. W.B. Saunders. 2016 Apr 1 [cited 2021 May 13];150(4):875–887.e9. Available from: https://doi.org/10.1053/j.gastro.2015.12.034
Zhan MX, Tang L, Lu YF, Wu HH, Guo Z Bin, Shi ZM, et al. Ulinastatin exhibits antinociception in rat models of acute somatic and visceral pain through inhibiting the local and central inflammation. J Pain Res. 2021;14:1201–14.
Zhang RX, Liu B, Wang L, Ren K, Qiao JT, Berman BM, et al. Spinal glial activation in a new rat model of bone cancer pain produced by prostate cancer cell inoculation of the tibia. Pain. 2005;118(1–2):125–36.
Zhao J, Li H, Shi C, Yang T, Xu B. Electroacupuncture inhibits the activity of astrocytes in spinal cord in rats with visceral hypersensitivity by inhibiting P2Y1 receptor-mediated MAPK/ERK signaling pathway. Evidence-based Complement. Altern Med. Hindawi Limited; 2020;2020:4956179.
Zielińska A, Sałaga M, Włodarczyk M, Fichna J. Focus on current and future management possibilities in inflammatory bowel disease-related chronic pain. Int J Color Dis. 2019;34(2):217–27.
Funding
This work was supported by operating grants from the Crohn’s and Colitis Canada (CCC) and the Canadian Institutes of Health Research (CIHR). MD holds a fellowship from the Alberta Children’s Hospital Research Institute (ACHRI). CA holds a Canada Research Chair in inflammatory pain.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2023 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Defaye, M., Altier, C. (2023). Neuron-Microglia Dynamic Duo in Chronic Abdominal Pain. In: Brierley, S.M., Spencer, N.J. (eds) Visceral Pain. Springer, Cham. https://doi.org/10.1007/978-3-031-25702-5_15
Download citation
DOI: https://doi.org/10.1007/978-3-031-25702-5_15
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-031-25701-8
Online ISBN: 978-3-031-25702-5
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)