
Abstract
Although elevated levels of lipoprotein(a) (Lp(a)) are increasingly recognized as a causal risk factor for the development of premature cardiovascular disease, the precise mechanisms by which Lp(a) exerts its harmful effects remain unclear. Studies using in vitro cell model systems—and a few in vivo studies in humans and animal models—show that Lp(a) and its unique constituent lipoprotein(a) (apo(a)) elicit pathological changes in vascular endothelial cells and smooth muscle cells, in circulating monocytes, and in macrophages. These effects together could promote the initiation and progression of atherosclerosis and favor the development of vulnerable plaques. Many of these changes are specifically mediated by the oxidized phospholipid that is covalently associated with kringle IV type 10 in apo(a). While in vitro and in vivo data are conflicting regarding a direct role of Lp(a) in inhibiting fibrinolysis, further studies are required to fully elaborate the role of Lp(a) in triggering thrombosis versus the development of thrombosis-prone atherosclerotic plaques. Lp(a) and apo(a) can also promote calcific aortic valve disease by promoting pro-osteogenic and proinflammatory responses in valve interstitial cells; again, the oxidized phospholipid in apo(a) plays a central role in this process.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- lipoprotein(a)
- apolipoprotein(a)
- atherosclerosis
- thrombosis
- calcific aortic valve disease
- oxidized phospholipids
- inflammation
- fibrinolysis
Introduction
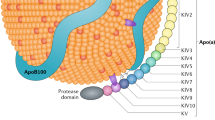
Although lipoprotein(a) (Lp(a)) was discovered almost 50 years ago (Berg and New 1963) and has been subsequently shown to be a causal and independent risk factor for atherosclerotic cardiovascular disease (ASCVD) and calcific aortic valve disease (CAVD) (Arsenault and Kamstrup 2022), the mechanisms by which Lp(a) mediates its pathogenic effects in vivo remain unclear. Lp(a) comprises an apoB-100-containing lipoprotein to which is attached the unique apolipoprotein(a) (apo(a)) moiety (Fig. 10.1). Amino acid analysis followed by complete sequencing of the human apo(a) cDNA in 1987 revealed a high level of sequence identity with the profibrinolytic enzyme plasminogen (Eaton et al. 1987; McLean et al. 1987). Apo(a) contains a series of tri-looped structures called kringles that are similar to the KIV domain of plasminogen, followed by sequences similar to the plasminogen KV and protease domains (Fig. 10.1). Due to several critical amino acid substitutions and a small deletion, the apo(a) protease-like domain has been shown to be catalytically inactive (Gabel and Koschinsky 1995).

Structure and functional domains of Lp(a). Lp(a) consists of apo(a) covalently linked to the apoB-100 moiety of an LDL-like lipoprotein particle. The lipid portion of the particle is a shell of phospholipids (PL) and free cholesterol (FC) surrounding a neutral lipid core of cholesteryl esters (CE) and triacylglycerols (TG). Apo(a) consists of ten types of KIV domains, a KV domain, and an inactive protease domain. KIV2 is repeated different numbers of times in different apo(a) isoforms. KIV5–KIV8 contain weak lysine-binding sites (wLBS), with those in KIV7 and KIV8 binding to specific lysine residues in apoB-1000 during the noncovalent step of Lp(a) assembly. KIV9 contains a single-free cysteine that mediates disulfide bond formation with apoB-100. KIV10 contains a strong lysine-binding site (sLBS) as well as covalently bound oxidized phospholipid (OxPL). The sLBS is required for OxPL addition, and together these features promote several pathogenic effects on vascular and immune cells. OxPL is also present noncovalently associated with the lipid moiety of Lp(a), and accounts for up to 50% of the total OxPL on Lp(a). EC endothelial cell, SMC smooth muscle cell, VIC valve interstitial cell
Apo(a) kringle IV sequences are present in ten types based on amino acid sequence; these have been designated KIV1–KIV10 (McLean et al. 1987; van der Hoek et al. 1993). The KIV2 sequence is present in a variable number of identically repeated copies (from 3 to greater than 40) which is a hallmark of Lp(a) and reflects allele size variation in LPA, the gene encoding apo(a) (Fig. 10.1) (Lackner et al. 1993; Marcovina et al. 1996). Interestingly, there is a strong inverse correlation between apo(a) size and Lp(a) plasma levels, which likely arises due to less efficient secretion of larger isoforms as a result of presecretory degradation of misfolded species in the endoplasmic reticulum (Boffa and Koschinsky 2022). The KIV9 domain houses the only unpaired cysteine in apo(a) and is involved in disulfide bond formation with a cysteine residue in the carboxyl-terminus of apoB-100 (Koschinsky et al. 1993). The KIV5–8 domains each contain a weak lysine-binding site (wLBS); the wLBS in KIV7 and KIV8 is required for intracellular noncovalent interaction between apo(a) and apoB that precedes extracellular disulfide bond formation (Fig. 10.1) (Becker et al. 2004; Youssef et al. 2022).
The apo(a) KIV10 domain contains a relatively strong lysine-binding site (sLBS) that has been studied extensively in attempts to understand the pathophysiology of Lp(a) in the vasculature. Lp(a) has been demonstrated to be the preferential lipoprotein carrier of proinflammatory oxidized phospholipids (OxPL), compared to LDL (Bergmark et al. 2008). These species are present both on the lipid portion of Lp(a) as well as covalently associated with apo(a) (Bergmark et al. 2008; Leibundgut et al. 2013). Interestingly, in this regard, it has been shown that the KIV10 sLBS is absolutely required for the covalent addition of oxidized phospholipid to this kringle, likely involving addition of the OxPL adduct to a histidine side chain through Michael reaction addition (Fig. 10.1) (Leibundgut et al. 2013; Scipione et al. 2015). The proinflammatory effect of the OxPL on KIV10 has been demonstrated in many studies, both in vitro and in vivo (Koschinsky and Boffa 2022; Dzobo et al. 2022). In vitro studies have shown the role of OxPL on KIV10 in promoting proinflammatory and phenotypes in a variety of vascular and inflammatory cells including valve interstitial cells (VICs) (see below).
Many studies using a variety of vascular cell types have shown that the apo(a) sLBS can compete with plasminogen for binding to cell surfaces, thereby inhibiting plasminogen activation to the active enzyme plasmin (Boffa 2022). Downstream effects on fibrin clot lysis have also been studied, with variable results, and the significance of Lp(a) in promoting thrombosis in the arterial and venous circulation remains controversial (Boffa 2022). Indeed, Lp(a) appears to confer risk for venous thromboembolism only in individuals with extremely high Lp(a) levels (Kamstrup et al. 2012). The role of Lp(a) in platelet function and coagulation, and in the lysis of platelet-rich clots, is not clear (Boffa 2022). Importantly, it is difficult to assess the contribution of Lp(a) to lysis of clots formed upon rupture of vulnerable atherosclerotic plaques (see below).
Despite the demonstration of elevated plasma Lp(a) levels as an independent and causal risk factor for ASCVD and CAVD, the mechanism of action of Lp(a) in these disease processes remains unclear. This reflects, in part, the complexity of the Lp(a) structure, as well as the lack of suitable animal models for Lp(a); together these present significant challenges to understanding the molecular and cellular basis of Lp(a) pathogenicity. Indeed, LPA is only present in Old World monkeys and apes and humans. Of note, the Old World species all lack a functional LBS in KIV10 preventing covalent OxPL addition to this kringle. Work on transgenic mice expressing human Lp(a) is progressing (Yeang et al. 2016), and ultimately should complement the significant insights that are being made on probing the effect of Lp(a) on human vascular and valve interstitial cell phenotypes (Fig. 10.2).

Overlapping pathogenic mechanisms of Lp(a) in atherosclerosis and calcific aortic valve disease. There are several common mechanisms mediated by Lp(a) in the two disorders. Compromised endothelial cell function leads to barrier permeability, infiltration of Lp(a) and monocytes, expression of endothelial cell surface receptors for monocytes, and mural thromboses. Lp(a) activates monocytes leading to cytokine secretion and enhanced potential for transendothelial migration. Within the vessel wall or valve, Lp(a) promotes macrophage foam cell formation and macrophage apoptosis, as well as stimulating the release of proinflammatory cytokines such as interleukin-8 (IL-8) from macrophages. Lp(a) promotes smooth muscle cell (SMC) migration and proliferation in the arterial intima. Lp(a) also promotes calcification of SMC in the atrial intima and osteogenic differentiation and calcification of valve interstitial cells. Many functions of Lp(a), indicated in red, are mediated by its bound oxidized phospholipid (OxPL). Lp(a) also transports the phospholipase D enzyme autotaxin into the aortic valve leaflet, where it catalyzed generation of the highly proinflammatory lysophosphatidic acid (lysoPA) using lysophosphatidylcholine (lysoPC) as a substrate. The OxPL on Lp(a) thus helps to explain why elevated Lp(a) is a causal risk factor for both atherothrombosis and aortic stenosis, despite the disease processes underlying each of these disorders being distinct. TNF-α tumor necrosis factor-α
Effect of Lp(a) on Vascular and Immune Cell Phenotype
Effects of Lp(a) on Vascular Endothelium
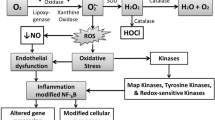
The vascular endothelial cell layer is critical in maintaining a nonpermeable barrier that protects the vessel wall from exposure to blood contents. As such, there have been a number of studies aimed at determining the effect of Lp(a) on endothelial function. Lp(a) deposition in the intimal layer of the arterial wall was reported nearly 30 years ago and suggested that Lp(a) can cross the endothelial cell layer and be preferentially retained in this milieu compared to LDL (Rath et al. 1989). In 2004, it was reported that the apo(a) component of Lp(a) elicits a dramatic rearrangement of the actin cytoskeleton characterized by increased central actin stress fiber formation, redistribution of focal adhesions, and VE-cadherin disruption; these effects are a consequence of apo(a)-mediated activation of the Rho-Rho kinase signaling pathway leading to increased myosin light chain phosphorylation (Pellegrino et al. 2004). A subsequent study showed that these effects are the product of increased phosphorylation of the myosin phosphatase regulatory subunit and hence inhibition of myosin phosphatase activity, that Lp(a) and apo(a) resulted in enhanced EC permeability, and that the KIV10 sLBS was required for these effects (Cho et al. 2008). Therefore, increasing EC permeability represents a mechanism by which Lp(a) can elicit a program of EC dysfunction in early atherosclerosis, facilitating deposition of Lp(a) and LDL in the vessel wall (Fig. 10.2). In a follow-up study, enhanced prostaglandin E2 synthesis and secretion were observed when cultured HUVECs were treated with apo(a) as a result of stimulation of β-catenin nuclear translocation and increased cyclooxygenase activity (Cho et al. 2013). Lp(a) and apo(a) were shown to activate a phosphatidylinositol-3 kinase and Akt-dependent pathway that resulted in phosphorylation and inhibition of GSK3β to promote β-catenin translocation; once again, these effects were attributable to the KIV10 domain of apo(a) (Cho et al. 2013).
Early studies demonstrated the ability of Lp(a) to elicit a proinflammatory response in HUVECS through enhanced expression of E-Selectin, VCAM1, and ICAM1 (Fig. 10.2) (Takami et al. 1998; Allen et al. 1998). The apo(a) component of Lp(a) binds to the β2-integrin Mac-1 in a lysine-dependent manner; this in turn promotes the adhesion of THP-1 monocytes to ECs and enhances their Mac-1-dependent transendothelial migration (Fig. 10.2) (Sotiriou et al. 2006). Interestingly, the Lp(a)-Mac-1 binding resulted in increased expression of tissue factor in these cells. Furthermore, the interaction between apo(a) and Mac-1 induces activation of the inflammatory transcription factor NFκB (Sotiriou et al. 2006). Taken together, these studies define a novel role for apo(a)/Lp(a) in promoting inflammatory cell recruitment, which may represent a novel mechanism for Lp(a) atherogenicity in vivo (Fig. 10.2). In a more a recent study, the oxPL on KIV10 was shown to activate human aortic endothelial cells, resulting in transendothelial migration of monocytes (Fig. 10.2) (Schnitzler et al. 2020). Transcriptome analysis of Lp(a)-stimulated ECs showed upregulation of inflammatory pathways related to monocyte adhesion and migration; these effects increased 6-phophofructo-2-kinase/fructose-2,6-biphosphatase (PFKFB)-3-mediated glycolysis and could be abolished by inhibition of PFKFB3 (Schnitzler et al. 2020).
Effects of Lp(a) on Vascular Smooth Muscle Cells
A number of early in vitro studies suggested that Lp(a) could contribute to cultured vascular SMC migration and proliferation through the ability of apo(a) to inhibit the plasmin-dependent activation of TGF-β (Fig. 10.2) (Grainger et al. 1993, 1994; O’Neil et al. 2004). More recently, the ability of OxPL on apo(a) KIV10 to stimulate the expression of Klf-4, an important factor in phenotypic switching of SMCs in atherosclerotic lesions, was attributed to the apo(a)-mediated activation of the long noncoding RNA MIAT (Fig. 10.2) (Fasolo et al. 2021). Lp(a) has also been implicated in the calcification of human aortic SMCs through Notch1-NFκB and Notch1-BMP2-Smad1/5/9 pathways (Peng et al. 2022). The Notch1-NFκB pathway resulted in increased osteopontin and inflammatory cytokine expression, while Lp(a)-mediated Notch1-BMP2-Smad1/5/9 activation also contributed to calcification of the cells. The ability of Lp(a) to increase VSMC calcification is another mechanism by which Lp(a) contributes to vascular disease (Fig. 10.2).
Lp(a) and Monocyte/Macrophage Phenotype
It is well-established that Lp(a) can contribute to macrophage foam cell formation (Fig. 10.2) (Keesler et al. 1996). However, the role of the OxPL on apo(a) in macrophage function is a relatively recent finding. The first direct evidence of the OxPL on Lp(a)/apo(a) mediating a proinflammatory response in macrophages was provided by Seimon and coworkers (Seimon et al. 2010); these investigators demonstrated that the OxPL was capable of inducing apoptosis in endoplasmic reticulum (ER)-stressed macrophages in a CD36/TLR2-dependent manner (Fig. 10.2). Lp(a)/apo(a) has also been shown to contribute to monocyte recruitment through enhancing secretion of chemokines I-309 and interleukin(IL)-8 from cultured macrophages (Fig. 10.2) (Scipione et al. 2015; Haque et al. 2000; Edelstein et al. 2003). In our own studies of the apo(a)-induced IL-8 expression in macrophages, we conclusively determined that this stimulatory effect was attributable to the covalent OxPL modification on apo(a) (Scipione et al. 2015). The apo(a)/OxPL-induced signaling cascade in our study also suggested a role for CD36/TLR2 and involved the JNK- and ERK-dependent activation of NFκB—a well-known series of molecular events in inflammatory pathways—in response to OxPL-containing apo(a). The OxPL moiety on apo(a) has also been implicated in the proinflammatory priming of human peripheral blood mononuclear cells (Fig. 10.2) (van der Valk et al. 2016). The same study used high-resolution in vivo imaging to show that monocytes from high Lp(a) individuals had a propensity of trafficking to the arterial wall, a result not seen in subjects with lower Lp(a) levels (van der Valk et al. 2016).
Lp(a) and Valve Interstitial Cell Phenotype
Recent studies have demonstrated a key role for Lp(a) in both the development and progression of aortic valve disease (Thanassoulis et al. 2013; Capoulade et al. 2015). This is likely mediated, in large part, by the ability of the OxPL component of apo(a) to modify the phenotype of valve interstitial cells to resulting in proinflammatory and pro-osteogenic responses in these cells (Fig. 10.2). In a recent study by Zheng and coworkers, treatment of VICs by Lp(a) or recombinant apo(a) resulted in osteogenic differentiation in these cells through the induction of IL6, BMP2, and RUNX2 expression (Zheng et al. 2019). The effects were attributed to the OxPL on Lp(a) and apo(a): the anti-oxPL antibody E06 blocked the effects of Lp(a) as did mutation of the KIV10 LBS which significantly reduced the effect of apo(a) (Zheng et al. 2019). OxPLs transported by Lp(a) also increase the load of reactive oxygen species in the aortic valve, loading to ROS-mediated activation of the NFκB pathway, and induction of a program of inflammatory gene expression (Bouchareb et al. 2015; Mathieu et al. 2017). Additionally, Lp(a) binds to autotaxin and delivers it to valves (Bouchareb et al. 2015; Mathieu et al. 2017); autotaxin promotes inflammation and osteogenic transdifferentiation of VICs through the production of LysoPA which in turn binds and signals through the lysoPA receptor (Fig. 10.2). Taken together, these studies suggest that Lp(a) can initiate a program of inflammation and osteoblastic differentiation in valvular interstitial cells which is a major contributing factor to AVS and CAVD. Lp(a) could also promote CAVD through promotion of valve endothelial cell dysfunction, immune cell infiltration, and foam cell formation (Fig. 10.2).
Effect of Lp(a) on Thrombosis and Thrombolysis
The homology of apo(a) and plasminogen revealed by cloning of a cDNA-encoding apo(a) immediately invited speculation of an antifibrinolytic role for Lp(a) (McLean et al. 1987). However, the decades of research that have ensued have yet to provide a definitive answer on this question (Boffa 2022). There is certainly a large body of evidence from in vitro studies pointing to the ability of Lp(a) and—especially—apo(a) to inhibit plasminogen activation and impede fibrinolysis (Fig. 10.3). The earliest studies showed that Lp(a) could compete with plasminogen for binding to fibrin, endothelial cells, and monocytes (Miles et al. 1989; Hajjar et al. 1989; Rouy et al. 1992). Subsequent studies showed that Lp(a) and/or apo(a) could inhibit lysis of fibrin clots and inhibit plasminogen activation on the surface of fibrin, fibrin degradation products, and platelets (Sangrar et al. 1995, 1997; Hancock et al. 2003; Ezratty et al. 1993). Definitive demonstration of inhibition on plasminogen activation on the cell surface was only recently provided by our group (Romagnuolo et al. 2014, 2018a, b). Apo(a) was shown to inhibit thrombolysis in rabbit jugular vein and mouse pulmonary embolism models (Biemond et al. 1997; Palabrica et al. 1995); notably, however, Lp(a) itself was not tested in these studies. Indeed, the available data from human epidemiological and genetic studies do not provide evidence for a direct antifibrinolytic/prothrombotic impact of elevated Lp(a) (Boffa 2022). Moreover, in a recent study using subjects undergoing Lp(a) lowering with antisense oligonucleotide therapy, we found that despite dramatic reductions in plasma Lp(a) concentrations, ex vivo plasma clot lysis time was not affected (Boffa et al. 2019). Furthermore, recombinant apo(a) had a potent antifibrinolytic effect whereas Lp(a) purified from human plasma lacked this effect (Boffa et al. 2019). Against this backdrop, we summarize below the clinical evidence with respect to Lp(a) and thrombosis and thrombolysis, and we outline areas for future studies of this issue.

Effects on imbalance between coagulation (formation of fibrin by thrombin cleavage of fibrinogen) and fibrinolysis (degradation of insoluble fibrin into soluble fibrin degradation products) can cause thrombosis. Lp(a) and apo(a) promote this imbalance by favoring coagulation (green mechanisms) and impeding fibrinolysis (red mechanisms). TFPI tissue factor pathway inhibitor
Is There Direct Evidence That Lp(a) Inhibits Fibrinolysis?
Earlier observational studies provided mixed results concerning whether elevated Lp(a) is a risk factor for the development of VTE (Boffa and Koschinsky 2016). This is notable in the sense that similar studies of atherosclerotic cardiovascular disease (ASCVD) quite consistently showed elevated Lp(a) to be an independent risk factor. With the advent of genetic approaches to study the association of genetically elevated Lp(a) with disease—including Mendelian randomization studies—the opportunity to assess a causal role for elevated Lp(a) in VTE and to eliminate confounding variables has arisen. Several large studies using genetic approaches have been published. All showed that genetically inherited elevated Lp(a) or genetical markers of high Lp(a) were not significant predictors of VTE (Kamstrup et al. 2012; Helgadottir et al. 2012; Danik et al. 2013; Larsson et al. 2020). Importantly, in several of these studies, a causal role for elevated Lp(a) in the development of atherothrombotic disorders was observed in the same population (Kamstrup et al. 2012; Helgadottir et al. 2012; Larsson et al. 2020). However, a posthoc observational study of one of these populations found that extremely high Lp(a) levels (≥95th percentile) were significantly associated with VTE (Kamstrup et al. 2012). A recent retrospective study of pulmonary embolism patients found no correlation between Lp(a) levels and the severity of pulmonary embolism (Gressenberger et al. 2022).
The general lack of association between elevated Lp(a) and risk for VTE (except for extremely high Lp(a) concentrations) is consistent with a minimal or absent antifibrinolytic ability of Lp(a). Venous thrombi are fibrin- and erythrocyte-rich and form as a consequence of blood stasis, hypercoagulability, and endothelial damage. Thrombi in the arterial tree are platelet-rich and fibrin-poor, and generally form as a sequela of atherosclerotic plaque erosion or rupture. Thus, mechanistic implications of associations between Lp(a) levels and ASCVD endpoints are confounded by the possibility that Lp(a) may contribute to atherosclerosis, thrombosis, or both. Interestingly, a consistent observation (albeit from relatively small sample sizes) has been the association between elevated Lp(a) levels and risk of ischemic stroke in children (Nowak-Gottl et al. 1999; Strater et al. 2002; Kenet et al. 2010; Goldenberg et al. 2013). These events are frequently seen in patients with inherited dispositions toward thrombophilia such as Factor V Leiden. That the events would occur in the absence of underlying atherosclerosis are clear from the young age of the patients, and this may speak to a prothrombotic or antifibrinolytic effect of Lp(a).
Early studies examined the proposition that elevated Lp(a) could reduce the efficacy of thrombolytic therapy. Most of these occurred in the setting of acute myocardial infarction (MBewu et al. 1994; Tranchesi et al. 1991; Armstrong et al. 1990; von Hodenberg et al. 1991; Brugemann et al. 1994), although one examined ischemic stroke (Ribo et al. 2004). None of these studies showed that Lp(a) levels are a significant predictor of successful recanalization, although all of them were limited by small sample sizes (and thus few patients with high Lp(a)) and/or having sampled blood for Lp(a) measurement in the postinfarct period where the acute phase response may have increased Lp(a) levels (Boffa 2022).
Taken together, the jury is still out on whether Lp(a) inhibits fibrinolysis in vivo. Further studies in animal models may be required to assess this question, and further assessment of the impact of Lp(a) on thrombolytic therapy in the setting of ischemic stroke, deep vein thrombosis, and pulmonary embolism may be warranted.
Could Lp(a) Promote Thrombosis Indirectly?
Elevated Lp(a) is clearly and consistently associated with ASCVD events, though less so with intermediate phenotypes such as intimal-medial thickness and coronary calcium scores (Kivimaki et al. 2011; Raitakari et al. 1999; Razavi et al. 2021; Mehta et al. 2022). This can be interpreted to mean that Lp(a) plays a more important role in precipitating atherothrombotic events, rather than in promoting the underlying atherosclerotic process. Two scenarios can be contemplated.
-
1.
Lp(a) could be promoting thrombus formation directly through an impact on the coagulation system or on platelet activation (Fig. 10.3). Little to no data on an effect of Lp(a) on coagulation have been published, although early studies showed that Lp(a) could exert a procoagulant effect by binding and inhibiting tissue factor pathway inhibitor (Fig. 10.3) (Caplice et al. 2001). We and others have shown that Lp(a) and apo(a) can impact fibrin clot structure, leading to a form resistant to lysis (Scipione et al. 2017; Skuza et al. 2019; Rowland et al. 2014) (Fig. 10.3); we also demonstrated that apo(a) could accelerate the rate of fibrin formation, which could also impact clot structure (Scipione et al. 2017). Lp(a) does bind to platelets (Ezratty et al. 1993; Martinez et al. 2001), and Lp(a) and apo(a) have been shown increase the proaggregant effect of certain agonists (such as the protease-activated receptor-1 ligand peptide SFLLRN and arachidonic acid) in washed platelets (Martinez et al. 2001; Rand et al. 1998). However, two recent studies in platelet-rich plasma showed that Lp(a) level did not predict the aggregation response to several agonists including ADP, collagen, or arachidonic acid (Salsoso et al. 2020; Kille et al. 2021).
-
2.
Lp(a) could be promoting a “vulnerable” plaque phenotype with a greater propensity to rupture and hence cause an atherothrombotic event (Fig. 10.3). Consistent with this scenario, it was reported in a carotid ultrasound study that Lp(a) level predicted the extent of stenosis but not total plaque area (Klein et al. 2008); the extent of stenosis could be interpreted to reflect ongoing rupture and thrombus formation. The proinflammatory effects of Lp(a) owing to its OxPL could result in a larger necrotic core and/or a thinner fibrous cap, both hallmarks of rupture-prone plaques. Very little direct study of this question has been attempted. In an immunohistochemical study of human coronary atherosclerotic plaques of varying phenotypes, apo(a) immunostaining was found in proximity to oxidation-specific epitopes—such as the OxPL recognized by E06—specifically in vulnerable or ruptured plaques (van Dijk et al. 2012).
It is clear from the above that, while some evidence for Lp(a)-promoting vulnerable plaque and/or arterial thrombosis exists, more research is necessary. This will require both work in animal models, such as transgenic mice expressing human Lp(a), as well as more imaging and biomarker studies in human patients. Such research is important because it may help to stratify risk in patients with high Lp(a) to identify those who would most benefit from emerging Lp(a)-lowering therapies.
Concluding Remarks
These are exciting times in the Lp(a) field, with burgeoning interest in this causal risk factor for CVD on from both basic researchers and clinicians. The quantitative importance of elevated Lp(a) as a risk factor is now widely accepted, although at present clinical adoption of Lp(a) measurement has lagged because of a lack, currently, of Lp(a)-lowering therapies. With Phase III cardiovascular outcomes trials in progress on an antisense oligonucleotide-targeting LPA mRNA, we are potentially on the cusp of having an effective treatment for lowering Lp(a) as well as definitive proof that elevated Lp(a) is harmful. At the same time, our understanding of the pathogenic mechanisms of Lp(a) continues to expand, with the role of Lp(a) as a proinflammatory mediator emerging as a key factor. Further studies of these mechanisms may lead to an alternative therapeutic strategy to mitigate the effect of elevated Lp(a)—interference with its pathogenic effects in the vasculature.
References
Allen S, Khan S, Tam S, et al. Expression of adhesion molecules by lp(a): a potential novel mechanism for its atherogenicity. FASEB J. 1998;12:1765–76.
Armstrong VW, Neubauer C, Schutz E, et al. Lack of association between raised serum Lp(a) concentration and unsuccessful thrombolysis after acute myocardial infarction. Lancet. 1990;336:1077.
Arsenault BJ, Kamstrup PR. Lipoprotein(a) and cardiovascular and valvular diseases: a genetic epidemiological perspective. Atherosclerosis. 2022;349:7–16.
Becker L, Cook PM, Wright TG, et al. Quantitative evaluation of the contribution of weak lysine-binding sites present within apolipoprotein(a) kringle IV types 6-8 to lipoprotein(a) assembly. J Biol Chem. 2004;279:2679–88.
Berg K, New A. Serum type system in man–the Lp system. Acta Pathol Microbiol Scand. 1963;59:369–82.
Bergmark C, Dewan A, Orsoni A, et al. A novel function of lipoprotein [a] as a preferential carrier of oxidized phospholipids in human plasma. J Lipid Res. 2008;49:2230–9.
Biemond BJ, Friederich PW, Koschinsky ML, et al. Apolipoprotein(a) attenuates endogenous fibrinolysis in the rabbit jugular vein thrombosis model in vivo. Circulation. 1997;96:1612–5.
Boffa MB. Beyond fibrinolysis: the confounding role of Lp(a) in thrombosis. Atherosclerosis. 2022;349:72–81.
Boffa MB, Koschinsky ML. Lipoprotein (a): truly a direct prothrombotic factor in cardiovascular disease? J Lipid Res. 2016;57:745–57.
Boffa MB, Koschinsky ML. Understanding the ins and outs of lipoprotein (a) metabolism. Curr Opin Lipidol. 2022;33:185–92.
Boffa MB, Marar TT, Yeang C, et al. Potent reduction of plasma lipoprotein (a) with an antisense oligonucleotide in human subjects does not affect ex vivo fibrinolysis. J Lipid Res. 2019;60:2082–9.
Bouchareb R, Mahmut A, Nsaibia MJ, et al. Autotaxin derived from lipoprotein(a) and valve interstitial cells promotes inflammation and mineralization of the aortic valve. Circulation. 2015;132:677–90.
Brugemann J, van der Meer J, Hillege HL, et al. Lipoprotein(a) levels in patients with myocardial infarction treated with anistreplase: no prediction of efficacy but inverse correlation with plasminogen activation in non-patency. Int J Cardiol. 1994;45:109–13.
Caplice NM, Panetta C, Peterson TE, et al. Lipoprotein (a) binds and inactivates tissue factor pathway inhibitor: a novel link between lipoproteins and thrombosis. Blood. 2001;98:2980–7.
Capoulade R, Chan KL, Yeang C, et al. Oxidized phospholipids, lipoprotein(a), and progression of calcific aortic valve stenosis. J Am Coll Cardiol. 2015;66:1236–46.
Cho T, Jung Y, Koschinsky ML. Apolipoprotein(a), through its strong lysine-binding site in KIV(10′), mediates increased endothelial cell contraction and permeability via a Rho/Rho kinase/MYPT1-dependent pathway. J Biol Chem. 2008;283:30503–12.
Cho T, Romagnuolo R, Scipione C, et al. Apolipoprotein(a) stimulates nuclear translocation of beta-catenin: a novel pathogenic mechanism for lipoprotein(a). Mol Biol Cell. 2013;24:210–21.
Danik JS, Buring JE, Chasman DI, et al. Lipoprotein(a), polymorphisms in the LPA gene, and incident venous thromboembolism among 21483 women. J Thromb Haemost. 2013;11:205–8.
Dzobo KE, Kraaijenhof JM, Stroes ESG, et al. Lipoprotein(a): an underestimated inflammatory mastermind. Atherosclerosis. 2022;349:101–9.
Eaton DL, Fless GM, Kohr WJ, et al. Partial amino acid sequence of apolipoprotein(a) shows that it is homologous to plasminogen. Proc Natl Acad Sci U S A. 1987;84:3224–8.
Edelstein C, Pfaffinger D, Hinman J, et al. Lysine-phosphatidylcholine adducts in kringle V impart unique immunological and potential pro-inflammatory properties to human apolipoprotein(a). J Biol Chem. 2003;278:52841–7.
Ezratty A, Simon DI, Loscalzo J. Lipoprotein(a) binds to human platelets and attenuates plasminogen binding and activation. Biochemistry. 1993;32:4628–33.
Fasolo F, Jin H, Winski G, et al. Long noncoding RNA MIAT controls advanced atherosclerotic lesion formation and plaque destabilization. Circulation. 2021;144:1567–83.
Gabel BR, Koschinsky MI. Analysis of the proteolytic activity of a recombinant form of apolipoprotein(a). Biochemistry. 1995;34:15777–84.
Goldenberg NA, Bernard TJ, Hillhouse J, et al. Elevated lipoprotein (a), small apolipoprotein (a), and the risk of arterial ischemic stroke in North American children. Haematologica. 2013;98:802–7.
Grainger DJ, Kirschenlohr HL, Metcalfe JC, et al. Proliferation of human smooth muscle cells promoted by lipoprotein(a). Science. 1993;260:1655–8.
Grainger DJ, Kemp PR, Liu AC, et al. Activation of transforming growth factor-beta is inhibited in transgenic apolipoprotein(a) mice. Nature. 1994;370:460–2.
Gressenberger P, Posch F, Pechtold M, et al. Lipoprotein(a) and pulmonary embolism severity-a retrospective data analysis. Front Cardiovasc Med. 2022;9:808605.
Hajjar KA, Gavish D, Breslow JL, et al. Lipoprotein(a) modulation of endothelial cell surface fibrinolysis and its potential role in atherosclerosis. Nature. 1989;339:303–5.
Hancock MA, Boffa MB, Marcovina SM, et al. Inhibition of plasminogen activation by lipoprotein(a): critical domains in apolipoprotein(a) and mechanism of inhibition on fibrin and degraded fibrin surfaces. J Biol Chem. 2003;278:23260–9.
Haque NS, Zhang X, French DL, et al. CC chemokine I-309 is the principal monocyte chemoattractant induced by apolipoprotein(a) in human vascular endothelial cells. Circulation. 2000;102:786–92.
Helgadottir A, Gretarsdottir S, Thorleifsson G, et al. Apolipoprotein(a) genetic sequence variants associated with systemic atherosclerosis and coronary atherosclerotic burden but not with venous thromboembolism. J Am Coll Cardiol. 2012;60:722–9.
Kamstrup PR, Tybjaerg-Hansen A, Nordestgaard BG. Genetic evidence that lipoprotein(a) associates with atherosclerotic stenosis rather than venous thrombosis. Arterioscler Thromb Vasc Biol. 2012;32:1732–41.
Keesler GA, Gabel BR, Devlin CM, et al. The binding activity of the macrophage lipoprotein(a)/apolipoprotein(a) receptor is induced by cholesterol via a post-translational mechanism and recognizes distinct kringle domains on apolipoprotein(a). J Biol Chem. 1996;271:32096–104.
Kenet G, Lutkhoff LK, Albisetti M, et al. Impact of thrombophilia on risk of arterial ischemic stroke or cerebral sinovenous thrombosis in neonates and children: a systematic review and meta-analysis of observational studies. Circulation. 2010;121:1838–47.
Kille A, Nuhrenberg T, Franke K, et al. Association of lipoprotein(a) with intrinsic and on-clopidogrel platelet reactivity. J Thromb Thrombolysis. 2021; https://doi.org/10.1007/s11239-021-02515-2.
Kivimaki M, Magnussen CG, Juonala M, et al. Conventional and Mendelian randomization analyses suggest no association between lipoprotein(a) and early atherosclerosis: the Young Finns Study. Int J Epidemiol. 2011;40:470–8.
Klein JH, Hegele RA, Hackam DG, et al. Lipoprotein(a) is associated differentially with carotid stenosis, occlusion, and total plaque area. Arterioscler Thromb Vasc Biol. 2008;28:1851–6.
Koschinsky ML, Boffa MB. Oxidized phospholipid modification of lipoprotein(a): Epidemiology, biochemistry and pathophysiology. Atherosclerosis. 2022;349:92–100.
Koschinsky ML, Cote GP, Gabel B, et al. Identification of the cysteine residue in apolipoprotein(a) that mediates extracellular coupling with apolipoprotein B-100. J Biol Chem. 1993;268:19819–25.
Lackner C, Cohen JC, Hobbs HH. Molecular definition of the extreme size polymorphism in apolipoprotein(a). Hum Mol Genet. 1993;2:933–40.
Larsson SC, Gill D, Mason AM, et al. Lipoprotein(a) in alzheimer, atherosclerotic, cerebrovascular, thrombotic, and valvular disease: mendelian randomization investigation. Circulation. 2020;141:1826–8.
Leibundgut G, Scipione C, Yin H, et al. Determinants of binding of oxidized phospholipids on apolipoprotein (a) and lipoprotein (a). J Lipid Res. 2013;54:2815–30.
Marcovina SM, Hobbs HH, Albers JJ. Relation between number of apolipoprotein(a) kringle 4 repeats and mobility of isoforms in agarose gel: basis for a standardized isoform nomenclature. Clin Chem. 1996;42:436–9.
Martinez C, Rivera J, Loyau S, et al. Binding of recombinant apolipoprotein(a) to human platelets and effect on platelet aggregation. Thromb Haemost. 2001;85:686–93.
Mathieu P, Arsenault BJ, Boulanger MC, et al. Pathobiology of Lp(a) in calcific aortic valve disease. Expert Rev Cardiovasc Ther. 2017;15:797–807.
MBewu AD, Durrington PN, Mackness MI, et al. Serum Lp(a) lipoprotein concentration and outcome of thrombolytic treatment for myocardial infarction. Br Heart J. 1994;71:316–21.
McLean JW, Tomlinson JE, Kuang WJ, et al. cDNA sequence of human apolipoprotein(a) is homologous to plasminogen. Nature. 1987;330:132–7.
Mehta A, Vasquez N, Ayers CR, et al. Independent association of lipoprotein(a) and coronary artery calcification with atherosclerotic cardiovascular risk. J Am Coll Cardiol. 2022;79:757–68.
Miles LA, Fless GM, Levin EG, et al. A potential basis for the thrombotic risks associated with lipoprotein(a). Nature. 1989;339:301–3.
Nowak-Gottl U, Strater R, Heinecke A, et al. Lipoprotein (a) and genetic polymorphisms of clotting factor V, prothrombin, and methylenetetrahydrofolate reductase are risk factors of spontaneous ischemic stroke in childhood. Blood. 1999;94:3678–82.
O’Neil CH, Boffa MB, Hancock MA, et al. Stimulation of vascular smooth muscle cell proliferation and migration by apolipoprotein(a) is dependent on inhibition of transforming growth factor-beta activation and on the presence of kringle IV type 9. J Biol Chem. 2004;279:55187–95.
Palabrica TM, Liu AC, Aronovitz MJ, et al. Antifibrinolytic activity of apolipoprotein(a) in vivo: human apolipoprotein(a) transgenic mice are resistant to tissue plasminogen activator-mediated thrombolysis. Nat Med. 1995;1:256–9.
Pellegrino M, Furmaniak-Kazmierczak E, LeBlanc JC, et al. The apolipoprotein(a) component of lipoprotein(a) stimulates actin stress fiber formation and loss of cell-cell contact in cultured endothelial cells. J Biol Chem. 2004;279:6526–33.
Peng J, Liu MM, Liu HH, et al. Lipoprotein (a)-mediated vascular calcification: population-based and in vitro studies. Metabolism. 2022;127:154960.
Raitakari OT, Adams MR, Celermajer DS. Effect of Lp(a) on the early functional and structural changes of atherosclerosis. Arterioscler Thromb Vasc Biol. 1999;19:990–5.
Rand ML, Sangrar W, Hancock MA, et al. Apolipoprotein(a) enhances platelet responses to the thrombin receptor-activating peptide SFLLRN. Arterioscler Thromb Vasc Biol. 1998;18:1393–9.
Rath M, Niendorf A, Reblin T, et al. Detection and quantification of lipoprotein(a) in the arterial wall of 107 coronary bypass patients. Arteriosclerosis. 1989;9:579–92.
Razavi AC, Cardoso R, Dzaye O, et al. Risk markers for limited coronary artery calcium in persons with significant aortic valve calcium (from the Multi-ethnic Study of Atherosclerosis). Am J Cardiol. 2021;156:58–64.
Ribo M, Montaner J, Molina CA, et al. Admission fibrinolytic profile predicts clot lysis resistance in stroke patients treated with tissue plasminogen activator. Thromb Haemost. 2004;91:1146–51.
Romagnuolo R, Marcovina SM, Boffa MB, et al. Inhibition of plasminogen activation by apo(a): role of carboxyl-terminal lysines and identification of inhibitory domains in apo(a). J Lipid Res. 2014;55:625–34.
Romagnuolo R, DeMarco K, Scipione CA, et al. Apolipoprotein(a) inhibits the conversion of Glu-plasminogen to Lys-plasminogen on the surface of vascular endothelial and smooth muscle cells. Thromb Res. 2018a;169:1–7.
Romagnuolo R, Scipione CA, Bazzi ZA, et al. Inhibition of pericellular plasminogen activation by apolipoprotein(a): roles of urokinase plasminogen activator receptor and integrins alphaMbeta2 and alphaVbeta3. Atherosclerosis. 2018b;275:11–21.
Rouy D, Koschinsky ML, Fleury V, et al. Apolipoprotein(a) and plasminogen interactions with fibrin: a study with recombinant apolipoprotein(a) and isolated plasminogen fragments. Biochemistry. 1992;31:6333–9.
Rowland CM, Pullinger CR, Luke MM, et al. Lipoprotein (a), LPA Ile4399Met, and fibrin clot properties. Thromb Res. 2014;133:863–7.
Salsoso R, Dalcoquio TF, Furtado RHM, et al. Relation of high lipoprotein (a) concentrations to platelet reactivity in individuals with and without coronary artery disease. Adv Ther. 2020;37:4568–84.
Sangrar W, Bajzar L, Nesheim ME, et al. Antifibrinolytic effect of recombinant apolipoprotein(a) in vitro is primarily due to attenuation of tPA-mediated Glu-plasminogen activation. Biochemistry. 1995;34:5151–7.
Sangrar W, Gabel BR, Boffa MB, et al. The solution phase interaction between apolipoprotein(a) and plasminogen inhibits the binding of plasminogen to a plasmin-modified fibrinogen surface. Biochemistry. 1997;36:10353–63.
Schnitzler JG, Hoogeveen RM, Ali L, et al. Atherogenic lipoprotein(a) increases vascular glycolysis, thereby facilitating inflammation and leukocyte extravasation. Circ Res. 2020;126:1346–59.
Scipione CA, Sayegh SE, Romagnuolo R, et al. Mechanistic insights into Lp(a)-induced IL-8 expression: a role for oxidized phospholipid modification of apo(a). J Lipid Res. 2015;56:2273–85.
Scipione CA, McAiney JT, Simard DJ, et al. Characterization of the I4399M variant of apolipoprotein(a): implications for altered prothrombotic properties of lipoprotein(a). J Thromb Haemost. 2017;15:1834–44.
Seimon TA, Nadolski MJ, Liao X, et al. Atherogenic lipids and lipoproteins trigger CD36-TLR2-dependent apoptosis in macrophages undergoing endoplasmic reticulum stress. Cell Metab. 2010;12:467–82.
Skuza AA, Polak M, Undas A. Elevated lipoprotein(a) as a new risk factor of cerebral venous sinus thrombosis: association with fibrin clot properties. J Thromb Thrombolysis. 2019;47:8–15.
Sotiriou SN, Orlova VV, Al-Fakhri N, et al. Lipoprotein(a) in atherosclerotic plaques recruits inflammatory cells through interaction with Mac-1 integrin. FASEB J. 2006;20:559–61.
Strater R, Becker S, von Eckardstein A, et al. Prospective assessment of risk factors for recurrent stroke during childhood—a 5-year follow-up study. Lancet. 2002;360:1540–5.
Takami S, Yamashita S, Kihara S, et al. Lipoprotein(a) enhances the expression of intercellular adhesion molecule-1 in cultured human umbilical vein endothelial cells. Circulation. 1998;97:721–8.
Thanassoulis, G, Campbell, CY, Owens, DS, et al., CHARGE Extracoronary Calcium Working Group. Genetic associations with valvular calcification and aortic stenosis. N Engl J Med. 2013;368:503–12.
Tranchesi B Jr, Chamone DF, Cobbaert C, et al. Coronary recanalization rate after intravenous bolus of alteplase in acute myocardial infarction. Am J Cardiol. 1991;68:161–5.
van der Hoek YY, Wittekoek ME, Beisiegel U, et al. The apolipoprotein(a) kringle IV repeats which differ from the major repeat kringle are present in variably-sized isoforms. Hum Mol Genet. 1993;2:361–6.
van der Valk FM, Bekkering S, Kroon J, et al. Oxidized phospholipids on lipoprotein(a) elicit arterial wall inflammation and an inflammatory monocyte response in humans. Circulation. 2016;134:611–24.
van Dijk RA, Kolodgie F, Ravandi A, et al. Differential expression of oxidation-specific epitopes and apolipoprotein(a) in progressing and ruptured human coronary and carotid atherosclerotic lesions. J Lipid Res. 2012;53:2773–90.
von Hodenberg E, Kreuzer J, Hautmann M, et al. Effects of lipoprotein (a) on success rate of thrombolytic therapy in acute myocardial infarction. Am J Cardiol. 1991;67:1349–53.
Yeang C, Cotter B, Tsimikas S. Experimental animal models evaluating the causal role of lipoprotein(a) in atherosclerosis and aortic stenosis. Cardiovasc Drugs Ther. 2016;30:75–85.
Youssef A, Clark JR, Marcovina SM, et al. Apo(a) and ApoB interact noncovalently within hepatocytes: implications for regulation of Lp(a) levels by modulation of ApoB secretion. Arterioscler Thromb Vasc Biol. 2022;42:289–304.
Zheng KH, Tsimikas S, Pawade T, et al. Lipoprotein(a) and oxidized phospholipids promote valve calcification in patients with aortic stenosis. J Am Coll Cardiol. 2019;73:2150–62.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2023 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Boffa, M.B., Koschinsky, M.L. (2023). Molecular Mechanisms of Lipoprotein(a) Pathogenicity: Tantalizing Clues and Unanswered Questions. In: Kostner, K., Kostner, G.M., Toth, P.P. (eds) Lipoprotein(a). Contemporary Cardiology. Humana, Cham. https://doi.org/10.1007/978-3-031-24575-6_10
Download citation
DOI: https://doi.org/10.1007/978-3-031-24575-6_10
Published:
Publisher Name: Humana, Cham
Print ISBN: 978-3-031-24574-9
Online ISBN: 978-3-031-24575-6
eBook Packages: MedicineMedicine (R0)