
Abstract
Angiotensin-(1–7) is a heptapeptide hormone of the renin-angiotensin system with anti-proliferative, anti-inflammatory, anti-oxidant, anti-angiogenic, and anti-fibrotic properties. This chapter summarizes published preclinical and clinical research assessing the use of angiotensin-(1–7) as a chemotherapy for cancer and cancer-related pathologies. Animal studies demonstrate that the heptapeptide hormone activates a unique angiotensin receptor mas1 to attenuate lung, breast, prostate, nasopharyngeal, and liver tumor growth by regulating multiple signaling pathways critical for carcinogenesis initiation and progression as well as reducing tumor-associated angiogenesis, inflammatory response, and fibrosis. Clinical benefit with limited side effects was observed in cancer patient trials assessing Ang-(1–7) alone or in combination with standard of care therapies. The published results thus far indicate that angiotensin-(1–7) may serve as a first-in-class, targeted monotherapy for the treatment of cancer or as an effective adjuvant to enhance reductions in cancer progression and reduce toxicity of current cancer therapeutic regimens.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
Introduction
Angiotensin-(1–7) [Ang-(1–7)] is a seven amino acid peptide hormone of the renin-angiotensin system (RAS), a primary regulator of blood pressure, electrolyte balance, cardiovascular and renal function, and cell growth [1,2,3,4]. In the RAS cascade, the non-functional precursor protein angiotensinogen is cleaved by the aspartyl protease renin to form the decapeptide angiotensin I (Ang I) (Fig. 24.1). Subsequently, one of three enzymes, neprilysin, thimet oligopeptidase, or prolylendopeptidase, convert Ang I to Ang-(1–7) depending on the physiological compartment. In a two-step process, Ang-(1–7) is produced by the cleavage of Ang I to the octapeptide Ang II by the dipeptidyl-carboxypeptidase angiotensin converting enzyme (ACE) or the endopeptidase chymase. The monocarboxypeptidase ACE2 then cleaves Ang II to Ang-(1–7). The heptapeptide hormone is ultimately degraded by two potential enzymes: ACE catabolizes Ang-(1–7) to the inactive degradation product Ang-(1–5) [5], while dipeptidyl peptidase 3 degrades the heptapeptide hormone to Ang-(3–7) or Ang-(5–7) (Fig. 24.1) [6, 7]. It is important to note that the RAS cascade is highly regulated with the proteolytic enzymes playing a critical role in maintaining the balance of tissue and circulating Ang II and Ang-(1–7) concentrations for normal physiological function.

Enzymatic synthesis and catabolism of Ang-(1–7)
The biological actions of peptide hormones are mediated by binding and activating cell-surface receptors, to initiate cascades of downstream reactions that modulate physiological processes. Ang II is the endogenous ligand for two G-protein coupled receptors (GPRCs), angiotensin 1 (AT1) and angiotensin 2 (AT2) receptors, while Ang-(1–7) binds and activates a unique 325 amino acid GPCR mas1, encoded by the MAS1 gene [1,2,3,4]. Ang-(1–7) interaction with the mas1 receptor promotes downstream signaling through generation of cyclic adenosine monophosphate (cAMP) [8, 9] to reduce cellular proliferation [8, 9]. Incubation with Rp-CAMPS, an inhibitor of the cAMP-dependent protein kinase A (PKA), in VSMCs abrogated the Ang-(1–7)-mediated reduction in cell proliferation, indicating that activation of PKA by cAMP is necessary for the heptapeptide hormone to exert inhibitory effects on cell growth [10]. Subsequent decades of research demonstrated that the heptapeptide hormone through activation of mas1 receptor counter-regulates the actions of mitogens, cytokines, and growth factors to prevent dysregulated vasoconstriction, cell proliferation, oxidative stress, fibrosis, inflammation, angiogenesis, and thrombosis, depending on the cell type and tissue (Fig. 24.2).

Ang-(1–7) blocks biological processes stimulated by cytokines, mitogens and growth factors. The background shows a breast tumor section stained for the proliferation marker Ki67 (red-brown stain)
Summarized below are published studies assessing the anti-cancer properties of Ang-(1–7). This article will focus on the in vivo preclinical research investigating the effect of the heptapeptide hormone on various types of cancer in animal models, the clinical trials reported using Ang-(1–7) as a chemotherapeutic drug, and the use of Ang-(1–7) in combination with standard of care therapies. The research described below highlight the use of Ang-(1–7) or viral vectors producing Ang-(1–7). In vivo studies that overexpress ACE2 to enhance production of Ang-(1–7) were not included as this enzyme has multiple physiological substrates complicating the ability to assess the precise molecular mechanism producing the observed effect in the animal or patient.
Ang-(1–7) and Cancer in Preclinical Animal Models
Lung Cancer
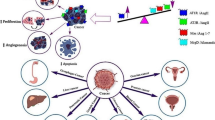
The first evidence that Ang-(1–7) inhibited cancer cell proliferation was reported in lung cancer cells by Gallagher and Tallant [11]. Sub-nanomolar concentrations of the heptapeptide hormone significantly reduced DNA synthesis and proliferation of human adenocarcinoma SK-LU-1 and A549 cells as well as non-small lung cancer SK-MES-1 cells as determined by attenuated serum-stimulated [3H]-thymidine incorporation, decreased cell number and ERK1/ERK2 dephosphorylation. Treatment with the heptapeptide hormone also decreased MAP kinase signaling and Akt and PI3K activation in platinum-resistant human lung cancer cells [12]. Delivery of Ang-(1–7) by lentiviral transduction resulted in reduced lung cell proliferation with an associated decrease in DNA synthesis and the replicative protein Cdc6 [13, 14], suggesting that the heptapeptide hormone regulates multiple proliferative signaling pathways. No reduction in lung cancer cell proliferation was observed following incubation with Ang I, Ang II, Ang-(2–8), Ang-(3–7) or Ang-(3–8) [11], demonstrating peptide specificity. Inhibition of proliferation by Ang-(1–7) was blocked by the mas1 receptor antagonist [D-Alanine7]-Ang-(1–7) (D-ala). Incubation with the Ang II type 1 receptor antagonist losartan or the Ang II type 2 receptor antagonist PD123319 did not attenuate the reduction in lung cancer cell proliferation by Ang-(1–7), supporting a selective receptor-mediated process.
Administration of the heptapeptide hormone by osmotic mini-pump [15], subcutaneous injection [16] or adenoviral vector delivery [13, 14] caused a significant reduction in lung tumor xenograft volume and wet weight as well as a decrease in the immunostaining of proliferation marker Ki67 in tumors from mice as compared to saline-treated control rodents. A similar attenuation of tumor volume and weight was observed following Ang-(1–7) treatment of mice harboring platinum resistant lung tumors [12]. No adverse reactions in the animals or tissue and organ abnormalities were observed with Ang-(1–7) administration, indicating that the heptapeptide hormone was well-tolerated and may have limited quality-of-life effects when administered to patients. A marked decrease in COX-2 mRNA and protein was observed in tumor tissue from Ang-(1–7) treated mice as compared to controls, suggesting that regulation of prostaglandin synthesis by the heptapeptide hormone may play a role in the attenuation of lung tumor growth. In support of these in vivo findings, Ang-(1–7) or the microRNA miR-513a that is upregulated by Ang-(1–7) inhibited COX-2 production and activity in serum-stimulated human A549 lung cancer cells with an associated decrease in the anti-inflammatory prostaglandin E2 [11, 17, 18].
Ang-(1–7) also inhibited angiogenesis in human A549 tumor xenografts [16] and platinum-resistant lung cancer xenografts [12] with a concomitant decrease in the pro-angiogenic cytokine VEGF. Treatment of the parent A549 lung cancer cells or platinum-resistant human lung cancer cells with the heptapeptide hormone also resulted in a reduction in VEGF protein and mRNA. Additionally, the heptapeptide hormone markedly reduced neovascularization of the chick chorioallantoic membrane and endothelial tubule branching, two models of angiogenesis. Collectively, these studies suggest that Ang-(1–7) could serve as an anti-angiogenic drug to reduce lung cancer growth.
Migration and invasion of human A549 lung cancer cells in vitro were attenuated significantly following treatment with nanomolar concentrations of Ang-(1–7) [19]. A concomitant inactivation of Akt/pKB and MAPK signaling and a decrease in metalloproteinase 2 and 9 activity was observed, suggesting that the heptapeptide hormone blocks migration and invasion by inhibiting the degradation of the extracellular matrix by these two enzymes. Lung cancer cell invasion was attenuated by Ang-(1–7) or miRNA-149-3p, a microRNA upregulated by the heptapeptide hormone, with an associated decrease in integrin subunit beta, a key protein in proliferation and extracellular matrix interaction [17, 18]. Knockdown or antagonist blockade of the mas1 receptor prevented the decrease in migration and invasion by Ang-(1–7), indicating a receptor-mediated process. In addition, epithelial-mesenchymal transition in which epithelial cells gain migratory properties was inhibited in human lung cancer cells following transfection with a lentiviral construct to over-express the heptapeptide hormone [13, 14]. The results of these studies suggest that Ang-(1–7) may inhibit multiple steps in the metastasis cascade (Fig. 24.3); however, animal studies are needed to determine whether the heptapeptide hormone effectively reduces lung cancer metastasis in vivo.

Summary of the characteristics of tumors reduced by Ang-(1–7) activation of the mas1 receptor
Breast Cancer
Ang-(1–7) inhibited the growth of human estrogen receptor positive tumors or human estrogen receptor positive, HER2-overexpressing tumors in the mammary fat pad of ovariectomized female mice receiving estrogen replacement therapy, as compared to tumors from control animals [20]. A concomitant decrease in interstitial and perivascular fibrosis and collagen I deposition was observed in tumors from mice infused with the heptapeptide hormone, in addition to the reduction in tumor volume and weight. This suggests that one mechanism whereby Ang-(1–7) attenuates tumor growth is by preventing the proliferation of growth-promoting cancer-associated fibroblasts [21]. In support, the heptapeptide hormone inhibited in vitro proliferation of fibroblasts isolated from orthotopic breast tumors with an associated reduction in the pro-fibrotic proteins fibronectin and transforming growth factor β (TGF-β) [20]. A decrease in MAP kinase phosphorylation and an accompanying increase in the phosphatase DUSP1 was observed in tumor-associated fibroblasts incubated with Ang-(1–7), suggesting an attenuation of MAP kinase signaling to reduce fibroblast proliferation.
Luo et al. [22] showed that treatment with tamoxifen caused an up-regulation of the mas1 receptor in estrogen-receptor positive MCF-7 cells but not triple-negative basal A MDA-MB-468 cells. Cell proliferation and invasion were inhibited but apoptosis was enhanced significantly in estrogen-receptor positive MCF-7 cells, triple-negative basal A MDA-MB-468 cells, and triple-negative basal B 4T1 cells incubated with Ang-(1–7). These effects were prevented in all cell lines by knockdown of mas1 with an siRNA. Further, increased tumor growth was observed following injection of 4T1 cells transfected with the mas1 siRNA as compared to 4T1 cells containing a control siRNA. Taken together, these results demonstrate the receptor-mediated action of Ang-(1–7) in the regulation of proliferation and suggest that mas1 may serve as a negative regulator of tumor growth (Fig. 24.3).
Ang-(1–7) also prevented the migration of human triple negative MDA-MB-231 cells; migration was equivalent to control levels following co-administration of the mas1 receptor antagonist A-779 with the heptapeptide hormone [23]. Incubation of MDA-MB-231 cells with Ang-(1–7) resulted in increased E-cadherin, a cancer metastasis suppressor, and reduced ZEB1, TWIST1 and Snail1, negative transcriptional regulators of E-cadherin, supporting the potential anti-metastatic actions of the heptapeptide hormone. Further, a marked decrease in NF-κB p65, a transcriptional regulator of Snail1, phosphorylation of PAK1, a regulator of Snail1 nuclear translocation, and the activity of the intracellular Ca++ regulator SOCE was observed in MDA-MB-231 cells after incubation with Ang-(1–7). Taken together, these results suggest that the heptapeptide hormone modulates the PAK1/NF-κB/Snail1 signaling pathways by decreasing SOCE-mediated Ca2+ influx to reduce metastasis.
Prostate Cancer
Krishnan et al. [24] demonstrated a marked reduction in the proliferation of human LNCaP prostate cancer cells with an associated decrease in secreted angiogenic factors VEGF and PlGF following incubation with Ang-(1–7). Further, the transcription factor HIF-1α, a primary regulator of VEGF family signaling, was also reduced, suggesting that the heptapeptide hormone could inhibit prostate tumor growth in part by attenuating angiogenesis. This was supported by in vivo studies in that the volume and weight of human LNCaP prostate xenograft tumors as well as tumor angiogenesis was diminished following infusion of Ang-(1–7) for 54 days. Ki67 and MAP kinase Erk1/2 phosphorylation were decreased in tumors from mice treated with the heptapeptide hormone, demonstrating the anti-proliferative actions of Ang-(1–7). In addition, tumors from mice administered Ang-(1–7) had reduced VEGF and PlGF protein and mRNA as well as VEGF receptors Flt-1 and Flk1 but enhanced concentrations of sFlt-1. SFlt-1 is a soluble decoy receptor that traps VEGF and PlGF to reduce circulating concentration of the angiogenic factors. Additionally, s-Flt-1binds to the membrane VEGF receptors and disrupts proliferative signaling. Taken together, these results suggest that Ang-(1–7) disrupts VEGF family signaling to reduce prostate tumor angiogenesis, leading to an attenuation of tumor growth (Fig. 24.3).
Ang-(1–7) attenuated the migration of human PC3 cells in vitro, an effect blocked by the mas receptor antagonist D-alanine-Ang-(1–7), suggesting that activation of mas by the heptapeptide hormone may block an early step in metastasis [25]. In support, infusion of Ang-(1–7) into mice one week prior to the injection of human PC3 cells into the aortic arch prevented the formation of tumors at metastatic sites, while 100% of the control mice developed tumors in the submandibular bone, the spinal column or the long bone of the leg. The heptapeptide hormone also prevented the growth of tumors produced by injecting prostate cancer cells directly into the tibia. The inhibition of tumor proliferation was associated with a decrease in osteoclastogenesis, indicating that the heptapeptide hormone inhibited the formation of osteolytic pits required for tumor cell engraft and grow in the bone microenvironment [26]. These studies suggest that Ang-(1–7) may serve as an effective anti-metastatic agent in men with prostate cancer.
Nasopharyngeal Carcinoma
The Ang-(1–7) receptor mas was increased in nasopharyngeal carcinoma cell lines and patient tumors as compared to an immortalized nasopharyngeal epithelial cell line and normal human nasopharyngeal epithelial tissue, suggesting that nasopharyngeal carcinomas may be susceptible to the anti-proliferative effects of the heptapeptide hormone [13]. Transduction of nasopharyngeal carcinoma cells with a lentiviral construct producing Ang-(1–7) significantly reduced cell proliferation and migration as compared to cells harboring the control construct. The effect was blocked by the mas receptor antagonist A-779. Similar to the observations of Krishnan et al. [25] in prostate tumors, the transduced nasopharyngeal carcinoma cells producing Ang-(1–7) had reduced phosphorylation of the MAP kinases ERK1/2 and p38, decreased angiogenic factors VEGF, PlGF, transcription factor Hif-1a, VEGF receptors Flt-1 and Flk-1, and increased the soluble decoy VEGF receptor sFlt-1. These data indicate that the heptapeptide hormone could attenuate nasopharyngeal carcinoma growth by inhibiting angiogenesis. Injection of an adenoviral vector construct that produced Ang-(1–7) significantly decreased the weight of nasopharyngeal xenograft tumors with an associated reduction in the proliferation marker Ki67. A significant reduction in vessel density was observed in the tumors from mice injected with the Ang-(1–7) adenoviral construct as well as a decrease in tumor VEGF and PlGF, HIF-1α, as well as Flt1 and Flk1 as compared to tumors from control mice. Conversely, the sFlt-1 was increased with the enhanced production of the heptapeptide hormone, suggesting that Ang-(1–7) attenuates nasopharyngeal cancer through inhibition of angiogenesis.
Daily subcutaneous injection of Ang-(1–7) also reduced nasopharyngeal carcinoma xenographs with a concomitant reduction in the proliferation marker Ki67 [27]. In addition, the phosphorylation of p38, Akt, p-GSK3-β, and mTOR was decreased significantly in tumors from mice treated with the heptapeptide hormone, while an increase in the autophagy proteins PI3K, LC3-II, and Becline-1 was observed. Co-administration of the mas1 receptor antagonist A779 prevented the attenuation of tumor growth, the inactivation of the PI3K/Akt/mTor and p38 signaling pathways and the induction of autophagy by the heptapeptide hormone. Ang-(1–7) reduced proliferation, migration, and invasion of nasopharyngeal carcinoma cells in vitro as well as activated autophagy, further supporting the in vivo observations. The results of the two studies published thus far indicate that the heptapeptide hormone may reduce nasopharyngeal carcinomas by multiple mechanisms and warrant further investigation (Fig. 24.3).
Liver Cancer
The growth of mouse hepatic tumor xenografts was inhibited significantly following subcutaneous infusion of Ang-(1–7) as assessed by tumor volume and weight [28]. The effect was blocked by the mas receptor antagonist A779, demonstrating a receptor-mediated process. The decrease in hepatic tumor proliferation was associated with an increase in apoptotic cells and caspase 3 activity. Administration of the heptapeptide hormone to tumor-bearing mice also markedly attenuated angiogenesis in the liver tumors with a concomitant reduction in VEGF mRNA and protein. Reduced proliferation and increased apoptosis following incubation with Ang-(1–7) was observed in hepatic H22 cancer cells. The effect was blocked by co-administration of A779, further supporting the in vivo results.
Mao et al. [29] also demonstrated attenuation of hepatic tumor growth in mice using adenoviral delivery of Ang-(1–7). A significant reduction in tumor size and weight was observed in mice administered the heptapeptide hormone as compared to animals receiving the control vector. Inhibition of cancer cell proliferation was demonstrated by a reduction in immunostaining of Ki67 and Cdc6 (cell division cycle 6 protein) in tissue sections of tumors from Ang-(1–7) treated mice as compared to controls. In addition, a marked decrease in the mRNA of the angiogenic factors VEGF and PlGF as well as the VEGF receptors Flt-1 and Flk-1 was observed in tumor samples from mice receiving the heptapeptide hormone. Taken together, these two studies demonstrate that Ang-(1–7) reduces liver cancer in mice by pleiotropic mechanisms and suggest that the heptapeptide hormone may effectively inhibit liver cancer in patients (Fig. 24.3).
Preclinical animal studies support a cytostatic mechanism of action for Ang-(1–7). While the heptapeptide hormone inhibits tumor growth as compared to the tumors from untreated control animals, the tumor weight or size is generally not decreased to baseline measures prior to initiation of Ang-(1–7) treatment [12, 15, 16, 20, 25, 27, 30, 31]. Thus, it may be possible to reduce doses of cytotoxic chemotherapeutics in combination with Ang-(1–7) and still obtain positive outcomes.
Clinical Trials in Cancer Patients Administered Ang-(1–7)
Subcutaneous injection of Ang-(1–7) in escalating doses (100, 200, 400, and 700 µg/Kg) was administered to cohorts of 3 patients with solid tumors refractory to standard of care therapy for five consecutive days of a 21 day cycle [32]. Serum concentrations of the heptapeptide hormone prior to study initiation were equivalent to healthy adults [33]. The mean half-life was between 25 and 37 min, similar to previous report by Rodgers et al. [34] in breast cancer patients, with a maximum bioavailability of 1 h post injection. Attenuation of tumor growth for at least three months was observed in four of the 15 evaluable patients, while one patient with metastatic sarcoma receiving the highest dose of Ang-(1–7) had a mixed response without disease progression for 10 months. The heptapeptide hormone was well tolerated with mild toxicities for most patients. The maximum tolerated dose (MTD) was defined as 400 μg/kg since serious adverse events, including vascular abnormalities or neuropathy, were observed in two of six patients at the 700 μg/kg dose. Prevention in tumor growth was associated with a decrease in circulating concentrations of the angiogenic factor PlGF [32, 35], suggesting a reduction in angiogenesis by Ang-(1–7).
In a Phase II trial, patients with advanced metastatic sarcoma self-injected 20 mg/day of Ang-(1–7) in a 21-day cycle [36]. This dose was approximately equivalent to the MTD of 400 µg/kg designated in the Phase I trial [32]. Response Evaluation Criteria in Solid Tumors (RECIST 1.1) [37, 38], an assessment of tumor burden, was used to evaluate patient response to the heptapeptide hormone. In 9 of 20 patients, tumor progression was attenuated for more than three months; two of these patients had disease stabilization for 10 and 19 months. No progressive decrease in plasma PlGF or VEGF with Ang-(1–7) administration was observed in any patient. One patient experienced a grade 3 deep vein thrombosis, which was resolved with anticoagulants. The primary endpoint of a 10% response rate (percentage of patient’s cancer reduction after treatment) was not reached with this small cohort.
Based on these two studies, further clinical trials with Ang-(1–7) as a monotherapy are warranted, as stabilization of tumor progression was observed in a significant percentage of the small number of patients assessed. Similar to animal studies described above, the clinical trial results described thus far suggest that the heptapeptide hormone has cytostatic actions and may be more efficacious when administered with cytotoxic therapeutics.
Combination Therapy with Ang-(1–7) in Preclinical Models
Summarized below are studies assessing Ang-(1–7) with radiation or standard of care chemotherapeutics. Cancer therapy regimens generally consist of multiple procedures, including surgery, radiation, and cancer drugs. Studies in animal models are vital to determine whether a combination therapy is more efficacious without the addition of side effects and there are no antagonistic actions.
Circulating white blood cells as well as bone marrow myeloid, erythroid and megakaryocyte progenitor cells were increased markedly following Ang-(1–7) administration to mice receiving total body irradiation [39]. Co-administration of A779, the mas1 receptor antagonist, blocked the improvement in hematopoietic recovery induced by the heptapeptide hormone. Surprisingly, treatment with Ang-(1–7) several days after total body irradiation still invoked an improvement in hematopoietic recovery [40]. In fact, a more pronounced enhancement of bone marrow progenitor cells was observed when Ang-(1–7) treatment was delayed until after cessation of the myelotoxic damage induced by irradiation. Treatment with the heptapeptide hormone also reduced pathological fibrosis induced by radiotherapy. Interstitial and perivascular fibrosis was increased significantly in the skeletal muscle of mice exposed to clinically equivalent doses irradiation in the hindlimb as compared to non-irradiated mice [41]. Infusion of Ang-(1–7) prior to radiation effectively prevented the pathological fibrotic effect. The radiation-induced increase in the profibrotic cytokines TGF-β and connective tissue growth factor CTGF in the soleus muscle was blocked by administration of the heptapeptide hormone prior to irradiation. These results suggest that Ang-(1–7) may serve as an effective prophylactic to prevent or recover tissue or organ damage caused by radiotherapy.
Many chemotherapeutic drugs also cause multi-lineage cytopenias and most hematopoietic agents do not provide protection for the various cell types found in the blood. Adjuvant treatment of Ang-(1–7) significantly increased early lineage bone marrow progenitor cells in mice following treatment with the myelosuppressive drugs 5-fluoruracil or cyclophosphamide [42, 43] or gemcitabine [44]. The combination drug treatment not only enhanced the number of bone marrow progenitor cells, circulating blood cells and platelets in myelosuppressive mice greater than the concentration observed with either agent alone but reduced concentrations of Neupogen or Epogen was required when in combination with Ang-(1–7). Collectively, these studies indicated that Ang-(1–7) may enhance recovery of bone marrow hematopoietic precursors and circulating blood cells and elements in patients following myelosuppressive chemotherapy.
Ager et al. [30, 31] published the first study showing that Ang-(1–7) effectively reduced liver metastasis induced by intrasplenic injection of colorectal cancer into mice. Combination therapy of the heptapeptide hormone with an ACE inhibitor or AT1 receptor blocker did not result in enhanced tumor inhibition. A subsequent study supported the attenuation of liver metastasis in mice by Ang-(1–7) or the ACE inhibitor Captopril and further showed that both treatments also caused a marked increase in hepatic macrophages at the tumor margins [30, 31]. This suggests that one mechanism whereby the heptapeptide hormone or ACE inhibitors reduce liver metastasis is through the immunomodulation of Kupffer cells, which secrete anti-proliferative factors.
Doxorubicin (Dox) is an effective, anti-cancer drug used to treat patients with breast, lung, ovarian, bladder, and pediatric cancers. Unfortunately, clinical administration is limited due to potential cumulative, dose-dependent drug toxicity, which may be transient or ultimately lead to heart failure. Clinically equivalent doses of Dox induced alterations in cardiac morphometry as well as global and diastolic dysfunction in male and female juvenile rats, similar to the pathologies observed in pediatric patients administered anthracycline therapy [45]. The Dox-mediated cardiac toxicity was blocked by Ang-(1–7) co-administration to the rats. The heptapeptide hormone attenuated the increase in NADPH oxidase 4 (Nox4), by-products of lipid peroxidation, malondialdehyde and 4-hydroxynonenal and the decrease in the antioxidant enzymes superoxide dismutase and catalase caused by Dox administration. Further, the heptapeptide hormone prevented the enhanced interstitial and coronary vessel fibrosis with an associated increase in inflammatory cardiac TGF-β1 and pSMAD2 in the hearts of juvenile rats of both sexes following Dox administration. Dox caused a significant increase in pulse wave velocity, a measure of arterial stiffness in rats of both sexes but the mechanism was distinct [46]. An increase in lumen diameter, wall thickness, media hypertrophy and reduced elasticity was observed in the aortic arches of male rats administered Dox, while in the juvenile female rats the anthracycline increased fibrosis. The Dox-mediated damage to the juvenile rat aortic arches was prevented by co-administrations of Ang-(1–7). The results of these two studies suggest that the heptapeptide hormone may serve as an effective preventative agent to reduce cardiovascular damage caused by anthracycline administration to cancer patients (Fig. 24.4).

Dox-induced cardiac damage that is prevented by Ang-(1–7) administration
Administration of Ang-(1–7) or sunitinib (a multi-targeted receptor tyrosine kinase inhibitor) reduced human clear cell renal cell (RCC) cancer growth in mice; however, treatment with both drugs in combination more effectively diminished tumor proliferation [47]. The mas1 receptor antagonist A779 blocked the heptapeptide hormone inhibition of RCC growth in mice, demonstrating a receptor-mediated process. Similar results were obtained with co-administration of Ang-(1–7) with axitinib, a combined PD-L1 (immune checkpoint inhibitor of the programmed death-ligand 1) and VEGF-TKI (VEGF receptor-tyrosine kinase inhibitor), a chemotherapy regimen used to treat resistant RCC. Monotherapy using Ang-(1–7) or the dual combination of VEGF-TKI and PD-L1 inhibitor significantly inhibited RCC tumor growth in mice compared to tumors in the control animals, while combination of the two drugs enhanced the reduction in tumor proliferation. These results suggest that this multiple drug regimen including Ang-(1–7) may be more efficacious in patients with RCC cancer than standard of care therapy.
Taken together, the result of these combination studies suggest that Ang-(1–7) may enhance the efficacy as well as prevent the toxicity of standard of care therapies, as no adverse outcomes were reported in the animals receiving combination therapy.
Clinical Trials in Cancer Patients Administered Ang-(1–7) With Standard of Care Therapy
Rodgers et al. [34] assessed the use of Ang-(1–7) as an adjuvant to Dox and cyclophosphamide chemotherapy administered in newly diagnosed breast cancer patients. This Phase I/II trial evaluated the toxicity and optimal biologic dose of Ang-(1–7) administered to breast cancer patients after surgery as well as before and during chemotherapy to mitigate multiple types of cytopenia. Two days after chemotherapy and at least 10 consecutive days in three consecutive cycles, patients received escalating doses of Ang-(1–7) by subcutaneous injection or filgrastim, an approved synthetic drug that stimulates bone marrow production of granulocyte colony-stimulating factor. Ang-(1–7) was administered daily two days after surgery for seven consecutive days followed by a 1 week drug holiday before the first cycle of chemotherapy to assess toxicity. No dose limiting toxicity was found for Ang-(1–7) and no patients administered with the heptapeptide hormone experienced a treatment-related SAE. While the dose required to mitigate cytopenia varied by hematological lineages, patients treated with the heptapeptide hormone following chemotherapy showed stabilized platelet concentration, reduced incidence of anemia, lymphopenia, and mucositis, as well as an accelerated recovery of leukocytes, lymphocytes, neutrophils and hemoglobin. This study suggests that Ang-(1–7) is safe and mitigated the multilineage cytopenias caused by chemotherapy, demonstrating the potential of Ang-(1–7) as an adjuvant for chemotherapy.
Similar results were obtained in a clinical trial evaluating the efficacy of Ang-(1–7) as an adjuvant therapy for patients with recurrent ovarian, Fallopian tube, or peritoneal carcinoma [48]. In this randomized, double-blind, placebo-controlled Phase II trial, patients received placebo or Ang-(1–7) with either intravenous cisplatin followed by gemcitabine or gemcitabine with carboplatin. The heptapeptide hormone in combination with myelosuppressive chemotherapy markedly enhanced platelet count and reduced Grade 4 thrombocytopenia in patients. Importantly, Ang-(1–7) was evaluated as a safe, tolerable drug with limited side effects. Unfortunately, the study was terminated early, due to low enrollment, lower than expected grade 3–4 thrombocytopenia in the placebo group, and a change in clinical practices to taxane-based chemotherapies. Nevertheless, the results of this trial support the use of Ang-(1–7) as a therapeutic to prevent cumulative myelotoxicity induced by chemotherapeutic agents.
Conclusions
The research data summarized above indicate that Ang-(1–7) attenuates the growth of multiple tumor types in mice by pleiotropic mechanisms, including altered or activated cellular signaling, reduced angiogenesis, tumor-associated fibrosis, and inflammation as well as increased apoptosis and autophagy. Ang-(1–7) activates mas1, a unique angiotensin receptor to mediate these anti-cancer properties, indicating that the heptapeptide hormone is a targeted therapy. The heptapeptide hormone has a broader spectrum of anti-cancer properties than current standard-of-care therapies with limited toxicity, indicating enhanced quality-of-life for patients. Thus, Ang-(1–7) may effectively serve as a first-in-class, targeted drug for the treatment of cancer and cancer-related pathologies or as an adjuvant to mitigate toxicity caused by existing treatment regimens. However, before Ang-(1–7) could be considered a marketable drug it will be necessary to overcome the limited stability of the heptapeptide hormone in the circulation as well as the lack of oral bioavailability and a patent-protected structure needed for drug development to proceed.
References
Santos RAS, Sampaio WO, Alzamora AC, Motta-Santos D, Alenina N, Bader M et al (2018) The ACE2/angiotensin-(1–7)/MAS axis of the renin-angiotensin system: focus on angiotensin-(1–7). Physiol Rev 98(1):505–553
Santos RAS, Oudit GY, Verano-Braga T, Canta G, Steckelings UM, Bader M (2019) The renin-angiotensin system: going beyond the classical paradigms. Am J Physiol Heart Circ Physiol 316(5):H958–H970
Paz Ocaranza M, Riquelme JA, García L, Jalil JE, Chiong M, Santos RAS et al (2020) Counter-regulatory renin-angiotensin system in cardiovascular disease. Nat Rev Cardiol 17(2):116–129
Khajehpour S, Aghazadeh-Habashi A (2021) Targeting the protective arm of the renin-angiotensin system: focused on angiotensin-(1–7). J Pharmacol Exp Ther 377(1):64–74
Chappell MC, Pirro NT, Sykes A, Ferrario CM (1998) Metabolism of angiotensin-(1–7) by angiotensin-converting enzyme. Hypertension 31(1 Pt 2):362–367
Marshall AC, Pirro NT, Rose JC, Diz DI, Chappell MC (2014) Evidence for an angiotensin-(1–7) neuropeptidase expressed in the brain medulla and CSF of sheep. J Neurochem 130(2):313–323
Cruz-Diaz N, Wilson BA, Pirro NT, Brosnihan KB, Marshall AC, Chappell MC (2016) Identification of dipeptidyl peptidase 3 as the Angiotensin-(1–7) degrading peptidase in human HK-2 renal epithelial cells. Peptides 83:29–37
Tallant EA, Diz DI, Ferrario CM (1999) State-of-the-Art lecture. Antiproliferative actions of angiotensin-(1–7) in vascular smooth muscle. Hypertension 34(4 Pt 2):950–957
McCollum LT, Gallagher PE, Ann TE (2012) Angiotensin-(1–7) attenuates angiotensin II-induced cardiac remodeling associated with upregulation of dual-specificity phosphatase 1. Am J Physiol Heart Circ Physiol 302(3):H801–H810
Tallant EA, Clark MA (2003) Molecular mechanisms of inhibition of vascular growth by angiotensin-(1–7). Hypertension 42(4):574–579
Gallagher PE, Tallant EA (2004) Inhibition of human lung cancer cell growth by angiotensin-(1–7). Carcinogenesis 25(11):2045–2052
Geng YL, Ding YJ, Ni L, Xu KD, Le VM, Ji R et al (2021) The role of angiotensin-(1–7) on acquired platinum resistance-induced angiogenesis in non-small cell lung cancer in vitro and in vivo. Neoplasma 68(4):770–779
Pei N, Wan R, Chen X, Li A, Zhang Y, Li J et al (2016) Angiotensin-(1–7) decreases cell growth and angiogenesis of human nasopharyngeal carcinoma xenografts. Mol Cancer Ther 15(1):37–47
Chen X, Chen S, Pei N, Mao Y, Wang S, Yan R et al (2017) AAV-Mediated angiotensin 1–7 overexpression inhibits tumor growth of lung cancer in vitro and in vivo. Oncotarget 8(1):354–363
Menon J, Soto-Pantoja DR, Callahan MF, Cline JM, Ferrario CM, Tallant EA et al (2007) Angiotensin-(1–7) inhibits growth of human lung adenocarcinoma xenografts in nude mice through a reduction in cyclooxygenase-2. Cancer Res 67(6):2809–2815
Soto-Pantoja DR, Menon J, Gallagher PE, Tallant EA (2009) Angiotensin-(1–7) inhibits tumor angiogenesis in human lung cancer xenografts with a reduction in vascular endothelial growth factor. Mol Cancer Ther 8(6):1676–1683
Silva BeO, Lima KF, Gonçalves LR, Silveira MB, Moraes KC (2016) MicroRNA profiling of the effect of the heptapeptide angiotensin-(1–7) in A549 lung tumor cells reveals a role for miRNA149–3p in cellular migration processes. PLoS ONE 11(9):e0162094
da Silveira MB, Lima KF, da Silva AR, Dos Santos RAS, Moraes KCM (2018) Mir-513a-3p contributes to the controlling of cellular migration processes in the A549 lung tumor cells by modulating integrin β-8 expression. Mol Cell Biochem 444(1–2):43–52
Ni L, Feng Y, Wan H, Ma Q, Fan L, Qian Y et al (2012) Angiotensin-(1–7) inhibits the migration and invasion of A549 human lung adenocarcinoma cells through inactivation of the PI3K/Akt and MAPK signaling pathways. Oncol Rep 27(3):783–790
Cook KL, Metheny-Barlow LJ, Tallant EA, Gallagher PE (2010) Angiotensin-(1–7) reduces fibrosis in orthotopic breast tumors. Cancer Res 70(21):8319–8328
Yoshida GJ, Azuma A, Miura Y, Orimo A (2019) Activated fibroblast program orchestrates tumor initiation and progression; molecular mechanisms and the associated therapeutic strategies. Int J Mol Sci 20(9)
Luo Y, Tanabe E, Kitayoshi M, Nishiguchi Y, Fujiwara R, Matsushima S et al (2015) Expression of MAS1 in breast cancer. Cancer Sci 106(9):1240–1248
Yu C, Tang W, Wang Y, Shen Q, Wang B, Cai C et al (2016) Downregulation of ACE2/Ang-(1–7)/Mas axis promotes breast cancer metastasis by enhancing store-operated calcium entry. Cancer Lett 376(2):268–277
Krishnan B, Smith TL, Dubey P, Zapadka ME, Torti FM, Willingham MC et al (2013) Angiotensin-(1–7) attenuates metastatic prostate cancer and reduces osteoclastogenesis. Prostate 73(1):71–82
Krishnan B, Torti FM, Gallagher PE, Tallant EA (2013) Angiotensin-(1–7) reduces proliferation and angiogenesis of human prostate cancer xenografts with a decrease in angiogenic factors and an increase in sFlt-1. Prostate 73(1):60–70
Chen F, Han Y, Kang Y (2021) Bone marrow niches in the regulation of bone metastasis. Br J Cancer 124(12):1912–1920
Lin YT, Wang HC, Chuang HC, Hsu YC, Yang MY, Chien CY (2018) Pre-treatment with angiotensin-(1–7) inhibits tumor growth via autophagy by downregulating PI3K/Akt/mTOR signaling in human nasopharyngeal carcinoma xenografts. J Mol Med (Berlin) 96(12):1407–1418
Liu Y, Li B, Wang X, Li G, Shang R, Yang J et al (2015) Angiotensin-(1–7) suppresses hepatocellular carcinoma growth and angiogenesis via complex interactions of Angiotensin II type 1 receptor, Angiotensin II type 2 receptor and mas receptor. Mol Med 21:626–636
Mao Y, Pei N, Chen X, Chen H, Yan R, Bai N et al (2018) Angiotensin 1–7 overexpression mediated by a Capsid-optimized AAV8 vector leads to significant growth inhibition of hepatocellular carcinoma. Int J Biol Sci 14(1):57–68
Ager EI, Wen SW, Chan J, Chong WW, Neo JH, Christophi C (2011) Altered efficacy of AT1R-targeted treatment after spontaneous cancer cell-AT1R upregulation. BMC Cancer 11:274
Wen SW, Ager EI, Neo J, Christophi C (2013) The renin angiotensin system regulates Kupffer cells in colorectal liver metastases. Cancer Biol Ther 14(8):720–727
Petty WJ, Miller AA, McCoy TP, Gallagher PE, Tallant EA, Torti FM (2009) Phase I and pharmacokinetic study of angiotensin-(1–7), an endogenous antiangiogenic hormone. Clin Cancer Res 15(23):7398–7404
Merrill DC, Karoly M, Chen K, Ferrario CM, Brosnihan KB (2002) Angiotensin-(1–7) in normal and preeclamptic pregnancy. Endocrine 18(3):239–245
Rodgers KE, Oliver J, diZerega GS (2006) Phase I/II dose escalation study of angiotensin 1–7 [A(1–7)] administered before and after chemotherapy in patients with newly diagnosed breast cancer. Cancer Chemother Pharmacol 57(5):559–568
Petty WJ, Aklilu M, Varela VA, Lovato J, Savage PD, Miller AA (2012) Reverse translation of phase I biomarker findings links the activity of angiotensin-(1–7) to repression of hypoxia inducible factor-1α in vascular sarcomas. BMC Cancer 12:404
Savage PD, Lovato J, Brosnihan KB, Miller AA, Petty WJ (2016) Phase II trial of angiotensin-(1–7) for the treatment of patients with metastatic sarcoma. Sarcoma 2016:4592768
Schwartz LH, Seymour L, Litière S, Ford R, Gwyther S, Mandrekar S et al (2016) RECIST 1.1 - Standardisation and disease-specific adaptations: perspectives from the RECIST working group. Eur J Cancer 62:138–145
Iannessi A, Beaumont H, Liu Y, Bertrand AS (2021) RECIST 1.1 and lesion selection: how to deal with ambiguity at baseline? Insights Imaging 12(1):36
Rodgers KE, Xiong S, diZerega GS (2002) Accelerated recovery from irradiation injury by angiotensin peptides. Cancer Chemother Pharmacol 49(5):403–411
Rodgers KE, Espinoza T, Roda N, Meeks CJ, Hill C, Louie SG et al (2012) Accelerated hematopoietic recovery with angiotensin-(1–7) after total body radiation. Int J Radiat Biol 88(6):466–476
Willey JS, Bracey DN, Gallagher PE, Tallant EA, Wiggins WF, Callahan MF et al (2016) Angiotensin-(1–7) attenuates skeletal muscle fibrosis and stiffening in a mouse model of extremity sarcoma radiation therapy. J Bone Joint Surg Am 98(1):48–55
Rodgers K, Xiong S, DiZerega GS (2003) Effect of angiotensin II and angiotensin(1–7) on hematopoietic recovery after intravenous chemotherapy. Cancer Chemother Pharmacol 51(2):97–106
Ellefson DD, diZerega GS, Espinoza T, Roda N, Maldonado S, Rodgers KE (2004) Synergistic effects of co-administration of angiotensin 1–7 and Neupogen on hematopoietic recovery in mice. Cancer Chemother Pharmacol 53(1):15–24
Rodgers KE, Espinoza TB, Roda N, Meeks CJ, diZerega GS (2013) Angiotensin-(1–7) synergizes with colony-stimulating factors in hematopoietic recovery. Cancer Chemother Pharmacol 72(6):1235–1246
Rahimi O, Kirby J, Varagic J, Westwood B, Tallant EA, Gallagher PE (2020) Angiotensin-(1–7) reduces doxorubicin-induced cardiac dysfunction in male and female Sprague-Dawley rats through antioxidant mechanisms. Am J Physiol Heart Circ Physiol 318(4):H883–H894
Rahimi O, Melo AC, Westwood B, Grier RDM, Tallant EA, Gallagher PE (2022) Angiotensin-(1–7) reduces doxorubicin-induced aortic arch dysfunction in male and female juvenile Sprague Dawley rats through pleiotropic mechanisms. Peptides 152:170784
Khanna P, Soh HJ, Chen C-H, Saxena R, Amin S, Naughton M et al. (2021) ACE2 abrogates tumor resistance to VEGFR inhibitors suggesting angiotensin-(1–7) as a therapy for clear cell renal cell carcinoma. Sci Transl Med 13(577):eabc0170
Pham H, Schwartz BM, Delmore JE, Reed E, Cruickshank S, Drummond L et al (2013) Pharmacodynamic stimulation of thrombogenesis by angiotensin (1–7) in recurrent ovarian cancer patients receiving gemcitabine and platinum-based chemotherapy. Cancer Chemother Pharmacol 71(4):965–972
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2023 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Melo, A.C., Tallant, E.A., Gallagher, P.E. (2023). Angiotensin-(1–7): A Prospective Cancer Therapeutic. In: Bhullar, S.K., Tappia, P.S., Dhalla, N.S. (eds) The Renin Angiotensin System in Cancer, Lung, Liver and Infectious Diseases. Advances in Biochemistry in Health and Disease, vol 25. Springer, Cham. https://doi.org/10.1007/978-3-031-23621-1_24
Download citation
DOI: https://doi.org/10.1007/978-3-031-23621-1_24
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-031-23620-4
Online ISBN: 978-3-031-23621-1
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)