
Abstract
The Renin-Angiotensin System (RAS) is associated with regulation of blood pressure, electrolyte balance, and hemostasis. It is implicated in cancer hallmarks because of its local expression in almost all of the body’s tissues. RAS has recently been implicated in the progression of colorectal cancer (CRC) and subsequent liver metastasis. This review summarizes various RAS-associated cellular signaling pathways in colon cancer.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Renin-angiotensin system
- Colorectal cancer
- Liver metastasis
- Angiotensin
- Angiotensin-converting enzyme inhibitors
- Angiotensin receptor blockers
Introduction
Colorectal cancer is the world's third most prevalent malignancy and the fourth leading cause of cancer-related deaths, with approximately 9,40,000 new cases and 5,00,000 deaths reported each year [1]. CRC develops from the epithelial cells that line the colon and rectum. Colon cells replicate at a very high rate, with 1010 epithelial cells changed every day. This rapid replication rate increases susceptibility of colon epithelium to mutation and, as a result, carcinogenesis [2]. CRC pathogenesis is a complex interaction of genetic predisposition and lifestyle factors. CRC subtypes are hereditary and non-hereditary, with the majority being sporadic and caused by a somatic mutation in response to environmental factors [3]. It can develop from a serrated hyperplastic polyp or an adenomatous polyp (AP) through the adenoma-carcinoma sequence, as well as from spontaneous mutations and inherited conditions. Although a single BRAF (B-Raf proto-oncogene serine/threonine kinase) mutation can cause CRC in serrated-type hyperplastic polyps, the bulk of colon cancers today are caused by an AP through the adenoma to carcinoma sequence [4]. Colonoscopy is used routinely by physicians to detect the presence of APs, and lesions can be studied by histological evaluation. Further, histological results are used to determine the cells’ malignant potential [5]. Surgical resection offers the best chance of recovery. But, only about 20–25% of patients are candidates for surgery, and recurrence rates range from 40 to 70%. For the vast majority of these patients, palliative systemic chemotherapy is the preferred treatment option [6]. The primary cause of death from CRC is metastasis to the liver, which accounts for more than 70% of all deaths. In up to 30–40% of individuals with advanced CRC, the liver is the primary site of metastasis. 20–25% of CRC patients have detectable synchronous liver metastasis while primary diagnosis and another 40–50% will develop metachronous liver metastasis within three years of primary tumor resection [7]. Recent studies revealed variations in the compositions of the intestinal microbiota between CRC patients and healthy individuals [8].
Components of Renin-Angiotensin System (RAS), are renin, angiotensinogen (AGT), angiotensin I (Ang I), angiotensin-converting enzyme (ACE), angiotensin II (Ang II), angiotensin II type 1 receptor (AT1R), and angiotensin II type 2 receptor (AT2R). Recently, counter-regulatory axis of RAS is identified which comprises of angiotensin 1–7 (Ang 1–7), MAS Receptor (MASR), and angiotensin-converting enzyme 2 (ACE-2) [9]. Overactivation of RAS, which causes hypertension, raises the risk of cancer, cancer progression, and mortality in cancer patients [10]. Chronic inflammation and high levels of Vascular Endothelial Growth Factor (VEGF) during hypertension lead to endothelial dysfunction and angiogenesis, which may then act as auxiliary factors during cancer development [11]. Several studies have been conducted to investigate the role of RAS in the pathophysiology of CRC [12, 13]. Many renin-angiotensin system inhibitors have been shown to reduce the risk of cancer [14]. ACE inhibitors (ACEIs)/angiotensin II receptor blockers (ARBs) are RAS inhibitors that are used to treat hypertension. However, as more studies on RAS inhibitors have been conducted, it has been discovered that RAS inhibitors play a role not only in the treatment of hypertension but also in the treatment of colorectal cancer [15, 16].
Renin-Angiotensin System-Associated Cellular Signaling Pathways in Colorectal Cancer
In terms of cancer etiology, the mechanism of cancer development is a complex multistage process that involves sequential mutational events that occur concurrently with cancer progression [17]. Mutations in signaling pathways and genes that interact with RAS are involved in several tasks, including cell growth and survival. As a result, in a cancerous cell, these events will be interrupted [18]. The EGFR/MAPK, PI3K, and Wnt/β-catenin signaling pathways affect a variety of biological activities, including cell proliferation, differentiation, angiogenesis, apoptosis, and survival. These are some of the primary signaling pathways through which RAS protein functions.
RAS and EGFR/MAPK in Colorectal Cancer
When a ligand binds to receptor tyrosine kinase, the pathway is activated for the first time. For example, epidermal growth factor (EGF), whose receptor is EGFR. EGF releases from cell surface before it can bind with EGFR. This is accomplished through the use of the TACE/ADAM-17 enzyme, which cleaves transforming growth factor-α and amphiregulin, two EGF family ligands [19]. Following ligand binding, the receptor dimerizes and becomes phosphorylated. Following that, inside a cell, a protein complex is formed, with growth factor receptor-bound protein 2 (GRB2) attaching to the receptor while being bound by son of sevenless (SOS) [20]. SOS exhibits guanine nucleotide exchange factor activity after being attached to RAS. The RAF proteins (A-RAF, B-RAF, and C-RAF) can be recruited to the cell surface by active GTP-RAS [20]. It has been proposed that the Ras–Raf–ERK signaling pathway regulates cell growth, differentiation, and survival [20]. Malignant transformation and tumor progression results, if this pathway is faulty. A mutation in the RAS protein is one of the most common ways that the MAPK is activated; mutations in the KRAS protein are found in approximately 40% of all colorectal cancer (CRC) cases, whereas NRAS mutations are less common, accounting for 5% of all CRC cases. The most common mutations in KRAS and NRAS are found at codons 12, 13, and 61. These mutations can be found in early adenomas as well as cells with little potential for malignancy. Silencing these mutant codons results in a diminution of the tumorigenic features of diseased cells, according to both in vitro and animal studies [17]. BRAF mutates in the MAPK pathway, which appears to occur in about 5% to 10% of all colon cancer patients. The V600E mutation is the most common BRAF mutation in all cancers, including colorectal cancer [21]. As previously stated, the EGFR/MAPK signaling pathway has been linked to oncogenic processes and thus plays an important role in tumor growth and CRC progression [22]. This pathway's aberrant expression has been identified as a potential target for CRC treatment.
RAS and PI3K in Colorectal Cancer
PI3K/Akt is a critical intracellular signal pathway that regulates a wide range of cellular activities including cell growth, proliferation, differentiation, and migration. PI3K is divided into three classes, I–III, established on structural and functional differences. Type class IA contains two PI3K subunits, one regulatory (p85) and one catalytic (p110). Three genes, PIK3R1, PIK3R2, and PIK3R3, encode different isoforms of p85 as well as different types of p110 such as alpha, beta, gamma, and delta, which produce PIK3CA, PIK3CB, and PIK3CD, respectively [23]. Stimulating Ras activation or extracellular factors through RTK, causes PI3K activation. Cancer has been treated by inhibiting the PI3K/Akt pathway [24]. Akt phosphorylation has been linked to the downregulation of cell proliferation and apoptosis in human CRCs. Akt controls downstream targets like mTOR. Phosphatase and tensin homologue protein (PTEN), a tumor suppressor molecule, dephosphorylates PIP3 and thus inhibits the PI3K pathway [23]. P110 is encoded by the gene PIK3CA. RAS mutations frequently coexist with PIK3CA exon 9 and 20 mutations. 15–25% of CRCs are thought to have PIK3CA mutations, which might lead to higher PI3K activity [25]. Overall, the PI3K signaling pathway has been shown to play a carcinogenic role in the initiation and progression of CRC. Several studies have found that inhibiting this pathway specifically causes a decrease in CRC cell growth and an increase in apoptosis [24].
RAS and Wnt/β-Catenin in Colorectal Cancer
The Wnt/β-catenin signaling pathway has shown its role in a variety of biological processes, including embryonic development, tissue homeostasis, and carcinogenesis [26]. Excess Wnt/β-catenin signaling causes abnormal cell proliferation and inhibits apoptosis, which cause progression of colorectal cancer [27]. β-Catenin degradation is usually mediated by the destruction complex, which is made up of the adenomatous polyposis coli protein (APC) and numerous other proteins in the absence of Wnts. Further, Wnt phosphorylation of LRP6 disrupts this destruction complex. The active β-catenin then translocates into the nucleus, where it binds to the TCF/LEF to trigger the production of target oncogenes namely cyclin D1 and c-Myc [28, 29]. Over 80% of CRCs have APC loss-of-function mutations, and roughly 5% have β-catenin activating mutations, which result in constitutive activation of the Wnt/β-catenin pathway and thus contribute to oncogenesis [30]. Recent research has shown that silencing Wnt3 significantly reduced the activation of the Wnt/β-catenin pathway and the proliferation of CRC cells with APC or β-catenin mutations [31]. The 350-amino-acid protein, (Pro)renin receptor ((P)RR) was first identified as a key component of the RAS and is widely expressed in the human body. Additionally, it performs the role of an adapter protein that associates with the Wnt receptor complex and aids in the activation of Wnt/β-catenin signaling independently of the renin-angiotensin system. Aberrant (P)RR expression, through the Wnt/β-catenin signaling pathway, enhances CRC carcinogenesis in the presence of constitutive pathway component mutations [32].
Role of Renin-Angiotensin System in Colorectal Cancer
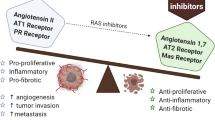
Novel physiologic functions of the renin-angiotensin system in the renal and cardiovascular systems have been well established. Recent research has looked into and identified RAS receptors and mediators in other tissues and organs of the body [33]. Components of the RAS system were also found in the lamina propria, lending credence to the hypothesis that the RAS also performs paracrine functions. AT2R is primarily stimulated at low Ang II concentrations, which stimulates colonic water and sodium absorption. When exposed to high levels of Ang II, the AT1R-mediated pathway is primarily activated, inhibiting sodium reabsorption and/or stimulating sodium secretion [34]. The activation of AT1R may also influence colonic motility. Losartan, an AT1R inhibitor, downregulates the effect competitively, requiring a higher concentration to produce the same contractile effect [35]. Ang II stimulates sodium and water absorption and secretion via the AT1R and AT2R, respectively. It was discovered that RAS components are dysregulated in CRC, indicating that RAS plays a role in CRC pathology (Fig. 22.1). The liver is the site of most CRC metastasis, and angiotensinogen production there is normal, raising the possibility of RAS and its role in CRC metastasis. ACE, Ang II, and MASR were found to be upregulated in CRC liver metastasis, while AT1R was found to be inhibited when compared to normal liver tissue. Angiotensinogen generated by the liver is converted by ACE into Ang II, which induces angiogenesis and metastasis via the AT1R by upregulating VEGF and TGF-β, respectively [34]. Ang II inhibits cell growth, invasion, and apoptosis in several CRC-derived cell lines. It has also been shown to increase cell growth and invitro invasion into type IV collagen while decreasing apoptosis in a dose-dependent manner. Although AT1R was found to be decreased in the liver during CRC metastasis, Kupffer cells (KCs) overexpressed AT1R, which promotes CRC liver metastasis [36]. AT1R deletion in CRC animal models resulted in decreased liver metastasis and downregulated TGF-β production in KCs, showing that KCs increases liver metastasis by inducing TGF-β production via AT1R signaling [36]. RAS inhibitors have been linked to lower risk and mortality from CRC. Treatment with ACEIs and ARBs was found to improve recurrence-free survival in early-stage CRC and left-sided CRC cases. Overexpression of the AGTR1 gene, on the other hand, was associated with poor recurrence-free survival in the advanced stages [37]. Furthermore, ARBs were able to improve both overall survival and progression-free survival (PFS) in individuals with metastatic CRC who received first-line chemotherapy [38].

The role of renin-angiotensin system components in colorectal cancer. CRC-colorectal cancer, MET- mesenchymal-epithelial transition, EMT- epithelial-mesenchymal transition, Ang I-angiotensin I, Ang II-angiotensin II, AT1R-angiotensin II type 1 receptor, AT2R-angiotensin II type 2 receptor, Ang 1–7-angiotensin 1–7, MASR-MAS Receptor
Renin-Angiotensin System in Colorectal Cancer Liver Metastasis
In up to 30–40% of individuals with advanced illness, the liver is typically the primary metastatic location for CRC, it may be the only site of spread (Fig. 22.2) [39]. In response to tissue injury and hypoxia, the local RAS is up-regulated in the liver [40]. Ang II promotes the expression of a variety of growth and pro-angiogenic factors, including VEGF. Other RAS components mediate counter-regulatory effects of Ang II/AT1R signaling, which has proliferative and angiogenic effects. Activation of the AT2R, which is expressed more than the AT1R in primary CRC, inhibits angiogenesis and cellular proliferation [41]. In liver injury, the expression of an ACE homologue, ACE2, is increased [42]. This enzyme directly produces the peptide Ang-(1–7) from Ang II and indirectly from Ang I. Ang-(1–7) antagonizes some Ang II-induced effects, via the MASR [43]. CRC cells with angiotensin-activating ability produce abundant Ang-2 from AGT in the liver and cause liver metastasis [44]. Several RAS components are expressed in primary CRC, and blocking the RAS inhibited tumor growth in a mouse model of CRC liver metastasis [45]. RAS expression changes significantly in the tumor-bearing captopril-treated liver and in CRC metastasis. Captopril treatment reduced ACE expression in CRC liver metastasis. Following captopril treatment, angiotensinogen expression is lower in CRC liver metastasis and lower in the liver surrounding tumors [1]. Treatment with captopril alters the spatial and temporal infiltration of tumor lymphocytes expressing CD3+ and CD4+. It modulates T cell subpopulations that infiltrate into tumor and liver tissues in different ways [46]. In a mouse model of CLM, both captopril and irbesartan significantly inhibited tumor growth [45]. Anti-hypertensive drugs that target the renin-angiotensin system (anti-RAS) in combination with bevacizumab have been shown to significantly improve anti-angiogenic efficacy in CRC liver metastasis. Anti-RAS blocks the contraction of fibroblasts and the deposition of ECM, which prevents liver metastasis from hardening and increasing bevacizumab’s anti-angiogenic effect [44]. Diabetes has been linked to angiotensin activation and the progression of CRC liver metastasis. The expression of renin and chymase in CRC cells provides an angiotensin activation mechanism. Cathepsin D produces Ang I from AGT instead of renin in cardiac myocytes, fibroblasts, and vascular smooth muscle cells. Renin expression was found in both HT29 and CT26 cells, and it was dose-dependently related to glucose concentration. Levels of intracellular Ang II are dramatically raised by a high glucose concentration in cardiac fibroblasts by increasing renin levels. Clinical data showed high tumoral renin concentrations, high tumoral Ang II concentrations, and liver metastasis in diabetic cancer patients [47].

Schematic representation of the role of the renin-angiotensin system in Colorectal cancer liver metastasis. Colon cancer metastasis to the liver occurs when tumor cells migrate and colonize from the colon to the liver, resulting in metastatic spread. The renin-angiotensin system is involved in liver metastasis, and inhibiting it reduces metastatic stiffness, hence boosting the anti-angiogenic effect
Renin-Angiotensin System Targeting Therapy for Colorectal Cancer
According to recent research, drugs that target paracrine hormone systems that promote tumor formation may give an alternate or supplementary therapeutic option in CRC patients. Long-term inhibition of the renin-angiotensin system in hypertensive patients is related to a lower incidence of numerous human malignancies [16]. The angiotensin I converting enzyme is a critical enzyme in the RAS, converting the physiologically inactive angiotensin (Ang) I precursor to Ang II, the RAS's primary effector peptide. Renin-angiotensin system inhibitors’ potential protective impact is gaining attention due to their potential involvement as chemopreventive medications against colorectal cancer. A variety of renin-angiotensin system inhibitors have been shown to reduce the risk of colorectal cancer (Table 22.1) [48, 49]. Recent research shows that Ang II/AT1R signaling has proliferative and angiogenic effects, while additional RAS components have counter-regulatory effects. Activation of the AT2R, which is expressed more than the AT1R in primary CRC, for example, suppresses angiogenesis and cellular proliferation [41].
The renin-angiotensin system has been widely studied in the context of liver and renal fibrosis. Ang II, the major effector of the RAS, has been demonstrated to stimulate TGF-β1 and NF-κB signaling pathways in inflammatory diseases such as hepatic and renal fibrosis [50]. Studies on renal fibrosis showed that multiple RAS genes in renal tissue are regulated by the Wnt/β-catenin pathway. Furthermore, ICG-001, a small molecule inhibitor of Wnt signaling, was able to reduce kidney fibrosis [51]. In a mice model of colorectal cancer liver metastasis, RAS inhibition by captopril could modify the Wnt/β-catenin pathway and EMT/MET in the context of liver regeneration following partial hepatectomy [52]. AT1R blocker, Telmisartan had antiproliferative and apoptotic effects in human colon cancer cells at therapeutic blood concentrations, and telmisartan had potency at least similar to pioglitazone, a complete PPARγ agonist. PPARγ blockade with GW9662 did not completely reverse pioglitazone's antiproliferative and apoptotic effects in human colon cancer cells. PPARγ inhibition boosted antiproliferative and apoptotic effects in the telmisartan-treated cells [53].
Conclusion and Future Perspectives
In most malignancies, the ACE/Ang II/AT1R axis plays a tumorigenic role, whereas the ACE-2/Ang 1–7/MASR axis plays an antitumorigenic role. Furthermore, ACEIs and ARBs have been shown to enhance colorectal cancer outcomes. RAS activation is a critical component of colorectal cancer for disease development and liver metastasis. In addition, Renin-angiotensin system blockade is a promising therapeutic for colorectal cancer liver metastases. Because of their potential role as chemopreventive drugs against CRC, RAS inhibitors’ potential protective impact is gaining attention, however, there are still few studies that examine this association. More research is needed to confirm the significance of the renin-angiotensin system in colorectal cancer and its potential as a therapeutic target.
References
Neo JH, Ager EI, Angus PW, Zhu J, Herath CB, Christophi C (2010) Changes in the renin angiotensin system during the development of colorectal cancer liver metastases. BMC Cancer 10(1):134
Komarova N (2005) Cancer, aging and the optimal tissue design. Semin Cancer Biol 15(6):494–505
Aran V, Victorino AP, Thuler LC, Ferreira CG (2016) Colorectal cancer: epidemiology, disease mechanisms and interventions to reduce onset and mortality. Clin Colorectal Cancer 15(3):195–203
Takahashi H, Iyoda I, Takeda K, Sasaki S, Okajima H, Yamasaki H et al (1984) Centrally-induced vasopressor responses to sodium-potassium adenosine triphosphatase inhibitor, ouabain, may be mediated via angiotensin II in the anteroventral third ventricle in the brain. Jpn Circ J 48(11):1243–1250
Cappell MS (2008) Pathophysiology, clinical presentation, and management of colon cancer. Gastroenterol Clin North Am 37(1):1–24
Faerden AE, Naimy N, Wiik P, Reiertsen O, Weyessa S, Trønnes S et al (2005) Total mesorectal excision for rectal cancer: difference in outcome for low and high rectal cancer. Dis Colon Rectum 48(12):2224–2231
Narayanan A, Wickremesekera SK, Van Schaijik B, Marsh RW, Brasch HD, Tan ST et al (2019) Cancer stem cells in liver metastasis from colon adenocarcinoma express components of the renin-angiotensin system. J Cancer Metastasis Treat 2019
Cheng Y, Ling Z, Li L (2020) The intestinal microbiota and colorectal cancer. Front Immunol 11
George AJ, Thomas WG, Hannan RD (2010) The renin–angiotensin system and cancer: old dog, new tricks. Nat Rev Cancer 10(11):745–759
Ishikane S, Takahashi-Yanaga F (2018) The role of angiotensin II in cancer metastasis: potential of renin-angiotensin system blockade as a treatment for cancer metastasis. Biochem Pharmacol 151:96–103
Sanidas E, Velliou M, Papadopoulos D, Fotsali A, Iliopoulos D, Mantzourani M et al (2020) Antihypertensive drugs and risk of cancer: between Scylla and Charybdis. Am J Hypertens 33(12):1049–1058
Makar GA, Holmes JH, Yang Y-X (2014) Angiotensin-converting enzyme inhibitor therapy and colorectal cancer risk. JNCI J Natl Cancer Inst 106(2)
Zheng X, Liu G, Cui G, Cheng M, Zhang N, Hu S (2017) Angiotensin-converting enzyme gene deletion polymorphism is associated with lymph node metastasis in colorectal cancer patients in a Chinese population. Med Sci Monit 23:4926–4931
Gullapalli N, Bloch MJ, Basile J (2010) Renin-angiotensin-aldosterone system blockade in high-risk hypertensive patients: current approaches and future trends. Ther Adv Cardiovasc Dis 4(6):359–373
Bangalore S, Kumar S, Kjeldsen SE, Makani H, Grossman E, Wetterslev J et al (2011) Antihypertensive drugs and risk of cancer: network meta-analyses and trial sequential analyses of 324 168 participants from randomised trials. Lancet Oncol 12(1):65–82
Lever AF, Hole DJ, Gillis CR, McCallum IR, McInnes GT, MacKinnon PL et al (1998) Do inhibitors of angiotensin-I-converting enzyme protect against risk of cancer? Lancet 352(9123):179–184
Fearon ER (2011) Molecular genetics of colorectal cancer. Annu Rev Pathol Mech Dis 6(1):479–507
Zenonos K (2013) RAS signaling pathways, mutations and their role in colorectal cancer. World J Gastrointest Oncol. 5(5):97
Merchant NB, Voskresensky I, Rogers CM, LaFleur B, Dempsey PJ, Graves-Deal R et al (2008) TACE/ADAM-17: a component of the epidermal growth factor receptor axis and a promising therapeutic target in colorectal cancer. Clin Cancer Res 14(4):1182–1191
Nandan MO, Yang VW (2011) An update on the biology of RAS/RAF mutations in colorectal cancer. Curr Colorectal Cancer Rep. 7(2):113–120
Tan YH, Liu Y, Eu KW, Ang PW, Li WQ, Tellez MS et al (2008) Detection of BRAF V600E mutation by pyrosequencing. Pathology 40(3):295–298
Hutchinson RA, Adams RA, McArt DG, Salto-Tellez M, Jasani B, Hamilton PW (2015) Epidermal growth factor receptor immunohistochemistry: new opportunities in metastatic colorectal cancer. J Transl Med 13(1):217
Papadatos-Pastos D, Rabbie R, Ross P, Sarker D (2015) The role of the PI3K pathway in colorectal cancer. Crit Rev Oncol Hematol 94(1):18–30
Courtney KD, Corcoran RB, Engelman JA (2010) The PI3K pathway as drug target in human cancer. J Clin Oncol 28(6):1075–1083
Baldus SE, Schaefer K-L, Engers R, Hartleb D, Stoecklein NH, Gabbert HE (2010) Prevalence and Heterogeneity of KRAS, BRAF, and PIK3CA mutations in primary colorectal adenocarcinomas and their corresponding metastases. Clin Cancer Res 16(3):790–799
Logan CY, Nusse R (2004) The WNT signaling pathway in development and disease. Annu Rev Cell Dev Biol 20(1):781–810
de Sousa EMF, Vermeulen L, Richel D, Medema JP (2011) Targeting Wnt signaling in colon cancer stem cells. Clin Cancer Res 17(4):647–653
Klaus A, Birchmeier W (2008) Wnt signalling and its impact on development and cancer. Nat Rev Cancer 8(5):387–398
He T-C, Sparks AB, Rago C, Hermeking H, Zawel L, da Costa LT et al (1998) Identification of c- MYC as a Target of the APC Pathway. Science (80- ) 281(5382):1509–1512
Goto T, Marusawa H, Chiba T (2013) Landscape of genetic aberrations detected in human colorectal cancers. Gastroenterology 145(3):686–688
Voloshanenko O, Erdmann G, Dubash TD, Augustin I, Metzig M, Moffa G et al (2013) Wnt secretion is required to maintain high levels of Wnt activity in colon cancer cells. Nat Commun 4(1):2610
Wang J, Shibayama Y, Zhang A, Ohsaki H, Asano E, Suzuki Y et al (2019) (Pro)renin receptor promotes colorectal cancer through the Wnt/beta-catenin signalling pathway despite constitutive pathway component mutations. Br J Cancer 120(2):229–237
Hirasawa K, Sato Y, Hosoda Y, Yamamoto T, Hanai H (2002) Immunohistochemical localization of Angiotensin II receptor and local renin-angiotensin system in human colonic mucosa. J Histochem Cytochem 50(2):275–282
Childers WK (2015) Interactions of the renin-angiotensin system in colorectal cancer and metastasis. Int J Colorectal Dis 30(6):749–752
Mastropaolo M, Zizzo MG, Mulè F, Serio R (2013) Angiotensin II contractile effects in mouse colon: role for pre- and post-junctional AT 1A receptors. Acta Physiol 207(2):337–345
Shimizu Y, Amano H, Ito Y, Betto T, Yamane S, Inoue T et al (2017) Angiotensin II subtype 1a receptor signaling in resident hepatic macrophages induces liver metastasis formation. Cancer Sci 108(9):1757–1768
Ozawa T, Hashiguchi Y, Yagi T, Fukushima Y, Shimada R, Hayama T et al (2019) Angiotensin I-converting enzyme inhibitors/angiotensin II receptor blockers may reduce tumor recurrence in left-sided and early colorectal cancers. Int J Colorectal Dis 34(10):1731–1739
Osumi H, Matsusaka S, Wakatsuki T, Suenaga M, Shinozaki E, Mizunuma N (2015) Angiotensin II type-1 receptor blockers enhance the effects of bevacizumab-based chemotherapy in metastatic colorectal cancer patients. Mol Clin Oncol. 3(6):1295–1300
Weiss L, Grundmann E, Torhorst J, Hartveit F, Moberg I, Eder M et al (1986) Haematogenous metastastic patterns in colonic carcinoma: An analysis of 1541 necropsies. J Pathol 150(3):195–203
Herath CB, Warner FJ, Lubel JS, Dean RG, Jia Z, Lew RA et al (2007) Upregulation of hepatic angiotensin-converting enzyme 2 (ACE2) and angiotensin-(1–7) levels in experimental biliary fibrosis. J Hepatol 47(3):387–395
Nakajima M, Hutchinson HG, Fujinaga M, Hayashida W, Morishita R, Zhang L et al (1995) The angiotensin II type 2 (AT2) receptor antagonizes the growth effects of the AT1 receptor: gain-of-function study using gene transfer. Proc Natl Acad Sci 92(23):10663–10667
Donoghue M, Hsieh F, Baronas E, Godbout K, Gosselin M, Stagliano N et al (2000) A novel angiotensin-converting enzyme–related carboxypeptidase (ACE2) converts angiotensin i to angiotensin 1–9. Circ Res 87(5)
Machado RDP, Santos RAS, Andrade SP (2001) Mechanisms of angiotensin-(1–7)-induced inhibition of angiogenesis. Am J Physiol Integr Comp Physiol. 280(4):R994-1000
Shen Y, Wang X, Lu J, Salfenmoser M, Wirsik NM, Schleussner N et al (2020) Reduction of liver metastasis stiffness improves response to bevacizumab in metastatic colorectal cancer. Cancer Cell 37(6):800-817.e7
Neo JH, Malcontenti-Wilson C, Muralidharan V, Christophi C (2007) Effect of ACE inhibitors and angiotensin II receptor antagonists in a mouse model of colorectal cancer liver metastases. J Gastroenterol Hepatol 22(4):577–584
Vallejo Ardila DL, Walsh KA, Fifis T, Paolini R, Kastrappis G, Christophi C et al (2020) Immunomodulatory effects of renin–angiotensin system inhibitors on T lymphocytes in mice with colorectal liver metastases. J Immunother Cancer 8(1):e000487
Shimomoto T, Ohmori H, Luo Y, Chihara Y, Denda A, Sasahira T et al (2012) Diabetes-associated angiotensin activation enhances liver metastasis of colon cancer. Clin Exp Metastasis 29(8):915–925
Chen X, Yi C, Ya K (2020) Renin–angiotensin system inhibitor use and colorectal cancer risk and mortality: a dose–response meta analysis. J Renin-Angiotensin-Aldosterone Syst 21(3):147032031989564
Cheung KS, Chan EW, Seto WK, Wong ICK, Leung WK (2020) ACE (Angiotensin-converting enzyme) inhibitors/angiotensin receptor blockers are associated with lower colorectal cancer risk. Hypertension 76(3):968–975
Ranjbar R, Shafiee M, Hesari A, Ferns GA, Ghasemi F, Avan A (2019) The potential therapeutic use of renin–angiotensin system inhibitors in the treatment of inflammatory diseases. J Cell Physiol 234(3):2277–2295
Zhou L, Liu Y (2016) Wnt/β-catenin signaling and renin–angiotensin system in chronic kidney disease. Curr Opin Nephrol Hypertens 25(2):100–106
Riddiough GE, Fifis T, Walsh KA, Muralidharan V, Christophi C, Tran BM et al (2021) Captopril, a renin-angiotensin system inhibitor, attenuates features of tumor invasion and down-regulates C-Myc expression in a mouse model of colorectal cancer liver metastasis. Cancers (Basel) 13(11):2734
Lee LD, Mafura B, Lauscher JC, Seeliger H, Kreis ME, Gröne J (2014) Antiproliferative and apoptotic effects of telmisartan in human colon cancer cells. Oncol Lett 8(6):2681–2686
Koh SL, Ager EI, Costa PLN, Malcontenti-Wilson C, Muralidharan V, Christophi C (2014) Blockade of the renin–angiotensin system inhibits growth of colorectal cancer liver metastases in the regenerating liver. Clin Exp Metastasis 31(4):395–405
Yang Y, Ma L, Xu Y, Liu Y, Li W, Cai J et al (2020) Enalapril overcomes chemoresistance and potentiates antitumor efficacy of 5-FU in colorectal cancer by suppressing proliferation, angiogenesis, and NF-κB/STAT3-regulated proteins. Cell Death Dis 11(6):477
Tabatabai E, Khazaei M, Asgharzadeh F, Nazari SE, Shakour N, Fiuji H et al (2021) Inhibition of angiotensin II type 1 receptor by candesartan reduces tumor growth and ameliorates fibrosis in colorectal cancer. EXCLI J 20:863–878
Ozeki K, Tanida S, Morimoto C, Inoue Y, Mizoshita T, Tsukamoto H et al (2013) Telmisartan inhibits cell proliferation by blocking nuclear translocation of ProHB-EGF C-terminal fragment in colon cancer cells. PLoS ONE 8(2):e56770
Hashemzehi M, Rahmani F, Khoshakhlagh M, Avan A, Asgharzadeh F, Barneh F et al (2021) Angiotensin receptor blocker Losartan inhibits tumor growth of colorectal cancer. EXCLI J 20:506
Hachiya K, Masuya M, Kuroda N, Yoneda M, Tsuboi J, Nagaharu K et al (2021) Irbesartan, an angiotensin II type 1 receptor blocker, inhibits colitis-associated tumourigenesis by blocking the MCP-1/CCR2 pathway. Sci Rep 11(1):19943
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2023 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Bhatt, L.K., Parihar, N., Prabhavalkar, K.S. (2023). Renin-Angiotensin System: A Potential Therapeutic Target for Colorectal Cancer. In: Bhullar, S.K., Tappia, P.S., Dhalla, N.S. (eds) The Renin Angiotensin System in Cancer, Lung, Liver and Infectious Diseases. Advances in Biochemistry in Health and Disease, vol 25. Springer, Cham. https://doi.org/10.1007/978-3-031-23621-1_22
Download citation
DOI: https://doi.org/10.1007/978-3-031-23621-1_22
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-031-23620-4
Online ISBN: 978-3-031-23621-1
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)