
Abstract
Cancer is one the most dreadful diseases in the world and its genetic origin has been undoubtedly proved and studied worldwide. Although, in recent decades, epigenetics has managed to shed light on many unknown mechanisms of carcinogenesis, being irrepressible by genetic laws. Epigenetic alterations represent a totality of heritable changes in gene expression that do not affect DNA sequence. Four major epigenetic alterations: DNA epigenetic alterations, histone post-translational modifications, remodeling complexes and non-coding RNAs are described in more detail in this chapter. In cancer, epigenetic deregulations are preceded by genetic mutations in genes of epigenetic machinery, and cause in result modifications of chromatin state. In this case, upregulation of oncogenes or downregulation of tumor suppressor gene expression became the worst outcome. In comparison with genetic mutations, epigenetic ones can be reversible. Therefore, different therapeutic strategies are developing to restore normal epigenetic landscape in tumor cells, pharmaceutical agents being classified by their main epigenetic targets. Common epigenetic regulators, such as HDAC inhibitors, DOT1L inhibitors, LSD inhibitors, EZH2 inhibitors and others are also described below. Besides of natural or synthetic regulators, epigenetic modifications can also be triggered by predisposition to different health conditions, onset of other non-cancerous diseases, virosis, aging or stress. Another background by which epigenetic profile is affected, and therefore can be reversed, includes different lifestyle factors such as environmental circumstances, diet or practicing exercises. For these reasons, we hope that this chapter will highlight the importance of epigenetic deregulations in cancer, and will also encourage further investigations of epigenetic mechanisms, validation of novel epigenetic biomarkers as well as development of new suitable epigenetic drug regulators in order to improve cancer therapy.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Cancer
- Epigenetics
- DNA modifications
- Histone modifications
- Remodeling complexes
- Non-coding RNAs
- Cancer therapy
- Epigenetic inhibitors
- Diet
- Environment
- Heritability
1 Carcinogenesis
Nowadays, cancer is one of the most frightening diseases, characterized by the uncontrolled growth of abnormal cells. Unfortunately, its power over human lives continues to lie in a variety of well-orchestrated and precise dysregulations of important intra- and intercellular pathways that control vital cellular processes such as nutrition, growth, proliferation, survival, and intercellular communication, and mortality. Once these dysregulations have been initiated, they are difficult to control because even the slightest trigger factor—roots its effects into a vast network of interconnected cascades [1, 2]. In defiance of the fact that clinicians are not yet able to completely eradicate them, cancer is becoming more and more vulnerable due to all the revealing information which we have continued to accumulate since the understandings of the first basic tumorigenesis mechanisms were outlined. In 2000, Hanahan and Weinberg defined these driving forces as hallmarks of cancer, where six essential mechanisms acquired by tumor cells were described: self-sufficiency in growth signals, insensitivity to anti-growth signals, tissue invasion, and metastasis, limitless replicative potential, sustained angiogenesis and evading apoptosis [1]. These hallmarks had been instantly acknowledged in cancer's existing status as a “genetic” disease and a plethora of studies have been accomplished to demonstrate and strengthen this concept. Numerous genetic mutations have been identified and associated with a malignant profile, being used therefore as biomarkers for distinguishing normal versus cancerous tissues [2, 3], as well as different tumor types and even different stages of disease within the same cancer type [4]. However, the general trait of any malignant cell has been established as genomic instability, prone to aberrant and abnormal survival. Therefore, such alterations as deletions, substitutions, or translocations always led, in one way or another, either to the activation or overexpression of oncogenes, such as MYC, RAS, RAF, AKT, BCL-XL, BCL-2, or to the inactivation or silencing of tumor suppressor genes such as P53, pRB, BAX, BAK. The first ones favored proliferation, invasion, metastasis, expression of growth factors, and angiogenic or antiapoptotic signals, conversely acting the last one’s [2,3,4].
Even though cancer development may have a genetic origin has been undoubtedly approved, there were still some discrepancies in terms of attempts to associate genotype with phenotype profile. Consequently, the studies of cancer mechanisms have continued so that at present, the data in the literature already list 14 hallmarks of cancer. The last 4 hallmarks, described with examples by Hanahan in 2022, are very different from the original ones and open much more horizons for a better understanding of complex tumor mechanisms than the first six originally described. One of these, which raise particularly increased interest in bringing to light the concept of the genetic-independent evolution of cancer, is termed “nonmutational epigenetic reprogramming” [3, 5].
2 Major Epigenetic Alterations in Cancer
Epigenetic alterations are in general related to normal biological processes such as aging or differentiation, being considered a reversible process. Alteration of epigenetic signatures by a wide range of factors (particularly environmental), along with genetic and transcriptomic alterations, are considered as driving events in several diseases including cancer [6]. Therefore, the identification of tumor-specific epigenetics and the factors that affect epigenetic patterns should be evaluated to unmask truly disease-specific alterations.
Epigenetics, which means “upon the genes”, represents the study of heritable changes in gene expression, that do not involve alterations in the DNA sequence, consequently involving phenotype changes without the genotype ones even in the cells which share identical genome. The term “epigenetics” appeared for the first time in 1942 when Conrad Waddington coined it to describe phenomena that do not follow “normal genetic rules” [7].
Epigenetic changes play an essential role in a series of normal biological processes, such as embryonic development, viral protection, genetic imprinting, and X-chromosome inactivation [8]; disruption of these epigenetic processes has been considered key mechanisms in a variety of pathologies including Beckwith-Wiedemann (BWS), Silver-Russell, Prader-Willi and Angelman syndromes as well as autoimmune diseases—systemic lupus erythematosus (SLE), rheumatoid arthritis (RA), systemic sclerosis (SSc), Sjogren’s syndrome (SS), autoimmune thyroid diseases (AITD) and different neurological diseases [9,10,11].
Although epigenetic reprogramming is officially assigned as the hallmark of cancer just recently, as mentioned above, having an important role in growth-related pathways gave the earliest clues about the connection of these two about 40 years ago. In 1983, Feinberg and Vogelstein enlightened the first epigenetic mechanism attributed to cancer cells. Already knowing that the methylation process is related to the silencing of certain genes in some pathologies, they used Southern blotting and methylation-sensitive enzymatic restriction to demonstrate that the genome of cancer cells is hypomethylated at CpG dinucleotides compared to normal cells, which have generally hypermethylated genome [12, 13]. Other milestones in the history of human cancer epigenetics can be found comprehensively described by Feinberg and Tycko in 2004 [14].
Initially, three main types of epigenetic mechanisms were distinguished: DNA methylation, loss of imprinting (LOI), and histone modifications [14]. Although they had already encompassed a significant amount of information regarding the most important epigenetic elements, nowadays four categories of epigenetic mechanisms have been established with more well-clarified mechanisms within one category. These epigenetic modifications include:
-
(1)
DNA epigenetic alterations,
-
(2)
histone post-translational modifications (PTMs),
-
(3)
remodeling chromatin complexes, and
-
(4)
noncoding RNAs regulation [10, 14,15,16,17], schematic representation presented in Fig. 1.
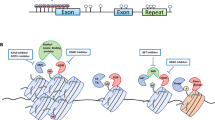
Fig. 1 
Schematic representation of three main key steps of epigenetic regulation in cancer cells. I. Genetic background of epigenetic mechanisms. Initially, genetic mutations of certain genes that are involved in the functioning of the epigenetic machinery occur. II. Epigenetic mechanisms. Endpoint products of these altered genes, being either enzymes, protein complexes, or RNAs, become under- or overexpressed and lead to epigenetic changes which include DNA (1) and histone (2) alterations, aberrant functioning of remodeling complexes (3), and noncoding RNAs (4). III. Phenotypic results. Acting separately or in combination with each other, epigenetic changes specifically modulate the accessibility of promoters for binding with transcription factors thus regulating the expression of oncogenes or tumor suppressor genes. Aberrant expression of these target genes induces dysregulations in pathways of growth, proliferation, survival, invasion, apoptosis, and senescence, which play a crucial role in tumorigenesis and cancer development. Abbreviations SUMO—Small Ubiquitin-like Modifier; SWI/SNF—switching defective/sucrose non-fermenting; ISWI—imitation-switch; NuRD—nucleosome remodeling and histone deacetylase; INO80—inositol 80; lncRNAs—long noncoding RNAs; sncRNAs—small noncoding RNAs; miRNAs—microRNAs; siRNAs—small small interfering RNAs; piRNAs—PIWI-interacting RNAs; snoRNAs—small nucleolar RNAs

All four epigenetic mechanisms determine either gene expression or silencing by controlling the chromatin state (condensed or decondensed). This is achieved through differential expression of enzymes that have abilities to change the accessibility of chromatin for binding of DNA transcription factors. A common way to identify and distinguish these enzymes is to assign them one of the statuses “writer”, “reader” or “eraser”. The enzymes which deposit such modifications by adding different chemical groups are called “writers”. They predominantly fall into the first two categories, contributing to DNA or histone modifications. The molecular marks which recruit chromatin remodelers and noncoding RNAs to mediate downstream effects, their interaction is controlled by proteins called ‘readers’. Additionally, chemical modifications can be removed by ‘erasers’, a fact that can label epigenetic alterations as reversible ones [18]. Currently, all four categories of epigenetic mechanisms may be involved, separately or in combination with each other, in the development of cancer. Further, the main mechanisms from each category are briefly discussed. Some representative examples are presented in Table 1.
2.1 DNA Epigenetic Alterations
DNA epigenetic alterations include methylation, demethylation, hydroxymethylation, and its oxidation derivatives. The concurrence of DNA methylation and demethylation is in general related to transcription regulation. For DNA methylation an important role is attributed to DNA methyltransferases (DNMT) and for demethylation to the ten-eleven translocation (TET) [19].
The first one is best-studied and refers to the addition of a methyl group (–CH3) at the fifth position on the pyrimidine ring of cytosine (5mC) within CpG dinucleotides. These are clustered together into “CpG islands” and are found in about 40–60% of human gene promoters and repetitive regions of the DNA [20]. Two aberrant forms of DNA methylation can be distinguished during tumor development and progression: global DNA hypomethylation and local hypermethylation. The first one implies an overall loss of 5-methyl-cytosine. The last one makes promoters inaccessible for binding with transcription factors (TFs) and therefore it is a significant mark of gene silencing associated with gene inactivation. In cancer, particularly tumor suppressor genes and certain regulatory antioncogenes have a hypermethylated promoter, that leads to important alterations responsible for the tumorigenesis [21]. The process of methylation is catalyzed by DNA methyltransferases (DNMTs) which transfer a methyl group from donor S-adenosyl-L-methionine (SAM) to the cytosine residue. There are three main types of DNMTs known in mammals: DNMT1, DNMT3a, and DNMT3b, each having a specific mechanism of action. DNMT1 maintains the existing methylation pattern following DNA replication, while DNMT3a and DNMT3b catalyze din novo unmethylated CpGs, especially at new-formed strands of DNA after replication. Another family member, DNMT-3L lacks intrinsic methyltransferase activity, instead, it interacts with DNMT3a and DNMT3b to facilitate the methylation of retrotransposons [22]. In addition to direct inhibition of gene expression, methylated sites can also recruit specific proteins from the methyl-binding domain (MBD) family, such as MBD1, MBD2, MBD3, and MBD4, which in their turn recruit histone-modifying enzymes and chromatin-remodeling complexes [23, 24]. This MBD1 inhibition in pancreatic cancer affected the antioxidant response element target genes through epigenetic regulation of KEAP1 [25]. MBD1 plays a role in tumorigenesis by repressing tumor suppressor genes like CDH1, RASSF1A, TIMP3, P14ARF, and Rb [26].
Somatic mutations in DNMTs and MBD proteins have been associated with deregulated pathways in different cancers, having oncogenic or tumor suppressor roles [27]. As effect of MBD epigenetic gene silencing mechanisms a wide range of transcriptional factors are released (Table 1).
In contrast to DNMTs, the ten-eleven translocation (TET 1–3) family of proteins catalyzes the oxidation of 5mC into 5-hydroxymethylcytosine (5hmC) and further products which modulate the DNA methylation landscape. TETs proteins have two cofactors, Fe(II) and 2-oxoglutarate (2OG), which are indispensable for successively 5mC oxidation. TETs loss-of-function is commonly observed in various cancers [28]. DNA hydroxymethylation facilitated by TET1 controlling the WNT signaling is a key factor in the tumor growth [29]. TETs’ role in cancer is considered as context-dependent tumor-suppressor genes and/or oncogenes in solid tumors [30]. The anti- or oncogenic roles are directly related to the combination of different signaling pathways in different tumors [31].
2.2 Histone Post-translational Modification
Another type of epigenetic alteration occurs at the chromatin level. Here, nucleosome forms the basic structural unit of chromatin. It consists of DNA wrapped around an octamer of histone proteins which are represented by two copies of proteins H2A, H2B, H3, and H4. Histone modifications can contribute to chromatin compaction, nucleosome dynamics, and transcription alteration regulated by a fine-tuning mechanism regulated by chromatin modifiers and histone modifications [6, 32].
Histones are characterized by N-terminal tails rich in positively charged lysine (K) which in combination with a negatively charged DNA backbone confer a tightly packed state of chromatin [33]. Epigenetic changes occur when histones undergo post-translational modifications (PTMs) which primarily involve the addition or removal of certain chemical groups by specific enzymes at their N-terminal tails. These modifications trigger conformational changes in the chromatin structure, conferring either condensed (heterochromatin) or relaxed (euchromatin) state. Tight nucleosomes can become loose when the positive lysine residues are neutralized, therefore the access of the transcriptional machinery to the adjacent promoter of the gene will be enabled and the gene will be expressed. Conversely, the addition of more positive residues or groups to the surface of histones can enforce a chromatin tightened state and increase gene repression, without involving any DNA alterations in both cases. Consequently, more types of PTMs can be distinguished depending on which chemical group was added. The most well-known modifications are methylation, acetylation, and phosphorylation. These modifications are also established as epigenetic histone marks.
2.3 Histone Methylation
Histone methylation is catalyzed by writer-enzymes, histone methyltransferases (HMTs). It typically includes the addition of methyl groups (–CH3) to lysine (K) and/or arginine (R) residues. Finale regulation (activation or repression of the gene) results not only from the process of methylation per se but also from the position of methylated amino acid and the number of methyl groups added. For example, one of the most recognizable histone marks is the addition of three methyl groups by lysine methyltransferases (KMTs) at lysine 9 of histone H3 (H3K9me3) which results in gene silencing. Meanwhile, mono- and dimethylation of the same residue (H3K9me and H3K9me2) has the opposite effect. Other activating and repressive marks are H3K4me3 and H3K27me3, respectively [34]. One specific methyltransferase is enhancer of zeste homolog 2 (EZH2), which is a catalytic component of the polycomb repressive complex 2 (PRC2) and plays the primary role in the trimethylation of H3K27. Therefore, it is one of the important epigenetic elements which is responsible for gene silencing, being usually overexpressed in cancers [35]. Removal of the methyl group is performed by histone demethylases (HDMs). The expression level of both HMTs and HDMs can be altered in different tumor types, more information is provided in Table 1.
2.4 Histone Acetylation
Histone acetylation is catalyzed by histone acetyltransferases (HATs) which add acetyl group (–CH3CO) and neutralize the positively charged histone thus leading to conformational changing in chromatin structure and activation of gene transcription. H3K27ac and H2BK5ac are some examples of histone marks that correspond with actively transcribed genes. Histone deacetylases (HDACs) act conversely, thus inducing a back shift to the repression state of the gene. HDACs bind to and deacetylate a diversity of protein targets including transcription factors, involved in the control of cell growth, differentiation, and apoptosis [36]. A particular type of HDACs is a highly conserved family of NAD(+)-dependent HDACs called sirtuins (SIRTs). Seven mammalian sirtuins (SIRT1–7) are known to be implicated in many cellular processes, especially in epithelial-mesenchymal transition (EMT), invasion, and metastases [37].
2.5 Histone Phosphorylation
Histone phosphorylation, performed by kinases, occurs mainly at serine (S), threonine (T), and tyrosine (Y) residues and is associated with accessible chromatin conformation. Many histone marks have been found mutually working together, for example, histone H3 phosphorylation at tyrosine41 (H3Y41) is enriched at active promoters close to transcription start-sites (TSS) together with the H3K4me3 mark [38], loss of the trimethylation of H4K20 (H4K20me3) and acetylation of H4K16 (H4K16Ac), along with DNA hypomethylation is labeled as the common hallmark of primary tumors [39] as well as reduced levels of lysine acetylation (H3K9ac, H3K18ac, H4K12ac) and methylation (H3K4me2, H4K20me3) and arginine methylation (H4R3me2) [39]. Other PTMs include ubiquitination, SUMO (small ubiquitin-like modifiers)-ylation, neddylation citrullination, deamination, formylation, biotinylation, O-GlcNAcylation, propionylation, butyrylation, crotonylation, proline isomerization, ADP-ribosylation and lactylation [32, 34].
Besides covalent histone modifications which affect directly the state of chromatin, some alterations involve the exchange of canonical histones in the nucleosome with histone variants. Histone variants arise from mutations in genes that encode histone proteins. They became considered potential drivers of cancer initiations, being either up or downregulated in different cancer types [40]. For instance, macroH2A (mH2A) is one of the most distinguishable known histone variants, due to its special macro domain with the 25-kDa-sized globular module. In malignant melanoma, mH2A2 turned out to be the downregulated [41]. In contrast with mH2A, overexpression of variant histone H2A.Z.2 isoform presented an oncogenic role and provided a proliferative effect in the same malignance [42]. Other studies showed that even different splice isoforms can discriminate between different stages of tumor development and show differential expression levels, such as maH2A1.1 which is downregulated in primary colorectal cancer samples compared to normal colon tissue, while mH2A1.2 is upregulated [43].
2.6 Remodeling Complexes
While PTMs of histone represent intrinsic epigenetic changes at the chromatin level, there is also an extrinsic way to manipulate the chromatin state which is performed by remodeling complexes. Chromatin remodeling is considered an important gateway to regulating gene transcription. Therefore, this mechanism has important implications for targeted cancer therapeutic strategies, considering that cancer can select a multi‐subunit remodeler proteome for oncogenic advantage [44]. Based on the different structures and enzymatic activity of these complexes, they are categorized into four major families: the switching defective/sucrose non-fermenting (SWI/SNF) family, the imitation-switch (ISWI) family, the nucleosome remodeling and histone deacetylase complex (NuRD), and the inositol 80 (INO80) families. Remodeling occurs when the interaction between DNA and histone proteins is reconfigured by specific ATP-dependent enzymes which make up the subunits of remodelers [45]. As result, remodelers can manipulate nucleosome sliding along DNA, create access to transcription factors to gene promoters, and eject or replace certain histone variants [46]. Families also have different domain structures, such as SANT domains, bromodomains, PHD domains, DNA-binding domains, and chromo-domains that assign them certain specificity. Having a pivotal role in transcriptional profile regulation, mutations in these remodeling complexes were immediately associated with cancer malignancies. The most studied is SWI/SNF complex. It activates predominantly in two forms, based on its constituent core subunits: BRG1-associated factors (BAF) and polybromo-associated BAF (PBAF). The first one, as results from the name, contains subunits as BRG1 or BRM, and ARID1A/ARID1B, while the last one contains BRG1 only, and ARID2 and BRD7. SWI/SNF complexes have been found to function close to promoter or enhancer regions and interact with transcription to modulate gene expression and contribute to lineage specification, differentiation, and development. Consequently, it has been recognized as tumor suppressor complexes [47], although recent data accumulate controversial evidence [48,49,50], most of the studies being related to the regulation of mitotic cell divisions and DNA repair mechanisms [49,50,51]. BRG1 is considered not only a prognostic marker but also a therapeutic target [50, 52]. Generally, genes that encode component subunits of BAF or PBAF are found to be mutated, especially downregulated or inactivated in a variety of cancers [47, 52]. In contrast with SWI/SNF complexes, elements of other complexes, such as INO80, have been elevated in some cancer types mediating oncogenic signaling and promoting tumor growth [52,53,54,55].
2.7 Noncoding RNAs
Noncoding RNAs took a step forward in the overall regulation of gene expression, interfering before transcription and translation levels as well. Varieties of noncoding RNAs are categorized into two major types, according to their length: small noncoding RNAs (sncRNAs, under 200 nucleotides) and long noncoding RNAs (lncRNAs, more than 200 nucleotides) [56,57,58]. The sncRNAs include small nucleolar RNAs (snoRNAs), PIWI-interacting RNAs (piRNAs), small interfering RNAs (siRNAs), and microRNAs (miRNAs), the last ones being the most studied and strongly correlated with cancer development. These non-coding RNA transcripts regulate gene expression via complementarily binding to the 3′ UTR of the target mRNA [16]. Several studies revealed important aspects of epigenetics directly connected to this noncoding RNA transcript, particularly small RNAs that can direct the cytosine methylation and histone modifications that are involved in gene expression regulation [59].
The length of miRNAs represents approximately 22 nucleotides. Their role lies in complementary binding to a specific sequence of target mRNA and thus inducing mRNA-silencing. Therefore, many miRNAs have been found overexpressed in different tumor types, primarily downregulating the expression of tumor suppressor genes [23, 58, 60]. Nevertheless, some studies correlate the function of specific miRNAs with tumor suppressor activity [57, 58]. Differential expression of miRNAs is strongly correlated with the epigenetic process of DNA methylation due to the specific location of miRNA-encoding genes associated with CpG islands in their promoter regions. Additionally, miRNA genes might be located in specific chromatin structures that predispose them even more to DNA methylation [61, 62]. For example, miR-200, which targets zinc finger transcription factor ZEB1 together with zinc finger homeobox protein ZEB2 and provides an inhibitory effect on the epithelial-mesenchymal transition (EMT) process, is subject to methylation and also trimethylation of H3K27 to favor EMT and promote cancer development [63] (Fig. 2a).

Representative examples of ncRNAs that are involved in gene expression modulation through interaction with cancer epigenomics. a miR-200; b lncRNAs PRNCR1 and PCGEM1; c lncRNA HOTAIR. Abbreviations mRNA—messenger RNA; ZEB1—zinc finger transcription factor; ZEB2—zinc finger homeobox protein; EMT—epithelial-mesenchymal transition; PRNCR1—prostate cancer noncoding RNA1; PCGEM1—prostate cancer gene expression marker 1; AR—androgen receptor; ARE—AR response element; DOT1L—disruptor of telomeric silencing 1-like; Pygo2—Pygopus2; HOTAIR—HOX Transcript Antisense Intergenic RNA; PRC2—polycomb repressive complex 2; EZH2—enhancer of zeste homolog 2; LSD1—lysine-specific demethylase 1; H3K27me3—trimethylated lysine 27 of histone 3; H3K27me0—unmethylated lysine 27 of histone 3; H3K4me2—dimethylated lysine 4 of histone 3; H3K4me0—unmethylated lysine 4 of histone 3
On the side of sncRNAs, lncRNAs encompass even more regulatory functions. They have been associated with modulation of mRNA processing, control of transcription in cis or trans, as well as of post-transcriptional process and protein activity, organization of nuclear domains, and interfering with chromatin remodeling complexes [58, 64]. Likewise, they have been positively correlated with both tumor suppression and tumorigenesis, being, therefore, up- and downregulated in a variety of cancers [58]. As aforementioned, lncRNAs can modulate epigenetic processes in multiple interconnected ways. For example, in prostate cancer—prostate cancer noncoding RNA1 (PRNCR1) binds to the acetylated enhancer of androgen receptor and recruits histone H3K79 methyltransferase—disruptor of telomeric silencing 1-like (DOT1L). Consequently, methylation of androgen receptor facilitates the recruitment of another lncRNA, prostate cancer gene expression marker 1 (PCGEM1), to its N-terminal region. PCGEM1-recruited Pygopus2 (Pygo2) recognizes histone mark H3K4me3 and provide selective looping of enhancer with promoter, thus modulating gene expression of target gene androgen receptor (AR). AR enhance G1–S progression of cell cycle and therefore cell proliferation [65, 66] (Fig. 2b). In breast cancer, as well as in a variety of other cancers, well-known and overexpressed lncRNA HOX Transcript Antisense Intergenic RNA (HOTAIR) interacts with polycomb repressive complex (PRC2) and lysine-specific demethylase 1 (LSD1) thus recruiting them to the target gene and inducing gene silencing via H3K27-methylation and H3K4-demethylation [67] (Fig. 2c).
Different noncoding RNAs can also synergistically or antagonistically interact one with another to modulate gene expression. lncRNAs and circRNAs might act as miRNAs sponges by directly binding to them and abolishing their function. A study on breast cancer identified the lncRNA FAM83H-AS1 secludes miR-136-5p and therefore encourages metadherin-induced proliferation, migration, and invasion [68].
3 Epigenetics Drugs for Cancer Therapy
Considering the important function of the epigenetic dysregulation towards the origin and progression of cancer is considered an important cancer hallmark, an important number of preclinical and clinical studies are involved in testing and validation as a therapeutic strategy to restore the reversible normal epigenetic landscape in cancer cells by inhibiting enzymes of the epigenetic machinery in a wide range of cancer types [17, 119]. Until present, a wide range of natural or synthetic chemical agents as epigenetic regulators are tested and classified based on the main epigenetic target, the common DNMT inhibitors, HDAC inhibitors, DOT1L inhibitors, LSD inhibitors [120], EZH2 inhibitors, BET inhibitors [17]. Some of these inhibitors have been approved by the US FDA for the treatment of diverse malignancies and an important number of these compounds are undergoing clinical trials.
Inhibitors of DNMT and histone acetyltransferases/deacetylases have been revealed to inhibit tumor growth by reactivating epigenetically silenced tumor suppressor genes and silencing oncogenes [121].
DNMTs inhibitors can reverse the DNA hypermethylation status of tumor suppressor genes, they have been divided into two classes cytosine analog inhibitors and non-nucleotide analog inhibitors [16]. The most common demethylating are 5-azacytidine and 5-aza-2′-deoxycytidine, already approved for cancer therapy and hematologic pathologies. The main issue related to this type of agent is related to the unspecific reactivation of methylated sequences of tumor suppressor genes CpG islands. In parallel, also a global genomic demethylation process that causes chromosomal instability was observed [24].
MBDs are considered a valuable target for cancer inhibition, to avoid problems related to genomic instability, by inhibiting DNA methylation per se [24].
HDAC proteins are related to multiple oncogenic steps, HDAC inhibitors are involved in the prevention of tumor suppressor genes if recruited to promoters together with fusion oncogenes such as PML-RARα. Another application of HDAC inhibitors is to prevent the expression of HDACs proteins, that are in general overexpressed in multiple solid tumors or hematological malignancies, with high expression levels being in general related to an unfavorable prognostic [122].
Several HDACi are already approved in the clinic (Vorinostat, romidepsin, panobinostat, and belinostat in hematological malignancies), meanwhile others are tested currently in phase I or II clinical trials including pracinostat, givinostat, resminostat, abexinostat, entinostat, quisinostat [16].
Vorinostat and romidepsin were the first drugs to be approved that influence epigenetic post-translational modification of histone proteins [123]. Suberoylanilide hydroxamic acid (SAHA; vorinostat) is a non-selective broad-spectrum HDACI that induces acetylation of histones, this was demonstrated to be relegated with the overexpression of p21 as the effect of activation of the acetylated histone H3 and H4 in bladder carcinoma and endometrial stromal sarcomas [16, 124]. MS-275 inhibits HDACs 1–3 and 9, generally, this inhibitor was tested in conjunction with other agents [125]. Generally, this class of compounds is used to impede oncogenesis by acting apoptosis and cell cycle arrest and affecting the DNA damage pathway [122].
Preclinical work with BET inhibitors was focused on the comprehension of the relationship of BET proteins in regulating the cell cycle [122]. BET inhibition is generally related to transcriptional repression and cell cycle arrest [122]. Transcription factors are implicated in a wide range of pathologies, in a large number of human diseases such as cancers [126]. Accumulating investigations reveal that repression of EZH2 by small molecular inhibitors or gene knockdown leads to a decreased cell proliferation and tumor formation capacity [127] (Table 2).
4 How Epigenetic Processes Can Be Manipulated for Cancer Patient Benefit
The epigenetic processes are composed of DNA methylation, chromatin remodeling, histone modification, and non-coding RNA regulation [148,149,150]. These processes have a significant role in genome function and are dynamic, which means that they can be modified during their whole life by different factors [151, 152]. Because epigenetic processes are reversible, different factors can be used for epigenetic manipulation that can be implemented in cancer research. Epigenetic changes take place during our whole life and some epigenetic changes can be transferred from generation to generation [153]. Another important fact is that environmental conditions affect the epigenetic processes that occur during our life. Fraga et al. observed that monozygotic twins have similar DNA methylation and histone acetylation patterns; while older twins have significant differences in these epigenetic patterns meaning that the environmental conditions and diet influence their epigenetic profile [154]. Epigenetic changes have been linked also to exercise and Denham et al. described in their review how exercise can change the epigenetic profile of people and how it can prevent several diseases. They also showed that the epigenetic changes induced by exercise are reversible [155]. Diet is also a very important factor in diseases and it was demonstrated that poor nutrition in mothers during pregnancy can be linked to different diseases in humans and mice and most of the mechanisms involved are epigenetic [156,157,158]. Another epigenetic factor that has been shown to have an important role in the heritability of health and diseases is non-coding RNAs, where psychological stress and low protein diet affect the expression level of sperm non-coding RNAs, and these alterations are transmitted to offspring [159,160,161,162]. Studies showed that aging and obesity can also modify the sperm epigenome in humans [163, 164]. Alcohol is an important factor that can dysregulate epigenetic mechanisms by inhibiting the activity of methionin synthase (MTR), methionine adenosyl transferase (MAT) and DNA methyltransferases (DNMTs) [165]. Rossi et la observed that alcohol consumption is correlated to colorectal cancer development [166]. Heterocyclic amines are known for their genotoxic effect, but the mechanism through which they activate carcinogenesis still is not well understood. A study on rats observed that 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP) tumors have a specific signature of dysregulated miRNAs including let-7 family, mir-21, mir-126, mir-29c, mir-145, and mir-215 [167]. Rodriguez-Miguel et al. observed that high corn-oil diet of rats can induce epigenetic changes that can be related to breast cancer progression [168].
Another important factor that can modulate epigenetic changes is chronic inflammation. One factor that can induce chronic inflammation is stress. It was observed that stress can induce chronic inflammation in patients with thyroid disease, and in some cases by epigenetic regulation can induce thyroid cancer [169]. He et al. observed that chronic inflammation of colon mucosa have different methylation patterns in genes involved in cancer development like PIK3CA, AKT, MAPK, Ras, Wnt or TGFb [170]. Chronic inflammation in cystic fibrosis or chronic obstructive pulmonary disease is related to epigenetic reprogramming of airways macrophages, which in turn favor tissue damaging and diseases progression [171]. Ahmad et al. observed that inflammation in COVID-19, lung cancer and other imflammatory lung diseases are regulated by different miRNAs and environmental induced inflammation is strictly regulated by epigenetic changes [172]. Also, oncogenic viruses have been shown to influence carcinogenesis through epigenetic modifications, including DNA methylation, chromatin remodeling histone modification, long noncoding RNA, microRNA, and circular RNA [173]. Rattan et al. discussed in their review the importance of gut microbiome and epigenetic changes in hepatocellular carcinoma [174].
5 Conclusions and Perspectives
Throughout an increasing amount of studies, epigenetics became a very intricate field in the overall understanding of cancer initiation and progress. The majority of epigenetic deregulations are the results of genetic mutations of certain genes that encode enzymes involved in the functioning of the epigenetic machinery. The final products of these mutations, being either solitarily enzymes, catalytic subunits from protein complexes, or noncoding RNAs, interconnections between them can regulate, through different specific mechanisms, the access of transcription factors to gene promoters, facilitating either expression or repression of particular target genes. On the other hand, this indirect regulation creates a “ladder of a multistep processes” and therefore gives the opportunity to additionally influence these “intermediate steps” before they reach the worse outcomes. Fortunately, in comparison with already established mutations that trigger altered functioning of genes that play roles in initiating events in the tumorigenesis cascade, epigenetic alterations can be reversible due to their increased plasticity and sensitivity to environmental factors.
Epigenetic changes, including DNA methylation, histone modification, and non-coding RNA expression, have also been reported in a wide range of solid tumors, emphasizing important alteration in cell proliferation, apoptosis, invasion, or metastasis. Epigenetics enables us to explore the potential mechanism underlying cancer phenotypes. Great effort has been devoted to understanding the role of these epigenetic alterations involved during development and cancer progression. Precise techniques should be developed and standardized for epigenetic evaluation in the genome or from a specific population of cells to hopefully a few or even a single cell.
An important role is related to the crosstalk between DNA methylation and histone modifications regulated by different nuclear factors. Pharmacological restoration of the epigenetic balance of gene expression is used in biomarker discovery and as a therapeutic target for human cancers.
Validation of novel epigenetics biomarkers will assist in diagnosis, prediction of drug response and eventually identifying the responsive patients. All in all, multidisciplinary field researchers need to work together to optimize the drug engineering process (novel compounds or drug derivatives from existing ones, in different combinations) to be tested in preclinical and clinical trials. The main aspiration is to translate epigenetic therapy into the clinic for the treatment of cancers and tailor effective strategies based on cancer types and epigenome-specific alterations. For this a better understanding of anticipatory processes in the living becomes a preliminary. We make reference here only to a suggestion originating from the biomolecular scientist Harry Rubin (communicated in Nadin, [175])—healthy cells keep cancer cells under control. Only when the anticipatory function is affected, does the cancerous cells get out of control. Louie [176] defined Nadin as the “anticipation guru”—enough for us to take his reference to cancer and anticipation at heart.
References
Hanahan, D., Weinberg, R.A.: The hallmarks of cancer. Cell 100(1), 57–70 (2000)
Morsia, E., Torre, E., Poloni, A., Olivieri, A., Rupoli, S.: Molecular Pathogenesis of myeloproliferative neoplasms: from molecular landscape to therapeutic implications. Int. J. Mol. Sci. 23(9) (2022)
Baylin, S.B., Jones, P.A.: Epigenetic determinants of cancer. Cold Spring Harb. Perspect. Biol. 8(9), a019505 (2016)
Tan, T., Shi, P., Abbas, M.N., Wang, Y., Xu, J., Chen, Y., Cui, H.: Epigenetic modification regulates tumor progression and metastasis through EMT (review). Int. J. Oncol. 60(6) (2022)
Hanahan, D.: Hallmarks of cancer: new dimensions. Cancer Discov. 12(1), 31–46 (2022)
Mancarella, D., Plass, C.: Epigenetic signatures in cancer: proper controls, current challenges and the potential for clinical translation. Genome Med. 13(1), 23 (2021)
Waddington, C.H.: The epigenotype, 1942. Int. J. Epidemiol. 41(1) 10–13 (2012)
Tsai, H.C., Baylin, S.B.: Cancer epigenetics: linking basic biology to clinical medicine. Cell Res. 21(3), 502–517 (2011)
Hattori, H., Hiura, H., Kitamura, A., Miyauchi, N., Kobayashi, N., Takahashi, S., Okae, H., Kyono, K., Kagami, M., Ogata, T. et al.: Association of four imprinting disorders and ART. Clin. Epigen. 11(1), 21 (2019)
Mazzone, R., Zwergel, C., Artico, M., Taurone, S., Ralli, M., Greco, A., Mai, A.: The emerging role of epigenetics in human autoimmune disorders. Clin. Epigenetics 11(1), 34 (2019)
Landgrave-Gómez, J., Mercado-Gómez, O., Guevara-Guzmán, R.: Epigenetic mechanisms in neurological and neurodegenerative diseases. Front. Cell. Neurosci. 9 (2015)
Feinberg, A.P., Vogelstein, B.: Hypomethylation of ras oncogenes in primary human cancers. Biochem. Biophys. Res. Commun. 111(1), 47–54 (1983)
Han, Z.G.: Epigenetic analysis in the search for tumor suppressor genes. Epigenomics 2(4), 489–493 (2010)
Feinberg, A.P., Tycko, B.: The history of cancer epigenetics. Nat. Rev. Cancer 4(2), 143–153 (2004)
Brueckner, B., Garcia, B.R., Siedlecki, P., Musch, T., Kliem, H.C., Zielenkiewicz, P., Suhai, S., Wiessler, M., Lyko, F.: Epigenetic reactivation of tumor suppressor genes by a novel small-molecule inhibitor of human DNA methyltransferases. Cancer Res. 65(14), 6305–6311 (2005)
Lu, Y., Chan, Y.-T., Tan, H.-Y., Li, S., Wang, N., Feng, Y.: Epigenetic regulation in human cancer: the potential role of epi-drug in cancer therapy. Mol. Cancer 19(1), 79–79 (2020)
Rodríguez-Paredes, M., Esteller, M.: Cancer epigenetics reaches mainstream oncology. Nat. Med. 17(3), 330–339 (2011)
Biswas, S., Rao, C.M.: Epigenetic tools (the writers, the readers and the erasers) and their implications in cancer therapy. Eur. J. Pharmacol. 837, 8–24 (2018)
Shi, J., Xu, J., Chen, Y.E., Li, J.S., Cui, Y., Shen, L., Li, J.J., Li, W.: The concurrence of DNA methylation and demethylation is associated with transcription regulation. Nat. Commun. 12(1), 5285 (2021)
Micevic, G., Theodosakis, N., Bosenberg, M.: Aberrant DNA methylation in melanoma: biomarker and therapeutic opportunities. Clin. Epigenetics 9(1), 34 (2017)
Moran, B., Silva, R., Perry, A.S., Gallagher, W.M.: Epigenetics of malignant melanoma. Semin Cancer Biol. 51, 80–88 (2018)
Kinney, S.R., Pradhan, S.: Regulation of expression and activity of DNA (cytosine-5) methyltransferases in mammalian cells. Prog. Mol. Biol. Transl. Sci. 101, 311–333 (2011)
Kanwal, R., Gupta, K., Gupta, S.: Cancer epigenetics: an introduction. Methods Mol. Biol. 1238, 3–25 (2015)
Lopez-Serra, L., Esteller, M.: Proteins that bind methylated DNA and human cancer: reading the wrong words. Br. J. Cancer 98(12), 1881–1885 (2008)
Zhang, B., Xu, J., Li, C., Shi, S., Ji, S., Xu, W., Liu, J., Jin, K., Liang, D., Liang, C., et al.: MBD1 is an epigenetic regulator of KEAP1 in pancreatic cancer. Curr. Mol. Med. 16(4), 404–411 (2016)
Xu, J., Liu, C., Yu, X.J., Jin, C., Fu, D.L., Ni, Q.X.: Activation of multiple tumor suppressor genes by MBD1 siRNA in pancreatic cancer cell line BxPC-3. Zhonghua Yi Xue Za Zhi 88(28), 1948–1951 (2008)
Mahmood, N., Rabbani, S.A.: DNA Methylation readers and cancer: mechanistic and therapeutic applications. Front. Oncol. 9 (2019)
An, J., Rao, A., Ko, M.: TET family dioxygenases and DNA demethylation in stem cells and cancers. Exp. Mol. Med. 49(4), e323–e323 (2017)
Neri, F., Dettori, D., Incarnato, D., Krepelova, A., Rapelli, S., Maldotti, M., Parlato, C., Paliogiannis, P., Oliviero, S.: TET1 is a tumour suppressor that inhibits colon cancer growth by derepressing inhibitors of the WNT pathway. Oncogene 34(32), 4168–4176 (2015)
Panjarian, S., Issa, J.J.: The roles of DNA demethylases in triple-negative breast cancer. Pharmaceuticals 14(7) (2021)
Liu, W., Wu, G., Xiong, F., Chen, Y.: Advances in the DNA methylation hydroxylase TET1. Biomark Res. 9(1), 76–76 (2021)
Zhao, Z., Shilatifard, A.: Epigenetic modifications of histones in cancer. Genome Biol. 20(1), 245 (2019)
Park, S.-Y., Kim, J.-S.: A short guide to histone deacetylases including recent progress on class II enzymes. Exp. Mol. Med. 52(2), 204–212 (2020)
Audia, J.E., Campbell, R.M.: Histone modifications and cancer. Cold Spring Harb. Perspect. Biol. 8(4), a019521 (2016)
Chase, A., Cross, N.C.P.: Aberrations of EZH2 in cancer. Clin. Cancer Res. 17(9), 2613–2618 (2011)
Glozak, M.A., Seto, E.: Histone deacetylases and cancer. Oncogene 26(37), 5420–5432 (2007)
Palmirotta, R., Cives, M., Della-Morte, D., Capuani, B., Lauro, D., Guadagni, F., Silvestris, F.: Sirtuins and cancer: role in the epithelial-mesenchymal transition. Oxid. Med. Cell. Longev. 2016, 3031459 (2016)
Dawson, M.A., Foster, S.D., Bannister, A.J., Robson, S.C., Hannah, R., Wang, X., Xhemalce, B., Wood, A.D., Green, A.R., Göttgens, B., et al.: Three distinct patterns of histone H3Y41 phosphorylation mark active genes. Cell. Rep. 2(3), 470–477 (2012)
Fraga, M.F., Ballestar, E., Villar-Garea, A., Boix-Chornet, M., Espada, J., Schotta, G., Bonaldi, T., Haydon, C., Ropero, S., Petrie, K., et al.: Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nat. Genet. 37(4), 391–400 (2005)
Vardabasso, C., Hasson, D., Ratnakumar, K., Chung, C.Y., Duarte, L.F., Bernstein, E.: Histone variants: emerging players in cancer biology. Cell. Mol. Life Sci. 71(3), 379–404 (2014)
Lei, S., Long, J., Li, J.: MacroH2A suppresses the proliferation of the B16 melanoma cell line. Mol. Med. Rep. 10(4), 1845–1850 (2014)
Vardabasso, C., Hake, S.B., Bernstein, E.: Histone variant H2A.Z.2: a novel driver of melanoma progression. Mol. Cell. Oncol. 3(2), e1073417 (2016)
Sporn, J.C., Jung, B.: Differential regulation and predictive potential of MacroH2A1 isoforms in colon cancer. Am. J. Pathol. 180(6), 2516–2526 (2012)
Nair, S.S., Kumar, R.: Chromatin remodeling in cancer: a gateway to regulate gene transcription. Mol. Oncol. 6(6), 611–619 (2012)
Sundaramoorthy, R.: Nucleosome remodelling: structural insights into ATP-dependent remodelling enzymes. Essays Biochem. 63(1), 45–58 (2019)
Lorch, Y., Kornberg, R.D.: Chromatin-remodeling for transcription. Q. Rev. Biophys. 50, e5 (2017)
Wang, X., Haswell, J.R., Roberts, C.W.: Molecular pathways: SWI/SNF (BAF) complexes are frequently mutated in cancer–mechanisms and potential therapeutic insights. Clin. Cancer Res. 20(1), 21–27 (2014)
Pyo, J.S., Son, B.K., Oh, D., Kim, E.K.: BRG1 is correlated with poor prognosis in colorectal cancer. Hum. Pathol. 73, 66–73 (2018)
Sobczak, M., Pietrzak, J., Płoszaj, T., Robaszkiewicz, A.: BRG1 activates proliferation and transcription of cell cycle-dependent genes in breast cancer cells. Cancers 12(2) (2020)
Bai, J., Mei, P., Zhang, C., Chen, F., Li, C., Pan, Z., Liu, H., Zheng, J.: BRG1 is a prognostic marker and potential therapeutic target in human breast cancer. PLoS ONE 8(3), e59772 (2013)
Sobczak, M., Pitt, A.R., Spickett, C.M., Robaszkiewicz, A.: PARP1 co-regulates EP300-BRG1-dependent transcription of genes involved in breast cancer cell proliferation and DNA repair. Cancers 11(10) (2019)
Kinoshita, F., Kohashi, K., Sugimoto, M., Takamatsu, D., Kiyozawa, D., Eto, M., Oda, Y.: The SWI/SNF chromatin-remodeling complex status in renal cell carcinomas with sarcomatoid or rhabdoid features. Virchows Arch. 477(5), 651–660 (2020)
Hu, J., Liu, J., Chen, A., Lyu, J., Ai, G., Zeng, Q., Sun, Y., Chen, C., Wang, J., Qiu, J., et al.: Ino80 promotes cervical cancer tumorigenesis by activating Nanog expression. Oncotarget 7(44), 72250–72262 (2016)
Zhou, B., Wang, L., Zhang, S., Bennett, B.D., He, F., Zhang, Y., Xiong, C., Han, L., Diao, L., Li, P., et al.: INO80 governs superenhancer-mediated oncogenic transcription and tumor growth in melanoma. Genes Dev. 30(12), 1440–1453 (2016)
Zhang, S., Zhou, B., Wang, L., Li, P., Bennett, B.D., Snyder, R., Garantziotis, S., Fargo, D.C., Cox, A.D., Chen, L., et al.: INO80 is required for oncogenic transcription and tumor growth in non-small cell lung cancer. Oncogene 36(10), 1430–1439 (2017)
Zimta, A.A., Tigu, A.B., Braicu, C., Stefan, C., Ionescu, C., Berindan-Neagoe, I.: An emerging class of long non-coding RNA with oncogenic role arises from the snoRNA host genes. Front. Oncol. 10, 389 (2020)
Irimie, A.I., Zimta, A.A., Ciocan, C., Mehterov, N., Dudea, D., Braicu, C., Berindan-Neagoe, I.: The unforeseen non-coding RNAs in head and neck cancer. Genes 9(3) (2018)
Irimie, A.I., Braicu, C., Sonea, L., Zimta, A.A., Cojocneanu-Petric, R., Tonchev, K., Mehterov, N., Diudea, D., Buduru, S., Berindan-Neagoe, I.: A looking-glass of non-coding RNAs in oral cancer. Int. J. Mol. Sci. 18(12) (2017)
Costa, F.F.: Non-coding RNAs, epigenetics and complexity. Gene 410(1), 9–17 (2008)
Braicu, C., Gulei, D., Raduly, L., Harangus, A., Rusu, A., Berindan-Neagoe, I.: Altered expression of miR-181 affects cell fate and targets drug resistance-related mechanisms. Mol. Aspects Med. 70, 90–105 (2019)
Weber, B., Stresemann, C., Brueckner, B., Lyko, F.: Methylation of human microRNA genes in normal and neoplastic cells. Cell Cycle 6(9), 1001–1005 (2007)
Loginov, V.I., Rykov, S.V., Fridman, M.V., Braga, E.A.: Methylation of miRNA genes and oncogenesis. Biochemistry (Mosc) 80(2), 145–162 (2015)
Davalos, V., Moutinho, C., Villanueva, A., Boque, R., Silva, P., Carneiro, F., Esteller, M.: Dynamic epigenetic regulation of the microRNA-200 family mediates epithelial and mesenchymal transitions in human tumorigenesis. Oncogene 31(16), 2062–2074 (2012)
Jurj, A., Zanoaga, O., Braicu, C., Lazar, V., Tomuleasa, C., Irimie, A., Berindan-Neagoe, I.: A comprehensive picture of extracellular vesicles and their contents. Mole. Transf. Cancer Cells Cancers (Basel) 12(2) (2020)
Bardhan, A., Banerjee, A., Basu, K., Pal, D.K., Ghosh, A.: PRNCR1: a long non-coding RNA with a pivotal oncogenic role in cancer. Hum. Genet. 141(1), 15–29 (2022)
Yang, L., Lin, C., Jin, C., Yang, J.C., Tanasa, B., Li, W., Merkurjev, D., Ohgi, K.A., Meng, D., Zhang, J., et al.: lncRNA-dependent mechanisms of androgen-receptor-regulated gene activation programs. Nature 500(7464), 598–602 (2013)
Tsai, M.C., Manor, O., Wan, Y., Mosammaparast, N., Wang, J.K., Lan, F., Shi, Y., Segal, E., Chang, H.Y.: Long noncoding RNA as modular scaffold of histone modification complexes. Science 329(5992), 689–693 (2010)
Han, C., Fu, Y., Zeng, N., Yin, J., Li, Q.: LncRNA FAM83H-AS1 promotes triple-negative breast cancer progression by regulating the miR-136-5p/metadherin axis. Aging 12(4), 3594–3616 (2020)
Saito, Y., Kanai, Y., Nakagawa, T., Sakamoto, M., Saito, H., Ishii, H., Hirohashi, S.: Increased protein expression of DNA methyltransferase (DNMT) 1 is significantly correlated with the malignant potential and poor prognosis of human hepatocellular carcinomas. Int. J. Cancer 105(4), 527–532 (2003)
Gu, Y., Yang, P., Shao, Q., Liu, X., Xia, S., Zhang, M., Xu, H., Shao, Q.: Investigation of the expression patterns and correlation of DNA methyltransferases and class I histone deacetylases in ovarian cancer tissues. Oncol. Lett. 5(2), 452–458 (2013)
Joensuu, E.I., Nieminen, T.T., Lotsari, J.E., Pavicic, W., Abdel-Rahman, W.M., Peltomäki, P.: Methyltransferase expression and tumor suppressor gene methylation in sporadic and familial colorectal cancer. Genes Chromosom. Cancer 54(12), 776–787 (2015)
Tzelepi, V., Logotheti, S., Efstathiou, E., Troncoso, P., Aparicio, A., Sakellakis, M., Hoang, A., Perimenis, P., Melachrinou, M., Logothetis, C., et al.: Epigenetics and prostate cancer: defining the timing of DNA methyltransferase deregulation during prostate cancer progression. Pathology 52(2), 218–227 (2020)
Ley, T.J., Ding, L., Walter, M.J., McLellan, M.D., Lamprecht, T., Larson, D.E., Kandoth, C., Payton, J.E., Baty, J., Welch, J., et al.: DNMT3A mutations in acute myeloid leukemia. N. Engl. J. Med. 363(25), 2424–2433 (2010)
Zhang, Y.-Y., Zhou, J.-D., Yang, D.-Q., He, P.-F., Yao, D.-M., Qian, Z., Yang, J., Xu, W.-R., Lin, J., Qian, J.: Intragenic hypomethylation of DNMT3A in patients with myelodysplastic syndrome. Clin. Chem. Labor. Med. (CCLM) 56(3), 485–491 (2018)
Gokul, G., Gautami, B., Malathi, S., Sowjanya, A.P., Rani Poli, U., Jain, M., Ramakrishna, G., Khosla, S.: DNA Methylation profile at the DNMT3L promoter: a potential biomarker for cervical cancer. Epigenetics 2(2), 80–85 (2007)
Filipczak, P.T., Leng, S., Tellez, C.S., Do, K.C., Grimes, M.J., Thomas, C.L., Walton-Filipczak, S.R., Picchi, M.A., Belinsky, S.A.: p53-suppressed oncogene TET1 prevents cellular aging in lung cancer. Cancer Res. 79(8), 1758–1768 (2019)
Guo, H., Zhu, H., Zhang, J., Wan, B., Shen, Z.: TET1 suppresses colon cancer proliferation by impairing β-catenin signal pathway. J. Cell Biochem. 120(8), 12559–12565 (2019)
Sang, Y., Cheng, C., Tang, X.F., Zhang, M.F., Lv, X.B.: Hypermethylation of TET1 promoter is a new diagnosic marker for breast cancer metastasis. Asian Pac. J. Cancer Prev. 16(3), 1197–1200 (2015)
Fu, H.L., Ma, Y., Lu, L.G., Hou, P., Li, B.J., Jin, W.L., Cui, D.X.: TET1 exerts its tumor suppressor function by interacting with p53-EZH2 pathway in gastric cancer. J. Biomed. Nanotechnol. 10(7), 1217–1230 (2014)
Elliott, E.K., Hopkins, L.N., Hensen, R., Sutherland, H.G., Haupt, L.M., Griffiths, L.R.: Epigenetic regulation of miR-92a and TET2 and their association in non-Hodgkin lymphoma. Front. Genet. 12, 768913 (2021)
Tefferi, A., Lim, K.H., Abdel-Wahab, O., Lasho, T.L., Patel, J., Patnaik, M.M., Hanson, C.A., Pardanani, A., Gilliland, D.G., Levine, R.L.: Detection of mutant TET2 in myeloid malignancies other than myeloproliferative neoplasms: CMML, MDS, MDS/MPN and AML. Leukemia 23(7), 1343–1345 (2009)
Cao, T., Pan, W., Sun, X., Shen, H.: Increased expression of TET3 predicts unfavorable prognosis in patients with ovarian cancer-a bioinformatics integrative analysis. J. Ovarian Res. 12(1), 101 (2019)
Ghaedamini, S., Nikbakht, M., Soleimani, M.: Assessment of Immuno-histochemical expression of MBD1 in colorectal adenocarcinoma and its correlations with prognostic factors. Middle East J. Dig. Dis. 12(1), 39–44 (2020)
Dai, S.-D., Wang, Y., Miao, Y., Zhao, Y., Zhang, Y., Jiang, G.-Y., Zhang, P.-X., Yang, Z.-Q., Wang, E.-H.: Cytoplasmic Kaiso is associated with poor prognosis in non-small cell lung cancer. BMC Cancer 9(1), 178 (2009)
Hu, A., Hong, F., Li, D., Jin, Y., Kon, L., Xu, Z., He, H., Xie, Q.: Long non-coding RNA ROR recruits histone transmethylase MLL1 to up-regulate TIMP3 expression and promote breast cancer progression. J. Transl. Med. 19(1), 95 (2021)
Watanabe, Y., Castoro, R.J., Kim, H.S., North, B., Oikawa, R., Hiraishi, T., Ahmed, S.S., Chung, W., Cho, M.-Y., Toyota, M., et al.: Frequent alteration of MLL3 frameshift mutations in microsatellite deficient colorectal cancer. Plos One 6(8), e23320 (2011)
Larsson, C., Cordeddu, L., Siggens, L., Pandzic, T., Kundu, S., He, L., Ali, M.A., Pristovšek, N., Hartman, K., Ekwall, K., et al.: Restoration of KMT2C/MLL3 in human colorectal cancer cells reinforces genome-wide H3K4me1 profiles and influences cell growth and gene expression. Clin. Epigenetics 12(1), 74 (2020)
Limberger, T., Schlederer, M., Trachtová, K., Garces de los Fayos Alonso, I., Yang, J., Högler, S., Sternberg, C., Bystry, V., Oppelt, J., Tichý, B., et al.: KMT2C methyltransferase domain regulated INK4A expression suppresses prostate cancer metastasis. Mole. Cancer 21(1), 89 (2022)
Varambally, S., Dhanasekaran, S.M., Zhou, M., Barrette, T.R., Kumar-Sinha, C., Sanda, M.G., Ghosh, D., Pienta, K.J., Sewalt, R.G., Otte, A.P., et al.: The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature 419(6907), 624–629 (2002)
Xu, C., Hou, Z., Zhan, P., Zhao, W., Chang, C., Zou, J., Hu, H., Zhang, Y., Yao, X., Yu, L., et al.: EZH2 regulates cancer cell migration through repressing TIMP-3 in non-small cell lung cancer. Med. Oncol. 30(4), 713 (2013)
Wang, Y., Sun, B., Zhang, Q., Dong, H., Zhang, J.: p300 Acetylates JHDM1A to inhibit osteosarcoma carcinogenesis. Artif. Cells Nanomed. Biotechnol. 47(1), 2891–2899 (2019)
Lu, D.H., Yang, J., Gao, L.K., Min, J., Tang, J.M., Hu, M., Li, Y., Li, S.T., Chen, J., Hong, L.: Lysine demethylase 2A promotes the progression of ovarian cancer by regulating the PI3K pathway and reversing epithelial-mesenchymal transition. Oncol. Rep. 41(2), 917–927 (2019)
Wagner, K.W., Alam, H., Dhar, S.S., Giri, U., Li, N., Wei, Y., Giri, D., Cascone, T., Kim, J.-H., Ye, Y., et al.: KDM2A promotes lung tumorigenesis by epigenetically enhancing ERK1/2 signaling. J. Clin. Investig. 123(12), 5231–5246 (2013)
Liu, Y., Wang, Y., Chen, C., Zhang, J., Qian, W., Dong, Y., Liu, Z., Zhang, X., Wang, X., Zhang, Z., et al.: LSD1 binds to HPV16 E7 and promotes the epithelial-mesenchymal transition in cervical cancer by demethylating histones at the Vimentin promoter. Oncotarget 8(7) (2016)
Hayami, S., Kelly, J.D., Cho, H.-S., Yoshimatsu, M., Unoki, M., Tsunoda, T., Field, H.I., Neal, D.E., Yamaue, H., Ponder, B.A.J., et al.: Overexpression of LSD1 contributes to human carcinogenesis through chromatin regulation in various cancers. Int. J. Cancer 128(3), 574–586 (2011)
Zhang, S., Liu, M., Yao, Y., Yu, B., Liu, H.: Targeting LSD1 for acute myeloid leukemia (AML) treatment. Pharmacol. Res. 164, 105335 (2021)
Xue, L., Hou, J., Wang, Q., Yao, L., Xu, S., Ge, D.: RNAi screening identifies HAT1 as a potential drug target in esophageal squamous cell carcinoma. Int. J. Clin. Exp. Pathol. 7(7), 3898–3907 (2014)
Fan, P., Zhao, J., Meng, Z., Wu, H., Wang, B., Wu, H., Jin, X.: Overexpressed histone acetyltransferase 1 regulates cancer immunity by increasing programmed death-ligand 1 expression in pancreatic cancer. J. Exp. Clin. Cancer Res. 38(1), 47 (2019)
Han, N., Shi, L., Guo, Q., Sun, W., Yu, Y., Yang, L., Zhang, X., Zhang, M.: HAT1 induces lung cancer cell apoptosis via up regulating fas. Oncotarget 8(52), 89970–89977 (2017)
Tang, W., Zhou, W., Xiang, L., Wu, X., Zhang, P., Wang, J., Liu, G., Zhang, W., Peng, Y., Huang, X., et al.: The p300/YY1/miR-500a-5p/HDAC2 signalling axis regulates cell proliferation in human colorectal cancer. Nat. Commun. 10(1), 663 (2019)
Choi, J.-R., Lee, S.-Y., Shin, K.S., Choi, C.Y., Kang, S.J.: p300-mediated acetylation increased the protein stability of HIPK2 and enhanced its tumor suppressor function. Sci. Rep. 7(1), 16136 (2017)
Ono, H., Basson, M.D., Ito, H.: P300 inhibition enhances gemcitabine-induced apoptosis of pancreatic cancer. Oncotarget 7(32) (2016)
Chen, M.-K., Cai, M.-Y., Luo, R.-Z., Tian, X., Liao, Q.-M., Zhang, X.-Y., Han, J.-D.: Overexpression of p300 correlates with poor prognosis in patients with cutaneous squamous cell carcinoma. Br. J. Dermatol. 172(1), 111–119 (2015)
Ishihama, K., Yamakawa, M., Semba, S., Takeda, H., Kawata, S., Kimura, S., Kimura, W.: Expression of HDAC1 and CBP/p300 in human colorectal carcinomas. J. Clin. Pathol. 60(11), 1205 (2007)
Li, M., Luo, R.-Z., Chen, J.-W., Cao, Y., Lu, J.-B., He, J.-H., Wu, Q.-L., Cai, M.-Y.: High expression of transcriptional coactivator p300 correlates with aggressive features and poor prognosis of hepatocellular carcinoma. J. Transl. Med. 9(1), 5 (2011)
Gao, Y., Geng, J., Hong, X., Qi, J., Teng, Y., Yang, Y., Qu, D., Chen, G.: Expression of p300 and CBP is associated with poor prognosis in small cell lung cancer. Int. J. Clin. Exp. Pathol. 7(2), 760–767 (2014)
Oh, J.H., Lee, J.-Y., Kim, K.H., Kim, C.Y., Jeong, D.S., Cho, Y., Nam, K.T., Kim, M.H.: Elevated GCN5 expression confers tamoxifen resistance by upregulating AIB1 expression in ER-positive breast cancer. Cancer Lett. 495, 145–155 (2020)
Mustachio, L.M., Roszik, J., Farria, A.T., Guerra, K., Dent, S.Y.: Repression of GCN5 expression or activity attenuates c-MYC expression in non-small cell lung cancer. Am. J. Cancer Res. 9(8), 1830–1845 (2019)
Yin, Y.W., Jin, H.J., Zhao, W., Gao, B., Fang, J., Wei, J., Zhang, D.D., Zhang, J., Fang, D.: The histone acetyltransferase GCN5 expression is elevated and regulated by c-Myc and E2F1 transcription factors in human colon cancer. Gene. Expr. 16(4), 187–196 (2015)
Chen, D.Q., Pan, B.Z., Huang, J.Y., Zhang, K., Cui, S.Y., De, W., Wang, R., Chen, L.B.: HDAC 1/4-mediated silencing of microRNA-200b promotes chemoresistance in human lung adenocarcinoma cells. Oncotarget 5(10), 3333–3349 (2014)
Sudo, T., Mimori, K., Nishida, N., Kogo, R., Iwaya, T., Tanaka, F., Shibata, K., Fujita, H., Shirouzu, K., Mori, M.: Histone deacetylase 1 expression in gastric cancer. Oncol. Rep. 26(4), 777–782 (2011)
Zhang, Z., Yamashita, H., Toyama, T., Sugiura, H., Ando, Y., Mita, K., Hamaguchi, M., Hara, Y., Kobayashi, S., Iwase, H.: Quantitation of HDAC1 mRNA expression in invasive carcinoma of the breast*. Breast Cancer Res. Treat. 94(1), 11–16 (2005)
Long, J., Fang, W.Y., Chang, L., Gao, W.H., Shen, Y., Jia, M.Y., Zhang, Y.X., Wang, Y., Dou, H.B., Zhang, W.J., et al.: Targeting HDAC3, a new partner protein of AKT in the reversal of chemoresistance in acute myeloid leukemia via DNA damage response. Leukemia 31(12), 2761–2770 (2017)
Gupta, M., Concepcion, C.P., Fahey, C.G., Keshishian, H., Bhutkar, A., Brainson, C.F., Sanchez-Rivera, F.J., Pessina, P., Kim, J.Y., Simoneau, A., et al.: BRG1 loss predisposes lung cancers to replicative stress and ATR dependency. Cancer Res. 80(18), 3841–3854 (2020)
Cheng, X., Zhao, J.-X., Dong, F., Cao, X.-C.: ARID1A mutation in metastatic breast cancer: a potential therapeutic target. Front. Oncol. 11 (2021)
Wang, T., Gao, X., Zhou, K., Jiang, T., Gao, S., Liu, P., Zuo, X., Shi, X.: Role of ARID1A in epithelial-mesenchymal transition in breast cancer and its effect on cell sensitivity to 5-FU. Int. J. Mol. Med. 46(5), 1683–1694 (2020)
Kim, Y.S., Jeong, H., Choi, J.W., Oh, H.E., Lee, J.H.: Unique characteristics of ARID1A mutation and protein level in gastric and colorectal cancer: a meta-analysis. Saudi J. Gastroenterol. 23(5), 268–274 (2017)
Wu, J., He, K., Zhang, Y., Song, J., Shi, Z., Chen, W., Shao, Y.: Inactivation of SMARCA2 by promoter hypermethylation drives lung cancer development. Gene 687, 193–199 (2019)
Li, Q., Birkbak, N.J., Gyorffy, B., Szallasi, Z., Eklund, A.C.: Jetset: selecting the optimal microarray probe set to represent a gene. BMC Bioinform. 12(1), 474 (2011)
Song, Y., Zhang, H., Yang, X., Shi, Y., Yu, B.: Annual review of lysine-specific demethylase 1 (LSD1/KDM1A) inhibitors in 2021. Eur. J. Med. Chem. 228, 114042 (2022)
Yan, B., Li, X., Johnson, A., Yang, Y., Jian, W., Qiu, Y.: Chapter 18—epigenetic drugs for cancer therapy. In: Huang, S., Litt, M.D., Blakey, C.A. (eds.) Epigenetic Gene Expression and Regulation, pp. 397–423. Academic Press, Oxford (2015)
Doroshow, D.B., Eder, J.P., LoRusso, P.M.: BET inhibitors: a novel epigenetic approach. Ann. Oncol. 28(8), 1776–1787 (2017)
Ganesan, A.: Multitarget drugs: an epigenetic epiphany. ChemMedChem 11(12), 1227–1241 (2016)
Hrzenjak, A., Moinfar, F., Kremser, M.L., Strohmeier, B., Petru, E., Zatloukal, K., Denk, H.: Histone deacetylase inhibitor vorinostat suppresses the growth of uterine sarcomas in vitro and in vivo. Mol. Cancer 9, 49 (2010)
Hess-Stumpp, H., Bracker, T.U., Henderson, D., Politz, O.: MS-275, a potent orally available inhibitor of histone deacetylases—the development of an anticancer agent. Int. J. Biochem. Cell. Biol. 39(7–8), 1388–1405 (2007)
Lambert, M., Jambon, S., Depauw, S., David-Cordonnier, M.-H.: Targeting transcription factors for cancer treatment. Molecules 23(6), 1479 (2018)
Gan, L., Yang, Y., Li, Q., Feng, Y., Liu, T., Guo, W.: Epigenetic regulation of cancer progression by EZH2: from biological insights to therapeutic potential. Biomark. Res. 6(1), 10 (2018)
Liu, N., Zhao, R., Ma, Y., Wang, D., Yan, C., Zhou, D., Yin, F., Li, Z.: The development of epigenetics and related inhibitors for targeted drug design in cancer therapy. Curr. Top. Med. Chem. 18(28), 2380–2394 (2018)
Cheng, J.C., Matsen, C.B., Gonzales, F.A., Ye, W., Greer, S., Marquez, V.E., Jones, P.A., Selker, E.U.: Inhibition of DNA methylation and reactivation of silenced genes by zebularine. J. Natl. Cancer Inst. 95(5), 399–409 (2003)
Winquist, E., Knox, J., Ayoub, J.P., Wood, L., Wainman, N., Reid, G.K., Pearce, L., Shah, A., Eisenhauer, E.: Phase II trial of DNA methyltransferase 1 inhibition with the antisense oligonucleotide MG98 in patients with metastatic renal carcinoma: a national cancer Institute of Canada Clinical Trials Group investigational new drug study. Invest. New Drugs 24(2), 159–167 (2006)
Amato, R.J., Stephenson, J., Hotte, S., Nemunaitis, J., Bélanger, K., Reid, G., Martell, R.E.: MG98, a second-generation DNMT1 inhibitor, in the treatment of advanced renal cell carcinoma. Cancer Invest. 30(5), 415–421 (2012)
Larsson, P., Ulfhammer, E., Magnusson, M., Bergh, N., Lunke, S., El-Osta, A., Medcalf, R.L., Svensson, P.A., Karlsson, L., Jern, S.: Role of histone acetylation in the stimulatory effect of valproic acid on vascular endothelial tissue-type plasminogen activator expression. PLoS ONE 7(2), e31573 (2012)
Du, L., Wang, D., Wei, X., Liu, C., Xiao, Z., Qian, W., Song, Y., Hou, X.: MS275 as class I HDAC inhibitor displayed therapeutic potential on malignant ascites by iTRAQ-based quantitative proteomic analysis. BMC Gastroenterol. 22(1), 29 (2022)
Richardson, P.G., Laubach, J.P., Lonial, S., Moreau, P., Yoon, S.S., Hungria, V.T., Dimopoulos, M.A., Beksac, M., Alsina, M., San-Miguel, J.F.: Panobinostat: a novel pan-deacetylase inhibitor for the treatment of relapsed or relapsed and refractory multiple myeloma. Expert Rev. Anticancer Ther. 15(7), 737–748 (2015)
Singh, A., Patel, V.K., Jain, D.K., Patel, P., Rajak, H.: Panobinostat as pan-deacetylase inhibitor for the treatment of pancreatic cancer: recent progress and future prospects. Oncol. Ther. 4(1), 73–89 (2016)
Sabnis, R.W.: Novel quinoline compounds as EZH2 inhibitors for treating cancer. ACS Med. Chem. Lett. 13(5), 755–756 (2022)
Wang, X., Cao, W., Zhang, J., Yan, M., Xu, Q., Wu, X., Wan, L., Zhang, Z., Zhang, C., Qin, X., et al.: A covalently bound inhibitor triggers EZH2 degradation through CHIP-mediated ubiquitination. Embo J. 36(9), 1243–1260 (2017)
Kong, X., Chen, L., Jiao, L., Jiang, X., Lian, F., Lu, J., Zhu, K., Du, D., Liu, J., Ding, H., et al.: Astemizole arrests the proliferation of cancer cells by disrupting the EZH2-EED interaction of polycomb repressive complex 2. J. Med. Chem. 57(22), 9512–9521 (2014)
Si, Y., Bon, C., Barbachowska, M., Cadet-Daniel, V., Jallet, C., Soresinetti, L., Boullé, M., Duchateau, M., Matondo, M., Agou, F., et al.: A novel screening strategy to identify histone methyltransferase inhibitors reveals a crosstalk between DOT1L and CARM1. RSC Chem. Biol. 3(4), 456–467 (2022)
Daigle, S.R., Olhava, E.J., Therkelsen, C.A., Majer, C.R., Sneeringer, C.J., Song, J., Johnston, L.D., Scott, M.P., Smith, J.J., Xiao, Y., et al.: Selective killing of mixed lineage leukemia cells by a potent small-molecule DOT1L inhibitor. Cancer Cell 20(1), 53–65 (2011)
Yang, L., Lei, Q., Li, L., Yang, J., Dong, Z., Cui, H.: Silencing or inhibition of H3K79 methyltransferase DOT1L induces cell cycle arrest by epigenetically modulating c-Myc expression in colorectal cancer. Clin. Epigenetics 11(1), 199 (2019)
Daigle, S.R., Olhava, E.J., Therkelsen, C.A., Basavapathruni, A., Jin, L., Boriack-Sjodin, P.A., Allain, C.J., Klaus, C.R., Raimondi, A., Scott, M.P., et al.: Potent inhibition of DOT1L as treatment of MLL-fusion leukemia. Blood 122(6), 1017–1025 (2013)
Song, Z., Wei, Z., Wang, Q., Zhang, X., Tao, X., Wu, N., Liu, X., Qian, J.: The role of DOT1L in the proliferation and prognosis of gastric cancer. Biosci. Rep. 40(1) (2020)
Barth, J., Abou-El-Ardat, K., Dalic, D., Kurrle, N., Maier, A.M., Mohr, S., Schütte, J., Vassen, L., Greve, G., Schulz-Fincke, J., et al.: LSD1 inhibition by tranylcypromine derivatives interferes with GFI1-mediated repression of PU.1 target genes and induces differentiation in AML. Leukemia 33(6), 1411–1426
Piboonprai, K., Khumkhrong, P., Khongkow, M., Yata, T., Ruangrungsi, N., Chansriniyom, C., Iempridee, T.: Anticancer activity of arborinine from Glycosmis parva leaf extract in human cervical cancer cells. Biochem. Biophys. Res. Commun. 500(4), 866–872 (2018)
Fang, Y., Yang, C., Teng, D., Su, S., Luo, X., Liu, Z., Liao, G.: Discovery of higenamine as a potent, selective and cellular active natural LSD1 inhibitor for MLL-rearranged leukemia therapy. Bioorg. Chem. 109, 104723 (2021)
Li, X., Song, L., Xu, S., Tippin, M., Meng, S., Xie, J., Uchio, E., Zi, X.: Kava root extracts hinder prostate cancer development and tumorigenesis by involvement of dual inhibition of MAO-A and LSD1. J. Transl. Genet. Genom. 5, 163–172 (2021)
Bird, A.: DNA methylation patterns and epigenetic memory. Genes Dev. 16(1), 6–21 (2002)
Kouzarides, T.: Chromatin modifications and their function. Cell 128(4), 693–705 (2007)
Morris, K.V., Mattick, J.S.: The rise of regulatory RNA. Nat. Rev. Genet. 15(6), 423–437 (2014)
Bernstein, B.E., Meissner, A., Lander, E.S.: The mammalian epigenome. Cell 128(4), 669–681 (2007)
Monk, D., Mackay, D.J.G., Eggermann, T., Maher, E.R., Riccio, A.: Genomic imprinting disorders: lessons on how genome, epigenome and environment interact. Nat. Rev. Genet. 20(4), 235–248 (2019)
Anway, M.D., Cupp, A.S., Uzumcu, M., Skinner, M.K.: Epigenetic transgenerational actions of endocrine disruptors and male fertility. Science 308(5727), 1466–1469 (2005)
Fraga, M.F., Ballestar, E., Paz, M.F., Ropero, S., Setien, F., Ballestar, M.L., Heine-Suñer, D., Cigudosa, J.C., Urioste, M., Benitez, J., et al.: Epigenetic differences arise during the lifetime of monozygotic twins. Proc. Natl. Acad. Sci. USA 102(30), 10604–10609 (2005)
Denham, J.: Exercise and epigenetic inheritance of disease risk. Acta Physiol. 222(1) (2018)
Radford, E.J., Ito, M., Shi, H., Corish, J.A., Yamazawa, K., Isganaitis, E., Seisenberger, S., Hore, T.A., Reik, W., Erkek, S., et al.: In utero effects. In utero undernourishment perturbs the adult sperm methylome and intergenerational metabolism. Science 345(6198), 1255903 (2014)
Martínez, D., Pentinat, T., Ribó, S., Daviaud, C., Bloks, V.W., Cebrià, J., Villalmanzo, N., Kalko, S.G., Ramón-Krauel, M., Díaz, R., et al.: In utero undernutrition in male mice programs liver lipid metabolism in the second-generation offspring involving altered Lxra DNA methylation. Cell. Metab. 19(6), 941–951 (2014)
Lee, H.S.: Impact of maternal diet on the epigenome during in utero life and the developmental programming of diseases in childhood and adulthood. Nutrients 7(11), 9492–9507 (2015)
Wu, L., Lu, Y., Jiao, Y., Liu, B., Li, S., Li, Y., Xing, F., Chen, D., Liu, X., Zhao, J., et al.: Paternal psychological stress reprograms hepatic gluconeogenesis in offspring. Cell. Metab. 23(4), 735–743 (2016)
Gapp, K., Jawaid, A., Sarkies, P., Bohacek, J., Pelczar, P., Prados, J., Farinelli, L., Miska, E., Mansuy, I.M.: Implication of sperm RNAs in transgenerational inheritance of the effects of early trauma in mice. Nat. Neurosci. 17(5), 667–669 (2014)
Rodgers, A.B., Morgan, C.P., Bronson, S.L., Revello, S., Bale, T.L.: Paternal stress exposure alters sperm microRNA content and reprograms offspring HPA stress axis regulation. J. Neurosci. Official J. Soc. Neurosci. 33(21), 9003–9012 (2013)
Carone, B.R., Fauquier, L., Habib, N., Shea, J.M., Hart, C.E., Li, R., Bock, C., Li, C., Gu, H., Zamore, P.D., et al.: Paternally induced transgenerational environmental reprogramming of metabolic gene expression in mammals. Cell 143(7), 1084–1096 (2010)
Atsem, S., Reichenbach, J., Potabattula, R., Dittrich, M., Nava, C., Depienne, C., Böhm, L., Rost, S., Hahn, T., Schorsch, M., et al.: Paternal age effects on sperm FOXK1 and KCNA7 methylation and transmission into the next generation. Hum. Mol. Genet. 25(22), 4996–5005 (2016)
Donkin, I., Versteyhe, S., Ingerslev, L.R., Qian, K., Mechta, M., Nordkap, L., Mortensen, B., Appel, E.V., Jørgensen, N., Kristiansen, V.B., et al.: Obesity and bariatric surgery drive epigenetic variation of spermatozoa in humans. Cell. Metab. 23(2), 369–378 (2016)
Varela-Rey, M., Woodhoo, A., Martinez-Chantar, M.L., Mato, J.M., Lu, S.C.: Alcohol, DNA methylation, and cancer. Alcohol Res. Curr. Rev. 35(1), 25–35 (2013)
Rossi, M., Jahanzaib Anwar, M., Usman, A., Keshavarzian, A., Bishehsari, F.: Colorectal cancer and alcohol consumption-populations to molecules. Cancers 10(2) (2018)
Chen, Y.S., Wang, R., Dashwood, W.M., Löhr, C.V., Williams, D.E., Ho, E., Mertens-Talcott, S., Dashwood, R.H.: A miRNA signature for an environmental heterocyclic amine defined by a multi-organ carcinogenicity bioassay in the rat. Arch. Toxicol. 91(10), 3415–3425 (2017)
Rodríguez-Miguel, C., Moral, R., Escrich, R., Vela, E., Solanas, M., Escrich, E.: The role of dietary extra virgin olive oil and corn oil on the alteration of epigenetic patterns in the rat DMBA-induced breast cancer model. PLoS ONE 10(9), e0138980 (2015)
Kyriacou, A., Tziaferi, V., Toumba, M.: Stress, Thyroid Dysregulation and Thyroid Cancer in Children and Adolescents. Proposed Impending Mechanisms. Hormone Research in Paediatrics (2022)
He, J., Han, J., Liu, J., Yang, R., Wang, J., Wang, X., Chen, X.: Genetic and epigenetic impact of chronic inflammation on colon mucosa cells. Front. Genet. 12, 722835 (2021)
Hey, J., Paulsen, M., Toth, R., Weichenhan, D., Butz, S., Schatterny, J., Liebers, R., Lutsik, P., Plass, C., Mall, M.A.: Epigenetic reprogramming of airway macrophages promotes polarization and inflammation in muco-obstructive lung disease. Nat. Commun. 12(1), 6520 (2021)
Ahmad, S., Manzoor, S., Siddiqui, S., Mariappan, N., Zafar, I., Ahmad, A., Ahmad, A.: Epigenetic underpinnings of inflammation: connecting the dots between pulmonary diseases, lung cancer and COVID-19. Seminars in cancer biology (2021)
Pietropaolo, V., Prezioso, C., Moens, U.: Role of virus-induced host cell epigenetic changes in cancer. Int. J. Mole. Sci. 22(15) (2021)
Rattan, P., Minacapelli, C.D., Rustgi, V.: The Microbiome and Hepatocellular Carcinoma. Liver Transpl. Official Publ. Am. Assoc. Study Liver Diseas. Int. Liver Transpl. Soc. 26(10), 1316–1327 (2020)
Nadin, M.: Reporting on anticipatory systems: a subject surviving opportunism and intolerance. Int. J. Gen. Syst. 46(2), 93–122 (2017)
Louie, A.H.: Intangible Life—Functorial Connections in Relational Biology. Springer (2017)
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2022 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Isacescu, E., Braicu, C., Pop, L., Berindan-Neagoe, I., Stefan, C. (2022). Epigenetics. In: Nadin, M. (eds) Epigenetics and Anticipation. Cognitive Systems Monographs, vol 45. Springer, Cham. https://doi.org/10.1007/978-3-031-17678-4_10
Download citation
DOI: https://doi.org/10.1007/978-3-031-17678-4_10
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-031-17677-7
Online ISBN: 978-3-031-17678-4
eBook Packages: MedicineMedicine (R0)