
Abstract
Vascular disease and its consequences remain the most common causes of mortality and significant lifelong disability in developed, and most developing, countries. Age is the most potent risk factor for the responsible vascular changes and although these are in part dependent on the “quality of the arterial tissue” we inherit, their trajectory can be impacted by the environment our vessels are exposed to. This chapter will review the molecular, cellular, structural, and functional changes associated with aging, the impact of these changes on cardiovascular outcomes, and lifestyle and pharmacologic interventions designed to modify them.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


1 Introduction
Longevity is a vascular question, which has been well expressed in the axiom that man is only as old as his arteries. To a majority of men death comes primarily or secondarily through this portal. The onset of what may be called physiological arterio-sclerosis depends, in the first place, on the quality of arterial tissue which the individual has inherited, and secondarily on the amount of wear and tear to which he has subjected it.—Sir William Osler, 1891 [1].
Sir William Osler’s famous quotation is as relevant today as it was in the late nineteenth century. Vascular disease and its consequences remain the most common causes of mortality and significant lifelong disability in developed, and most developing, countries. Age is the most potent risk factor for the responsible vascular changes and although these are in part dependent on the “quality of the arterial tissue” we inherit, their trajectory can be impacted by the environment our vessels are exposed to. This chapter will review the molecular, cellular, structural, and functional changes associated with aging, the impact of these changes on cardiovascular outcomes, and lifestyle and pharmacologic interventions designed to modify them.
2 Molecular and Cellular Mechanisms of Vascular Aging
The vasculature consists of two primary cell types: endothelial cells and vascular smooth muscle cells. The endothelium is the innermost, lumen-facing layer of the vasculature [2]. Endothelial cells communicate with vascular smooth muscle cells via a variety of mechanisms including paracrine mediators such as endothelial nitric oxide synthase-derived nitric oxide, extracellular vesicles, and microRNAs, as well as interactions via the extracellular matrix and direct cell-cell pathways [3, 4]. Endothelial cell function is considered a “barometer” of vascular health because it is a driver of the development, progression, and clinical manifestations of cardiovascular and cerebrovascular diseases. Endothelial dysfunction is considered a major risk factor for cardiovascular disease, the leading cause of mortality and loss of independence in older Americans [5]. The number of older individuals in the USA is rapidly growing with a projected increase in those >65 years of age from 56.2 million in 2021 to 80.8 million in 2040 [6]. Thus, there is an unmet need to understand responsible mechanisms, which can inform the development and testing of novel treatments for the amelioration of the age-associated increased cardiovascular risk.
Several mechanisms are involved in vascular aging. Among them are reduced nitric oxide bioavailability, increased oxidative stress, mitochondrial dysfunction, sterile inflammation or “inflammaging”, genomic/epigenetic alterations, telomere shortening, and stem cell depletion.
The following sections highlight aging-associated vascular cell changes, and their contribution to the pathogenesis of both microvascular and macrovascular diseases (Fig. 1).

Conceptual model for the role of cell-autonomous and noncell-autonomous mechanisms in vascular aging. The model predicts that circulating progeronic (e.g., inflammatory cytokines, renin-angiotensin system [RAS], and aldosterone) and antigeronic factors (e.g., IGF-1 [insulin-like growth factor 1], mediators of caloric restriction, estrogen) derived from the brain, the endocrine system, cells of the immune system, and the adipose tissue orchestrate aging processes simultaneously in the endothelial and smooth muscle cells within the large vessels and microcirculation. The hierarchical regulatory cascade for vascular aging involves modulation of cell-autonomous cellular and molecular aging processes. The resulting functional dysregulation of vascular cells (i.e., impaired vasomotor, barrier, secretory, and transport functions of the vasculature, as well as adverse structural remodeling) promotes the development of a wide range of age-related vascular pathologies. AMI acute myocardial infarction, GDF11 growth differentiation factor 11, PAD peripheral artery disease. (From: Circulation Research. 2018;123:849–867)
2.1 Role of Oxidative and Nitrative Stress
Nitric oxide (NO) bioavailability and its endocrine and paracrine effects are fundamentally important for orchestration of endothelial cell and related vascular function [3]. Additionally, NO exerts potent anti-inflammatory actions, i.e., reduced leukocyte adhesion and antithrombotic effects, and the reduction in NO bioavailability present in the aging vascular phenotype promotes a pro-inflammatory and pro-atherogenic milieu [4]. Increased production of reactive oxygen species (ROS) from a variety of sources, e.g. reduced NADPH (nicotinamide adenine dinucleotide phosphate) oxidases, and mitochondria likely contribute to endothelial dysfunction and resulting large artery stiffening in animal models of advancing age and humans [7]. Increased ROS production has a variety of effects on vascular function through oxidation of critical proteins and induction of redox-sensitive transcription factors; however, one of its most important effects is impairing NO production and activity leading to an age-related imbalance of endothelium-dependent vasorelaxation and vasoconstriction and resulting dysregulation of tissue perfusion [8]. Some important examples of NO-deficiency-related end-organ dysfunction in the aging organism are related to myocardial ischemia and neurovascular uncoupling resulting from impaired coronary artery and cerebral vascular dilatation in response to increases in oxygen and nutrient demand [9, 10]. Furthermore, alteration in the activation of the key enzyme for NO production, endothelial nitric oxide synthase (eNOS), by means of reduced substrate (l-arginine) and co-factor (BH4) activity, as well as increased endothelin-1 (vasoconstricting factor) and overall reduction in eNOS protein expression likely play fundamental roles in the age-related reduction in NO bioavailability [11]. The reaction of NO and superoxide yields the reactive metabolite oxidant peroxynitrite, which is present in aging vascular endothelial cells and is the result of vascular oxidative stress. Peroxinitrite exerts proinflammatory effects via enhancing the redox-sensitive NF-κB (nuclear factor kappa-light-chain-enhancer of activated B cells), which triggers pro-inflammatory cytokine expression. Furthermore, peroxinitrite exerts direct cytotoxic effects and impairs mitochondrial function of aged vascular cells [12, 13]. Another impact of vascular oxidative stress is vascular stiffening, which may be related to the development of vascular aneurysms. Oxidative stress is linked to MMP (matrix metalloproteinases) activation leading to disruption of the structural integrity of aged arteries [14]. The ROS-MMP axis is associated with a variety of age-related changes in the cerebral micro- and macro-circulation, e.g., development of cerebral microhemorrhages leading to cognitive decline [15]. Finally, anti-oxidant interventions decrease vascular stiffness in preclinical models [16].
2.2 Mitochondrial Function
Similar to NO-bioavailability, orchestrated mitochondrial activity plays a fundamental role in normal vascular function and impaired mitochondrial function related to aging leads to diminished respiratory chain function and electron leakage associated with increased ROS-production and resulting reduced energy production, e.g. reduced ATP [17]. Further, mitochondria-derived H2O2 is associated with low-grade vascular inflammation via inducing NF-κB activation [18]. Increases in mitochondrial ROS (mtROS) and the associated impaired electron transport mechanism, can be further exaggerated by peroxynitrite-mediated nitration and inhibition of MnSOD (manganese-dependent superoxide dismutase), reduced cellular glutathione content, and impaired Nrf2 (nuclear factor [erythroid-derived 2]-like 2)-mediated antioxidant defense responses [19]. Additionally, models of hypertension-induced mtROS production in aged vascular smooth muscle cells (VSMCs) increased MMP activation in the vascular wall, resulted in disruption of the structural integrity of aged arteries and led to formation of cerebral microhemorrhages [20]. Targeted mitochondrial antioxidant interventions with the mitochondrial antioxidant MitoQ, resveratrol, and the tetrapeptide SS-31, were demonstrated to attenuate ROS production and improve endothelial function in arteries from rodent models of aging and specifically treatment with SS-31 improved neurovascular coupling and improved cognitive function in these models [21, 22].
Normal aging increases mutations and deletions in mitochondrial DNA (mtDNA) leading to decreased mitochondrial energy production. Furthermore, mitochondrial oxidative stress results in mtDNA damage. MtDNA is subject to an accelerated mutation rate due to a variety of factors, including proximity to sites of ROS production in the mitochondria, a lack of protective histone coverage in the mtDNA, and limited efficiency of mtDNA repair mechanisms [23, 24]. The impact of such changes in aging was explored in a limited body of literature to date. Mitochondrial mutations likely play a causal role in atherogenesis in rodent models of atherosclerosis. For example, apolipoprotein E knock-out mice have an mtDNA polymerase (polG [DNA polymerase subunit gamma]) deficient in proof-reading activity, leading to accumulating mutations in mtDNA and demonstrate dysfunctional proliferation and apoptosis of vascular smooth muscle cells and accelerated atherosclerosis [25].
Vascular mitochondrial function depends on the NAD+ (nicotinamide adenine dinucleotide)-dependent pro-survival enzyme SIRT1 (sirtuin1) for mitochondrial biogenesis and cellular energy metabolism, as well as on controlling mtROS production and sequestration of damaged mitochondria by autophagy. Studies have demonstrated that treatment with the NAD+ intermediate nicotinamide mononucleotide through activation of sirtuin-mediated pathways can improve age-related functional alterations in the rodent aorta and reverse the age-related decline in mitochondrial function. Additional, and potential related mechanisms contributing to impaired bioenergetics in aged vascular cells include oxidation or nitration of mitochondrial proteins, impairment of the different electron transport chain complexes, and impaired mitochondrial autophagy (mitophagy) [26].
2.3 Vascular Inflammation
Age-associated inflammation, or “inflammaging”, is associated with macrovascular and microvascular pathologies, ranging from atherogenesis and aneurysm formation to microvascular dysfunction, blood-brain barrier disruption, and Alzheimer pathologies. The sterile, low-grade inflammation in aging individuals impacts the microenvironment in the vascular wall and promotes vascular dysfunction. Several mechanisms have been proposed including a pro-inflammatory shift in the gene expression profile of vascular endothelial and smooth muscle cells leading to the production of a variety of inflammatory cytokines and mediators [27]. Additional mechanisms contributing to vascular inflammation in aging will be discussed in the following paragraph.
Aged endothelial and smooth muscle cells exhibit significant activation of the inflammatory “gatekeeper” NF-κB [12, 13]. Additional mediators of pro-inflammatory pathways described in aging or senescent endothelium and leading to the production and release of a wide range of inflammatory cytokines and chemokines are p38MAPK, the DNA damage response pathway, and GATA4 (transcription factor GATA-4) [28, 29]. As previously discussed, increases in ROS mediate proinflammatory signaling pathways, including NF-κB activation leading to vascular endothelial activation and expression of proinflammatory paracrine mediators and ultimately promote atherogenesis. Impaired resilience to oxidative stress in aging also exacerbates vascular inflammation induced by cardiovascular risk factors, e.g. hypertension, obesity, and metabolic syndrome [30, 31]. NF-κB inhibition decreases systemic inflammation and extends health span [32]. Additionally, NF-κB-mediated cytokine production is a potent activator of cellular oxidative stress (e.g., TNF-α activates NADPH oxidases). Pharmacological activators of SIRT1 have anti-inflammatory effects in aging rodents [33].
Activation of the innate immune system by TLRs (toll-like receptors) and the NLRP3 (NACHT, LRR, and PYD domains-containing protein 3) inflammasome complex is associated with the sterile inflammation in the vascular wall [34]. Activation of TLR4-mediated, MyD88-dependent signaling pathways in aging VSMCs is associated with the production of several proinflammatory paracrine mediators (e.g., IL-1, IL-6, IL-8, and TNF-α). Furthermore, the canonical Nlrp3 inflammasome contributes to systemic inflammation in aged rodent models [35].
Finally, the interaction between aging and environmental inflammatory factors (e.g., particulate exposure) has been proposed to exacerbate vascular inflammation. For example, bacterial breakdown products entering the circulation through a leaky intestinal barrier or by chronic infection with viruses that exhibit endothelial tropism likely play a role in vascular inflammation. Some viruses, such as the cytomegalovirus, replicate in vascular endothelial cells and long-term exposure and chronic infection are shown to predict increased incidences of frailty and mortality [36].
2.4 Genomic Instability
Experimental evidence indicates that a variety of genetic lesions accumulate within aged cells, including somatic mutations, chromosomal aneuploidies, copy number variations, and telomere shortening. Age-related oxidative stress-induced DNA damage is often proposed as an interconnecting mechanism between the oxidative stress hypothesis of aging and the genomic instability associated with vascular aging [37]. Endothelial cells have impaired DNA repair pathways compared to other cell types and interventions that cause DNA damage (e.g. radiation) result in phenotypic and functional changes in endothelial cells, decrease microvascular density, impair vasodilation, and promote proinflammatory changes, mimicking several aspects of the aging phenotype. Several rodent models have been developed to further investigate the impact of genetic stability changes in aging. For example, one model with defective nucleotide excision repair genes (ERCC1 and XPD) results in an aging-like vascular phenotypes, including endothelial cell dysfunction, increased vascular stiffness and senescence cell count, as well as hypertension [38]. Further, a second model of genetic deficiency in the spindle assembly checkpoint protein BubR1 (budding uninhibited by benzimidazole-related 1), also shows aging-like vascular phenotypes, with a phenotype similar to those outlined above [39]. However, both models also exhibit a short life span associated with severe functional deficits in multiple organ systems and the relevance of this model to normal aging has been questioned. Finally, telomere shortening and associated cellular senescence (see following paragraph below for more details) have been proposed as an important mechanism in vascular aging [40].
2.5 Epigenetic Alterations
Vascular aging is associated with a variety of cellular epigenetic alterations such as DNA methylation changes, posttranslational modification of histones, and dysregulation of microRNAs (miRNAs). Among these, modification in DNA methylation is the central regulator of genomic function and aging is associated with adverse changes in vascular cell methylation patterns [41]. Furthermore, interventions such as caloric restriction can partially reverse alterations in the aging-associated methylation pattern of several organ systems and altered methylation of genes important for vascular function have been observed in a rodent model [42]. Next, post-translational histone modifications are regulated by histone acetyltransferases and histone deacetylases. Changes in expression and reduced activity of class III histone deacetylases, which are the NAD+ utilizing sirtuin family, contribute to vascular endothelial cell aging [43]. Most human protein coding genes are controlled via posttranscriptional repression by noncoding micro RNAs. Vascular cell micro RNAs contribute to the regulation of important biological processes, such as angiogenesis, atherogenesis, aging related impairment of the angiogenic processes, decreased cellular stress resilience, and atherosclerotic plaque formation and destabilization and are all associated with dysregulation of miRNA expression in vascular endothelial and VSMCs [44]. More specifically insulin-like growth factor 1 (IGF-1) deficiency early in life can lead to adverse changes in posttranscriptional miRNA-mediated control of vascular function associated genes and may contribute to the known adverse cardiovascular late-life effects observed in IGF-deficiency [45]. Finally, several of these epigenetic changes are reversible and epigenome-influencing interventions may prove to be successful in limiting, preventing, or even reversing aging related vascular dysfunction [46].
2.6 Vascular Cell Senescence
As part of the aging process, vascular endothelial and smooth muscle cells, similar to other cell types, can permanently withdraw from the cell cycle in response to endogenous and exogenous stressors such as oxidative stress, dysfunctional telomeres, DNA damage, and a variety of paracrine signals, and undergo distinctive phenotypic alterations, including changes to the proinflammatory secretome. This process is called cellular senescence and particular endothelial cell senescence contributes to endothelial dysfunction in aging and pathophysiological conditions associated with accelerated vascular aging [47, 48]. Additionally, studies have demonstrated that elimination of senescent cells can extend the life span in rodents, suggesting that cellular senescence plays a fundamental role in the physiological decline associated with aging [49]. Accelerating this process in a model of irradiation-induced, DNA damage-mediated senescence in a neurovascular rodent model was associated with significant cerebral-microvascular dysfunction, simulating the vascular aging phenotype [50]. Furthermore, genetic and pharmacologic interference to eliminate senescent cells in an LDL receptor knockout mouse model of atherosclerosis resulted in marked changes in atherosclerotic plaque morphology and suggested a role of senescent cells in facilitating plaque instability, e.g., promoting inflammation, and upregulation of MMPs [49]. Different pharmacologic senolytic interventions to clear senescent cells have been shown to improve endothelial cell function and are proposed to exhibit an atheroprotective effect. Finally, vascular senescence-associated secretory pathways can induce paracrine senescence, and alter the function of neighboring endothelial cells and VSMCs, adversely impacting vascular function [51].
2.7 The Renin–Angiotensin–Aldosterone System (RAAS)
Alterations of the RAAS via the angiotensin converting enzyme (ACE) homologue acn-1 in a model organism, genetic or pharmacological interference with the RAAS in a rodent model, and pharmacological ACE inhibition were shown to have a regulatory effect on the life span and may reverse age-related phenotypic and functional changes in the aged vasculature, such as reducing arterial stiffness [52]. Further, human data in elderly subjects suggest that upregulation of tissue RAAS can lead to intimal thickening and remodeling in large conduit arteries associated with an aging vascular phenotype [53]. Additionally, infusion of angiotensin II in young rats accelerates vascular aging, e.g., carotid media thickening and intima infiltration by VSMCs [54]. A variety of other aging-related changes have been observed in experimental models of RAAS upregulation in the vascular wall, e.g. inducing low-grade inflammatory and oxidative stress responses, development of cerebral microhemorrhages, and disruption of the blood-brain barrier [53, 55]. More specifically, local expression of mineralocorticoids and their receptors in the vasculature, e.g., aldosterone, promotes vascular inflammatory changes and leads to adverse vascular remodeling of VSMCs [56]. Overall, the overwhelming data of RAAS involvement in aging emphasizes the importance of the enzyme system and associated peptide hormones in regulating fundamental aging processes.
2.8 Extracellular Matrix (ECM) Remodeling in Vascular Aging
Aging of the vasculature is associated with a variety of changes to the ECM of the subendothelial basement membrane, intima, media, adventitia, and interstitial matrix. Some examples of these changes are decreases in ECM biosynthesis, post-translational modifications of ECM components, and alterations in cell-matrix interactions [57]. ECM integrity is fundamentally important for vascular integrity as it provides mechanical scaffolding as well as mediates the signal transduction required for vascular homeostasis, morphogenesis, and cell differentiation. Aging is associated with changes in the expression of growth factors that regulate ECM biosynthesis and decreased synthesis of several elasticity and resilience-associated components such as elastin, which renders the vasculature more susceptible to wall tension changes related to pulsatile pressure waves and can result in structural vascular damage [58]. Furthermore, collagen synthesis is impacted by aging-associated increased transforming growth factor-β (TGF-β) leading to vascular fibrosis and arterial stiffening [59]. Next, changes in the secretory phenotype of vascular cells, e.g. endothelial and smooth muscle cells, are observed with aging. An increase in MMP secretion and increased MMP activation related to oxidative stress further impair structural integrity and promote pathological remodeling, which can lead to stiffening, hypertension, and aneurysm formation [60]. Several lines of evidence suggest that age-associated activation of the RAAS as well as a decline in IGF-1 are also involved and impact the ECM changes observed in aging [61]. Finally, the described alterations in the biomechanical properties of large arteries associated with age-related ECM remodeling likely also affect microvascular transport mechanism, barrier function, and vein function.
2.9 Progenitor Cell Exhaustion
Exhaustion of the vascular progenitor cell pool and an inability to replenish the pool with functional and differentiated endothelial cells and VSMCs compromise the biological functions and impair neovasculariztion of the aged vasculature. Additionally, age-associated sterile inflammation, oxidative stress, and activation of the RAAS alter the function of circulating EPCs by decreasing differentiation, migration, and survival factors [62, 63]. Animal studies highlighted the importance of circulating factors, e.g., serum from young rats improved function in EPCs previously isolated from old rats [64]. Additionally, studies in ApoE knockout mice demonstrated that bone marrow derived EPCs from young rats slowed atherosclerosis progression whereas EPCs from older rats was ineffective [63]. However, to date, human data are conflicting, and additional data are needed to more clearly define the contribution of changes in EPC number and functionality to the aging vasculature phenotype.
3 Structural Changes Accompanying Age-Associated Increased Stiffness
One of the most important age-associated vascular changes is an increase in central vascular, primarily aortic, stiffness, which alters the pressure and flow characteristics of the wave as it travels through the central, to the peripheral, vessels (Fig. 2) [65]. With the contraction of the left ventricle, ejection of blood from the heart creates a pressure wave that travels along the aorta at a velocity dependent on the stiffness and thickness of the artery. As the pressure wave moves forward, it encounters resistance due to the branching of the arterial tree. This creates a reflected pressure wave that returns at a velocity-dependent, as well, on the stiffness and other properties of the arterial wall. In healthy young individuals, the reflected pressure wave returns to the central aorta during diastole. The expansion of the aorta during systole dampens the pressure at that time and forward stroke volume is reduced by the volume within the expanded aorta. Recoil during diastole increases diastolic pressure and forward stroke volume at that time. The elastic aorta in young individuals thus dampens central pressure during systole, increases it during diastole, and converts the pulsatile flow generated by the heart during systole to a more continuous flow throughout the cycle. However, with advancing age the large elastic arteries stiffen, and therefore expand less, and the left ventricle must generate more systolic pressure for any given stroke volume. In addition, the lower recoil during diastole results in lower central pressure and less forward flow during diastole. The increased “pulsatility” causes microvascular damage, particularly in high flow dependent organs such as the brain, heart, and kidneys.

Compliance, distensibility and PWV. (a) Typical relationship between intra-arterial pressure and lumen area when varying the pressure over a sufficiently large range. The area compliance (red line) is the slope to the pressure-area relationship, which can be calculated at any pressure level. At low pressure, the load is mainly taken by elastin, and the artery has a high compliance. As pressure increases, the load is progressively shifted to stiffer collagen fibers, leading to a functionally lower compliance. Vascular smooth muscle tone also affects compliance, particularly in distal aortic segments and intermediate-sized arteries. Normalizing area compliance to the local radius yields the distensibility coefficient (see Table 1). (b) For a homogenous tube, the distensibility coefficient (Dist coeff) is theoretically linked to the PWV via the Bramwell-Hill equation (where ρ is the density of the blood) in an inverse, non-linear fashion; an increase in PWV by a factor 2 (which is about the change observed in humans from the age of 20 to the age of 70) implies a decrease in distensibility by a factor 4. An alternative formulation is the Moens-Korteweg equation, linking PWV to the stiffness of the wall material (incremental elastic modulus, Einc), the wall thickness (h) and lumen diameter (D). (From: J Am Coll Cardiol. 2019 Sep 3;74(9):1237–1263)
The major components of the aorta are elastin and collagen and the significant changes both undergo with advancing age are responsible for the increase in stiffness [65,66,67,68]. There is a decrease and fraying of elastin that results from increased activity of matrix metallo proteinases and other enzymes regulated, in part, by pro-inflammatory cytokines [68]. Increased activity of the RAAS also increases elastin loss and decreases elastin synthesis. Accumulation of collagen, also stimulated by the RAAS, is accompanied by cross-links between collagen molecules and condensates of glucose produced by nonenzymatic reactions. These advanced glycation end-products form cross-links between collagen molecules that increase stiffness and are resistant to enzymatic degradation [69]. Increased stiffness decreases stretch-induced increases in nitric oxide (NO) bioavailability and by interacting with receptors on immune and other cells increases oxidant stress and upregulates cytokines, stimulating vascular inflammation. There is, as well, proliferation and migration of vascular smooth muscle cells to the intima and osteogenic transdifferentiation of the cells, which increases the mineralization of the extracellular matrix [70, 71]. These changes are not passive, but rather dynamic processes triggered by cellular senescence, autophagy, and mediated, at least in part, by oxidative stress, inflammation, RAAS, and angiotensin II signaling [72]. Furthermore, the consequent higher systolic and pulse pressures due to stiffening may stimulate the production of these factors, resulting in further stiffening and a repeating cycle.
3.1 Measures of Aortic Stiffness
Aortic stiffness is best defined by the change in intra-aortic distending pressure relative to the change in volume. The invasive nature of this assessment, however, precludes its use in large studies of healthy individuals. More easily assessed indices include brachial pulse pressure, the pulse pressure/stroke volume ratio, and carotid and radial tonometry-derived central aortic waveforms. The latter can be used to assess the augmentation index, defined by the increased pressure accompanying the reflected wave divided by the pulse pressure, with higher values indicating increased stiffness. One of the first, and remarkably prescient, studies were reported by Bramwell and Hill in The Lancet 100 years ago [73]. They studied healthy individuals and measured pulse wave velocity using the time between the arrival of the pulse in the carotid and radial arteries and the estimated distance the pulse traveled through the aorta by subtracting the distance between the sternoclavicular joint and the carotid artery from the distance between the sternoclavicular joint and the radial artery. A similar non-invasive technique is still used with the exception that the femoral, rather than the radial, pulse is recorded, and the distance is obtained by subtracting the distance between the carotid measurement site and the sternal notch from the distance between the sternal notch and the femoral measurement site. Reference values for pulse wave velocity in 11,092 individuals without known cardiovascular disease and diabetes and who were not receiving anti-hypertensive or lipid lowering therapies are presented in Table 1 [74
]. Optimal blood pressure was defined as <120/80, normal as ≥120/80 and <130/85; high normal as ≥130/85 and <140/90, grade 1 hypertension as ≥140/90 and <160/100, and Grade II/III hypertension as ≥160/100 mmHg.
3.2 Consequences of Increased Stiffness
3.2.1 Cardiovascular Performance
The relationship between central vascular stiffness and left ventricular contractility is termed arterial-ventricular coupling. It can be quantified using the ratio of effective arterial elastance (Ea) to end-systolic left ventricular elastance (Elv) [75, 76] and estimated non-invasively. A ratio close to 0.5 is associated with optimal cardiovascular efficiency [77, 78]. The Baltimore Longitudinal Study of Aging reported longitudinal changes in left ventricular volumes as assessed by repeated multigated blood pool scans and blood pressures over an average follow-up period of 12.2 years in 129 individuals without evidence of coronary or other cardiac disease [79]. The projected trajectories of Ea, Elv, and the ratio Ea/Elv in different age groups are presented in Fig. 3a, b. Ea increases with age and the rate of increase increases with advancing age. Elv also increases with aging; however, the increase in Elv is less than that of Ea, which results in an age-associated increase in Ea/Elv indicating progressive AV uncoupling, due primarily to an increase in vascular stiffness. This was associated with left ventricular remodeling characterized by increased end-systolic volume and reduced stroke and end-diastolic volumes, a pattern consistent with that present in individuals with heart failure with a preserved ejection fraction.

(a) Age-group trajectories generated from averaged individual, model-predicted longitudinal trajectories in Ea, Elv, and Ea/Elv, projected over 20 years follow-up. (b) Age-group average rates of changes. (From: Geroscience. 2021 Apr;43(2):551–561)
In addition to these left ventricular structural changes, increased central vascular stiffness impacts left ventricular performance, primarily by increasing afterload during left ventricular ejection as the pressure required to eject a given volume increases with a progressively stiffer aorta. Due to the increase in pulse wave velocity associated with the stiffer aorta, the reflected wave returns earlier, in systole rather than diastole, further increasing systolic pressure and left ventricular afterload. In addition, the lower diastolic pressure due to both lower “recoil” during diastole, and the absence of the reflected wave at that time, decreases coronary perfusion pressure, which, when combined with the increased myocardial oxygen demand due to the higher afterload, may impact left ventricular performance in the setting of obstructive disease.
3.2.2 Clinical Outcomes
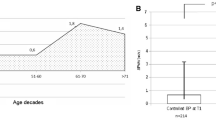
In clinical outcome studies, central vascular stiffness is usually assessed as pulse pressure, pulse wave velocity, or by using radial or carotid tonometry. Although these measures do not precisely define relative changes in aortic volume with aortic pressure, they are non-invasive, safe, predictive of outcomes, and, in many clinical centers, readily available. The Framingham investigators reported the association between stiffness as indexed by carotid-femoral pulse wave velocity on cardiovascular and other events in 7283 Framingham Study participants over a median follow-up period of 15 years [80]. In adjusted analysis, each standard deviation increase in the carotid-femoral pulse wave velocity was associated with an increased risk of hypertension (HR 1.32), diabetes (HR 1.32), chronic kidney disease (HR 1.19), dementia (HR 1.27) coronary heart disease (HR 1.37), transient ischemic attack or stroke (HR 1.24), and death (HR 1.29) (see Fig. 4). In a pooled analysis from 16 studies examining the impact of aortic pulse wave velocity on cardiac outcomes in 17,635 participants [81], for every one standard deviation increase in loge aortic pulse wave velocity, there was a significant 54% increased 5 year stroke risk for those 61–70 years of age and a 37% increase for those over 70 years of age. Similarly, there was a 31% increase in coronary heart disease events for those 61–70 years and a 14% increase for those over 70 years of age. In a community of adults over 70 years of age, the Health ABC Study investigators reported that over a 4.6 year follow-up aortic pulse wave velocity quartiles were significantly associated with cardiovascular mortality, with a relative risk 2.1, 3.0, and 2.3 for quartiles 2, 3, and 4 versus the first quartile 1 [82].

Relations of carotid-femoral pulse wave velocity (CFPWV) and incidence of outcome events dichotomized by median age (50 years for analyses combining FOS [Framingham Offspring Study] and Gen 3 [Third Generation; incidence of hypertension, diabetes, chronic kidney disease (CKD)b, cardiovascular disease (CVD), CVD subtypes, and death]; 60 years for analyses limited to FOS [incidence of CKDa and dementia]). Hazards ratios are per SD increment in CFPWV from age-stratified models adjusting for model 3 covariates (sex, current smoking status, total cholesterol concentration in blood/HDL (high-density lipoprotein) cholesterol, diabetes [or fasting blood glucose, for incident diabetes], systolic blood pressure, and antihypertensive treatment). Interaction p values are from models pooling age groups. CKDa and CKDb refer to definitions of incident disease based on reduced estimated glomerular filtration rate (eGFR) and increased urine albumin to creatinine ratio vs reduced eGFR alone, respectively. CHD coronary heart disease, TIA transient ischemic attack. (From: Hypertension. 2022;79:1045–1056)
Hypertension is the most common risk factor in the older population and is strongly related to stroke and heart failure risk (see Chap. 1). In the Atherosclerosis Risk in Communities study, pulse wave velocity was an independent predictor of incident hypertension [83]. Arterial stiffness was assessed by ultrasound detected change in carotid arterial diameter, adjusted for the diastolic and pulse pressures. After adjustment for traditional risk factors, there was a significant increase in the incidence of hypertension over a mean follow-up of 3 years with rates of 9.6% in the least elastic and 6.7% in the most elastic quartiles. For every one standard deviation decrease in elasticity there was a 15% greater risk of hypertension. Najjar et al. reported the impact of pulse wave velocity measured using carotid and femoral tonometry in 306 Baltimore Longitudinal Study of Aging participants who were normotensive at baseline on the development of hypertension over 4 years [84]. Thirty-four percent developed hypertension and for every 1 m/s increase in PWV, the hazard of developing hypertension was 1.10 (CI 1.00–1.30).
Age also impacts the association between different blood pressure indices and coronary heart disease risk. The Framingham Heart Study compared the importance of systolic, diastolic, and pulse pressure blood pressures to the risk for coronary artery disease over a mean follow-up period of 17 years (Table 2) [85]. Diastolic pressure was the strongest predictor for those under 50 years of age and the components were equally predictive for those 50–59 years. However, for those 60 years of age and older, while systolic blood pressure remained significant (hazard ratio 1.17 (1.11–1.24, p < 0.001)), the strongest predictor was pulse pressure, with a hazard ratio of 1.24 (1.16–1.33, p < 0.001) for every 10 mmHg increase after adjustment for diabetes and other cardiovascular risk factors.
Stiffness, as indexed by pulse pressure, is also related to the development of congestive heart failure (CHF). In a study of 1621 participants 65 years or older, with a mean age of 77.9 years, there was a 14% increase in the risk of CHF for every 10-mmHg increase in pulse pressure over a mean follow-up period of 3.8 years and for those in the highest quartile, the risk was 55% higher than for those in the lowest quartile [86]. In Framingham Heart Study participants 50–79 years of age, there was a 55% increased risk for the development of heart failure over a 17.4 year mean follow-up period for every 16 mmHg increase in pulse pressure [87]. Increased central vascular stiffness is also believed to play an important pathophysiologic role in patients with heart failure and a preserved ejection fraction, a common form of heart failure in older patients [88]. Abnormal central vascular hemodynamics are particularly evident during exercise [89]. In a study comparing measures of cardiac function and arterial parameters, at rest and during exercise in 98 patients with heart failure and a preserved ejection fraction (mean age 68 years) and 22 control patients with hypertension but without heart failure (mean age 62 years), parameters of central stiffness did not differ at rest. At a common 20-W supine cycle exercise workload and adjusted for age and body mass index, stiffness assessed by end-systolic central blood pressure divided by the stroke volume index was significantly higher in the heart failure with preserved ejection fraction group (3.26 ± 0.92) than in the control group (2.64 ± 0.71, p < 0.004). The higher arterial stiffness correlated with higher pulmonary capillary wedge pressures and lower cardiac outputs during the exercise period. Peak exercise capacity was about 40% lower, and stiffness about 30% higher in the heart failure with preserved ejection fraction group. Thus, there is a significant association between central vascular stiffness and cardiac function indices of exercise performance in patients with heart failure and a preserved ejection fraction, which is independent of hypertension.
Measures of vascular stiffness are also associated with incident atrial fibrillation, cognitive decline, and renal failure. In Framingham Heart Study offspring and third-generation cohorts all older than 45 years and with a mean age of 61 years, each standard deviation of augmentation index and of central pulse pressure assessed with tonometry wave forms, adjusted for age, sex, and hypertension, was associated with atrial fibrillation hazard ratios of 1.16 (95% CI 1.02–1.32, p = 0.02) and 1.14 (95% CI 1.02–1.28 p = 0.02) for augmentation index and central pulse pressure respectively [90]. Cognitive changes and central vascular stiffness were assessed in 1101 Framingham Offspring participants, all of whom were 60 years of age or older, over a 10-year follow-up period. Stiffness, defined by the carotid-femoral pulse wave velocity was significantly associated with an increased risk of mild cognitive impairment (HR 1.40, CI 1.13–1.73), all cause dementia (HR 1.45, CI 1.13–1.87) and Alzheimer’s Disease (1.41, CI 1.06–1.67) [91]. In a 356 participant subset of the Cardiovascular Health Study, with a mean age of 77.8 years, for whom carotid femoral pulse wave velocity was measured and incident dementia assessed over a 15-year follow-up period, the risk of incident dementia for those in the highest quartile was 57% higher than for those in the lowest quartile, adjusted for conventional risk factors and anti-hypertensive medications [92]. Longitudinal changes in brain MRI scans over a mean 4.9-year follow-up period were analyzed in 278 individuals with normal or mild cognitive impairments from the Vanderbilt Memory and Aging Project. Increased pulse wave velocity was associated with an increase in white matter intensity, likely related to ischemia, and a decrease in gray matter [93]. Measures of arterial stiffness are also associated with decline in kidney function and renal failure in studies of older individuals [94, 95]. The MESA study examined the association of vascular stiffness indexed by pulse pressure and radial artery tonometry and changes in renal function in 4853 persons, all of whom had an initial estimated GFR of >60 mL/min/1.73 m2 [95]. Higher pulse pressure and vascular stiffness were associated with faster rates of kidney decline. Compared to those with a pulse pressure of 40–50 mmHg the decline in eGFR was 0.29, 0.56 and 0.91 mL/min/1.73 m2/year faster among those with pulse pressures of 50–60, 60–70, and >71 mmHg respectively, all p < 0.01.
3.3 Intervention Strategies
The above studies indicating an association between increased central vascular stiffness and adverse cardiovascular outcomes prompted studies of non-pharmacologic and pharmacologic interventions designed to decrease stiffness. Although the underlying mechanisms cannot be directly assessed, the consequences in terms of stiffness indices can be. The primary non-pharmacologic approaches are changes in dietary pattern and exercise. The benefits of the Dietary Approaches to Stop Hypertension (DASH) and Mediterranean diets on cardiovascular health are well established [96, 97]. The principal components of many of these diets include increased cocoa, coffee, extra virgin olive oil, fermented foods, fiber, fish, fruits, nuts, vegetables, and whole grains along with decreased intake of high glycemic carbohydrates, processed foods, and sugar, and are associated with deceased vascular stiffness [98, 99]. The responsible mechanisms are multifactorial and include impacts on secreted cytokines, function of immune cells, adipose tissue, skeletal muscle, pancreas, and liver. These are believed to be associated with activation of SIRT-1 and AMPK pathways, depression of mTOR, protein catabolism, fatty acid oxidation, autophagy, mitochondrial homeostasis, and protection against oxidative stress and inflammation. In pre-clinical models, caloric restriction is associated with decreased central vascular stiffness, [100] thought to be related to activation of anti-oxidant/and anti-inflammatory pathway systems and of endogenous cellular stress resistance pathways, SIRT-1, AMPK, and mTOR [100]. In addition to the types of foods, the intake of certain elements is also associated with vascular function. Thus, calcium, potassium, and magnesium may be beneficial, while sodium intake is associated with injury to the vessel wall [99]. Gates et al. conducted a double-blind cross-over study demonstrating that decreasing sodium chloride intake improved large artery elastic compliance in older adults [101]. They assessed the impact of two weeks of decreased sodium intake, to about 60 mmol/day, consistent with the DASH diet, on carotid artery compliance in 12 individuals with stage 1 hypertension. In addition to a significant decrease in systolic pressure, carotid artery compliance increased by 46%, % augmentation index decreased from 40 ± 2 to 29 ± 2 (p < 0.05) and carotid artery pulse pressure decreased from 50 ± 6 to 40 ± 6 (p < 0.05). Potential mechanisms for sodium’s detrimental effects include direct injury to the vessel wall, vascular smooth muscle cell hypertrophy, and increased reactive oxygen species, inflammation and resultant fibrosis.
Weight loss reduces arterial stiffness in obese and overweight adults [102, 103]. Following a 12-week 1200–1500 kcal/day diet, 25 individuals with an average body mass index of 30.0 ± 0.6 kg/m2, lost 7.1 ± 0.7 kg. Carotid-femoral pulse wave velocity decreased by 187 ± 29 cm/s and the decrease was correlated with reduced total body and abdominal obesity [103]. The long-term success of caloric restriction, per se, is low because of difficult adherence. In addition, there are concerns regarding caloric restriction including loss of skeletal muscle mass, decreased bone mineral density, and decreased intake of important nutrients. Potential alternatives include caloric restriction for a few days each week and limiting feeding to a limited number of hours (e.g. 8 h) each day.
Physical activity decreases central vascular stiffness and age-related increases in stiffness can be mitigated by exercise in both men and women [104, 105]. The Baltimore Longitudinal Study of Aging measured VO2 max in rigorously phenotyped adults aged 21–96 years of age [106]. With increasing age, in the entire cohort, augmentation index and aortic PWV increased out of proportion to the blood pressure increase. However, these measures of aortic stiffness were lower in endurance-trained male athletes (defined by a VO2 max 1 standard deviation above their age-matched non-trained controls), compared with sedentary individuals (defined as less than 20 min of aerobic exercise three times weekly) of similar age. Similarly, while augmentation index and pulse wave velocity were higher in sedentary post-menopausal women than comparable pre-menopausal women, these measures were similar in physically active pre- and post-menopausal active women defined by performing endurance training, or actively competing in running races, an average of 6 ± 1 h of activity per week [106].
3.3.1 Drug Effects on Vascular Stiffness
Some pharmacologic agents also decrease measures of vascular stiffness. The majority of the studies are with anti-hypertensive agents. Those which decrease stroke volume will decrease expansion required of the rigid aortic elements during ejection and therefore pressure. Thus, pulse wave velocity may change but there is no change in the pressure/volume relationship per se. Vascular smooth muscle vasodilation induced by ACE-inhibitors and angiotensin receptor II receptor blockers decrease stiffness measures. The benefit of these may extend beyond an impact related to an acute effect on blood pressure due to a decrease in vascular smooth muscle cell proliferation and vascular calcification. The impact of drugs on pressure and stiffness may also differ. Thus, lisinopril and metoprolol reduced blood pressure similarly but the angiotensin converting enzyme inhibitor improved compliance while the beta blocker did not [107]. In general, and in the few studies which have been performed, ACE-inhibitors, angiotensin II receptor blockers, and calcium antagonists improve compliance relative to beta blockers (with the possible exception of those with vasodilating properties), despite similar blood pressure reductions [108]. It should be emphasized, however, that no randomized study has determined whether decreasing compliance independent of blood pressure decreases adverse cardiovascular events.
3.3.2 The Impact of Urban Environments on Age-Associated Vascular Properties
The importance of the environment to which our vasculature is exposed in mediating the age associated vascular changes is also illustrated in studies comparing these properties in rural and urban dwellers. When compared with urban societies, those in isolated rural communities that have been slow to assimilate into urban societies have very low rates of prevalent hypertension and lower age-associated increases in pressure with increasing age.
The prevalence of hypertension and longitudinal changes in blood pressure were studied in 2248 adults aged 20–90 years among the Tsimane, forager-horticulturalists in the Bolivian Amazon [109]. In the entire group the prevalence of hypertension was only 3.9% in women and 5.2% in men. The increases in systolic, diastolic, and pulse pressure per decade, respectively, were 2.86, 0.95, and 1.95 mmHg for women and 0.91, 0.93, and −0.02 mmHg for men, all of which are significantly lower than the same profiles in the United States [110]. In a study comparing pulse wave velocity (PWV), matched for age and blood pressure, PWV in the arm was significantly lower in rural Guangzhou (PWV = 0.61 (age) + 817) than in Beijing (PWV = 4.8 (age) + 998) [111]. One of the most salient studies examining this question was the Yi migrant study, which examined blood pressures in 8241 Yi farmers, 2675 Yi who had migrated from the rural to an urban environment, and 3689 urban residents [112]. The prevalence of borderline or definite hypertension in the farmer population was only 2.69% in men and 2.42% in women 65 years of age or older, and rose to 30.77% in men and 55.55% in women migrants and to 57.57% in men and 70.0% women urban dwellers. For men 65 years of age or older, the mean systolic pressures expressed as mean ± SD were 110.8 ± 12.3 in the farmers, 130.4 ± 14.0 in the migrants, and 136.9 ± 23.6 mmHg in the urban dwellers. For women, the respective numbers were 111.2 ± 12.6, 135.0 ± 17.2 and 146.5 ± 25.6 mmHg. Furthermore, the change in systolic pressure with age, expressed as the mean ± SE mmHg/year was also significantly lower in the farmers (0.13 ± 0.01) than in the migrants (0.33 ± 0.03) and the urban dwellers (0.36 ± 0.02). Migration was associated with decreased physical activity and increased intake of animal products, sugar, salt, fat, and total calories. Furthermore, adjusting for BMI reduced much of the hypertension risk. Although these studies suggest that a significant component of the age-associated increase in measures of central vascular stiffness can be mitigated by lifestyle factors, it is not practical for most currently living in an urban/suburban environment to reproduce the lifestyle practiced in these remote rural areas.
4 Vascular Endothelial Cell Function Testing
The previous sections describe in detail the molecular and cellular mechanisms of vascular aging with an emphasis on vascular endothelial cell function as well as the structural changes accompanying age associated increased stiffness and interventions to improve vascular stiffness. The following paragraphs focus on vascular function testing and the clinical implications of age associated impairments in vascular endothelial cell function.
Endothelial dysfunction is associated with a plethora of cardiovascular diseases and can be observed in larger conduit arteries (macrovascular dysfunction) as well as smaller resistance vessels (microvascular dysfunction). Furthermore, the presence of endothelial dysfunction is a clinical prognosticator for cardiovascular events and associated mortality. Vascular endothelial function testing is a useful tool in selecting therapies and assessing the response to interventions. For this purpose, several invasive as well as non-invasive modalities have emerged for clinical and research purposes and strengths and limitations of these techniques will be discussed in the following sections.
4.1 Invasive Coronary Endothelial Function Assessment
Coronary endothelial dysfunction is a reliable predictor of cardiovascular events and associated mortality and an important vascular bed in studies of vasoreactivity [113]. Endothelial dysfunction diagnosed by invasive methods is prevalent in a variety of cardiometabolic diseases and is associated with future atherosclerotic cardiovascular events [114, 115]. Furthermore, responses to risk factor modifying therapies targeting endothelial cell dysfunction improve vascular function [116].
The gold standard for coronary endothelial functional assessment uses invasive quantitative angiography [117]. Blood flow velocity, measured using a Doppler wire, and epicardial diameter are used to calculate coronary blood flow [118]. To assess endothelial cell-dependent function an endothelial-dependent vasodilator, e.g. acetylcholine, is infused and the response to the acetylcholine-induced increased production of nitric oxide is recorded [119]. The normal, functional, coronary vasculature response to low dose acetylcholine infusion is coronary arterial vasodilation and increased blood flow (by >50%). In the setting of vascular endothelial cell dysfunction, the increases in coronary flow and dimension are less and, at times, even paradoxical vasoconstriction occurs [120]. Other, less commonly used agents for invasive endothelial-dependent coronary vascular function testing are bradykinin, papaverine, Substance P and adenosine [121, 122]. Non-pharmacologic endothelial-dependent stressors to assess coronary vascular function include cold pressor testing [123] and exercise testing using an ergometer [124].
Advantages of the invasive, catheter-based, coronary endothelial assessment include the precision and accuracy of direct rather than surrogate measures of coronary arterial function assessment. These are, however, balanced by the potential risks of vascular access, coronary vessel engagement with the catheter, exposure to radiation and contrast agents, and potential systemic, hemodynamic effects of the pharmacologic intervention. These concerns limit repeated assessments of invasive coronary endothelial function testing outside of clinically indicated procedures.
4.2 Non-invasive Coronary Artery and Peripheral Arterial Endothelial Function Assessment
There are a variety of non-invasive, imaging, modalities to assess endothelial-dependent coronary- and peripheral vascular function including magnetic resonance imaging (MRI) [125], positron emission tomography (PET)/computed tomography (CT) [126, 127], computed tomography angiography (CTA) [128] and ultrasound to assess brachial artery flow - mediated dilatation (FMD) [129]. The following section will focus on the two most used techniques at our institution: MRI for coronary artery endothelial function testing and FMD for peripheral vascular function testing.
4.2.1 Non-invasive Evaluation of Epicardial Coronary Endothelial Function and Microvascular Function Testing: MRI
MRI in combination with isometric handgrip exercise (IHE), a known endothelial-dependent stressor, is a non-invasive, and reproducible method to assess coronary endothelial function (CEF). [3, 125] CEF reflects coronary endothelial NO release and, as such, can be used to test pathophysiologic contributors to cardiovascular diseases [130]. The normal response to IHE is an increase in NO bioavailability with subsequent coronary vascular dilation and increases in coronary flow velocity and flow. Furthermore, our studies showed impaired CEF in patients with CAD [131] and separately in people living with HIV compared to otherwise risk-factor-matched control participants [125]. The coronary artery area and flow responses to stressors as assessed by MRI have been validated when compared to invasive measures using quantitative coronary angiography with Doppler techniques [132]. This MRI technique therefore provides an opportunity to monitor the impact of interventions to improve CEF and therefore decrease the development and progression of atherosclerosis during the early stages of coronary disease [133]. MRI techniques can also be used to interrogate microvascular function. Stress perfusion cardiovascular magnetic resonance (CMR), distinct from the coronary vasoreactivity approaches outlined above, typically use vasodilator stress (e.g., adenosine) to detect changes in myocardial blood flow in response to stress. Several studies support the use of stress perfusion CMR to investigate myocardial perfusion reserve, which reflects microvascular function in the absence of significant CAD [134, 135]. Like MRI for CEF testing, the prognostic value of stress CMR was similar to that of invasive strategies [136].
4.2.2 Peripheral Endothelial Function Assessment: Brachial Artery Flow Mediated Dilatation and Peripheral Artery Tonometry
Flow-mediated vasodilatation (FMD) of the brachial artery measures the response of a focal segment of the vessel to endothelial-dependent NO release induced by a hyperemic stimulus, usually at the time of increased blood flow following a 5 min blood pressure cuff occlusion of flow. FMD is defined as the change in arterial diameter post-stimulus compared to the baseline diameter, measured manually or with edge-detection software [129, 137]. Furthermore, baseline and hyperemic (maximal) blood flows are calculated from the onset of one arterial waveform to the beginning of the next waveform using the time-averaged pulsed Doppler spectral trace time-velocity integral. FMD assessed vascular endothelial cell function is a validated method to detect endothelial dysfunction, which predicts future cardiovascular events [138].
Peripheral artery tonometry (PAT) is also used to assess endothelial function. PAT uses a pneumatic finger probe to measure the arterial pulse wave amplitude at baseline and with reactive hyperemia. There is a poor correlation between the FMD and PAT techniques [139], and it was proposed that PAT is a more accurate assessment of micro- rather than macrovascular, function [140].
4.3 Endothelial Function in the Coronary and Peripheral Vasculature
As outlined in the prior sections, endothelial function assessed by invasive and non-invasive modalities is an important predictor of vascular health and cardiovascular outcomes. However, some studies show only a modest association between systemic and coronary endothelial function assessments [141]. Furthermore, there may be significant variability of the endothelial cell function measures within a vascular territory or even within the coronary vasculature of the same person [131]. A variety of mechanisms may explain these differences, including differences in local shear stress, the resistance of downstream vessels, and neurohormonal regulation. Finally, endothelial function assessment in the coronary and systemic vasculatures, with individual strengths and limitations, may provide complementary insights into vascular endothelial cell function [142].
References
Martin CF. Osler as clinician and teacher. Can Med Assoc J. 1920;10(Spec Issue):82–6.
Gimbrone MA Jr, Garcia-Cardena G. Endothelial cell dysfunction and the pathobiology of atherosclerosis. Circ Res. 2016;118(4):620–36.
Hays AG, et al. Coronary vasomotor responses to isometric handgrip exercise are primarily mediated by nitric oxide: a noninvasive MRI test of coronary endothelial function. Am J Physiol Heart Circ Physiol. 2015;308(11):H1343–50.
Jones SP, Bolli R. The ubiquitous role of nitric oxide in cardioprotection. J Mol Cell Cardiol. 2006;40(1):16–23.
Virani SS, et al. Heart disease and stroke statistics-2020 update: a report from the American Heart Association. Circulation. 2020;141(9):e139–596.
Vespa J, Medina L, Armstrong DM. Demographic turning points for the United States: population projections for 2020 to 2060. Current Population Reports. Washington, DC: Census Bureau; 2020. p. P25–1144.
Rizvi F, et al. Effects of aging on cardiac oxidative stress and transcriptional changes in pathways of reactive oxygen species generation and clearance. J Am Heart Assoc. 2021;10(16):e019948.
Finkel T, Holbrook NJ. Oxidants, oxidative stress and the biology of ageing. Nature. 2000;408(6809):239–47.
Csiszar A, et al. Aging-induced phenotypic changes and oxidative stress impair coronary arteriolar function. Circ Res. 2002;90(11):1159–66.
Toth P, et al. Resveratrol treatment rescues neurovascular coupling in aged mice: role of improved cerebromicrovascular endothelial function and downregulation of NADPH oxidase. Am J Physiol Heart Circ Physiol. 2014;306(3):H299–308.
Yang YM, et al. eNOS uncoupling and endothelial dysfunction in aged vessels. Am J Physiol Heart Circ Physiol. 2009;297(5):H1829–36.
Donato AJ, et al. Direct evidence of endothelial oxidative stress with aging in humans: relation to impaired endothelium-dependent dilation and upregulation of nuclear factor-kappaB. Circ Res. 2007;100(11):1659–66.
Csiszar A, et al. Inflammation and endothelial dysfunction during aging: role of NF-kappaB. J Appl Physiol (1985). 2008;105(4):1333–41.
Wang M, et al. Matrix metalloproteinases promote arterial remodeling in aging, hypertension, and atherosclerosis. Hypertension. 2015;65(4):698–703.
Ungvari Z, et al. Cerebral microhemorrhages: mechanisms, consequences, and prevention. Am J Physiol Heart Circ Physiol. 2017;312(6):H1128–43.
Fleenor BS, et al. Superoxide signaling in perivascular adipose tissue promotes age-related artery stiffness. Aging Cell. 2014;13(3):576–8.
Ungvari Z, Sonntag WE, Csiszar A. Mitochondria and aging in the vascular system. J Mol Med (Berl). 2010;88(10):1021–7.
Ungvari Z, et al. Increased mitochondrial H2O2 production promotes endothelial NF-kappaB activation in aged rat arteries. Am J Physiol Heart Circ Physiol. 2007;293(1):H37–47.
Ungvari Z, et al. Age-associated vascular oxidative stress, Nrf2 dysfunction, and NF-{kappa}B activation in the nonhuman primate Macaca mulatta. J Gerontol A Biol Sci Med Sci. 2011;66(8):866–75.
Springo Z, et al. Aging exacerbates pressure-induced mitochondrial oxidative stress in mouse cerebral arteries. J Gerontol A Biol Sci Med Sci. 2015;70(11):1355–9.
Gioscia-Ryan RA, et al. Mitochondria-targeted antioxidant (MitoQ) ameliorates age-related arterial endothelial dysfunction in mice. J Physiol. 2014;592(12):2549–61.
Pearson KJ, et al. Resveratrol delays age-related deterioration and mimics transcriptional aspects of dietary restriction without extending life span. Cell Metab. 2008;8(2):157–68.
Nissanka N, Moraes CT. Mitochondrial DNA damage and reactive oxygen species in neurodegenerative disease. FEBS Lett. 2018;592(5):728–42.
Bratic A, Larsson NG. The role of mitochondria in aging. J Clin Invest. 2013;123(3):951–7.
Yu E, et al. Mitochondrial DNA damage can promote atherosclerosis independently of reactive oxygen species through effects on smooth muscle cells and monocytes and correlates with higher-risk plaques in humans. Circulation. 2013;128(7):702–12.
Sack MN, Finkel T. Mitochondrial metabolism, sirtuins, and aging. Cold Spring Harb Perspect Biol. 2012;4(12):a013102.
Wang M, et al. Proinflammation: the key to arterial aging. Trends Endocrinol Metab. 2014;25(2):72–9.
Kang C, et al. The DNA damage response induces inflammation and senescence by inhibiting autophagy of GATA4. Science. 2015;349(6255):aaa5612.
Freund A, Patil CK, Campisi J. p38MAPK is a novel DNA damage response-independent regulator of the senescence-associated secretory phenotype. EMBO J. 2011;30(8):1536–48.
Bailey-Downs LC, et al. Aging exacerbates obesity-induced oxidative stress and inflammation in perivascular adipose tissue in mice: a paracrine mechanism contributing to vascular redox dysregulation and inflammation. J Gerontol A Biol Sci Med Sci. 2013;68(7):780–92.
Tucsek Z, et al. Aging exacerbates obesity-induced cerebromicrovascular rarefaction, neurovascular uncoupling, and cognitive decline in mice. J Gerontol A Biol Sci Med Sci. 2014;69(11):1339–52.
Hasegawa Y, et al. Blockade of the nuclear factor-kappaB pathway in the endothelium prevents insulin resistance and prolongs life spans. Circulation. 2012;125(9):1122–33.
Gano LB, et al. The SIRT1 activator SRT1720 reverses vascular endothelial dysfunction, excessive superoxide production, and inflammation with aging in mice. Am J Physiol Heart Circ Physiol. 2014;307(12):H1754–63.
Karasawa T, Takahashi M. Role of NLRP3 inflammasomes in atherosclerosis. J Atheroscler Thromb. 2017;24(5):443–51.
Song Y, et al. Aging enhances the basal production of IL-6 and CCL2 in vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 2012;32(1):103–9.
Wang GC, et al. Cytomegalovirus infection and the risk of mortality and frailty in older women: a prospective observational cohort study. Am J Epidemiol. 2010;171(10):1144–52.
Shah AV, Bennett MR. DNA damage-dependent mechanisms of ageing and disease in the macro- and microvasculature. Eur J Pharmacol. 2017;816:116–28.
Durik M, et al. Nucleotide excision DNA repair is associated with age-related vascular dysfunction. Circulation. 2012;126(4):468–78.
Matsumoto T, et al. Aging-associated vascular phenotype in mutant mice with low levels of BubR1. Stroke. 2007;38(3):1050–6.
Liu J, et al. Roles of telomere biology in cell senescence, replicative and chronological ageing. Cells. 2019;8(1):54.
Zhang W, et al. Epigenetic modifications in cardiovascular aging and diseases. Circ Res. 2018;123(7):773–86.
Gensous N, et al. The impact of caloric restriction on the epigenetic signatures of aging. Int J Mol Sci. 2019;20(8):2022.
Kida Y, Goligorsky MS. Sirtuins, cell senescence, and vascular aging. Can J Cardiol. 2016;32(5):634–41.
de Lucia C, et al. microRNA in cardiovascular aging and age-related cardiovascular diseases. Front Med (Lausanne). 2017;4:74.
Tarantini S, et al. IGF-1 deficiency in a critical period early in life influences the vascular aging phenotype in mice by altering miRNA-mediated post-transcriptional gene regulation: implications for the developmental origins of health and disease hypothesis. Age (Dordr). 2016;38(4):239–58.
Lopez-Otin C, et al. The hallmarks of aging. Cell. 2013;153(6):1194–217.
Campisi J, d’Adda di Fagagna F. Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol. 2007;8(9):729–40.
Erusalimsky JD. Vascular endothelial senescence: from mechanisms to pathophysiology. J Appl Physiol (1985). 2009;106(1):326–32.
Childs BG, et al. Senescent intimal foam cells are deleterious at all stages of atherosclerosis. Science. 2016;354(6311):472–7.
Ungvari Z, et al. Ionizing radiation promotes the acquisition of a senescence-associated secretory phenotype and impairs angiogenic capacity in cerebromicrovascular endothelial cells: role of increased DNA damage and decreased DNA repair capacity in microvascular radiosensitivity. J Gerontol A Biol Sci Med Sci. 2013;68(12):1443–57.
Roos CM, et al. Chronic senolytic treatment alleviates established vasomotor dysfunction in aged or atherosclerotic mice. Aging Cell. 2016;15(5):973–7.
Kumar S, Dietrich N, Kornfeld K. Angiotensin converting enzyme (ACE) inhibitor extends Caenorhabditis elegans life span. PLoS Genet. 2016;12(2):e1005866.
Wang M, et al. Proinflammatory profile within the grossly normal aged human aortic wall. Hypertension. 2007;50(1):219–27.
Wang M, et al. Angiotensin II activates matrix metalloproteinase type II and mimics age-associated carotid arterial remodeling in young rats. Am J Pathol. 2005;167(5):1429–42.
Wang M, et al. Aging increases aortic MMP-2 activity and angiotensin II in nonhuman primates. Hypertension. 2003;41(6):1308–16.
McCurley A, Jaffe IZ. Mineralocorticoid receptors in vascular function and disease. Mol Cell Endocrinol. 2012;350(2):256–65.
Bonnans C, Chou J, Werb Z. Remodelling the extracellular matrix in development and disease. Nat Rev Mol Cell Biol. 2014;15(12):786–801.
Toth P, et al. Aging exacerbates hypertension-induced cerebral microhemorrhages in mice: role of resveratrol treatment in vasoprotection. Aging Cell. 2015;14(3):400–8.
Fleenor BS, et al. Arterial stiffening with ageing is associated with transforming growth factor-beta1-related changes in adventitial collagen: reversal by aerobic exercise. J Physiol. 2010;588(Pt 20):3971–82.
Jacob MP. Extracellular matrix remodeling and matrix metalloproteinases in the vascular wall during aging and in pathological conditions. Biomed Pharmacother. 2003;57(5–6):195–202.
Tarantini S, et al. Insulin-like growth factor 1 deficiency exacerbates hypertension-induced cerebral microhemorrhages in mice, mimicking the aging phenotype. Aging Cell. 2017;16(3):469–79.
Keymel S, et al. Impaired endothelial progenitor cell function predicts age-dependent carotid intimal thickening. Basic Res Cardiol. 2008;103(6):582–6.
Rauscher FM, et al. Aging, progenitor cell exhaustion, and atherosclerosis. Circulation. 2003;108(4):457–63.
Zhu G, et al. Young environment reverses the declined activity of aged rat-derived endothelial progenitor cells: involvement of the phosphatidylinositol 3-kinase/Akt signaling pathway. Ann Vasc Surg. 2009;23(4):519–34.
Chirinos JA, et al. Large-artery stiffness in health and disease: JACC state-of-the-art review. J Am Coll Cardiol. 2019;74(9):1237–63.
Paneni F, et al. The aging cardiovascular system: understanding it at the cellular and clinical levels. J Am Coll Cardiol. 2017;69(15):1952–67.
Angoff R, Mosarla RC, Tsao CW. Aortic stiffness: epidemiology, risk factors, and relevant biomarkers. Front Cardiovasc Med. 2021;8:709396.
Fritze O, et al. Age-related changes in the elastic tissue of the human aorta. J Vasc Res. 2012;49(1):77–86.
Zieman SJ, Melenovsky V, Kass DA. Mechanisms, pathophysiology, and therapy of arterial stiffness. Arterioscler Thromb Vasc Biol. 2005;25(5):932–43.
Monk BA, George SJ. The effect of ageing on vascular smooth muscle cell behaviour—a mini-review. Gerontology. 2015;61(5):416–26.
Pescatore LA, Gamarra LF, Liberman M. Multifaceted mechanisms of vascular calcification in aging. Arterioscler Thromb Vasc Biol. 2019;39(7):1307–16.
Ungvari Z, et al. Mechanisms of vascular aging. Circ Res. 2018;123(7):849–67.
Bramwell JC, Hill AV. The velocity of pulse wave in man. Proc R Soc Lond Ser B Biol Sci. 1922;93(652):298–306.
Reference Values for Arterial Stiffness Collaboration. Determinants of pulse wave velocity in healthy people and in the presence of cardiovascular risk factors: ‘establishing normal and reference values’. Eur Heart J. 2010;31(19):2338–50.
Najjar SS, et al. Age and gender affect ventricular-vascular coupling during aerobic exercise. J Am Coll Cardiol. 2004;44(3):611–7.
Redfield MM, et al. Age- and gender-related ventricular-vascular stiffening: a community-based study. Circulation. 2005;112(15):2254–62.
De Tombe PP, et al. Ventricular stroke work and efficiency both remain nearly optimal despite altered vascular loading. Am J Phys. 1993;264(6 Pt 2):H1817–24.
Segers P, Stergiopulos N, Westerhof N. Relation of effective arterial elastance to arterial system properties. Am J Physiol Heart Circ Physiol. 2002;282(3):H1041–6.
AlGhatrif M, et al. Longitudinal uncoupling of the heart and arteries with aging in a community-dwelling population. Geroscience. 2021;43(2):551–61.
Vasan RS, et al. Arterial stiffness and long-term risk of health outcomes: the Framingham Heart Study. Hypertension. 2022;79(5):1045–56.
Ben-Shlomo Y, et al. Aortic pulse wave velocity improves cardiovascular event prediction: an individual participant meta-analysis of prospective observational data from 17,635 subjects. J Am Coll Cardiol. 2014;63(7):636–46.
Sutton-Tyrrell K, et al. Elevated aortic pulse wave velocity, a marker of arterial stiffness, predicts cardiovascular events in well-functioning older adults. Circulation. 2005;111(25):3384–90.
Liao D, et al. Arterial stiffness and the development of hypertension. The ARIC study. Hypertension. 1999;34(2):201–6.
Najjar SS, et al. Pulse wave velocity is an independent predictor of the longitudinal increase in systolic blood pressure and of incident hypertension in the Baltimore Longitudinal Study of Aging. J Am Coll Cardiol. 2008;51(14):1377–83.
Franklin SS, et al. Does the relation of blood pressure to coronary heart disease risk change with aging? The Framingham Heart Study. Circulation. 2001;103(9):1245–9.
Chae CU, et al. Increased pulse pressure and risk of heart failure in the elderly. JAMA. 1999;281(7):634–9.
Haider AW, et al. Systolic blood pressure, diastolic blood pressure, and pulse pressure as predictors of risk for congestive heart failure in the Framingham Heart Study. Ann Intern Med. 2003;138(1):10–6.
Omote K, Verbrugge FH, Borlaug BA. Heart failure with preserved ejection fraction: mechanisms and treatment strategies. Annu Rev Med. 2022;73:321–37.
Reddy YNV, et al. Arterial stiffening with exercise in patients with heart failure and preserved ejection fraction. J Am Coll Cardiol. 2017;70(2):136–48.
Shaikh AY, et al. Relations of arterial stiffness and brachial flow-mediated dilation with new-onset atrial fibrillation: the Framingham Heart Study. Hypertension. 2016;68(3):590–6.
Pase MP, et al. Aortic stiffness and the risk of incident mild cognitive impairment and dementia. Stroke. 2016;47(9):2256–61.
Cui C, et al. Aortic stiffness is associated with increased risk of incident dementia in older adults. J Alzheimers Dis. 2018;66(1):297–306.
Bown CW, et al. Elevated aortic pulse wave velocity relates to longitudinal gray and white matter changes. Arterioscler Thromb Vasc Biol. 2021;41(12):3015–24.
Yannoutsos A, et al. Clinical relevance of aortic stiffness in end-stage renal disease and diabetes: implication for hypertension management. J Hypertens. 2018;36(6):1237–46.
Peralta CA, et al. Association of pulse pressure, arterial elasticity, and endothelial function with kidney function decline among adults with estimated GFR >60 mL/min/1.73 m(2): the Multi-Ethnic Study of Atherosclerosis (MESA). Am J Kidney Dis. 2012;59(1):41–9.
Appel LJ, et al. A clinical trial of the effects of dietary patterns on blood pressure. DASH Collaborative Research Group. N Engl J Med. 1997;336(16):1117–24.
Menotti A, Puddu PE. How the Seven Countries Study contributed to the definition and development of the Mediterranean diet concept: a 50-year journey. Nutr Metab Cardiovasc Dis. 2015;25(3):245–52.
LaRocca TJ, Martens CR, Seals DR. Nutrition and other lifestyle influences on arterial aging. Ageing Res Rev. 2017;39:106–19.
Mozaffarian D, Wu JHY. Flavonoids, dairy foods, and cardiovascular and metabolic health: a review of emerging biologic pathways. Circ Res. 2018;122(2):369–84.
Wang M, et al. Calorie restriction curbs proinflammation that accompanies arterial aging, preserving a youthful phenotype. J Am Heart Assoc. 2018;7(18):e009112.
Gates PE, et al. Dietary sodium restriction rapidly improves large elastic artery compliance in older adults with systolic hypertension. Hypertension. 2004;44(1):35–41.
Figueroa A, et al. Effects of diet and/or low-intensity resistance exercise training on arterial stiffness, adiposity, and lean mass in obese postmenopausal women. Am J Hypertens. 2013;26(3):416–23.
Dengo AL, et al. Arterial destiffening with weight loss in overweight and obese middle-aged and older adults. Hypertension. 2010;55(4):855–61.
Santos-Parker JR, LaRocca TJ, Seals DR. Aerobic exercise and other healthy lifestyle factors that influence vascular aging. Adv Physiol Educ. 2014;38(4):296–307.
Lopes S, et al. Exercise training reduces arterial stiffness in adults with hypertension: a systematic review and meta-analysis. J Hypertens. 2021;39(2):214–22.
Vaitkevicius PV, et al. Effects of age and aerobic capacity on arterial stiffness in healthy adults. Circulation. 1993;88(4 Pt 1):1456–62.
De Cesaris R, et al. Forearm arterial distensibility in patients with hypertension: comparative effects of long-term ACE inhibition and beta-blocking. Clin Pharmacol Ther. 1993;53(3):360–7.
Dudenbostel T, Glasser SP. Effects of antihypertensive drugs on arterial stiffness. Cardiol Rev. 2012;20(5):259–63.
Gurven M, Blackwell AD, Rodríguez DE, Stieglitz J, Kaplan H. Does blood pressure inevitably rise with age?: Longitudinal evidence among forager-horticulturalists. Hypertension. 2012 Jul;60(1):25-33. doi: https://doi.org/10.1161/HYPERTENSIONAHA.111.189100.. Epub 2012 May 21. PMID: 22700319; PMCID: PMC3392307.
Tsao CW, Aday AW, Almarzooq ZI, Alonso A, Beaton AZ, Bittencourt MS, Boehme AK, Buxton AE, Carson AP, Commodore-Mensah Y, MSV E, Evenson KR, Eze-Nliam C, Ferguson JF, Generoso G, Ho JE, Kalani R, Khan SS, Kissela BM, Knutson KL, Levine DA, Lewis TT, Liu J, Loop MS, Ma J, Mussolino ME, Navaneethan SD, Perak AM, Poudel R, Rezk-Hanna M, Roth GA, Schroeder EB, Shah SH, Thacker EL, LB VW, Virani SS, Voecks JH, Wang NY, Yaffe K, Martin SS. Heart Disease and Stroke Statistics-2022 Update: A Report From the American Heart Association. Circulation. 2022;145(8):e153–639. https://doi.org/10.1161/CIR.0000000000001052. Epub 2022 Jan 26. Erratum in: Circulation. 2022 Sep 6;146(10):e141
Avolio AP, Deng FQ, Li WQ, Luo YF, Huang ZD, Xing LF, O’Rourke MF. Effects of aging on arterial distensibility in populations with high and low prevalence of hypertension: comparison between urban and rural communities in China. Circulation. 1985 Feb;71(2):202-210. doi: https://doi.org/10.1161/01.cir.71.2.202.
He J, Klag MJ, Whelton PK, Chen JY, Mo JP, Qian MC, Mo PS, He GQ. Migration, blood pressure pattern, and hypertension: the Yi Migrant Study. Am J Epidemiol. 1991;134(10):1085–101. https://doi.org/10.1093/oxfordjournals.aje.a116012.
Halcox JP, et al. Prognostic value of coronary vascular endothelial dysfunction. Circulation. 2002;106(6):653–8.
Houghton JL, et al. Effect of African-American race and hypertensive left ventricular hypertrophy on coronary vascular reactivity and endothelial function. Hypertension. 1997;29(3):706–14.
Cosson E, et al. Impaired coronary endothelium-dependent vasodilation is associated with microalbuminuria in patients with type 2 diabetes and angiographically normal coronary arteries. Diabetes Care. 2006;29(1):107–12.
Treasure CB, et al. Beneficial effects of cholesterol-lowering therapy on the coronary endothelium in patients with coronary artery disease. N Engl J Med. 1995;332(8):481–7.
Schachinger V, Britten MB, Zeiher AM. Prognostic impact of coronary vasodilator dysfunction on adverse long-term outcome of coronary heart disease. Circulation. 2000;101(16):1899–906.
Doucette JW, et al. Validation of a Doppler guide wire for intravascular measurement of coronary artery flow velocity. Circulation. 1992;85(5):1899–911.
Zeiher AM, et al. Modulation of coronary vasomotor tone in humans. Progressive endothelial dysfunction with different early stages of coronary atherosclerosis. Circulation. 1991;83(2):391–401.
Ludmer PL, et al. Paradoxical vasoconstriction induced by acetylcholine in atherosclerotic coronary arteries. N Engl J Med. 1986;315(17):1046–51.
Niccoli G, Scalone G, Crea F. Coronary functional tests in the catheterization laboratory - pathophysiological and clinical relevance. Circ J. 2015;79(4):676–84.
Randaccio P. A program for optimizing radiation beam parameters in radiotherapy (author’s transl). Radiol Med. 1979;65(10):741–6.
Nabel EG, et al. Dilation of normal and constriction of atherosclerotic coronary arteries caused by the cold pressor test. Circulation. 1988;77(1):43–52.
Gordon JB, et al. Atherosclerosis influences the vasomotor response of epicardial coronary arteries to exercise. J Clin Invest. 1989;83(6):1946–52.
Leucker TM, et al. Coronary endothelial dysfunction is associated with elevated serum PCSK9 levels in people with HIV independent of low-density lipoprotein cholesterol. J Am Heart Assoc. 2018;7(19):e009996.
Beanlands RS, et al. Noninvasive quantification of regional myocardial flow reserve in patients with coronary atherosclerosis using nitrogen-13 ammonia positron emission tomography. Determination of extent of altered vascular reactivity. J Am Coll Cardiol. 1995;26(6):1465–75.
Taqueti VR, Di Carli MF. Coronary microvascular disease pathogenic mechanisms and therapeutic options: JACC state-of-the-art review. J Am Coll Cardiol. 2018;72(21):2625–41.
George RT, et al. Quantification of myocardial perfusion using dynamic 64-detector computed tomography. Investig Radiol. 2007;42(12):815–22.
Corretti MC, et al. Guidelines for the ultrasound assessment of endothelial-dependent flow-mediated vasodilation of the brachial artery: a report of the International Brachial Artery Reactivity Task Force. J Am Coll Cardiol. 2002;39(2):257–65.
Sara JD, et al. Prevalence of coronary microvascular dysfunction among patients with chest pain and nonobstructive coronary artery disease. JACC Cardiovasc Interv. 2015;8(11):1445–53.
Hays AG, et al. Noninvasive visualization of coronary artery endothelial function in healthy subjects and in patients with coronary artery disease. J Am Coll Cardiol. 2010;56(20):1657–65.
Hundley WG, et al. Assessment of coronary arterial flow and flow reserve in humans with magnetic resonance imaging. Circulation. 1996;93(8):1502–8.
Leucker TM, et al. Evolocumab, a PCSK9-monoclonal antibody, rapidly reverses coronary artery endothelial dysfunction in people living with HIV and people with dyslipidemia. J Am Heart Assoc. 2020;9(14):e016263.
Shufelt CL, et al. Cardiac magnetic resonance imaging myocardial perfusion reserve index assessment in women with microvascular coronary dysfunction and reference controls. Cardiovasc Diagn Ther. 2013;3(3):153–60.
Dorbala S, et al. Coronary microvascular dysfunction is related to abnormalities in myocardial structure and function in cardiac amyloidosis. JACC Heart Fail. 2014;2(4):358–67.
Kelle S, et al. A bi-center cardiovascular magnetic resonance prognosis study focusing on dobutamine wall motion and late gadolinium enhancement in 3,138 consecutive patients. J Am Coll Cardiol. 2013;61(22):2310–2.
Haluska B, et al. Automated edge-detection technique for measurement of brachial artery reactivity: a comparison of concordance with manual measurements. Ultrasound Med Biol. 2001;27(9):1285–9.
Matsuzawa Y, et al. Prognostic value of flow-mediated vasodilation in brachial artery and fingertip artery for cardiovascular events: a systematic review and meta-analysis. J Am Heart Assoc. 2015;4(11):e002270.
Allan RB, et al. A comparison of flow-mediated dilatation and peripheral artery tonometry for measurement of endothelial function in healthy individuals and patients with peripheral arterial disease. Eur J Vasc Endovasc Surg. 2013;45(3):263–9.
Bonetti PO, et al. Noninvasive identification of patients with early coronary atherosclerosis by assessment of digital reactive hyperemia. J Am Coll Cardiol. 2004;44(11):2137–41.
Iantorno M, et al. Simultaneous noninvasive assessment of systemic and coronary endothelial function. Circ Cardiovasc Imaging. 2016;9(3):e003954.
Minhas AS, et al. Imaging assessment of endothelial function: an index of cardiovascular health. Front Cardiovasc Med. 2022;9:778762.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2023 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Leucker, T.M., Goldenberg, J., Gerstenblith, G. (2023). Aging of the Vasculature. In: Leucker, T.M., Gerstenblith, G. (eds) Cardiovascular Disease in the Elderly. Contemporary Cardiology. Humana, Cham. https://doi.org/10.1007/978-3-031-16594-8_4
Download citation
DOI: https://doi.org/10.1007/978-3-031-16594-8_4
Published:
Publisher Name: Humana, Cham
Print ISBN: 978-3-031-16593-1
Online ISBN: 978-3-031-16594-8
eBook Packages: MedicineMedicine (R0)