
Abstract
Malaria is among the most prevalent parasite infections caused by Plasmodium genus. According to the most recent data available for the year 2020, the disease killed about 627,000 people, the majority (67%) of whom were children under the age of 5. Resistance, notably in Plasmodium falciparum, has been a foremost factor in the doubling of malaria-related child mortality in eastern and southern Africa. Additionally, antimalarial drug resistance is the utmost likely cause of malaria’s global recurrence in the last three decades. Plasmodium falciparum and Plasmodium vivax have been found to be resistant to currently available antimalarial medicines. Plasmodium falciparum parasite has evolved resistance to practically all antimalarial agents in use, while Plasmodium vivax exhibited resistance to primaquine and chloroquine in some areas. Understanding the statistics of distinct classes of antimalarial drug resistance and introducing strategies that can postpone the emergence of resistance are crucial for making predictions on the onset and spread of resistance to existing antimalarial drugs and recently introduced molecules. Furthermore, understanding the mechanism of resistance and finding particular genetic loci linked to this phenotype are critical for antimalarial resistance surveillance and containment. This chapter summarizes the mechanisms and molecular markers of antimalarial drug resistance and emerging strategies to counter its resistance.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Drug resistance
- Antimalarial drugs
- Molecular markers
- Plasmodium falciparum
- Plasmodium vivax
- Malaria vaccine
1 Introduction
Malaria is among the most prevalent parasite infections caused by Plasmodium genus (i.e., Plasmodium falciparum, Plasmodium vivax, Plasmodium ovale, Plasmodium malariae, and Plasmodium knowelsi), which is spread through a mosquito vector’s bite. Despite the knowledge that malaria is preventable and treatable, it continues to have a devastating effect on people’s quality of life and economy all over the world [1]. As per the most recent WHO report, malaria resulted in approximately 241 million infections and 0.627 million deaths in 2020, with the majority of these cases and fatalities occurring in WHO African Region nations [2, 3]. Total resources required to tackle malaria were estimated at US$ 6800 million in 2020 [3, 4]. Among Plasmodium parasites, P. falciparum and Plasmodium vivax are the most common [5]. Malaria prevention, control, and eradication are malaria-endemic nations’ long-term goals (MECs). About every endemic country is presently striving to eliminate malaria, with the ultimate goal of eradicating this deadly illness from the planet. However, antimalarial medication resistance is the foremost obstacle to achieving the goal [3, 4]. The multidrug resistance observed in the forests of Southeast Asia and South America is most likely explained by established and severe drug pressure mixed with inadequate antiparasitic immunity. Nonetheless, resistance levels may vary depending on location and time [6, 7]. The parasite P. falciparum has acquired resistance to practically all presently used antimalarial medicines, whereas P. vivax has shown resistance to chloroquine and primaquine in some areas [6, 7]. The emergence of resistance has been linked to a significant increase in malaria mortality among residents of some endemic areas [6]. Understanding the statistics of distinct classes of antimalarial drug resistance and introducing strategies that can postpone the emergence of resistance are crucial for predicting the onset and spread of resistance to existing antimalarial drugs and recently introduced molecules [8]. Furthermore, understanding the mechanism of resistance and finding particular genetic loci linked to this phenotype are critical for antimalarial resistance surveillance and containment. This chapter summarizes the resistance mechanisms to current antimalarial drugs, molecular marker of antimalarial drug resistance, and emerging strategies to fight against antimalarial resistance.
2 Spread of Antimalarial Drug Resistance
Although southeast Asia’s Greater Mekong Subregion (GMS) only accounts for a minor portion of the worldwide malaria burden, it has been a hotspot for antimalarial drug resistance [9]. The first instances of chloroquine-resistant P. falciparum appeared in the Thai-Cambodian border, and spread of chloroquine-resistant parasites emerged from two centers, South America and Southeast Asia [10, 11]. Within 20 years, chloroquine-resistant P. falciparum had spread throughout the tropical and subtropical regions of the world [12]. Eventually, chloroquine was replaced with sulfadoxine-pyrimethamine to treat chloroquine-resistant P. falciparum; however, sulfadoxine-pyrimethamine resistance developed quickly [13,14,15]. The global health situation, particularly in sub-Saharan Africa, was serious by the end of the 1990s, with more than three million people dying each year with malaria [16]. Many drugs were developed as alternate agents based on the chemistry of existing drugs. These include chloroquine and quinine analogs such as amodiaquine, mefloquine piperaquine, pyronaridine, and atovaquone [6, 17]. However, drug-resistant parasites were identified against these drugs within a short span of time [17]. Novartis created and introduced artemether and lumefantrine fixed-dose combination, a first artemisinin-based combination treatment (ACT) [18]. The widespread use of this novel medication in South Africa around the year 2000 resulted in an 87.5% reduction in malaria mortality in less than a year [19]. In 2001, WHO advised ACT as first-line therapy for uncomplicated malaria in locations where the parasites were resistant to conventional medications, such as mefloquine [20]. Following the introduction of ACTs, the first reports of P. falciparum resistant to artemisinin were found in western Cambodia in 2006–2007 and then in GMS countries [20,21,22]. The global distribution of resistant P. falciparum is now varied, reflecting changes in the treatment use and transmitting intensity. In southeast Asia, resistance to ACTs, dihydroartemisinin–piperaquine and artesunate–mefloquine, has also been reported [24,25,26,27,28,29,30,31]. Artemisinin partial resistance (clonal expansion of PfKelch13 mutations) is now identified in African countries such as Rwanda (R561H), Uganda (C469Y and A675V), and the Horn of Africa (R622I). However, PfKelch13 mutations related to artemisinin partial resistance have become common in the GMS [3]. Figure 1 shows the timeline of antimalarial resistance.

Timeline of antimalarial drug resistance
Antimalarial resistance has slowly emerged in P. vivax and P. ovale, which is usually attributed to smaller parasitic organism count in humans and thus fewer mutation events. For example, P. vivax and Plasmodium ovale have the ability to bypass erythrocyte schizonticides by exoerythrocytic stage in the liver. Chloroquine resistance in P. vivax was first discovered in Australian visitors to Papua New Guinea in the late 1980s, while chloroquine resistance in Plasmodium malariae was discovered in the early 2000s [23,24,25]. Chloroquine resistance was developed internationally according to the genetic markers found in P. vivax [26]. Global distribution of malarial resistance is shown in Table 1 [27].
3 Mechanism of Antimalarial Drug Resistance
WHO defines drug resistance as the “ability of a parasite strain to survive and/or multiply despite the administration and absorption of a drug given in doses equal to or higher than those usually recommended but within tolerance of the subject” [28]. Antimalarial drug resistance ascends due to genetic mutations that occur by a change in single nucleotide polymorphisms or gene amplifications [29]. Table 2 summarizes the mechanism of action and associated genetic markers of antimalarial drug resistance, while Fig. 2 depicts the mechanism of resistance. Certain other factors of concern are overall parasite load, drug of choice for treating the disease, strength of the drug, improper dosing, and counterfeit drugs. These contribute to antimalarial drug resistance and higher risk of recurrence [30, 31].

Mechanisms of resistance to major antimalarial drugs. Heme detoxification in the digestive vacuole, tetrahydrofolate synthesis in the cytosol, and electron transport in the mitochondrion are all often targeted biological pathways. Resistance to 4-aminoquinolines [chloroquine (CQ), amodiaquine (AQ)], aryl-amino alcohols [quinine (QN), lumefantrine (LMF), and mefloquine (MFQ) is mostly determined by point mutations in the transporters pfcrt and pfmdr1. The key marker of parasite resistance to ART is variant forms of K13 (pfK13). Resistance to sulfadoxine (SD) and both pyrimethamine (PM) and proguanil (PG) can be conferred by mutations in two critical enzymes of the folate biosynthesis pathway, dihydropteroate synthetase (DHPS) and dihydrofolate reductase (DHFR). Atovaquone (AVQ) inhibits mitochondrial electron transport, and resistance to the medicine can be conferred by a single point mutation in the cytochrome b subunit (cytb) of the bc1 complex. QND includes 4-aminoquinolines, arylamino alcohols, and Mannich base
3.1 Chloroquine and Related Compounds
Since chloroquine’s introduction in the late 1940s, it has been the cornerstone of attempts to treat and manage malaria. The effectiveness, relative safety of prescribed therapeutic dosages and low cost of chloroquine have contributed to its importance [32]. Chloroquine is a diprotic weak base, and at physiological pH, it exists in three forms: unprotonated, monoprotonated, and diprotonated [33]. The only membrane-permeable form of chloroquine is uncharged chloroquine, which readily diffuses into the digesting vacuole (DV) of the parasite [34]. Chloroquine molecule become protonated, accumulate into the DV, bind to hematin, and prevent the formation of hemozoin crystal [34,35,36,37,38,39]. The free hematin appears to interfere with the plasmodium detoxifying mechanisms and eventually results in parasite death [33, 34, 38]. Quinine, chloroquine, amodiaquine, primaquine, mefloquine, quinoline methanols, quinine (QN), and quinidine (QND) work by these mechanisms [38, 39]. Recent research has revealed that chloroquine-sensitive parasites collect far more chloroquine in the digestive vacuole than chloroquine-resistant variants [35, 37, 40]. Chloroquine-resistant cultures may efflux chloroquine from the digestive vacuole up to 40 times quicker than chloroquine-sensitive strains [35]. Single nucleotide polymorphisms in the pfcrt gene have been linked to the lower chloroquine accumulation in the plasmodium vacuole of resistant strains [40]. Phenotypes found to carry the pfcrt mutation are K76T or K76N or K76I. The mutation of K76T amino acid in pfcrt is believed to interact with positively charged chloroquine. This allows its exit from the vacuole and thus reduces the concentration of chloroquine in the digestive vacuole [41,42,43,44,45,46,47]. There are studies which suggest that a single amino acid change S16R will introduce a positive charge in the vacuole and block the leak to restore chloroquine [48, 49]. Amodiaquine, a 4-aminoquinoline chemically similar to chloroquine, has been widely used as a monotherapy and is currently being utilized as a companion agent in ACTs [50, 51]. The single nucleotide polymorphisms in Pfcrt and Pfmdr1 are linked with lower sensitivity to chloroquine and amodiaquine [50, 51]. In vitro experiments show that the presence of pfcrt codon 72 to 76 corresponds to significant levels of resistance in desethylamodiaquine [51]. Resistance to mefloquine (4-methanolquinoline) is also mediated by amplification of pfmdr1 [52, 53]. However, mutations in Pfmdr1-N86 variant have been linked to lowered sensitivity to lumefantrine (hydrophobic arylamino alcohol antimalarials) in Africa and Asia [54,55,56], and single nucleotide polymorphisms at Y191H and A437S in pfmrp were found to be linked with quinine resistance [57]. Furthermore, pfnhe1 mutation may also cause quinine resistance [47].
3.2 Antifolate Compounds
Antifolate agents are segmented into two classes, dihydropteroate synthase (DHPS) inhibitors and dihydrofolate reductase (DHFR) inhibitors [58, 59]. These combinations are recommended for the synergistic effect. Inhibition of enzymes can produce a lethal effect on the parasite. Sulfadoxine inhibits the dihydropteroate synthetase (DHPS), whereas proguanil, cycloguanil, and pyrimethamine inhibit dihydrofolate reductase (DHFR) [58, 60, 61]. Pyrimethamine resistance occurs due to mutation at dhfr domain of pfdhfr-ts, at 108, 59, and 51 codons [62]. Similarly, mutation in P. falciparum dihydropteroate synthetase (Pfdhps) gene compromises sulfadoxine at codon A437G/K540E, A437G/A581G, or A437G/K540E/A581G [63].
3.3 Atovaquone
Atovaquone is a lipophilic hydroxynaphthoquinone analogue that possesses similar structural properties to coenzyme Q. It hampers the membrane potential by disrupting respiration and pyrimidine biosynthesis. Atovaquone is a competitive inhibitor of ubiquinol and hinders the mitochondrial electron transport chain at bc1 [23]. In comparison with artemisinin and chloroquine, atovaquone exhibits slow parasite death. However, these compounds are active in liver-stage malaria. Mutation at the ubiquinol binding site confers resistance. Y268N/S/C mutations in the codon of the cytb gene are associated with atovaquone resistance, though in combination with proguanil, the resistance is retarded [64, 65].
3.4 Artemisinin
Artemisinin is the backbone of antimalarial therapy and has a distinct endoperoxide bridge, which is necessary for its action [66]. The mechanism of artemisinin is unknown, but the prevalent notion is the cleaved endoperoxide bridge, resulting in generation of free radicals or reactive oxygen species (ROS) that alkylate important biomolecules [67]. A report revealed that artesunate exerts its antimalarial effect by breaking DNA double strands in P. falciparum by ROS generation. Single nucleotide polymorphisms in pfatp6 and ubp1 have been linked to increased artesunate resistance in French Guyana field isolates and P. chabaudi (rodent malaria parasite), respectively [68, 69]. Furthermore, mutations in the K13 protein’s propeller region at Y493H, R539T, I543T, and C580Y have been identified as a significant predictor of artemether and artesunate resistance in P. falciparum [70]. Artesunate, dihydroartemisinin, and artesunate are prescribed in combination with other antimalarial drugs (i.e., lumefantrine, mefloquine, amodiaquine, sulfadoxine–pyrimethamine, piperaquine, and pyronaridine) to extend the life span of artemisinin therapies by limiting the establishment of resistant plasmodium [20].
3.5 Doxycycline
Doxycycline, from the class of tetracycline antibiotics, acts by inhibiting bacterial protein synthesis. Recent studies showed that doxycycline inhibits nucleotides and deoxynucleotides in falciparum. It is used as prophylaxis in regions where there is chloroquine and multidrug-resistant P. falciparum malaria. Resistance to doxycycline has not been reported [71].
3.6 Clindamycin
In 1967, clindamycin was first reported to have antimalarial activity and work by binding onto 50s ribosome of P. falciparum [72, 73]. For MDR P. falciparum malaria, a combination of clindamycin with quinine is preferred [74].
4 Molecular Marker of Antimalarial Drug Resistance
In the fight against malaria, keeping track of and identifying drug-resistant P. falciparum strains are critical. In vitro and/or in vivo drug susceptibility testing have traditionally been used to identify resistant parasite strains. These approaches, while useful in detecting resistant strains, are expensive and time-consuming [75]. Identifying the molecular markers (mutations/single nucleotide polymorphism in malaria parasite genomics) of resistance that are associated with resistance to a particular antimalarial drug is another cost-effective, time-saving, high-throughput, and robust method that shows a lot of promise in the identification of resistant parasite strains [75].
4.1 Molecular Markers for Drug Resistance in P. falciparum
4.1.1 pfcrt Gene
The PfCRT belongs to the drug transporter superfamily and is found in the plasmodium DV [43, 47]. Drug susceptibility tests revealed that pfcrt polymorphisms (notably the K76T mutation) have a role in chloroquine resistance, which was later verified as a reliable predictor of chloroquine treatment response [76, 77]. The pfcrt K76T polymorphism has also been considered as an indication of amodiaquine resistance. Other antimalarial medicines, such as halofantrine, mefloquine, lumefantrine, artemisinin, and quinine, are also affected by pfcrt polymorphisms [77, 78].
4.1.2 pfmdr1 Gene
The PfMDR1 is a protein found in the digesting vacuole membrane. Pfmdr1 functions as a general importer, sequestering xenobiotics and medicines into the DV [79]. It may also have an indirect effect on drug flow by influencing intracellular ion gradients such as Cl- or pH. The pfmdr1 gene is linked to antimalarial drug resistance through amplification of pfmdr1 copy number or by single nucleotide polymorphism in the gene. The polymorphisms of pfmdr1 alter the substrate specificity and result in resistance to different antimalarial drugs [81]. The pfmdr1 N86Y polymorphism has been associated with chloroquine and amodiaquine treatment failure, whereas pfmdr1 D1246Y is involved in resistance to amodiaquine and desethylamodiaquine [80]. The pfmdr1 N86-F184-D1246 and pfmdr1 N1042D are linked with resistance to lumefantrine in Africa and Thai-Myanmar border, respectively [82,83,84]. The pfmdr1 S1034C/N1042D/D1246Y polymorphism is associated with decreased potency of quinine [64]. Amplification of pfmdr1 copy number of pfmdr1 has been linked with reduced sensitivity to dihydroartemisinin, halofantrine, mefloquine, quinine, and artesunate [80].
4.1.3 pfmrp Gene
PfMRP is a protein that regulates transport. Resistance to antimalarial medications such as quinine and chloroquine has been linked to mutations in the pfmrp gene [57]. In Asia and the Americas, Y191H and A437S have been associated with chloroquine resistance, while in the Americas, Y191H and A437S have been linked to quinine resistance. Pfmrp may operate as a secondary determinant in the modulation of parasite resistance to primaquine, piperaquine, and artemisinin [40, 85].
4.1.4 Pfnhe-1 Gene
The pfnhe-1 gene of P. falciparum was discovered on chromosome 13 of the parasite genome. The pfnhe aggressively effluxes protons to keep the parasite’s pH around 7.4 [86]. Increased DNNND repeat number in microsatellite ms4670 has been linked to lower quinine sensitivity and might be used as a viable marker for quinine resistance [87].
4.1.5 pfdhps and pfdhfr Gene
Sulfadoxine inhibits pfdhps, and pyrimethamine and cycloguanil have been shown to affect pfdhfr function [88]. Resistance to sulfadoxine-pyrimethamine combination treatment has been linked to point mutations in both pfdhfr and pfdhps. S108N, N51I, C59R, and I164L are important polymorphisms in pfdhfr that cause pyrimethamine resistance, but pfdhfr A16V/S108T confers resistance to cycloguanil [89,90,91]. Sulfadoxine resistance is significantly linked to the pfdhps S436A/F, A437G, K540E, A581G, and A613S/T polymorphisms [92].
4.1.6 pfatp6 and Kelch 13 Gene
The pfatp6 gene is a molecular marker that has been linked to partial resistance to artemisinin and its derivatives. In D10 parasite strains, pfatp6 L263E mutation has been linked to higher artemisinin and dihydroartemisinin IC50 values. Artemether IC50 values were observed to be high in clinical isolates from France with the pfatp6 S769N mutation [93]. Kelch 13 gene is another gene that has been linked to artemisinin and its derivatives. Y493H, R539T, I543T, F446L, P574L, and C580Y are all mutations in Kelch 13 gene that have been linked to artemisinin resistance [70, 94].
4.1.7 Pfcytb Gene
Atovaquone is a potent inhibitor of cytochrome bc1 complex [95]. Resistance to atovaquone has been linked to pfcytb Y268S/C/N mutations [23]. As a result, the cytochrome b gene can be used as a molecular marker to track atovaquone resistance.
4.2 Molecular Markers for Drug Resistance in P. vivax
In P. vivax, orthologs of pfcrt and pfmdr1 have been identified as pvcrt-o and pvmdr1, respectively. Sulfadoxine-pyrimethamine susceptibility has been connected to changes in dhfr and dhps genes in P. vivax, while chloroquine resistance has been linked to P. vivax isolates with pvmdr1 Y976F mutation [96].
5 Emerging Strategies to Combat Antimalarial Resistance
Looking at the current scenario, many researchers are recommending artemisinin-based combination therapies (ACT) for multiresistant parasites [94, 97]. The emergence and dissemination of resistance to existing therapies indicate the requirement for the development of new antimalarial treatments [98]. Ultimately new advancement is introduced for drug discovery, and even older drugs are revived for the development of new antimalarial drugs [99, 100]. A list of antimalarials at different stages of drug discovery and development pipeline is shown in Table 3.
5.1 Advancement on Existing Antimalarial
Furtherance on the existing treatment can be established by modifying the dose or combining the drug with another one having different mechanisms of actions [101, 102]. Increasing the total dose of chloroquine given to patients is one potential advancement that can be performed. Another strategic change that can be brought is to maintain a daily dose of the drug course prescribed but extend treatment from 3 days to 5–7 days. Many researchers have gone through a triple-drug combination of various infectious diseases to overcome their resistance. The same concept can be applied over here in malaria, and the usual combination of ACT may also be implemented over here [103].
Piperaquine/dihydroartemisinin, artesunate-mefloquine, piperaquine/dihydroartemisinin plus mefloquine, artemether–lumefantrine, and artemether–lumefantrine plus amodiaquine have all recently been examined in large-scale trials. Another approach to be considered is resistance reversal agents. Here, the resistance of antimalarial drugs is being inhibited with the help of agents, which are likely to reverse the resistance generated by parasites against the agents. Proven resistance reversal agents are calcium channel blockers, chlorpheniramine, and primaquine against P. falciparum to chloroquine [104, 105]. The next strategy can be to reintroduce drugs withdrawn for some years. It has been proven that withdrawal of chloroquine from many countries such as Malawi [106] and Tanzania [107] for a couple of years due to resistance generated and then introducing it again reestablished the potency of the drug against parasites. This recovery could aid in the development of chloroquine-based combination medications as an alternative to sulfadoxine-pyrimethamine as an intermittent preventive treatment for pregnant women, since the public health benefits are diminishing due to sulfadoxine-pyrimethamine resistance.
5.2 New Drug Discovery and Development
Additional approaches to developing strong antimalarial medications include rational targeting of critical parasite activities (target-based) and whole-cell phenotypic screening for chemicals that have desired effect on parasite cells [108]. The crucial targets include metabolite biosynthesis, membrane transport, and signaling system, and the hemoglobin degradation processes [109, 110]. KAI407, 0KAF156, plasmodium phosphatidylinositol-4-OH kinase inhibitors, KAE609, P. falciparum P-type ATPase 4 inhibitors, and DDD107498 are some of the new antimalarial compounds discovered using a cell-based approach [111,112,113]. In contrast, Plasmodium DHODH inhibitor, DSM265, a compound with multistage antimalarial activity and the inhibitor of DHFR and P218 are the potent parasite inhibitors found from target-based screening techniques [114].
5.3 Malaria Vaccine
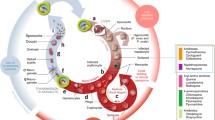
The vaccine is considered to realize malaria eradication apart from traditional antimalarial activity and mosquito control measures. As a result, vaccine development has become a long-term priority for many researchers. Vaccines that target one or more stages of the parasite life cycle are currently being tested. Depending on the stage of the target, there are preerythrocytic vaccinations, blood-stage vaccines, and transmission-blocking vaccines [115]. WHO recommends widespread use of RTS,S/AS01 (RTS,S) malaria vaccine among children in regions with moderate to high P. falciparum malaria transmission. Many next-generation vaccines are in Phase 2 trials, which mainly target all stages of P. falciparum malaria parasite life cycle (Fig. 3, Table 4) [92, 115, 116].

Life cycle of P. falciparum in human showing blood and liver stages
6 Conclusion
Although the area of drug research has advanced in recent years, a new effective antimalarial medicine is still required. The use of modern empirical and theoretical research in population genetics can help build a strategy to postpone the calamity caused by drug-resistance evolution against newly launched antimalarial medications. Furthermore, drug research programs may now benefit from the assays available to uncover medications with a more extensive range of activity, which will help to limit disease transmission and resistance spread. Another requirement is the development of quick, dependable diagnostic tools for detecting the existence of mutations imparting medication resistance concurrently with infection diagnosis.
References
Rasmussen C, Alonso P, Ringwald P. Current and emerging strategies to combat antimalarial resistance. Expert Rev Anti-Infect Ther. 2021;0(0):1–20. Available from:. https://doi.org/10.1080/14787210.2021.1962291.
Takumida M, Takumida H, Anniko M. Localization of histamine (H1, H2, H3 and H4) receptors in mouse inner ear. Acta Otolaryngol. 2016;136(6):537–44. https://doi.org/10.3109/00016489.2015.1136433.
Ippolito MM, Moser KA, Kabuya J-BB, Cunningham C, Juliano JJ. Antimalarial Drug Resistance and Implications for the WHO Global Technical Strategy. Curr Epidemiol Rep. 2021;8(2):46–62. Available from:. https://doi.org/10.1007/s40471-021-00266-5.
Leang R, Taylor WRJ, Bouth DM, Song L, Tarning J, Char MC, et al. Evidence of Plasmodium falciparum malaria multidrug resistance to Artemisinin and Piperaquine in Western Cambodia: Dihydroartemisinin-Piperaquine Open-Label Multicenter Clinical Assessment. Antimicrob Agents Chemother. 2015;59(8):4719–26. https://doi.org/10.1128/AAC.00835-15.
Amelo W, Makonnen E. Efforts made to eliminate drug-resistant malaria and its challenges. Biomed Res Int. 2021;2021:5539544. https://doi.org/10.1155/2021/5539544.
Blasco B, Leroy D, Fidock DA. Antimalarial drug resistance: linking Plasmodium falciparum parasite biology to the clinic. Nat Med. 2017;23(8):917–28. https://doi.org/10.1038/nm.4381.
Chu CS, White NJ. The prevention and treatment of Plasmodium vivax malaria. PLoS Med. 2021;18(4):e1003561. https://doi.org/10.1371/journal.pmed.1003561.
Siddiqui FA, Liang X, Cui L. Plasmodium falciparum resistance to ACTs: emergence, mechanisms, and outlook. Int J Parasitol Drugs Drug Resist. 2021;16:102–18. Available from: https://www.sciencedirect.com/science/article/pii/S2211320721000269
Rathmes G, Rumisha SF, Lucas TCD, Twohig KA, Python A, Nguyen M, et al. Global estimation of antimalarial drug effectiveness for the treatment of uncomplicated Plasmodium falciparum malaria 1991–2019. Malar J. 2020;19(1):374. Available from:. https://doi.org/10.1186/s12936-020-03446-8.
Moore DV, Lanier JE. Observations on two Plasmodium falciparum infections with an abnormal response to chloroquine. Am J Trop Med Hyg. 1961;10:5–9. https://doi.org/10.4269/ajtmh.1961.10.5.
Harinasuta T, Suntharasamai P, Viravan C. Chloroquine-resistant falciparum malaria in Thailand. Lancet (London, UK). 1965;2(7414):657–60. https://doi.org/10.1016/s0140-6736(65)90395-8.
Payne D. Spread of chloroquine resistance in Plasmodium falciparum. Parasitol Today. 1987;3(8):241–6. https://doi.org/10.1016/0169-4758(87)90147-5.
Rønn AM, Msangeni HA, Mhina J, Wernsdorfer WH, Bygbjerg IC. High level of resistance of Plasmodium falciparum to sulfadoxine-pyrimethamine in children in Tanzania. Trans R Soc Trop Med Hyg. 1996;90(2):179–81. https://doi.org/10.1016/s0035-9203(96)90129-7.
Clyde DF, Shute GT. Resistance of Plasmodium falciparum in Tanganyika to pyrimethamine administered at weekly intervals. Trans R Soc Trop Med Hyg. 1957;51(6):505–13. https://doi.org/10.1016/0035-9203(57)90039-1.
Clyde DF. Observations on monthly pyrimethamine (daraprim) prophylaxis in an East African village. East Afr Med J. 1954;31(2):41–6.
Breman JG, Alilio MS, Mills A. Conquering the intolerable burden of malaria: what’s new, what’s needed: a summary. Am J Trop Med Hyg. 2004;71(2 Suppl):1–15.
Bosman A, Mendis KN. A major transition in malaria treatment: the adoption and deployment of artemisinin-based combination therapies. Am J Trop Med Hyg. 2007;77(6 Suppl):193–7.
Na-Bangchang K, Karbwang J. Pharmacology of antimalarial drugs, current anti-malarials. In: Kremsner PG, Krishna S, editors. Encyclopedia of Malaria. New York: Springer; 2019. p. 1–82. Available from:. https://doi.org/10.1007/978-1-4614-8757-9_149-1.
Snow RW, Amratia P, Kabaria CW, Noor AM, Marsh K. The changing limits and incidence of malaria in Africa: 1939–2009. Adv Parasitol. 2012;78:169–262. https://doi.org/10.1016/B978-0-12-394303-3.00010-4.
Dondorp AM, Nosten F, Yi P, Das D, Phyo AP, Tarning J, et al. Artemisinin resistance in Plasmodium falciparum malaria. N Engl J Med. 2009;361(5):455–67. https://doi.org/10.1056/NEJMoa0808859.
Rogers WO, Sem R, Tero T, Chim P, Lim P, Muth S, et al. Failure of artesunate-mefloquine combination therapy for uncomplicated Plasmodium falciparum malaria in southern Cambodia. Malar J. 2009;8:10. https://doi.org/10.1186/1475-2875-8-10.
van der Pluijm RW, Imwong M, Chau NH, Hoa NT, Thuy-Nhien NT, Thanh NV, et al. Determinants of dihydroartemisinin-piperaquine treatment failure in Plasmodium falciparum malaria in Cambodia, Thailand, and Vietnam: a prospective clinical, pharmacological, and genetic study. Lancet Infect Dis. 2019;19(9):952–61. https://doi.org/10.1016/S1473-3099(19)30391-3.
Korsinczky M, Chen N, Kotecka B, Saul A, Rieckmann K, Cheng Q. Mutations in Plasmodium falciparum cytochrome b that are associated with atovaquone resistance are located at a putative drug-binding site. Antimicrob Agents Chemother. 2000;44(8):2100–8. https://doi.org/10.1128/AAC.44.8.2100-2108.2000.
Whitby M, Wood G, Veenendaal JR, Rieckmann K. Chloroquine-resistant Plasmodium vivax, vol. 2. Lancet (London, England). England; 1989. p. 1395. https://doi.org/10.1016/s0140-6736(89)92002-3.
Maguire JD, Sumawinata IW, Masbar S, Laksana B, Prodjodipuro P, Susanti I, et al. Chloroquine-resistant Plasmodium malariae in south Sumatra, Indonesia. Lancet (London, UK). 2002;360(9326):58–60. https://doi.org/10.1016/S0140-6736(02)09336-4.
Hupalo DN, Luo Z, Melnikov A, Sutton PL, Rogov P, Escalante A, et al. Population genomics studies identify signatures of global dispersal and drug resistance in Plasmodium vivax. Nat Genet. 2016;48(8):953–8. Available from:. https://doi.org/10.1038/ng.3588.
Showler AJ, Kain KC, Boggild AK. Protozoan diseases: Malaria clinical features, management, and prevention, International Encyclopedia of Public Health, vol. 5. 2nd ed. Elsevier; 2016. 103–113 p. Available from:. https://doi.org/10.1016/B978-0-12-803678-5.00360-X.
Reyburn H. New WHO guidelines for the treatment of malaria. Vol. 340, BMJ (Clinical research ed.). England; 2010. p. c2637. https://doi.org/10.1136/bmj.c2637
Paget-McNicol S, Saul A. Mutation rates in the dihydrofolate reductase gene of Plasmodium falciparum. Parasitology. 2001;122(Pt 5):497–505. https://doi.org/10.1017/s0031182001007739.
Guyant P, Corbel V, Guérin PJ, Lautissier A, Nosten F, Boyer S, et al. Past and new challenges for malaria control and elimination: the role of operational research for innovation in designing interventions. Malar J. 2015;14(1):279. Available from:. https://doi.org/10.1186/s12936-015-0802-4.
Newton PN, Green MD, Mildenhall DC, Plançon A, Nettey H, Nyadong L, et al. Poor quality vital anti-malarials in Africa – an urgent neglected public health priority. Malar J. 2011;10(1):352. Available from:. https://doi.org/10.1186/1475-2875-10-352.
Roux AT, Maharaj L, Oyegoke O, Akoniyon OP, Adeleke MA, Maharaj R, et al. Chloroquine and Sulfadoxine-Pyrimethamine resistance in Sub-Saharan Africa—a review. Front Genet. 2021;12:668574. https://doi.org/10.3389/fgene.2021.668574.
Homewood CA, Warhurst DC, Peters W, Baggaley VC. Lysosomes, pH and the anti-malarial action of chloroquine. Nature. 1972;235(5332):50–2. https://doi.org/10.1038/235050a0.
Yayon A, Cabantchik ZI, Ginsburg H. Identification of the acidic compartment of Plasmodium falciparum-infected human erythrocytes as the target of the antimalarial drug chloroquine. EMBO J. 1984;3(11):2695–700.
Chinappi M, Via A, Marcatili P, Tramontano A. On the mechanism of chloroquine resistance in Plasmodium falciparum. PLoS One. 2010;5(11):e14064. https://doi.org/10.1371/journal.pone.0014064.
Bossi R, Braga PC, Centanni S, Legnani D, Moavero NE, Allegra L. Antitussive activity and respiratory system effects of levodropropizine in man. Arzneimittelforschung. 1988;38(8):1159–62.
Browning DJ. Pharmacology of Chloroquine and Hydroxychloroquine. In: Hydroxychloroquine and Chloroquine Retinopathy. New York: Springer; 2014. p. 35–63. https://doi.org/10.1007/978-1-4939-0597-3_2
Bray PG, Janneh O, Raynes KJ, Mungthin M, Ginsburg H, Ward SA. Cellular uptake of chloroquine is dependent on binding to ferriprotoporphyrin IX and is independent of NHE activity in Plasmodium falciparum. J Cell Biol. 1999;145(2):363–76. https://doi.org/10.1083/jcb.145.2.363.
Bray PG, Mungthin M, Ridley RG, Ward SA. Access to Hematin: the basis of chloroquine resistance. Mol Pharmacol. 1998;54(1):170–9. Available from: https://molpharm.aspetjournals.org/content/54/1/170
Ecker A, Lehane AM, Clain J, Fidock DA. PfCRT and its role in antimalarial drug resistance. Trends Parasitol. 2012;28(11):504–14. https://doi.org/10.1016/j.pt.2012.08.002.
Bray PG, Martin RE, Tilley L, Ward SA, Kirk K, Fidock DA. Defining the role of PfCRT in Plasmodium falciparum chloroquine resistance. Mol Microbiol. 2005;56(2):323–33. https://doi.org/10.1111/j.1365-2958.2005.04556.x.
Wellems TE, Plowe CV. Chloroquine-resistant malaria. J Infect Dis. 2001;184(6):770–6. https://doi.org/10.1086/322858.
Martin RE, Kirk K. The malaria parasite’s chloroquine resistance transporter is a member of the drug/metabolite transporter superfamily. Mol Biol Evol. 2004;21(10):1938–49. https://doi.org/10.1093/molbev/msh205.
Cooper RA, Lane KD, Deng B, Mu J, Patel JJ, Wellems TE, et al. Mutations in transmembrane domains 1, 4 and 9 of the Plasmodium falciparum chloroquine resistance transporter alter susceptibility to chloroquine, quinine and quinidine. Mol Microbiol. 2007;63(1):270–82. https://doi.org/10.1111/j.1365-2958.2006.05511.x.
Doucette PA, Whitson LJ, Cao X, Schirf V, Demeler B, Valentine JS, et al. Dissociation of human copper-zinc superoxide dismutase dimers using chaotrope and reductant. Insights into the molecular basis for dimer stability. J Biol Chem. 2004;279(52):54558–66. https://doi.org/10.1074/jbc.M409744200.
Cooper RA, Hartwig CL, Ferdig MT. pfcrt is more than the Plasmodium falciparum chloroquine resistance gene: a functional and evolutionary perspective. Acta Trop. 2005;94(3):170–80. https://doi.org/10.1016/j.actatropica.2005.04.004.
Fidock DA, Nomura T, Talley AK, Cooper RA, Dzekunov SM, Ferdig MT, et al. Mutations in the P. falciparum digestive vacuole transmembrane protein PfCRT and evidence for their role in chloroquine resistance. Mol Cell. 2000;6(4):861–71. https://doi.org/10.1016/s1097-2765(05)00077-8.
Cooper RA, Ferdig MT, Su X-Z, Ursos LMB, Mu J, Nomura T, et al. Alternative mutations at position 76 of the vacuolar transmembrane protein PfCRT are associated with chloroquine resistance and unique stereospecific quinine and quinidine responses in Plasmodium falciparum. Mol Pharmacol. 2002;61(1):35–42. https://doi.org/10.1124/mol.61.1.35.
Lehane AM, Kirk K. Chloroquine resistance-conferring mutations in pfcrt give rise to a chloroquine-associated H+ leak from the malaria parasite’s digestive vacuole. Antimicrob Agents Chemother. 2008;52(12):4374–80. https://doi.org/10.1128/AAC.00666-08.
Beshir K, Sutherland CJ, Merinopoulos I, Durrani N, Leslie T, Rowland M, et al. Amodiaquine resistance in Plasmodium falciparum Malaria in Afghanistan is associated with the pfcrt SVMNT Allele at Codons 72 to 76. Antimicrob Agents Chemother. 2010;54(9):3714–6. https://doi.org/10.1128/AAC.00358-10.
Ndung’u L, Langat B, Magiri E, Ng’ang’a J, Irungu B, Nzila A, et al. Amodiaquine resistance in Plasmodium berghei is associated with PbCRT His95Pro mutation, loss of chloroquine, artemisinin and primaquine sensitivity, and high transcript levels of key transporters. Wellcome Open Res. 2017;2:44. https://doi.org/10.12688/wellcomeopenres.11768.2.
Price RN, Uhlemann A-C, Brockman A, McGready R, Ashley E, Phaipun L, et al. Mefloquine resistance in Plasmodium falciparum and increased pfmdr1 gene copy number. Lancet (London, UK). 2004;364(9432):438–47. https://doi.org/10.1016/S0140-6736(04)16767-6.
Preechapornkul P, Imwong M, Chotivanich K, Pongtavornpinyo W, Dondorp AM, Day NPJ, et al. Plasmodium falciparum pfmdr1 amplification, mefloquine resistance, and parasite fitness. Antimicrob Agents Chemother. 2009;53(4):1509–15. https://doi.org/10.1128/AAC.00241-08.
Baliraine FN, Rosenthal PJ. Prolonged selection of pfmdr1 polymorphisms after treatment of falciparum malaria with artemether-lumefantrine in Uganda. J Infect Dis. 2011;204(7):1120–4. https://doi.org/10.1093/infdis/jir486.
Windle ST, Lane KD, Gadalla NB, Liu A, Mu J, Caleon RL, et al. Evidence for linkage of pfmdr1, pfcrt, and pfk13 polymorphisms to lumefantrine and mefloquine susceptibilities in a Plasmodium falciparum cross. Int J Parasitol Drugs Drug Resist. 2020;14:208–17. Available from: https://www.sciencedirect.com/science/article/pii/S2211320720300403
Pickard AL, Wongsrichanalai C, Purfield A, Kamwendo D, Emery K, Zalewski C, et al. Resistance to antimalarials in Southeast Asia and genetic polymorphisms in pfmdr1. Antimicrob Agents Chemother. 2003;47(8):2418–23. https://doi.org/10.1128/AAC.47.8.2418-2423.2003.
Mu J, Ferdig MT, Feng X, Joy DA, Duan J, Furuya T, et al. Multiple transporters associated with malaria parasite responses to chloroquine and quinine. Mol Microbiol. 2003;49(4):977–89. https://doi.org/10.1046/j.1365-2958.2003.03627.x.
Gregson A, Plowe CV. Mechanisms of resistance of malaria parasites to Antifolates. Pharmacol Rev. 2005;57(1):117–45. Available from: https://pharmrev.aspetjournals.org/content/57/1/117
Yuthavong Y. Basis for antifolate action and resistance in malaria. Microbes Infect. 2002;4(2):175–82. https://doi.org/10.1016/s1286-4579(01)01525-8.
Nzila A. The past, present and future of antifolates in the treatment of Plasmodium falciparum infection. J Antimicrob Chemother. 2006;57(6):1043–54. Available from:. https://doi.org/10.1093/jac/dkl104.
Sridaran S, McClintock SK, Syphard LM, Herman KM, Barnwell JW, Udhayakumar V. Anti-folate drug resistance in Africa: meta-analysis of reported dihydrofolate reductase (dhfr) and dihydropteroate synthase (dhps) mutant genotype frequencies in African Plasmodium falciparum parasite populations. Malar J. 2010;9(1):247. Available from:. https://doi.org/10.1186/1475-2875-9-247.
Japrung D, Leartsakulpanich U, Chusacultanachai S, Yuthavong Y. Conflicting requirements of Plasmodium falciparum dihydrofolate reductase mutations conferring resistance to pyrimethamine-WR99210 combination. Antimicrob Agents Chemother. 2007;51(12):4356–60. https://doi.org/10.1128/AAC.00577-07.
Jiang T, Chen J, Fu H, Wu K, Yao Y, Eyi JUM, et al. High prevalence of Pfdhfr–Pfdhps quadruple mutations associated with sulfadoxine–pyrimethamine resistance in Plasmodium falciparum isolates from Bioko Island, Equatorial Guinea. Malar J. 2019;18(1):101. Available from:. https://doi.org/10.1186/s12936-019-2734-x.
Reed MB, Saliba KJ, Caruana SR, Kirk K, Cowman AF. Pgh1 modulates sensitivity and resistance to multiple antimalarials in Plasmodium falciparum. Nature. 2000;403(6772):906–9. https://doi.org/10.1038/35002615.
Corrêa Soares JBR, Menezes D, Vannier-Santos MA, Ferreira-Pereira A, Almeida GT, Venancio TM, et al. Interference with hemozoin formation represents an important mechanism of schistosomicidal action of antimalarial quinoline methanols. PLoS Negl Trop Dis. 2009;3(7):e477. https://doi.org/10.1371/journal.pntd.0000477.
Eastman RT, Fidock DA. Artemisinin-based combination therapies: a vital tool in efforts to eliminate malaria. Nat Rev Microbiol. 2009;7(12):864–74. https://doi.org/10.1038/nrmicro2239.
Bray PG, Ward SA, O’Neill PM. Quinolines and artemisinin: chemistry, biology and history. Curr Top Microbiol Immunol. 2005;295:3–38. https://doi.org/10.1007/3-540-29088-5_1.
Jambou R, Legrand E, Niang M, Khim N, Lim P, Volney B, et al. Resistance of Plasmodium falciparum field isolates to in-vitro artemether and point mutations of the SERCA-type PfATPase6. Lancet (London, UK). 2005;366(9501):1960–3. https://doi.org/10.1016/S0140-6736(05)67787-2.
Hunt P, Afonso A, Creasey A, Culleton R, Sidhu ABS, Logan J, et al. Gene encoding a deubiquitinating enzyme is mutated in artesunate- and chloroquine-resistant rodent malaria parasites. Mol Microbiol. 2007;65(1):27–40. https://doi.org/10.1111/j.1365-2958.2007.05753.x.
Ariey F, Witkowski B, Amaratunga C, Beghain J, Langlois A-C, Khim N, et al. A molecular marker of artemisinin-resistant Plasmodium falciparum malaria. Nature. 2014;505(7481):50–5. https://doi.org/10.1038/nature12876.
Yeh E, DeRisi JL. Chemical rescue of malaria parasites lacking an apicoplast defines organelle function in blood-stage Plasmodium falciparum. PLoS Biol. 2011;9(8):e1001138. https://doi.org/10.1371/journal.pbio.1001138.
Lell B, Kremsner PG. Clindamycin as an antimalarial drug: review of clinical trials. Antimicrob Agents Chemother. 2002;46(8):2315–20. https://doi.org/10.1128/AAC.46.8.2315-2320.2002.
Obonyo CO, Juma EA. Clindamycin plus quinine for treating uncomplicated falciparum malaria: a systematic review and meta-analysis. Malar J. 2012;11:2. https://doi.org/10.1186/1475-2875-11-2.
Ramharter M, Oyakhirome S, Klein Klouwenberg P, Adégnika AA, Agnandji ST, Missinou MA, et al. Artesunate-clindamycin versus quinine-clindamycin in the treatment of Plasmodium falciparum malaria: a randomized controlled trial. Clin Infect Dis an Off Publ Infect Dis Soc Am. 2005;40(12):1777–84. https://doi.org/10.1086/430309.
Hodoameda P. P. falciparum and its molecular markers of resistance to antimalarial drugs. In: Tyagi RK, editor. Plasmodium species and drug resistance. Rijeka: IntechOpen; 2021. Available from:. https://doi.org/10.5772/intechopen.98372.
Djimdé A, Doumbo OK, Cortese JF, Kayentao K, Doumbo S, Diourté Y, et al. A molecular marker for chloroquine-resistant falciparum malaria. N Engl J Med. 2001;344(4):257–63. https://doi.org/10.1056/NEJM200101253440403.
Menard D, Yapou F, Manirakiza A, Djalle D, Matsika-Claquin MD, Talarmin A. Polymorphisms in pfcrt, pfmdr1, dhfr genes and in vitro responses to antimalarials in Plasmodium falciparum isolates from Bangui, Central African Republic. Am J Trop Med Hyg. 2006;75(3):381–7.
Echeverry DF, Holmgren G, Murillo C, Higuita JC, Björkman A, Gil JP, et al. Short report: polymorphisms in the pfcrt and pfmdr1 genes of Plasmodium falciparum and in vitro susceptibility to amodiaquine and desethylamodiaquine. Am J Trop Med Hyg. 2007;77(6):1034–8.
Ferreira PE, Holmgren G, Veiga MI, Uhlén P, Kaneko A, Gil JP. PfMDR1: mechanisms of transport modulation by functional polymorphisms. PLoS One. 2011;6(9):e23875. https://doi.org/10.1371/journal.pone.0023875.
Wilson CM, Volkman SK, Thaithong S, Martin RK, Kyle DE, Milhous WK, et al. Amplification of pfmdr 1 associated with mefloquine and halofantrine resistance in Plasmodium falciparum from Thailand. Mol Biochem Parasitol. 1993;57(1):151–60. https://doi.org/10.1016/0166-6851(93)90252-s.
Picot S, Olliaro P, de Monbrison F, Bienvenu A-L, Price RN, Ringwald P. A systematic review and meta-analysis of evidence for correlation between molecular markers of parasite resistance and treatment outcome in falciparum malaria. Malar J. 2009;8:89. https://doi.org/10.1186/1475-2875-8-89.
Dokomajilar C, Nsobya SL, Greenhouse B, Rosenthal PJ, Dorsey G. Selection of Plasmodium falciparum pfmdr1 alleles following therapy with artemether-lumefantrine in an area of Uganda where malaria is highly endemic. Antimicrob Agents Chemother. 2006;50(5):1893–5. https://doi.org/10.1128/AAC.50.5.1893-1895.2006.
Sisowath C, Petersen I, Veiga MI, Mårtensson A, Premji Z, Björkman A, et al. In vivo selection of Plasmodium falciparum parasites carrying the chloroquine-susceptible pfcrt K76 allele after treatment with artemether-lumefantrine in Africa. J Infect Dis. 2009;199(5):750–7. https://doi.org/10.1086/596738.
Anderson TJC, Nair S, Qin H, Singlam S, Brockman A, Paiphun L, et al. Are transporter genes other than the chloroquine resistance locus (pfcrt) and multidrug resistance gene (pfmdr) associated with antimalarial drug resistance? Antimicrob Agents Chemother. 2005;49(6):2180–8. https://doi.org/10.1128/AAC.49.6.2180-2188.2005.
Raj DK, Mu J, Jiang H, Kabat J, Singh S, Sullivan M, et al. Disruption of a Plasmodium falciparum multidrug resistance-associated protein (PfMRP) alters its fitness and transport of antimalarial drugs and glutathione. J Biol Chem. 2009;284(12):7687–96. https://doi.org/10.1074/jbc.M806944200.
Bosia A, Ghigo D, Turrini F, Nissani E, Pescarmona GP, Ginsburg H. Kinetic characterization of Na+/H+ antiport of Plasmodium falciparum membrane. J Cell Physiol. 1993;154(3):527–34. https://doi.org/10.1002/jcp.1041540311.
Ferdig MT, Cooper RA, Mu J, Deng B, Joy DA, Su X, et al. Dissecting the loci of low-level quinine resistance in malaria parasites. Mol Microbiol. 2004;52(4):985–97. https://doi.org/10.1111/j.1365-2958.2004.04035.x.
Hyde JE. Exploring the folate pathway in Plasmodium falciparum. Acta Trop. 2005;94(3):191–206. https://doi.org/10.1016/j.actatropica.2005.04.002.
Cowman AF, Morry MJ, Biggs BA, Cross GA, Foote SJ. Amino acid changes linked to pyrimethamine resistance in the dihydrofolate reductase-thymidylate synthase gene of Plasmodium falciparum. Proc Natl Acad Sci USA. 1988;85(23):9109–13. https://doi.org/10.1073/pnas.85.23.9109.
Thaithong S, Ranford-Cartwright LC, Siripoon N, Harnyuttanakorn P, Kanchanakhan NS, Seugorn A, et al. Plasmodium falciparum: gene mutations and amplification of dihydrofolate reductase genes in parasites grown in vitro in presence of pyrimethamine. Exp Parasitol. 2001;98(2):59–70. https://doi.org/10.1006/expr.2001.4618.
Urdaneta L, Plowe C, Goldman I, Lal AA. Point mutations in dihydrofolate reductase and dihydropteroate synthase genes of Plasmodium falciparum isolates from Venezuela. Am J Trop Med Hyg. 1999;61(3):457–62. https://doi.org/10.4269/ajtmh.1999.61.457.
Wernsdorfer WH, Noedl H. Molecular markers for drug resistance in malaria: use in treatment, diagnosis and epidemiology. Curr Opin Infect Dis. 2003;16(6):553–8. https://doi.org/10.1097/00001432-200312000-00007.
Krishna S, Pulcini S, Fatih F, Staines H. Artemisinins and the biological basis for the PfATP6/SERCA hypothesis. Trends Parasitol. 2010;26(11):517–23. https://doi.org/10.1016/j.pt.2010.06.014.
Ashley EA, Dhorda M, Fairhurst RM, Amaratunga C, Lim P, Suon S, et al. Spread of artemisinin resistance in Plasmodium falciparum malaria. N Engl J Med. 2014;371(5):411–23. https://doi.org/10.1056/NEJMoa1314981.
Barton V, Fisher N, Biagini GA, Ward SA, O’Neill PM. Inhibiting Plasmodium cytochrome bc1: a complex issue. Curr Opin Chem Biol. 2010;14(4):440–6. https://doi.org/10.1016/j.cbpa.2010.05.005.
Hawkins VN, Joshi H, Rungsihirunrat K, Na-Bangchang K, Sibley CH. Antifolates can have a role in the treatment of Plasmodium vivax. Trends Parasitol. 2007;23(5):213–22. https://doi.org/10.1016/j.pt.2007.03.002.
Lu F, Culleton R, Zhang M, Ramaprasad A, von Seidlein L, Zhou H, et al. Emergence of indigenous artemisinin-resistant Plasmodium falciparum in Africa. New England J Med USA. 2017;376:991–3. https://doi.org/10.1056/NEJMc1612765.
Devine SM, MacRaild CA, Norton RS, Scammells PJ. Antimalarial drug discovery targeting apical membrane antigen 1. MedChemComm. 2017;8(1):13–20. https://doi.org/10.1039/c6md00495d.
Cowell AN, Istvan ES, Lukens AK, Gomez-Lorenzo MG, Vanaerschot M, Sakata-Kato T, et al. Mapping the malaria parasite druggable genome by using in vitro evolution and chemogenomics. Science. 2018;359(6372):191–9. https://doi.org/10.1126/science.aan4472.
Renslo AR. Antimalarial drug discovery: from Quinine to the Dream of Eradication. ACS Med Chem Lett. 2013;4(12):1126–8. https://doi.org/10.1021/ml4004414.
Davis TME, Karunajeewa HA, Ilett KF. Artemisinin-based combination therapies for uncomplicated malaria. Med J Aust. 2005;182(4):181–5. https://doi.org/10.5694/j.1326-5377.2005.tb06650.x.
Kimani J, Phiri K, Kamiza S, Duparc S, Ayoub A, Rojo R, et al. Efficacy and safety of Azithromycin-Chloroquine versus Sulfadoxine-Pyrimethamine for intermittent preventive treatment of Plasmodium falciparum malaria infection in pregnant women in Africa: an open-label, randomized trial. PLoS One. 2016;11(6):e0157045. https://doi.org/10.1371/journal.pone.0157045.
Coulibaly B, Pritsch M, Bountogo M, Meissner PE, Nebié E, Klose C, et al. Efficacy and safety of triple combination therapy with artesunate-amodiaquine-methylene blue for falciparum malaria in children: a randomized controlled trial in Burkina Faso. J Infect Dis. 2015;211(5):689–97. https://doi.org/10.1093/infdis/jiu540.
Rosenthal PJ. Antimalarial drug discovery: old and new approaches. J Exp Biol. 2003;206(Pt 21):3735–44. https://doi.org/10.1242/jeb.00589.
Sidhu ABS, Verdier-Pinard D, Fidock DA. Chloroquine resistance in Plasmodium falciparum malaria parasites conferred by pfcrt mutations. Science. 2002;298(5591):210–3. https://doi.org/10.1126/science.1074045.
Kublin JG, Cortese JF, Njunju EM, Mukadam RAG, Wirima JJ, Kazembe PN, et al. Reemergence of chloroquine-sensitive Plasmodium falciparum malaria after cessation of chloroquine use in Malawi. J Infect Dis. 2003;187(12):1870–5. https://doi.org/10.1086/375419.
Mohammed A, Ndaro A, Kalinga A, Manjurano A, Mosha JF, Mosha DF, et al. Trends in chloroquine resistance marker, Pfcrt-K76T mutation ten years after chloroquine withdrawal in Tanzania. Malar J. 2013;12:415. https://doi.org/10.1186/1475-2875-12-415.
Drinkwater N, McGowan S. From crystal to compound: structure-based antimalarial drug discovery. Biochem J. 2014;461(3):349–69. https://doi.org/10.1042/BJ20140240.
Comer E, Beaudoin JA, Kato N, et al. Diversity-oriented synthesis-facilitated medicinal chemistry: toward the development of novel antimalarial agents. J Med Chem. 2014;57(20):8496–502. https://doi.org/10.1021/jm500994n.
Deu E. Proteases as antimalarial targets: strategies for genetic, chemical, and therapeutic validation. FEBS J. 2017;284(16):2604–28. https://doi.org/10.1111/febs.14130.
McNamara CW, Lee MC, Lim CS, Lim SH, Roland J, Simon O, et al. Targeting Plasmodium PI(4)K to eliminate malaria. Nature. 2013;504(7479):248–53. https://doi.org/10.1038/nature12782.
Rottmann M, McNamara C, Yeung BKS, Lee MCS, Zou B, Russell B, et al. Spiroindolones, a potent compound class for the treatment of malaria. Science. 2010;329(5996):1175–80. https://doi.org/10.1126/science.1193225.
Baragaña B, Hallyburton I, Lee MCS, Norcross NR, Grimaldi R, Otto TD, et al. A novel multiple-stage antimalarial agent that inhibits protein synthesis. Nature. 2015;522(7556):315–20. https://doi.org/10.1038/nature14451.
Phillips MA, Lotharius J, Marsh K, White J, Dayan A, White KL, et al. A long-duration dihydroorotate dehydrogenase inhibitor (DSM265) for prevention and treatment of malaria. Sci Transl Med. 2015;7(296):296ra111. https://doi.org/10.1126/scitranslmed.aaa6645.
Coelho CH, Doritchamou JYA, Zaidi I, Duffy PE. Advances in malaria vaccine development: report from the 2017 malaria vaccine symposium. NPJ Vaccin. 2017;2:34. https://doi.org/10.1038/s41541-017-0035-3.
Han L, Hudgens MG, Emch ME, Juliano JJ, Keeler C, Martinson F, et al. RTS,S/AS01 malaria vaccine efficacy is not modified by seasonal precipitation: results from a phase 3 randomized controlled trial in Malawi. Sci Rep. 2017;7(1):7200. https://doi.org/10.1038/s41598-017-07533-w.
Author information
Authors and Affiliations
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2023 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Patel, C.A., Pande, S., Shukla, P., Ranch, K., Al-Tabakha, M.M., Boddu, S.H.S. (2023). Antimalarial Drug Resistance: Trends, Mechanisms, and Strategies to Combat Antimalarial Resistance. In: Shegokar, R., Pathak, Y. (eds) Malarial Drug Delivery Systems. Springer, Cham. https://doi.org/10.1007/978-3-031-15848-3_3
Download citation
DOI: https://doi.org/10.1007/978-3-031-15848-3_3
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-031-15847-6
Online ISBN: 978-3-031-15848-3
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)