
Abstract
According to the American Diabetes Association, in 2015, 9.4% of the United States population had diabetes and about 50% of these patients will or already have developed peripheral neuropathy. Furthermore, peripheral neuropathy is detectable in about 30% of subjects with pre-diabetes and impaired glucose tolerance. The only treatment recognized for diabetic peripheral neuropathy is glycemic control, which slows progression in patients with type 1 diabetes but is less effective in subjects with type 2 diabetes. With the occurrence of obesity and type 2 diabetes at epidemic levels there is a critical need of a treatment. Diabetic peripheral neuropathy has a complex etiology with at least six major pathways involved in its development: metabolic, vascular, immunologic, neurohormonal growth factor deficiency, genetic, and extracellular matrix remodeling. In light of this complicated etiology any effective treatment for diabetic peripheral neuropathy will likely require a combination of lifestyle and therapeutic interventions. However, before an effective treatment strategy can be developed a more comprehensive understanding of the factors contributing to neurovascular and neural dysfunction in diabetes is needed. This article will address some of the major mechanisms including aldose reductase pathway, non-enzymatic glycation, hexosamine and protein kinase C pathways, oxidative and nitrosative stress, inflammatory stress, and proteases thought to contribute to the development and progression of diabetic peripheral neuropathy.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Diabetic peripheral neuropathy
- Epineurial arterioles
- Oxidative stress
- Nitrosative stress
- Inflammatory stress
- Non-enzymatic glycation
- Hexosamine
- Protein kinase C
- Angiotensin converting enzyme
- Neutral endopeptidase
1 Introduction
Diabetic peripheral neuropathy is a heterogenous condition that can manifest as many different symptoms and are the most common complications of diabetes mellitus with an estimated prevalence ranging up to 50% and possibly higher depending on the diagnostic criteria, whether the subjects have type 1 or type 2 diabetes and duration of diabetes [1,2,3]. Diabetic peripheral neuropathy affects both sensorimotor and autonomic parts of the peripheral nervous system [1]. The most common clinically recognized form is diabetic distal symmetric sensorimotor polyneuropathy which is characterized by the progressive loss of nerve fibers, both large and small [1, 4]. In this progressive disorder the most distal nerve segments of the feet and hands are affected first and involve retraction of terminal sensory axons in the periphery with relative preservation of the perikarya. This phenomenon is often referred to as “dying back syndrome” or the “stocking and glove” pattern reflects damage to the longest sensory axons first and thus is considered a length-dependent neuropathy [4, 5]. This decrease in sensory perception is the most common and earliest form of diabetic peripheral neuropathy and is gradual with symptoms of tingling, pain, and loss of sensation in the toes [6, 7]. Clinical evidence of motor dysfunction is less prevalent with only 1–6% of diabetic patients displaying clinical symptoms and generally occurs in patients with established diabetic peripheral neuropathy [7]. A decrease in motor nerve conduction velocity early in diabetic animal models is a common finding but there is also evidence in animal models and humans of a decrease in compound muscle action potential amplitudes and reduced muscle strength but this has been much less studied in the pre-clinical or clinical setting [7, 8]. The Diabetes Control and Complications Trial (DCCT) and United Kingdom Prospective Diabetes Study (UKPDS) that focused on type 1 and type 2 diabetes, respectively, demonstrated that hyperglycemia is an important contributing factor to the onset and progression of nerve damage especially in those subjects with type 1 diabetes [9, 10]. However, other reports/studies have shown that good glycemic control provides little benefit in towards peripheral neuropathy in those with type 2 diabetes and recent evidence also suggests that small nerve fiber damage occurs in individuals with impaired glucose tolerance, independent of chronic hyperglycemia, and the diagnosis of diabetes [11,12,13,14]. Thus, other conditions, in addition to hyperglycemia, must contribute to the onset and progression of diabetic peripheral neuropathy in subjects with type 2 diabetes. Because of the multiple clinical manifestations associated with diabetic peripheral neuropathy that can include pain in 15–30% of diabetic patients with neuropathy determining the pathophysiology and therapy for diabetic neuropathies is challenging [15].
Studies using animal models of both type 1 and type 2 diabetes have resulted in the identification of wide array of pathological mechanisms as contributing to diabetes peripheral neuropathy. Diabetic peripheral neuropathy has been described by some investigators to be a disease of the vasculature leading to nerve ischemia and altered nerve function [16,17,18,19,20]. Other investigators have proposed that diabetic peripheral neuropathy is caused by a combination of metabolic defects associated with an increased flux of glucose through the aldose reductase pathway leading to a defect in Na+/K+-ATPase and protein kinase C activities and an alteration of signal transduction pathways in the nerve [21, 22]. Additional pathologic contributors to diabetic peripheral neuropathy have been reported to include increased formation of advanced glycation endproducts, hexosamine pathway dysregulation, reduced neurotrophic support, increased inflammatory, and oxidative stress and dyslipidemia [23, 24]. Overall, these mechanisms and likely others cause damage to neurons, Schwann cells, and the vasculature. Ultimately, relentless damage to the nerve complex and surrounding vasculature leads to diabetic peripheral neuropathy. Given the complex etiology of diabetic peripheral neuropathy a successful treatment will likely require a combination of early detection, lifestyle changes, and pharmaceutical interventions targeting the mechanisms deemed most responsible for the pathogenesis. Before this can occur additional studies are needed to determine the most relevant and targetable causes of diabetic peripheral neuropathy.
The present review will focus on a number of the mechanisms introduced above with an emphasis on those impacted by metabolic dysregulation.
2 Role of Aldose Reductase and Polyol Pathway
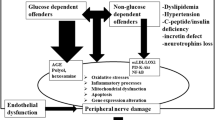
The polyol pathway is catalyzed by two enzymes. Aldose reductase, the first enzyme of this pathway, is believed to have a primary role in early metabolic damage to peripheral nerves [25]. In this initial step glucose is reduced to sorbitol by a reaction that requires NADPH, which indirectly contributes to an increase in oxidative stress. The second step is the oxidation of sorbitol to form fructose a reaction catalyzed by NAD-dependent sorbitol dehydrogenase (see Fig. 1). During periods of excess glucose such as diabetes tissues independent of insulin for glucose transport accumulate glucose intracellularly leading to an increased metabolism of glucose by this pathway and the accumulation of sorbitol causing osmotic stress and a decrease the intracellular levels of myo-inositol and taurine. The decrease in myo-inositol and taurine occurs in response to the excess intracellular accumulation of the osmolyte sorbitol and is referred to the compatible osmolyte hypothesis (to be discussed below). Other negative consequences of this pathway when activated are the excess generation of fructose, which is a more potent glycation agent than glucose leading to an increase in glycative stress.

Illustration of polyol pathway with blue ovals representing conditions that contributes to diabetic peripheral neuropathy
The polyol pathway remains an interesting therapeutic target for the treatment of diabetic peripheral neuropathy even though early clinical trials were unsuccessful in achieving meaningful improvement in human subjects in spite of wide spread success of pre-clinical studies [25,26,27]. Pre-clinical studies with a variety of diabetic animal models using multiple approaches including inhibitors of both aldose reductase and sorbitol dehydrogenase and gene and dietary manipulation have provided overwhelming evidence of an important role for this pathway in diabetes complications including peripheral neuropathy. In rodent studies a wide variety of aldose reductase inhibitors have been shown to improve multiple endpoints associated with diabetic peripheral neuropathy. In this article I will focus on results from pre-clinical and clinical studies performed with three different aldose reductase inhibitors; sorbinil, epalrestat and ranirestat.
Sorbinil was one of the earliest aldose reductase inhibitors to undergo extensive pre-clinical and clinical studies for diabetic peripheral neuropathy. In studies with type 1 diabetic rats treated with streptozotocin treatment with sorbinil prevented as well as reversed defective axonal transport and slowing of motor nerve conduction velocity [28, 29]. Sorbinil treatment also normalized myo-inositol levels in the sciatic nerve in these studies. Another group of investigators demonstrated that treating streptozotocin-induced diabetic rats with sorbinil corrected both motor and sensory nerve conduction velocity as well as endoneurial nutritive blood flow of the sciatic nerve, metabolic abnormalities that included the NAD+/NADH redox imbalance and energy deficiency and improved oxidative stress [30]. Cameron et al. [31] in studies also using streptozotocin-diabetic rats demonstrated that sorbinil treatment protected axon growth retardation.
In early studies of the sciatic nerve untreated diabetic rats were found to have a decrease in Na+/K+ ATPase activity and myo-inositol content that was corrected following sorbinil treatment [32, 33]. All these studies reported that sorbinil treatment reduced tissue sorbitol levels. These studies led to the compatible osmolyte hypothesis that stated myo-inositol depletion and abnormal signaling by inositol phospholipids contribute to a decrease in Na+/K+ ATPase activity in the sciatic nerve, which generates the transmembrane sodium and potassium potentials necessary for nerve impulse conduction and the sodium gradient needed for sodium-dependent uptake of substrates [32]. The compatible osmolyte hypothesis was derived from studies demonstrating that sorbitol accumulation caused by hyperglycemia/diabetes and activation of the polyol pathway leads to a corresponding decrease in myo-inositol and taurine levels in peripheral nerves [34]. The diabetes-induced decrease in myo-inositol and taurine in peripheral nerves was preventable by treatment with an aldose reductase inhibitor such as sorbinil that prevented the increase in sorbitol [35, 36]. This hypothesis as being a contributing factor to peripheral neuropathy is supported by additional studies demonstrating the replenishing myo-inositol or taurine through the diet partially reversed slowing of motor nerve conduction velocity in diabetic rats [34, 37, 38]. Using another approach my laboratory has shown that treating rats with a diet containing a high concentration of l-fucose caused a slowing of nerve conduction velocity and decrease sciatic nerve Na+/K+ ATPase activity and myo-inositol content [39]. l-Fucose is a potent competitive inhibitor of myo-inositol transport by neural and endothelial cells [40,41,42]. Restoring myo-inositol levels through the diet of rats fed the l-fucose diet restored nerve function and Na+/K+ ATPase activity [39]. My laboratory has also demonstrated that treating streptozotocin-induced diabetic rats with sorbinil or myo-inositol improved endoneurial blood flow, motor nerve conduction velocity, and vascular function of epineurial arterioles of the sciatic nerve [43]. The ladder finding is important because we had previously demonstrated that decrease of vascular relaxation to acetylcholine by epineurial arterioles precedes the slowing of nerve conduction velocity indicating that vascular dysregulation is a contributing factor to diabetes-induced nerve dysfunction [44]. Adding to the theory that myo-inositol depletion contributes to diabetic peripheral neuropathy a recent study has demonstrated that mRNA and protein expression of myo-inositol cotransporters in the sciatic nerve are significantly decreased in experimental diabetes [45].
Other animal studies that lend support of the accumulation of polyols causing slowing of motor nerve conduction velocity are galactose fed rats. Feeding rats a diet enriched with galactose leads to a large accumulation of the polyol, galactitol and slowing of motor nerve conduction velocity [46]. Interestingly in this study the authors were not able to demonstrate a decrease in sensory nerve conduction velocity with galactose intoxication. In contrast, in streptozotocin-induced diabetic rats both motor and sensory nerve conduction velocity was decreased in this study [46]. The effects of galactose intoxication on polyol accumulation and myo-inositol depletion are prevented by and aldose reductase inhibitor [47, 48]. Studies have also been done using an inhibitor of sorbitol dehydrogenase the second enzyme in the polyol pathway that converts sorbitol to fructose (see Fig. 1). In those studies it has been found that blocking sorbitol’s conversion to fructose exacerbated sympathetic autonomic neuropathy in streptozotocin-induced diabetic rats and Zucker diabetic rats [49, 50]. The effects of the sorbitol dehydrogenase inhibitor were prevented by the addition of sorbinil. Lastly, in mice overexpressing human aldose reductase compared to wild type mice the induction of diabetes using streptozotocin caused a significantly greater increase in sorbitol and fructose in peripheral nerves even though both sets mice had comparable levels of hyperglycemia [51]. Both the diabetic wild type and aldose reductase transgenic mice had defective nerve conduction velocities but this was significantly more severe in the diabetic aldose reductase transgenic mice. Treating these mice with an aldose reductase inhibitor significantly prevented the accumulation of sorbitol and slowing of nerve conduction velocity [51]. In contrast, studies of diabetes in mice deficient in aldose reductase revealed significantly lower levels of sorbitol compared to diabetic wild type mice and protection from slowing of nerve conduction velocities [52]. Furthermore, aldose reductase deficiency in diabetic mice significantly reduced several markers of oxidative stress that were significantly increased in diabetic wild type mice [52].
The effect of sorbinil has also been extensively studied in human subjects with diabetes. The design of these studies varied as did the number of subjects enrolled, treatment period, dose, duration, and endpoints examined. Many of the clinical studies conducted with sorbinil were limited to a small number of subjects and duration of treatment of a year or less. The results from these studies were mixed with some studies reporting a significant improvement in nerve conduction velocity and axonal atrophy [53,54,55] while other studies reported limited to no benefit [56,57,58]. The results from these trials was summarized nicely by Pfeifer et al. [59] stating that “future trials should be designed with adequate statistical power, with consideration of the variability of the endpoint measurements for long enough duration, and with rigorous quality control to definitively confirm the utility of aldose reductase inhibitors in the treatment of diabetic distal symmetrical polyneuropathy and autonomic neuropathy.” This review was written in 1997 and the same problems are still a challenge for adequate clinical trials for diabetic peripheral neuropathy in 2021.
Epalrestat is another aldose reductase inhibitor that has an extensive history but unlike sorbinil it is being used clinically for treatment of diabetic peripheral neuropathy primarily in Japan and recent pre-clinical and clinical studies provide evidence that it may also be beneficial for diabetic nephropathy [60,61,62]. In pre-clinical studies conducted in rats using a combination of a high fat and high carbohydrate diet followed by a low dose of streptozotocin, a model for type 2 diabetes, it was found that epalrestat treatment protected the diabetic rats from peripheral neuropathy through inhibition of the polyol pathway and by alleviating oxidative stress [63]. Results from several clinical studies report that epalrestat treatment of 150 mg/day may improve motor and sensory nerve conduction velocity and subjective neuropathy symptoms with minimal side effects [62, 64].
In collaboration with the Obrosova laboratory we have also demonstrated that inhibition of aldose reductase with fidarestat of type 1 diabetic rats attenuates oxidative-nitrosative stress and activation of poly(ADP-ribose) polymerase while improving multiple endpoints associated with diabetic peripheral neuropathy [65]. This energized studies in my laboratory to examine the effect of the combination of α-lipoic acid, an antioxidant, and fidarestat on vascular and neural complications in a type 1 diabetic rat model [66]. The results from this study demonstrated that the combination therapy of α-lipoic acid and fidarestat was more efficacious in preventing diabetes-induced vascular and neural dysfunction than monotherapy that required higher doses to be equally effective. Our studies attributed this to the combination therapy allowing for the increased conversion of α-lipoic acid to the more effective antioxidant dihydrolipoic acid, which provides a greater protection from oxidative damage than does glutathione (GSH) (Fig. 2 [67]). Fidarestat blocked aldose reductase activity thereby conserving NADPH levels, which were then available for the production of dihydrolipoic acid whose availability is important for the production of GSH [67]. Interestingly, several groups have reported that the combination of α-lipoic acid and epalrestat is better than monotherapy clinically for diabetic peripheral neuropathy and improvement of motor and sensory nerve conduction velocity [68, 69].

Lipoic acid (LA) and dihydrolipoic acid (DHLA) metabolism (modified from [67])
Ranirestat is one of the more recent aldose reductase inhibitors to be tested pre-clinically and clinically for diabetic peripheral neuropathy. Ranirestat is an uncompetitive/reversible inhibitor of aldose reductase [70]. In long-term studies with streptozotocin-diabetic rats ranirestat reduced sorbitol accumulation in the sciatic nerve and improved the decrease in motor nerve conduction velocity [71]. Treatment with ranirestat also prevented the deformity of myelinated fibers and the decrease in their axonal and myelin areas (atrophy) in sural nerves as well as the changes in the size frequency histogram of myelinated fibers [70]. In another independent study with a study design more relevant to clinical practice ranirestat treatment was started 12 weeks after the onset of hyperglycemia using streptozotocin-treated rats. At the time of treatment both motor and sensory nerve conductions were decreased and the untreated diabetic rats were hypoalgesic in response to a thermal stimulus [72]. Following only 6 weeks of treatment both motor and sensory nerve conduction was improved as was the foot withdrawal latency. Ranirestat treatment also improved the intraepidermal nerve fiber density. The authors of this study concluded that ranirestat has the potential for regeneration in the peripheral nervous system [72]. Ota et al. [73] conducted studies evaluating the effect of ranirestat compared to epalrestat treatment on peripheral neuropathy and cataract formation in spontaneously diabetic torii rats. In this study they found that ranirestat and epalrestat prevented diabetic neuropathy but only ranirestat prevented cataract formation. There have been several clinical studies performed using ranirestat. Sekiguchi et al. [74] found that 52 weeks of treatment with ranirestat (40 mg/day) was well tolerated and improved nerve conduction velocity but was not able to detect any improvement in symptoms and signs. In two separate studies led by Dr. Vera Bril with ranirestat treatment for up to 52–60 weeks with dosing at 20–40 mg/day it was found treatment improved motor nerve conduction velocity and sensory nerve conduction velocity in one study but not the other [74, 75]. In both studies ranirestat was reported to be well tolerated with no differences compared to placebo in adverse events. Ranirestat is the furthest advanced aldose reductase inhibitor for clinical trials except for epalrestat and reproducibly exhibits some degree of measurable objective beneficial outcomes [76]. Its favorable safety profile makes it an attractive choice for further exploration.
3 Non-enzymatic Glycation
Another common pathway that was recognized early to contribute to diabetic peripheral neuropathy was non-enzymatic glycation and the formation of advanced glycation endproducts. Advanced glycation endproducts are created from non-enzymatic reactions of reducing sugars such as glucose or fructose with free amino groups of proteins, lipids, or nucleic acids to initially form Schiff bases or Amadori adducts that in the early stages are reversible (Fig. 3) [77,78,79]. Overtime these products continue to undergo reactions that include dehydration, fragmentation, and cross-linking to form irreversible advanced glycation endproducts [77]. The accumulation of these compounds is associated with many disease states including diabetes. Their accumulation causes structural damage to tissues and organs including components of peripheral nerves such as Schwann cells and cytoskeletal proteins; tubulin, neurofilaments, and actin [80, 81]. The accumulating damage to these tissues culminates in abnormal nerve function including slowed axonal transport, atrophy and degeneration, and slowing of nerve conduction velocity [77, 82]. Advanced glycation endproducts can also indirectly affect peripheral nerves by altering vascular structure and function and ultimately affecting blood flow causing localized ischemia. Advanced glycation endproducts can also elicit their effects through binding of the receptor for advanced glycation endproducts (RAGE) [83]. RAGE is expressed in endothelial cells and Schwann cells [84]. Activation of RAGE has been shown to stimulate NF-κB and increase oxidative stress [85,86,87]. Advanced glycation endproducts have also been shown to induce basement membrane hypertrophy in endoneurial microvessels and disrupt the blood-nerve barrier [88]. In epineurial arterioles, resistance size blood vessels that provide circulation to the sciatic nerve, endothelium-dependent vascular relaxation is mediated in part by acetylcholine via nitric oxide generating mechanism important for regulating endoneurial blood flow [89]. We and others have demonstrated that endoneurial blood flow is improved with inhibitors of advanced glycation endproduct formation as well as oxidative stress; thereby linking these two potential mechanisms of diabetes vascular and neural complications [43, 90]. Studies with mice deficient in RAGE and use of a competitive decoy for advanced glycation endproducts, soluble RAGE (sRAGE), have contributed to our understanding of the role of advanced glycation endproducts may have in the development of diabetic peripheral neuropathy [91,92,93]. In diabetic RAGE-null mice nerve conduction velocity was improved as was nerve generation compared to diabetic wild type mice [94, 95]. Also, treating diabetic wild type mice with sRAGE with the intent to sequester RAGE ligands also improved diabetic peripheral neuropathy [95]. Interestingly, it has been shown that RAGE is expressed in about 30% of all nerve fibers in normal, healthy human subjects, and expression is higher in those subjects with peripheral neuropathy [96].

Pathogenic mechanisms associated with non-enzymatic glycation (modified from [77])
The studies above imply that molecules that may inhibit the action of advanced glycation endproducts may be effective approach for treatment of diabetic peripheral neuropathy. One of the earlier compounds used to inhibit the effects of advanced glycation endproducts was aminoguanidine. Several laboratories including my own have shown that treating diabetic rats with aminoguanidine improve peripheral neuropathy with mechanisms contributing to inhibition of free radical formation and improvement in vascular function [22, 43, 97,98,99,100,101]. Pyridoxamine and analogues have also been shown to be effective inhibitors of advanced glycation endproduct formation and treatment for diabetes complications including diabetic peripheral neuropathy [90, 102, 103].
Much like the study of the polyol pathway in animal models of diabetes there is considerable support from pre-clinical studies with diabetic rodents for a pathologic role of advanced glycation endproducts in the development and progression of diabetic peripheral neuropathy. Unfortunately, there have been few clinical trials testing the effects of inhibitors of advanced glycation endproducts on diabetic peripheral neuropathy.
4 Hexosamine and Protein Kinase C
The hexosamine pathway contributes to an increase in oxidative stress and has been implicated in the pathogenesis of diabetes complications including diabetic peripheral neuropathy [104]. Under conditions of elevated intracellular glucose and flux into glycolysis, excess, fructose-6-phosphate is diverted from glycolysis to produce glucosamine-6-phosphate, which in turn converts into uridine diphosphate-N-acetyl glucosamine, a substrate for the formation of proteoglycans and other glycoproteins [105]. The diphosphate-N-acetyl glucosamine can also increase glycosylation of proteins such as endothelial nitric oxide synthase causing impaired enzyme activity, reduced formation of nitric oxide and vascular dysfunction [106]. It is not entirely clear what kinds of peripheral nerve proteins may be modified by the hexosamine pathway during diabetes, although there is emerging evidence that imbalances in the glycolated state of nervous system proteins is associated with neurodegenerative disease [107]. One of the more promising treatments based on animal studies of the hexosamine pathway is benfotiamine, a fat-soluble derivative of thiamine [108]. In diabetic rats benfotiamine was found to relieve inflammatory and neuropathic pain [109]. In a phase III trial clinical trial of 165 patients with symmetrical, distal diabetic polyneuropathy benfotiamine treatment for 6 weeks reported a significant improvement in the Neuropathy Symptom Score with best results obtained with the high dose according to pain patient-reported symptoms [110].
Protein kinase C (PKC) is another potential target for diabetic peripheral neuropathy treatment. Protein kinase C comprises a superfamily of isoenzymes, many of which are activated by 1,2-diacylglycerol (DAG) [111]. Increased PKC activity is another pathogenic mechanism that has been linked to diabetic vascular disease via animal studies using PKC inhibitors. In the vasculature increased production of diacylglycerol (DAG) leads to activation of PKC isoforms (α, β1 and 2, δ) [111]. Increased PKC activity has been associated with changes in blood flow, basement membrane thickening, extracellular matrix expansion, increased vascular permeability, and changes in enzyme activities including Na+-K+ ATPase, phosphatidylinositol 3-kinase, and mitogen activated protein kinase [111]. In the nerve DAG levels are decreased with diabetes and no consistent change in activity of PKC isoforms has been observed [111, 112]. Nonetheless, inhibition of PKC β of diabetic rats has been reported to correct reduced nerve blood flow and slowing of nerve conduction velocity suggesting a major role of the neurovasculature in diabetic peripheral neuropathy [111, 113,114,115]. This is further supported by studies showing that treatment of diabetic rats with a PKCβ inhibitor also improved vascular dysfunction of mesenteric arteries to acetylcholine [115]. Furthermore, the benefits of PKC inhibition on nerve blood flow and nerve conduction velocity were attenuated by a nitric oxide synthase inhibitor [116]. Studies of diabetic peripheral neuropathy in human subjects treated with a PKC β inhibitor, ruboxistaurin, for 6 months to 1 year reported mixed effects with improvement in skin microvascular blood flow in diabetes subjects with neuropathy but limited improvement in sensory symptoms, vibration detection threshold and Neuropathy Total Symptom Score-6 [117,118,119].
5 Oxidative and Nitrosative Stress
Oxidative stress is a condition resulting from an imbalance between the generation of reactive oxygen species (ROS) and the ability of antioxidant mechanisms to neutralize these compounds [120]. The most common forms of ROS are superoxide (O2−), hydrogen peroxide (H2O2), hydroxyl radical (OH−), and peroxynitrite (ONOO−) [121]. Enzymes and pathways located throughout the cell, including the plasma membrane, cytosol, mitochondria, and peroxisomes have the ability to generate these compounds under both normal and pathological conditions [121]. Superoxide is the most biologically important ROS. It can be produced by the electron transport chain of the mitochondria, and by NADH oxidase, NAD(P)H oxidase, xanthine oxidase, cyclooxygenase, lipoxygenase, cytochrome P-450, and, during periods of tetrahydrobiopterin deficiency, by nitric oxide synthase [121]. Superoxide can spontaneously acquire an electron to form H2O2. The formation of H2O2 from O2− can also occur via a reaction catalyzed by superoxide dismutase (SOD) of which there is three isoforms: Mn-SOD, which is located in the mitochondria and two isoforms of Cu,Zn-SOD, which are located either in the cytosol or extracellularly [121]. Hydrogen peroxide can be converted to water by the action of catalase or by glutathione peroxidase in the presence of reduced glutathione [121]. However, in the presence of trace metals such as Fe, H2O2 can form OH− via a process known as the Fenton reaction [121]. The formation of ONOO−, which is the result of a reaction between O2− and NO, is pathologically important in vascular disease and has been demonstrated to be enhanced in diabetes [121, 122]. It is ONOO− that is responsible for much of the cytotoxicity of nitric oxide (NO) that contributes to nitrosative stress [123]. Peroxynitrite has a short half-life but is able to diffuse across cell membranes and depending on the cell environment can cause a nitrosylation of proteins, which generally reduces enzyme activity, oxidation of glutathione, an important antioxidant, and increased peroxidation of lipids [123].
As discussed above, endothelial dysfunction contributes significantly to diabetic vascular disease, which is an important factor in the development of diabetic peripheral neuropathy. This is supported by studies such as Cameron and Cotter who demonstrated that reduced nerve perfusion is a contributing factor in the etiology of diabetic peripheral neuropathy [124]. Free radicals such as O2− and OH− cause vascular endothelial damage and reduced NO-mediated vasodilation. Inhibition of advanced glycosylation and autoxidation, major sources of free radicals, by aminoguanidine and transition metal chelators, or antioxidants and free radical scavengers have been demonstrated to improve the diabetes-induced decrease in endoneurial blood flow and improve neural dysfunction such as slowed nerve conduction velocity [3, 124,125,126,127,128,129,130,131,132]. The endothelium, via the release of vasodilators and vasoconstrictors, controls the vascular tone. The three major factors produced by the endothelium that contribute to the regulation of vascular relaxation are NO, prostacyclin and the as yet an unidentified factor referred to as endothelium-derived hyperpolarizing factor (EDHF). Impaired endothelium-dependent vasodilation has been demonstrated in various vascular beds of animal models of diabetes and humans with type 1 and type 2 diabetes [133, 134]. The mechanisms induced by hyperglycemia/diabetes considered to contribute to endothelial dysfunction as discussed above are the activation of PKC, increased activity of the polyol pathway, increased formation of advanced glycation endproducts, and increased oxidative stress. Interestingly, studies by Brownlee and colleagues have suggested hyperglycemia-induced production of O2− by mitochondria of endothelial cells as the common link for mechanisms of diabetes-induced vascular dysfunction [135, 136]. Fernyhough has described mitochondrial dysfunction in diabetic neuropathy as a “series of unfortunate metabolic events” [137]. Other chapters in this publication will review the impact of mitochondrial dysfunction, nutrient stress and loss of insulin-dependent growth factor support. Our studies conducted with intact vascular tissue consisting of epineurial arterioles of the sciatic nerve lend support to the studies by Brownlee and colleagues conducted with cultured endothelial cells [138]. Studies by my laboratory have provided evidence that the generation of oxidative stress through the production of O2− and ONOO− impairs vascular function and endothelium-dependent vascular relaxation of epineurial arterioles of the sciatic nerve from diabetic rats, which precedes the slowing of motor nerve conduction velocity [43, 44, 125, 139]. Studies designed to investigate the source of O2− formation provided results suggesting that complex I of the mitochondrial electron transport chain and possibly NAD(P)H oxidase are responsible for the increase in O2− formation observed with epineurial arterioles from the sciatic nerve [138]. It was shown that pretreating epineurial arterioles from diabetic rats with the PKC inhibitor bisindolylmaleimide (GF 109203X) improved acetylcholine-mediated vascular relaxation but did not prevent the increase in O2− formation suggesting that activation of PKC by oxidative stress is downstream of O2− formation [138]. We have also demonstrated that treating diabetic rats with three different types of antioxidants prevented the diabetes-induced increase in O2− and ONOO− formation in aorta and epineurial arterioles of the sciatic nerve and diabetes-induced vascular and neural dysfunction, thereby providing additional evidence that increased oxidative stress contributes to diabetes-induced vascular and neural disease [125, 126]. Studies from other laboratories have provided further evidence that antioxidants may prevent vascular complications in diabetes. Treating diabetic rats with tempol, a stable superoxide dismutase mimic compound, abolished the diabetes-induced increase in vascular O2−, malondialdehyde and 8-epi-prostaglandin F(2α), and also the impairment in relaxation of aortic rings to acetylcholine [125]. Cameron and colleagues have demonstrated that treating diabetic rats with α-lipoic acid or the metal chelators hydroxyethyl starch deferoxamine or trientine prevented the diabetes-induced impairment in vascular relaxation associated with hyperalgesia and neurovascular deficits [128, 129, 140,141,142]. In addition, Keegan et al. demonstrated that treating diabetic rats with α-lipoic acid improved endothelium-dependent vascular relaxation of corpus cavernosum smooth muscle [131]. In another study we demonstrated that diabetic peripheral neuropathy in a rat model of type 2 diabetes was improved by treatment with mitoquinone (Mito-Q) but interestingly in this study treatment did not improve vascular reactivity by epineurial arterioles to acetylcholine [143]. Because metals chelators and OH− scavengers have also been demonstrated to be effective in preventing diabetes-induced vascular and neural dysfunction it is likely that the formation of OH− may also contribute to impairment of vascular reactivity and nerve function in diabetes [124, 128, 129, 132, 140, 141, 144].
As discussed above in regard to improving diabetes impaired vascular function prevention of oxidative stress is a promising approach for intervention and halting of diabetic peripheral neuropathy. A variety of antioxidants including vitamin E have been demonstrated to have beneficial effects in treating neuropathy in diabetes patients and diabetic animal models [145,146,147]. More recently α-lipoic acid has shown promise as a potential antioxidant treatment for diabetic neuropathy [148, 149]. Our studies have demonstrated that α-lipoic acid provides good protection against oxidative stress in diabetic rats of 4–6 week duration [125]. The treatment of diabetic rats with α-lipoic acid significantly improved diabetes-induced decrease in endoneurial blood flow, endothelium-dependent vascular relaxation in arterioles that provide circulation to the sciatic nerve, and motor nerve conduction velocity. α-Lipoic acid treatment also reduced the production of O2− by the aorta and O2− and ONOO− by epineurial arterioles. Treating diabetic rats with α-lipoic acid prevented the diabetes-induced increase in thiobarbituric acid reactive substances in serum and significantly improved lens glutathione levels. α-Lipoic acid is a good metal chelator and is capable of scavenging hydroxyl radicals, hypochlorous acid and singlet oxygen, but not O2− or peroxyl radicals [149,150,151,152]. However, in its reduced form, as dihydrolipoic acid, it is a good scavenger of O2− and prevents initiation of lipid peroxidation [149,150,151,152]. In vivo, α-lipoic acid can be converted into dihydrolipoic acid [149, 150] (Fig. 2). This reaction requires NADPH, which is reduced in diabetes due to the increased flux of glucose through the aldose reductase pathway [120, 153]. Therefore, one potential form of combination therapy for the treatment of diabetic neuropathy may be combining an aldose reductase inhibitor with α-lipoic acid [66]. This combination should promote the formation of dihydrolipoic acid, thereby enhancing the antioxidant potential of α-lipoic acid and possibly providing a synergistic effect. In a study by Nakamura et al. of diabetic neuropathy in streptozotocin-induced diabetic rats, they found that treating diabetic rats with the aldose reductase inhibitor NZ-314 improved nerve function and reduced oxidative stress [154]. They concluded that the efficacious effect of aldose reductase inhibition on diabetic neuropathy may be mediated by decreasing oxygen free radicals. This would agree with our studies described above demonstrating that treating diabetic rats with fidarestat and α-lipoic acid was a better treatment for diabetic peripheral neuropathy than either monotherapy [66]. Efficacy of α-lipoic acid for diabetic peripheral neuropathy has also been examined in human subjects. In a review article by Papanas and Ziegler [155] that examined placebo-controlled clinical trials and meta-analyses they concluded that there is evidence supporting α-lipoic acid as an efficacious and safe treatment for diabetic neuropathy.
The development of superoxide dismutase mimetics are another class of antioxidants with potential for treatment of diabetes complications including diabetic peripheral neuropathy [126, 156, 157]. Because of limitations associated with enzyme therapies these non-peptidyl compounds may offer advantages resulting in better clinical therapies and outcomes for diseases mediated by O2− radicals such as diabetes [158]. In our studies we demonstrated that treating diabetic rats with M40403 inhibited the generation of O2− by aorta and epineurial vessels of the sciatic nerve, the formation of ONOO− by epineurial vessels of the sciatic nerve, the reduction in endoneurial blood flow, the slowing of motor nerve conduction velocity and impairment of endothelium-dependent vasodilation of epineurial arterioles. It also improved the diabetes induced increase in serum TBARS and sciatic nerve conjugated diene level, two additional markers of oxidative stress [126]. M40403 is a prototypic example of a stable, low molecular weight, manganese-containing, non-peptidic molecule possessing the function and catalytic rate of native SOD enzymes, but with the advantage of being a much smaller molecule (molecular weight 483 vs. 30,000 for M40403 and the native enzyme, respectively) [126, 158, 159].
Another pathway for targeting as a potential treatment for diabetic peripheral neuropathy is nitrosative stress and the activation of poly(ADP-ribose) polymerase (PARP) [160, 161]. Studies using peroxynitrite decomposition catalysts as treatments of diabetic rodents have shown improvement in nerve conduction velocity as well as other sensory related neuropathic deficits and improvement of vascular relaxation by epineurial, coronary, and mesenteric arterioles [162,163,164,165]. These treatments were also shown to reduce nitrotyrosine and PARP immunofluorescence in the sciatic nerve and dorsal root ganglion neurons [165]. Others studies have demonstrated that the PARP inhibitor 3-aminobenzamide reduced nitrotyrosine immunoreactivity in vascular tissue and improved vascular reactivity of epineurial arterioles and neural function [166,167,168]. Studies from my laboratory that lend support to PARP activation and metabolic stress contributing to diabetic peripheral neuropathy have shown that treating dietary obese or type 2 diabetic mice with nicotinamide riboside improved multiple endpoints associated with peripheral neuropathy [169].
6 Additional Mechanisms and Treatment Options
Other pathways and downstream mediators of pathology associated with diabetic peripheral neuropathy include mitogen activated protein kinase (MAPK), nuclear factor κB (NF-κB), cyclooxygenase 2 (COX-2), 12/15-lipoxygenase (12/15-LOX), and Na+/H+-exchanger-1 (NHE-1). These downstream mediators of neural impairment have in common their activation via oxidative/inflammatory stress. Activation of the MAPK pathway has been documented in neuron and Schwann cells by hyperglycemia as well as in sciatic nerve of diabetic rodents and human subjects with type 1 and type 2 diabetes [170, 171]. Different treatment modalities have been employed in pre-clinical studies for treatment of diabetic peripheral neuropathy showing improvement in neural endpoints as well as reduction in activation of MAPK [172,173,174]. These studies indicate a potential therapeutic approach for treatment of diabetic peripheral neuropathy through inhibition of MAPK pathway.
Activation of PARP and MAPK can contribute to regulation of gene expression through activation of NF-κB [175]. Nuclear factor-2 erythroid related factor-2 (Nrf2) and NF-κB pathways are potential therapeutic targets for diabetic neuropathy [176]. Pre-clinical studies therapeutically targeting Nrf2 and NF-κB have demonstrated by inhibiting NF-κB and/or activating Nrf2 can reduce inflammation and oxidative stress and improve endpoints related to diabetic peripheral neuropathy [177,178,179,180,181,182].
Other potential targets for treatment of diabetic peripheral neuropathy include inhibition of Na+/K+ exchangers [183]. Treating streptozotocin-induced diabetic rats with cariporide reversed multiple endpoints associated with diabetic peripheral neuropathy and interestingly also reduced markers linked to increased oxidative and nitrosative stress and advanced glycation endproducts suggesting that the pathology of activation of Na+/K+ exchangers in diabetes occurs upstream of these common mediators of diabetic peripheral neuropathy.
Impairment in lipid metabolism is another potential therapeutic target for treatment of diabetic peripheral neuropathy [184]. Cyclooxygenase-2 pathway is upregulated in the peripheral nerves and dorsal root ganglia neurons in experimental diabetes and chemical inhibition or gene deletion improves peripheral nerve function including nerve conduction velocity and loss of epidermal nerve fibers while reducing oxidative stress and inflammation [185, 186]. Treating diabetic mice with the cyclooxygenase-2 inhibitor meloxicam has been shown to reduce neuropathic pain [187]. Other anti-inflammatory or anti-oxidative stress reagents thymoquinone and sulforaphane have been shown to improve in pre-clinical studies diabetic peripheral neuropathy while reducing cyclooxygenase-2 expression in Schwann cells or sciatic nerves [188, 189]. 12/15-Lipoxygenase overexpression is evident in the sciatic nerve and spinal cord of diabetic rodents [190]. Pharmacological inhibition of 12/15-lipoxygenase or gene deficiency has been shown to alleviate some but not all pathological changes associated with diabetic peripheral neuropathy of large and small fibers while reducing MAPK activation and relieving oxidative and inflammatory stress [191,192,193].
In recent years my laboratory has focused on two additional pathways of intervention as a therapeutic target for diabetic peripheral neuropathy. In the next two sections I will briefly review our studies of the effect of inhibition of neutral endopeptidase or neprilysin and nutritional supplementation of omega-3 polyunsaturated fatty acids (PUFA) primarily found in fish oil eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA) and their metabolites. I have previously provided reviews for both of these topics [194, 195].
Angiotensin converting enzyme (ACE), dipeptidyl peptidase IV (DPP-IV), and neprilysin (neutral endopeptidase) are all proteases. As a group these proteases can contribute to disease conditions through the breakdown of important vasoactive and neuroprotective peptides as well as other peptides such as glucagon-like peptide 1. DPP-IV inhibitors (gliptins) are a class of drugs approved by the FDA for hyperglycemia-related treatment of type 2 diabetes [196]. Pre-clinical studies have demonstrated that dipeptidyl peptidase IV inhibitors are also an effective treatment for diabetic peripheral neuropathy [197,198,199,200]. The renin-angiotensin system (RAS) plays a major role in the pathophysiology of cardiovascular and renal disease [201,202,203]. Most known effects of angiotensin II are mediated via activation of the AT1-receptor [202]. Activation of the AT1-receptor is involved in vasoconstriction, inactivation of bradykinin, water and salt homeostasis, control of neuro-humoral systems, reactive oxygen species production, cellular hypertrophy, and hyperplasia and apoptosis [203]. ACE inhibitors and angiotensin receptor blockers are well-known treatments for hypertension, atherosclerosis, congestive heart failure, stroke, and myocardial infarction including those patients with diabetes [201, 204]. In diabetic animal models we and others have demonstrated that ACE inhibitors and to a lesser extent angiotensin receptor blockers can alleviate many deficits associated with diabetic peripheral neuropathy as well as vascular dysfunction of epineurial arterioles of the sciatic nerve [205,206,207,208,209,210]. In addition to pre-clinical studies in animal models of diabetes there are also results from three small clinical trials demonstrating that diabetic neuropathy can be improved by treatment of patients with quinapril, trandolapril or lisinopril [211,212,213].
The other lesser known protease that is included in this discussion is neprilysin. Neprilysin is found in many tissues including vascular and renal tissue and its activity is increased by fatty acids and glucose in human microvascular cells [214,215,216,217]. Interestingly, neprilysin activity has been shown to be activated by protein kinase C, which is increased in vascular tissues by diabetes including endothelial cells [218, 219]. Importantly for this discussion neprilysin degrades vasoactive neuropeptides including; natriuretic peptides, adrenomedullin, bradykinin, endothelin, glucagon-like peptide 1, substance P, and calcitonin gene-related peptide [220, 221]. Thus, inhibition of this protease provides great potential for the treatment of diabetes complications. To test this question several pharmaceutical companies created what was termed vasopeptidase inhibitors. At the time these inhibitors were a new class of drug designed to simultaneously inhibit neprilysin and ACE activity [222]. They were first developed for treating hypertension and heart failure [223, 224]. In regard to diabetes complications it has been shown that vascular conductance in the femoral artery of streptozotocin-induced diabetic rats was improved by a vasopeptidase inhibitor [225]. Furthermore, it has been demonstrated that vasopeptidase inhibitors are neuroprotective and prevent nephropathy in ZDF rats [226, 227]. Vasopeptidase inhibitors have also been reported to decrease matrix metalloproteinases and AGE accumulation/formation in type 2 diabetes and improve wound healing [228, 229]. However, there was no information available about the potential benefits of vasopeptidase inhibitors in diabetic peripheral neuropathy until we performed our studies. Using the vasopeptidase inhibitor ilepatril (AVE7688) we demonstrated in both pre-diabetic and type 1 or type 2 diabetic mice and rats that the combined inhibition of ACE and neprilysin was more effective than monotherapy in improving neurovascular function and slowing/reversing motor and sensory peripheral nerve deficits including diabetes-induced decrease in innervation and sensitivity of corneal nerves [207, 230,231,232,233,234]. Furthermore, we demonstrated that deletion of the neprilysin gene protected mice from the development of neuropathologic deficits and loss of corneal nerves upon induction of diabetes [235]. However, despite greater efficacy for hypertension than monotherapy, development of vasopeptidase inhibitors was halted due to significant off-target effects in some cohorts, most notably increased frequency of angioedema in hypertensive subjects [236]. This led to the development of LCZ696 by Novartis a combination of sacubitril (neprilysin inhibitor) and valsartan (angiotensin receptor blocker) (Entresto). In 2015 the FDA approved the use of sacubitril/valsartan in patients with heart failure with reduced ejection fraction [237]. We obtained this drug to study its effect on vascular and neural complications in a rat model of type 2 diabetes [238]. Using an early and late intervention study design the results demonstrated efficacy of sacubitril/valsartan in improving vascular and neural function that was superior to valsartan alone. In the early intervention protocol, sacubitril/valsartan treatment was found to slow progression of these deficits and, with late intervention treatment, was found to stimulate restoration of vascular reactivity, motor and sensory nerve conduction velocities, and sensitivity/regeneration of sensory nerves of the skin and cornea. We concluded that sacubitril/valsartan may be an effective treatment for diabetic peripheral neuropathy and due to its safety profile and FDA approval could advance quickly to clinical trials.
Recently we have also been interested in the potential for omega-3 PUFA as a treatment for diabetic peripheral neuropathy. Since inflammatory stress is considered to be a primary mechanism for diabetic peripheral neuropathy and omega-3 PUFA and their metabolites are known to have anti-inflammatory properties it was reasonable to expect that fish oil, a common source for omega-3 PUFA, may be an effective treatment [239, 240]. Although trials using treatment with omega-3 PUFA have primarily focused on cardiovascular disease risk reduction [241,242,243,244,245,246], there is significant evidence of their benefits beyond cardiovascular disease. For instance, several recent studies have reported potential benefits of omega-3 PUFA consumption on several chronic inflammatory and autoimmune diseases, stroke, muscle atrophy, and neurodegenerative disease [247,248,249,250,251,252,253,254]. Nonetheless, the consumption of omega-3 PUFA remains low in the Western diet due to historically increased consumption of omega-6 PUFA.
In our own most recent pre-clinical studies, we found that long-chain omega-3 PUFA, primarily EPA and DHA derived from fish (menhaden) oil, were effective in reversing impaired nerve conduction velocities and nerve fiber density and sensitivity in the skin and cornea [255,256,257]. Furthermore, our studies have demonstrated that E and D series resolvins (resolution-phase interaction products), metabolites of EPA and DHA, respectively, (see Fig. 4) which are known to have anti-inflammatory and neuroprotective properties, may be the active mediator of the EPA and DHA effects on diabetic peripheral neuropathy and nerve regeneration [255, 258, 259]. Thus, optimizing production of these metabolites may be an effective approach to increasing the efficacy of EPA and DHA supplements in the diet, and thus ensuring a better outcome for diabetic peripheral neuropathy. In that regard we have demonstrated that the combination of menhaden oil and salsalate, a highly effective agent in blocking proinflammatory chemokines and cytokines, with a large margin of safety and low costs [260, 261] increase production of resolvin D1 in vivo vs. menhaden oil alone and this combination is more efficacious toward improving peripheral neuropathy in diabetic rodents [259, 262]. Another important evidence, supporting future clinical trials of omega-3 PUFA is that our pre-clinical studies have found that the circulating levels of EPA and DHA in rats receiving menhaden oil was comparable to the EPA and DHA serum levels of human subjects taking 4 g of EPA + DHA per day (fish oil capsules) for 4 weeks [257, 263]. For instance, the omega-3 PUFA levels in serum increased from 5.6% to 14.4% and the EPA to AA ratio increased from 0.12 to 0.88 in human participants treated with 4 g of fish oil for 4 weeks [263]. This is similar to the 3.9% to 15.4% increase in the serum omega-3 PUFA levels and 0.04 to 0.54 increase in the EPA to AA ratio we found in our study of diabetic rats treated with menhaden oil [256]. Thus, increasing the circulatory levels of EPA and DHA, and favorably altering the omega-3 index to a healthy range of 8–12% could be an effective disease modifying therapy for treating diabetic peripheral neuropathy [264].

Metabolism of EPA and DHA
In humans, a proof-of-concept study that tested the effects of 12 months of ~2 g daily omega-3 PUFA supplementation on the progression of diabetic peripheral neuropathy in patients with type 1 diabetes, reported a 29% significant increase in corneal nerve fiber length (primary outcome), but no effect on sensory nerve conduction velocity [265]. Unfortunately, it is unclear if omega-3 PUFA therapeutic levels were achieved in these participants given that serum omega-3 index or omega-3 PUFA metabolites were not assessed. However, the above change in corneal fiber length was comparable to the increase in corneal fiber length we observed in type 1 diabetes rats treated with menhaden oil and provides encouraging information to support future clinical trials using omega-3 PUFA and salsalate as an intervention for diabetic peripheral neuropathy [256].
7 Conclusion
Over the years our knowledge and understanding of the different mechanisms that contribute to the etiology of diabetic peripheral neuropathy has expanded considerably. However, this information has not provided us with a successful treatment. There are numerous reasons why this may be the case with some of this being covered in other chapters. From my point of view some of the reasons that may factor into these past failures are that animals are not suitable models for diabetic neuropathy as it occurs in humans, many of the human studies that have been conducted to date are flawed with patients being to advance in disease, treatment periods to short and endpoints not adequate to detect early status of disease and reversibility. However, a primary reason I believe for the failure of finding an effective treatment for diabetic peripheral neuropathy is the monotherapy approach that has been taken for many of the studies. It has been repeatedly demonstrated and reported that the etiology of diabetic peripheral neuropathy is complex with many mechanisms, some discussed in this chapter as well as in other chapters, contributing to the development and progression of the disease. Our cardiology colleagues routinely treat their patients with hypertension by advising them to consider lifestyle and dietary changes as well as prescribing several different drugs that target the different potential mechanisms responsible for high blood pressure. Thus, it seems irrational to think that anything other than a combination of lifestyle changes that include increased exercise and better dietary choices and a combination of interventional drugs will be needed to successfully treat diabetic peripheral neuropathy. I have provided you evidence of this from different animal studies in this chapter. What investigators and clinicians need to be asking are what the primary targets are for successfully treating diabetic peripheral neuropathy and what the best drugs to use for these targets are?
References
Zenker J, Ziegler D, Chrast R. Novel pathogenic pathways in diabetic neuropathy. Trends Neurosci. 2013;36:439–49.
Vinik A. Clinical practice. Diabetic sensory and motor neuropathy. N Engl J Med. 2016;374:1455–64.
Pop-Busui R, Boulton A, Feldman E, et al. Diabetic neuropathy: a position statement by the American Diabetes Association. Diabetes Care. 2017;40:136–54.
Perkins B, Bril V. Electrophysiology testing in diabetic neuropathy. Handb Clin Neurol. 2014;126:235–48.
Feldman E, Callaghan B, Pop-Busui R, et al. Diabetic neuropathy. Nat Rev Dis Primers. 2019;5:41.
Kobayashi M, Zochodne DW. Diabetic neuropathy and the sensory neuron: new aspects of pathogenesis and their treatment implications. J Diabetes Investig. 2018;9:1239–54.
Wilson NM, Wright DE. Experimental motor neuropathy in diabetes. Handb Clin Neurol. 2014;126:461–7.
Zochodne DW, Ramji N, Toth C. Neuronal targeting in diabetes mellitus: a story of sensory neurons and motor neurons. Neuroscientist. 2008;14:311–8.
Writing team for the Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications Research Group. Effect of intensive therapy on the microvascular complications of type 1 diabetes mellitus. JAMA. 2002;287:2563–9.
Stratton I, Adler AI, Neil H, et al. Association of glycaemia with macrovascular and microvascular complications of type 2 diabetes (UKPDS 35): prospective observational study. BMJ. 2000;321:405–12.
Bönhof GJ, Herder C, Strom A, Papanas N, Roden M, Ziegler D. Emerging biomarkers, tools, and treatments for diabetic polyneuropathy. Endocr Rev. 2019;40:153–92.
Zochodne D. Diabetes mellitus and the peripheral nervous system: manifestations and mechanisms. Muscle Nerve. 2007;36:144–66.
Tavakoli M, Mojaddidi M, Fadavi H, Malik R. Pathophysiology and treatment of painful diabetic neuropathy. Curr Pain Headache Rep. 2008;12:192–7.
Smith A, Singleton J. Impaired glucose tolerance and neuropathy. Neurologist. 2008;14:23–9.
Malik R. Wherefore art thou, O treatment for diabetic neuropathy? Int Rev Neurobiol. 2016;127:287–317.
Cameron NE, Cotter MA, Low PA. Nerve blood flow in early experimental diabetes in rats: relation to conduction deficits. Am J Phys. 1991;261:E1–8.
Nukada H, Dyck PJ. Microsphere embolization of nerve capillaries and fiber degeneration. Am J Pathol. 1984;115:275–87.
Cameron NE, Cotter MA, Dines KC, Maxfield EK, Carey F, Mirrlees DJ. Aldose reductase inhibition, nerve perfusion, oxygenation and function in streptozotocin-diabetic rats: dose-response considerations and independence from a myo-inositol mechanism. Diabetologia. 1994;37:651–63.
Cameron NE, Cotter MA, Archibald V, Dines KC, Maxfield EK. Anti-oxidant and pro-oxidant effects on nerve conduction velocity, endoneurial blood flow and oxygen tension in non-diabetic and streptozotocin-diabetic rats. Diabetologia. 1994;37:449–59.
Kles KA, Vinik AI. Pathophysiology and treatment of diabetic peripheral neuropathy: the case for diabetic neurovascular function as an essential component. Curr Diabetes Rev. 2006;2:131–45.
Cotter MA, Dines KC, Cameron NE. Prevention and reversal of motor and sensory peripheral nerve conduction abnormalities in streptozotocin-diabetic rats by the prostacyclin analogue iloprost. Naunyn Schmiedebergs Arch Pharm. 1993;347:534–40.
Cameron NE, Cotter MA, Dines KC, Love A. Effects of aminoguanidine on peripheral nerve function and polyol pathway metabolites in streptozotocin-diabetic rats. Diabetologia. 1992;35:946–50.
Sima AA. Pathological mechanisms involved in diabetic neuropathy: can we slow the process? Curr Opin Investig Drugs. 2006;7:324–37.
Pop-Busui R, Sima A, Stevens M. Diabetic neuropathy and oxidative stress. Diabetes Metab Res Rev. 2006;22:257–73.
Gabbay KH. Aldose reductase inhibition in the treatment of diabetic neuropathy: where are we in 2004? Curr Diab Rep. 2004;4:405–8.
Oates PJ. Aldose reductase, still a compelling target for diabetic neuropathy. Curr Drug Targets. 2008;9:14–36.
Chalk C, Benstead TJ, Moore F. Aldose reductase inhibitors for the treatment of diabetic polyneuropathy. Cochrane Database Syst Rev. 2007;CD004572.
Tomlinson DR, Moriarty RJ, Mayer JH. Prevention and reversal of defective axonal transport and motor nerve conduction velocity in rats with experimental diabetes by treatment with the aldose reductase inhibitor Sorbinil. Diabetes. 1984;33:470–6.
Willars GB, Tomlinson DR, Robinson JP. Studies of sorbinil on axonal transport in streptozotocin-diabetic rats. Metabolism. 1986;35(4 Suppl 1):66–70.
Obrosova IG, Huysen CV, Fathallah L, Cao XC, Greene DA, Stevens MJ. As aldose reductase inhibitor reverses early diabetes-induced changes in peripheral nerve function, metabolism, and antioxidative defense. FASEB J. 2002;16:123–5.
Cameron NE, Leonard MB, Ross IS, Whiting PH. The effects of sorbinil on peripheral nerve conduction velocity, polyol concentrations and morphology in the streptozotocin-diabetic rat. Diabetologia. 1986;29:168–74.
Greene DA, Lattimer SA. Recent advances in the therapy of diabetic peripheral neuropathy by means of a aldose reductase inhibitor. Am J Med. 1985;79:13–7.
Greene DA. A sodium-pump defect in diabetic peripheral nerve corrected by sorbinil administration: relationship to myo-inositol metabolism and nerve conduction slowing. Metabolism. 1986;35(4 Suppl 1):60–5.
Stevens MJ, Lattimer SA, Kamijo M, Van Huysen C, Sima AA, Greene DA. Osmotically-induced nerve taurine depletion and the compatible osmolyte hypothesis in experimental diabetic neuropathy in the rat. Diabetologia. 1993;36:608–14.
Greene DA, Mackway AM. Decreased myo-inositol content and Na+-K+-ATPase activity in superior cervical ganglion of STZ-diabetic rat and prevention by aldose reductase inhibition. Diabetes. 1986;35:1106–8.
Finegold D, Lattimer SA, Nolle S, Bernstein M, Greene DA. Polyol pathway activity and myo-inositol metabolism. A suggested relationship in the pathogenesis of diabetic neuropathy. Diabetes. 1983;32:988–92.
Gillon KR, Hawthorne JN, Tomlinson DR. Myo-inositol and sorbitol metabolism in relation to peripheral nerve function in experimental diabetes in the rat: effect of aldose reductase inhibition. Diabetologia. 1983;25:365–71.
Pop-Busui R, Sullivan KA, Van Huysen CV, et al. Depletion of taurine in experimental diabetic neuropathy: implications for nerve metabolic, vascular, and functional deficits. Exp Neurol. 2001;168:259–72.
Yorek MA, Wiese TJ, Davidson EP, et al. Reduced motor nerve conduction velocity and Na(+)-K(+)-ATPase activity in rats maintained on L-fucose diet. Reversal by myo-inositol supplementation. Diabetes. 1993;42:1401–6.
Yorek MA, Stefani MR, Dunlap JA, Ro KS, Davidson EP. Trans-hydroxyl group configuration on carbons 2 and 3 of glucose. Responsible for acute inhibition of myo-inositol transport? Diabetes. 1991;40:1016–23.
Yorek MA, Dunlap JA, Stefani MR, Davidson EP. L-fucose is a potent inhibitor of myo-inositol transport and metabolism in cultured neuroblastoma cells. J Neurochem. 1992;58:1626–36.
Stefani MR, Dunlap JA, Yorek MA. Effect of L-fucose on proliferation and myo-inositol metabolism in cultured cerebral microvessel and aortic endothelial cells. J Cell Physiol. 1992;153:321–31.
Coppey LJ, Gellett JS, Davidson EP, Dunlap JA, Yorek MA. Effect of treating streptozotocin-induced diabetic rats with sorbinil, myo-inositol or aminoguanidine on endoneurial blood flow, motor nerve conduction velocity and vascular function of epineurial arterioles of the sciatic nerve. Int J Diabetes Res. 2002;3:21–36.
Coppey LJ, Davidson EP, Dunlap JA, Lund DD, Yorek MA. Slowing of motor nerve conduction velocity in streptozotocin-induced diabetic rats is preceded by impaired vasodilation in arterioles that overlie the sciatic nerve. Int J Exp Diabetes Res. 2000;1:131–43.
Farias VX, Uchoa PN, Aquino CP, et al. Expression of myo-inositol cotransporters in the sciatic nerve and dorsal root ganglia in experimental diabetes. Braz J Med Biol Res. 2019;52:e8589.
Kalichman MW, Dines KC, Bobik M, Mizisin AP. Nerve conduction velocity, laser Doppler flow, and axonal caliber in galactose and streptozotocin diabetes. Brain Res. 1998;810:130–7.
Dvornik E, Simard-Duquesne N, Krami M, et al. Polyol accumulation in galactosemic and diabetic rats: control by an aldose reductase inhibitor. Science. 1973;182:1146–8.
Kamijo M, Basso M, Cherian PV, Hohman TC, Sima AA. Galactosemia produces ARI-preventable nodal changes similar to those of diabetic neuropathy. Diabetes Res Clin Pract. 1194;25:117–29.
Schmidt RE, Dorsey DA, Beaudet LN, et al. A potent sorbitol dehydrogenase inhibitor exacerbates sympathetic autonomic neuropathy in rats with streptozotocin-induced diabetes. Exp Neurol. 2005;192:407–19.
Schmidt RE, Dorsey DA, Beaudet LN, et al. Inhibition of sorbitol dehydrogenase exacerbates autonomic neuropathy in rats with streptozotocin-induced diabetes. J Neuropathol Exp Neurol. 2001;60:1153–69.
Yagihashi S, Yamagishi SI, Wada R, et al. Neuropathy in diabetic mice overexpressing human aldose reductase and effects of aldose reductase inhibitor. Brain. 2001;124:2448–58.
Ho ECM, Lam KSL, Chen YS, et al. Aldose reductase-deficient mice are protected from delayed motor nerve conduction velocity, increased c-Jun NH2-terminal kinase activation, depletion of reduced glutathione, increased superoxide accumulation, and DNA damage. Diabetes. 2006;55:1946–53.
Sima AA, Bril V, Nathaniel V, et al. Regeneration and repair of myelinated fibers in sural-nerve biopsy specimens from patients with diabetic neuropathy treated with sorbinil. N Engl J Med. 1988;319:548–55.
Sima AA, Prashar A, Nathaniel V, Bril V, Werb MR, Greene DA. Overt diabetic neuropathy: repair of axo-glial dysjunction and axonal atrophy by aldose reductase inhibition and its correlation to improvement in nerve conduction velocity. Diabet Med. 1993;10:115–21.
Gieron MA, Malone JI, Lowitt S, Korthals JK. Improvement in peripheral nerve function after one year of Sorbinil. Neuroreport. 1991;2:348–50.
Lewin IG, O’Brien IA, Morgan MH, Corrall RJ. Clinical and neurophysiological studies with the aldose reductase inhibitor, sorbinil, in symptomatic diabetic neuropathy. Diabetologia. 1984;26:445–8.
O’Hare JP, Morgan MH, Alden P, Chissel S, O’Brien IA, Corrall RJ. Aldose reductase inhibition in diabetic neuropathy: clinical and neurophysiological studies of one year’s treatment with sorbinil. Diabet Med. 1988;5:537–42.
Fagius J, Brattberg A, Jameson S, Berne C. Limited benefit of treatment of diabetic polyneuropathy with an aldose reductase inhibitor: a 24-week controlled trial. Diabetologia. 1985;28:323–9.
Pfeifer MA, Schumer MP, Gelber DA. Aldose reductase inhibitors: the end of an era or the need for different trial designs? Diabetes. 1997;46(Suppl 2):S82–9.
He J, Gao H-X, Yang N, et al. The aldose reductase inhibitor epalrestat exerts nephritic protection on diabetic nephropathy in db/db mice through metabolic modulation. Acta Pharmacol Sin. 2019;40:86–97.
Hotta N, Kawamori R, Fukuda M, Shigeta Y. Long-term clinical effects of epalrestat, an aldose reductase inhibitor, on progression of diabetic neuropathy and other microvascular complications: multivariate epidemiological analysis based on patient background factors and severity of diabetic neuropathy. Diabet Med. 2012;29:1529–33.
Ramirez MA, Borja NL. Epalrestat: an aldose reductase inhibitor for the treatment of diabetic neuropathy. Pharmacotherapy. 2008;28:646–55.
Li Q-R, Wang Z, Zhou W, et al. Epalrestat protects against diabetic peripheral neuropathy by alleviating oxidative stress and inhibiting polyol pathway. Neural Regen Res. 2016;11:345–51.
Hotta N, Akanuma Y, Kawamori R, et al. Long-term clinical effects of epalrestat, an aldose reductase inhibitor, on diabetic peripheral neuropathy: the 3-year, multicenter, comparative Aldose Reductase Inhibitor-Diabetes Complications Trial. Diabetic Care. 2006;29:1538–44.
Obrosova IG, Pacher P, Szabo C, et al. Aldose reductase inhibition counteracts oxidative-nitrosative stress and poly(ADP-ribose) polymerase activation in tissue sites for diabetes complications. Diabetes. 2005;54:234–42.
Yorek MA, Coppey LJ, Gellett JS, Davidson EP, Lund DD. Effect of fidarestat and alpha-lipoic acid on diabetes-induced epineurial arteriole vascular dysfunction. Exp Diabesity Res. 2004;5:123–35.
Rochette L, Ghibu S, Muresan A, Vergely C. Alpha-lipoic acid: molecular mechanisms and therapeutic potential in diabetes. Can J Physiol Pharmacol. 2015;93:1021–7.
Wang X-T, Lin H-X, Xu S-A, Lu Y-K. Lipoic acid combined with epalrestat versus lipoic acid in treating diabetic peripheral neuropathy: a meta-analysis. Zhongguo Yi Xue Ke Xue Yuan Xue Bao. 2017;39:656–64.
Zhao M, Chen J-Y, Chu Y-D, Zhu Y-B, Luo L, Bu S-Z. Efficacy of epalrestat plus α-lipoic acid combination therapy versus monotherapy in patients with diabetic peripheral neuropathy: a meta-analysis of 20 randomized controlled trials. Neural Regen Res. 2018;13:1087–95.
Matsumoto T, Ono Y, Kurono M, Kuromiya A, Nakamura K, Bril V. Ranirestat (AS-3201), a potent aldose reductase inhibitor, reduces sorbitol levels and improves motor nerve conduction velocity in streptozotocin-diabetic rats. J Pharmacol Sci. 2008;107:231–7.
Matsumoto T, Ono Y, Kurono M, Kuromiya A, Toyosawa K, Ueda Y, Bril V. Long-term treatment with ranirestat (AS-3201), a potent aldose reductase inhibitor, suppresses diabetic neuropathy and cataract formation in rats. J Pharmacol Sci. 2008;107:340–8.
Asano S, Himeno T, Hayami T, et al. Ranirestat improved nerve conduction velocities, sensory perception, and intraepidermal nerve fiber density in rats with overt diabetic polyneuropathy. J Diabetes Res. 2019;2019:2756020.
Ota A, Kakehashi A, Toyoda F, et al. Effects of long-term treatment with ranirestat, a potent aldose reductase inhibitor, on diabetic cataract and neuropathy in spontaneously diabetic torii rats. J Diabetes Res. 2013;2013:175901.
Sekiguchi K, Kohara N, Baba M, et al. Aldose reductase inhibitor ranirestat significantly improves nerve conduction velocity in diabetic polyneuropathy: a randomized double-blind placebo-controlled study in Japan. J Diabetes Investig. 2019;10:466–74.
Bril V, Hirose T, Tomioka S, Buchanan R. Ranirestat for the management of diabetic sensorimotor polyneuropathy. Diabetes Care. 2009;32:1256–60.
Giannoukakis N. Evaluation of ranirestat for the treatment of diabetic neuropathy. Expert Opin Drug Metab Toxicol. 2014;10:1051–9.
Sima AAF, Sugimoto K. Experimental diabetic neuropathy: an update. Diabetologia. 1999;42:773–88.
Brownlee M, Cerami A, Vlassara H. Advanced glycosylation end products in tissue and the biochemical basis of diabetic complications. N Engl J Med. 1988;318:1315–21.
Zochodne D. Mechanisms of diabetic neuron damage: molecular pathways. Handb Clin Neurol. 2014;126:379–99.
Ryle C, Leow CK, Donaghy M. Nonenzymatic glycation of peripheral and central nervous system proteins in experimental diabetes mellitus. Muscle Nerve. 1997;20:577–84.
Cullum NA, Mahon J, Stringer K, McLean WG. Glycation of rat sciatic nerve tubulin in experimental diabetic mellitus. Diabetologia. 1991;34:387–9.
Ozturk G, Sekeroglu MR, Erdogan E, Ozturk M. The effect of non-enzymatic glycation of extracellular matrix proteins on axonal regeneration in vitro. Acta Neuropathol. 2006;112:627–32.
Forbes J, Cooper M. Mechanisms of diabetic complications. Physiol Rev. 2013;93:137–88.
Wada R, Yagihashi S. Role of advanced glycation end products and their receptors in development of diabetic neuropathy. Ann N Y Acad Sci. 2005;1043:598–604.
Bekircan-Kurt CB, Tan E, Ozdamar SE. The activation of RAGE and NF-κB in nerve biopsies of patients with axonal and vasculitic neuropathy. Noro Psikiyatr Ars. 2015;52:279–82.
Yamagishi S, Nakamura K, Matsui T, Noda Y, Imaizumi T. Receptor for advanced glycation end products (RAGE): a novel therapeutic target for diabetic vascular complication. Curr Pharm Des. 2008;14:487–95.
Yamagishi S, Nakamura K, Matsui T, Ueda S, Fukami K, Okuda S. Agents that block advanced glycation end product (AGE)-RAGE (receptor for AGEs)-oxidative stress system: a novel therapeutic strategy for diabetic vascular complications. Expert Opin Investig Drugs. 2008;17:983–96.
Shimizu F, Sano Y, Haruki H, Kanda T. Advanced glycation end-products induce basement membrane hypertrophy in endoneurial microvessels and disrupt the blood-nerve barrier by stimulating the release of TGF-β and vascular endothelial growth factor (VEGF) by pericytes. Diabetologia. 2011;54:1517–26.
Terata K, Coppey LJ, Davidson EP, Dunlap JA, Gutterman DD, Yorek MA. Acetylcholine-induced arteriolar dilation is reduced in streptozotocin-induced diabetic rats with motor nerve dysfunction. Br J Pharmacol. 1999;128:837–43.
Cameron NE, Gibson TM, Nangle MR, Cotter MA. Inhibitors of advanced glycation end product formation and neurovascular dysfunction in experimental diabetes. Ann N Y Acad Sci. 2005;1043:784–92.
Sugimoto K, Yasujima M, Yagihashi S. Role of advanced glycation end products in diabetic neuropathy. Curr Pharm Des. 2008;14:953–61.
Lukic I, Humpert P, Nawroth P, Bierhaus A. The RAGE pathway: activation and perpetuation in the pathogenesis of diabetic neuropathy. Ann N Y Acad Sci. 2008;1126:76–80.
Toth C, Martinez J, Zochodne D. RAGE, diabetes, and the nervous system. Curr Mol Med. 2007;7:766–76.
Juranek JK, Geddis MS, Song F, et al. RAGE deficiency improves postinjury sciatic nerve regeneration in type 1 diabetic mice. Diabetes. 2013;62:931–43.
Bierhaus A, Humpert PM, Morcos M, et al. Understanding RAGE, the receptor for advanced glycation end products. J Mol Med. 2005;83:876–86.
Juranek JK, Kothary P, Mehra A, Hays A, Brannagan TH, Schmidt AM. Increased expression of the receptor for advanced glycation end-products in human peripheral neuropathies. Brain Behav. 2013;3:701–9.
Yagihashi S, Kamijo M, Baba M, Yagihashi N, Nagai K. Effect of aminoguanidine on functional and structural abnormalities in peripheral nerve of STZ-induced diabetic rats. Diabetes. 1992;41:47–52.
Miyauchi Y, Shikama H, Takasu T, et al. Slowing of peripheral motor nerve conduction was ameliorated by aminoguanidine in streptozotocin-induced diabetic rats. Eur J Endocrinol. 1996;134:467–73.
Sugimoto K, Yagihashi S. Effects of aminoguanidine on structural alterations of microvessels in peripheral nerve of streptozotocin diabetic rats. Microvasc Res. 1997;53:105–12.
Kihara M, Schmelzer JD, Poduslo JF, Curran GL, Nickander KK, Low PA. Aminoguanidine effects on nerve blood flow, vascular permeability, electrophysiology, and oxygen free radicals. Proc Natl Acad Sci U S A. 1991;88:6107–11.
Cameron NE, Cotter MA. Rapid reversal by aminoguanidine of the neurovascular effects of diabetes in rats: modulation by nitric oxide synthase inhibition. Metabolism. 1996;45:1147–52.
Metz TO, Alderson NL, Thorpe SR, Baynes JW. Pyridoxamine, an inhibitor of advanced glycation and lipoxidation reactions: a novel therapy for treatment of diabetic complications. Arch Biochem Biophys. 2003;419:41–9.
Nagai R, Schirakawa JI, Ohno RI, Moroishi N, Nagai M. Inhibition of AGEs formation by natural products. Amino Acids. 2014;46:261–6.
Du XL, Edelstein D, Rossetti L, et al. Hyperglycemia-induced mitochondrial superoxide overproduction activates the hexosamine pathway and induces plasminogen activator inhibitor-1 expression by increasing Sp1 glycosylation. Proc Natl Acad Sci U S A. 2000;97:12222–6.
Román-Pintos LM, Villegas-Rivera G, Rodríguez-Carrizalez AD, Miranda-Díaz AG, Cardona-Muñoz EG. Mellitus: inflammation, oxidative stress, and mitochondrial function. J Diabetes Res. 2016;3425617
Yagihashi S. Glucotoxic mechanisms and related therapeutic approaches. Int Rev Neurobiol. 2016;127:121–49.
Heath JM, Sun Y, Yuan K, et al. Activation of AKT by O-linked N-acetylglucosamine induces vascular calcification in diabetes mellitus. Circ Res. 2014;114:1094–102.
Hammes H, Du X, Edelstein D, et al. Benfotiamine blocks three major pathways of hyperglycemic damage and prevents experimental diabetic retinopathy. Nat Med. 2003;9:294–9.
Sanchez-Ramirez GM, Caram-Salas NL, Rocha-Ganzalez HI, et al. Benfotiamine relieves inflammatory and neuropathic pain in rats. Eur J Pharmacol. 2006;530:48–53.
Stracke H, Gaus W, Achenbach U, Federlin K, Bretzel R. Benfotiamine in diabetic polyneuropathy (BEN-DIP): results of a randomized, double-blind, placebo-controlled clinical study. Exp Clin Endocrinol Diabetes. 2008;116:600–5.
Eichberg J. Protein kinase C changes in diabetes: is the concept relevant to neuropathy? Int Rev Neurobiol. 2002;50:61–82.
Evcimen ND, King GL. The role of protein kinase C activation and the vascular complications of diabetes. Pharmacol Res. 2007;55:498–510.
Sasase T, Morinaga H, Abe T, et al. Protein kinase C beta inhibitor prevents diabetic peripheral neuropathy, but not histopathological abnormalities of retina in spontaneously diabetic Torii rat. Diabetes Obes Metab. 2009;11:1084–7.
Nakamura J, Kato K, Hamada Y, et al. A protein kinase C-beta-selective inhibitor ameliorates neural dysfunction in streptozotocin-induced diabetic rats. Diabetes. 1999;48:2090–5.
Cotter MA, Jack AM, Cameron NE. Effects of the protein kinase C beta inhibitor LY333531 on neural and vascular function in rats with streptozotocin-induced diabetes. Clin Sci. 2002;103:311–21.
Cameron NE, Cotter MA, Jack AM, Basso MD, Hohman TC. Protein kinase C effects on nerve function, perfusion, Na(+),K(+)-ATPase activity and glutathione content in diabetic rats. Diabetologia. 1999;42:1120–30.
Casellini CM, Barlow PM, Rice AL, et al. A 6-month randomized, double-blind-masked, placebo-controlled study evaluating the effects of the protein kinase C-beta inhibitor ruboxistaurin on skin microvascular blood flow and other measures of diabetic peripheral neuropathy. Diabetes Care. 2007;30:896–902.
Brooks B, Delaney-Robinson C, Molyneaux L, Yue DK. Endothelial and neural regulation of skin microvascular blood flow in patients with diabetic peripheral neuropathy: effect of treatment with the isoform-specific protein kinase C beta inhibitor, ruboxistaurin. J Diabetes Complicat. 2008;22:88–95.
Vinik AI, Bril V, Kempler P, et al. Treatment of symptomatic diabetic peripheral neuropathy with the protein kinase C beta-inhibitor ruboxistaurin mesylate during a 1-year randomized, placebo-controlled, double-blind clinical trial. Clin Ther. 2005;27:1164–80.
van Dam PS. Oxidative stress and diabetic neuropathy: pathophysiological mechanisms and treatment perspectives. Diabetes Metab Res Rev. 2002;18:176–84.
Schnackenberg CG. Physiological and pathophysiological roles of oxygen radicals in the renal microvasculature. Am J Phys. 2002;282:R335–42.
Radi R, Peluffo G, Alvarez MN, Naviliat M, Cayota A. Unraveling peroxynitrite formation in biological systems. Free Radic Biol Med. 2001;30:463–88.
Fredstrom S. Nitric oxide, oxidative stress, and dietary antioxidants. Nutrition. 2002;18:537–9.
Cameron NE, Cotter MA. Metabolic and vascular factors in the pathogenesis of diabetic neuropathy. Diabetes. 1997;46:S31–7.
Coppey LJ, Gellett JS, Davidson EP, Dunlap JA, Lund DD, Yorek MA. Effect of antioxidant treatment of streptozotocin-induced diabetic rats on endoneurial blood flow, motor nerve conduction velocity and vascular reactivity of epineurial arterioles of the sciatic nerve. Diabetes. 2001;50:1927–37.
Coppey LJ, Gellett JS, Davidson EP, et al. Effect of M40403 treatment of diabetic rats on endoneurial blood flow, motor nerve conduction velocity and vascular function of epineurial arterioles of the sciatic nerve. Br J Pharmacol. 2001;134:21–9.
Yorek MA, Coppey LJ, Gellett JS, et al. Effect of treatment of diabetic rats with Dehydroepiandrosterone (DHEA) on vascular and neural function. Am J Phys. 2002;283:E1067–75.
Cameron NE, Cotter MA, Maxfield EK. Antioxidant treatment prevents the development of peripheral nerve dysfunction in streptozotocin-diabetic rats. Diabetologia. 1993;36:299–304.
Cameron NE, Cotter MA. Neurovascular dysfunction in diabetic rats: potential contribution of autoxidation and free radicals examined using transition metal chelating agents. J Clin Invest. 1995;96:1159–63.
Cameron NE, Cotter MA. Effects of antioxidants on nerve and vascular dysfunction in experimental diabetes. Diabetes Res Clin Pract. 1999;45:137–46.
Keegan A, Cotter MA, Cameron NE. Effects of diabetes and treatment with the antioxidant α-lipoic acid on endothelial and neurogenic responses of corpus cavernosum in rats. Diabetologia. 1999;42:343–50.
Karasu C, Dewhurst M, Stevens EJ, Tomlinson DR. Effects of anti-oxidant treatment on sciatic nerve dysfunction in streptozotocin-diabetic rats; comparison with essential fatty acids. Diabetologia. 1995;38:129–34.
Pieper GM, Siebeneich W. Diabetes-induced endothelial dysfunction is prevented by long-term treatment with the modified iron chelator, hydroxyethyl starch conjugated-deferoxamine. J Cardiovasc Pharmacol. 1997;30:734–8.
De Vriese AS, Verbeuren TJ, Van de Voorde J, Lameire NH. Vanhoutte PM endothelial dysfunction in diabetes. Br J Pharmacol. 2000;130:963–74.
Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature. 2001;414:813–20.
Nishikawa T, Edelstein D, Du XL, et al. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature. 2000;404:787–90.
Fernyhough P. Mitochondrial dysfunction in diabetic neuropathy: a series of unfortunate metabolic events. Curr Diab Rep. 2015;15:89.
Coppey LJ, Gellett JS, Davidson EP, Yorek MA. Preventing superoxide formation in epineurial arterioles of the sciatic nerve from diabetic rats restores endothelium-dependent vasodilation. Free Radic Res. 2003;37:33–40.
Nassar T, Kadery B, Lotan C, Da’as N, Kleinman Y, Haj-Yehia A. Effects of the superoxide dismutase-mimetic compound tempol on endothelial dysfunction in streptozotocin-induced diabetic rats. Eur J Pharmacol. 2002;436:111–8.
Cameron NE, Jack AM, Cotter MA. Effect of alpha-lipoic acid on vascular responses and nociception in diabetic rats. Free Radic Biol Med. 2001;31:125–35.
Cameron NE, Cotter MA. Effects of an extracellular metal chelator on neurovascular function in diabetic rats. Diabetologia. 2001;44:621–8.
Inkster ME, Cotter MA, Cameron NE. Effects of trientine, a metal chelator, on defective endothelium-dependent relaxation in the mesenteric vasculature of diabetic rats. Free Radic Res. 2002;36:1091–9.
Fink B, Coppey L, Davidson E, et al. Effect of mitoquinone (Mito-Q) on neuropathic endpoints in an obese and type 2 diabetic rat model. Free Radic Res. 2020;54:311–8.
Cameron NE, Tuck Z, McCabe L, Cotter MA. Effect of the hydroxyl radical scavenger, dimethylthiourea, on peripheral nerve tissue perfusion, conduction velocity and nociception in experimental diabetes. Diabetologia. 2001;44:1161–9.
Manzella D, Barbieri M, Ragno E, Paolisso G. Chronic administration of pharmacologic doses of vitamin E improves the cardiac autonomic nervous system in patients with type 2 diabetes. Am J Clin Nutr. 2001;73:1052–7.
van Dam PS, Bravenboer B, van Asbeck BS, Marx JJ, Gispen WH. High rat food vitamin E content improves nerve function in streptozotocin-diabetic rats. Eur J Pharmcol. 1999;376:217–22.
Nicklander KK, Schmelzer JD, Rohwer DA, Low PA. Effect of alpha-tocopherol deficiency on indices of oxidative stress in normal and diabetic peripheral nerve. J Neurol Sci. 1994;126:6–14.
Kocak G, Aktan F, Canbolat O, et al. Alpha-lipoic acid treatment ameliorates metabolic parameters, blood pressure, vascular reactivity and morphology of vessels already damaged by streptozotocin-diabetes. Diabetes Nutr Metab. 2000;13:308–18.
Coleman MD, Eason RC, Bailey CJ. The therapeutic use of lipoic acid in diabetes: a current perspective. Environ Toxicol Pharmacol. 2001;10:167–72.
Packer L, Kraemer K, Rimbach G. Molecular aspects of lipoic acid in the prevention of diabetes complications. Nutrition. 2001;17:888–95.
Dincer Y, Telci A, Kayah R, Yilmaz IA, Cakatay U, Akcay T. Effect of α-lipoic acid on lipid peroxidation and anti-oxidant enzyme activities in diabetic rats. Clin Exp Pharmacol Physiol. 2002;29:281–4.
Jones W, Li X, Zhi-Chao Q, Perriott L, Whitesell RR, May JM. Uptake, recycling , and antioxidant actions of α-lipoic acid in endothelial cells. Free Rad Biol Med. 2002;33:83–93.
Bravenboer B, Kappelle AC, Hamers FPR, van Buren T, Erkelens DW, Gispen WH. Potential use of glutathione for the prevention and treatment of diabetic neuropathy in the streptozotocin-induced diabetic rat. Diabetologia. 1992;35:813–7.
Nakamura J, Hamada Y, Chaya S, et al. Transition metals and polyol pathway in the development of diabetic neuropathy in rats. Diabetes Metab Res Rev. 2002;18:395–402.
Papanas N, Ziegler D. Efficacy of α-lipoic acid in diabetic neuropathy. Expert Opin Pharmacother. 2014;15:2721–31.
Cuzzocrea S, Riley DP, Caputi AP, Salvemini D. Antioxidant therapy: a new pharmacological approach in shock, inflammation, and ischemia/reperfusion injury. Pharmacol Rev. 2001;53:135–59.
Doggrell SA. Therapeutic potential of selective superoxide dismutase mimetics. Drugs Future. 2002;27:385–90.
Salvemini D, Wang ZQ, Zweier JL, et al. A nonpeptidyl mimic of superoxide dismutase with therapeutic activity in rats. Science. 1999;286:304–6.
Salvemini D, Riley DP. M40403. Drugs Fut. 2002;25:1027–33.
Pacher P, Obrosova IG, Mabley JG, Szabo C. Role of nitrosative stress and peroxynitrite in the pathogenesis of diabetic complications. Emerging new therapeutical strategies. Curr Med Chem. 2005;12:267–75.
Cowell RM, Russell JW. Nitrsative injury and antioxidant therapy in the management of diabetic neuropathy. J Investig Med. 2004;52:33–44.
Vareniuk I, Pavlov IA, Drel VR, et al. Nitrosative stress and peripheral diabetic neuropathy in leptin-deficient (ob/ob) mice. Exp Neurol. 2007;205:425–36.
Obrosova IG, Drel VR, Oltman CL, et al. Role of nitrosative stress in early neuropathy and vascular dysfunction in streptozotocin-diabetic rats. Am J Physiol Endocrinol Metab. 2007;293:E1645–55.
Obrosova IG, Mabley JG, Zsengeller Z, et al. Role for nitrosative stress in diabetic neuropathy: evidence from studies with a peroxynitrite decomposition catalyst. FASEB J. 2005;19:401–3.
Drel VR, Pacher P, Vareniuk I, et al. A peroxynitrite decomposition catalyst counteracts sensory neuropathy in streptozotocin-diabetic mice. Eur J Pharmacol. 2007;569:48–58.
Obrosova IG, Drel VR, Pacher P, et al. Oxidative-nitrosative stress and poly(ADP-ribose) polymerase (PARP) activation in experimental diabetic neuropathy: the relation is revisited. Diabetes. 2005;54:3435–41.
Obrosova IG, Li F, Abatan OI, et al. Role of poly(ADP-ribose) polymerase activation in diabetic neuropathy. Diabetes. 2004;53:711–20.
Ilnytska O, Lyzogubov VV, Stevens MJ, et al. Poly(ADP-ribose) polymerase inhibition alleviates experimental diabetic sensory neuropathy. Diabetes. 2006;55:1686–94.
Trammell SAJ, Weidemann BJ, Chadda A, et al. Nicotinamide riboside opposes type 2 diabetes and neuropathy in mice. Sci Rep. 2016;6:26933.
Purves T, Middlemas A, Agthong S, et al. A role for mitogen-activated protein kinases in the etiology of diabetic neuropathy. FASEB J. 2001;15:2508–14.
Zhu H, Yu W-J, Le Y, et al. High glucose levels increase the expression of neurotrophic factors associated with p-p42/p44 MAPK in Schwann cells in vitro. Mol Med Rep. 2012;6:179–84.
Stavniichuk R, Drel VR, Shevalye H, et al. Baicalein alleviates diabetic peripheral neuropathy through inhibition of oxidative-nitrosative stress and p38 MAPK activation. Exp Neurol. 2011;230:106–13.
Zhou J, Du X, Long M, et al. Neuroprotective effect of berberine is mediated by MAPK signaling pathway in experimental diabetic neuropathy in rats. Eur J Pharmacol. 2016;774:87–94.
Sweitzer SM, Medicherla S, Almirez R, et al. Antinociceptive action of a p38alpha MAPK inhibitor, SD-282, in a diabetic neuropathy model. Pain. 2004;109:409–19.
Kovacs K, Vaczy A, Fekete K, et al. PARP inhibitor protects against chronic hypoxia/reoxygenation-induced retinal injury by regulation of MAPKs, HIF1α, Nrf2, and NFκB. Invest Ophthalmol Vis Sci. 2019;60:1478–90.
Yerra VG, Negi G, Sharma SS, Kumar A. Potential therapeutic effects of the simultaneous targeting of the Nrf2 and NF-κB pathways in diabetic neuropathy. Redox Biol. 2013;1:394–7.
Li J, Hu X, Liang F, et al. Therapeutic effects of moxibustion simultaneously targeting Nrf2 and NF-κB in diabetic peripheral neuropathy. Appl Biochem Biotechnol. 2019;189:1167–82.
Shay KP, Moreau RF, Smith EJ, Smith AR, Hagen TM. Alpha-lipoic acid as a dietary supplement: molecular mechanisms and therapeutic potential. Biochim Biophys Acta. 2009;1790:1149–60.
Agca CA, Tuzcu M, Hayirli A, Sahin K. Taurine ameliorates neuropathy via regulating NF-κB and Nrf2/HO-1 signaling cascades in diabetic rats. Food Chem Toxicol. 2014;71:116–21.
Ma J, Shi M, Zhang X, et al. GLP-1R agonists ameliorate peripheral nerve dysfunction and inflammation via p38 MAPK/NF-κB signaling pathways in streptozotocin-induced diabetic rats. Int J Mol Med. 2018;41:2977–85.
Kumar A, Negi G, Sharma SS. Suppression of NF-κB and NI-κB regulated oxidative stress and neuroinflammation by BAY11-7082 (IκB phosphorylation inhibitor) in experimental diabetic neuropathy. Biochimie. 2012;94:1158–65.
Kumar A, Sharma SS. NF-kappaB inhibitory action of resveratrol: a probable mechanism of neuroprotection in experimental diabetic neuropathy. Biochem Biophys Res Commun. 2010;394:360–5.
Lupachyk S, Watcho P, Shevalye H, et al. Na+/K+ exchanger 1 inhibition reverses manifestation of peripheral diabetic neuropathy in type 1 diabetic rats. Am J Physiol Endocrinol Metab. 2013;305:E396–404.
Kellogg AP, Cheng HT, Pop-Busui R. Cyclooxygenase-2 pathway as a potential therapeutic target in diabetic peripheral neuropathy. Curr Drug Targets. 2008;9:68–76.
Kellogg AP, Pop-Busui R. Peripheral nerve dysfunction in experimental diabetes is mediated by cyclooxygenase-2 and oxidative stress. Antioxid Redox Signal. 2005;7:1521–9.
Kellogg AP, Wiggin TD, Larkin DD, Hayes JM, Stevens MJ, Pop-Busui R. Protective effects of cyclooxygenase-2 gene inactivation against peripheral nerve dysfunction and intraepidermal nerve fiber loss in experimental diabetes. Diabetes. 2007;56:2997–3005.
Kimura S, Kontani H. Demonstration of antiallodynic effects of the cyclooxygenase-2 inhibitor meloxicam on established diabetic neuropathic pain in mice. J Pharmacol Sci. 2009;110:213–7.
Chen L, Li B, Chen B, et al. Thymoquinone alleviates the experimental diabetic peripheral neuropathy by modulation of inflammation. Sci Rep. 2016;6:31656.
Moustafa P, Abdelkader NF, El-Awdan SA, El-Shabrawy OA, Zaki HF. Extracellular matrix remodeling and modulation of inflammation and oxidative stress by sulforaphane in experimental diabetic peripheral neuropathy. Inflammation. 2018;41:1460–76.
Stavniichuk R, Drel VR, Shevalye H, et al. Role of 12/15-lipoxygenase in nitrosative stress and peripheral prediabetic and diabetic neuropathies. Free Radic Biol Med. 2010;49:1036–45.
Stavniichuk R, Obrosov AA, Drel VR, Nadler JL, Obrosova IG, Yorek MA. 12/15-Lipoxygenase inhibition counteracts MAPK phosphorylation in mouse and cell culture models of diabetic peripheral neuropathy. J Diabetes Mellitus. 2013;3:33015. https://doi.org/10.4236/jdm.2013.33015.
Obrosova IG, Stavniichuk R, Drel VR, et al. Different roles of 12/15-lipoxygenase in diabetic large and small fiber peripheral and autonomic neuropathies. Am J Pathol. 2010;177:1436–47.
Stavniichuk R, Shevalye H, Hirooka H, Nadler JL, Obrosova IG. Interplay of sorbitol pathway or glucose metabolism, 12/15-lipoxygenase, and mitogen-activated protein kinase in the pathogenesis of diabetic peripheral neuropathy. Biochem Pharmacol. 2012;83:932–40.
Yorek MA. The potential role of angiotensin converting enzyme and vasopeptidase inhibitors in the treatment of diabetic neuropathy. Curr Drug Targets. 2008;9:77–84.
Yorek MA. Is fish oil a potential treatment for diabetic peripheral neuropathy? Curr Diabetes Rev. 2018;14:339–49.
Deacon CF. Dipeptidyl peptidase 4 inhibitors in the treatment of type 2 diabetes mellitus. Nat Rev Endocrinol. 2020;16:642–53.
Jin HY, Liu WJ, Park JH, Baek HS, Park TS. Effect of dipeptidyl peptidase-IV (DPP-IV) inhibitor (Vildagliptin) on peripheral nerves in streptozotocin-induced diabetic rats. Arch Med Res. 2009;40:536–44.
Tsubi K, Mizukami H, Inaba W, Baba M, Yagihashi S. The dipeptidyl peptidase IV inhibitor vildagliptin suppresses development of neuropathy in diabetic rodents: effects on peripheral sensory nerve function, structure and molecular changes. J Neurochem. 2016;136:859–70.
Bianchi R, Cervellini C, Porretta-Serapiglia C, et al. Beneficial effects of PKF275-055, a novel, selective, orally bioavailable, long-lasting dipeptidyl peptidase IV inhibitor in streptozotocin-induced diabetic peripheral neuropathy. J Pharmacol Exp Ther. 2012;340:64–72.
Davidson EP, Coppey LJ, Dake B, Yorek MA. Treatment of streptozotocin-induced diabetic rats with alogliptin: effect on vascular and neural complications. Exp Diabetes Res. 2011;2011:810469.
Volpe M, Savoia C, De Paolis P, Ostrowska B, Tarasi D, Rubattu S. The renin-angiotensin system as a risk factor and therapeutic target for cardiovascular and renal disease. J Am Soc Nephrol. 2002;13:S173–8.
Nickenig G, Ostergren J, Struijker-Boudier H. Clinical evidence for the cardiovascular benefits of angiotensin receptor blockers. J Renin-Angiotensin-Aldosterone Syst. 2006;7(Suppl 1):S1–7.
Mogensen CE. Microalbuminuria and hypertension with focus on type 1 and type 2 diabetes. J Intern Med. 2003;254:45–66.
Li EC, Heran BS, Wright JM. Angiotensin converting enzyme (ACE) inhibitors versus angiotensin receptor blockers for primary hypertension. Cochrane Database Syst Rev. 2014;2014:CD009096.
Cameron NE, Cotter MA, Robertson S. Angiotensin converting enzyme inhibition prevents development of muscle and nerve dysfunction and stimulates angiogenesis in streptozotocin-diabetic rats. Diabetologia. 1992;35:12–8.
Coppey LJ, Davidson EP, Rinehart TW, et al. ACE inhibitor or angiotensin II receptor antagonist attenuates diabetic neuropathy in streptozotocin-induced diabetic rats. Diabetes. 2006;55:341–8.
Davidson EP, Coppey LJ, Holmes A, Yorek MA. Effect of inhibition of angiotensin converting enzyme and/or neutral endopeptidase on vascular and neural complications in high fat fed/low dose streptozotocin-diabetic rats. Eur J Pharmacol. 2012;677:180–7.
Manschot SM, Gispen WH, Kappelle LJ, Biessels GJ. Nerve conduction velocity and evoked potential latencies in streptozotocin-diabetic rats: effects of treatment with an angiotensin converting enzyme inhibitor. Diabetes Metab Res Rev. 2003;19:469–77.
Aggarwal M, Singh J, Sood S, Arora B. Effects of lisinopril on streptozotocin-induced diabetic neuropathy in rats. Methods Find Exp Clin Pharmacol. 2001;23:131–4.
Oltman CL, Davidson EP, Coppey LJ, Kleinschmidt TL, Lund DD, Yorek MA. Attenuation of vascular/neural dysfunction in Zucker rats treated with enalapril or rosuvastatin. Obesity. 2008;16:82–9.
Didangelos T, Tziomalos K, Margaritidis C, et al. Efficacy of administration of an angiotensin converting enzyme inhibitor for two years on autonomic and peripheral neuropathy in patients with diabetes mellitus. J Diabetes Res. 2017;2017:6719239.
Malik RA, Williamson S, Abbott C, et al. Effect of angiotensin-converting-enzyme (ACE) inhibitor trandolapril on human diabetic neuropathy: randomized double-blind controlled trial. Lancet. 1998;352:1978–81.
Reja A, Tesfaye S, Harris ND, Ward JD. Is ACE inhibition with lisinopril helpful in diabetic neuropathy? Diabet Med. 1995;12:307–9.
Ebihara F, Di Marco GS, Juliano MA, Casarini DE. Neutral endopeptidase expression in mesangial cells. J Renin-Angiotensin-Aldosterone Syst. 2003;4:228–33.
Muangman P, Spenny ML, Tamura RN, Gibran NS. Fatty acids and glucose increase neutral endopeptidase activity in human microvascular endothelial cells. Shock. 2003;19:508–12.
Edwards RM, Pullen M, Nambi P. Distribution of neutral endopeptidase activity along the rat and rabbit nephron. Pharmacology. 1999;59:45–50.
Gonzalez W, Soleilhac JM, Fournie-Zaluski MC, Roques BP, Michel JB. Characterization of neutral endopeptidase in vascular cells, modulation of vasoactive peptide levels. Eur J Pharmacol. 1998;345:323–31.
Kikkawa F, Shibata K, Suzuki T, et al. Signal pathway involved in increased expression of neutral endopeptidase 24.11 by gonadotropin releasing hormone in choriocarcinoma cells. Placenta. 2004;25:176–83.
Suzki T, Ino K, Kikkawa F, et al. Neutral endopeptidase/CD10 expression during phorbol ester-induced differentiation of choriocarcinoma cells through the protein kinase C- and extracellular signal-regulated kinase-dependent signalling pathway. Placenta. 2002;23:475–82.
Pu Q, Schiffrin EL. Effect of ACE/NEP inhibition on cardiac and vascular collagen in stroke-prone spontaneously hypertensive rats. Am J Hypertens. 2001;14:1067–72.
Plamboeck A, Holst JJ, Carr RD, Deacon CF. Neutral endopeptidase 24.11 and dipeptidyl peptidase IV are both mediators of the degradation of glucagon-like peptide 1 in the anaesthetised pig. Diabetologia. 2005;48:1882–90.
Weber M. Emerging treatments for hypertension: potential role for vasopeptidase inhibition. Am J Hypertens. 1999;12:139S–47S.
Trindade PT, Rouleau JL. Vasopeptidase inhibitors: potential role in the treatment of heart failure. Heart Fail Monit. 2001;2:2–7.
Nathisuwan S, Talbert RL. A review of vasopeptidase inhibitors: a new modality in the treatment of hypertension and chronic heart failure. Pharmacotherapy. 2002;22:27–42.
Arbin V, Claperon N, Fournie-Zaluski MC, Roques BP, Peyroux J. Effects of combined neutral endopeptidase 24-11 and angiotensin-converting enzyme inhibition on femoral vascular conductance in streptozotocin-induced diabetic rats. Br J Pharmacol. 2000;130:1297–304.
Schafer S, Schmidts H-L, Bleich M, Busch AE, Linz W. Nephroprotection in Zucker diabetic fatty rats by vasopeptidase inhibition is partly bradykinin B2 receptor dependent. Br J Pharmacol. 2004;143:27–32.
Schäfer S, Linz W, Vollert H, et al. The vasopeptidase inhibitor AVE7688 ameliorates type 2 diabetic nephropathy. Diabetologia. 2004;47:98–103.
Spenny ML, Muangman P, Sullivan SR, et al. Neutral endopeptidase inhibition in diabetic wound repair. Wound Repair Regen. 2002;10:295–301.
Wihler C, Schafer S, Schmid K, et al. Renal accumulation and clearance of advanced glycation end-products in type 2 diabetic nephropathy: effect of angiotensin-converting enzyme and vasopeptidase inhibition. Diabetologia. 2005;48:1645–53.
Oltman CL, Davidson EP, Coppey LJ, Kleinschmidt TL, Dake B, Yorek MA. Role of the effect of inhibition of neutral endopeptidase on vascular and neural complications in streptozotocin-induced diabetic rats. Eur J Pharmacol. 2001;650:556–62.
Coppey L, Davidson E, Lu B, Gerard C, Yorek M. Vasopeptidase inhibitor ilepatril (AVE7688) prevents obesity- and diabetes-induced neuropathy in C57Bl/6J mice. Neuropharmacology. 2011;60:259–66.
Davidson EP, Kleinschmidt TL, Oltman CL, Lund DD, Yorek MA. Treatment of streptozotocin-induced diabetic rats with AVE7688, a vasopeptidase inhibitor: effect on vascular and neural disease. Diabetes. 2007;56:355–62.
Oltman CL, Davidson EP, Coppey LJ, Kleinschmidt TL, Yorek MA. Treatment of Zucker diabetic fatty rats with AVE7688 improves vascular and neural dysfunction. Diabetes Obes Metab. 2009;11:223–33.
Davidson EP, Coppey LJ, Yorek MA. Early loss of innervation of cornea epithelium in streptozotocin-induced type 1 diabetic rats: improvement with ilepatril treatment. Invest Ophthalmol Vis Sci. 2012;53:8067–74.
Yorek MS, Obrosov A, Lu B, Gerard C, Kardon RH, Yorek MA. Effect of inhibition or deletion of neutral endopeptidase on neuropathic endpoints in high fat fed/low dose streptozotocin-treated mice. J Neuropathol Exp Neurol. 2016;75:1072–80.
von Lueder TG, Atar D, Krum H. Current role of neprilysin inhibitors in hypertension and heart failure. Pharmacol Ther. 2014;144:41–9.
Sangaralingham LR, Sangaralingham SJ, Shah ND, Yao X, Dunlay SM. Adoption of sacubitril/valsartan for the management of patients with heart failure. Circ Heart Fail. 2018;11:e004302.
Davidson EP, Coppey LJ, Shevalye H, Obrosov A, Yorek MA. Vascular and neural complications in type 2 diabetic rats: improvement by sacubitril/valsartan greater than valsartan alone. Diabetes. 2018;67:1616–26.
Serhan CN, Chiang N, Dalli J. New pro-resolving n-3 mediators bridge resolution of infectious inflammation to tissue regeneration. Mol Asp Med. 2018;64:1–17.
Serhan CN, Yacoubian S, Yang R. Anti-inflammatory and proresolving lipid mediators. Annu Rev Pathol. 2008;3:279–312.
Grisold A, Callaghan BC, Feldman EL. Mediators of diabetic neuropathy: is hyperglycemia the only culprit? Curr Opin Endocrinol Diabetes Obes. 2017;24:103–11.
Bittner V. Implications for REDUCE IT in clinical practice. Prog Cardiovasc Dis. 2019;62:395–400.
Kris-Etherton PM, Richter CK, Bowen KJ, et al. Recent clinical trials shed new light on the cardiovascular benefits of omega-3 fatty acids. Methodist Debakey Cardiovasc J. 2019;15:171–8.
Shaikh SR, Kinnun JJ, Leng X, Williams JA, Wassall SR. How polyunsaturated fatty acids modify molecular organization in membranes: insight from NMR studies of model systems. Biochim Biophys Acta. 2015;1848:211–9.
Simopoulos AP. The importance of the omega-6/omega-3 fatty acid ratio in cardiovascular disease and other chronic diseases. Exp Biol Med. 2008;233:674–88.
Gil A, Serra-Majem L, Calder PC, Uauy R. Systematic reviews of the role of omega-3 fatty acids in the prevention and treatment of disease. Br J Nutr. 2012;107:S1–2.
Kaliannan K, Li X-Y, Wang B, et al. Multi-omic analysis in transgenic mice implicates omega-6/omega-3 fatty acid imbalance as a risk factor for chronic disease. Commun Biol. 2019;2:276.
Lanchais K, Capel F, Tournadre A. Could omega-3 fatty acids preserve muscle health in rheumatoid arthritis? Nutrients. 2020;12:E223.
Ueno Y, Miyamoto N, Yamashiro K, Tanaka R, Hattori N. Omega-3 polyunsaturated fatty acids and stroke burden. Int J Mol Sci. 2019;20:E5549.
Li X, Bi X, Wang S, Zhang Z, Li F, Zhao AZ. Therapeutic potential of ω-3 polyunsaturated fatty acids in human autoimmune diseases. Front Immunol. 2019;10:2241.
Marton LT, Goulart RA, Carvalho ACA, Barbalho SM. Omega fatty acids and inflammatory bowel diseases: an overview. Int J Mol Sci. 2019;20:E4851.
McGlory C, Calder PC, Nunes EA. The influence of omega-3 fatty acids on skeletal muscle protein turnover in health, disuse, and disease. Front Nutr. 2019;6:144.
Avallone R, Vitale G, Bertolotti M. Omega-3 fatty acids and neurodegenerative diseases: new evidence in clinical trials. Int J Mol Sci. 2019;20:E4256.
Abdolmaleki F, Kovanen PT, Mardani R, Gheibi-Hayat SM, Bo S, Sahebkar A. Resolvins: emerging players in autoimmune and inflammatory diseases. Clin Rev Allergy Immunol. 2020;58:82–91.
Shevalye H, Yorek MS, Coppey LJ, et al. Effect of enriching the diet with menhaden oil or daily treatment with resolvin D1 on neuropathy in a mouse model of type 2 diabetes. J Neurophysiol. 2015;114:199–208.
Coppey LJ, Davidson EP, Obrosov A, Yorek MA. Enriching the diet with menhaden oil improves peripheral neuropathy in streptozotocin-induced type 1 diabetic rats. J Neurophysiol. 2015;113:701–8.
Coppey LJ, Holmes A, Davidson EP, Yorek MA. Partial replacement with menhaden oil improves peripheral neuropathy in high fat fed low dose streptozotocin type 2 diabetic rat. J Nutr Metab. 2012;2012:950517.
Obrosov A, Coppey LJ, Shevalye H, Yorek MA. Effect of fish oil vs. resolvin D1, E1, methyl esters of resolvins D1 or D2 on diabetic peripheral neuropathy. J Neurol Neurophysiol. 2017;8(453)
Yorek MS, Coppey LJ, Shevalye H, Obrosov A, Kardon RH, Yorek MA. Effect of treatment with salsalate, menhaden oil, combination of salsalate and menhaden oil, or resolvin D1 of C57Bl/6J type 1 diabetic mouse on neuropathic endpoints. J Nutr Metab. 2016;2016:5905891.
Goldfine AB, Fonseca V, Jablonski KA, et al. Salicylate (salsalate) in patients with type 2 diabetes: a randomized trial. Ann Intern Med. 2013;159:1–12.
Goldfine AB, Fonseca V, Jablonski KA, et al. The effects of salsalate on glycemic control in patients with type 2 diabetes: a randomized trial. Ann Intern Med. 2010;152:346–57.
Davidson EP, Coppey LJ, Shevalye H, Obrosov A, Yorek MA. Effect of dietary content of menhaden oil with or without salsalate on neuropathic endpoints in high-fat-fed/low-dose streptozotocin-treated Sprague Dawley rats. J Diabetes Res. 2018;2018:2967127.
Superko HR, Superko SM, Nasir K, Agatston A, Garrett BC. Omega-3 fatty acid blood levels. Circulation. 2013;128:2154–61.
Laidlaw M, Holub BJ. Effects of supplementation with fish oil-derived n-3 fatty acids and alpha-linolenic acid on circulating plasma lipids and fatty acid profiles in women. Am J Clin Nutr. 2003;77:37–42.
Lewis EJH, Perkins BA, Lovblom LE, Bazinet RP, Wolever TMS, Bril V. Effect of omega-3 supplementation on neuropathy in type 1 diabetes: a 12-month pilot trial. Neurology. 2017;88:2294–301.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2023 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Yorek, M. (2023). Metabolic Mechanisms in Diabetic Neuropathy. In: Tesfaye, S., Gibbons, C.H., Malik, R.A., Veves, A. (eds) Diabetic Neuropathy. Contemporary Diabetes. Humana, Cham. https://doi.org/10.1007/978-3-031-15613-7_15
Download citation
DOI: https://doi.org/10.1007/978-3-031-15613-7_15
Published:
Publisher Name: Humana, Cham
Print ISBN: 978-3-031-15612-0
Online ISBN: 978-3-031-15613-7
eBook Packages: MedicineMedicine (R0)